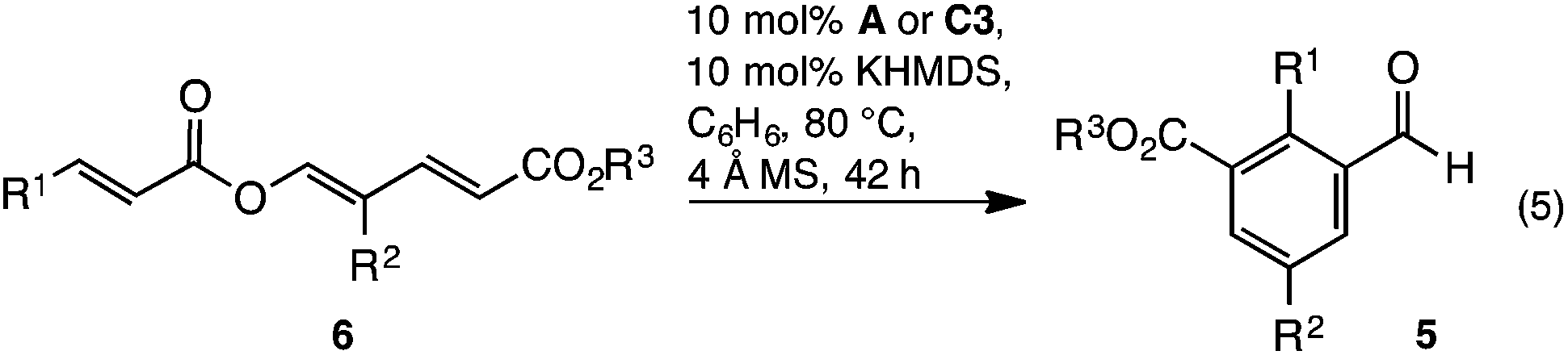DOI:
10.1039/C4SC03726J
(Edge Article)
Chem. Sci., 2015,
6, 2366-2370
N-Heterocyclic carbene catalysed redox isomerisation of esters to functionalised benzaldehydes†
Received
2nd December 2014
, Accepted 22nd January 2015
First published on 26th January 2015
Abstract
N-Heterocyclic carbene catalysed redox isomerisation with reduction about the carbonyl has been developed in the transformation of trienyl esters to tetrasubstituted benzaldehydes. The reaction proceeds in good to excellent yield, and in cases that provide 2,2′-biaryls, enantioselectivity is observed. Mechanistic studies demonstrate the intermediacy of a cyclohexenyl β-lactone, while implicating formation of the homoenolate as turnover limiting.
Introduction
Beyond N-heterocyclic carbene (NHC) catalysed formation of acyl anions,1 advances in the field have revealed subsequent umpolung events that provide acyl azoliums, their enols, and enolates.2 Collectively these redox isomerisation reactions have emerged as the dominant paradigm, allowing aldehyde containing substrates (1) to be converted to functionalised, and often enantioenriched, esters (2) (Fig. 1, eqn (1)).2–7 In contrast, to the best of our knowledge, the reverse, in which esters (3) undergo redox isomerisation to give aldehydes (4), are unknown (eqn (2)). Herein, we report an NHC catalysed redox isomerisation with ester reduction (3 → 4).
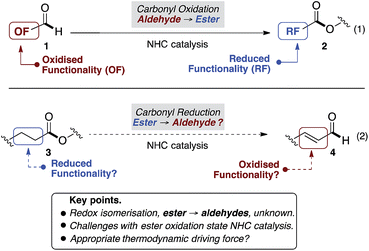 |
| | Fig. 1 Conceptual background. | |
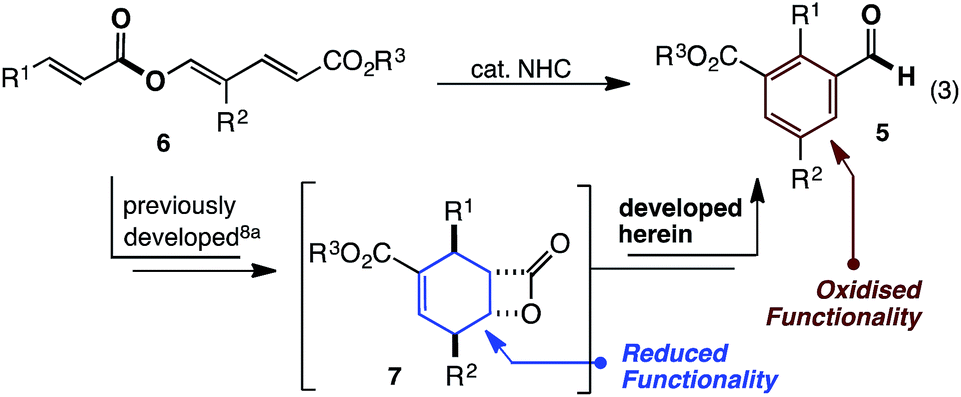
Over the last 5 years, studies from our group have revealed NHC catalysed (4 + 2) annulations which define an approach to cyclohexenes orthogonal to the Diels–Alder reaction.8 Mechanistically, these reactions commence with a vinylogous Michael/aldol/lactonisation cascade to produce cyclohexenyl β-lactones.8c These can decarboxylate,8b be trapped with nucleophilic reagents8c and, in some cases, be isolated.8a Within this family of reactions we recently developed an enantioselective cycloisomerisation of triene 6 to cyclohexenyl β-lactone 7 (Fig. 2).8a Whilst developing this transformation, a remarkable switch in reactivity was observed through subtle changes to catalyst and solvent. Specifically, this allowed the conversion of trienyl ester 6 to benzaldehyde 5via the previously described cascade, coupled with a redox isomerisation resulting in reduction about the carbonyl group (eqn (3)). This type of redox isomerisation is rare. Chi et al. has reported related reactivity, although in their studies a subsequent isomerisation returned ester containing products.9f,i Aside from the novelty of this redox isomerisation, discovery of this reaction adds to the limited family of known NHC-catalysed reactions with ester substrates,2k,8–11 while also defining a new approach to heavily substituted benzaldehyde derivatives.12 In this edge article, we report the development, scope and mechanistic study of this reaction.
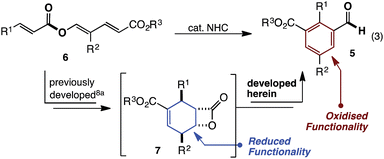 |
| | Fig. 2 Developed herein. | |
Results and discussion
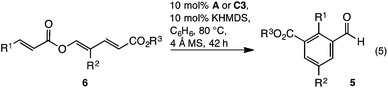
Reaction development commenced with triene 6a. Upon heating in THF, with an NHC derived from precatalyst A, the desired redox isomerised aldehyde 5a formed as a minor component, along with β-lactone 7a and cyclohexadiene 8a (Table 1, entry 1). Formation of benzaldehyde 5a generated water, which may protonate the NHC. To eliminate this pathway, a desiccant was introduced thereby improving the yield of 5a to 43% (Table 1, entry 2). The reaction displayed significant sensitivity to solvent. Thus while dioxane disfavoured benzaldehyde formation (Table 1, entry 3), toluene and benzene both improved selectivity, with the latter giving 5a in 64% isolated yield when heated at reflux (Table 1, entries 4 and 5). Unfortunately, longer reaction times favoured decarboxylation, and the yield of 5a was not increased. To improve the outcome, and avoid decarboxylation, modification of the catalyst was examined. While Ender's TPT precatalyst B and N-tBu or N-Mes morpholinone precatalysts C1 or C2 failed to improve the outcome (Table 1, entries 6–8), 2,6-dimethoxyphenyl C3 gave aldehyde 5a in a moderately improved yield (Table 1, entry 9).13 In contrast to the reaction with A, conducting the reaction with C3 for an extended period improved the outcome; with aldehyde 5a isolated in 87% yield without appreciable decarboxylation (Table 1, entry 10).
Table 1 Selected optimisations
Reaction scope
The generality of the reaction was examined with the transformation of trienyl esters 6a–n (Table 2) using precatalysts A and C3. While commercially available IMes precatalyst A was adequate in most cases, the yield was generally enhanced using C3. For example, while examining the impact of electronics about the cinnamoyl portion, it was found that electron neutral, rich, or moderately poor trienes 6a–d gave the expected aldehydes 5a–d in 68–91% yield using C3 (Table 2, entries 1a, 2a, 3a and 4a) and 11–26% lower yield using IMes A (Table 2, entries 1b, 2b, 3b and 4b). In contrast, the highly electron poor p-NO2C6H4 triene 6e only reacted with IMes precatalyst A (Table 2, entry 5). The reaction's capacity to tolerate heteroaromatic substituents was examined using furan containing triene 6f, which in-turn provided benzaldehyde 5f in acceptable yield (Table 2, entry 6). Next, modification of the R2 substituent within the diene was investigated. A benzyl group was tolerated, with benzaldehydes 5g and h prepared in 80 and 85% yield respectively (Table 2, entries 7 and 8). Similarly, ethyl benzaldehydes 5i and j could be prepared, albeit in modest yields (Table 2, entries 9 and 10). The isopropyl group was not tolerated, with only traces of the expected aldehyde 5k observed (Table 2, entry 11). Finally, the reaction was found to be insensitive to the nature of the ester, with methyl and isopropyl esters reacting smoothly to give the expected benzaldehydes 6l, m and n in good yield (Table 2, entries 12–14).
Table 2 Scope
|
|
| Entry |
Precat |
5/6 |
R1 |
R2 |
R3 |
Yielda |
|
Isolated yield following flash column chromatography.
|
| 1a |
C3
|
a
|
Ph |
CH3 |
Et |
87% |
| b |
A
|
a
|
Ph |
CH3 |
Et |
65% |
| 2a |
C3
|
b
|
p-CH3C6H4 |
CH3 |
Et |
91% |
| b |
A
|
b
|
p-CH3C6H4 |
CH3 |
Et |
65% |
| 3a |
C3
|
c
|
p-CH3OC6H4 |
CH3 |
Et |
71% |
| b |
A
|
c
|
p-CH3OC6H4 |
CH3 |
Et |
51% |
| 4a |
C3
|
d
|
p-BrC6H4 |
CH3 |
Et |
68% |
| b |
A
|
d
|
p-BrC6H4 |
CH3 |
Et |
57% |
| 5 |
A
|
e
|
p-NO2C6H4 |
CH3 |
Et |
43% |
| 6 |
C3
|
f
|
2-Furyl |
CH3 |
Et |
56% |
| 7 |
C3
|
g
|
Ph |
Bn |
Et |
80% |
| 8 |
C3
|
h
|
p-CH3C6H4 |
Bn |
Et |
85% |
| 9 |
C3
|
i
|
Ph |
Et |
Et |
52% |
| 10 |
C3
|
j
|
p-CH3C6H4 |
Et |
Et |
59% |
| 11 |
C3
|
k
|
Ph |
iPr |
Et |
Trace |
| 12 |
C3
|
l
|
Ph |
CH3 |
CH3 |
77% |
| 13 |
C3
|
m
|
p-CH3C6H4 |
CH3 |
CH3 |
72% |
| 14 |
C3
|
n
|
p-CH3C6H4 |
CH3 |
iPr |
81% |
Enantioselectivity and mechanistic studies
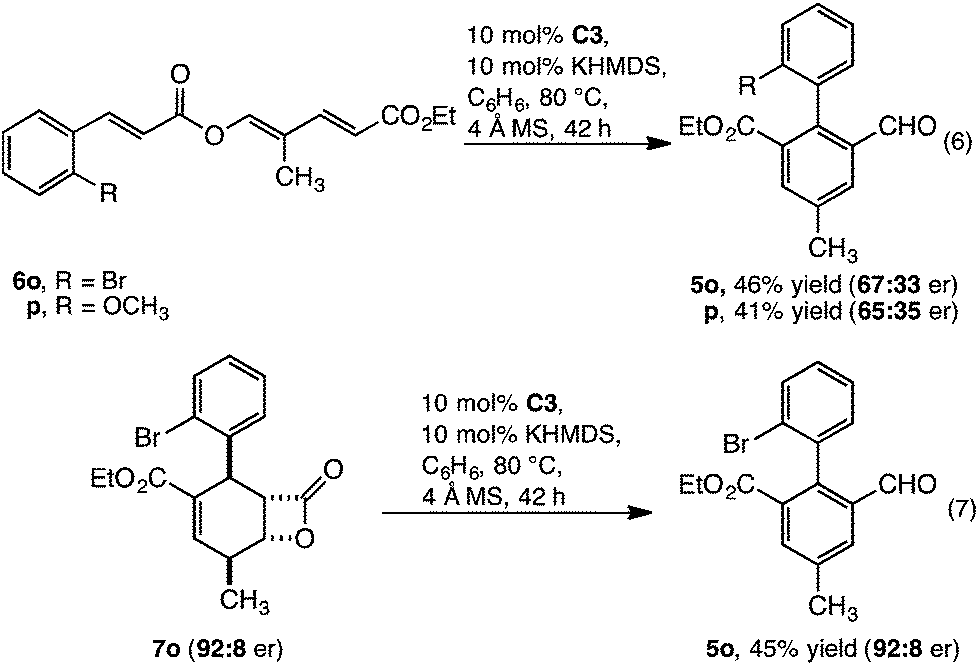
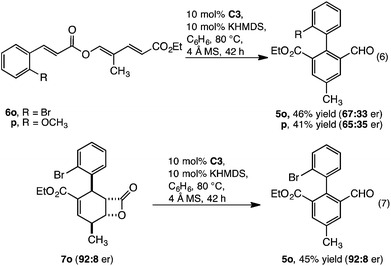
The transformation, as catalysed by homochiral C3, introduces a potential approach to enantioenriched axially chiral products. This was realised, with 6o and p converted to enantioenriched 2,2′ biaryls 5o and p with moderate enantioselectivity and yield (eqn (6)). Although considerable effort was directed towards improving the level of enantioselectivity, this was not possible. To understand this limitation, we investigated whether the low enantioselectivity was connected to limitations in (i) point to axial chirality relay14 or (ii) establishing point chirality. Thus, when the enantioenriched β-lactone intermediate 7o (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 er) was prepared8a and subsequently converted to 5o by catalyst C3 complete stereoretention was observed (eqn (7)). This result indicates that the limitations are likely linked to accessing enantioenriched β-lactone intermediate 7 with catalyst C3 (Scheme 1). Supporting this observation, when conversion of 6o to 5o was monitored (eqn (6)), the enantiopurity of the intermediate β-lactone 7o was found to be low (∼60:40 er).15 Thus, the challenge in realising this reaction as a highly enantioselective process is centred on developing a catalyst that allows both (i) a highly enantioselective β-lactone synthesis and (ii) redox isomerisation. On-going studies are focused on addressing this challenge.
:8 er) was prepared8a and subsequently converted to 5o by catalyst C3 complete stereoretention was observed (eqn (7)). This result indicates that the limitations are likely linked to accessing enantioenriched β-lactone intermediate 7 with catalyst C3 (Scheme 1). Supporting this observation, when conversion of 6o to 5o was monitored (eqn (6)), the enantiopurity of the intermediate β-lactone 7o was found to be low (∼60:40 er).15 Thus, the challenge in realising this reaction as a highly enantioselective process is centred on developing a catalyst that allows both (i) a highly enantioselective β-lactone synthesis and (ii) redox isomerisation. On-going studies are focused on addressing this challenge.
 |
| | Scheme 1 | |
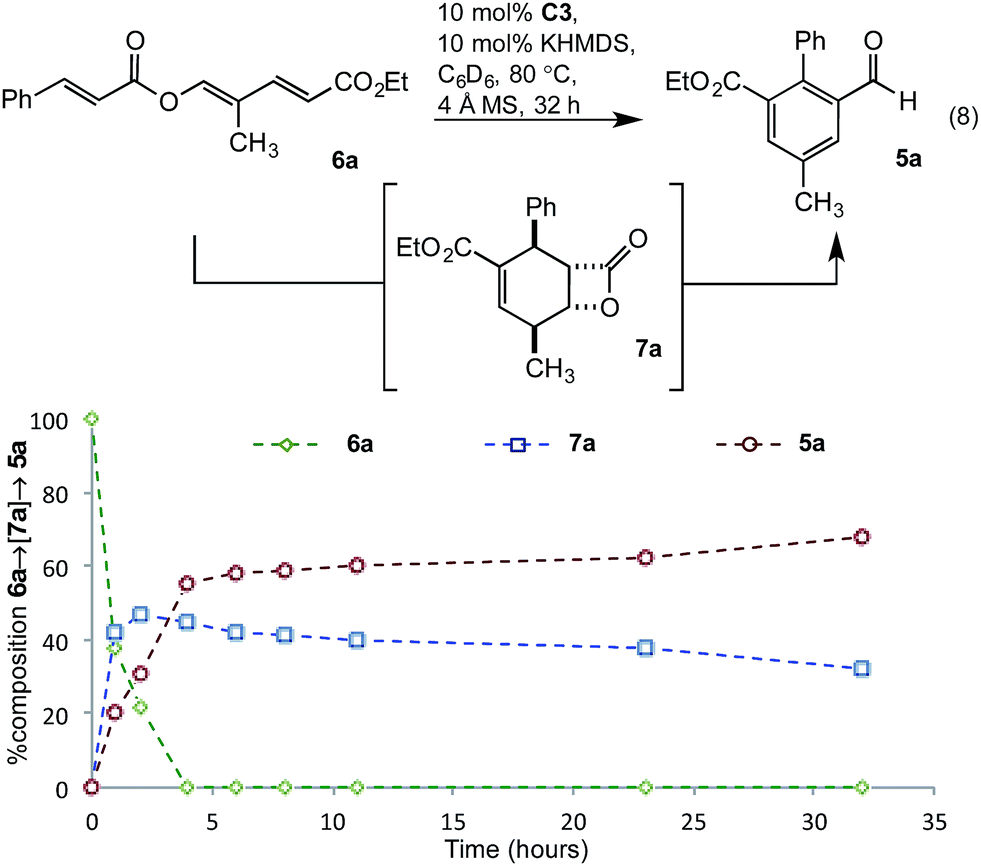
To gain greater insight into the mechanism of the transformation, studies probing the intermediacy of the β-lactone and the nature of the turnover-limiting step were undertaken. Thus, conversion of triene 6a to benzaldehyde 5a was monitored by in situ1H-NMR spectroscopic analysis in deuterated benzene (eqn (8)). After two hours, extensive consumption of triene 6a, along with formation of β-lactone 7a as the major product and benzaldehyde 5a as a minor component, was observed (Fig. 3). By the fourth hour, benzaldehyde 5a was the major product and levels of β-lactone 7a had decreased, consistent with the β-lactone serving as an intermediate en route to 5a. In contrast to the reaction in undeuterated solvents (Tables 1 and 2), this transformation was slower and failed to progress beyond 32 hours. Thus, all subsequent kinetic investigations were terminated after 32 hours.
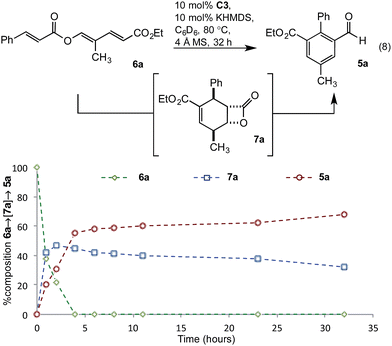 |
| | Fig. 3 Intermediacy of β-lactone 7a. | |
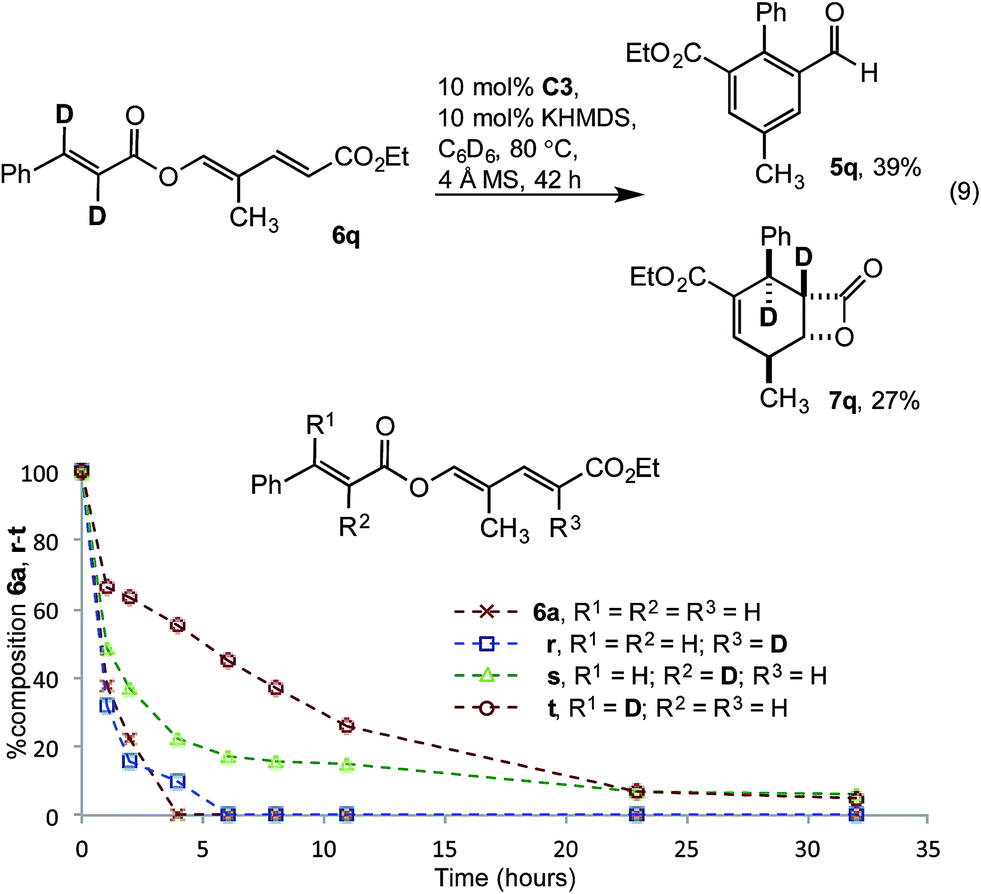
The impact of deuteration on reaction rate was examined with dideuterated cinnamate 6q. Using the standard reaction conditions aldehyde 5q was isolated in a modest 39% yield, along with 27% β-lactone 7q (eqn (9)). The magnitude of the rate decrease implicated impedance of the redox isomerisation by a primary kinetic isotope effect (KIE). To deconvolute these results, monodeuterated substrates 6r, s and t were prepared. 1H-NMR spectroscopic monitoring of the consumption of deutero-diene 6r and α-deutero cinnamate 6s showed similar rates of consumption to 6a. However, conversion of β-deutero cinnamate 6t was significantly retarded (Fig. 4). While a full kinetic analysis is required to eliminate possible involvement of a secondary KIE, the results were consistent with turnover limiting proton transfer to form the homoenolate (vide infra).
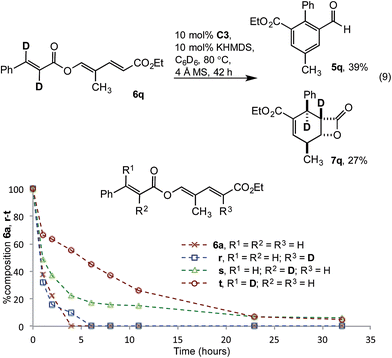 |
| | Fig. 4 Impact of substrate deuteration. | |
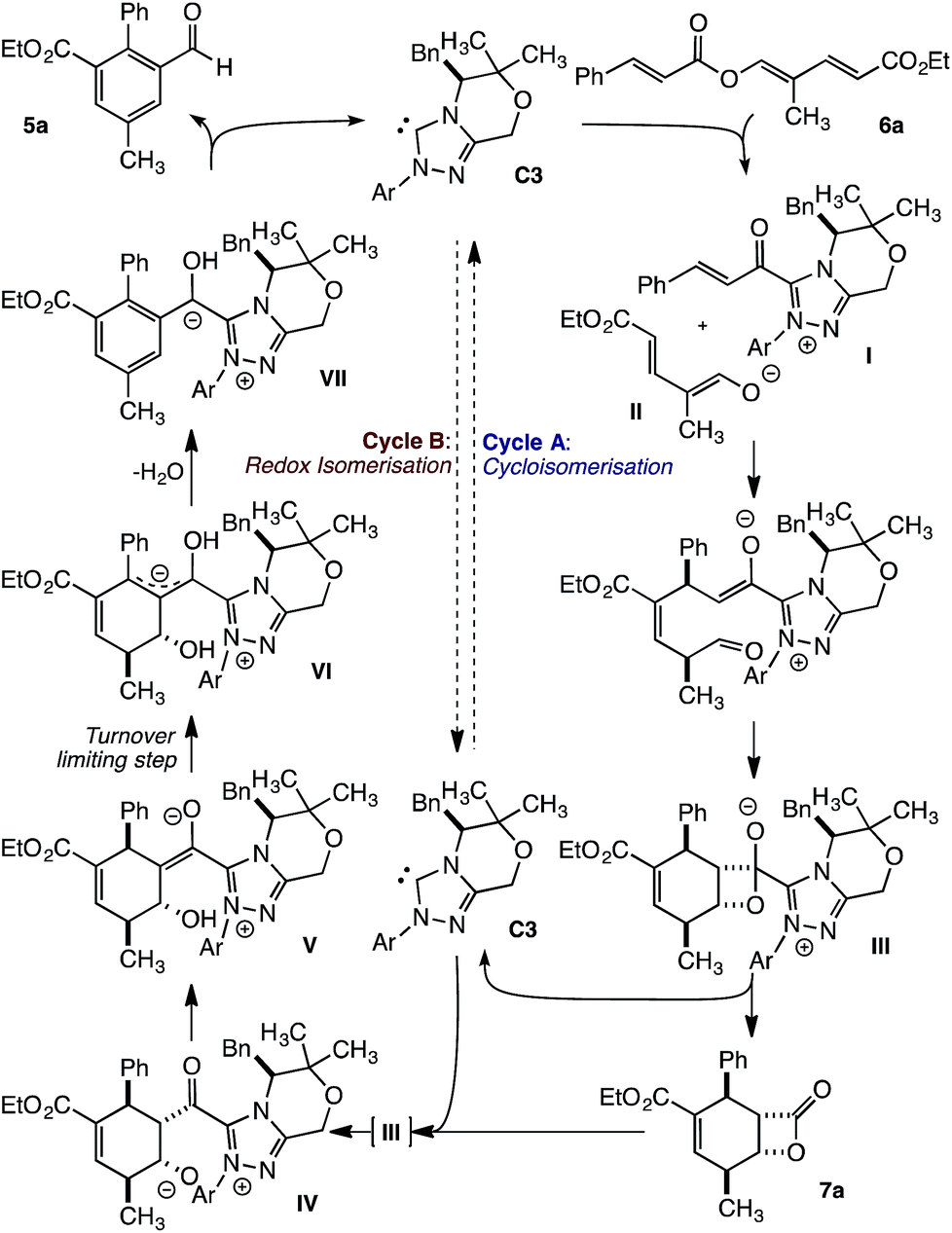
From these studies, a mechanism that involves two linked catalytic cycles can be proposed. The transformation begins with fragmentation of trienyl ester 6a to give α,β-unsaturated acyl azolium I and dienolate II (Fig. 5). These unite in a vinylogous Michael addition, followed by a pseudoconcerted (2 + 2) cycloaddition8c to provide β-lactone hemiacetal III. Loss of the catalyst provides β-lactone 7a to complete cycle A. This pathway is dominant early in the transformation. As β-lactone 7a accumulates, and triene 6a is consumed, addition of the NHC to 7a regenerates IIIen route to acyl azolium IV. Subsequent proton transfer provides acyl azolium enolate V and ultimately homoenolate VI, in the turnover limiting event, by a β-deprotonation first described by Chi.9f,i Finally, elimination of water gives aromatic intermediate VII, while proton transfer and loss of the NHC liberates benzaldehyde 5a and regenerates the NHC.
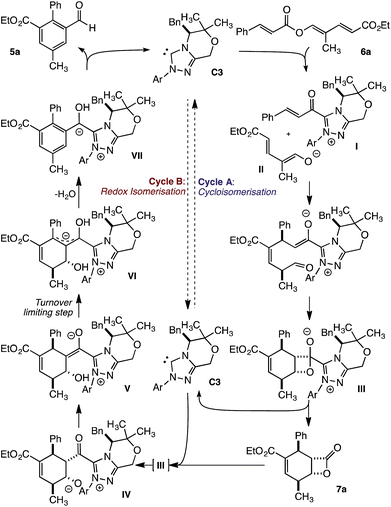 |
| | Fig. 5 Potential reaction mechanism. | |
Conclusions
The capacity of NHCs to access acyl anions en route to acyl azolium intermediates has been pivotal to modern studies in NHC organocatalysis. Herein, we describe a reaction that occurs in the opposite direction. Key to achieving this has been the observation that β-lactones can undergo fragmentation rather than decarboxylation, and the use of aromatisation as a driving force to allow unusual reaction pathways. These discoveries demonstrate proof of concept for redox isomerisation with carbonyl reduction, while providing a novel synthesis of benzaldehydes 5. Presumably, other substrates bearing internally oxidisable functionality or the use of chemoselective reducing agents,16 should enable related transformations characterised by redox isomerisation with reduction at the carbonyl. Many questions remain with this reaction. Particularly intriguing is the reaction's remarkable sensitivity to solvent polarity and catalyst nucleophilicity and the surprising absence of benzoin condensation pathways. On-going mechanistic studies are focused on these questions.
Acknowledgements
The authors acknowledge financial support from the Australian Research Council through the Discovery (DP120101315) and Future Fellowship (FT110100319) programs.
Notes and references
-
(a) T. Ukai, R. Tanaka and T. Dokawa, J. Pharm. Soc. Jpn., 1943, 63, 296 CAS;
(b) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS;
(c) H. Stetter and M. Schreckenberg, Angew. Chem., Int. Ed. Engl., 1973, 12, 81 CrossRef.
- For a selection of recent reviews on NHC catalysis see:
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; For homoenolate chemistry see:
(b) V. Nair, R. S. Menon, A. T. Biju, C. R. Sinu, R. R. Paul, A. Jose and V. Sreekumar, Chem. Soc. Rev., 2011, 40, 5336 RSC; For acyl azolium enolates see:
(c) H. U. Vora, P. Wheeler and T. Rovis, Adv. Synth. Catal., 2012, 354, 1617 CrossRef CAS PubMed;
(d) J. Douglas, G. Churchill and A. D. Smith, Synthesis, 2012, 44, 2295 CrossRef CAS PubMed; For cascade catalysis see:
(e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; For acyl anion chemistry see:
(f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; For applications in total synthesis see:
(g) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 11686 CrossRef CAS PubMed; For acyl anion free catalysis see:
(h) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906 RSC; For catalysis under oxidative conditions see:
(i) S. De Sarkar, A. Biswap, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664 CrossRef CAS PubMed; For acyl azoliums and enol azoliums see:
(j) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed; For reactions with esters see:
(k) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS PubMed; For an introduction to NHCs see:
(l) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed.
- Early examples:
(a) K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126 CrossRef CAS PubMed;
(b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS PubMed;
(c) S. S. Sohn and J. W. Bode, Org. Lett., 2005, 7, 3873 CrossRef CAS PubMed.
- Early examples:
(a) N. T. Reynolds, J. R. de Alaniz and T. Rovis, J. Am. Chem. Soc., 2004, 126, 9518 CrossRef CAS PubMed;
(b) N. T. Reynolds and T. Rovis, J. Am. Chem. Soc., 2005, 127, 16406 CrossRef CAS PubMed.
- An early example: C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS PubMed.
- An early example: A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905 CrossRef CAS PubMed.
- An early example: K. Zeitler, Org. Lett., 2006, 8, 637 CrossRef CAS PubMed.
-
(a) L. Candish, A. Levens and D. W. Lupton, J. Am. Chem. Soc., 2014, 136, 14397 CrossRef CAS PubMed. For related (4 + 2) annulations see:
(b) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694 CrossRef CAS PubMed;
(c) S. J. Ryan, A. Stasch, M. N. Paddon-Row and D. W. Lupton, J. Org. Chem., 2012, 77, 1113 CrossRef CAS PubMed.
- For work from our group with ester substrates see:
(a) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2009, 131, 14176 CrossRef CAS PubMed;
(b) L. Candish and D. W. Lupton, Org. Lett., 2010, 12, 4836 CrossRef CAS PubMed;
(c) L. Candish and D. W. Lupton, Org. Biomol. Chem., 2011, 9, 8182 RSC;
(d) L. Candish and D. W. Lupton, Chem. Sci., 2012, 3, 380 RSC;
(e) M. Kowalczyk and D. W. Lupton, Angew. Chem., Int. Ed., 2014, 53, 5314 CrossRef CAS PubMed and ref. 8. For selected examples from the Chi group see:
(f) L. Hao, Y. Du, H. Lv, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154 CrossRef CAS PubMed;
(g) L. Hao, S. Chen, J. Xu, B. Tiwari, Z. Fu, T. Li, J. Lim and Y. R. Chi, Org. Lett., 2013, 15, 4956 CrossRef CAS PubMed;
(h) S. Chen, L. Hao, Y. Zhang, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 5822 CrossRef CAS PubMed;
(i) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835 CrossRef CAS PubMed;
(j) J. Xu, Z. Jin and Y. R. Chi, Org. Lett., 2013, 15, 5028 CrossRef CAS PubMed.
- For anhydrides see:
(a) A. Lee, A. Younai, C. K. Price, J. Izquierdo, R. K. Mishra and K. A. Scheidt, J. Am. Chem. Soc., 2014, 136, 10589 CrossRef CAS PubMed;
(b) X.-Y. Chen, Z.-H. Gao, C.-Y. Song, C.-L. Zhang, Z.-X. Wang and S. Ye, Angew. Chem., Int. Ed., 2014, 53, 11611 CrossRef CAS PubMed;
(c) Z. Jin, S. Chen, Y. Wang, P. Zheng, S. Yang and Y. R. Chi, Angew. Chem., Int. Ed., 2014, 53, 13506 CrossRef CAS PubMed.
- The use of β-lactones as substrates for NHC catalysis to our knowledge is unreported, although many reactions culminate in their formation, see:
(a) C. Burstein, S. Tschan, X. Xie and F. Glorius, Synthesis, 2006, 2418 CAS;
(b) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS PubMed;
(c) P.-C. Chiang, J. Kaeobamrung and J. W. Bode, J. Am. Chem. Soc., 2007, 129, 3520 CrossRef CAS PubMed;
(d) M. Wadamoto, E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 10098 CrossRef CAS PubMed;
(e) M. He and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 418 CrossRef CAS PubMed;
(f) J. Kaeobamrung and J. W. Bode, Org. Lett., 2009, 11, 677 CrossRef CAS PubMed;
(g) E. M. Phillips, J. M. Roberts and K. A. Scheidt, Org. Lett., 2010, 12, 2830 CrossRef CAS PubMed;
(h) D. T. Cohen, C. C. Eichman, E. M. Phillips, E. R. Zarefsky and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 51, 7309 CrossRef CAS PubMed.
- For an alternate approach to aromatic materials using NHC catalysis see: T. Zhu, P. Zheng, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, Nat. Commun., 2014, 5, 5027 CrossRef CAS PubMed.
- For discussions on the impact of NHC electronics on reaction outcome see:
(a) T. Rovis, Chem. Lett., 2008, 37, 2 CrossRef CAS;
(b) J. Mahatthananchai and J. W. Bode, Chem. Sci., 2012, 3, 192 RSC. For studies with catalyst C3:
(c) F. Liu, X. Bugaut, M. Schedler, R. Fröhlich and F. Glorius, Angew. Chem., 2011, 50, 12626 CrossRef CAS PubMed;
(d) L. Candish, C. M. Forsyth and D. W. Lupton, Angew. Chem., Int. Ed., 2013, 52, 9149 CrossRef CAS PubMed and references therein.
- For selected examples of point to axial chirality generation in biaryl synthesis see:
(a) A. I. Meyers and D. G. Wettlaufer, J. Am. Chem. Soc., 1984, 106, 1135 CrossRef CAS;
(b) M. Shindo, K. Koga and K. Tomioka, J. Am. Chem. Soc., 1992, 114, 8732 CrossRef CAS;
(c) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18 CrossRef CAS PubMed;
(d) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458 CrossRef CAS PubMed.
- Exact enantiopurity determination was not possible due to contamination with the benzaldehyde product and partial co-elution by HPLC.
- Early examples of reagent based generation of acyl azoliums see:
(a) J. Castells, H. Llitjos and M. Moreno-Mañas, Tetrahedron Lett., 1977, 18, 205 CrossRef. For recent studies see:
(b) B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Org. Lett., 2007, 9, 371 CrossRef CAS PubMed;
(c) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H and 13C NMR of new materials. See DOI: 10.1039/c4sc03726j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence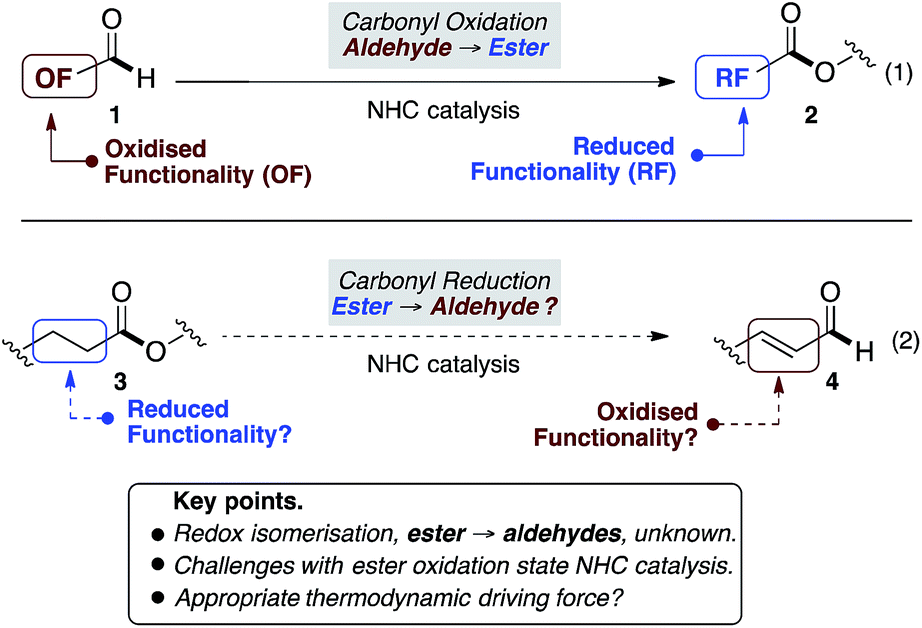
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
: