
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the impact of different liposomal formulations on the plasma protein corona composition might give hints on the targeting capability of nanoparticles†
Esther
Imperlini‡
a,
Luisa
Di Marzio‡
 b,
Armando
Cevenini
cd,
Michele
Costanzo
cd,
Nicola d'Avanzo
ef,
Massimo
Fresta
fg,
Stefania
Orrù
*dh,
Christian
Celia
*bijk and
Francesco
Salvatore
*cd
b,
Armando
Cevenini
cd,
Michele
Costanzo
cd,
Nicola d'Avanzo
ef,
Massimo
Fresta
fg,
Stefania
Orrù
*dh,
Christian
Celia
*bijk and
Francesco
Salvatore
*cd
aDepartment for Innovation in Biological, Agrofood and Forest Systems, University of Tuscia, Viterbo, 01100, Italy
bDepartment of Pharmacy, University of Chieti – Pescara “G. d’Annunzio”, Via dei Vestini 31, 66100, Chieti, Italy. E-mail: c.celia@unich.it; Tel: +39 0871 3554711
cDepartment of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, Naples, 80131, Italy. E-mail: salvator@unina.it; Tel: +39 3356069177
dCEINGE-Biotecnologie Avanzate Franco Salvatore, Naples, 80145, Italy. E-mail: stefania.orru@uniparthenope.it; Tel: +39 081 3737880
eDepartment of Experimental and Clinical Medicine, University of Catanzaro “Magna Graecia”, Viale “S. Venuta”, 88100, Catanzaro, Italy
fDepartment of Experimental and Clinical Medicine, Research Center “ProHealth Translational Hub”, “Magna Graecia” University of Catanzaro, Campus Universitario “S. Venuta”—Building of BioSciences, Viale S. Venuta, 88100, Catanzaro, Italy
gDepartment of Health Sciences, University of Catanzaro “Magna Graecia”, Viale “S. Venuta”, 88100, Catanzaro, Italy
hDepartment of Medical, Movement and Wellness Sciences, University of Naples Parthenope, Naples, 80133, Italy
iLithuanian University of Health Sciences, Laboratory of Drug Targets Histopathology, Institute of Cardiology, A. Mickeviciaus g. 9, LT-44307, Kaunas, Lithuania
jInstitute of Nanochemistry and Nanobiology, School of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, China
kUdA-TechLab, Research Center, University of Chieti-Pescara “G. D'Annunzio”, 66100, Chieti, Italy
First published on 2nd July 2024
Abstract
Nanoparticles (NPs) interact with biological fluids after being injected into the bloodstream. The interactions between NPs and plasma proteins at the nano-bio interface affect their biopharmaceutical properties and distribution in the organ and tissues due to protein corona (PrC) composition, and in turn, modification of the resulting targeting capability. Moreover, lipid and polymer NPs, at their interface, affect the composition of PrC and the relative adsorption and abundance of specific proteins. To investigate this latter aspect, we synthesized and characterized different liposomal formulations (LFs) with lipids and polymer-conjugated lipids at different molar ratios, having different sizes, size distributions and surface charges. The PrC composition of various designed LFs was evaluated ex vivo in human plasma by label-free quantitative proteomics. We also correlated the relative abundance of identified specific proteins in the coronas of the different LFs with their physicochemical properties (size, PDI, zeta potential). The evaluation of outputs from different bioinformatic tools discovered protein clusters allowing to highlight: (i) common as well as the unique species for the various formulations; (ii) correlation between each identified PrC and the physicochemical properties of LFs; (iii) some preferential binding determined by physicochemical properties of LFs; (iv) occurrence of formulation-specific protein patterns in PrC. Investigating specific clusters in PrC will help decode the multivalent roles of the protein pattern components in the drug delivery process, taking advantage of the bio-nanoscale recognition and identification for significant advances in nanomedicine.
1. Introduction
Liposomes are versatile, biocompatible, biodegradable, low toxic and poorly immunogenic drug delivery systems, which are able to encapsulate both hydrophilic and lipophilic compounds and enhance tumor targeting.1,2 Lipid composition, average size, surface charge and shape affect biopharmaceutical properties of liposomes as well as their biodistribution and targeting.3–7 Liposomal formulations (LFs) can improve the safety and efficiency of therapeutics by (i) reducing systemic side effects, (ii) preventing early degradation of payloads, and (iii) targeting specific cells and tissues.8,9The incorporation of polyethylene glycol (PEG) polymers into the liposome bilayer (PEGylation) or the modification of the surface properties with other polymeric macromolecules (or surfactants) extend liposomal blood circulation and decrease the clearance mediated by the mononuclear phagocyte system (MPS), hence avoiding their accumulation in the liver and spleen.10
MPS activation is triggered by the opsonization process, which is involved in the neutralization of non-self-antigens11 and is able to reduce liposome biodistribution and circulation half-life.12 In this scenario, PEGylation of liposomes has long been considered an efficient anti-opsonization strategy to improve long circulation and passive targeting of nanoparticles (NPs).13,14
PEG has been widely used to improve the stability and stealth properties of liposomes as well as other lipid-based nanocarriers. To date, PEG with a molecular weight of 2 kDa is currently used to make liposomes that are used in the clinic, such as Doxil® and Onivyde®, and more recently siRNA/mRNA-loaded lipid nanoparticles, such as Onpattro® and Comirnaty®.15
However, recent studies demonstrated that PEG polymers, at medium and high molecular weights, conjugated with therapeutics, can activate complement cascades and the relative immune system mediators after multiple systemic injections;16 on the other hand, they cannot completely prevent the adsorption of plasma proteins on the liposome surface, thus allowing the formation of a coating called protein corona (PrC).12,17,18
PrC formation depends on the high free energy of the NP surface, which by interacting with biological fluid components, is coated by various macromolecules, like proteins but also lipids.18,19 The composition of PrC depends on the intrinsic NP properties, such as the material, size, shape and surface charge.18,20 The amount of adsorbed proteins is positively correlated with NP size and depends on the curvature of NPs; in fact, the largest particles have a smaller surface bend that allows the proteins to interact more freely with a greater surface area.21
PrC formation determines a new biological complex, namely the PrC-NP, with a dynamic structure ruled by affinity interactions (proteins/NP surface, protein/protein).22 At the nano-bio interface, plasma proteins are continuously desorbed and adsorbed on the NP surface according to the “Vroman effect”.23 In particular, the most tightly bound layer of proteins, the so-called “hard corona”,24 seems to play a fundamental role in the biological interactions of NPs and in the recognition of PrC-NP complexes with the target tissue.25 In fact, PrC affects the biodistribution of NPs since the plasma proteins show different affinities for different tissues, thus representing an example of active natural targeting.22,26 As a consequence, PrC is also instrumental in the pharmacokinetics of NPs and their delivered payloads.11
PrC composition plays a key role in the stability of the NPs by inducing or preventing their aggregation, together with the NP surface charge and steric hindrances of interacting NPs.27,28 This knowledge is pivotal to rationally design NPs of different charges and/or steric hindrances whose specific PrC composition could determine, in turn, a specific distribution in vivo.29
Macromolecule-conjugated lipids like ganglioside-monosialic acid (GM1), with stealth properties and low immune responses at specific molar ratios and molecular weight,30,31 allow a prolonged circulation of NPs and prevent their MPS uptake and clearance from the bloodstream equal to PEG-phospholipids.32 Sialic acid residues of GM1 and the oligosaccharide residues conjugated to ceramide lipids have different structures and flexibility33 compared to polyethylene oxide units of PEG. Differences in the backbone structure of macromolecule-conjugated lipids provide different binding affinity and interactions with circulating proteins that are adsorbed on the NP surface, thereby changing its features although the stealth properties are maintained.34 These properties further affect the circulation time of NPs after systemic injection, and thus, their clinical outcomes.35
In this scenario, LFs represent a versatile class of drug delivery systems able to address new therapeutic opportunities, as mentioned above. Hence, we designed and characterized five different LFs containing: (i) anionic phospholipids (LF1 and LF2); (ii) anionic phospholipids and PEGylated lipids (LF3 and LF4); (iii) anionic phospholipids and GM1 glycolipids (LF5). Each of them was extruded at two different sizes, thus resulting in ten different LFs to be investigated. After incubation in human plasma (HuP), we profiled their PrC composition to study the impact of the variety of chemical and biological moieties at the nano-bio interfaces of NPs. This formulation/size-based multiplexed mapping allowed us to identify specific protein patterns possibly instrumental for interaction with diverse biological tissues.36,37 The combined data from PrC profiling and physicochemical characterization of liposomes can be used as an effective approach to obtain NPs with optimal properties for their specific purpose.
2. Methods
2.1. Chemical and reagents
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine monohydrate (DPPC), 1,2-dipalmitoyl-sn-glycero-3(phospho-L-serine), sodium salt (DPPS), 1,2-dimyristoyl-sn-glycero-3-phospho-(1-rac-glycerol), sodium salt (DMPG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-750], ammonium salt (DSPEmPEG750), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-5000], ammonium salt (DSPEmPEG5000), ganglioside, brain, ovine–sodium salt (GM1) and cholesterol (Chol) were obtained from Avanti® Polar Lipids (Alabaster, AL, USA).N-2-Hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES), Whatman filters, DL-dithiothreitol minimum 99% titration, modified trypsin, Laemmli buffer, dithiothreitol (DTT), iodoacetamide (IAA), and ammonium bicarbonate (AMBIC) were purchased from Sigma Aldrich (Milano, Italy). Gelcode® blue stain reagent was obtained from Thermo Fisher Scientific (Waltham, MA, US). Tris–HCl, sodium dodecyl sulfate (SDS), acrylamide, bis-acrylamide, ammonium persulfate (APS), tetramethylethylenediamine (TEMED), tris/glycine/SDS (TGS) and tris/glycine (TG) buffers were obtained from Bio-Rad Laboratories Srl (Segrate, Italy). HPLC grade acetonitrile (ACN) and formic acid were purchased from J. T. Baker (Deventer, Netherlands).
2.2. Liposome preparation
Liposomal formulations (LFs) were prepared by using the thin layer evaporation method and extrusion technique.38 Briefly, lipids (different molar ratios of DPPC, DPPS, DMPG, DSPE-mPEG750, DSPE-mPEG5000, GM1 and Chol, as reported in Table 1) were dissolved in chloroform/methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Then, the organic solvent was removed by using a rotary evaporator (model Laborota 4000 type Heidolph, Delchimica, Naples, Italy) at 50 °C to remove any solvent residues, and the lipid film was left under the hood overnight at room temperature. The resulting multilamellar liposomes were obtained by hydrating the dried lipid film with HEPES (10 mM, pH = 7.4) and by alternating three cycles of heating at 60 °C and of continuous stirring for 3 min. LFs were extruded by using a Lipex ExtruderTM device (Northern Lipids Inc., BC, Canada) with polycarbonate membranes (Whatman Inc., NJ, USA) having pore sizes of 200 nm and 100 nm. Large LFs (LF-L) and small LFs (LF-S) were collected and used for further experiments.
:1, v/v). Then, the organic solvent was removed by using a rotary evaporator (model Laborota 4000 type Heidolph, Delchimica, Naples, Italy) at 50 °C to remove any solvent residues, and the lipid film was left under the hood overnight at room temperature. The resulting multilamellar liposomes were obtained by hydrating the dried lipid film with HEPES (10 mM, pH = 7.4) and by alternating three cycles of heating at 60 °C and of continuous stirring for 3 min. LFs were extruded by using a Lipex ExtruderTM device (Northern Lipids Inc., BC, Canada) with polycarbonate membranes (Whatman Inc., NJ, USA) having pore sizes of 200 nm and 100 nm. Large LFs (LF-L) and small LFs (LF-S) were collected and used for further experiments.
| LFs | DPPCa | DPPSb | DMPGc | DSPE-mPEG750d | DSPE-mPEG5000e | GM1f | CHOLg |
|---|---|---|---|---|---|---|---|
| a 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine monohydrate. b 1,2-Dipalmitoyl-sn-glycero-3(phospho-L-serine). c 1,2-Dimyristoyl-sn-glycero-3-phospho-(1-rac-glycerol). d 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-750]; 750 is low-MW PEG. e 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene-glycol)-5000]; 5000 is high-MW PEG. f Monosialotetrahexosylganglioside. g Cholesterol. | |||||||
| LF1 | 6 | 1 | — | — | — | — | 3 |
| LF2 | 6 | — | 1 | — | — | — | 3 |
| LF3 | 6 | — | — | 1 | — | — | 3 |
| LF4 | 6 | — | — | — | 1 | — | 3 |
| LF5 | 6 | — | — | — | — | 0.5 | 3.5 |
2.3. Physicochemical characterization of liposomes
The average size (nm) (hydrodynamic radius) and polydispersity index (PDI) of LFs were measured by using dynamic light scattering (DLS).39 Zeta potential (mV) was determined as electrophoretic mobility by laser doppler electrophoresis (LDE). To avoid multi-scattering phenomena, samples were diluted (1:30 v/v) in HEPES (10 mM, pH = 7.4) and then analyzed by using Zetasizer Nano ZS90 (Malvern Panalytical, United Kingdom). Measurements were performed in three different replicates; each of them was repeated at least five times, and data were reported as average ± standard deviation (SD).
2.4. Human plasma preparations and incubation with liposomes
Human plasma (HuP) samples were obtained at CEINGE – Biotecnologie Avanzate Franco Salvatore (Naples, Italy) from healthy adult volunteers (30–50 years) in accordance with the relevant laws and guidelines existing in Italy with the declaration of Helsinki. Informed consents were obtained from human participants (blood plasma donors) of this study, and the experiments were performed in accordance with the guidelines of the declaration of Helsinki and approved by the Ethics Committee of the University of Naples Federico II (approval number 318/20).Blood samples were collected by using the BDTMP100 Blood Collection System in the presence of K2EDTA anticoagulant and protease inhibitor cocktail. HuP samples were prepared as previously reported.18 Briefly, after clot formation, HuP was collected by centrifugation at 1000g for 5 min.
To reduce inter-individual variations, we pooled HuP samples from the different donors and stored their aliquots at −80 °C. Afterwards, HuP aliquots were thawed at 4 °C and warmed at room temperature.
The HuP samples were incubated (1:1, v/v) with LFs (10 mg ml−1) at 37 °C for 1 h. After incubation, PrC-NP complexes were isolated by centrifugation at 15000g for 10 min and washed with PBS twice. At each washing step, samples were transferred into new Protein LoBind tubes and then resuspended in Laemmli buffer.
2.5. Protein corona electrophoresis and in-gel digestion
Proteins suspended in Laemmli buffer were first denatured at 95 °C for 5 min and then fractionated by 12% (v/v) SDS-PAGE. The protein bands were stained using Gelcode® blue stain reagent (Thermo Fisher Scientific, USA) according to the manufacturing procedures. Molecular weights (MW) of protein bands were estimated by using Precision Plus All Blue protein standards (Bio-Rad). The gel image was acquired using the scanner GS-800 Calibrated Densitometer (Bio-Rad).The whole gel lanes were manually cut into 2 mm gel slices. Each slice was washed with ACN and 50 mM AMBIC as previously reported.40 The protein bands were then reduced with 10 mM DTT in 50 mM AMBIC at 56 °C for 45 min and then alkylated in 55 mM IAA in 50 mM AMBIC at room temperature, in the dark for 30 min. After washing with ACN and 50 mM AMBIC, each slice was incubated with 10 ng μL−1 trypsin at 4 °C as previously described.41 Peptide mixtures were extracted and resuspended in 0.2% (v/v) formic acid.
2.6. LC-MS/MS analysis for protein identification and quantification
Protein identification was carried out using a liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based proteomics pipeline.42,43 LC-MS/MS analyses were performed using the LC/MSD Trap XCT Ultra (Agilent Technologies, Palo Alto, CA) equipped with a 1100 HPLC system and a chip cube (Agilent Technologies) as previously described.44Raw MS/MS data files were submitted to Mascot software (Matrix Science, UK) for protein identification.45 Precisely, the following search parameters were applied: NCBI as protein database; trypsin as specific proteolytic enzyme; Homo sapiens as taxonomy; one missed cleavage; S-carbamidomethyl cysteine as fixed modification; oxidized methionine and N-terminal pyroglutamic as variable modifications; 300 ppm and 0.6 Da as mass tolerance on precursor ions and product ions, respectively. Finally, a Mascot individual ion score >43 was considered for an unambiguous data interpretation (p value <0.05).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository46 with the dataset identifier PXD052701.
Quantitative analysis of LC-MS/MS data was performed by label-free quantification, subjecting the Mascot format text files to Proteome Discoverer v1.4 (Thermo Fisher Scientific, USA). Spectral count (SpC) values and Normalized Spectral Abundance Factor (NSAF) were used as quantitative parameters for estimating protein abundance.47 Briefly, the SpC of each protein was divided by its length, defining the spectral abundance factor (SAF), and normalized to the total sum of SpC in a given lane, obtaining the Normalized Spectral Abundance Factor (NSAF). To compare the expression of the same protein between different LFs, the log ratio of SpC (Rsc) was calculated as previously reported.40
2.7. Bioinformatics analysis
The identified proteins were classified according to the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 (https://www.david.abcc.ncifcrf.gov/).48 Based on Fisher's exact test, the DAVID tool can measure the protein enrichment in Gene Ontology (GO) annotation terms (p-value ≤0.05). In addition, the Search Tool for the Retrieval of Interacting Genes (STRING) v11 (https://www.string-db.org/) was used to analyze the most statistically significant and non-redundant biological processes among the differentially represented proteins from the label-free proteomic analysis.49 Only annotations of biological processes with a false discovery rate (FDR) ≤0.05 are considered significant.Proteome data were further analyzed using Perseus software v1.6.15.0 for hierarchical clustering through heatmap visualization and profile plot analysis.50,51 All the clustering analyses were carried out by choosing Euclidean distance and preprocessing with the k-means algorithm, allowing spontaneous grouping without preserving sample order. Moreover, STRING or the STRINGapp through Cytoscape v3.9 software was also used to build protein–protein interaction (PPI) networks for the proteins of interest.52,53
3. Results and discussion
Nanotechnology and proteomics coupled with bioinformatics have been implemented to investigate the biological effect of PrC adsorbed on different multicomponent LFs, and hence, to shed light on their possible ability to target specific tissues.3.1. Physicochemical characteristics of liposomes with different formulations
Liposomes, with different lipid compositions, were studied to evaluate the effect of the net charge of phospholipid polar head groups and the surface properties of nanocarriers on the protein corona adsorption and composition. PrC composition mainly depends on liposome surface properties, although payload drug molecules can further affect these properties.54 Currently, PEG is the gold standard polymer that confers long-circulating properties to liposomes.55 However, evidence showed that PEG activates the immune system after multiple injections,56 and alternative PEG-like polymers with stealth properties are needed for systemic injection to avoid the immune system activation by liposomes. In this scenario, gangliosides such as GM1 represent a suitable alternative to PEG owing to their stealth properties, stability in biological fluids, and immune system avoidance.16 GM1 was recently used as a neuroprotective agent for the treatment of brain disorders,57,58 and GM1-liposomes are currently in phase I clinical trial for the treatment of Parkinson's disease.59 We recently demonstrated that GM1-liposomes are stable in human plasma and like PEGylated liposomes, long-circulate after systemic injection in murine models and decrease neuronal inflammation and stroke in animal models.60Given such premises, five liposomal formulations (from LF1 to LF5) were synthetized by self-assembling different molar ratios of some lipids (DPPC, DPPS, DMPG, DSPE-mPEG750, DSPE-mPEG5000, GM1) and cholesterol, as shown in Table 1. All LFs contained zwitterionic DPPC, whose safe nature and low immunogenic properties make the liposomes suitable for nanomedicine applications. LFs designed for this study contained: (i) anionic phospholipids (LF1 and LF2);61 (ii) anionic phospholipids and PEGylated lipids (LF3 and LF4); (iii) anionic phospholipids and GM1 glycolipids (LF5). Each LF was extruded through polycarbonate filters with large (200 nm; LF-L) and small (100 nm; LF-S) pore sizes, thus obtaining a total of 10 formulations.
All these formulations were characterized by using DLS and LDE analyses; average size, polydispersity (PDI) and zeta potential of bare LFs were used as references for their physicochemical characterization. As reported in Fig. 1a, the average size of all bare LFs was in the range from 116.0 ± 1.8 nm to 200.1 ± 1.5 nm. LF1 showed the highest average size among all LFs (p value < 0.005, Fig. 1b). Conversely, the average size of LF3 was the smallest of all LFs-L and LFs-S, respectively (p value < 0.005, Fig. 1b). Among PEG-coating formulations, LF4 showed significantly increased average size compared to LF3 (p value < 0.005, Fig. 1b). These results could depend on the different MW of PEG chains bound to DSPE in LF4 and LF3 that, by rearranging differently on the liposomal surface, affected the final packing of the liposomal bilayer. In fact, the greater flexibility of high MW PEG compared to low MW PEG might affect the self-assembling of lipid derivatives into the liposomal bilayer.62,63 Nonetheless, PEG chains (up to 5 kDa MW) conjugated to DSPE have been widely used to synthesize long-circulating liposomes for drug delivery.64 Only LF5-L showed a similar average size compared to LF4-L with a non-significant difference (Fig. 1b). These results could be explained by the similar spatial distribution and rearrangement of polyethylene oxide chains of DSPE-PEG5000 and monosialotetrahexosyl chains of GM1, and by the amphipathic nature of both macromolecules and their relative steric barrier activity.65 Moreover, the amphipathic properties of DSPE-PEG5000 and GM1 allow micelle-like structures depending on the binding energy of curvature radius and the self-assembling energy-dependent process.66,67
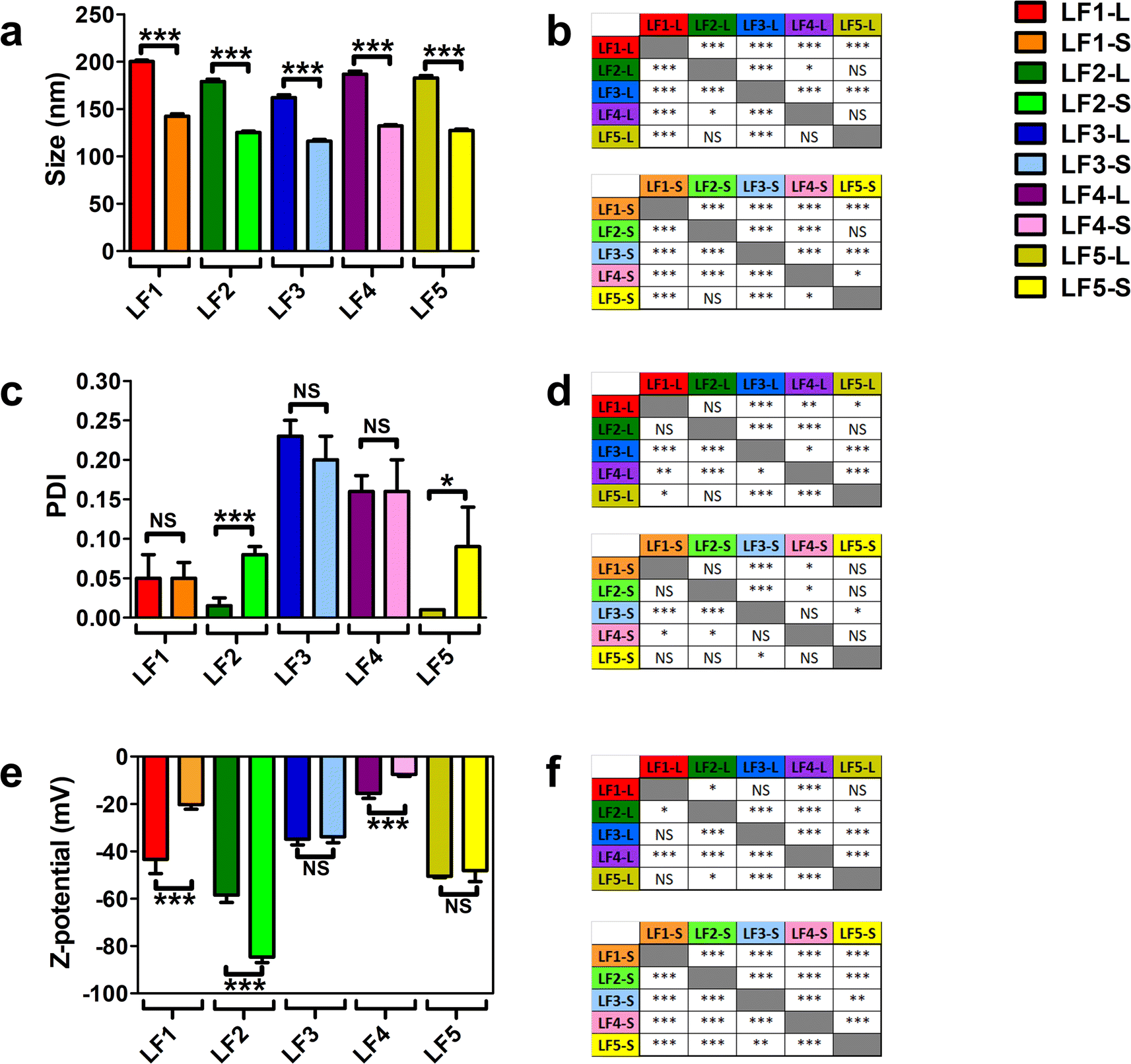 | ||
| Fig. 1 Dynamic light scattering (DLS) and laser doppler electrophoresis (LDE) analyses of LFs. LFs were produced using five different compositions, as reported in Table 1. For each composition, the vesicles were extruded at two different sizes through polycarbonate filters with large (200 nm) and small (100 nm) pore size membrane filters, thus finally obtaining ten different LFs. LFs extruded through 200 nm and 100 nm pore-size membrane filters were named LF-L and LF-S, respectively. (a) Measurements of average size (nm), (c) polydispersity index (PDI) and (e) zeta potential (mV) of different bare LFs. Results are the mean of three different replicates for which five technical repeats were considered and data were expressed as average ± SD. (b, d and f) Statistical significance was calculated by one-way T-test for each pairwise comparison among the LFs for the differences in average size, PDI and Z-potential, respectively. * = p value < 0.05; *** = p value < 0.005; NS = not significant. | ||
All the LFs showed PDI <0.25, thus suggesting that liposomes were stable with narrow size distribution68 (Fig. 1c). PEG-containing LFs (LF3 and LF4) showed higher PDI values compared to the others (Fig. 1d), likely because of PEG chains on the liposome structure. Interestingly, by comparing PDI values between large and small LFs, only LF2-L (p < 0.005) and LF5-L (p < 0.05) showed significant differences compared to their small counterparts. We can speculate that the highest net negative charge of LF2 and LF5, due to DMPG and GM1, generated a diverse charge distribution on their surface, characterized by a significantly different colloidal interface area. Such conditions may change the net curvature radius of liposomes, increasing their final values of PDI after extrusion through polycarbonate membrane at a specific pressure.
The analyses of zeta potential showed negative values for all LFs with a net negative surface charge below −15 mV (Fig. 1e), thus suggesting the synthesis of stable liposomes. LF2 showed the most negative zeta potential compared to all LFs (p < 0.005 and p < 0.05, Fig. 1f). The MW of PEG affects the zeta potential of LF3 and LF4, with LF3 more negatively charged than LF4 (Fig. 1f). Differences in the zeta potential values can depend on the number of polyethylene oxide chains that are present in the backbone structure of DSPE-PEG5000 and DSPE-PEG750, suggesting that low MW PEG has a lower shielding capability than high MW PEG.69 Different from PEG, GM1 had a lower shielding effect on LF5 (Fig. 1e). Finally, all LFs, except for LF3 and LF5, also showed statistically significant differences among LFs-L versus LFs-S (p < 0.005, Fig. 1e).
3.2. Quali-quantitative composition of liposome PrC and correlations with LF physicochemical properties
HuP was incubated with LFs to allow the formation of PrC-NP complexes. After the elution from LFs, adsorbed HuP proteins were separated by SDS-PAGE (ESI Fig. S1†), and the bands were analyzed by LC-MS/MS for protein identification. The PrC components associated with each LF (from LF1 to LF5) are listed in ESI Tables S1–S5,† including the details of MS analysis.We identified a total of 72 unique proteins adsorbed on and shared by the different LFs; in particular, MS analysis assigned 36 protein species for LF1-S, 42 for LF1-L, 39 for LF2-S, 43 for LF2-L, 41 for LF3-S, 47 for LF3-L, 29 for LF4-S, 24 for LF4-L, 34 for LF5-S and 43 for LF5-L (ESI Tables S1–S5†). Based on MS spectral counts, the relative abundances of these identified proteins were expressed as normalized spectral abundance factor (NSAF) and then evaluated and compared in all LFs. A total of 23 different proteins identified in the corona of at least eight out of ten LFs are listed in Table 2. Among them, 11 species were found to bind all the LFs with different abundances. Immunoglobulin kappa constant (IGKC) and albumin (ALB) were the most represented species in all formulations, showing 24.0 ± 5.5% and 19.0 ± 6.3% as average abundance, respectively. Some other species had a preferential binding based on the size of a specific LF (for example, band 3 anion transport protein, SLC4A1, for LF5-L) or were undetected in specific formulations, regardless of the size (for example, haptoglobin, HP, and transferrin, TF) for LF4.
| Gene | NSAF LF1-S | NSAF LF1-L | NSAF LF2-S | NSAF LF2-L | NSAF LF3-S | NSAF LF3-L | NSAF LF4-S | NSAF LF4-L | NSAF LF5-S | NSAF LF5-L |
|---|---|---|---|---|---|---|---|---|---|---|
| FCN3 | 9.1% | 6.3% | 2.7% | 0.8% | 4.4% | 3.5% | 6.1% | 5.4% | 5.7% | 4.8% |
| ALB | 13.2% | 15.9% | 24.5% | 20.3% | 26.5% | 23.9% | 7.9% | 12.4% | 20.2% | 25.1% |
| F5 | 1.3% | 3.7% | 1.5% | 1.2% | 0.7% | 0.4% | 1.1% | 0.8% | 0.1% | 0.5% |
| VTN | 1.2% | 2.0% | 0.7% | 1.6% | 0.6% | 0.8% | 1.2% | 1.2% | 0.4% | 0.5% |
| SERPINA1 | 1.4% | 1.4% | 2.2% | 1.7% | 1.9% | 1.9% | 0.9% | 1.1% | 1.8% | 1.4% |
| FGG | 0.3% | 0.6% | 1.3% | 0.8% | 1.3% | 1.5% | 0.7% | 0.8% | 1.0% | 2.0% |
| C1QC | 1.2% | 2.5% | 2.2% | 1.0% | 1.1% | 2.9% | 2.9% | 2.7% | 1.7% | 1.2% |
| IGKC | 26.3% | 18.7% | 19.5% | 16.2% | 25.1% | 19.4% | 26.6% | 30.8% | 33.2% | 23.8% |
| CD5L | 2.3% | 2.7% | 1.5% | 0.8% | 0.8% | 1.3% | 1.4% | 1.1% | 0.4% | 0.4% |
| IGHM | 14.2% | 13.0% | 6.1% | 14.4% | 6.7% | 11.0% | 10.8% | 14.1% | 3.0% | 4.5% |
| SLC4A1 | 0.4% | 1.1% | 2.2% | 1.3% | 1.2% | 2.3% | 1.7% | 0.7% | — | 0.1% |
| C3 | 0.5% | 0.4% | 0.8% | 0.8% | 0.6% | 0.3% | — | 0.2% | 1.3% | 0.8% |
| A2M | 0.1% | 0.4% | 0.8% | 0.7% | 0.6% | 1.0% | — | 0.1% | 1.4% | 1.7% |
| APOA1 | 2.3% | 2.6% | 0.9% | 1.4% | 1.0% | 1.9% | 0.6% | — | 1.9% | 0.7% |
| C1QB | 2.8% | 3.5% | — | 1.1% | 1.8% | 0.7% | 4.9% | 0.7% | 1.6% | 1.3% |
| C1QA | 1.2% | 0.9% | 0.3% | 0.7% | 1.1% | 0.7% | 1.3% | 0.4% | — | — |
| HP | 1.2% | 1.5% | 1.5% | 0.7% | 1.5% | 0.8% | — | — | 0.7% | 2.2% |
| TF | 0.8% | 0.9% | 1.4% | 1.4% | 1.2% | 0.5% | — | — | 1.1% | 1.3% |
| FGA | 0.2% | 0.1% | 0.1% | — | 0.2% | 0.2% | 0.2% | 0.7% | — | 0.1% |
| IGLC2 | — | — | 5.0% | 5.1% | 7.3% | 3.8% | 7.6% | 2.7% | 10.0% | 7.8% |
| IGHG2 | — | — | 10.2% | 13.3% | 7.7% | 8.7% | 13.8% | 9.5% | 4.2% | 7.5% |
| IGHA1 | — | — | 1.4% | 2.3% | 1.0% | 1.5% | 2.3% | 1.6% | 1.2% | 0.3% |
The functional/pathway analysis performed on this specific set of proteins revealed that 15 out of 23 clustered in a high-interconnected network related to the “complement and coagulation cascades” pathway (FDR = 8.09 × 10−21), also including a subnetwork of the complement C1q subunits (C1QA, C1QB and C1QC), (ESI Fig. S2†).
To assess whether the relative abundance of this set of 23 species could be related to their physicochemical properties, we determined the Pearson's coefficients between protein-specific MS spectral counts and the average size, or PDI or zeta potential of the bare LFs (Fig. 2). As shown in Fig. 2a, only vitronectin (VTN) exhibited a significant positive association with the average size of LFs. Interestingly, high levels of VTN were observed in the PrC of lipidic NPs, showing a preferential accumulation in tumor tissues.70 This finding highlights the importance of VTN-enriched corona on liposomes in terms of their putative capability to target cancer cells over-expressing the integrin receptor.
 | ||
| Fig. 2 Correlation between the relative abundance of PrC components and physicochemical properties of LFs. Protein abundance based on MS spectral counts was correlated with (a) the average size, (b) PDI and (c) zeta potential of bare LFs by calculating Pearson's coefficients. The proteins are reported with the names/acronyms of their coding genes. | ||
As for PDI, only C1QC and TF proteins showed significant Pearson's R values: C1QC showed a positive association, thus implicating that the higher amount matched more polydisperse liposomes, whereas TF showed a negative correlation, suggesting a binding preference to more homogeneous LFs (Fig. 2b). C1QC is a well-known protein of the C1q family, which carries out an essential role in innate immunity, contributing to nonspecific host defense.71,72 On the other hand, TF is an abundant human serum iron-binding glycoprotein with multi-task functions,73 well known for its natural targeting ability; as a matter of fact, it is used as a ligand for functionalizing NPs to actively target brain74 and/or cancer cells.75,76
Interestingly, we found several significant correlations between the protein relative abundances of PrC components and zeta potential (Fig. 2c). Indeed, ficolin-3 (FCN3), C1QA and C1QB were positively associated, thus implicating that their preferred binding was toward the less negative NPs. On the other hand, the relative abundances of ALB, alpha-1-antitrypsin (SERPINA1), complement C3 (C3), alpha-2-macroglobulin (A2M), HP and TF were negatively associated with zeta potential, meaning that higher contents correlated with lower zeta potential (Fig. 2c).
3.3. Functional classification of plasma proteins in the corona of LFs
The enrichment of Gene Ontology (GO) terms was performed on all the identified proteins to characterize them functionally according to Molecular Function (MF), Cellular Component (CC) and Biological Process (BP) categories (Table 3). The “immunoglobulin receptor binding” MF is the most represented GO term among the different LFs, except for LF5. Similarly, the most significant CC and BP categories included “fibrinogen complex” and “protein polymerization”, being the PrCs highly enriched in fibrinogen proteins (Table 3). In line with previous findings,12,77 fibrinogen and immunoglobulins mediate the opsonization phenomena, by which liposomes are marked to be eliminated by MPS. Interestingly, “complement activation” BP, to which opsonins belong, was not significantly represented in the coronas of PEGylated liposomes (Table 3), thus endorsing the anti-opsonization effect of PEG coating of NPs.13 Accordingly, also apolipoproteins, another class of opsonin whose adsorption is able to modulate liposome biodistribution,78 clustered in GO terms that are significantly represented in the coronas of PEG-free LFs (Table 3).| LFsLF | GO terms | Functional categories | p-Value | Proteins |
|---|---|---|---|---|
| a Molecular function. b Cellular component. c Biological process. | ||||
| LF1-L | Immunoglobulin receptor binding | MFa | 3.5 × 10−7 | IGHA2, IGHG3, IGHM, IGKC, IGJ |
| Fibrinogen complex | CCb | 7.5 × 10−7 | FGA, FGB, FGG, THBS1 | |
| Protein polymerization | BPc | 3.2 × 10−6 | FGA, FGB, FGG, VTN | |
| LF1-S | Immunoglobulin receptor binding | MFa | 1.7 × 10−7 | IGHA2, IGHG3, IGHM, IGKC, IGJ |
| Protein polymerization | BPc | 1.9 × 10−6 | FGA, FGB, FGG, VTN | |
| Fibrinogen complex | CCb | 1.1 × 10−4 | FGA, FGB, FGG | |
| Very-low-density lipoprotein particle | CCb | 5.9 × 10−4 | APOB, APOE, APOA1 | |
| LF2-L | Complement activation | BPc | 6.8 × 10−13 | C1QA, C1QB, C1QC, C3, C4A, CFB, FCN3, IGHG2, IGKC, IGLC2 |
| Immunoglobulin receptor binding | MFa | 4.2 × 10−9 | IGHA1, IGHG2, IGHM, IGKC, IGLC2, IGJ | |
| Fibrinogen complex | CCb | 1.8 × 10−4 | FGB, FGG, THBS1 | |
| Very-low-density lipoprotein particle | CCb | 9.6 × 10−4 | APOA1, APOE, APOL1 | |
| LF2-S | Immunoglobulin receptor binding | MFa | 2.8 × 10−9 | IGHA1, IGHG2, IGHM, IGKC, IGLC2, IGJ |
| Protein polymerization | BPc | 3.0 × 10−6 | FGA, FGB, FGG, VTN | |
| LF3-L | Immunoglobulin receptor binding | MFa | 6.7 × 10−9 | IGHA1, IGHG2, IGHM, IGKC, IGLC2, IGJ |
| Fibrinogen complex | CCb | 1.2 × 10−6 | FGA, FGB, FGG, THBS1 | |
| LF3-S | Immunoglobulin receptor binding | MFa | 3.6 × 10−9 | IGHA1, IGHG2, IGHM, IGKC, IGLC2, IGJ |
| Fibrinogen complex | CCb | 8.1 × 10−7 | FGA, FGB, FGG, THBS1 | |
| LF4-L | Immunoglobulin receptor binding | MFa | 2.6 × 10−8 | IGHA1, IGHG2, IGHM, IGKC, IGLC2 |
| Protein polymerization | BPc | 5.5 × 10−7 | FGA, FGB, FGG, VTN | |
| LF4-S | Immunoglobulin receptor binding | MFa | 7.6 × 10−8 | IGHA1, IGHG2, IGHM, IGKC, IGLC2 |
| Fibrinogen complex | CCb | 2.7 × 10−7 | FGA, FGB, FGG, THBS1 | |
| Protein polymerization | BPc | 1.2 × 10−6 | FGA, FGB, FGG, VTN | |
| LF5-L | Phagocytosis, engulfment | BPc | 2.0 × 10−8 | GSN, IGHA1, IGHG2, IGHM, IGKC, IGLC2 |
| Fibrinogen complex | CCb | 8.8 × 10−7 | FGA, FGB, FGG, THBS1 | |
| Protein polymerization | BPc | 3.8 × 10−6 | FGA, FGB, FGG, VTN | |
| LF5-S | Complement activation | BPc | 1.3 × 10−14 | C1QB, C1QC, C3, C4A, CFB, FCN2, FCN3, IGHG2, IGKC, IGLC2 |
| Phagocytosis, engulfment | BPc | 6.6 × 10−9 | GSN, IGHA1, IGHG2, IGHM, IGKC, IGLC2 | |
| Phosphatidylcholine-sterol O-acyltransferase activator activity | MFa | 5.5 × 10−5 | APOA1, APOA4, APOE | |
| Protein polymerization | BPc | 2.9 × 10−4 | FGB, FGG, VTN | |
3.4. Differentially represented plasma proteins in the corona of LFs
| Protein | Gene | R SC |
|---|---|---|
|
a
R
sc, log2 ratio between the protein expression levels in the corona of LF1-L versus LF1-S. Proteins in grey with Rsc ≥1.40 or ≤−1.40 were considered differentially represented.
|
||
| Thrombospondin-1 | THBS1 | 4.27 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | 3.42 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 | 3.21 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | 2.97 |
| Spectrin beta chain, erythrocytic | SPTB | 2.68 |
| Spectrin alpha chain, erythrocytic 1 | SPTA1 | 2.68 |
| Ankyrin-1 | ANK1 | 2.32 |
| Ceruloplasmin | CP | 1.83 |
| Coagulation factor V | F5 | 1.32 |
| Alpha-2-macroglobulin | A2M | 1.31 |
| Band 3 anion transport protein | SLC4A1 | 1.22 |
| Immunoglobulin J chain | IGJ | 1.10 |
| Complement C1q subcomponent subunit C | C1QC | 0.737 |
| Histidine-rich glycoprotein | HRG | 0.645 |
| Vitronectin | VTN | 0.458 |
| Fibrinogen gamma chain | FGG | 0.441 |
| Clusterin | CLU | 0.441 |
| Immunoglobulin alpha-2 heavy chain | IGHA2 | 0.377 |
| Ig gamma-3 chain C region | IGHG3 | 0.341 |
| Actin, cytoplasmic 1 | ACTB | 0.044 |
| Haptoglobin | HP | 0.016 |
| Complement C1q subcomponent subunit B | C1QB | −0.001 |
| Serum albumin | ALB | −0.028 |
| CD5 antigen-like | CD5L | −0.087 |
| Apolipoprotein A–I | APOA1 | −0.141 |
| Serotransferrin | TF | −0.167 |
| Alpha-1-antitrypsin | SERPINA1 | −0.301 |
| Ficolin-2 | FCN2 | −0.352 |
| Immunoglobulin heavy constant mu | IGHM | −0.484 |
| Fibrinogen alpha chain | FGA | −0.496 |
| Lipopolysaccharide-binding protein | LBP | −0.497 |
| Complement C1q subcomponent subunit A | C1QA | −0.643 |
| Complement C3 | C3 | −0.667 |
| Immunoglobulin kappa variable 3–15 | IGKV3–15 | −0.786 |
| Immunoglobulin heavy variable 1–3 | IGHV1–3 | −0, 786 |
| Immunoglobulin kappa constant | IGKC | −0.826 |
| Ficolin-3 | FCN3 | −0.875 |
| Apolipoprotein E | APOE | −0.905 |
| Fibrinogen beta chain | FGB | −1.01 |
| C4b-binding protein alpha chain | C4BPA | −1.14 |
| Hemopexin | HPX | −1.39 |
| Vitamin D-binding protein | GC | −1.58 |
| Alpha-1-antichymotrypsin | SERPINA3 | −2.37 |
| Apolipoprotein B-100 | APOB | −3.30 |
| Protein | Gene | R sc |
|---|---|---|
|
a
R
sc, log2 ratio between the protein expression levels in the corona of LF2-L versus LF2-S. Proteins in grey with Rsc ≥1.40 or ≤−1.40 were considered differentially represented.
|
||
| Spectrin beta chain, erythrocytic | SPTB | 5.08 |
| Alpha-1-acid glycoprotein 1 | ORM1 | 4.42 |
| Complement C1q subcomponent subunit B | C1QB | 3.93 |
| Ankyrin-1 | ANK1 | 3.93 |
| Histidine-rich glycoprotein | HRG | 3.72 |
| Ceruloplasmin | CP | 3.72 |
| Thrombospondin-1 | THBS1 | 3.48 |
| Apolipoprotein L1 | APOL1 | 2.83 |
| C4b-binding protein alpha chain | C4BPA | 2.83 |
| Alpha-1B-glycoprotein | A1BG | 2.34 |
| Immunoglobulin J chain | IGJ | 2.10 |
| Alpha-1-acid glycoprotein 1 | ORM1 | 4.42 |
| Complement C1q subcomponent subunit B | C1QB | 3.93 |
| Ankyrin-1 | ANK1 | 3.93 |
| Histidine-rich glycoprotein | HRG | 3.72 |
| Ceruloplasmin | CP | 3.72 |
| Thrombospondin-1 | THBS1 | 3.48 |
| Apolipoprotein L1 | APOL1 | 2.83 |
| C4b-binding protein alpha chain | C4BPA | 2.83 |
| Alpha-1B-glycoprotein | A1BG | 2.34 |
| Immunoglobulin J chain | IGJ | 2.10 |
| Immunoglobulin heavy constant mu | IGHM | 1.39 |
| Vitronectin | VTN | 1.23 |
| Complement C1q subcomponent subunit A | C1QA | 0.867 |
| Ig alpha-1 chain C region | IGHA1 | 0.741 |
| Apolipoprotein A–I | APOA1 | 0.568 |
| Complement factor B | CFB | 0.503 |
| Ig gamma-2 chain C region | IGHG2 | 0.426 |
| Vitamin D-binding protein | GC | 0.380 |
| Alpha-1-antichymotrypsin | SERPINA3 | 0.380 |
| Ig lambda-2 chain C regions | IGLC2 | 0.016 |
| Spectrin alpha chain, erythrocytic 1 | SPTA1 | 0.016 |
| Complement C4-A | C4A | 0.016 |
| Complement C3 | C3 | −0.028 |
| Serotransferrin | TF | −0.045 |
| Alpha-2-macroglobulin | A2M | −0.091 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | −0.226 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | −0.226 |
| Immunoglobulin kappa constant | IGKC | −0.273 |
| Peroxiredoxin-2 | PRDX2 | −0.274 |
| Alpha-1-antitrypsin | SERPINA1 | −0.353 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 | −0.383 |
| Serum albumin | ALB | −0.402 |
| Coagulation factor V | F5 | −0.407 |
| Fibrinogen gamma chain | FGG | −0.600 |
| Band 3 anion transport protein | SLC4A1 | −0.727 |
| CD5 antigen-like | CD5L | −0.840 |
| Actin, cytoplasmic 1 | ACTB | −0.935 |
| Haptoglobin | HP | −1.05 |
| Complement C1q subcomponent subunit C | C1QC | −1.05 |
| Apolipoprotein E | APOE | −1.11 |
| Fibrinogen beta chain | FGB | −1.52 |
| Ficolin-3 | FCN3 | −1.66 |
| Catalase | CAT | −2.31 |
| Clusterin | CLU | −2.79 |
| Fibrinogen alpha chain | FGA | −2.79 |
| Immunoglobulin heavy variable 3–9 | IGHV3 | −3.16 |
| Serum amyloid P-component | APCS | −3.45 |
| Ficolin-2 | FCN2 | −4.24 |
| Protein | Gene | R sc |
|---|---|---|
|
a
R
sc, log2 ratio between the protein expression levels in the corona of LF3-L versus LF3-S. Proteins in grey with Rsc ≥1.40 or ≤−1.40 were considered differentially represented.
|
||
| Ficolin-2 | FCN2 | 3.60 |
| Complement factor H-related protein 1 | CFHR1 | 3.35 |
| Histidine-rich glycoprotein precursor | HRG | 3.35 |
| Desmoplakin I | DSP | 3.35 |
| Annexin A1 | ANXA1 | 2.21 |
| Alpha-1-antichymotrypsin | SERPINA3 | 2.21 |
| Heat shock protein HSP 90-alpha | HSP90AA1 | 2.21 |
| Spectrin beta chain, erythrocytic | SPTB | 1.78 |
| Thrombospondin-1 | THBS1 | 1.49 |
| Complement C1q subcomponent subunit C | C1QC | 1.25 |
| Immunoglobulin J chain | IGJ | 1.11 |
| Complement factor H | CFH | 1.11 |
| Fibrinogen beta chain | FGB | 0.939 |
| Band 3 anion transport protein | SLC4A1 | 0.905 |
| Ankyrin-1 | ANK1 | 0.848 |
| Apolipoprotein A-I | APOA1 | 0.717 |
| Alpha-2-macroglobulin | A2M | 0.687 |
| Immunoglobulin heavy constant mu | IGHM | 0.676 |
| CD5 antigen-like | CD5L | 0.584 |
| Hemopexin | HPX | 0.542 |
| Ig alpha-1 chain C region | IGHA1 | 0.449 |
| Vitronectin | VTN | 0.439 |
| Actin, cytoplasmic 1 | ACTB | 0.420 |
| Fibrinogen gamma chain | FGG | 0.088 |
| Ig gamma-2 chain C | IGHG2 | 0.087 |
| Spectrin alpha chain, erythrocytic 1 | SPTA1 | 0.048 |
| Alpha-1-antitrypsin | SERPINA1 | −0.033 |
| Fibrinogen alpha chain | FGA | −0.113 |
| Serum albumin | ALB | −0.394 |
| Ficolin-3 | FCN3 | −0.454 |
| Ceruloplasmin | CP | −0.477 |
| Immunoglobulin kappa constant | IGKC | −0.497 |
| Complement C1q subcomponent subunit A | C1QA | −0.647 |
| Coagulation factor V | F5 | −0.708 |
| Pre-serum amyloid P component | APCS | −0.851 |
| Haptoglobin | HP | −0.971 |
| Complement C3 | C3 | −0.979 |
| Ig lambda-2 chain C regions | IGLC2 | −1.01 |
| Erythrocyte band 7 integral membrane protein | STOM | −1.01 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 | −1.20 |
| Serotransferrin | TF | −1.25 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | −1.25 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | −1.34 |
| Complement C1q subcomponent subunit B | C1QB | −1.34 |
| Complement component C4A | C4A | −2.33 |
| Gelsolin | GSN | −2.44 |
| Apolipoprotein B-100 | APOB | −3.58 |
| Protein | Gene | R sc |
|---|---|---|
|
a
R
sc, log2 ratio between the protein expression levels in the corona of LF4-L versus LF4-S. Proteins in grey with Rsc ≥1.40 or ≤−1.40 were considered differentially represented.
|
||
| Immunoglobulin heavy variable 3–66 | IGHV3-66 | 3.64 |
| Histidine-rich glycoprotein | HRG | 3.64 |
| Complement component C3 | C3 | 3.64 |
| Alpha-2-macroglobulin precursor | A2M | 3.15 |
| Fibrinogen alpha chain | FGA | 1.73 |
| Immunoglobulin kappa variable 3D-11 | IGKV3D-11 | 1.32 |
| Immunoglobulin kappa variable 3–20 | IGKV3-20 | 1.12 |
| Fibrinogen beta chain | FGB | 0.951 |
| Serum albumin | ALB | 0.807 |
| Immunoglobulin heavy constant mu | IGHM | 0.491 |
| Alpha-1-antitrypsin | SERPINA1 | 0.380 |
| Fibrinogen gamma chain | FGG | 0.295 |
| Immunoglobulin kappa constant | IGKC | 0.255 |
| Vitronectin | VTN | −0.001 |
| Complement C1q subcomponent subunit C | C1QC | −0.026 |
| Ficolin-3 | FCN3 | −0.166 |
| CD5 antigen-like | CD5L | −0.259 |
| Ig alpha-1 chain C region | IGHA1 | −0.441 |
| Coagulation factor V | F5 | −0.462 |
| Ig gamma-2 chain C region | IGHG2 | −0.581 |
| Band 3 anion transport protein | SLC4A1 | −1.19 |
| Complement C1q subcomponent subunit A | C1QA | −1.30 |
| Ig lambda-2 chain C regions | IGLC2 | −1.35 |
| Peroxiredoxin-2 | PRDX2 | −1.50 |
| Apolipoprotein A-I | APOA1 | −1.99 |
| Thrombospondin-1 | THBS1 | −2.00 |
| Ankyrin 1 | ANK1 | −2.36 |
| Complement C1q subcomponent subunit B | C1QB | −2.46 |
| Erythrocyte band 7 integral membrane protein | STOM | −2.65 |
| Actin, cytoplasmic 1 | ACTB | −2.89 |
| Spectrin alpha chain, erythrocytic 1 | SPTA1 | −2.89 |
| Spectrin beta chain, erythrocytic | SPTB | −3.28 |
| Protein | Gene | R sc |
|---|---|---|
|
a RSC, log2 ratio between the protein expression levels in the corona of LF5-L versus LF5-S. Proteins in grey with RSC ≥1.40 or ≤−1.40 were considered differentially represented.
|
||
| Immunoglobulin kappa variable 2–30 | IGKV2-30 | 3.12 |
| Immunoglobulin heavy variable 3–72 | IGHV3-72 | 2.91 |
| Histidine-rich glycoprotein | HRG | 2.67 |
| Band 3 anion transport protein | SLC4A1 | 2.38 |
| Immunoglobulin kappa variable 3–20 | IGKV3-20 | 2.38 |
| Fibrinogen alpha chain | FGA | 2.38 |
| Thrombospondin-1 precursor | THBS1 | 2.01 |
| Plasma protease C1 inhibitor precursor | SERPING1 | 2.01 |
| Coagulation factor V | F5 | 1.91 |
| Alpha-2-HS-glycoprotein | AHSG | 1.53 |
| Complement factor H | CFH | 1.53 |
| Haptoglobin | HP | 1.46 |
| Apolipoprotein E | APOE | 1.18 |
| Fibrinogen gamma chain | FGG | 0.757 |
| Ig gamma-2 chain C region | IGHG2 | 0.693 |
| Hemopexin | HPX | 0.562 |
| Immunoglobulin heavy constant mu | IGHM | 0.440 |
| Serum albumin | ALB | 0.245 |
| Alpha-2-macroglobulin | A2M | 0.139 |
| Vitronectin | VTN | 0.123 |
| Serotransferrin | TF | 0.054 |
| Zinc-alpha2-glycoprotein | AZGP1 | 0.053 |
| Complement factor B | CFB | 0.053 |
| CD5 antigen-like | CD5L | −0.143 |
| Inter-alpha-trypsin inhibitor heavy chain ITIH1 | ITIH1 | −0.180 |
| Complement component C4A | C4A | −0.291 |
| Fibrinogen beta chain | FGB | −0.301 |
| Serum amyloid P-component | APCS | −0.311 |
| Ficolin-3 | FCN3 | −0.443 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 | −0.495 |
| Complement C1q subcomponent subunit B | C1QB | −0.525 |
| Ig lambda-2 chain C regions | IGLC2 | −0.531 |
| Alpha-1-antitrypsin | SERPINA1 | −0.564 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 | −0.640 |
| Complement C1q subcomponent subunit C | C1QC | −0.657 |
| Immunoglobulin kappa constant | IGKC | −0.694 |
| Gelsolin | GSN | −0.797 |
| Alpha-1-antichymotrypsin | SERPINA3 | −0.798 |
| Complement C3 | C3 | −0.932 |
| Apolipoprotein A–I | APOA1 | −1.63 |
| Ceruloplasmin | CP | −1.77 |
| Ig alpha-1 chain C region | IGHA1 | −2.25 |
| Ficolin-2 | FCN2 | −2.55 |
| Apolipoprotein A-IV | APOA4 | −3.12 |
 | ||
| Fig. 3 Venn diagram of differentially represented proteins in the corona. (a) Venn diagram shows the over-represented (red arrow) and under-represented (green arrow) proteins specific for each of the five pairwise comparisons (listed in the box) and those common to at least three pairwise comparisons. (b) Within each pairwise comparison, proteins shared by LFs were reported together with their differential representation (LFs-L versus LFs-S). Only proteins with Rsc of ≥1.40 or ≤−1.40 were included in the diagram. n.d. = not determined (the protein was not identified in both LF-L and LF-S formulations); / = below the threshold, i.e., Rsc < 1.40 or >−1.40. | ||
Moreover, other proteins were found in PrC as common species to at least three LFs as reported in the Venn diagram of Fig. 3a; their binding preferences (LFs-L versus LFs-S) are shown in Fig. 3b. For example, thrombospondin-1 (THBS1) was shared by all the LFs; whereas spectrin beta chain, erythrocytic (SPTB) by all LFs except for GM1-coated LFs and ankyrin-1 (ANK1) only by PEG-free LFs (LF1 and LF2) and PEGylated LF4. Interestingly, all these common proteins were, in general, over-represented in LFs-L versus LFs-S except for PEGylated LF4 (Fig. 3b).
Among the investigated LFs, PEG- and GM1-bearing LFs (LF3–5) are suitable NPs for applications of in vivo delivery in terms of extended circulation time and immune/reticuloendothelial system avoidance;79–82 moreover, GM1-NPs are also able to overcome the brain barrier (BBB).83,84 Hence, we pointed out the composition-specific patterns shared by small and large LF5 in comparison with the corresponding PEGylated LFs (Fig. 4a).
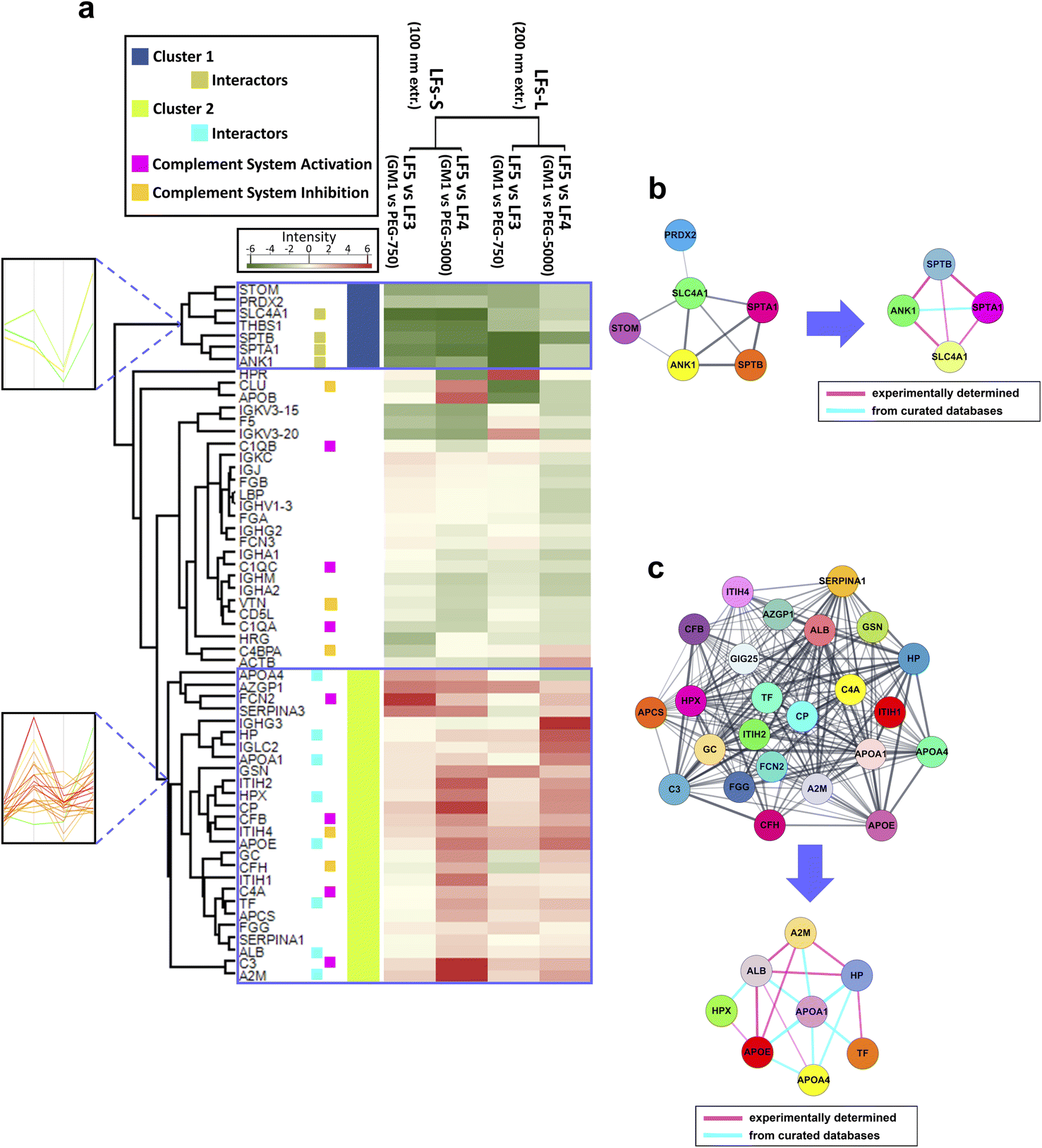 | ||
| Fig. 4 Proteomic and functional characterization of GM1-incorporating LFs. (a) Heatmap visualization of corona protein relative abundance of LF5-S and LF5-L (GM1-LFs at 100 nm and 200 nm extrusion, respectively) in comparison with LF3 (PEG750-LFs) or LF4 (PEG5000-LFs). Protein abundance based on Rsc is reported as color intensity ranging from green to red. Two clusters were identified, namely cluster 1 and cluster 2, with marked opposite NP-binding preference. Profile plots of the selected clusters are also highlighted (left inserts) (a magnification of these profile plots is contained in ESI Fig. S3,† where most of the protein names are also indicated). Functional annotation was reported for selected proteins in the heatmap. (b and c) Results of protein–protein interaction networks retrieved by STRING analysis for the proteins of cluster 1 and 2, respectively. From each of the two networks, subnetworks were generated to highlight the species known to physically interact with each other. The thickness of edges (connecting lines) is proportional to interaction confidence. | ||
Interestingly, the comparison between LF5 and LF3/LF4 corona allowed us to identify two dense groups of proteins, termed cluster 1 and cluster 2, which showed a marked opposite NP-binding preference (Fig. 4a). In fact, cluster 1 proteins preferentially bind PEGylated liposomes (LF3 and LF4), while cluster 2 proteins preferentially adsorb onto GM1 liposomes (LF5). The trends of relative abundance of cluster 1 and 2 proteins are highlighted by a profile plot analysis in Fig. 4a, left inserts. A magnification of these profile plots, including protein names, is shown in ESI Fig. S4.†
Cluster 1 is constituted by 7 proteins, namely erythrocyte band 7 integral membrane protein (STOM), peroxiredoxin-2 (PRDX2), SLC4A1, THBS1, SPTB, spectrin alpha chain, erythrocytic 1 (SPTA1), ANK1, and is more represented in the PrC of PEGylated LFs (LF3 and LF4) than in GM1-incorporating ones (LF5). All these proteins, except THBS1, cluster together, as revealed by STRING analysis (Fig. 4b). Among them, SLC4A1, SPTB, SPTA1, and ANK1 physically interact with each other. Interestingly, similar proteins were identified in vitro and in vivo in the PrC of PEGylated formulations, enforcing a putative role of these species in the nano-bio interfaces between HuP and NPs.18,85,86 However, the effects of such corona proteins remain to be understood. As for THBS1, this secreted glycoprotein interacts with components of the extracellular matrix and various cell surface receptors. It is present in plasma, acting as a regulator of blood pressure and hemostasis, and it also plays roles in regulating immune responses. THBS1 can bridge phagocytic immune cells with other cell types, including platelets and apoptotic cells, thereby promoting the activity of professional phagocytic cells.87 Hence, we may speculate that the presence of this protein on the NP surface might enhance the MPS clearance. Here, THBS1, as the other cluster1 proteins, preferentially binds LF3 and LF4 rather than LF5, suggesting that the supposed enhanced phagocytic effect could be higher in PEGylated LFs than in GM1 LFs. Moreover, THBS1 showed size-dependent differential binding with all the LFs (Fig. 3a and b), and it is more represented in the corona of LFs-L than LFs-S, except in the case of LF4 (bearing long PEG chains). This additional result could imply that the phagocytic effect could be higher for LF-L that for LF-S, except for high MW PEGylated liposomes.
Cluster 2 is comprised of 26 proteins, of which 24 species cluster together upon STRING analysis (Fig. 4c). Among them, 6 proteins are regulators or factors of the complement system (ficolin-2, FCN2, complement factor B, CFB, ITIH4, complement factor H, CFH, complement C4-A, C4A, C3).71,88 In addition, within Cluster 2, apolipoprotein A-IV (APOA4), HP, apolipoprotein A–I (APOA1), hemopexin (HPX), apolipoprotein E (APOE), TF, ALB, and A2M constitute a network of interacting species (Fig. 4c), suggesting a possible mechanism of cooperative adsorption of proteins.89,90
Some proteins belonging to cluster 2 have been previously reported in PrC studies and here they are shared by most of the investigated LFs (Table 2). In fact, besides ALB, whose functions as NP components are well-known,18,74 TF, APOE and APOA1 are used to functionalize liposomes or other NPs for active targeting. Conjugation of NPs with TF is extensively used for drug delivery purposes since such strategy exploits the ability to specifically bind to transferrin receptors (TFR); in fact, many types of tumor cells and endothelial cells over-express TFR, thereby allowing TF-conjugated NPs to take advantage of TFR-mediated endocytosis.91 Indeed, liver cells highly overexpress TFRs92 and, recently, TF-conjugated liposomes were used to effectively target liver cancer.93 However, a large body of evidence indicates that TF-functionalized liposomes are particularly effective for blood–brain barrier (BBB) targeting and drug delivery to the central nervous system (CNS)94–96 since TFRs are abundant on the plasmatic membrane of brain endothelial cells and neurons.95,96
APOE and APOA1 apolipoproteins are considered (together with ALB) dysopsonins78,89 that can inhibit monocyte uptake74 and confer stealth properties to NPs.97 As part of the delicate balance between opsonins and dysopsonins that determine the fate of NPs,78 the preferential presence on the LF5 corona of APOE and APOA1, together with ALB, could represent a molecular label that potentially makes GM1-liposomes more suitable than PEG-liposomes for drug delivery applications. Interestingly, APOE conjugation with liposomes is widely used as a means to enhance delivery to the CNS98–100 and liver.101 In fact, APOE functions as a ligand for low density lipoprotein (LDL) receptors (LDLR) that are expressed at high levels by normal and tumoral hepatocytes, brain endothelial cells and neuronal cells.98 The presence of APOE on the surface of NPs enhances the ability to overcome the BBB through LDLR-mediated transcytosis/endocytosis, thereby favoring the accumulation of NPs or their payload to brain tumors.98
APOA1 is a main protein constituent of high-density lipoprotein (HDL) and can actively bind to HDL receptors and mainly to scavenger receptor type I (SR-B1), which are highly expressed on the membrane of hepatocytes.102,103 For this reason, APOA1-conjugated liposomes/NPs have been widely adopted in liver-targeted drug delivery strategies.102,103
Taken together, the co-presence of TF, APOE and APOA1 on the PrC of GM1-containing liposomes could be a signature characterizing NPs able to target specific tissues, such as the brain and the liver.
4. Conclusions
This study consisted of a formulation/size-based multiplexed mapping of PrC composition that allowed us to highlight (i) the common as well as the unique species for all formulations; (ii) the correlation between each identified corona protein and various physicochemical properties (size, PDI or zeta potential); (iii) some preferential binding determined by the size within the same formulation, or by the formulation within the two extrusion sizes; (iv) the striking different corona composition between PEG- and GM1-containing LFs. In fact, we identified two clusters of plasma proteins with a preferential binding to PEG-containing liposomes (cluster 1) or GM1-containing formulations (cluster 2).Currently, some authors recognize it as instrumental to distinguish protein patterns within PrC constituted by a quali-quantitative variety of species organized in a sort of multifaceted protein domains. These may represent a structural/functional tridimensional scaffold motif that mediates cell targeting and uptake.36,37 In agreement with the novel concept of bionanosynapsis, representing the interaction between PrC-NPs and biological tissues able to trigger a cellular response,36,37 we suggest cluster 1 and 2 as multifaceted protein domains that specifically contribute to the novel biological identity of PEG- and GM1-coated liposomes, respectively.
The results obtained in this work take into account the possibility of controlling the PrC composition by modulating physicochemical properties and tuning surface features of LFs to the possible future aim of targeting active molecules to the specific tissue of action. Investigating clusters in PrC will help to decode the multivalent roles of the protein pattern components in the drug delivery process, taking advantage of the bionanoscale recognition and identity for significant advances in nanomedicine.
However, to reach such future progress, there is an urgent need for innovative approaches, including the implementation of artificial intelligence and machine learning algorithms aimed at disentangling the heterogeneous composition of NP coronas, such as the metabolite corona.104,105 Indeed, the low molecular weight metabolites may further influence the bio-nano interactions, and hence, other specific biological insights.
Author contributions
CC, EI, LDM, SO: conceptualization, methodology, investigation, writing the original draft. FS and MF: overall supervision and project planning and administration, writing, editing, and revision. CC, LDM and NdA performed the experiments for the synthesis and physicochemical characterization of liposomes, analyzed the resulting data and discussed the relative results. EI performed proteomic analyses. EI, AC and MC analyzed the proteomic data, performed bioinformatic analyses and correlations, and discussed the relative results. SO supervised proteomic and bioinformatic analyses. FS and MF drafted and supervised the editing of the manuscript and critically revised the results and the whole content of the paper. All the authors participated in the writing and revision of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository46 with the dataset identifier PXD052701.Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
This work was supported by: Ministero della Salute [RF-2010-23183729] to F. S.; Grants from Regione Campania [CIRO project: infrastructures and scientific instrumentation to CEINGE (Coordinator: Prof. Francesco Salvatore) D. D. n.366/2018; SATIN “Neoplasia studies” POR Campania FESR 2014/2020; D. D. Regione Campania n.459/2018; “Predictive Medicine in neoplasia”, L. Regione Campania n.752/2019 and n.38/2020; “Studi sulla Lotta alle Malattie Neoplastiche” (BURC: L.752/2019; Legge n.38/2020 art.16, D. D. Regione Campania n.48/2021; D. D. Regione Campania n.359/2022; D. D. Regione Campania n.9/2023) ] all to F. S.; Ministero dell‘Università e della Ricerca (MUR) [FAR 2018 (D56C18000780005), FAR 2019 (D54I19002790005)]; the European Union – NextGenerationEU, under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 – M4C2, Investment 1.5 – Call for tender No. 3277 of 30.12.2021, Italian Ministry of University, Award Number: ECS00000041, Project Title: “Innovation, digitalization and sustainability for the diffused economy in Central Italy”, Concession Degree No. 1057 of 23.06.2022 adopted by the Italian Ministry of University. CUP: D73C22000840006 (grant holder: Luisa Di Marzio and Christian Celia); Overseas Visiting Fellow Program 2022, University of Shanghai, China (grant holder: Christian Celia). We are grateful to Jean Ann Gilder (Scientific Communication s.r.l., Naples, Italy), a native English expert, for editing the text language.References
- E. Beltrán-Gracia, A. López-Camacho, I. Higuera-Ciapara, J. B. Velázquez-Fernández and A. A. Vallejo-Cardona, Cancer Nanotechnol., 2019, 10, 11 CrossRef.
- G. Bozzuto and A. Molinari, Int. J. Nanomed., 2015, 975 CrossRef CAS.
- H. Nsairat, D. Khater, U. Sayed, F. Odeh, A. Al Bawab and W. Alshaer, Heliyon, 2022, 8, e09394 CrossRef CAS.
- X. Wang, F. Wang, S. Li, G. Yin and X. Pu, Curr. Drug Delivery, 2022, 19, 940–948 CrossRef CAS PubMed.
- F. Gong, Z. Wang, R. Mo, Y. Wang, J. Su, X. Li, C. T. Q. Omonova, A. M. Khamis, Q. Zhang, M. Dong and Z. Su, J. Controlled Release, 2022, 349, 940–953 CrossRef CAS.
- T. Shehata, K. Ogawara, K. Higaki and T. Kimura, Int. J. Pharm., 2008, 359, 272–279 CrossRef CAS.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- K. Yang, K. Tran and A. Salvati, Biomolecules, 2022, 13, 59 CrossRef.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- Y. Takakura and Y. Takahashi, J. Controlled Release, 2022, 350, 486–493 CrossRef CAS PubMed.
- C. Corbo, R. Molinaro, A. Parodi, N. E. Toledano Furman, F. Salvatore and E. Tasciotti, Nanomedicine, 2016, 11, 81–100 CrossRef CAS.
- F. Giulimondi, L. Digiacomo, D. Pozzi, S. Palchetti, E. Vulpis, A. L. Capriotti, R. Z. Chiozzi, A. Laganà, H. Amenitsch, L. Masuelli, G. Peruzzi, M. Mahmoudi, I. Screpanti, A. Zingoni and G. Caracciolo, Nat. Commun., 2019, 10, 3686 CrossRef CAS.
- S. Palchetti, V. Colapicchioni, L. Digiacomo, G. Caracciolo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 189–196 CrossRef CAS.
- A. Cevenini, C. Celia, S. Orrù, D. Sarnataro, M. Raia, V. Mollo, M. Locatelli, E. Imperlini, N. Peluso, R. Peltrini, E. De Rosa, A. Parodi, L. Del Vecchio, L. Di Marzio, M. Fresta, P. A. Netti, H. Shen, X. Liu, E. Tasciotti and F. Salvatore, Pharmaceutics, 2020, 12, 559 CrossRef CAS.
- Y. Gao, M. Joshi, Z. Zhao and S. Mitragotri, Bioeng. Transl. Med., 2023, 9, e10600 CrossRef PubMed.
- N. D'Avanzo, C. Celia, A. Barone, M. Carafa, L. Di Marzio, H. A. Santos and M. Fresta, Adv. Ther., 2020, 3, 1900170 CrossRef.
- E. Tasciotti, R. Molinaro, F. Taraballi, N. Toledano Furman, M. Sherman, A. Parodi, F. Salvatore and C. Corbo, Int. J. Nanomed., 2016, 11, 3049–3063 CrossRef.
- E. Imperlini, C. Celia, A. Cevenini, A. Mandola, M. Raia, M. Fresta, S. Orrù, L. Di Marzio and F. Salvatore, Nanoscale, 2021, 13, 5251–5269 RSC.
- S. Yang, Y. Liu, Y. Wang and A. Cao, Small, 2013, 9, 1635–1653 CrossRef CAS PubMed.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS.
- T. Cedervall, I. Lynch, M. Foy, T. Berggård, S. C. Donnelly, G. Cagney, S. Linse and K. A. Dawson, Angew. Chem., Int. Ed., 2007, 46, 5754–5756 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS.
- L. Vroman, Nature, 1962, 196, 476–477 CrossRef CAS PubMed.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, J. Controlled Release, 2012, 161, 164–174 CrossRef CAS.
- L. Wang, J. Li, J. Pan, X. Jiang, Y. Ji, Y. Li, Y. Qu, Y. Zhao, X. Wu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17359–17368 CrossRef CAS PubMed.
- M. Kendall, P. Ding and K. Kendall, Nanotoxicology, 2011, 5, 55–65 CrossRef CAS PubMed.
- M. A. Wells, A. Abid, I. M. Kennedy and A. I. Barakat, Nanotoxicology, 2012, 6, 837–846 CrossRef CAS.
- K. Yang, B. Mesquita, P. Horvatovich and A. Salvati, Acta Biomater., 2020, 106, 314–327 CrossRef CAS PubMed.
- M. Pannuzzo, S. Esposito, L.-P. Wu, J. Key, S. Aryal, C. Celia, L. di Marzio, S. M. Moghimi and P. Decuzzi, Nano Lett., 2020, 20, 4312–4321 CrossRef CAS.
- L. Wei, D. Zhao, W. Sun, L. Lin, D. Sui, W. Li, Y. Gui, J. Wang, Y. Deng and Y. Song, Eur. J. Pharm. Biopharm., 2023, 184, 50–61 CrossRef CAS PubMed.
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS.
- S. Arumugam, S. Schmieder, W. Pezeshkian, U. Becken, C. Wunder, D. Chinnapen, J. H. Ipsen, A. K. Kenworthy, W. Lencer, S. Mayor and L. Johannes, Nat. Commun., 2021, 12, 3675 CrossRef CAS.
- J. W. Shreffler, J. E. Pullan, K. M. Dailey, S. Mallik and A. E. Brooks, Int. J. Mol. Sci., 2019, 20(23), 6056 CrossRef CAS.
- L. Wang, X. Ji, D. Guo, C. Shi and J. Luo, Mol. Pharm., 2021, 18, 2349–2359 CrossRef CAS.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, Nat. Rev. Mater., 2023, 8, 422–438 CrossRef PubMed.
- K. A. Dawson and Y. Yan, Nat. Nanotechnol., 2021, 16, 229–242 CrossRef CAS.
- J. Wolfram, B. Scott, K. Boom, J. Shen, C. Borsoi, K. Suri, R. Grande, M. Fresta, C. Celia, Y. Zhao, H. Shen and M. Ferrari, Curr. Drug Delivery, 2016, 13, 711–719 CrossRef CAS.
- G. N. Roviello, G. Roviello, D. Musumeci, D. Capasso, S. Di Gaetano, M. Costanzo and C. Pedone, RSC Adv., 2014, 4, 28691–28698 RSC.
- E. Imperlini, I. Colavita, M. Caterino, P. Mirabelli, D. Pagnozzi, L. Del Vecchio, R. Di Noto, M. Ruoppolo and S. Orrù, J. Cell. Biochem., 2013, 114, 2577–2587 CrossRef CAS.
- E. Imperlini, A. Mancini, S. Spaziani, D. Martone, A. Alfieri, M. Gemei, L. Del Vecchio, P. Buono and S. Orrù, Proteomics, 2010, 10, 3165–3175 CrossRef CAS.
- M. Costanzo, M. Caterino, A. Cevenini, L. Kollipara, O. Shevchuk, C. D. L. Nguyen, A. Sickmann and M. Ruoppolo, in NATO Science for Peace and Security Series A: Chemistry and Biology, Springer Science and Business Media B.V., 2020, pp. 221–223 Search PubMed.
- M. Costanzo, A. Cevenini, L. Kollipara, M. Caterino, S. Bianco, F. Pirozzi, G. Scerra, M. D'Agostino, L. M. Pavone, A. Sickmann and M. Ruoppolo, Cell Biosci., 2024, 14, 63 CrossRef CAS PubMed.
- E. Imperlini, S. Orrù, C. Corbo, A. Daniele and F. Salvatore, J. Neurochem., 2014, 129, 1002–1012 CrossRef CAS PubMed.
- F. Prisco, D. De Biase, G. Piegari, F. Oriente, I. Cimmino, V. De Pasquale, M. Costanzo, P. Santoro, M. Gizzarelli, S. Papparella and O. Paciello, Pathogens, 2021, 10, 463 CrossRef CAS PubMed.
- Y. Perez-Riverol, J. Bai, C. Bandla, D. García-Seisdedos, S. Hewapathirana, S. Kamatchinathan, D. J. Kundu, A. Prakash, A. Frericks-Zipper, M. Eisenacher, M. Walzer, S. Wang, A. Brazma and J. A. Vizcaíno, Nucleic Acids Res., 2022, 50, D543–D552 CrossRef CAS PubMed.
- D. Drongitis, M. Caterino, L. Verrillo, P. Santonicola, M. Costanzo, L. Poeta, B. Attianese, A. Barra, G. Terrone, M. B. Lioi, S. Paladino, E. Di Schiavi, V. Costa, M. Ruoppolo and M. G. Miano, Hum. Mol. Genet., 2022, 31, 1884–1908 CrossRef CAS PubMed.
- B. T. Sherman, M. Hao, J. Qiu, X. Jiao, M. W. Baseler, H. C. Lane, T. Imamichi and W. Chang, Nucleic Acids Res., 2022, 50, W216–W221 CrossRef CAS.
- V. Manganelli, I. Salvatori, M. Costanzo, A. Capozzi, D. Caissutti, M. Caterino, C. Valle, A. Ferri, M. Sorice, M. Ruoppolo, T. Garofalo and R. Misasi, Cells, 2021, 10, 3394 CrossRef CAS.
- M. Costanzo, M. Caterino, A. Cevenini, V. Jung, C. Chhuon, J. Lipecka, R. Fedele, I. C. Guerrera and M. Ruoppolo, Data Brief, 2020, 33, 106453 CrossRef.
- M. Costanzo, M. Caterino, I. Salvatori, V. Manganelli, A. Ferri, R. Misasi and M. Ruoppolo, Data Brief, 2022, 41, 107843 CrossRef CAS.
- M. Gonzalez Melo, A. O. Fontana, D. Viertl, G. Allenbach, J. O. Prior, S. Rotman, R. G. Feichtinger, J. A. Mayr, M. Costanzo, M. Caterino, M. Ruoppolo, O. Braissant, F. Barbey and D. Ballhausen, Mol. Genet. Metab., 2021, 134, 287–300 CrossRef CAS PubMed.
- L. Santorelli, M. Caterino and M. Costanzo, OMICS: J. Integr. Biol., 2022, 26, 633–649 CrossRef CAS.
- E. Quagliarini, L. Digiacomo, S. Renzi, D. Pozzi and G. Caracciolo, Nano Today, 2022, 47, 101657 CrossRef CAS.
- W. Fan, H. Peng, Z. Yu, L. Wang, H. He, Y. Ma, J. Qi, Y. Lu and W. Wu, Acta Pharm. Sin. B, 2022, 12, 2479–2493 CrossRef CAS PubMed.
- B.-M. Chen, T.-L. Cheng and S. R. Roffler, ACS Nano, 2021, 15, 14022–14048 CrossRef CAS PubMed.
- C. Finsterwald, S. Dias, P. J. Magistretti and S. Lengacher, Front. Pharmacol., 2021, 12, 653842 CrossRef CAS PubMed.
- A. Benady, D. Freidin, C. G. Pick and V. Rubovitch, Sci. Rep., 2018, 8, 13340 CrossRef.
- InnoMedica Schweiz AG and Bern, Svizzera, Safety Evaluation of Intravenous Talineuren (TLN) in Parkinson’s Disease-affected Patients, ClinicalTrials.gov ID: NCT04976127, 2021, https://www.clinicaltrials.gov/study/NCT04976127?term=Talineuren%26rank=1.
- N. d’Avanzo, D. Paolino, A. Barone, L. Ciriolo, A. Mancuso, M. C. Christiano, A. M. Tolomeo, C. Celia, X. Deng and M. Fresta, Drug Delivery Transl. Res., 2024 DOI:10.1007/s13346-024-01556-3.
- D. Prasad and K. Muniyappa, Biochemistry, 2019, 58, 1295–1310 CrossRef CAS.
- J. Morgenstern, P. Baumann, C. Brunner and J. Hubbuch, Int. J. Pharm., 2017, 519, 408–417 CrossRef CAS.
- K. Abe, K. Higashi, K. Watabe, A. Kobayashi, W. Limwikrant, K. Yamamoto and K. Moribe, Colloids Surf., A, 2015, 474, 63–70 CrossRef CAS.
- P. Resnier, E. Lepeltier, A. L. Emina, N. Galopin, J. Bejaud, S. David, C. Ballet, T. Benvegnu, F. Pecorari, I. Chourpa, J.-P. Benoit and C. Passirani, RSC Adv., 2019, 9, 27264–27278 RSC.
- A. Mori, A. L. Klibanov, V. P. Torchilin and L. Huang, FEBS Lett., 1991, 284, 263–266 CrossRef CAS PubMed.
- P. Grad, L. Gedda and K. Edwards, J. Colloid Interface Sci., 2020, 578, 281–289 CrossRef CAS PubMed.
- M. E. Haque and B. R. Lentz, Biochemistry, 2004, 43, 3507–3517 CrossRef CAS.
- E. Jaradat, E. Weaver, A. Meziane and D. A. Lamprou, Mol. Pharm., 2023, 20, 6184–6196 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS.
- A. A. Sebak, I. E. O. Gomaa, A. N. ElMeshad, M. H. Farag, U. Breitinger, H.-G. Breitinger and M. H. AbdelKader, Int. J. Nanomed., 2020, 15, 9539–9556 CrossRef CAS PubMed.
- D. Ricklin, G. Hajishengallis, K. Yang and J. D. Lambris, Nat. Immunol., 2010, 11, 785–797 CrossRef CAS.
- M. Chen, M. Ding, Y. Li, X. Zhong, S. Liu, Z. Guo, X. Yin, S. Fu and J. Ye, Dev. Comp. Immunol., 2018, 87, 98–108 CrossRef CAS PubMed.
- P. T. Gomme, K. B. McCann and J. Bertolini, Drug Discovery Today, 2005, 10, 267–273 CrossRef CAS.
- R. Wang, Z. Zhang, B. Liu, J. Xue, F. Liu, T. Tang, W. Liu, F. Feng and W. Qu, Biomater. Sci., 2021, 9, 3621–3637 RSC.
- H. Li and Z. M. Qian, Med. Res. Rev., 2002, 22, 225–250 CrossRef CAS.
- M. R. Sepand, M. Ghavami, S. Zanganeh, S. Stacks, F. Ghasemi, H. Montazeri, C. Corbo, H. Derakhshankhah, S. N. Ostad, M. H. Ghahremani and M. Mahmoudi, Nanoscale, 2020, 12, 4935–4944 RSC.
- O. K. Kari, J. Ndika, P. Parkkila, A. Louna, T. Lajunen, A. Puustinen, T. Viitala, H. Alenius and A. Urtti, Nanoscale, 2020, 12, 1728–1741 RSC.
- E. Papini, R. Tavano and F. Mancin, Front. Immunol., 2020, 11, 567365 CrossRef CAS PubMed.
- D. Liu, Q. Hu and Y. K. Song, Biochim. Biophys. Acta, Biomembr., 1995, 1240, 277–284 CrossRef.
- E. Nogueira, A. Loureiro, P. Nogueira, J. Freitas, C. R. Almeida, J. Härmark, H. Hebert, A. Moreira, A. M. Carmo, A. Preto, A. C. Gomes and A. Cavaco-Paulo, Faraday Discuss., 2013, 166, 417 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. B. Haroon, A. C. Hunter, Z. S. Farhangrazi and S. M. Moghimi, Adv. Drug Delivery Rev., 2022, 188, 114396 CrossRef CAS.
- D. Zou, W. Wang, D. Lei, Y. Yin, P. Ren, J. Chen, T. Yin, B. Wang, G. Wang and Y. Wang, Int. J. Nanomed., 2017, 12, 4879–4889 CrossRef CAS.
- M. Mora, M.-L. Sagristá, D. Trombetta, F. P. Bonina, A. De Pasquale and A. Saija, Pharm. Res., 2002, 19, 1430–1438 CrossRef CAS.
- M. Hadjidemetriou, S. McAdam, G. Garner, C. Thackeray, D. Knight, D. Smith, Z. Al-Ahmady, M. Mazza, J. Rogan, A. Clamp and K. Kostarelos, Adv. Mater., 2019, 31, e1803335 CrossRef PubMed.
- M. Hadjidemetriou, Z. Al-Ahmady, M. Mazza, R. F. Collins, K. Dawson and K. Kostarelos, ACS Nano, 2015, 9, 8142–8156 CrossRef CAS PubMed.
- S. Kaur and D. D. Roberts, Semin. Cell Dev. Biol., 2024, 155, 22–31 CrossRef CAS.
- R. Pihl, R. K. Jensen, E. C. Poulsen, L. Jensen, A. G. Hansen, I. B. Thøgersen, J. Dobó, P. Gál, G. R. Andersen, J. J. Enghild and S. Thiel, Sci. Adv., 2021, 7, eaba7381 CrossRef CAS.
- S. Panico, S. Capolla, S. Bozzer, G. Toffoli, M. Dal Bo and P. Macor, Pharmaceutics, 2022, 14, 2605 CrossRef CAS PubMed.
- R. L. Pinals, D. Yang, D. J. Rosenberg, T. Chaudhary, A. R. Crothers, A. T. Iavarone, M. Hammel and M. P. Landry, Angew. Chem., Int. Ed., 2020, 59, 23668–23677 CrossRef CAS.
- H. Iqbal, T. Yang, T. Li, M. Zhang, H. Ke, D. Ding, Y. Deng and H. Chen, J. Controlled Release, 2021, 329, 997–1022 CrossRef CAS PubMed.
- S. Li, H. Zhao, X. Mao, Y. Fan, X. Liang, R. Wang, L. Xiao, J. Wang, Q. Liu and G. Zhao, Pharm. Res., 2019, 36, 168 CrossRef CAS.
- A. Yang, Z. Sun, R. Liu, X. Liu, Y. Zhang, Y. Zhou, Y. Qiu and X. Zhang, Front. Oncol., 2021, 11, 727605 CrossRef CAS PubMed.
- T. Koneru, E. McCord, S. Pawar, K. Tatiparti, S. Sau and A. K. Iyer, ACS Omega, 2021, 6, 8727–8733 CrossRef CAS.
- S. Andrade, M. J. Ramalho, J. A. Loureiro and M. C. Pereira, Int. J. Pharm., 2022, 626, 122167 CrossRef CAS PubMed.
- S. Andrade, J. A. Loureiro and M. C. Pereira, Pharmaceutics, 2022, 14, 2163 CrossRef CAS.
- R. Bilardo, F. Traldi, A. Vdovchenko and M. Resmini, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2022, 14, e1788 CAS.
- M. Ismail, W. Yang, Y. Li, T. Chai, D. Zhang, Q. Du, P. Muhammad, S. Hanif, M. Zheng and B. Shi, Biomaterials, 2022, 287, 121608 CrossRef CAS PubMed.
- Y.-C. Kuo, I.-W. Ng and R. Rajesh, Mater. Sci. Eng., C, 2021, 127, 112233 CrossRef CAS.
- N. Grafals-Ruiz, C. I. Rios-Vicil, E. L. Lozada-Delgado, B. I. Quiñones-Díaz, R. A. Noriega-Rivera, G. Martínez-Zayas, Y. Santana-Rivera, G. S. Santiago-Sánchez, F. Valiyeva and P. E. Vivas-Mejía, Int. J. Nanomed., 2020, 15, 2809–2828 CrossRef CAS.
- P. Dalhaimer, B. Florey and S. Isaac, ACS Nano, 2023, 17, 837–842 CrossRef CAS.
- W. Xu, Y. Niu, X. Ai, C. Xia, P. Geng, H. Zhu, W. Zhou, H. Huang and X. Shi, Biomedicines, 2022, 10, 900 CrossRef CAS PubMed.
- R. Kuai, D. Li, Y. E. Chen, J. J. Moon and A. Schwendeman, ACS Nano, 2016, 10, 3015–3041 CrossRef CAS.
- G. J. Jang, J. Y. Jeong, H. Joung and S. Y. Han, Colloids Surf., B, 2023, 230, 113488 CrossRef CAS PubMed.
- A. J. Chetwynd, W. Zhang, J. A. Thorn, I. Lynch and R. Ramautar, Small, 2020, 16, e2000295 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00345d |
| ‡ These authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2024 |