DOI:
10.1039/C3RA47001F
(Paper)
RSC Adv., 2014,
4, 12321-12329
Influence of Ce substitution on the order-to-disorder structural transition, thermal expansion and electrical properties in Sm2Zr2−xCexO7 system
Received
25th November 2013
, Accepted 13th January 2014
First published on 14th January 2014
Abstract
Cerium substituted rare earth zirconates of the form Sm2Zr2−xCexO7 (x = 0, 0.1, 0.2, 0.3, 0.4, 0.5) were synthesized via solid state reaction route. Structural, morphological and electrical characterizations were conducted using high temperature X-ray diffraction, FT Raman spectroscopy, thermo-mechanical analysis, scanning electron microscopy and impedance spectroscopy techniques. Addition of Ce ions into the crystal induced structural disorder to both cationic and anionic sublattices, gradually transforming the unit cell from an ordered pyrochlore to a defect fluorite structure. Substitution of bigger Ce ions to the Zr sites led to an increase in the lattice parameter and a decrease in thermal expansion coefficient of the material. The ionic disorder led to a decreased energy barrier for the thermally activated conduction process thereby increasing the overall conductivity of the materials. The maximum value of total conductivity observed in this study is 3.93 × 10−3 S cm−1 at 1023 K, which is higher than the recently reported values in a similar Sm2Zr2O7 system. Beyond x = 0.4, the ion–ion interaction in the disordered lattice began to dominate, leading to an increased activation energy and decreased total conductivity. The results demonstrate that the thermal expansion of these oxides is predominantly influenced by the B–O bond energy rather than the Madelung binding energy.
1. Introduction
Pyrochlore structured compounds of the general form A2B2O6O′ have been so important in various facets of materials science due to their interesting physical and chemical properties. Many pyrochlore oxides are reported to have very good oxide ionic conductivity and are promising candidates to be used as solid oxide fuel cell electrolytes.1 There are two different cationic sites in a pyrochlore structure. With the six-coordinated B cation site as the origin (denoted by Wyckoff symbol 16c), the A cations occupy an eight coordinated 16d site (0.5, 0.5, 0.5). There are three different anion sites; the 48f (x, 0.125, 0.125), which has two A and two B nearest neighbours, the 8a (0.125, 0.125, 0.125), which has four B nearest neighbours and the 8b (0.375, 0.375. 0.375), which has four A nearest neighbours. The 8a site is normally vacant in an ideal ordered pyrochlore.
One interesting property of pyrochlore type compounds is the structural transition from their ordered form to a disordered defective fluorite structure with variation in chemical composition,2 temperature3 or pressure. Moreover, this disordering occurs simultaneously and independently for cations and anions.4,5 In a fluorite structure, there are only two crystallographically different lattice sites; Wyckoff site 4a (0, 0, 0) occupied by cations and 8c (0.25, 0.25, 025) occupied by anions. The ratio of the ionic radii of the cations involved, rA/rB, plays a crucial role in the stability of the pyrochlore structure.6 Pyrochlores are reported to be stable in the rA/rB ratio range of 1.46 to 1.78.2,7,8 Controlling this ratio and thereby inducing order–disorder transition to get improved ionic conduction in pyrochlore oxides has been attempted widely.9
Pyrochlore structured Sm2Zr2O7 is reported to possess good conducting properties comparable to other low-temperature ionic conductors.10 Attempts to enhance its electrical properties by homovalent and aliovalent substitution of cations are widely reported.11,12 In a Sm2−xLaxZr2O7 (0 ≤ x ≤ 1.0) system, La didn't alter the crystal structure of the parent lattice and its overall influence on the electrical conductivity was to decrease it.13 In independent works, Xia et al. reported that degree of structural disorder in (Sm1−xYx)2Zr2O7, (Sm1−xEux)2Zr2O7 and (Sm1−xMgx)2Zr2O7−x systems gradually increases with an increase in the substitution of ions, which also influenced the total ionic conductivity.14–16 The transformation from ordered pyrochlore to a disordered defect fluorite structure has been found to be influential in the ionic conductivities of many other chemical systems like Y2Sn2−xZrxO7 and Nd2−yHoyZr2O7 also.17,18 As far as the conducting properties of pyrochlore type crystals are concerned, the 48f oxygen site is reported to play a crucial role in the ion diffusion mechanism in the lattice.19,20 This 48f oxygen is also related to the thermal expansion properties of the crystal via the Madelung energy in zirconate pyrochlores.21,22 In industrial applications like fuel cells both conducting properties and the thermal expansion properties of the materials are important factors to be considered. Attempts to establish the correlation among unit cell properties and the electrical and thermal expansion behaviours in some pyrochlore systems have already been reported from our group.23,24
Here in this work an attempt has been made to tailor disordering in the pyrochlore type Sm2Zr2O7 via B-site substitution by Ce4+ and to study its influence on the total ionic conductivity of the system, while keeping track of the change in unit cell and thermal expansion properties.
2. Experimental
The Sm2Zr2−xCexO7 compositions were prepared via a conventional solid–state reaction route.23 Commercially available Sm2O3 (Alfa Aesar, 99.99%), ZrO2 (Aldrich, 99%) and CeO2 (Aldrich, 99.9%) were chosen as the starting materials. Thoroughly mixed reactants were calcined at 1300 °C for 6 hours. Calcined pellets were ground and recalcined at 1600 °C for 6 hours. Green cylindrical pellets prepared by uniaxial compaction at 25 MPa were sintered at 1650 °C for 24 hours. The phase analysis was done using an X-ray powder diffractometer (X'pert Pro, PANalytical). Detailed analysis of XRD data was carried out by Rietveld refinement using commercial X'Pert HighScore Plus software. The surface morphology of the surfaces of the sintered pellets was analyzed via electron microscopic imaging using a scanning electron microscope (JEOL JSM-5600LV). FT Raman spectra of the powdered samples were recorded using a Bruker RFS100/S spectrometer employing a standard InGaAs detector. The lattice thermal expansion properties were analyzed by the high temperature XRD technique in the range of 298 K to 1273 K using an Anton Paar HTK attachment to the X'pert Pro diffractometer. Thermal expansion of bulk samples was studied using a thermo mechanical analyzer (SII, TMA/SS7300) in the range of 323 K to 773 K. Electrical characterization of the sintered pellets was carried out using an Impedance analyzer (Solartron SI 1260) with a dielectric interface (Solartron 1296) in the temperature range 473 K to 1023 K.
3. Results and discussion
The Sm2Zr2−xCexO7 compositions with x = 0, 0.1, 0.2, 0.3, 0.4 and 0.5 are labelled as SZC 0, SZC 1, SZC 2, SZC 3, SZC 4 and SZC 5 respectively in the coming sections.
3.1. X-ray diffraction
The XRD patterns of Sm2Zr2−xCexO7 are shown in Fig. 1. It can be seen that the structure of the crystal lattice is dependent on the fraction of Ce4+ present. A gradual structural change happens as we go from x = 0 to x = 0.5. The pyrochlore structure is a superstructure of the parent fluorite structure characterized by the ordering of ions in the lattice due to the difference in ionic radii of the cations involved. The diffraction pattern of pyrochlore is basically that of the fluorite structure plus a set of low intensity diffraction peaks at around the 2θ values of 14° (111), 28° (311), 37° (331) 45° (511) and 51° (531), which correspond to the ordering in the lattice. The sample x = 0 is crystallized in a pyrochlore structure as can be seen in Fig. 1. The superstructure peaks of the ordered lattice are marked by a Py label in the figure. The ionic radii of Sm3+, Zr4+ and Ce4+ are 1.079 Å (VIII), 0.72 Å (VI) and 0.87 Å (VI) respectively25 where the roman numerals within the parentheses indicate the coordination number. Since Ce4+ has a larger ionic radius than Zr4+, its addition to the lattice will decrease the cationic size difference (rA/rB) thus disrupting the lattice order. This disordering of the lattice is manifested as the disappearance of the superstructure peaks in the XRD pattern as seen in the figure.
 |
| | Fig. 1 Powder XRD patterns of Sm2Zr2−xCexO7 compositions. | |
Fig. 2 shows the (111) and (311) superstructure peaks at various concentrations of Ce4+, from x = 0 to x = 0.5. It clearly indicates that addition of Ce4+ to the lattice disrupts the ionic order, which results in the disappearance of superlattice peaks at x = 0.5 and that it is crystallized in a disordered fluorite structure with Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. Fig. 3 shows a close examination of the (311)F/(622)Pydiffraction peak. It can be seen that with the addition of Ce4+, the peak shows a shift towards lower angles of diffraction indicating an increase in the lattice spacing resulting from the incorporation of comparatively larger Ce4+ ions in place of the Zr4+ ions.
m space group. Fig. 3 shows a close examination of the (311)F/(622)Pydiffraction peak. It can be seen that with the addition of Ce4+, the peak shows a shift towards lower angles of diffraction indicating an increase in the lattice spacing resulting from the incorporation of comparatively larger Ce4+ ions in place of the Zr4+ ions.
 |
| | Fig. 2 Variation of (111) and (311) superstructure peaks with Ce4+ substitution. | |
 |
| | Fig. 3 Shift in (311)F/(622)Py peak with Ce4+ doping (the smaller peak represents the corresponding Kα2 reflection). | |
A detailed structural analysis on XRD data was carried out by Rietveld refinement method using X'Pert HighScore Plus software. The stoichiometry used and the XRD patterns observed were used to construct initial structural models of various samples. A pseudo-Voigt profile function was used to fit the diffraction pattern. The background polynomial was refined with respect to a flat background and two other coefficients. Other profile functions including Caglioti parameters and the asymmetry parameter along with structural parameters like lattice constant and the oxygen x-parameter were refined. The refined parameters and their values corresponding to the best fit are listed in Table 1.
Table 1 Rietveld refined parameters
| Sample |
SZC 0 |
SZC 1 |
SZC 2 |
SZC 3 |
SZC 4 |
SZC 5 |
| Phase |
Pyrochlore |
Pyrochlore |
Pyrochlore |
Pyrochlore |
Pyrochlore |
Fluorite |
| Unit cell |
Cubic |
Cubic |
Cubic |
Cubic |
Cubic |
Cubic |
| Space group |
Fd3m |
Fd3m |
Fd3m |
Fd3m |
Fd3m |
Fm3m |
| Lattice constant (Å) |
10.5962(1) |
10.6041(1) |
10.6187(2) |
10.6387(4) |
10.6562(6) |
5.3441(1) |
| Specimen displacement |
−0.046091 |
−0.031777 |
−0.053971 |
−0.020307 |
0.01278 |
−0.037034 |
| Flat background |
94.27321 |
88.72801 |
86.85529 |
81.68981 |
73.78641 |
77.2878 |
| Coefficient 1 |
−29.82546 |
−22.04726 |
−21.53201 |
−22.47241 |
−17.51294 |
−25.57006 |
| Coefficient 2 |
33.30337 |
22.97596 |
21.18307 |
24.12136 |
23.62647 |
25.88489 |
| Scale factor |
0.000003 |
0.000003 |
0.000003 |
0.000002 |
0.000002 |
0.000127 |
| Oxygen x parameter |
0.3393(9) |
0.342(1) |
0.3448(9) |
0.347(1) |
0.351(1) |
— |
| |
| Profile parameters |
| U |
0.053718 |
0.063701 |
0.111902 |
0.745046 |
0.829743 |
0.087138 |
| V |
−0.020268 |
−0.027337 |
−0.052835 |
−0.266091 |
−0.331451 |
−0.0127 |
| W |
0.00713 |
0.008477 |
0.013785 |
0.051403 |
0.058735 |
0.005131 |
| Rexp |
8.65 |
9.05 |
9.15 |
9.33 |
9.85 |
9.66 |
| Rp |
7.58 |
7.66 |
7.24 |
7.85 |
11.08 |
10.21 |
| Rwp |
9.86 |
10.01 |
9.5 |
10.15 |
14.18 |
13.56 |
| GOF |
1.3 |
1.22 |
1.07 |
1.18 |
2.07 |
1.96 |
Samples from x = 0 to 0.4 were best fit using the Fd3m space group and x = 0.5 was best fit using the Fm3m space group. The rA/rB ratio for x = 0, 0.1, 0.2, 0.3, 0.4 and 0.5 samples are 1.5, 1.48, 1.47, 1.45, 1.44 and 1.42 respectively. Although the commonly observed lower limit of the pyrochlore radius ratio is 1.46,2 Sm2Zr2−xCexO7 sustains a pyrochlore structure to an rA/rB value of 1.44. The results of Rietveld refinement for two typical samples are shown in Fig. 4. The observed and calculated patterns along with the difference patterns are displayed there. The plots show good agreement between observed and calculated diffraction profiles.
 |
| | Fig. 4 Rietveld simulation and refinement of representative samples SZC 1 and SZC 5. | |
Rietveld analysis yielded accurate values for the lattice parameter, which confirmed the observation in Fig. 3. In Table 1 we can see that the addition of Ce ions leads to an increase in lattice parameter. The calculated lattice parameters from Table 1 as a function of Ce4+ content are shown in Fig. 5, wherein the lattice parameter of the fluorite type x = 0.5 sample has been doubled to fit in the graph. It can be seen that the lattice parameter increases almost linearly with an increase in Ce content. This is in agreement with Vegard's law26 according to which the lattice parameter of a mixture will have a linear relation with those of the components via the composition. This is applicable to the substitution of larger Ce4+ in place of smaller Zr4+ ions.
 |
| | Fig. 5 Variation of lattice parameter with Ce4+ substitution. | |
One characteristic of the pyrochlore structure is that the oxygen ion at the 48f position has a variable x-coordinate, typically in the range 0.3125 to 0.375.27,28 For the disordered fluorite structure this oxygen has an x-coordinate 0.375 at a tetrahedral position corresponding to an ideal cubic array of anions. As the 8a site in the pyrochlore unit cell is vacant, the 48f oxygen tends to shift from its ideal tetrahedral position towards the two nearby B cations leading to a decrease in net B–O bond length. But as the lattice undergoes a disordering, the shift of oxygen towards the B cation decreases. This will lead to an increase in the x-parameter as well as in the B–O bond length. Fig. 6 shows a plot of oxygen x-parameter and B–O bond length as a function of cerium content. The trend of increase in both the parameters shows the disorder induced by the incorporation of Ce4+ in the lattice. Dickson et al.29 showed that the position of 48f oxygen strongly influences the (111) Bragg reflection from the crystal and the intensity of the (111) reflection tends to be zero as the x-parameter increases towards 0.375. We have already observed this in Fig. 1.
 |
| | Fig. 6 Variation of oxygen x-parameter and B–O bond length with Ce4+ substitution. | |
3.2. FT Raman spectroscopy
Glerup et al.30 used Raman spectroscopy as an effective tool for distinguishing an ordered pyrochlore structure from a disordered defect fluorite structure. Since X-ray diffraction is much more sensitive to the disorder in the cationic sublattice while Raman spectroscopy is primarily sensitive to the vibrations of the cation–oxygen bonds, the latter can be more instrumental for the study of local disorder. The expected normal modes of vibrations of pyrochlore and fluorite structure are determined using factor group analysis and are widely reported in literature.30–33 Cubic pyrochlores of the general formula A2B2O6O′ belong to Fd3m space group with Z = 8 and the corresponding factor group is Oh. The site symmetry for A and B ions is D3d, for O it is C2v and for O′ it is Td.31 This is a highly ordered lattice where there are three crystallographically different anion sites. Factor group analysis has shown that pyrochlore structure possess six Raman active modes of vibration whose irreducible representations are given as| | |
Γ(Raman) = A1g + Eg + 4T2g
| (1) |
The origin of these modes and the corresponding wavenumbers are listed in Table 2, adapted from ref. 28.
Table 2 Various Raman modes of pyrochlore structure
| Symmetry |
Mode of vibration |
Wavenumber (cm−1) |
| Eg |
B–O6 bending |
302 |
| T2g |
Mostly B–O stretching with mixture of A–O stretching and O–B–O bending vibration |
400 |
| T2g |
Mostly O–B–O bend with mixture of B–O stretching |
507 |
| A1g |
Mostly O–B–O bending |
520 |
| T2g |
Mostly B–O stretching |
585 |
| T2g |
Mostly B–O stretching |
750 |
Cubic fluorites with the general formula AO2 belong to the Fm3m space group with Z = 4 and the corresponding factor group is Oh. The site symmetry for an A ion is Oh and for O it is Td.33 This is a disordered lattice where all the anions are randomly distributed among the eight total anion sites available in the unit cell. Hence all the O ions in a fluorite structure are in crystallographically identical lattice sites. Due to this the Raman spectrum of a fluorite structure is reduced to a broad continuum of density of states. Only one Raman active mode (T2g) is known in a fluorite structure.34
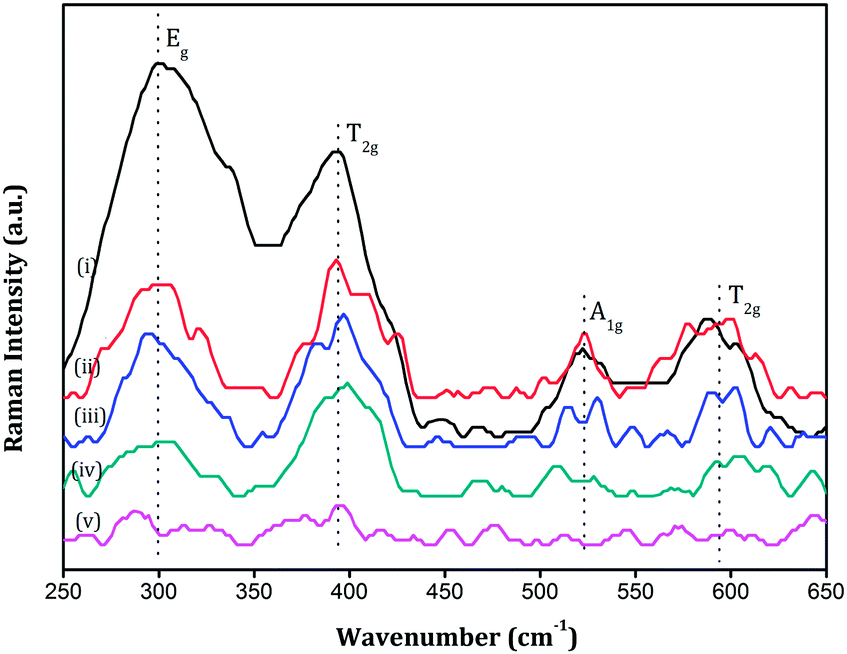
The FT Raman spectra of various Sm2Zr2−xCexO7 samples (x = 0.1, 0.2, 0.3, 0.4, 0.5) in the range 250–650 cm−1 are shown in Fig. 7. The peaks in the spectra are generally broad, they are characteristic of the ordered pyrochlore structure due to inherent deviations from translational symmetry in the lattice.35,36 This broadness is not due to small particle size because the sharp peaks in the XRD diagram and also the SEM analysis had shown that the particle size falls in the micrometer range in the present study. The Raman peaks were assigned frequencies following the work of Vandenborre.37 One notable difference in the present work is that we observe a broad peak at around 520 cm−1 in some samples whereas Vandenborre had observed two separate peaks at 523 and 532 cm−1. This superposition of two peaks to appear as a single unresolved broad peak has already been observed at the same position by B. P. Mandal et al.28 in the case of the Nd2−yYyZr2O7 system. The predominant bands in the spectra are the Eg band at ∼300 cm−1, the T2g bands at ∼394 cm−1 and ∼594 cm−1 and the A1g band at ∼523 cm−1 and other bands were very weak in intensity. The Eg band at ∼300 cm−1 and the T2g modes at ∼594 cm−1 are related to the 48f oxygen in pyrochlore structures whereas the T2g mode at ∼394 cm−1 is related to the 8b oxygen.38 Intensity of all these bands tends to diminish with an increase in the Ce4+ content in the lattice. This implies that addition of Ce4+ into the lattice disturbs the arrangement of both the 48f and 8b oxygen ions leading to an overall disorder of the anionic sublattice. Qu38 and Mandal28 in their independent works reported disorder in the zirconate systems with A-site substitution. They observed from Raman analysis that only the 48f oxygen undergoes considerable disorder and the 8b oxygen is almost intact with the substitution. But here in the present work, Raman analysis shows that with B-site substitution 8b oxygen also undergoes disorder. This may be due to the fact that Raman modes of vibrations in pyrochlores are mostly related to B–O bonds rather than to A–O bonds and B-site substitution makes it more evident in the Raman spectrum.
 |
| | Fig. 7 FT Raman spectra of (i) SZC 1 (ii) SZC 2 (iii) SZC 3 (iv) SZC 4 and (v) SZC 5. | |
3.3. Scanning electron microscopy
Typical SEM micrographs of thermally etched polished surfaces and the fractured cross sections of three representative samples, x = 0, 0.2 and 0.5 after sintering at 1650 °C for 8 hours are shown in Fig. 8. Visibly, the Sm2Zr2−xCexO7 samples are relatively dense with a small amount of porosity. The overall morphologies of all the samples are more or less similar. The average grain size falls in the range of a few micrometers. Distinct grain boundaries can also be identified. A table containing the bulk density of the samples expressed in terms of the percentage of their respective theoretical densities is shown below (Table 3) .
 |
| | Fig. 8 Surface morphology of sintered pellets of representative samples (a) SZC 0 (b) SZC 2 and (c) SZC 5, (d), (e) and (f) being the fractured surfaces of the same in order. | |
Table 3 Bulk density of samples
| Composition |
Bulk density (% of theoretical density) |
| SZC 0 |
86.2 |
| SZC 1 |
82.3 |
| SZC 2 |
85.8 |
| SZC 3 |
86.6 |
| SZC 4 |
87.1 |
| SZC 5 |
86.9 |
3.4. Thermal expansion studies
Influence of temperature on the crystallographic properties of materials is an important factor to be considered if they are to be used for high temperature applications like fuel cells. Structural stability of a solid sample with respect to temperature is best reflected in its thermal expansion coefficient (TEC). In the present work two methods have been adopted to study the thermal expansion of the samples; high temperature XRD and thermo mechanical analysis (TMA). High temperature XRD study was performed on powder samples at intervals of 200 degrees in a temperature range from 298 K to 1273 K. The expanded view of the (222) Bragg reflection of a representative sample SZC 1 to illustrate the evident effect of temperature on unit cell properties is shown in Fig. 9. The recorded XRD patterns of each sample at all temperatures were fitted by Le-bail fitting method using commercial X'pert HighScore plus software and the values of the lattice parameter were calculated. All the samples retained their respective crystal structures without undergoing any space group transformation. The lattice parameter of all the samples showed a linear increase with an increase in temperature, which is also qualitatively evident in the shift of the (222) peak in Fig. 9 towards lower angle side. From the plots of lattice parameter versus absolute temperature, the lattice thermal expansion coefficients were calculated using the equation| | |
αXRD = 1/a298(da/dT)K−1
| (2) |
where αXRD is the lattice thermal expansion coefficient, a298 is the value of lattice parameter at room temperature and da/dT is the slope of the temperature dependence of the lattice parameter.
 |
| | Fig. 9 Expanded view of the (222) reflection of representative sample SZC 1 at various temperatures. | |
The variation of the lattice thermal expansion coefficient, αXRD, with the doping level of Ce4+ is shown in Fig. 10.
 |
| | Fig. 10 Variation of the lattice thermal expansion coefficient with Ce4+ substitution as yielded by high temperature XRD. | |
Results of high temperature XRD studies give us an idea about the response of the crystal lattice to temperature in the microscopic level, in terms of the unit cell parameters. In order to confirm this trend, a macroscopic approach to the same has been performed by employing thermo mechanical analysis. Bulk samples in the form of sintered cylindrical pellets were loaded in the TMA apparatus and the variation in the linear dimension of the pellets with respect to temperature was studied. The coefficient of expansion was calculated using the equation
This equation is in essence similar to eqn (2) except that it deals with the macroscopic measure of a physical dimension of the bulk sample. A plot of αTMA against Ce4+ content is shown in Fig. 11. In this plot too, the thermal expansion coefficient is found to decrease with an increase in the amount of Ce4+ in the lattice.
 |
| | Fig. 11 Variation of bulk thermal expansion coefficient with Ce4+ substitution as yielded by thermo mechanical analysis on sintered pellets. | |
Both the high temperature XRD and TMA results go hand in hand with respect to this trend of a decrease in the thermal expansion coefficient with Ce4+ substitution. This result is opposite to the commonly reported trend of an increase in TEC with a decrease in Madelung binding energy.21,23 An increase in the x-parameter of the 48f oxygen negatively affects the Madelung energy39 and hence the trend in Fig. 6 should imply an increase in TEC with respect to an increase in Ce4+ content. The reverse trend in this system could be attributed to the change in diatomic bond energies of the B–O bonds. The bond energy of the Ce–O bond is (795 ± 8) kJ mol−1 whereas that of the Zr–O bond is (776 ± 13.4) kJ mol−1 (ref. 40) implying that the diatomic bond strength of the Ce–O bond is greater than that of the Zr–O bond. Hence the effect of substitution of Zr4+ by Ce4+ in the Sm2Zr2O7 lattice is to increase the ionization energy of the electrons participating in the B–O bond formation. A positive correlation between the exponential of the ionization energy of the bond-forming electrons and the square of the phonon frequency has been theoretically established by A. Arulsamy41 using the renormalization group method. This leads to a conclusion that the presence of Ce4+ in this system increases the phonon frequency resulting in increased rigidity of the lattice and hence lowering the thermal expansion. It can also be inferred that for Sm2Zr2−xCexO7 systems, the B–O bond energy dominates over the Madelung energy in deciding the thermal expansion properties.
3.5. Impedance spectroscopy
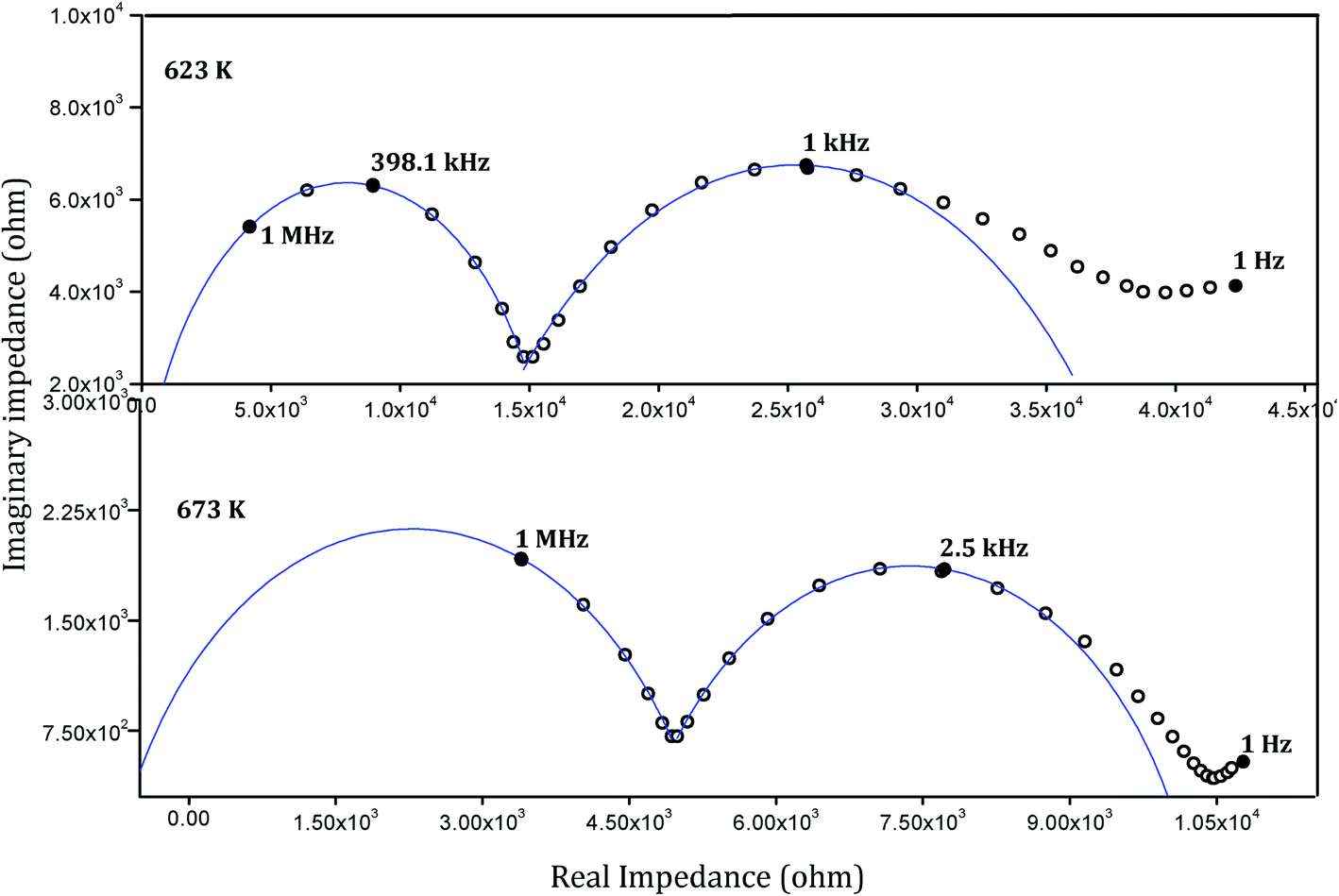
The complex plane representation of the imaginary part of the ac impedance of the sintered pellets against the real part (Cole–Cole plot) yielded semicircular arcs characteristic of most of the solid electrolyte systems. Typical impedance spectra of a representative sample SZC 3 at 623 K and 673 K are shown in Fig. 12. Calculating the capacitance values associated with the semicircular arcs is a way of understanding the nature of conduction associated with it. For grain conduction the capacitance values are in pF order and for grain boundary conduction they are in nF order.42 Here it could be seen that the high frequency arcs correspond to grain conduction and the low frequency arcs to grain boundary conduction. A slight deviation from the ideal semicircular behaviour is seen at the low frequency region of the plot. This behaviour is characteristic of certain ionic conductors and is the result of a blocking double layer capacitance at the electrode–sample interface.43 The capacitance values associated with these low frequency points were found to typically be in the microfarad order, which establishes this point. The intercept of the bulk and grain boundary arcs with the real axis would respectively yield grain resistance (Rg) and grain boundary resistance (Rgb) offered by the samples to the flow of charge across them. The total resistance RT will be the sum of these two resistive components (RT = Rg + Rgb). The total conductivity can then be calculated using the equation,
where l and A represent the thickness and the electroding surface area of the samples, respectively. The temperature dependence of the total conductivity of the samples is shown in Fig. 13 wherein ln(σT) is plotted against 1000/T. The plots could be fit with straight lines using the least square fitting method they are displayed in the figure using dotted lines. This implies that the conduction mechanism in the materials obey the Arrhenius equation,
σ = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/kBT) exp(−Ea/kBT) |
here Ea is the activation energy for the conduction process at the absolute temperature T and kB is the Boltzmann constant and σ0 is the pre-exponential factor.
 |
| | Fig. 12 Cole–Cole plots of representative sample SZC 3 at 623 K and 673 K. | |
 |
| | Fig. 13 Arrhenius plots of Sm2Zr2−xCexO7 compositions and their linear fit. | |
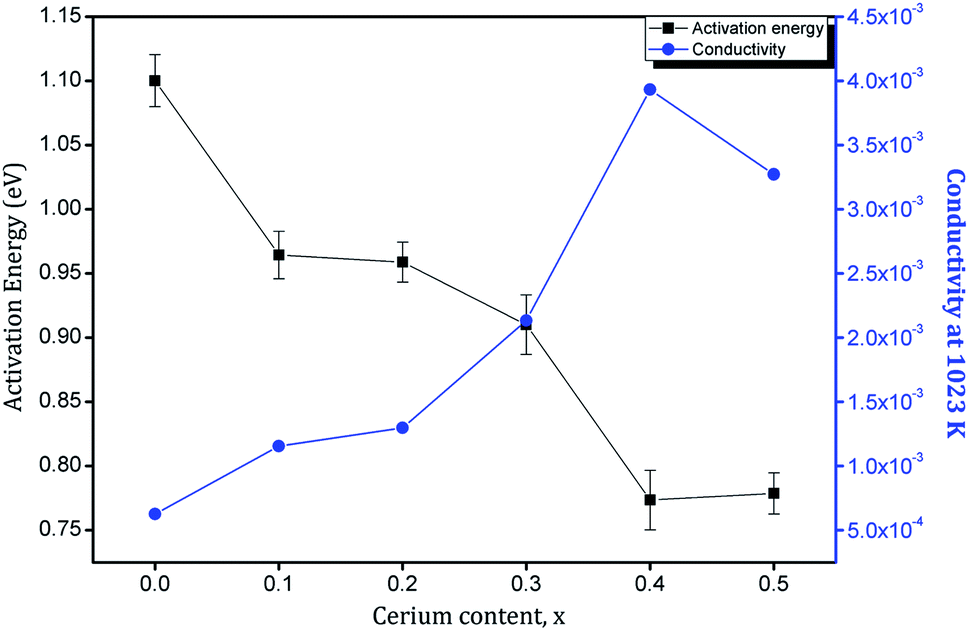
The slopes of the fitted straight lines give an idea about the activation energy involved. The conduction process by diffusion of oxide ions in Sm2Zr2−xCexO7 samples is evidently thermally activated with an activation energy that depends on the amount Ce4+ doped. Fig. 14 shows the plots of activation energy Ea and total conductivity at 1023 K against the cerium content in the Sm2Zr2O7 lattice. As the Ce4+ substitution increases, the activation energy shows a decrease from x = 0 to x = 0.4 and beyond that, it begins to increase again. The variation in total conductivity is evidently in accordance with this trend. Conductivity increases with an increase in Ce4+ content and shows a maximum at x = 0.4. Recalling the discussion on crystal structure, this can be correlated with the lattice disorder introduced by Ce4+ in the cubic pyrochlore structure of Sm2Zr2O7. In pyrochlore systems most of the ionic conduction is carried out by a vacancy hopping mechanism of the 48f oxygen.44,45 This conduction process is greatly influenced by the rA/rB ratio and the lattice constant.46 The decrease in the rA/rB ratio tends to disorder in the lattice and in turn the oxygen vacancy, which enables easy hoping of oxide ions due to creation of equivalent anion sites, which minimizes the activation energy for the oxygen hoping in the lattice. In addition, the increase in lattice constant also increases lattice free space (volume). The disordering process relieves the ions off the stringent conditions of Coulombic interactions with the neighbouring cations. This will introduce a decreased energy barrier for the mobile ion transport. Hence both these contributions are accompanied by a decrease in activation energy of the oxide ions and it accounts for the considerable decrease in activation energy (0.75–0.8 eV) observed in the present system. But disorder beyond a certain degree can prove to be detrimental to the conductivity as it can increase the ion–ion interaction within the lattice.9 This is evident as the increase in activation energy beyond x = 0.4. It can also be noted that this concentration corresponds to the boundary of the transition from an ordered pyrochlore to disordered fluorite structure.
 |
| | Fig. 14 Variation of activation energy and conductivity at 1023 K with Ce4+ substitution. | |
4. Conclusions
Sm2Zr2−xCexO7 system was prepared by solid state reaction route. Addition of Ce4+ to the ordered pyrochlore type Sm2Zr2O7 lead to the transition to a disordered defect fluorite structure. This is indicated by the disappearance of characteristic pyrochlore superlattice peaks in the XRD diagram. Raman spectroscopic studies revealed that the disorder is happening to both cationic and anionic sublattices. The anionic disorder involved both the 48f and 8b oxygen sites. High temperature XRD and thermo mechanical analysis showed that the substitution also leads to a decrease in the thermal expansion coefficient. Impedance spectroscopic analysis revealed that the conduction process in the system is thermally activated obeying the Arrhenius equation. Increasing Ce4+ content increased the total conductivity of the system with lattice disorder up to the pyrochlore–fluorite phase boundary and later it decreased for completely disordered structures.
Acknowledgements
One of the authors, Vaisakhan Thampi D S, would like to acknowledge the University Grants Commission (UGC) of India for the financial support.
References
- M. P. van Dijk, K. J. de Vries and A. J. Burggraaf, Solid State Ionics, 1983, 9–10(2), 913–919 CrossRef CAS.
- G. Aravamudan, G. V. Subba Rao and M. A. Subramanian, Prog. Solid State Chem., 1983, 15, 55–173 CrossRef.
- J. Wang, A. Nakamura and M. Takeda, Solid State Ionics, 2003, 164, 185–191 CrossRef CAS PubMed.
- R. C. Ewing, W. J. Weber and J. Lian, J. Appl. Phys., 2004, 95, 5949–5971 CrossRef CAS PubMed.
- J. Lian, L. Wang, J. Chen, K. Sun, R. C. Ewing, J. Matt Farmer and L. A. Boatner, Acta Mater., 2003, 51, 1493–1502 CrossRef CAS.
- F. X. Zhang, B. Manoun and S. K. Saxena, Mater. Lett., 2006, 60, 2773–2776 CrossRef CAS PubMed.
- B. C. Chakoumakos, J. Solid State Chem., 1984, 53, 120–129 CrossRef CAS.
- L. Minervini, R. W. Grimes and K. E. Sickafus, J. Am. Ceram. Soc., 2000, 83, 1873–1878 CrossRef CAS.
- J. Garcia-Barriocanal, A. Rivera-Calzada, M. Varela, Z. Sefrioui, M. R. Díaz-Guillén, K. J. Moreno, J. A. Díaz-Guillén, E. Iborra, A. F. Fuentes, S. J. Pennycook, C. Leon and J. Santamaria, ChemPhysChem, 2009, 10, 1003–1011 CrossRef CAS PubMed.
- K. Shinozaki, M. Miyauchi, K. Kuroda, O. Sakurai, N. Mizutani and M. Kato, J. Am. Ceram. Soc., 1979, 62, 538–539 CrossRef CAS.
- Z.-G. Liu, J.-H. Ouyang, K.-N. Sun and X.-L. Xia, J. Power Sources, 2010, 195, 7225–7229 CrossRef CAS PubMed.
- X.-L. Xia, J.-H. Ouyang and Z.-G. Liu, J. Power Sources, 2009, 189, 888–893 CrossRef CAS PubMed.
- Z.-G. Liu, J.-H. Ouyang, Y. Zhou, J. Xiang and X.-M. Liu, Mater. Des., 2011, 32, 4201–4206 CrossRef CAS PubMed.
- X.-L. Xia, J.-H. Ouyang, Z.-G. Liu, S. Gao and S. Li, J. Electrochem. Soc., 2010, 157, B470–B476 CrossRef CAS PubMed.
- J.-H. O. Xiao-Liang Xia, Z.-G. Liu and S. Li, Sci. Adv. Mater., 2010, 2, 497–502 CrossRef PubMed.
- X. L. Xia, Z. G. Liu, J. H. Ouyang and Y. Zheng, Fuel Cells, 2012, 12, 624–632 CrossRef CAS.
- M. de los Reyes, K. R. Whittle, Z. Zhang, S. E. Ashbrook, M. R. Mitchell, L.-Y. Jang and G. R. Lumpkin, RSC Adv., 2013, 3, 5090–5099 RSC.
- F. N. Sayed, D. Jain, B. P. Mandal, C. G. S. Pillai and A. K. Tyagi, RSC Adv., 2012, 2, 8341–8351 RSC.
- P. J. Wilde and C. R. A. Catlow, Solid State Ionics, 1998, 112, 173–183 CrossRef CAS.
- A. J. Burggraaf, T. van Dijk and M. J. Verkerk, Solid State Ionics, 1981, 5, 519–522 CrossRef CAS.
- K. Shimamura, T. Arima, K. Idemitsu and Y. Inagaki, Int. J. Thermophys., 2007, 28, 1074–1084 CrossRef CAS PubMed.
- K. V. G. Kutty, S. Rajagopalan, C. K. Mathews and U. V. Varadaraju, Mater. Res. Bull., 1994, 29, 759–766 CrossRef.
- A. N. Radhakrishnan, P. P. Rao, K. S. M. Linsa, M. Deepa and P. Koshy, Dalton Trans., 2011, 40, 3839–3848 RSC.
- A. N. Radhakrishnan, P. Prabhakar Rao, S. K. Mahesh, D. S. Vaisakhan Thampi and P. Koshy, Inorg. Chem., 2012, 51, 2409–2419 CrossRef CAS PubMed.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Vegard, Z. Phys., 1921, 5, 17–26 CrossRef CAS.
- B. J. Wuensch, K. W. Eberman, C. Heremans, E. M. Ku, P. Onnerud, E. M. E. Yeo, S. M. Haile, J. K. Stalick and J. D. Jorgensen, Solid State Ionics, 2000, 129, 111–133 CrossRef CAS.
- B. P. Mandal, P. S. R. Krishna and A. K. Tyagi, J. Solid State Chem., 2010, 183, 41–45 CrossRef CAS PubMed.
- F. J. Dickson, K. D. Hawkins and T. J. White, J. Solid State Chem., 1989, 82, 146–150 CrossRef CAS.
- M. Glerup, O. F. Nielsen and F. W. Poulsen, J. Solid State Chem., 2001, 160, 25–32 CrossRef CAS.
- R. A. McCauley, J. Appl. Phys., 1980, 51, 290–294 CrossRef CAS PubMed.
- F. W. Poulsen, M. Glerup and P. Holtappels, Solid State Ionics, 2000, 135, 595–602 CrossRef CAS.
- R. W. B. N. Kjerulf-Jensen and F. W. Poulsen, 2nd European Solid Oxide Fuel Cell Forum, 1996 Search PubMed.
- F. N. Sayed, V. Grover, K. Bhattacharyya, D. Jain, A. Arya, C. G. S. Pillai and A. K. Tyagi, Inorg. Chem., 2011, 50, 2354–2365 CrossRef CAS PubMed.
- B. P. Mandal, A. Banerji, V. Sathe, S. K. Deb and A. K. Tyagi, J. Solid State Chem., 2007, 180, 2643–2648 CrossRef CAS PubMed.
- C. Wan, Z. Qu, A. Du and W. Pan, J. Am. Ceram. Soc., 2011, 94, 592–596 CrossRef CAS.
- M. T. Vandenborre, E. Husson, J. P. Chatry and D. Michel, J. Raman Spectrosc., 1983, 14, 63–71 CrossRef CAS.
- Z. Qu, C. Wan and W. Pan, Chem. Mater., 2007, 19, 4913–4918 CrossRef CAS.
- M. A. Subramanian, G. Aravamudan and G. V. Subba Rao, Prog. Solid State Chem., 1983, 15, 55–173 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, 2005 Search PubMed.
- A. D. Arulsamy, Ann. Phys., 2011, 326, 541–565 Search PubMed.
- Z. G. Liu, J. H. Ouyang and K. N. Sun, Fuel Cells, 2010, 10, 1050–1056 CrossRef CAS.
- K. S. Sibi, A. N. Radhakrishnan, M. Deepa, P. Prabhakar Rao and P. Koshy, Solid State Ionics, 2009, 180, 1164 CrossRef CAS PubMed.
- M. Pirzada, R. W. Grimes, L. Minervini, J. F. Maguire and K. E. Sickafus, Solid State Ionics, 2001, 140, 201–208 CrossRef CAS.
- J. Chen, J. Lian, L. M. Wang, R. C. Ewing, R. G. Wang and W. Pan, Phys. Rev. Lett., 2002, 88, 105901 CrossRef CAS.
- H. Yamamura, H. Nishino, K. Kakinuma and K. Nomura, J. Ceram. Soc. Jpn., 2003, 111, 902–906 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.