Electrochemical synthesis of palladium nanoparticles in PVP solutions and their catalytic activity in Suzuki and Heck reactions in aqueous medium
Paula M. Ubermana,
Luis A. Pérezb,
Sandra E. Martín*a and
Gabriela I. Lacconi*b
aINFIQC (CONICET-Universidad Nacional de Córdoba), Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Haya de la Torre y Medina Allende, Ciudad Universitaria, X5000HUA, Córdoba, Argentina. E-mail: martins@fcq.unc.edu.ar; Fax: +54-351-5353867; Tel: +54-351-5353866
bINFIQC (CONICET-Universidad Nacional de Córdoba), Departamento de Fisicoquímica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Haya de la Torre y Medina Allende, Ciudad Universitaria, X5000HUA, Córdoba, Argentina. E-mail: Gabriela.Lacconi@gmail.com
First published on 18th February 2014
Abstract
Small palladium nanoparticles (PdNPs) with large-surface area were synthesized in aqueous media in the presence of poly(N-vinylpyrrolidone) (PVP) using a simple electrochemical method. Electroreduction of Pd(II) species on a vitreous carbon electrode was performed by application of a current density pulse and the resulting PVP–Pd dispersion was formed. Highly concentrated PdNPs suspensions, stabilized with PVP molecules were obtained in a short time at room temperature. The size and morphology of the nanostructures obtained depend on the current density (30 to 8 nm PdNPs, for current densities in the −100 and −350 mA cm−2 range). Nanoparticle structures with large-surface area were investigated by transmission electron microscopy. The surface area of the PdNPs notably increased when the current density is more negative. The resulting PVP-stabilized PdNPs showed a very high catalytic activity when they were used in Suzuki and Heck reactions. With elevated TON and TOF, and in aqueous medium, this catalytic system proved to be an environmental friendly alternative to the conventional Pd catalyst.
Introduction
Over the last decade, the use of transition metal nanoparticles (NPs) in catalysis has expanded considerably and led to remarkable applications. Palladium nanoparticles (PdNPs) are nowadays widely used in catalysis and have become a strategic tool for organic transformation, mainly due to the wide range of applications and the high catalytic activity that they exhibit.1–4 Electrochemical methods provides a simple, low cost and effective approaches to obtain Pd nanostructured materials at large scale under different experimental conditions.5–10 Moreover, some advantages of using electrochemical procedures for obtaining NPs can be summarized as follows: (i) high reproducibility; (ii) they can be performed in aqueous solution; (iii) the size and morphology of the NPs can be controlled by changing the electrochemical conditions; (iv) colloidal solutions are produced in a short time; (v) it is a simple experimental procedure performed at room temperature and (vi) the process could be carried out in environmentally benign conditions.Usually, the electrodeposition process is addressed to obtain metallic nanoparticles supported on solid substrates. The amount of particles deposited is restricted because they are fixed to the electrode surface, consideration which must be taken into account when they are used as catalysts in solution. For this reason, in many cases a colloidal suspension is preferred.11,12
On the other hand, since the catalytic activity of PdNPs is strongly influenced by the size and geometry, many studies on nucleation, growth mechanisms and structural characterization have been reported.13,14 The Pd(II) species are commonly complexed in aqueous solutions and several additives are normally incorporated to the bath, in order to modify the characteristics of the deposited crystallites, such as the microstructure, geometry, size and aggregation degree.15,16
It was demonstrated that poly(N-vinylpyrrolidone) (PVP) molecules produce a diminution of the metal deposition rate, thereby monodispersed NPs can be electrochemically synthesized.17–19 Moreover, it was showed that PVP prevents the formation of aggregates among the formed NPs.19 Taking into account this issue, the electrochemical synthesis of monodisperse-Pd suspension in the presence of PVP is a viable alternative that allows the simple formation of NPs on a large scale production.19,20
We have reported the synthesis of stable PdNPs by electrochemical methods using platinum electrodes at room temperature via the direct electroreduction of H2PdCl4 aqueous solution in the presence of PVP.21 These PdNPs exhibited highly efficient catalytic activity in Suzuki coupling reaction in aqueous medium. Particularly high turnover numbers (TON up to 104 to 105) were achieved with aryl iodides and bromides. It is important to note that such a very low Pd loading was exceptionally observed with other PdNPs in cross coupling reactions, and in these cases the term “homeopathic” doses was used.22,23
Considering that the size and morphology of NPs can be controlled by changing the conditions of preparation, we decided to explore other experimental conditions for the electrochemical synthesis of PdNPs. It is by using a different electrode material, with the aim to improve the catalytic activity. In the present work relevant issues for the preparation of concentrated suspensions of PdNPs by application of a simple potential-time perturbation to vitreous carbon electrode is provided. The advantage of using vitreous carbon is not only by the material cost but also its working window in potential is much wider than for platinum. The electrochemical strategy for the PdNPs synthesis was based on a fast charge transfer on the electrode surface, where an efficient movement of the electrolyte allowed that Pd particles (nucleated at the electrode–electrolyte interface) were carried away from the electrode, being stabilized with PVP molecules in the solution bulk. The study involved the effect of the current density of the galvanostatic pulse on the dimensions and catalytic activity of the PdNPs.
The interest in the use of NPs as catalysts has considerably increased given that nanocatalysis appears to be one of the most promising solutions towards efficient reactions under mild, and in some cases, environmentally benign conditions in the context of green chemistry.1,2,24–27 Accordingly, Pd-based NPs have shown excellent catalytic performance for coupling reactions.3,28 Particularly, the Suzuki reaction belongs to an indispensable set of Pd-catalyzed coupling reactions and it is nowadays one of the most important methods for the formation of biaryls.29–34 Biaryls backbone plays an important role as organic intermediates for different applications, including the preparation of biologically active molecules.35–37 The Heck reaction is a well-established method for the coupling of aryl halides with olefins.38–40 As mentioned above, in both reactions PdNPs stabilized by polymers, micelles, and ligands have been used as catalysts,3,28 just as in other C–C coupling reactions.41–43 However, regardless of advantages of electrochemical method to prepared NPs, only few examples of the use of electrochemically obtained PdNPs as catalysts in cross coupling reactions were reported.10,24
In this work, PdNPs with different size were produced by galvanostatic pulses using vitreous carbon as electrode. The comparative efficiencies of these catalysts were established for coupling reactions. There is also an attempt to develop more efficient and simple catalysts for the Suzuki and Heck coupling reactions, and the possibility to perform these reactions in aqueous medium is attractive to the development of sustainable chemistry. In such way, herein we reported the electrochemical synthesis and catalytic applications of PdNPs in aqueous medium for Suzuki and Heck reactions.
Experimental section
Materials and methods
Electrolytes for the electrochemical experiments contain KNO3, H2PdCl4 and poly(N-vinylpyrrolidone) polymer (Mw PVP = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da). Solutions with different concentration of PVP were prepared from analytical grade chemicals (16 and 28 g L−1) and Milli-Q-Millipore water.
000 Da). Solutions with different concentration of PVP were prepared from analytical grade chemicals (16 and 28 g L−1) and Milli-Q-Millipore water.
HCl 35%, iodobenzene, bromobenzene, p-iodoanisole, p-iodoacetophenone, p-bromoacetophenone, p-iodobenzonitrile, p-chlorobenzotrifluoride, phenylboronic acid, styrene, K3PO4, Na2CO3, K2CO3, EtOH 98% and anhydrous Na2SO4 were used without purification. All solvents were analytical grade and distilled before use. All reactions were carried out under atmosphere of nitrogen. Silica gel (0.063–0.200 mm) was used in column chromatography.
Potentiodynamic experiments of the vitreous carbon electrode between −1.25 and 1.0 V at 0.05 V s−1 in quiescent aqueous solutions with different composition were carried out in order to establish the electrochemical conditions.44 PdNPs dispersions were prepared from Pd(II) ions electroreduction by applying a current density pulse at the vitreous carbon electrode, in the presence of PVP as the stabilizing agent, under solution stirring. An Autolab PGSTAT100 (ECO CHEMIE) potentiostat/galvanostat was used for both, the potentiodynamic experiments and the galvanostatic pulses (synthesis of PdNPs).
The PdNPs were characterized by Transmission Electron Microscopy (TEM) using a JEM-Jeol 1120 microscope operating at 80 kV, available at the Research Institute IFFIVE by INTA in Córdoba, Argentina. The total content of palladium was determined by Atomic Absorption in a Perkin Elmer Analyst 600, using ET (electro thermal mode with graphite furnace) at the Institute ISIDSA, Universidad Nacional de Córdoba, in Córdoba, Argentina.
Gas chromatographic analysis were performed on a gas chromatograph with a flame ionization detector, and equipped with the following columns: HP-5 25 m × 0.20 mm × 0.25 μm column. 1H NMR and 13C NMR were conducted on a High Resolution Spectrometer Bruker Advance 400, in CDCl3 as solvent. Gas Chromatographic/Mass Spectrometer analyses were carried out on a GC/MS QP 5050 spectrometer equipped with a VF-5 ms, 30 m × 0.25 mm × 0.25 μm column. Melting points were performed with an electrical instrument. MW-induced reactions were performed in a CEM Focused Microwave TM Synthesis System, Model Discover single mode instrument.
Synthesis of Pd nanoparticles suspension
Electrochemical experiments were carried out in a three-electrode glass cell at room temperature. The working electrode was a vitreous carbon (geometric area = 0.085 cm2) with a platinum foil and a saturated calomel electrode (SCE) as counter and reference electrodes, respectively. All potentials are reported with respect to SCE. The vitreous carbon surface was mechanically polished using alumina–water slurries (0.3 and 0.05 μm) on a polishing cloth. Electro-formation of PdNPs on the carbon electrode was performed in KNO3 (0.1 M) and H2PdCl4 (0.5 × 10−3 M) solutions (pH 3.0), at various current density pulses (−100, −150, −250, −350 and −450 mA cm−2) during 600 s, with PVP stabilizers (16 and 28 g L−1). In order to avoid the electrochemical reduction of oxygen, the electrolyte was saturated with high purity nitrogen during 15 min prior to each experiment. Strong stirring of the solution (1000 rpm) with a magnetic stirrer was kept during the synthesis. Under these conditions, an abrupt color change from yellow to a dark brown was observed since the first seconds of the galvanostatic pulse, indicating the formation of PVP-stabilized PdNPs. After completion of the reaction the aqueous dispersion of PdNPs was placed in a 25 mL volumetric flask to be used as catalyst solution for coupling reactions without further purification.Characterization of Pd nanoparticles
Average dimensions and shape of PdNPs were determined by transmission electron microscope (TEM) images. Samples for morphological characterization were prepared by depositing a drop of the colloidal suspension on a 300 mesh formvar-carbon coated copper grid and dried at room temperature. Size distribution of PdNPs was established by the average over 100 NPs from different places at each image of every sample. Distribution plots of the size were resolved by fitting data with a Gaussian behavior.The total content of palladium in the colloidal suspensions was determined by atomic absorption using ET (electro thermal mode with graphite furnace) without previous digestion.
Representative procedure for the Suzuki coupling reaction
Into a 25 mL Schlenk tube with Teflon screw-cap septum equipped with a magnetic stirrer and a nitrogen inlet, aryl halide (1a–f) (0.5 mmol), phenyl boronic acid (2a) (0.75 mmol) and K3PO4 (1.5 mmol) were added, and three cycles vacuum/nitrogen were performed to change to nitrogen atmosphere. Then, ethanol (1 mL) and water (to obtain a total volume of 3 mL taking into account the volume of PdNPs solution) were added. Finally, the required volume of the PdNPs dispersion was added. At this stage, the formation of a white precipitated was observed in the reaction mixture. The reaction mixture was heated for the required time in an oil bath at 90 °C, and the solid was dissolved within a few minutes of heating. After being cooled to room temperature, the mixture was opened to the air and diluted with water. Then the mixture was extracted three times with diethyl ether (30 mL each). The biaryl product was purified by silica-gel column chromatography after being dried with anhydrous Na2SO4.The products were characterized by 1H NMR, 13C NMR, and GC-MS. All the spectroscopic data were in agreement with those previously reported for the following compounds: biphenyl (3a),45 4-methoxybiphenyl (3b),46 1-(biphenyl-4-yl)ethanone (3c),47 and 4-ciano-biphenyl (3d).47
Representative procedure for the Heck coupling reaction
Into a 25 mL Schlenk tube with Teflon screw-cap septum equipped with a magnetic stirrer and a nitrogen inlet, aryl halide (1c, 1f) (0.5 mmol), and K2CO3 (1.5 mmol) were added, and three cycles vacuum/nitrogen were performed to change to nitrogen atmosphere. Then, styrene (0.75 mmol), ethanol (1 mL) and water (to obtain a total volume of 3 mL taking into account the volume of PdNPs solution) were added. Finally, the required volume of the PdNPs dispersion was added. At this stage, the formation of a white precipitated was observed in the reaction mixture. The reaction mixture was heated for the required time in an oil bath at 90 °C, and the solid was dissolved within a few minutes of heating. After being cooled to room temperature, the mixture was opened to the air and diluted with water. Then the mixture was extracted three times with diethyl ether (30 mL each). The stilbene product was purified by silica-gel column chromatography after being dried with anhydrous Na2SO4.The products were characterized by 1H NMR, 13C NMR, and GC-MS. All the spectroscopic data were in agreement with those previously reported for the (E)-1-(4-styrylphenyl)ethanone (4) compound.48
Microwave assisted Heck coupling reactions
Into a 10 mL MW tube equipped with a magnetic stirrer, aryl halide (1c, 1f) (0.5 mmol), K2CO3 (1.5 mmol), styrene (0.75 mmol), ethanol (1 mL) and water (to obtain a total volume of 3 mL taking into account the volume of PdNPs solution) were added. Finally, the required volume of the PdNPs dispersion was added. Then, the reaction tube was sealed with a rubber cap and heated to 130 °C for 20 min under MW irradiation (Fixed Power, 50 W) using air cooling. After that, the reaction was cooled to room temperature and extracted three times with diethyl ether (30 mL each). The stilbene product 4 was purified by silica-gel column chromatography after being dried with anhydrous Na2SO4.Results and discussion
Electrochemical behavior
The electrochemical behavior of vitreous carbon electrode in the solution used for the galvanostatic synthesis of PdNPs was determined by experiments of Pd electrodeposition in quiescent electrolyte. The voltammetric response of the electrode (only the negative potential scans are shown) in 0.1 M KNO3 and 0.5 mM H2PdCl4 solution in the presence and absence of PVP at 0.05 V s−1 are shown in Fig. 1. As can be seen in the absence of PVP, a current peak due to the electroreduction of Pd(II) ions was defined at −0.59 V and at potentials more negative than −0.9 V, the increase in the current was due to the hydrogen evolution reaction on the carbon electrode (Fig. 1a). When the solution contained the aqueous soluble PVP polymer, the potentiodynamic profile showed an onset potential for Pd(II) electroreduction around −0.34 V and the reduction peak defined at −0.65 V (Fig. 1b). Cathodic current due to the proton-electroreduction on the Pd crystallites was noticed at potentials below −0.9 V. It is well-known that Pd electrodeposition on vitreous carbon is an irreversible and diffusion-controlled process.44 On the other side, when PVP was present in the electrolyte, the cathodic peak potential was notably shifted to more negative values and the charge under the curve was markedly diminished (Qa and Qb in Fig. 1 caption). | ||
| Fig. 1 Potentiodynamic profiles at the negative potential scans of vitreous carbon electrode in quiescent electrolytes: (a) 0.1 M KNO3 + 0.5 mM H2PdCl4 (Qa = −6.8 mC cm−2) and (b) 0.1 M KNO3 + 0.5 mM H2PdCl4 + 16 g L−1 PVP (Qb = −4.1 mC cm−2). Potential scan rate: 0.05 V s−1. | ||
These features were directly related to the inhibition of the process given by blocking of sites on the surface with PVP molecules. However, the onset potential for the hydrogen evolution reaction produced on the deposited particles was not modified when PVP was present.
Synthesis at constant current
A large-scale production of very small Pd particles with high stability was feasible when the electrochemical synthesis of PdNPs was carried out in 0.1 M KNO3 + 0.5 mM H2PdCl4 + 16 g L−1 PVP aqueous solutions. Application of a current density constant pulse to the vitreous carbon electrode for a period of time under strong stirring of the electrolyte lead to the electroreduction of Pd(II) ions at the electrode–solution interface. During the electrochemical synthesis, the metallic NPs were constantly expelled from the electrode and driven toward the solution bulk by convective motion and, the pale yellow electrolyte became obscured.Addition of the water-soluble polymer PVP was performed to stabilize the Pd particles in solution. As it was shown in the previous section, the presence of PVP produces inhibition of Pd electrodeposition on the electrode, which was an advantage for obtaining smaller particles. In addition, the polymer coating layer around particles favors the formation of a well-dispersed solution, preventing coalescence of particles.19 Thus, in order to obtain a colloidal suspension with well-dispersed NPs, PdNPs formed at the interface vitreous carbon–electrolyte should not remain deposited on the surface. Immediately after the first nuclei are formed on the electrode surface, they should be readily transferred to the bulk solution, where they can be stabilized with a capping agent. It has been shown that under suitable conditions of mechanical agitation of the solution, this experimental requirement could be achieved.18–21
The electrochemical response (potential vs. time transients) recorded during application of different current pulses at a fixed PVP concentration (16 g L−1) was compared. Fig. 2a shows that all curves recorded during 600 s after applying the galvanostatic pulse had a similar overall behavior. The beginning of the PdNPs synthesis took place at different potentials, which were more negative for pulses at lower current densities. After 100–150 s, the potential tended asymptotically to a constant value reached approximately in 600 s. In all cases, the final potential was more positive than the initial one. The potential reached after 600 s of the synthesis depended linearly with the value of the current density applied and the slope of this plot was a direct measure of the solution resistance (Fig. 2b).
 | ||
| Fig. 2 (a) Potential–time curves for Pd(II) ions electroreduction on vitreous carbon electrode by galvanostatic pulses at different current densities j (−450 < j < −100 mA cm−2). Electrolyte: 0.1 M KNO3 + 0.5 mM H2PdCl4 + 16 g L−1 PVP10 (except in curve (*) where it is 28 g L−1). Solution stirring rate: 1000 rpm. (b) Resistive behavior between the potential values reached after 600 s and the current density applied from data (a); red line corresponds to the linear fitting and (*) belongs to the potential value for 28 g L−1 PVP. | ||
It is important to notice that at the observed potential values, the mass transport occurred by diffusion and convection movement in the solution. In fact, there was a competition between different simultaneous reactions at the vitreous carbon–electrolyte interface: (a) electrodeposition of the Pd0 nuclei, (b) formation of stabilized Pd particles in the solution, and (c) hydrogen evolution reaction at the interface. On Scheme 1 it is shown a graphical summary with all the reactions occurring during the colloidal PdNPs suspension synthesis.
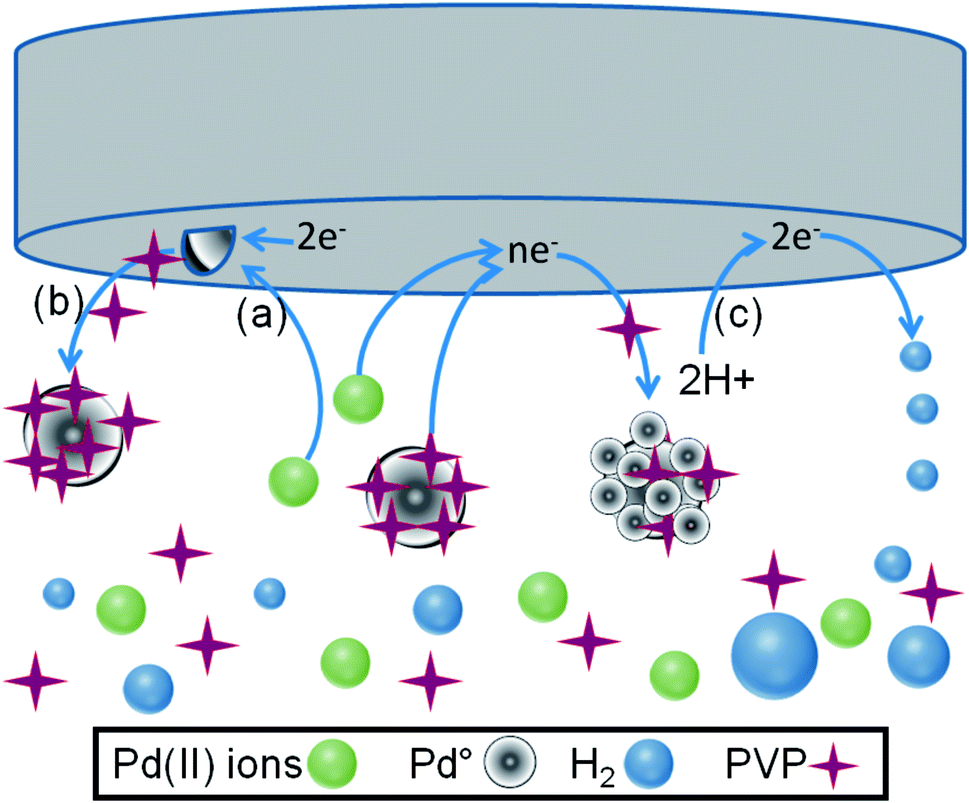 | ||
| Scheme 1 Representation of the electrochemical formation of PVP-stabilized PdNPs by considering the following reactions during the synthesis. (a) Pd(II) ions electroreduction to obtain Pd0 clusters; (b) stabilization of Pd clusters in solution with PVP; (c) formation of molecular H2 from H+ electroreduction. | ||
The first process occurs by electroreduction of Pd(II) ions and the nuclei are generated on the electrode surface. The second process is also consequence of the charge transfer performed to obtain small Pd0 clusters, which are expelled out from the interface being stabilized in the solution with PVP molecules.21,49–51 Finally, the third process corresponds to the formation of molecular hydrogen, which produces large changes of pH in the vicinity of the electrode. In addition, the movement of small bubbles can facilitate the departure of the particles to the solution bulk. Moreover, Pd particles could approach to the electrode–solution interface again and incorporate tiny Pd particles covering the surface. Another important aspect is the hydrogen absorption by the Pd particles.
It is well-known that the growth of a new phase on metallic electrodes is very irregular, producing clusters with dendritic structures when they are performed far to the thermodynamic equilibrium situation.52 Under the present experimental conditions (very negative potential values), it would be predictable the formation of anisotropic NPs. Moreover, retention of clusters on the electrode surface, could also affect the production of particles in solution, since they do not only provide catalytic centers to continue the Pd(II) electroreduction but also they are blocking part of the vitreous carbon surface. Due to these features, the synthesis yield can be restricted and the dispersion of particles size would be increased. After galvanostatic experiments, the electrode was observed with a microscope and very few Pd clusters remained on the surface, consequently, they were considered negligible compared to the amount of particles produced in the aqueous dispersion. On the other hand, the effect of the stabilizer was also considered by increasing the polymer concentration in the electrolyte. The potential–time behavior in curve (*) in Fig. 2a, for 28 g L−1 PVP is practically the same of that containing 16 g L−1 PVP for the same current pulse (at −350 mA cm−2). There was only a slight shifting of the potential to more negative values. This feature was explained by considering that higher stabilizer concentration facilitates the stripping of particles from the surface in a manner equivalent to the conditions given by a more negative current pulse. Moreover, stability of all PdNPs suspensions considering individual structures given by the PVP capping agent was demonstrated after several months, where no aggregation was observed.
It is well known that the properties of disperse deposits depend on the potential at which they were electrodeposited. In the galvanostatic experiments, the apparent current density is constant and the surface area of the deposit (if it remains on the electrode) increases leading to diminution of the real current density and the potential of electrodeposition. It is shown in our experiments (Fig. 2) that the potential did not increase much because the electrode surface was practically uncovered, since very few particles remained on the surface. All this is a clear indication that the deposited area was not large increased and the real current density was a little less than that applied.
Characterization of PVP–Pd nanoparticles
Due to the Pd(II) ions reduction rate in the electrochemical synthesis was proportional to the current density, the particle dimensions were strongly influenced by this parameter. In order to analyze the differences, PVP–PdNPs were obtained at various current densities. Fig. 3 displays typical low-magnification TEM images of the PdNPs obtained by galvanostatic pulses at −100, −150, −250 and −350 mA cm−2 during 600 s, using a vitreous carbon electrode in 0.1 M KNO3 + 0.5 mM H2PdCl4 + 16 g L−1 PVP10 solution.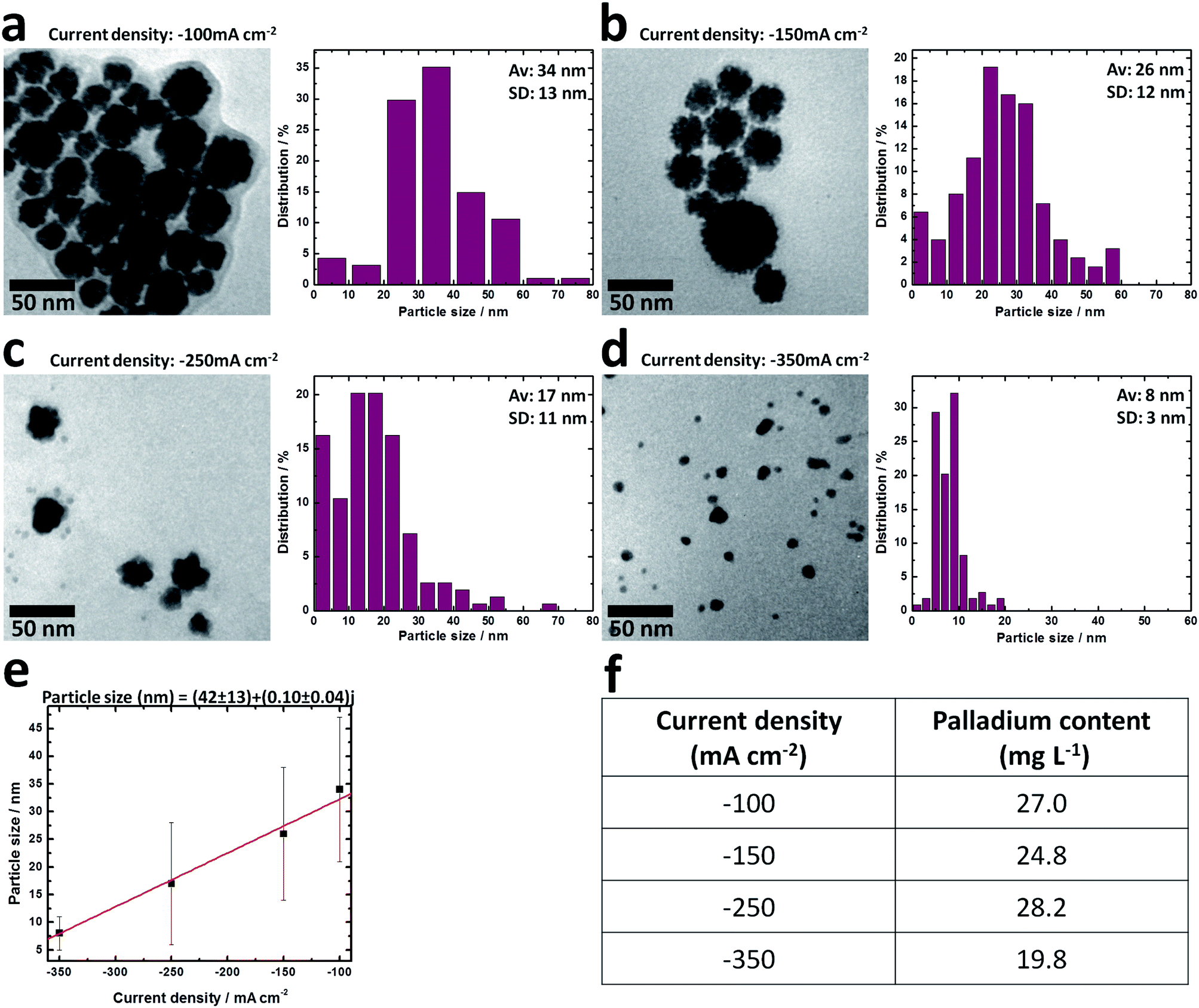 | ||
| Fig. 3 TEM images and size distribution diagrams of PdNPs obtained by galvanostatic pulses at −100 mA cm−2 (a), −150 mA cm−2 (b), −250 mA cm−2 (c) and −350 mA cm−2 (d), applied during 600 s at 25 °C using a vitreous carbon electrode in a stirred electrolyte (1000 rpm). Electrolyte: 0.1 M KNO3 + 0.5 mM H2PdCl4 + 16 g L−1 PVP10. (e) Dependence of the average particle size with the current density applied (red line corresponds to the linear fitting). (f) Palladium content determined by atomic absorption of the colloidal dispersion. | ||
PdNPs had tridimensional characteristics with hemispherical morphology, the average size varied between (34 ± 13) and (8 ± 3) nm when the current density was more negative from −100 mA cm−2 and −350 mA cm−2, respectively (Fig. 3). When the applied current density was −100 or −150 mA cm−2 (Fig. 3a and b), PdNPs with irregular rounded form and high dispersion in size were obtained. In addition, some Pd protrusions were covering the particle surface. They appeared to be decorated by tiny Pd particles on the surface. However, in Fig. 3c and d the fine surface structure on the particles disappeared and smaller particles with a very smooth surface (i.e. at −350 mA cm−2) were formed, as the cathodic current density was more negative. Insets in Fig. 3 show the size distribution diagrams for the respective particles. It is obvious that the average size gradually decreased when the galvanostatic pulse was more negative. The narrowest interval of dispersion in sizes was obtained at −350 mA cm−2 with an average diameter of particles of (8 ± 3) nm. Fig. 3e shows a linear dependence between the particle size and the current density.21 This dependence is simply explained taking into account the particles formation mechanism, where dimensions of particles were the result of the competence between the nucleation and growth rates. In the present case, reduction of Pd(II) ions occurred very fast because the high current densities applied, and nucleation was predominating over the nanoparticles growth.
Thinking that optimized dimensions of PdNPs could be improved by modifying the stabilizer concentration, experiments to the same current density at two different PVP concentrations were performed. Fig. 4 shows the comparative results obtained after applying −350 mA cm−2 during 600 s to the vitreous carbon electrode in solutions containing (a) 16 and (b) 28 g L−1 PVP.53 Distribution of particles size as well as the potential–time response for different PVP concentrations were not greatly modified (see Fig. 2–4).
 | ||
| Fig. 4 TEM images and size distribution diagrams of PdNPs obtained by a galvanostatic pulse at −350 mA cm−2 applied during 600 s at 25 °C using a vitreous carbon electrode in a stirred electrolyte (1000 rpm). Electrolyte: 0.1 M KNO3 + 0.5 mM H2PdCl4 + (a) 16 g L−1 or (b) 28 g L−1 PVP. | ||
Particles with similar size and morphology were obtained even when the amount of stabilizer molecules was highly increased. Possibly under these experimental conditions, the particles were so small that they were immediately capped with PVP molecules. Moreover, the fine structure of tiny particles decorating the Pd backbone could be consequence of the reducing action from hydrogen simultaneously generated in large quantities, which was accompanied by a noticeable change in local pH.54 When the PVP concentration decreased the PdNPs were quite unstable.9 More research is needed to understand the mechanism of decorated Pd particles formation, for example considering kinetics of PVP adsorption, diffusion rate of particles and temperature.
Generally, in the procedures for preparation of palladium nanoclusters it is assumed that all Pd(II) ions present in the original electrolyte bulk have been fully transformed to PdNPs. However, at the final moment of the synthesis, a black deposit was observed on the working electrode surface, presumably from the formation of a layer of palladium deposited.19 In order to approximate the results to a more real value, analytical determinations of the palladium content in the colloidal solution by atomic absorption measurements were carried out. The results included in Fig. 3f show that the recovery of total Pd was close to 60% of yield for −100, −150 and −250 mA cm−2 current densities.
Catalytic performance of PVP–PdNPs on the Suzuki coupling reaction
To develop useful catalysts for C–C coupling reactions, the PVP–PdNPs nanocatalysts were further investigated as potential catalysts for both the Suzuki and the Heck coupling reactions. In the last decades, the use of nanocatalysts in these reactions brought new insights in their scope.3,4 Moreover, the use of metal NPs allowed the development of sustainable methodologies that admit the use of more environmental friendly reaction medium such as the use of water, due to the preparation of stable NPs in this medium.55,56 In such way, it was of interest to evaluate the catalytic activity of the as-electrochemically prepared PVP–PdNPs in the Suzuki and Heck coupling reactions in aqueous medium.As it was well established that the size and shape of the nanocatalyst had an important effect on catalytic performance, and in order to explore the application of all the PVP–PdNPs electrochemically synthesized, we evaluated the catalytic activity of them in the Suzuki reaction of iodobenzene (1a) with phenylboronic acid (2a). All these reactions were carried out with the aqueous dispersions of PdNPs directly obtained from the electrochemical synthesis, without further purification. Based on our previous results,21 H2O–EtOH (3:1) was chosen as the solvent and K3PO4 as the base. The reactions were performed with very low loading of PdNPs (0.0008 mol% of Pd) at 90 °C under nitrogen atmosphere. In this aqueous medium all reagents were soluble when the mixture warmed up to 60 °C. The results are shown on Table 1.
|
||||
|---|---|---|---|---|
| Entry | Pd source | Time (h) | Yieldb (%) | TOFc |
| a Reaction conditions: 0.5 mmol of PhI, 0.75 mmol of PhB(OH)2, 1.5 mmol of K3PO4, 4 mL of the mixed H2O–EtOH 3:1, at 90 °C and under nitrogen atmosphere.b GC yields. The yields reported represent at least the average of two reactions.c TOF (turnover frequency, mol substrate converted per mol of Pd per hour).d The PdNPs were prepared in the presence of 28 g L−1 of PVP. |
||||
| 1 | PdNPs34nm |
24 | 65 | 2850 |
| 2 | PdNPs26nm |
24 | 83 | 3640 |
| 3 | PdNPs17nm |
24 | 95 | 4167 |
| 4 | PdNPs8nm |
2 | 94 | 61875 |
| 5d | PdNPs8nm |
2 | 69 | 36313 |
All the PVP–PdNPs proved to be very active as catalysts in the coupling reaction between 1a and boronic acid 2a. As general trend, the catalytic activities of the electrochemically prepared PVP–PdNPs have shown an enhanced activity as the size of the NPs was lowering, which is in agreement with a leaching mechanism.23,57 With the PdNPs34nm after 24 hours of reaction 65% of yield of product 3a was obtained (entry 1, Table 1). In the case of the PdNPs26nm, the biphenyl product was formed in 83% of yield (entry 2, Table 1). Completed conversion was achieved only after 24 hours when the PdNPs17nm were used as catalysts (entry 3, Table 1). The PdNPs8nm were by far much more active than the others PVP–Pd catalysts, accomplished completed conversion only after 2 hours in the same reactions conditions (entry 4, Table 1). The PdNPs8nm showed an impressive catalytic efficiency (TOF 61875 h−1) and high stability (TON 123750). Even more, this TOF value of 61875 is between 10 and 20 times superior to the others PVP–Pd catalysts.
In addition, the effect of the stabilizer concentration was evaluated employing the PVP–PdNPs prepared with 28 g L−1 PVP. In this case, the biphenyl 3a was obtained in 69% of yield, remaining nearly a 35% of substrate after 2 hours of reaction (entry 5, Table 1). These NPs had approximately the same size and were morphologically similar to those obtained in the presence of 16 g L−1 PVP. Thus, the decrease of the reactivity can be assigned to the higher amount of stabilizer present in these particles that hindered the interaction between the Pd and the organic reagents.
The PVP–PdNPs electrochemically obtained using Pt electrode, recently reported by our group,21 exhibited an outstanding catalytic activity for the Suzuki coupling in aqueous solvent with high TON (TON up to 104 to 105), although with not-so-high TOFs (higher TOF 20625 h−1). In order to compare the catalytic efficiencies of both types of PdNPs (Pt vs. vitreous carbon electrodes synthesized NPs), the Suzuki coupling reaction of iodobenzene (1a) with phenylboronic acid (2a) under the optimized reaction conditions with the complete family of NPs was carried out. The resulting catalytic activity was expressed as the TOFs as a function of NPs size and the obtained yield represented as bars in Fig. 5.
 | ||
| Fig. 5 Comparison of the catalytic activity between PdNPs prepared with Pt electrode (blue)21 and vitreous carbon electrode (red) as a function of NPs size for the Suzuki coupling reaction of iodobenzene (1a) with phenylboronic acid (2a) under the reaction conditions described on Table 1. | ||
The graphic clearly pointed out the higher catalytic activity of the newly synthesized PVP–PdNPs vs. the previous ones. In catalysis, preferably monodisperse NPs dispersions are used and the size and morphology of them can drastically modify the efficiency on the catalytic activity. Herein, it was established that the high surface roughness of PdNPs, electrochemically obtained with vitreous carbon electrode, even if they present different activities, proved to have higher activity as catalysts in Suzuki coupling reactions. Furthermore, the PdNPs8nm demonstrated an outstanding enhanced activity, although the reaction was achieved with “homeopathic” quantities of NPs.22 Likewise, the obtained TOF value is considerably high for an aqueous Suzuki coupling reaction catalyzed by PdNPs and allows us to conclude that PdNPs8nm were the best catalysts prepared.23,58
Once the catalytic activity of the PVP-stabilized PdNPs for the Suzuki coupling reaction was established, the scope of this reaction was investigated. For this reason, we screened several aryl halides with the most activity catalysts, the PdNPs8nm, in the previously optimized conditions using aqueous solvent and K3PO4 as base, as shown on Table 2. Electron-rich iodides and electron-deficient ones afforded the corresponding biphenyls with very good yields. When the reaction of p-iodoanisol (1b) with phenylboronic acid (2a) was carried out in the presence of PVP–PdNPs (0.0008 mol% of Pd) 4-methoxybiphenyl (3b) was achieved in 92% yield (entry 2, Table 2). With this substrate, which contains a deactivate group, 24 hours were required for total conversion. In addition, when the reaction with anisole 1b was performed with 0.003 mol% of Pd the total conversion was achieved in 6 hours (entry 3, Table 2). An excellent yield of biphenyl product 3c (90%) was achieved in the coupling of phenylboronic acid with p-iodoacetophenone (1c), with very high TON in 4 hours (entry 4, Table 2). It is noticeable that the catalyst loading could be reduced to 0.0005 mol%, allowing the formation of 90% of the biphenyl 3c with a TON of 198000 (entry 5, Table 2). Moreover, in the coupling reaction with p-iodobenzonitrile (1d), usually proven to be a challenging substrate, the biaryl 3d was achieved in 73% of yield (entry 6, Table 2). It is well known that with substrates containing nitrile group, the hydrolysis of the nitrile group could take place during the coupling reaction, producing the corresponding amide as byproduct.59 However, the Suzuki reaction to obtain the desired biaryl product 3d with the PdNPs8nm in aqueous medium was successfully carried out. The only side product observed in a significant amount was the one arriving from the dehalogenation of the substrates (15% of p-benzonitrile). The dehalogenation of aryl halides as a side reaction in Suzuki coupling has been reported.60,61 In these cases, the hydrogen rises from a primary or secondary alcohol used as solvent. As a final point, it is important to point out that the side product coming from the homocoupling reaction was not observed in any case.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halides | PdNPs (mol%) | Time (h) | Product yieldb (%) | Conversionc (%) | TON,d TOFd (h−1) |
| a Reaction conditions: 0.5 mmol of ArX, 0.75 mmol of PhB(OH)2, 1.5 mmol of K3PO4, 4 mL of the mixed H2O–EtOH 3:1, at 90 °C and under nitrogen atmosphere.b GC yields. The yields reported represent at least the average of two reactions.c Determine in relationship to the amount of initial substrate.d TON (turnover number, mol substrate converted per mol of Pd). TOF (turnover frequency, mol substrate converted per mol of Pd per hour). |
||||||
| 1 |  |
0.0008 | 2 | 94 (3a) | 99 | 123750, 61875 |
| 2 |  |
0.0008 | 24 | 92 (3b) | 99 | 123750, 5156 |
| 3 | 0.003 | 6 | 87 (3b) | 98 | 32667, 5444 |
|
| 4 |  |
0.0008 | 4 | 90 (3c) | 99 | 123750, 30938 |
| 5 | 0.0005 | 24 | 90 (3c) | 99 | 198000, 8250 |
|
| 6 |  |
0.0008 | 24 | 73 (3d) | 99 | 123750, 5156 |
| 7 |  |
0.003 | 24 | 78 (3a) | 80 | 26667, 1111 |
| 8 | 0.03 | 24 | 86 (3a) | 99 | 3300, 138 | |
| 9 |  |
0.006 | 12 | 90 (3c) | 99 | 16500, 1375 |
| 10 | 0.01 | 0.08 | 97 (3c) | 99 | 9900, 123750 |
|

In order to further explore the scope of the PdNPs as catalyst, the coupling reaction of substituted aryl bromides 1e–f with phenylboronic acid was carried out. When bromobenzene was employed, a higher loading of catalyst was necessary to reach an appreciable conversion. In such way, when the reaction was performed with 0.003 mol% of Pd 78% of yield of biphenyl 3a was obtained (entry 7, Table 2). Total conversion was achieved when we used a higher catalyst loading and the desired biaryl product 3a was obtained in higher yields (entry 8, Table 2). Otherwise, changing the palladium load to 0.006 mol% the reaction of 4-bromoacetophenone (1f) and phenylboronic acid afforded the biphenyl product 3c in 90% yields with a TOF value of 1375 h−1 (entry 9, Table 2), corresponding to one of the highest reached for this substrate.58 Additionally, when the coupling reaction of bromoacetophenone 1f was carried out with 0.01 mol% of Pd a white solid precipitate appears in the reaction mixture only after 5 minutes. This white solid corresponded to the biphenyl product 3c. Thus, only after 5 minutes product 3c was formed in 97% of yield, and a very high TOF value of 123750 h−1 was accomplished, being higher than other informed in literature (entry 10, Table 2).62,63 The ability of this catalyst to activate C–Cl bond was also evaluated in the reaction with p-chlorobenzotrifluoride. Unfortunately, no reaction was observed, and the chloroarene was completely recovered.
Finally, and as a proof of concept, the reusability of the PdNPs8nm catalyst was evaluated in the Suzuki coupling reaction of p-iodoacetophenone (1c) and phenyl boronic acid (2a). The PVP–PdNPs aqueous dispersion proved to be highly stable, and the PdNPs cannot be separated by a physical technique, as a centrifugation. Due to this, in order to evaluate the reuse of this catalyst, after 4 hours when the reaction was completed, another batch of reagents and base was added to the reaction tube. After that, a GC analysis was performed and product 3c was quantified in 89% of yield. It clearly indicates that the catalyst did not lose activity since in the first cycle the product 3c was obtained in 90% of yield.
In summary, all the PdNPs proved to be highly active catalyst for Suzuki coupling. Moreover, it was observed an important enhanced activity of the smaller PdNPs8nm catalyst, providing remarkable high turnover numbers (TON up to 104 to 105) and turnover frequency numbers (TOF also up to 104 to 105). It should be mentioned that no precipitate was observed when the aqueous NPs were stored in air for months. Additionally, the stabilizer concentration was also evaluated, and even so in the preparation of the PdNPs no appreciable differences were distinguished. However, in the catalytic process a large negative effect was observed when the stabilizer concentration was increased, possibly due to the capping effect on the particles surface.
Catalytic performance of PVP–PdNPs on the Heck coupling reaction
Several PVP-stabilized PdNPs have proven their effectiveness to catalyze C–C coupling reactions.3,4 The majority of the reports of the literature, however, consist in their application in the Suzuki reactions. Little attention has been given to their application in Heck couplings.64–66 In view of the success of our PVP-stabilized PdNPs on the Suzuki coupling reaction, and with the aim to improve catalyst applications of them, the performance of the NPs was briefly examined in Heck reaction. Results are summarized on Table 3.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Aryl halides | Base | PdNPs (mol%) | Time (h) | Yield of 4b (%) | Conversionc (%) | TON,d TOFd (h−1) |
| a Reaction conditions: 0.5 mmol of ArX, 0.75 mmol of styrene, 1.0 mmol of K2CO3, 4 mL of the mixed H2O–EtOH 3:1, at 90 °C and under nitrogen atmosphere.b GC yields. The yields reported represent at least the average of two reactions.c Relative to the amount of initial substrate.d TON (turnover number, mol substrate converted per mol of Pd). TOF (turnover frequency, mol substrate converted per mol of Pd per hour).e Reaction carried out 130 °C MW at fixed power of 50 W in a sealed-vessel.f 8% of acetophenone was detected. |
|||||||
| 1 |  |
K3PO4 | 0.006 | 24 | 55 | 57 | 9500, 396 |
| 2 | Na2CO3 | 0.006 | 24 | 58 | 72 | 1200, 500 | |
| 3 | K2CO3 | 0.006 | 24 | 62 | 70 | 11667, 486 |
|
| 4 | K2CO3 | 0.08 | 24 | 60 | 80 | 1000, 42 | |
| 5 | K2CO3 | 0.2 | 24 | 95 | 99 | 495, 24 | |
| 6e,f | K2CO3 | 0.2 | 0.33 | 85 | 99 | 495, 1500 | |
| 7e | K2CO3 | 0.2 | 0.17 | 97 | 99 | 495, 2912 | |
| 8 |  |
K2CO3 | 0.2 | 24 | 39 | — | — |
| 9e | K2CO3 | 0.2 | 0.33 | 84 | 99 | 495, 1500 | |
| 10e | K2CO3 | 0.2 | 0.17 | 98 | 99 | 495, 2912 | |
The p-iodoacetophenone (1c) and styrene were chosen as coupling reagents. At first, the reaction was carried out under the same conditions for the Suzuki reaction, using K3PO4 as base and H2O–EtOH (3:1) as the solvent at 90 °C. Under this condition and in the presence of 0.006 mol% of Pd, only 55% of yield the E-stilbene 4 was achieved (entry 1, Table 3). In an attempt to improve the conversion of substrate 1c, other inorganic bases were explored (entries 1–3, Table 3). The best results were obtained when K2CO3 was used, thus the K2CO3 was chosen as base for the Heck reaction with the PdNPs8nm catalyst. In order to obtain total conversion catalyst load was increased to 0.08 mol% of Pd, however, despite an increase in the conversion, no improvement in the yield of product 4 was observed (entry 4, Table 3). Only when 0.2 mol% of Pd were added, complete consumption of substrate 1c was accomplished and 97% of E-stilbene 4 was exclusively obtained, showing the high selectivity of the reaction.
In an effort to reduce the reaction time, microwave (MW) irradiation was explored as alternative to conventional heating. Direct and rapid heating by microwave irradiation in many cases enables reactions to be carried out in a fraction of the time generally required using conventional heating, and its application in several cases has led of reaction rate enhancement, and improvement of yield and selectivity.67 MW irradiation has been used in Pd-catalyzed coupling reactions,68 and particularly in Heck reactions.69,70 Thus, the MW assisted coupling reaction of 1c and styrene catalyzed by PdNPs8nm was evaluated under different conditions. The best results were obtained by heating the reaction mixture at 130 °C in a sealed-vessel at fixed power. After MW irradiation for 20 minutes completed conversion of substrate was observed, and product 4 was obtained in 85% of yield, and 8% of acetophenone was detected (entry 6, Table 3). However, decreasing the reaction time to 10 minutes resulted in a remarkable increase of the E-stilbene 4, without the formation of any other byproduct and with a TOF of 2912 h−1 (entry 7, Table 3).
Furthermore, the Heck reaction with p-bromoacetophenone (1f) was studied. As expected, this substrate was less reactive than the corresponding iodo derivative. When the reaction was performed under conventional heating, after 24 hours and in presence of 0.2 mol% of Pd, only 39% yield of product 4 was obtained (entry 8, Table 3). Likewise that with aryl iodides 1c, when the reaction was performed under MW irradiation for 20 minutes, styrene 4 was formed in 84% of yield (entry 9, Table 3). In this case, total conversion of the substrate was accomplished and 15% of acetophenone was detected. However, when the reaction was left for only 10 minutes, the E-stilbene 4 was obtained in 98% of yield (entry 10, Table 3).
In conclusion, it was found that PVP–PdNPs were effective and usefulness catalysts for Heck reaction with aryl iodides and aryl bromides under mild conditions in an environmental friendly solvent system. Good conversions and selectivity of the coupling products were obtained with conventional heating, however, it was found that the use of microwave heating in the Heck reaction allowed to achieve higher conversion, accelerated the reactions and excellent selectivities were achieved.
Conclusions
A galvanostatic deposition method was employed to produce PdNPs in aqueous solutions. By application of a current density pulse to the vitreous carbon electrode under strong stirring of the electrolyte containing PVP, a very high concentrated dispersion of PdNPs between 30 and 8 nm were obtained. It was established that the presence of the soluble polymer PVP avoid the particle coalescence, resulting in a very stable colloidal dispersion for a long time. Size and morphology of the dispersed-PdNPs depend on the value of current density applied, the efficient stirring of the solution and the appropriate concentration of the stabilizing agent. The increase of PVP concentration did not produce noticeable changes on the average dimensions and the potential-time transient. In addition, TEM images analysis evidences the presence of a fine structure (tiny particles) decorating the Pd backbone when they are obtained at current densities higher than −250 mA cm−2, which may be consequence of the simultaneous occurrence of hydrogen evolution reaction on the electrode surface.Although precise control of the uniform size of the nanoparticles can not be reached with electrochemical synthesis methods, the obtained PVP–PdNPs dispersions have a very high catalytic activity in Suzuki and Heck coupling reactions in aqueous media. This catalyst allowed performing the coupling reaction in aqueous medium, under no harsh conditions, in short reaction time, with very low catalyst loading and with very good yields and selectivity for the coupling products. Even deactivated aryl bromides can be used, and outstanding very high TOFs values were obtained. It is important to point out that the PVP–PdNPs catalyst were employ as-prepared, without further purification, and they can be stored for several months since no precipitation was observed. Finally, this catalytic system could be reused and proved to be an environmental friendly alternative to conventional Pd catalyst.
Acknowledgements
The research was supported by CONICET, FONCyT and SECyT-UNC. L.A.P. and P.M.U. are very grateful to CONICET for the fellowships.References
- U. Heiz and U. Landman, in Nanocatalysis, Springer, Berlin, 2007 Search PubMed.
- D. Peral, F. Gómez-Villarraga, X. Sala, J. Pons, J. C. Bayón, J. Ros, M. Guerrero, L. Vendier, P. Lecante, J. García-Antón and K. Philippot, Catal. Sci. Technol., 2013, 3, 475–589 CAS.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS PubMed.
- Y. Gimeno, A. Hernández Creus, P. Carro, S. González, R. C. Salvarezza and A. J. Arvia, J. Phys. Chem. B, 2002, 106, 4232–4244 CrossRef CAS.
- F. Li, B. Zhang, S. Dong and E. Wang, Electrochim. Acta, 1997, 42, 2563–2568 CrossRef CAS.
- M. T. Reetz and W. Helbig, J. Am. Chem. Soc., 1994, 116, 7401–7402 CrossRef CAS.
- D. Bera, S. C. Kuiry and S. Seal, J. Phys. Chem. B, 2004, 108, 556–562 CrossRef CAS.
- W. Pan, X. Zhang, H. Ma and J. Zhang, J. Phys. Chem. C, 2008, 112, 2456–2461 CAS.
- S. S. Shendage, U. B. Patil and J. M. Nagarkar, Tetrahedron Lett., 2013, 54, 3457–3461 CrossRef CAS PubMed.
- J. García-Martínez, R. W. J. Scott and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 11190–11191 CrossRef PubMed.
- N. Toshima, Y. Shiraishi, T. Teranishi, M. Miyake, T. Tominaga, H. Watanabe, W. Brijoux, H. Bönnemann and G. Schmid, Appl. Organomet. Chem., 2001, 15, 178–196 CrossRef CAS.
- D. Bera, S. C. Kuiry and S. Seal, J. Phys. Chem. B, 2004, 108, 556–562 CrossRef CAS.
- F. Li, B. Zhang, S. Dong and E. Wang, Electrochim. Acta, 1997, 42, 2563–2568 CrossRef CAS.
- G. Corthey, A. A. Rubert, A. L. Picone, G. Casillas, L. J. Giovanetti, J. M. Ramallo-López, E. Zelaya, G. A. Benitez, F. G. Requejo, M. J. Yacamán, R. C. Salvarezza and M. H. Fonticelli, J. Phys. Chem. C, 2012, 116, 9830–9837 CAS.
- T. R. Soreta, J. Strutwolf, O. Henry and C. K. O'Sullivan, Langmuir, 2010, 26, 12293–12299 CrossRef CAS PubMed.
- X.-L. Tang, P. Jiang, G.-L. Ge, M. Tsuji, S.-S. Xie and Y.-J. Guo, Langmuir, 2008, 24, 1763–1768 CrossRef CAS PubMed.
- A. B. Patil, S. R. Lanke, K. M. Deshmukh, A. B. Pandit and B. M. Bhanage, Mater. Lett., 2012, 79, 1–3 CrossRef CAS PubMed.
- B. Yin, H. Ma, S. Wang and S. Chen, J. Phys. Chem. B, 2003, 107, 8898–8904 CrossRef CAS.
- M. T. Reetz, R. Breinbauer and K. Wanninger, Tetrahedron Lett., 1996, 37, 4499–4502 CrossRef CAS.
- P. M. Uberman, L. A. Pérez, G. I. Lacconi and S. E. Martín, J. Mol. Catal. A: Chem., 2012, 363–364, 245–253 CrossRef CAS PubMed.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- C. Deraedt, L. Salmon, L. Etienne, J. Ruiz and D. Astruc, Chem. Commun., 2013, 49, 8169–8171 RSC.
- K. M. Deshmukh, Z. S. Qureshi, K. D. Bhatte, K. A. Venkatesan, T. G. Srinivasan, P. R. V. Raob and B. M. Bhanage, New J. Chem., 2011, 35, 2747–2751 RSC.
- J. Liu, F. He, T. M. Gunn, D. Zhao and C. B. Roberts, Langmuir, 2009, 25, 7116–7128 CrossRef CAS PubMed.
- M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS.
- B. F. G. Johnson, Top. Catal., 2003, 24, 147–159 CrossRef CAS.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- N. T. S. Phan, M. van der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS PubMed.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed.
- A. N. Cammidge and K. V. L. Crépy, Tetrahedron, 2004, 60, 4377–4386 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- K. Sambasivarao and M. Kalyaneswar, Chem.–Asian J., 2009, 4, 354–362 CrossRef PubMed.
- N. Miyaura, Top. Curr. Chem., 2002, 219, 11–59 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2005, 61, 11771–11835 CrossRef CAS PubMed.
- J. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170–1214 CrossRef PubMed.
- D. M. Cartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122–5150 RSC.
- G. Viau, R. Brayner, L. Poul, N. Chakroune, L. E. F. Fievet-Vincent and F. Fievet-Vincent, Chem. Mater., 2003, 15, 486–494 CrossRef CAS.
- A. Ohtaka, Y. Tamaki, Y. Igawa, K. Egami, O. Shimomura and R. Nomura, Tetrahedron, 2010, 66, 5642–5646 CrossRef CAS PubMed.
- C. Ornelas, A. K. Diallo, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2009, 351, 2147–2154 CrossRef CAS.
- E. A. Alvarez and D. R. Salinas, Electrochim. Acta, 2010, 55, 3714–3720 CrossRef PubMed.
- D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1979, 101, 4992–4998 CrossRef CAS.
- F.-Y. Tsai, D.-N. Lin, M.-J. Chen, C.-Y. Mou and S.-T. Liu, Tetrahedron, 2007, 63, 4304–4309 CrossRef CAS PubMed.
- J. V. Kingston and J. G. Verkade, J. Org. Chem., 2007, 72, 2816–2822 CrossRef CAS PubMed.
- B. O. A. Tasch, L. Bensch, D. Antovic and T. J. J. Müller, Org. Biomol. Chem., 2013, 11, 6113–6118 CAS.
- M. Rezaei, S. H. Tabaian and D. F. Haghshenas, Electrochim. Acta, 2012, 59, 360–366 CrossRef CAS PubMed.
- E. A. Alvarez and D. R. Salinas, Electrochim. Acta, 2010, 55, 3714–3720 CrossRef PubMed.
- L. Rodriguez-Sanchez, M. C. Blanco and M. A. Lopez-Quintela, J. Phys. Chem. B, 2000, 104, 9683–9688 CrossRef CAS.
- L. D. Rafailović and D. M. Minić, Hem. Ind., 2009, 63, 557–569 Search PubMed.
- PVP solubility in water = 100 g L−1 from Sigma-Aldrich product specifications.
- M. Rezaei, S. H. Tabaian and D. F. Haghshenas, Electrochim. Acta, 2013, 87, 381–387 CrossRef CAS PubMed.
- Z. Du, W. Zhou, F. Wang and J.-X. Wang, Tetrahedron, 2011, 67, 4914–4918 CrossRef CAS PubMed.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS PubMed.
- Z. Zheng, H. Li, T. Liu and R. Cao, J. Catal., 2010, 270, 268–274 CrossRef CAS PubMed.
- Ö. Metin, F. Durap, M. Aydemir and S. Özkara, J. Mol. Catal. A: Chem., 2011, 337, 39–44 CrossRef PubMed.
- A. Ishizuka, Y. Nakazaki and T. Oshiki, Chem. Lett., 2009, 38, 360–362 CrossRef CAS.
- R.-Y. Lai, C.-L. Chen and S.-T. Liu, J. Chin. Chem. Soc., 2006, 53, 979–985 CAS.
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly III and S. P. Nolan, J. Org. Chem., 2006, 71, 685–692 CrossRef CAS PubMed.
- D. Dey, T. Bhattacharya, B. Majumdar, S. Mandani, B. Sharma and T. K. Sarma, Dalton Trans., 2013, 42, 13821–13825 RSC.
- A. B. Patil, D. S. Patil and B. M. Bhanage, J. Mol. Catal. A: Chem., 2012, 365, 146–153 CrossRef CAS PubMed.
- C. Evangelisti, N. Panziera, A. D'Alessio, L. Bertinetti, M. Botavina and G. Vitulli, J. Catal., 2010, 272, 246–252 CrossRef CAS PubMed.
- F. Durap, Ö. Metin, M. Aydemir and S. Özkara, Appl. Organomet. Chem., 2009, 23, 498–503 CrossRef CAS.
- A. Gniewek, A. M. Trzeciak, J. J. Ziołkowski, L. Kepiński, J. Wrzyszcz and W. Tylus, J. Catal., 2005, 229, 332–343 CrossRef CAS PubMed.
- T. N. Glasnov, S. Findenig and C. O. Kappe, Chem.–Eur. J., 2009, 15, 1001–1010 CrossRef CAS PubMed.
- V. P. Mehta and E. V. van der Eycken, Chem. Soc. Rev., 2011, 40, 4925–4936 RSC.
- D. L. Martins, H. M. Alvarez, L. C. S. Aguiar and O. A. C. Antunes, Appl. Catal., A, 2011, 408, 47–53 CrossRef CAS PubMed.
- A. L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and R. Kia, Adv. Synth. Catal., 2013, 355, 957–972 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |