Reprocessing and recycling of thermosetting polymers based on bond exchange reactions
Kai Yuab,
Philip Tayntonc,
Wei Zhangc,
Martin L. Dunnd and
H. Jerry Qi*ab
aDepartment of Mechanical Engineering, University of Colorado at Boulder, Boulder, CO 80309-0427, USA. E-mail: qih@me.gatech.edu
bThe George Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA 30332-0405, USA
cDepartment of Chemistry and Biochemistry, University of Colorado at Boulder, Boulder, CO 80309-0427, USA
dSingapore University of Technology and Design, Singapore
First published on 20th January 2014
Abstract
It was recently reported that thermally malleable polymers with bond exchange reactions (BERs) can rearrange their microscopic network topology without impairing the network integrity. Besides, two pieces of polymers were shown to be able to weld together upon heating without introducing additional monomers or chemicals on the interface, enabling thermoforming, self-healing, and reprocessing of the polymer, which has drawn significant interest in the study of this type of polymers. This new breakthrough also opens an avenue to recycle thermoset polymers that traditionally are very difficult to recycle. This paper experimentally studies the reprocessing and recycling ability of a thermoset polymer (an epoxy thermosetting network) with exchangeable bonds in a pulverous state. Due to the large surface area and small size of polymer particles, their handling, transportation, and storage can offer convenience in comparison with traditional polymer scrap in bulk form, which can advance their immediate industrial applications in thermoset recycling, structure rehabilitations and component rework. A detailed investigation was performed to study the effect of reprocessing conditions (such as time and pressure) on the material mechanical performance. The reprocessing routine was also repeated multiple times (representing multiple generations of recycling) to manufacture complex objects and to join two separate bulk polymers.
1. Introduction
Reprocessing and recycling of thermosetting polymers is inherently difficult for traditional technology because of their chemically cross-linked nature. Disposal of such polymer waste typically requires landfills, or high temperature or toxic chemicals, which lead to significant environmental concerns. While thermoplastic polymers can flow and be easily reprocessed by heat, their solvent resistance, thermal and mechanical properties at high temperature are unsatisfactory, and hence cannot replace thermosets in many engineering applications, especially those requiring high-performance such as the aircraft and automotive industry.1In view of the increasing amount of thermosetting polymer waste generated, new reprocessing and recycling routes have been successively proposed. One approach consists of reutilizing finely ground thermoset scrap as reinforcements for other elastomers,2–4 ceramics5 and concrete6–9 to improve the related properties or extend their service lifetime. However, such operation on polymer wastes usually involves surface chemical modifications before usage to facilitate their combination with the matrix,2 which is costly and less viable commercially for large scale production. From both environmental and economic points of view, a preferable technology would enable us to simply weld thermosets and repair their components in service to improve the sustainability, or reprocess and recycle the thermoset into valuable resources without environmental impact. Tremendous efforts were made in the past to this end, such as surface interlocking,10,11 inter-diffusion of network defects12 and adding linear thermoplastic chains at the interface.13–15 For a more robust and efficient welding effect, reversible reactions based on covalent chemistry16–23 were developed where networks with bonds capable of breaking and reforming21,22 or exchanging pairs of atoms23 can as a result flow when heated. Another method consists of using radicals to initiate exchange reactions and create plasticity in thermosets.24–28 Although these two strategies offer possibilities to combine the reprocessability, reparability and excellent welding performance of thermoset polymers, their immediate widespread application is limited either because of the abrupt viscosity drop of the thermoset network during the operation,29,30 or the unavoidable termination reactions.1
In contrast, recently reported thermally malleable thermosetting epoxy polymers using bond exchange reactions (BER) by Leibler and co-workers1 are based on a different molecular mechanism. The network topology can be rearranged due to the internal transesterification at high temperature, and frozen upon cooling. The polymer viscosity is gradually changed versus temperature following an Arrhenius law during the operation,1 and hence the network integrity is maintained. In addition, since no additional monomers or termination reactions are introduced into the system, the numbers of links and average functionality of polymer chains are unchanged. Leibler and co-workers have experimentally demonstrated that the thermally induced BER can release the internal stress of a thermoset polymer, and allow the material to be reshaped, welded together, and reprocessed,1 which has drawn significant interests in the study of this type of polymers.31–34 This new breakthrough also opens an avenue to recycle thermoset polymers that traditionally are very difficult to recycle.1
In this paper, we experimentally study the reprocessing and recycling ability of such epoxy with exchangeable bonds in a pulverous state. Due to the large surface area and small size of polymer particles, their handling, transportation, and storage can offer convenience in comparison with traditional polymer scrap in bulk form, which can promote their incorporation into industrial applications in thermoset recycling, structure rehabilitations and component rework. Leibler and co-workers1 demonstrated that the epoxy glassy polymer (hard networks) essentially maintains its stress–strain behavior after being ground to a fine powder and injection molded back into a cohesive solid. But in this paper, we are more interested in further investigating the effect of reprocessing conditions (such as temperature, time, and pressure), as well conducting an investigation of the mechanical performance of the material through multiple generations of recycling. In addition, the reprocessing routine was repeated multiple times to manufacture complex objects and to join two separated bulk polymers.
2. Experimental methods
Fig. 1 shows schematically the overall procedure of recycling and reprocessing. The bulk polymer is first pulverized into powders, which are then compacted into a mold by applying a pressure. The compacted powders are then heated to weld them into a bulk polymer. This process is repeated several times and the bulk polymer is evaluated for its mechanical properties after each processing. The details are described below.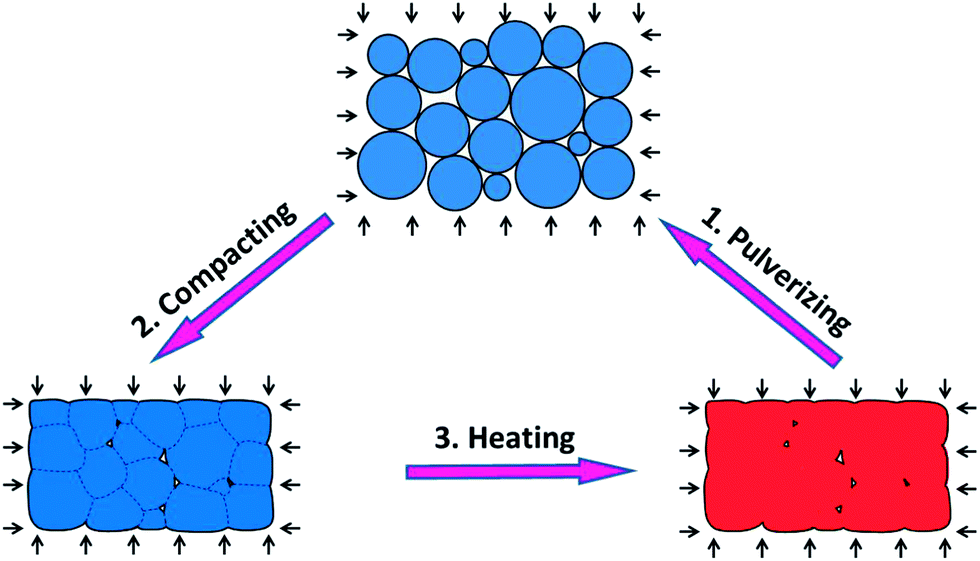 | ||
| Fig. 1 The schematic graphs for a typical reprocessing and recycling routine. (1) Bulk polymer is firstly pulverized into powders. (2) The powders were then compacted by applying pressure. (3) The compacted powder is heated under pressure. Due to the internal bond exchange reaction (BER), the polymer particles are welded and interfaces are disappeared. This process is repeated several times. | ||
2.1 Material
The thermosetting epoxy network polymer with exchangeable bonds used in this study was prepared following the method used by Leibler and coworkers.1 It was synthesized by using the reaction of diglycidyl ether of bisphenol A (DGEBA) and a mixture of fatty acids (Pripol 1040). 5 mol% metal catalyst (Zn(Ac)2) was added to facilitate the BER. Pripol 1040 was provided by Uniqema Inc. (Paterson, NJ, USA). DGEBA and metal catalyst were purchased from Sigma Aldrich (St. Louis, MO, USA). Detailed information about the structures of each chemical used is listed in Table 1.| Chemical | Structure | MW (g mol−1) | Provider |
|---|---|---|---|
| DGEBA | 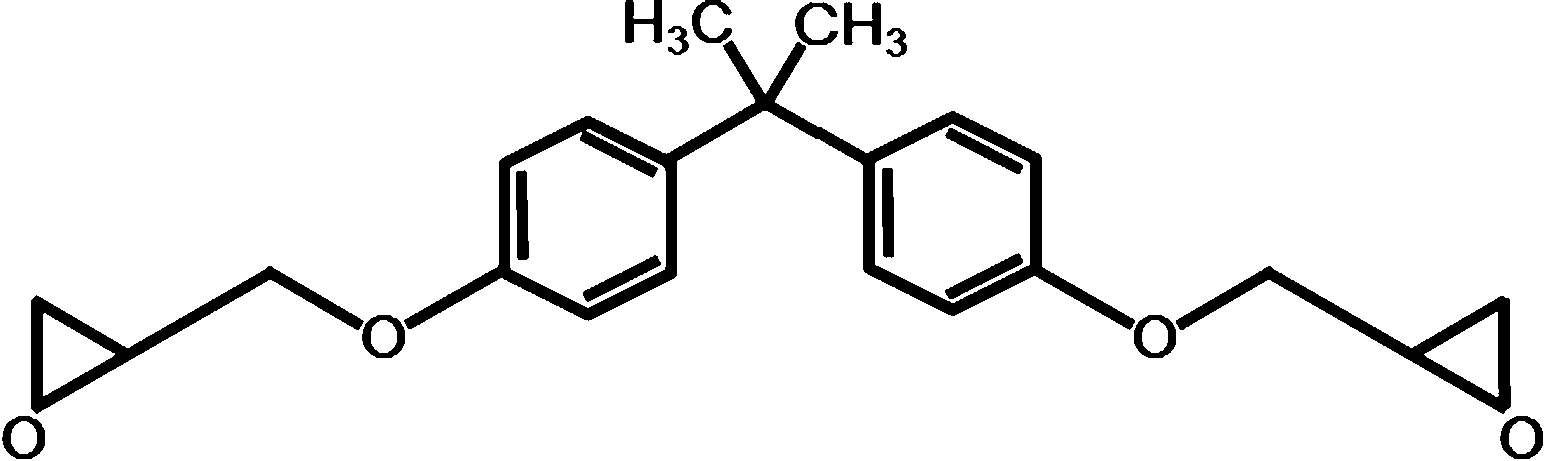 |
174 per epoxy | Sigma Aldrich |
| Pripol 1040 | 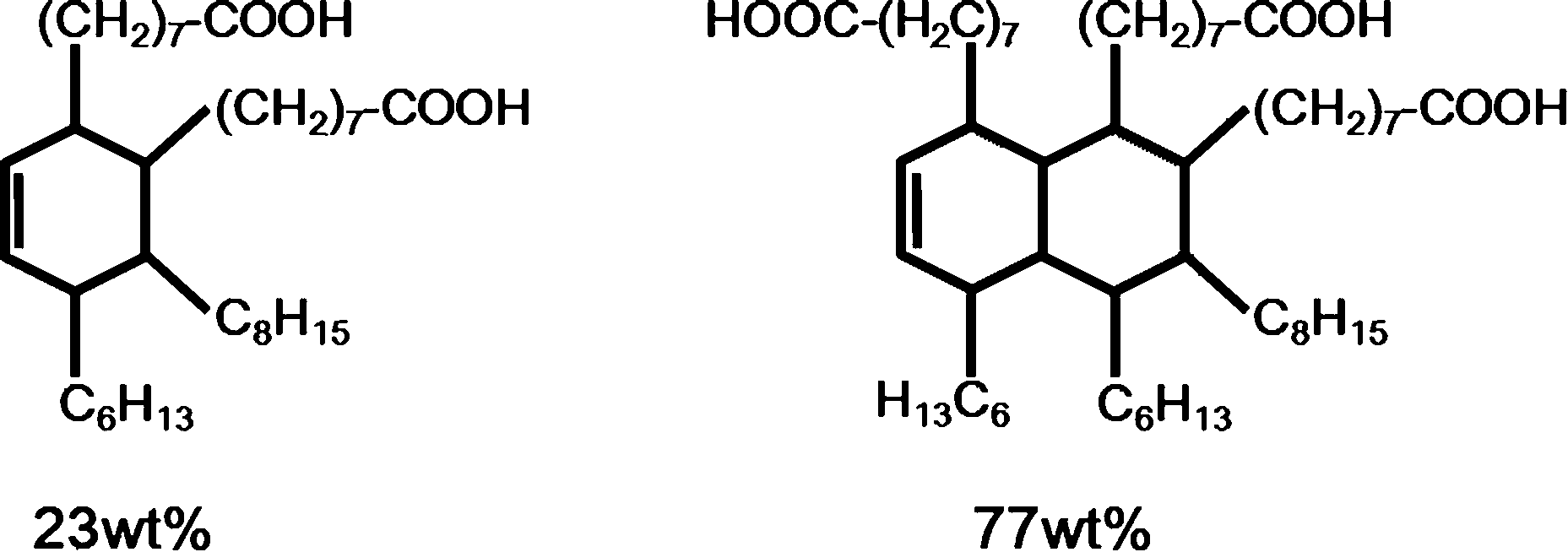 |
296 per acid | Uniqema Inc |
| Catalyst (Zn(Ac)2) | 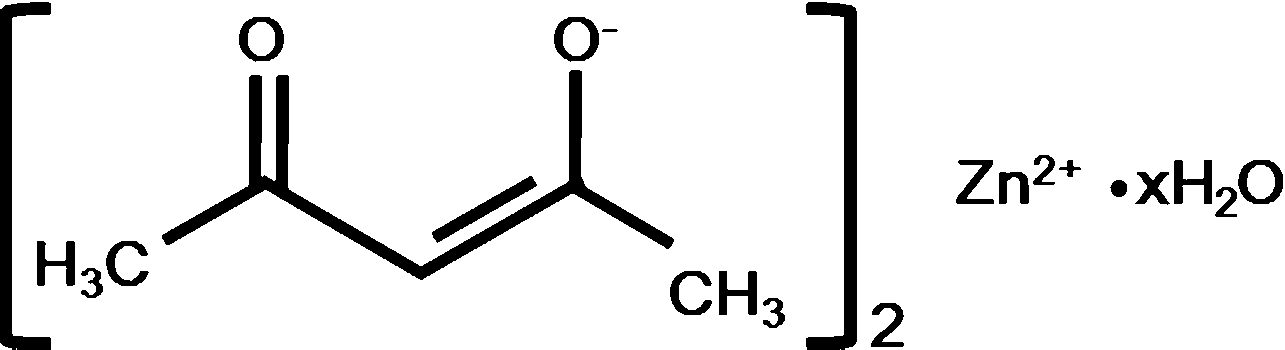 |
263.61 | Sigma Aldrich |
For polymer synthesis, the catalyst was mixed with fatty acids (5 mol% to COOH groups) in a round-bottom flask and heated under vacuum. Temperatures was gradually increased from 100 °C to 180 °C, and then hold at 180 °C until no gas evolution was observed and catalyst particles were fully solubilized (1 h). DGEBA in solid state was heated up to 130 °C until totally melted, and then manually stirred at 130 °C until the mixture became homogeneous and translucent (stoichiometry between COOH and epoxy is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Finally, the mixture was poured into a PTFE mold, and covered with a PTFE lid, and placed in an oven under a 500 g weight for 6 h at 130 °C.
:1). Finally, the mixture was poured into a PTFE mold, and covered with a PTFE lid, and placed in an oven under a 500 g weight for 6 h at 130 °C.
2.2 DMA and stress relaxation tests
The glass transition behavior of the prepared epoxy-acid thermoset network was tested by dynamic mechanical analysis (DMA). A bulk polymer sample, with the dimensions of 10 mm × 5 mm × 1.0 mm was tested by a DMA tester (Model Q800, TA Instruments, New Castle, DE, USA) on the tensile mode. The sample was firstly heated to 100 °C and stabilized for 20 minutes to reach thermal equilibrium, and then a preload of 1 kPa and an initial strain with a magnitude of 0.1% were applied. During the experiment, the strain was oscillated at a frequency of 1 Hz with a peak-to-peak amplitude of 0.1% while the temperature was decreased from 100 °C to −50 °C at a rate of 1 °C min−1. The temperature in the chamber was held at −50 °C for 30 minutes and then increased to 100 °C at the same rate. This procedure was repeated multiple times and the data from the last cooling step was reported. Fig. 2a shows the experimental results of the DMA experiments. The temperature corresponding to the peak of the tanδ curve (30.1 °C) was taken to be the glass transition temperature Tg. It should be noted that these experimental results are slightly different from the work of Leibler et al.35 where a different heating rate was adopted during the tests. The storage modulus of each sample was also plotted as a function of temperature.
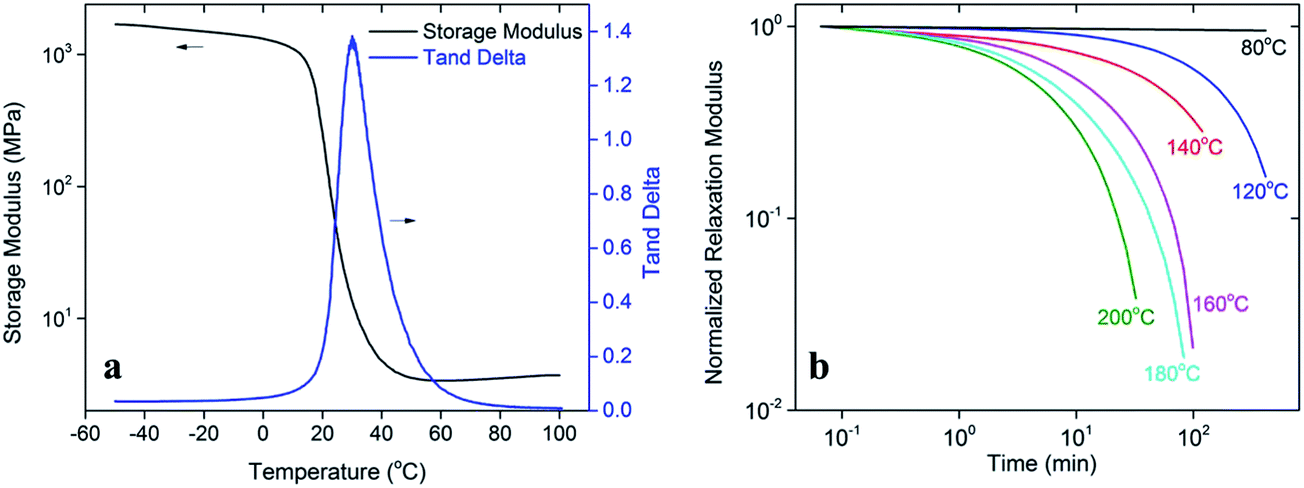 | ||
| Fig. 2 Thermomechanical characterization of the epoxy-acid thermosetting polymer. (a) Glass transition behavior of the prepared thermoset polymer. (b) The stress relaxation behavior of the thermoset polymer at different temperatures. | ||
The time and temperature dependent relaxation modulus of the epoxy-acid thermoset network was also tested on the DMA tester. During the test, the polymer sample with the same dimension mentioned above was firstly preloaded by 1 × 10−3 N force to maintain the straightness. After reaching the testing temperature, it was allowed 30 min for the thermal equilibrium. The specimen was stretched by 1% on the DMA tester and the deformation was maintained during the following tests. The decrease of stress was recorded and the stress relaxation modulus was calculated. Fig. 2b depicts the results of relaxation tests at 6 different temperatures (80 °C, 120 °C, 140 °C, 160 °C, 180 °C and 200 °C) on a double logarithmic plot. Specially, at 180 °C, the bond exchange reaction (BER) is initiated and the normalized relaxation modulus is decreased from 1 to 0.15 within 30 min, indicating an 85% release of the internal stress within the thermoset polymer. But under 80 °C, the material without internal BER involved only behaviors like elastomer with nearly constant relaxation modulus.
The stress relaxation behavior shown in Fig. 2 is attributed to the BERs,1,19 which is schematically shown in Fig. 3. In an exchange reaction, an active group connected to a dangling polymer chain moves around and adds to an existing bond. This will form an intermediate structure, and subsequently reduce to a new bond and a new active group. The collective effect of such exchange reactions is to rearrange the topology of the network structure while preserving the bond density. When mechanical stress is exerted on the network, the network topology rearrangement allows gradual relaxation of the stress. During this process, because the bond density is preserved, this material is insoluble and maintains macroscopic structural integrity even at high temperatures. In addition, when two pieces of this material are brought into contact, molecular bonds can be established across the contacting surfaces due to the exchange reactions, which is utilized in this study to achieve self-healing and interface welding among the polymer particles.
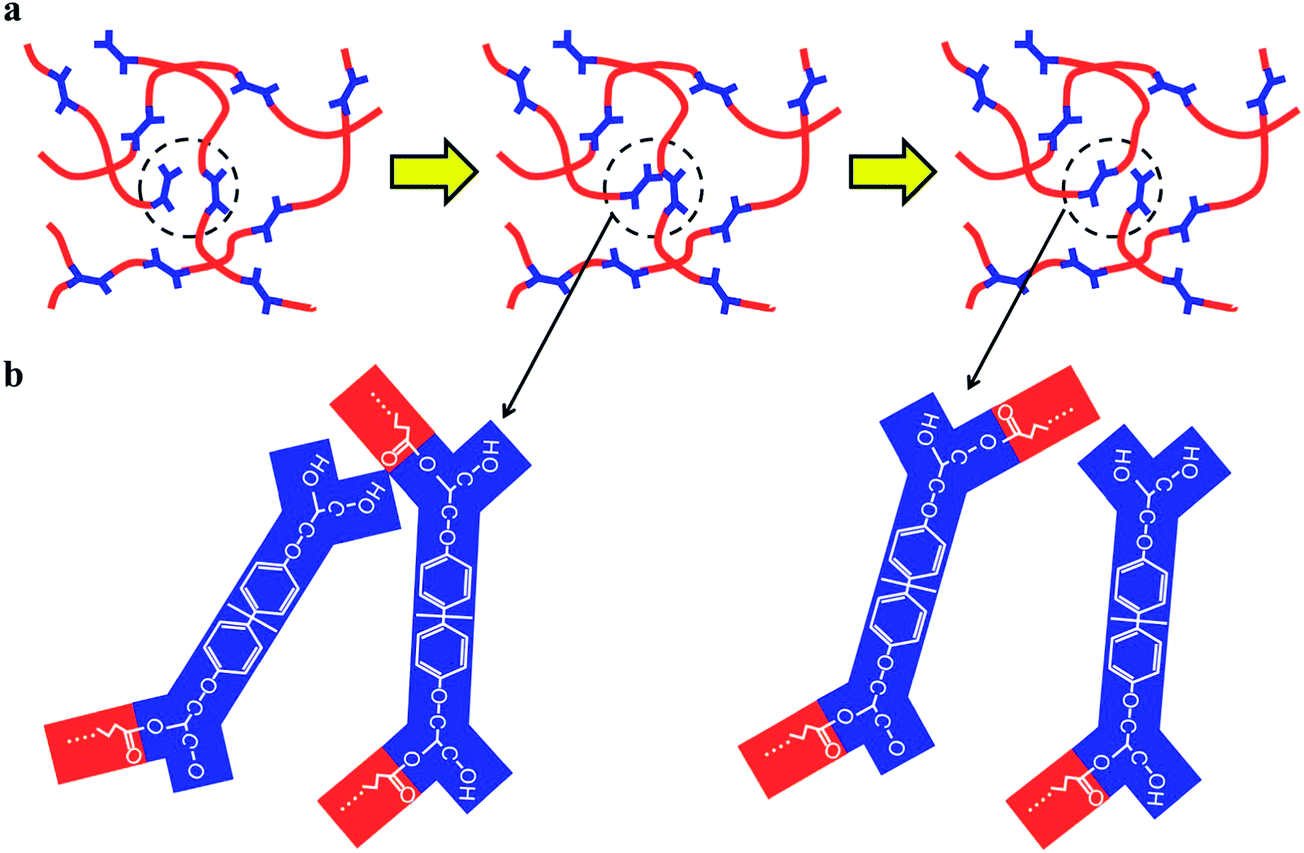 | ||
| Fig. 3 Schematic of the bond exchange reaction: (a) from left to right: before exchange, intermediate state and after exchange.1 (b) Structure change of the active groups during a bond exchange reaction. | ||
2.3 Tension tests
The DMA tester was used to carry out tension tests at room temperature (23 °C). The loading rate was chosen to be a small value (5% per min for all tests) to minimize viscoelastic effects. The samples were trimmed uniformly into the size of 12 mm × 3 mm × 1.1 mm for testing. For each tension test, at least four samples were tested and the average values were reported.2.4 Pulverous sample preparation
The bulk sample was manually abraded by a sandpaper to render the pulverous state. The isolated polymer particles were observed by using an optical microscope and shown in Fig. 4a. From the microscopic morphology, it is seen the particles are irregularly shaped in profile. The particle size and the distribution were analyzed using software Imagine J. As shown in Fig. 4b, the outline of each polymer particle was extracted. The particle diameter is taken to be the diameter of the circle with equal area of each corresponding particle profile. The analysis result is shown in the inset view of Fig. 4b. It is seen that the diameter of most particles (over 77%) are distributed within 25–120 μm.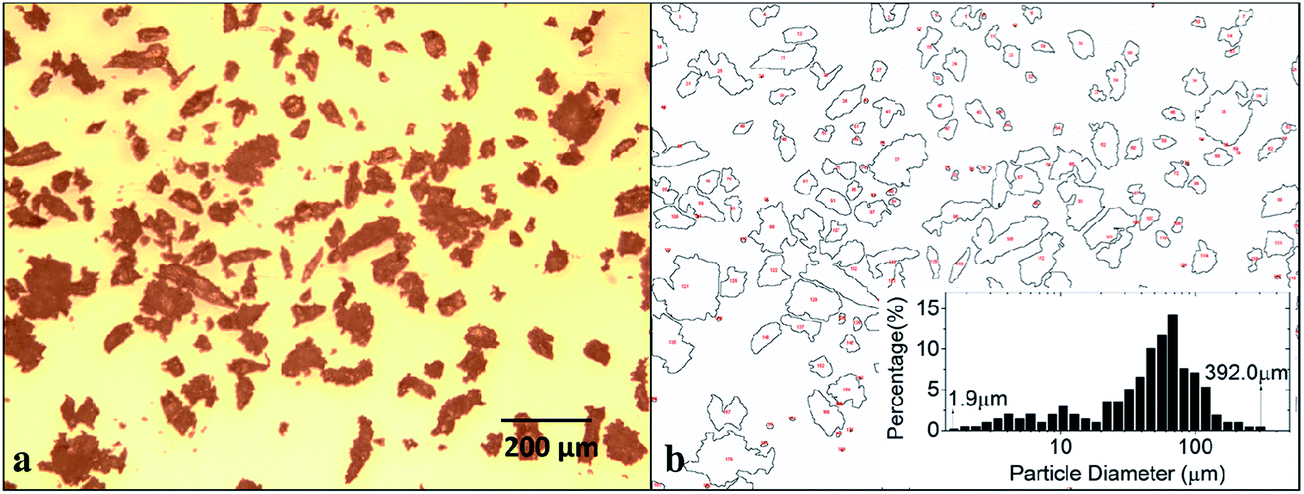 | ||
| Fig. 4 Analysis of particle sizes. (a) Microscopic observation of the isolated polymer particles. (b) Particle size and distribution analysis in Imagine J. | ||
2.5 Experimental setup for reprocessing
An aluminum punch mold (Fig. 5a and b) was machined. Since the rate of BER is thermally sensitive, it is important to precisely control the sample temperature during the tests. In this paper, we adopt the method that was previously developed by the authors.36 The specially designed aluminum punch mold is shown in Fig. 5a, where three hollow slots were machined in the platen to improve the thermal convective properties and temperature distribution during the heating. After filling the polymer powders into the mold (Fig. 5b), it was then transferred into a customized thermal chamber for heating, while the applied pressure was controlled by a universal material testing machine (MTS, Model Insight 10, Eden Prairie, MN, USA). The temperature in the thermal chamber manufactured by Thermcraft (Model LBO, Winston Salem, NC, USA) was controlled with a Eurotherm controller (Model Euro 2404, N. Chesterfield, VA, USA) where a built-in electrical heater with a fan and an externally attached tank of liquid nitrogen provided the heat and cooling. In the work of Leibler et al.,35 a 1 MPa in pressure was introduced to promote the surface welding effect. Indeed, for a polymer in the elastic state, such a pressure would not noticeably affect the thermodynamics and reactions of polymer chains until it reaches to an extremely high value (in GPa ranges) when the free volume of single polymer chain is collapsed.37–39 In this paper, the applied pressure was also chosen to be small enough (90 kPa in maximum) to avoid affecting the transesterification reactions. | ||
| Fig. 5 Experimental setup for the reprocessing and recycling test of thermoset polymer. (a) Aluminum punch mold. (b) Polymer powder is placed into the mold. (c) The heating is conducted in a customized thermal chamber with pressure applied via the MTS machine. | ||
3. Results and discussions
3.1 Time and pressure dependent reprocessing quality of the thermoset polymer
The reprocessing ability of the thermoset polymers is demonstrated in this section, with considerations on the influence of reprocessing time and pressure. A fresh bulk thermoset sample is shown in Fig. 6a for comparison. Fig. 6b shows the appearance of the polymer from the pulverous state to the finished bulk state in a typical case with a 45 kPa pressure. We limit ourselves in this paper to a constant temperature (180 °C) during the operation, and record the heating time immediately after the sample is placed in an oven at 180 °C. Due to the BER, the polymer powder starts surface welding and self-assembly. At 10 min (middle figure of Fig. 6b), isolated pulverous polymer tends to be in a compacted and consolidated form. Further heating of the sample under pressure for 30 min renders an essentially complete welding effect where the sample appears transparent and resembles the fresh sample (shown in the right figure of Fig. 6b), indicating that the interfaces among particles have disappeared.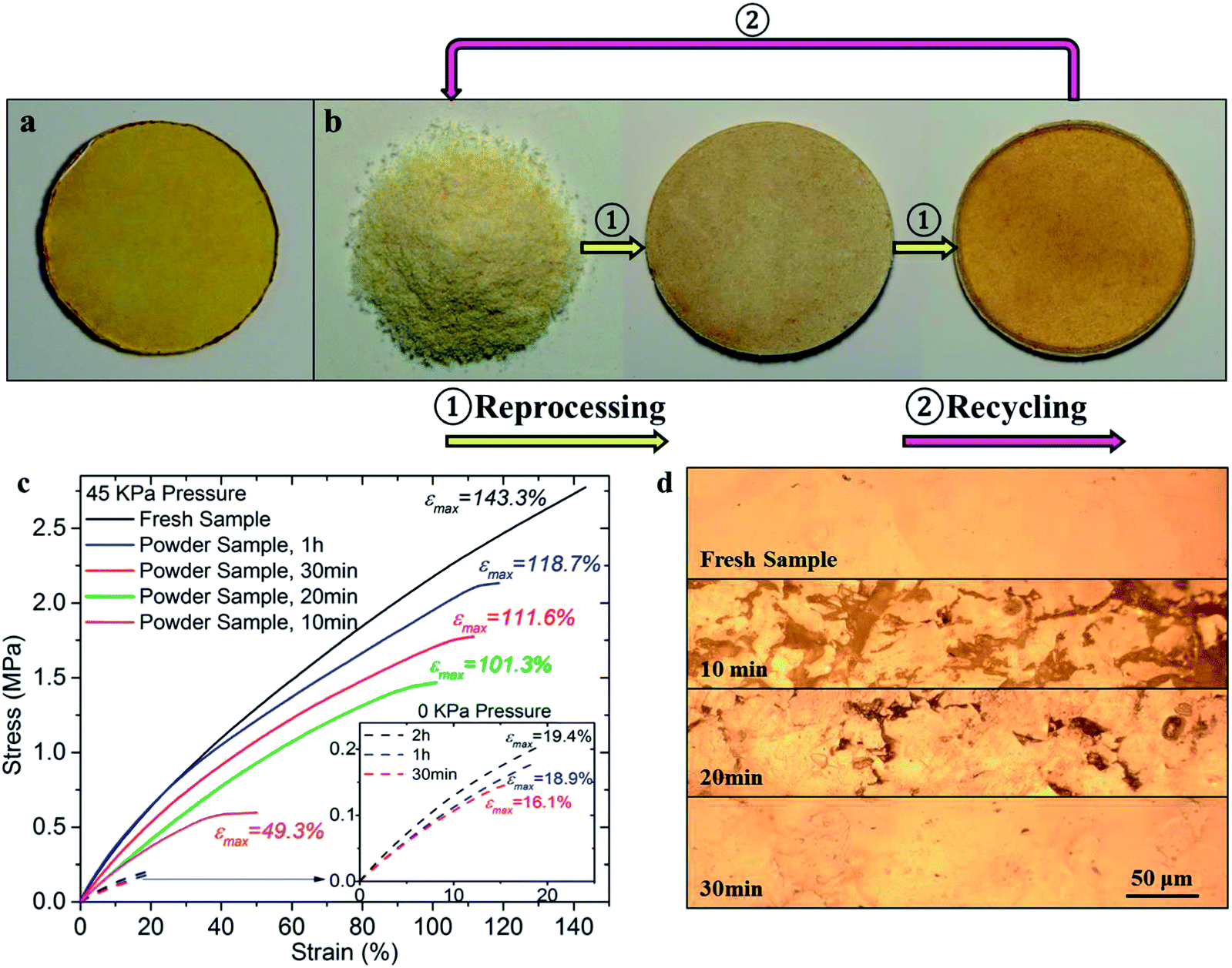 | ||
| Fig. 6 The typical reprocessing and recycling routine for the thermoset polymer and the stress–strain behavior after different reprocessing times. (a) A fresh bulk thermoset polymer. (b) The typical reprocessing and recycling routine (180 °C in temperature and 45 kPa in pressure). From left to right: pulverous polymer after milling, polymer powder after being heated for 10 min and 30 min respectively. (c) Stress–strain curves of the reprocessed sample after being heated for different time and with 45 kPa pressure applied. Note: the fresh sample is also heated at 180 °C for 1 h to normalize the comparison. Inset view: stress–strain curves of powder sample without pressure applied. (d) Surface morphology of both fresh sample and powder samples at different heating time. | ||
The time and pressure dependent welding effect of the pulverous thermoset polymer was quantified by the ultimate stretch (the stretch at the break) and initial elastic modulus (measured over the initial 15% stretch) of the reprocessed samples. The bulk thermoset polymer was reprocessed with different pressures (0 kPa, 2 kPa, 10 kPa, 45 kPa and 90 kPa, respectively) applied during the heating process. The stress–strain behavior of the thermoset samples reprocessed at 45 kPa is shown in Fig. 6c. A fresh polymer sample was also tested after being heated at 180 °C for 1 h to normalize the comparison.† For the pulverous sample heated for 10 min at 180 °C, we observed a relatively low initial elastic modulus of 2.2 MPa. The sample fails quickly with an ultimate stretch of ∼49.3%. With an increase of heating time, both the elastic modulus and ultimate stretch are improved in the reprocessed thermoset samples. With 30 min heating, the initial modulus and the ultimate stretch are 2.9 MPa and 111.6%, respectively (or 76.7% and 77.9% of those in the referential fresh sample, respectively). Such an increase in the material stretch ability is due to the gradually welded interfaces and increased connections among the polymer particles, as shown in the microscopic observations in Fig. 6d. Before the pulverous thermoset is totally welded, numerous voids (the dark regions in Fig. 6d with 10 min and 20 min of heating) exist within the matrix, which can propagate cracks under external loads and compromise stretch ability. At 30 min, the polymer powder is seen to be remolded into a coherent solid essentially resembling the fresh sample. The stress–strain curves in Fig. 6c also reveal that the welding process is largely completed within 30 min when 45 kPa pressure is applied. From 30 min to 1 h, the ultimate stretch of the reprocessed sample is increased by ∼7%. The inset in Fig. 6c also shows the case when there is no pressured applied. The incompact polymer powder can only be partially welded, as evidenced by the very limited stretchability of less than 20% with an initial modulus around 1 MPa, even after being heated for 2 h.
The ultimate stretch and elastic modulus of the reprocessed samples under different reprocessing pressure and time are summarized in Fig. 7a–d, respectively. Generally, both increasing pressure and increasing heating time can improve the final properties of the reprocessed samples. First, for the cases studied in this paper, both ultimate stretch and elastic modulus of the reprocessed samples can achieve the levels of fresh samples, with a clear trend where increasing either heating time or pressure can accelerate this process. Second, increasing pressure is more effective in recovering the elastic modulus than in recovering the ultimate stretch. For example, at 90 kPa, the elastic modulus reaches the fresh sample level within ∼20–30 min (Fig. 7b and d); but within the same time period, the ultimate stretch only reaches 120% stretch, which is ∼80% of that of the fresh samples (Fig. 7a and c). Third, the elongated heating time can recover both the ultimate stretch and the elastic modulus to the fresh sample levels, as long as the pressure is larger than a critical value. For example, even with 2 kPa pressure, both ultimate stretch and elastic modulus almost achieve the fresh sample levels if heating at 720 min (6 h). Certainly, from the manufacturing point of view, heating for 720 min is not energy efficient; increasing the pressure can shorten the heating time and thus help the design of an optimized reprocessing procedure, which deserves future studies.
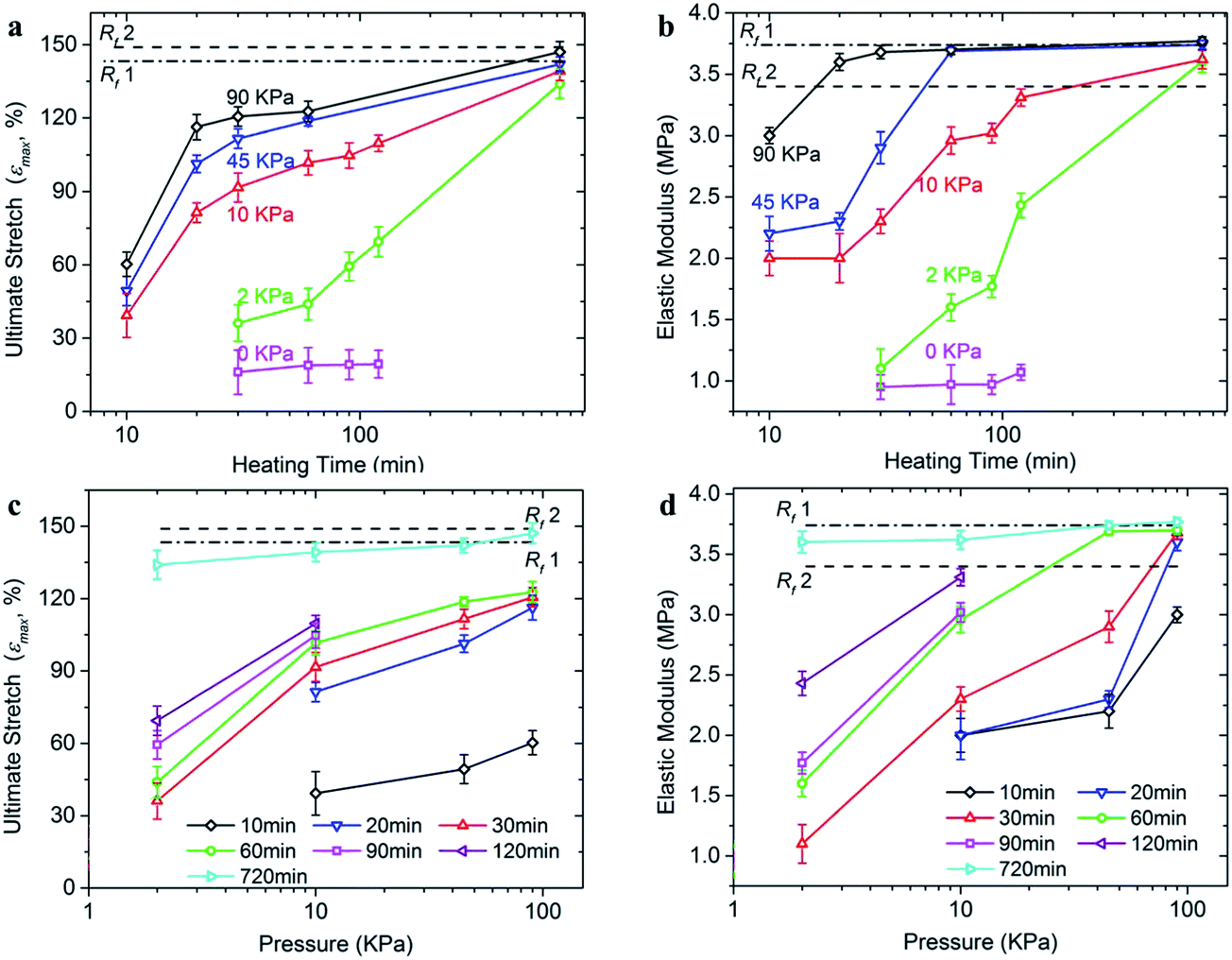 | ||
| Fig. 7 The time and pressure dependent ultimate stretch and initial modulus of the reprocessed thermoset polymer. (a) Ultimate stretch (maximum strain before fracture) plot of the reprocessed sample as a function of heating time. (b) Initial elastic modulus (within the first 15% stretch) as a function of heating time. (c) Ultimate stretch as a function of applied pressure. (d) Initial elastic modulus as a function of applied pressure. Note: Rf1 denotes the referential property of a fresh bulk sample after being heating at 180 °C, while Rf2 denotes that of the fresh sample without any heat treatment. | ||
3.2 Recycling ability of the thermoset polymer
Since the polymer chains in the thermoset are only exchanged but not consumed during the recycling operation, several cycles might be achieved. A set of aluminum molds with digital-shape grooves was designed to test the recycling ability of the thermoset polymer. During the experiments, each reprocessed sample was pulverized into powder by using the same grade of sandpaper, and then recycled to the next generation of the object. During each reprocessing cycle, the applied pressure is set to be 45 kPa. It should be noted that the influence of pressure on the healing efficiency of the pulverous polymer is related to the particle size. When compact the polymer powder at high temperatures, small-sized particles have larger contact surfaces and can quickly close the voids when a small pressure is applied. While for the large-sized particles, the applied pressure should be high enough to squeeze them together. Particles may also deform their shapes accordingly to close the voids before healing on the interfaces, which requires additional reprocessing time. Therefore, it is necessary to examine the possible alternation in particle size and distribution after the powder samples being reprocessed.Fig. 8a shows the microscopic observation of the isolated polymer particles after being reprocessed for four times. The particle size distribution is summarized in Fig. 8b and compared with that used in the first reprocessing routine (shown in Fig. 4). It is seen that after 4 cycles, the particles size is slightly decreased. For the freshly abraded polymer powder, most particles (over 77%) are distributed within 25–120μm, while this range is changed to 16–87 μm after the 4th cycle. This is because when we abrade a polymer sample with reprocessing history, in addition to the newly formed crack surfaces, some of the previously healed surfaces with lower strength may also break, which leads a smaller particle size distribution. However, since the change is not significant, we assume in the following study that the particle size does not vary during the recycling processes, and hence the interplay between particle size and pressure is negligible.
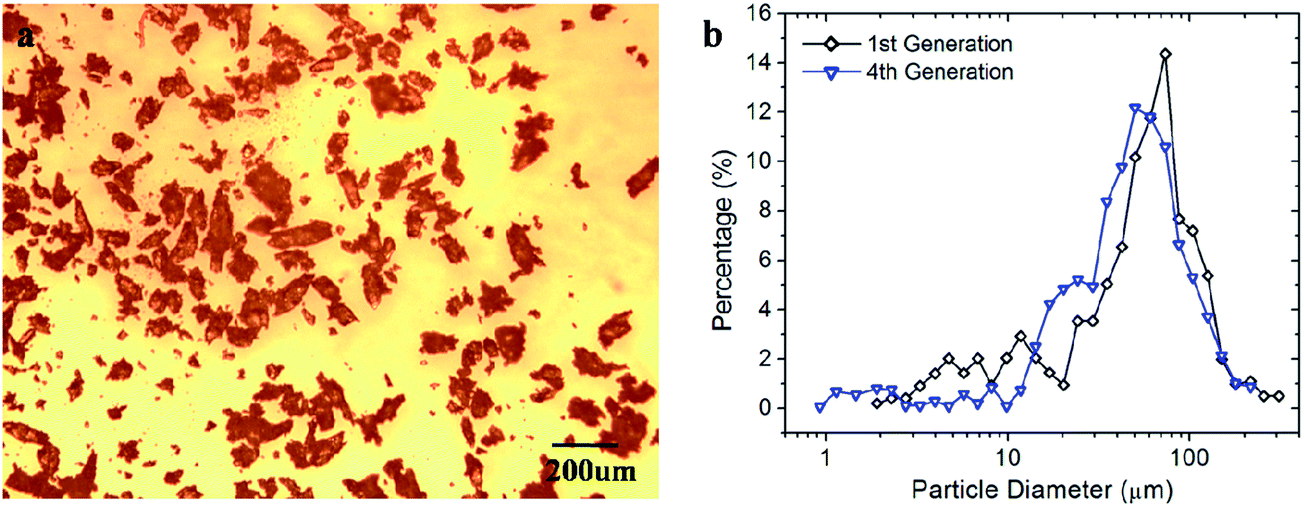 | ||
| Fig. 8 Analysis of particle sizes after being reprocessed for four times. (a) Microscopic observation of the isolated polymer particles. (b) Particle size and distribution analysis in Imagine J. Note: the black line indicated the particle size distribution shown in Fig. 4. | ||
With the same temperature (180 °C) and pressure (45 kPa) applied, all the pulverous thermosets were heated for 30 min. The manufactured sample in each cycle is shown in Fig. 9a, where the digit numbers indicate recycling generation. It is seen that pulverous thermoset polymer possesses a good recycling ability in exhibiting transparent samples in different shapes. The glass transition temperature becomes slightly higher at 35.7 °C after the 4th generation of reprocessing, as compared to 30.1 °C of the fresh sample. Besides, the rubber modulus (as seen in the storage modulus curve after 60 °C) is decreased after the 4th reprocessing. This is because during the pulverization of the polymer material, some permanently cross-linked strands of the polymer network are broken on the fracture surfaces34 and cannot be recovered during the subsequent healing process, which leads to a decreased crosslinking density and modulus in the rubber state. The stress–strain curves are shown in Fig. 9c. It is remarkable to note that even after four times of reprocessing that sample still can achieve an ultimate stretch of 91.8% stretch. Fig. 9d shows the evolution of the ultimate stretch and elastic modulus as a function of the number of recycling. Another set of recycling test are also presented in the figure where the heating time in each cycle is 2 h. It is clear that although the properties of recycled material decay over the number of recycling, they stay in a reasonably good range. Also, the amount of decay decreases as the number of recycling increases, indicating more cycles of recycling is possible. In addition, the elongated heating time will improve the properties of recycled material significantly. As shown in Fig. 9c, a 6 hour heating of the 4th generation sample leads to the ultimate stretch of 137.7%, which is comparable to that of the fresh sample without recycling history.
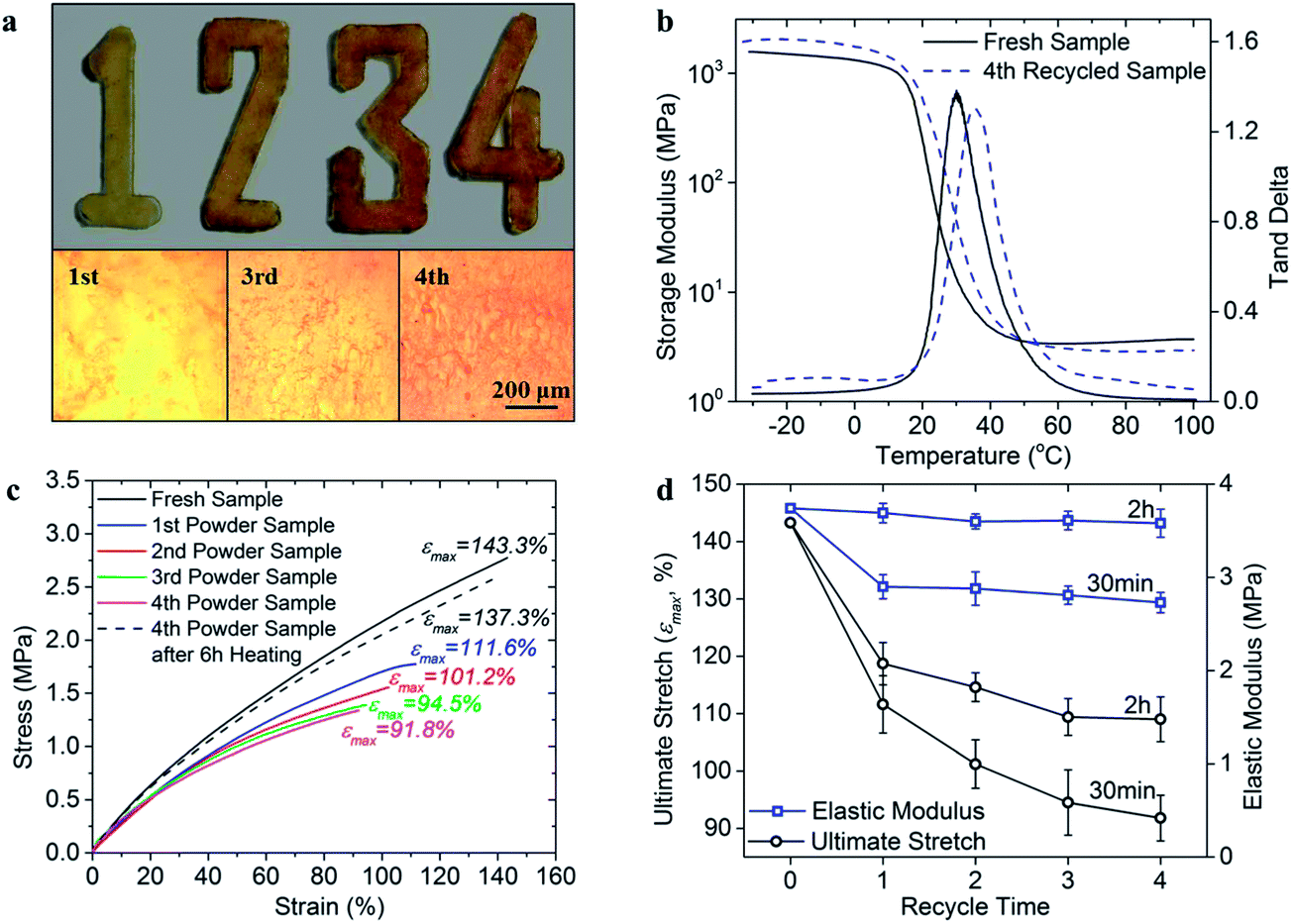 | ||
| Fig. 9 The recycling ability of the thermoset polymer. (a) Top view: sample manufactured by using polymer powder after being recycled for multiple times. Digit numbers indicate the recycle generation. Bottom view: the surface roughness observation on the 1st, 3rd and 4th recycled samples (b). Stress–strain behavior of the polymer powder sample after being recycled for different times. The heating time is 30 min in each cycle. (c) Glass transition behavior of the fresh sample and sample recycled for four times. (d) Ultimate stretch and initial elastic modulus plots as a function of recycle generations. Two sets of experiment are presented in the figure where the heating time in each reprocessing cycle is 30 min and 2 h respectively. | ||
3.3 Repair bulk polymers by using the powder medium
In most engineering applications, the structural thermoset polymers are susceptible to damage in the form of cracks, fractures and abrasions. Isolated interfaces might not be easily brought into contact again for welding. Structural patching using a bulk polymer is also not practical as it will potentially change the dimensions of polymer components. In view of these facts, the use of a pulverous thermoset as a repair medium is potentially a better choice. We therefore extended our study on the reprocessing and recycling ability of thermoset polymers to this particular engineering application, namely utilizing polymer powder to reconnect the bulk material. As shown in Fig. 10a, two strips of the bulk thermoset polymer were placed on a glass slide, with a large gap in between. Then the gap was filled with the polymer powder and sandwiched between two glass slides (Fig. 10b). Utilizing the standard heating condition (30 min at 180 °C with a pressure of 45 kPa), the two polymer pieces were reconnected as shown in Fig. 10c. The sample was further trimmed into a rectangular shape (Fig. 10d) for tension tests.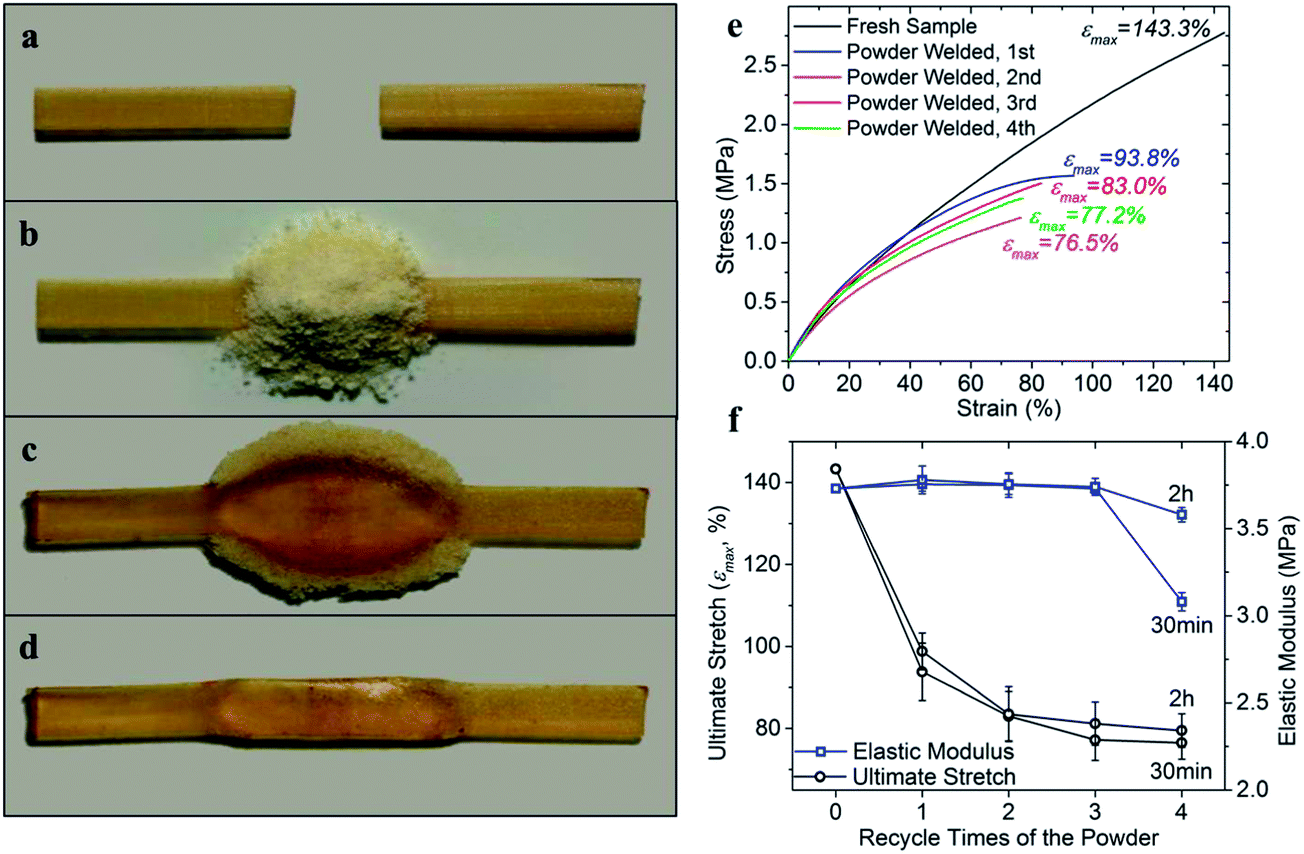 | ||
| Fig. 10 Welding isolated bulk thermoset strip by using the pulverous polymer. (a) Two separated pieces of the thermoset polymer are placed on a glass slide. (b) The gap is filled with the polymer powder. (c) The separated thermoset polymer is healed and assembled together after being heated at 180 °C for 30 min (45 kPa in pressure). (d) The sample is further trimmed into rectangular shape for tension test. (e) The stress–strain curves of the welded sample by using polymer powder recycled for different times. The heating time is 30 min in each test. (f) Ultimate stretch and initial elastic modulus plots as a function of recycle times of the applied polymer powder. Two sets of experiment are presented in the figure where the heating time in each test is 30 min and 2 h respectively. | ||
The welding procedure mentioned above was also repeated with thermoset powder with recycling history (up to four generations), while the bulk polymer strips were always fresh samples. The stress–strain curves are then plotted in Fig. 10e. It is seen that except for the 4th recycled powder healed sample, the initial elastic moduli of the other three samples are typically close to the fresh sample, while the ultimate stretch is lower than that of the reprocessed samples studied above. All the failures in the tested samples are located on the welded interface between the bulk polymer and the powder assembles. By using the powder that has been reprocessed four times, the ultimate stretch is decreased to 76.5% stretch. We conducted another set of experiments with 2 h of heating time in each operation, and the mechanical properties are summarized in Fig. 10f for comparison. Since the polymer materials were sandwiched between glass slides with pressure, increasing heating time would not significantly improve the material's stress-bearing ability, which is limited by in-plane pressure during the operation, and subsequently the interface strength between the bulk polymer and powder assembly.
4. Conclusions
We demonstrated that after being grinded into a pulverous state of micrometer size particles and recycled multiple times, the thermoset polymer with exchangeable bonds could be assembled again into a coherent solid with mechanical properties comparable to the fresh bulk polymer. Considering the efficient and robust welding effect among polymer particles, as well as the maintained merits of thermoset polymer as stress-bearing materials during the healing operation, this strategy is suitable for repair of polymer structures in service, as well as reprocessing and recycling of thermoset waste in engineering applications. Although the material studied in this paper is limited to an epoxy-acid network, one can extend the concept of bond exchange and particle recycling to other thermoset polymers via the incorporation of an appropriate catalytic bond exchange system.Acknowledgements
The authors gratefully acknowledge the financial support of a NSF grant CMMI-1404627, Program Manager Mary Toney. MLD acknowledges support from the Singapore Ministry of Education.References
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968 CrossRef CAS PubMed
.
- J. I. Kim, S. H. Ryu and Y. W. Chang, Mechanical and dynamic mechanical properties of waste rubber powder/HDPE composite, J. Appl. Polym. Sci., 2000, 77(12), 2595–2602 CrossRef CAS
- A. M. Shanmugharaj, J. K. Kim and S. H. Ryu, UV surface modification of waste tire powder: Characterization and its influence on the properties of polypropylene/waste powder composites, Polym. Test., 2005, 24(6), 739–745 CrossRef CAS
- A. M. Shanmugharaj, J. K. Kim and S. H. Ryu, Modification of rubber powder with peroxide and properties of polypropylene/rubber composites, J. Appl. Polym. Sci., 2007, 104(4), 2237–2243 CrossRef CAS
- A. A. Babar, et al., Performance of High-Permittivity Ceramic–Polymer Composite as a Substrate for UHF RFID Tag Antennas, Int. J. Antenn. Propag., 2012, 1–8 CrossRef
- F. Hernandez-Olivares, et al., Static and dynamic behaviour of recycled tyre rubber-filled concrete, Cem. Concr. Res., 2002, 32(10), 1587–1596 CrossRef CAS
- C. Albano, et al., Influence of scrap rubber addition to Portland I concrete composites: Destructive and non-destructive testing, Compos. Struct., 2005, 71(3–4), 439–446 CrossRef
- R. Siddique and T. R. Naik, Properties of concrete containing scrap-tire rubber – an overview, Waste Manage., 2004, 24(6), 563–569 CrossRef CAS PubMed
- G. Q. Li, et al., Development of waste tire modified concrete, Cem. Concr. Res., 2004, 34(12), 2283–2289 CrossRef CAS
- J. O. Outwater and D. J. Gerry, On Fracture Energy, Rehealing Velocity and Refracture Energy of Cast Epoxy Resin, J. Adhes., 1969, 1, 290–298 CrossRef
- M. A. M. Rahmathullah and G. R. Palmese, Crack-Healing Behavior of Epoxy-Amine Thermosets, J. Appl. Polym. Sci., 2009, 113(4), 2191–2201 CrossRef CAS
- M. Yamaguchi, S. Ono and M. Terano, Self-repairing property of polymer network with dangling chains, Mater. Lett., 2007, 61(6), 1396–1399 CrossRef CAS
- S. A. Hayes, et al., A self-healing thermosetting composite material, Composites, Part A, 2007, 38(4), 1116–1120 CrossRef
- X. F. Luo, et al., A Thermoplastic/Thermoset Blend Exhibiting Thermal Mending and Reversible Adhesion., ACS Appl. Mater. Interfaces, 2009, 1(3), 612–620 CAS
- A. M. Peterson, et al., Investigation of interpenetrating polymer networks for self-healing applications, Compos. Sci. Technol., 2012, 72(2), 330–336 CrossRef CAS
- S. J. Rowan, et al., Dynamic covalent chemistry, Angew. Chem., Int. Ed., 2002, 41(6), 898–952 CrossRef
- J. M. Lehn, Dynamers: dynamic molecular and supramolecular polymers, Prog. Polym. Sci., 2005, 30(8–9), 814–831 CrossRef CAS
- T. Maeda, H. Otsuka and A. Takahara, Dynamic covalent polymers: Reorganizable polymers with dynamic covalent bonds, Prog. Polym. Sci., 2009, 34(7), 581–604 CrossRef CAS
- C. J. Kloxin, et al., Covalent Adaptable Networks (CANS): A Unique Paradigm in Cross-Linked Polymers, Macromolecules, 2010, 43(6), 2643–2653 CrossRef CAS PubMed
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Using the dynamic bond to access macroscopically responsive structurally dynamic polymers, Nat. Mater., 2011, 10(1), 14–27 CrossRef CAS PubMed
- M. S. Green and A. V. Tobolsky, A New Approach to the Theory of Relaxing Polymeric Media, J. Appl. Phys., 1946, 17(5), 407–407 Search PubMed
- M. Rubinstein and A. N. Semenov, Thermoreversible gelation in solutions of associating polymers. 2. Linear dynamics, Macromolecules, 1998, 31(4), 1386–1397 CrossRef CAS
- L. Leibler, M. Rubinstein and R. H. Colby, Dynamics of Telechelic Ionomers – Can Polymers Diffuse Large Distances without Relaxing Stress, J. Phys. II, 1993, 3(10), 1581–1590 CrossRef CAS
- H. Otsuka, et al., Polymer scrambling: Macromolecular radical crossover reaction between
the main chains of alkoxyamine-based dynamic covalent polymers, J. Am. Chem. Soc., 2003, 125(14), 4064–4065 CrossRef CAS PubMed
- T. F. Scott, et al., Photoinduced plasticity in cross-linked polymers, Science, 2005, 308(5728), 1615–1617 CrossRef CAS PubMed
- R. Nicolay, et al., Responsive Gels Based on a Dynamic Covalent Trithiocarbonate Cross-Linker, Macromolecules, 2010, 43(9), 4355–4361 CrossRef CAS
- B. Ghosh and M. W. Urban, Self-Repairing Oxetane-Substituted Chitosan Polyurethane Networks, Science, 2009, 323(5920), 1458–1460 CrossRef CAS PubMed
- K. Imato, et al., Self-Healing of Chemical Gels Cross-Linked by Diarylbibenzofuranone-Based Trigger-Free Dynamic Covalent Bonds at Room Temperature, Angew. Chem., Int. Ed., 2012, 51(5), 1138–1142 CrossRef CAS PubMed
- B. J. Adzima, et al., Rheological and Chemical Analysis of Reverse Gelation in a Covalently Cross-Linked Diels-Alder Polymer Network, Macromolecules, 2008, 41(23), 9112–9117 CrossRef CAS PubMed
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Thermally Self-Healing Polymeric Materials: The Next Step to Recycling Thermoset Polymers?, Macromolecules, 2009, 42(6), 1906–1912 CrossRef CAS
- R. Long, H. J. Qi and M. L. Dunn, Modeling the mechanics of covalently-adaptable polymer networks with temperature-dependent bond exchange reactions, Soft Matter, 2013, 9(15), 4083–4096 RSC
- Y. Lu and Z. Guan, Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of Strong Carbon–Carbon Double Bonds, J. Am. Chem. Soc., 2012, 134(34), 14226–14231 CrossRef CAS PubMed
- Y. Lu, et al., Making Insoluble Polymer Networks Malleable via Olefin Metathesis, J. Am. Chem. Soc., 2012, 134(20), 8424–8427 CrossRef CAS PubMed
- E. B. Stukalin, et al., Self-Healing of Unentangled Polymer Networks with Reversible Bonds, Macromolecules, 2013, 46(18), 7525–7541 CrossRef CAS PubMed
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets, J. Am. Chem. Soc., 2012, 134(18), 7664–7667 CrossRef CAS PubMed
- K. K. Westbrook, et al., Improved testing system for thermomechanical experiments on polymers using uniaxial compression equipment, Polym. Test., 2010, 29(4), 503–512 CrossRef CAS
- A. S. Benjamin, et al., Acoustic properties of Kel F-800 copolymer up to 85 GPa, J. Chem. Phys., 2012, 137(1), 014514 CrossRef PubMed
- L. L. Stevens, et al., Brillouin-scattering determination of the acoustic properties and their pressure dependence for three polymeric elastomers, J. Chem. Phys., 2007, 127(10), 104906 CrossRef PubMed
- Z. A. Dreger, et al., Effect of high pressure on acoustic properties of several polymers: Use of impulsive stimulated light scattering method, J. Appl. Phys., 2011, 109(8), 083507 CrossRef
- L. I. Komarova, et al., A Study of the Infrared Absorption Spectra of Polymers based on Bisphenols and Formaldehyde during Hardening adn Thermal Degradation, Zh. Prikl. Spektrosk., 1967, 7(4), 611–614 CAS
- C. L. Beyler and M. M. Hirschler, Thermal Decomposition of Polymers, in SFPE Handbook of Fire Protection Engineering, ed. P. J. DiNenno, NFPA, 3rd edn, Quincy, 2001 Search PubMed
Footnote |
| † After being heated for 1 h at 180 °C, which is ∼150 °C above the glass transition temperature of the thermoset polymer, a slight thermal induced hardening40,41 was observed in the material. The stretch ability was decreased by 3.8% (from 149.0% to 143.3%) and the initial modulus within the 15% stretch was increased by 5.7% (3.5 MPa to 3.7 MPa). Both of them are presented as reference values in Fig. 7a–d. |
| This journal is © The Royal Society of Chemistry 2014 |