
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variations around the presence and position of sulfur in sugar-derived cyclic monomers: influence on polymerisation thermodynamics, polymer sequence and thermal properties†‡
Craig
Hardy
 a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*a
a,
Gabriele
Kociok-Köhn
b and
Antoine
Buchard
*a
aCentre for Sustainable and Circular Technologies, Department of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
bMaterials and Chemical Characterisation Facility (MC2), University of Bath, UK
First published on 23rd December 2022
Abstract
Introducing sulfur atoms into a polymer backbone is an interesting way to valorise an abundant by-product of the chemical industry and to modulate polymerisation thermodynamics towards polymer recycling to monomer. For bio-derived polymers, which are often highly oxygenated, replacing oxygen for sulfur atoms can also be a way to fine-tune their properties and widen their application scope. Herein, we report a series of 6-membered cyclic carbonate and thiocarbonate (xanthate, thionocarbonate and monothionocarbonate) monomers made from carbohydrate derivative D-glucal, in which the number and position of sulfur atoms in the polymerisable ring have been varied. Their organocatalysed ring-opening polymerisation (ROP) is demonstrated and contrasted in terms of ROP thermodynamics, polymer sequence, regioregularity and thermal properties. While the influence of the thiocarbonyl function varied across the series, a C–S bond in the polymerisable ring reduced ring strain and maximum monomer conversion at equilibrium. Sulfur did not induce crystallinity in the polymers studied, but the onset of thermal degradation and the glass transition temperature decreased with the amount of sulfur in the polymer linkages, regardless of its position. Degradation of the polymers under UV light was also explored. This work provides fundamental insight for the design of future sulfur-containing renewable polymeric materials.
Introduction
Incorporating sulfur atoms into polymer backbones has some benefits in terms of waste valorisation. Indeed, elemental sulfur is an abundant by-product of the oil and chemical industry,1–3 (over 70 million tonnes produced annually). Besides, the presence of sulfur has been shown to enhance the thermal (e.g., increased crystallinity), physical (e.g., affinity for metals4,5), mechanical and optical properties (e.g., high refractive index6,7), of the resulting polymers.8–12 In the context of sustainability, sulfur can also impart desirable features to polymers such as recyclability to monomer,13–17 degradability18 and photodegradability.19,20 For bio-derived polymers, which are often highly oxygenated, replacing oxygen atoms with sulfur ones can result in novel properties and broader applications. Recently, as part of our wider research programme on synthetic carbohydrate polymers,21–30 we have for example reported the polymerisation of a xanthate cyclic monomer derived from tri-O-acetyl-D-glucal (a commercial derivative of D-glucose) and CS2 (Fig. 1), and shown that when copolymerised with lactide, the resulting polymer chains can be cleaved using UV light.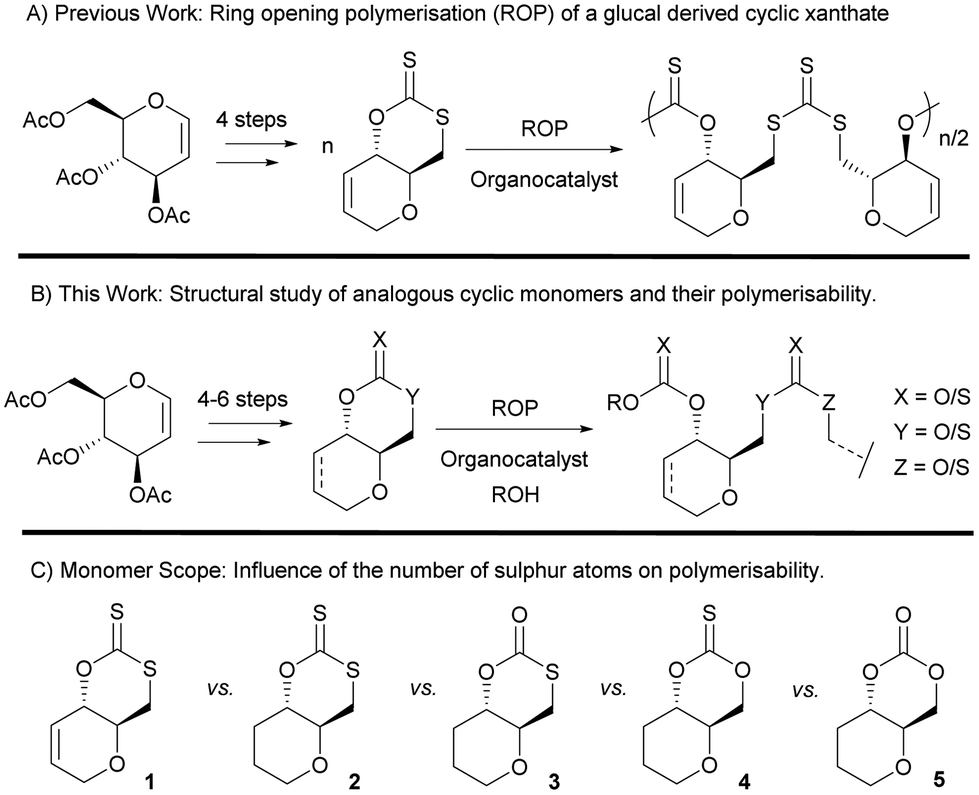 | ||
| Fig. 1 (A) Organocatalysed ring opening polymerisation of a cyclic xanthate; (B) organocatalysed ring opening polymerisation of novel cyclic monomers and investigation of polymerisability and the properties of resultant polymers; (C) concept of exploring quantity and arrangement of sulfur atoms within the carbonyl/thiocarbonyl linkage. | ||
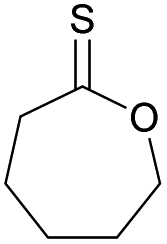
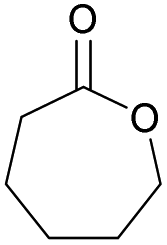
Table 1 shows selected examples of sulfur-containing cyclic monomers and their oxygenated counterparts, alongside some of their thermal properties. Even if not comprehensive, this table demonstrates the mounting interest in this area. It also highlights that trends remain difficult to identify, and that studies comparatively exploring the structure–property relationship of sulfur containing polymers and their oxygenated analogues are scarce,31 in particular with regards to the polymerisability of the monomers, depending on the number and position of sulfur atoms. However, this knowledge is of crucial importance. Recently Hong and coworkers32 thus showed that γ-thionobutyrolactone is easier to polymerise than γ-butyrolactone17 and γ-thiobutyrolactone.33,34
| Sulfur monomer | Ref. | Thermal properties | Oxygen monomer | Ref. | Thermal properties |
|---|---|---|---|---|---|
|
38 | T m = 9 °C |
|
39 | T g = −65–60 °C |
| T m = 56–60 °C | |||||
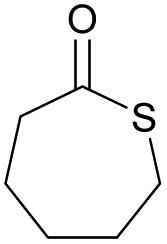
|
20 and 40 | T g = −40 °C |
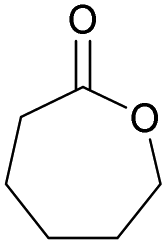
|
39 | T g = −65–60 °C |
| T m = 105 °C | T m = 56–60 °C | ||||
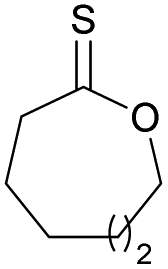
|
41 | T m = 19 °C |
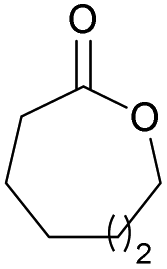
|
41 | T m = 61 °C |
|
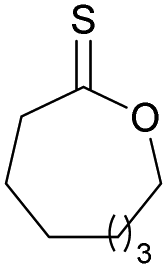
41 | T m = 0 °C |
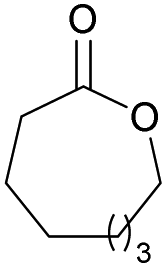
|
41 | T m = 70 °C |
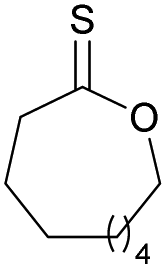
|
41 | T m = 62 °C |
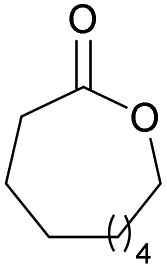
|
41 | T m = 97 °C |
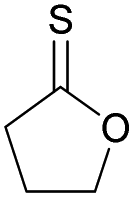
|
32 | T g = −49 °C |
|
17 | T g = −42–51 °C |
| T m = 100 °C | T m = 52–63 °C | ||||
|
31 | T g = 18 °C |
|
42 | T g = 50–60 °C |
|
15 | T g = 59 °C |
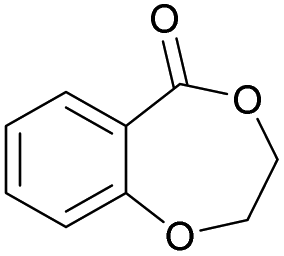
|
43 | T m = 78 °C |
| T m = 130–150 °C | |||||
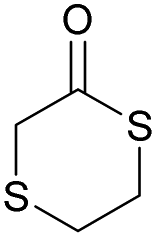
|
44 | T g = −34 °C |
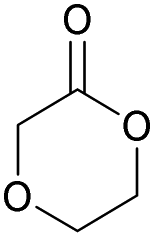
|
45 | T g = −6 °C |
| T m = 88 °C | T m = 108 °C | ||||
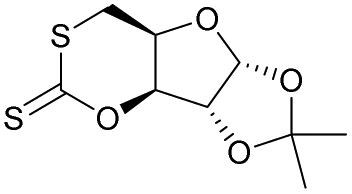
|
27 | T g = 114 °C |
|
28 | T g = 128 °C |
Herein we report the synthesis and polymerisation of a series of cyclic (thio)carbonate monomers derived from tri-O-acetyl-D-glucal, featuring different sulfur/oxygen configurations (Fig. 1). Monosaccharides are promising abundant, structurally diverse and highly functionalisable renewable building blocks for polymer synthesis,35,36 with Wooley and coworkers being the first to exploit tri-O-acetyl-D-glucal as a precursor towards cyclic monomers amenable to ROP.37 The molecular structures of each monomer are studied and discussed in relation to their polymerisability. The resulting polymer structures and thermal properties are compared, with trends being drawn around the influence of the number of sulfur atoms and their positions in the cyclic monomers.
Results and discussion
Synthesis
A series of sugar-based bicyclic monomers, with different sulfur/oxygen configurations within the carbonyl functional group, were synthesised from tri-O-acetyl-D-glucal (Scheme 1).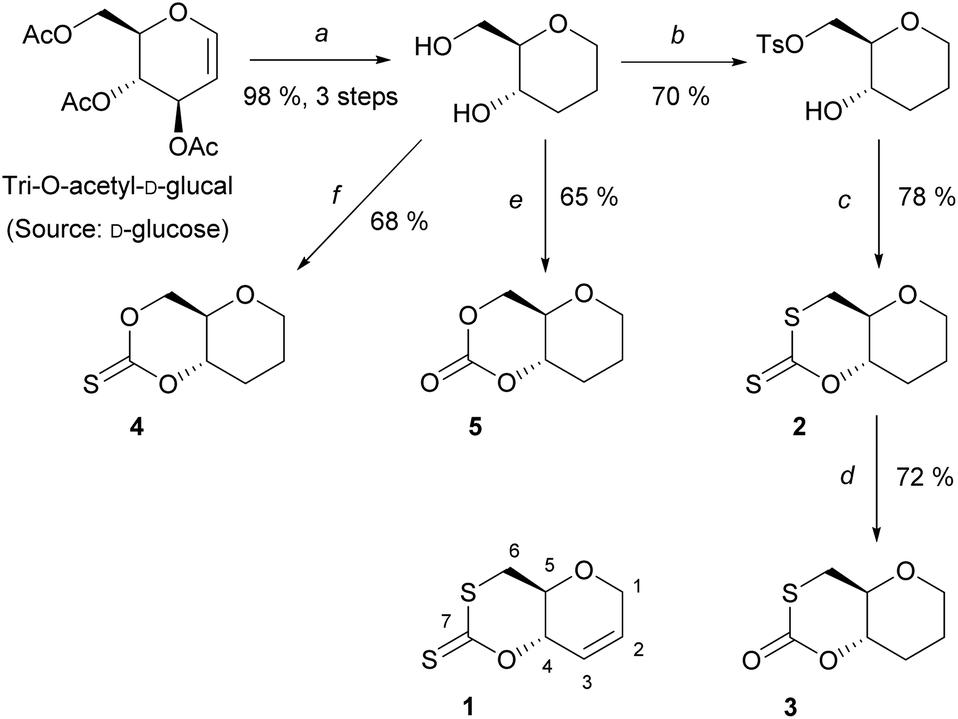 | ||
| Scheme 1 Synthesis of monomers, 1–5, from tri-O-acetyl-D-glucal; (a) (i) Et3SiH, BF3OEt2, DCM, 0 °C, 3 h, (ii) NaOMe (cat.), MeOH, rt, 2 h, (iii) Pd/C, MeOH, rt, 20 h (98%, 3 steps); (b) TsCl, pyridine, rt, 20 h, 70%; (c) CS2, t-BuOK, THF, 0 °C, 3 h, 78%; (d) O3, DCM, −78 °C, 72%; (e) CDI, DCM, 0 to 20 °C, 3 h, 65%; (f) TCDI, DCM, 0 to 20 °C, 3 h, 68%. Monomer 1 was synthesised according to literature.21 | ||
Building from strategies developed by Wooley and coworkers37 and by our team,21 each monomer synthesis begins with the Ferrier rearrangement of tri-O-acetyl-D-glucal with triethylsilane in the presence of a Lewis acid (BF3·EtO2), followed by the subsequent de-protection using sodium methoxide under Zemplén conditions. While the Ferrier rearrangement may add a functional alcohol onto the anomeric position, here a proton was inserted to remove the possibility of anomers and simplify analysis. The hydrogenation of the pseudo-glucal using Pd/C under H2 (1 atm)46 then produces a saturated sugar-derived diol, with almost quantitative yield (98%) over 3 steps.
Similar to the procedure reported for the unsaturated xanthate monomer, 1,21 tosylation at the C-6 position followed by cyclisation using CS2 produce saturated xanthate, 2 (see ESI, section 1‡). 2 is purified by recrystallisation using ethanol, and isolated as pale-yellow crystals. Next, ozonolysis can be used to cleave the thiocarbonyl group of xanthate 2 and replace it by a carbonyl group. Following a procedure by Zard and coworkers, under a stream of O3 in oxygen, full conversion of 2 into monothiocarbonate 3 is observed within minutes.473 is subsequently purified by column chromatography and isolated as white crystals. It is worth noting that this procedure is incompatible with xanthate 1, with the alkene functionality also being capable of undergoing ozonolysis. Lastly, thionocarbonate and carbonate monomers, 4 and 5 are synthesised from the diol using 1,1′-thiocarbonyldiimidazole (TCDI) and 1,1′-carbonyldiimidazole (CDI), respectively. 4 and 5 are purified by recrystallisation from ethanol and diethyl ether, respectively, and isolated as white crystals.
All novel monomers were fully characterised, including by NMR and FTIR spectroscopies, and their solid-state structures were elucidated by single crystal X-ray diffraction analysis (Fig. 2). The 13C{1H} NMR chemical shift of the (thio)carbonyl group was primarily used to identify each monomer. Like 1, 2 displays a signal at δC ≈ 208 ppm, characteristic of the C(S)SO linkage of a cyclic xanthate. 3 shows a signal at δC ≈ 165 ppm, consistent with a thiocarbonate (C(O)SO), while 4 features a signal at 189 ppm, corresponding to a thionocarbonate (C(S)O2). The quaternary carbon of the cyclic carbonate 5 displays a resonance at significantly lower chemical shift (δC ≈ 148 ppm) to the other monomers. Sulfur being more electropositive than oxygen, as the quantity of sulfur within and around the carbonyl functional group increases, so does the associated chemical shift. In addition, due to the adjacent electronegative oxygen atom, the C6 signal in 4 and 5 is also significantly de-shielded, appearing at higher chemical shifts in both 1H and 13C NMR spectroscopy (δH ≈ 4.51–4.15, δC ≈ 68 ppm) compared to 1–3 (δH ≈ 3.15–3.02, δC ≈ 32–34 ppm).37,48 The 3JH4−H5 coupling constant (9.5–9.8 Hz) for each compound is also consistent with trans-fused cyclic monomers.
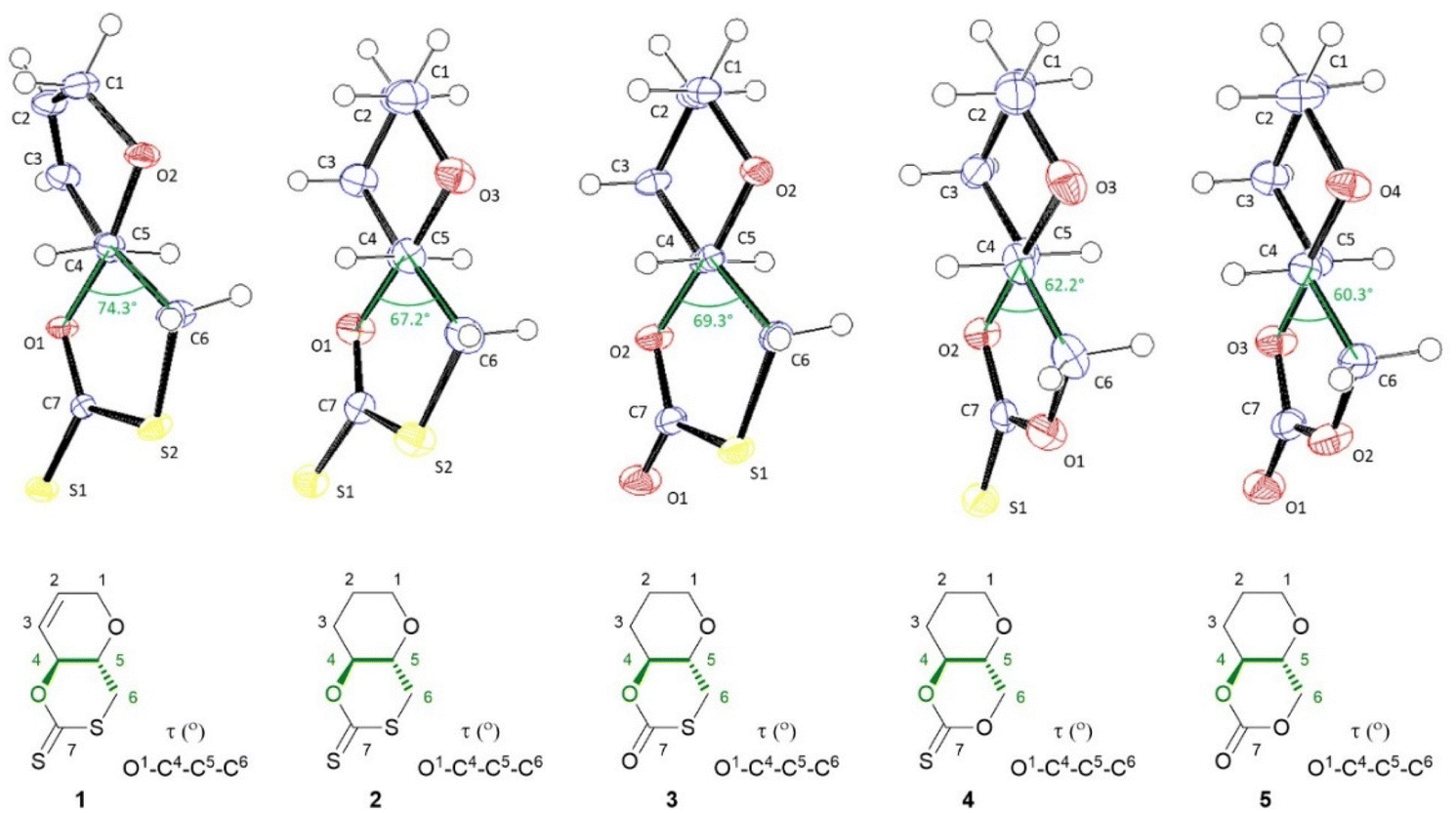 | ||
| Fig. 2 ORTEP drawings of the crystal structure of 1–5 with thermal ellipsoids at the 50% probability level (see ESI‡). For each crystal structure the O–C4–C5–C6 torsion angle is drawn and highlighted in the accompanying structures below. Single crystals were obtained for each monomer by layering chloroform with n-hexane. | ||
The molecular structure and stereochemistry of each monomer was further corroborated by X-ray diffraction crystallography (XRC) of single crystals obtained for each monomer by layering chloroform with n-hexane (Fig. 2). Fig. 2 also illustrates how the dihedral angle O1–C4–C5–C6 varies between monomers 1–5, a feature that will later be linked to ring strain and monomer polymerisability (vide infra).
Ring-opening polymerisation
The ring-opening polymerisation (ROP) of 2–5 was successfully conducted at room temperature in dichloromethane (DCM), using 4-methylbenzyl alcohol (4-MeBnOH) as initiator, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as catalyst, and 1–2 mol L−1 initial monomer concentration, unless stated otherwise (Table 2). Size exclusion chromatography (SEC) was used to confirm the polymeric nature of the products (vide infra). The polymerisation procedure was selected based on our previous study of the ROP of 1,21 which showed that TBD was efficient in polymerising cyclic xanthates, giving good polymerisation control and limiting the number of cyclic species formed by direct nucleophilic initiation.27,49,50| Entry | M | [M]0 | [M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[TBD]0:[I]0b :[TBD]0:[I]0b |
Time (h) | Conv.c (%) | M n,calc (kg mol−1) | M n,NMR (kg mol−1) | M n,SEC [ĐM] (kg mol−1) |
|---|---|---|---|---|---|---|---|---|
| a Polymerisations were carried out at room temperature, under an argon atmosphere, in anhydrous CH2Cl2 solvent with initial [M]0 = 1–2 mol L−1 (M = monomer), unless stated otherwise. b I = 4-methylbenzylalcohol. c Monomer conversion to polymer, calculated based on the relative integration of the monomer proton signals and polymer signals, in the 1H NMR spectrum. d Number-average molar mass as calculated using Mr(I) + (Mr(monomer) × [monomer]0/[I]0 × conv/100%). e Number-average molar mass as calculated using NMR spectroscopy. f Number-average molar mass and Dispersity (Mn,SEC, Mw,SEC, Đ), calculated by SEC relative to polystyrene standards in THF eluent. g Number-average molar mass and Dispersity (Mn,SEC, Mw,SEC, Đ), calculated by SEC relative to polystyrene standards in chloroform eluent. h Bimodal polymer distribution observed by GPC analysis. | ||||||||
| 1 | 2 | 1.0 | 100:1:1 |
6 | 28 | 5.5 | 3.9 | 3.6 [1.39] |
| 2 | 2 | 2.0 | 50:1:1 |
6 | 60 | 4.9 | 6.8 | 5.5 [1.54] |
| 3 | 2 | 2.0 | 100:1:1 |
6 | 54 | 8.7 | 7.5 | 6.4 [1.60] |
| 4 | 2 | 2.0 | 200:1:1 |
6 | 40 | 12.8 | 9.2 | 7.7 [1.57] |
| 5 | 2 | 3.0 | 100:1:1 |
6 | 59 | 11.4 | 5.7 | 5.6 [1.80] |
| 6 | 2 | 4.0 | 100:1:1 |
6 | 65 | 12.5 | 6.3 | 6.4 [1.80] |
| 7 | 3 | 1.0 | 100:1:1 |
6 | 15 | 2.7 | 1.9 | 1.9 [1.23]g |
| 8 | 3 | 2.0 | 50:1:1 |
6 | 36 | 3.8 | 2.0 | 2.6 [1.50]g,h |
| 9 | 3 | 2.0 | 100:1:1 |
6 | 42 | 6.4 | 2.3 | 2.8 [1.46]g,h |
| 10 | 3 | 2.0 | 200:1:1 |
6 | 26 | 9.2 | 1.2 | 1.8 [1.34]g,h |
| 11 | 4 | 0.3 | 100:1:1 |
0.15 | 38 | 6.7 | 5.2 | 4.0 [1.19] |
| 12 | 4 | 1.0 | 50:1:1 |
1 | >99 | 8.7 | 6.5 | 5.7 [1.38] |
| 13 | 4 | 1.0 | 100:1:1 |
1 | >99 | 17.4 | 9.3 | 7.1 [1.32] |
| 14 | 4 | 1.0 | 200:1:1 |
1 | >99 | 34.6 | 12.5 | 8.2 [1.31] |
| 15 | 4 | 2.0 | 100:1:1 |
1 | >99 | 17.4 | 9.6 | 7.9 [1.45] |
| 16 | 5 | 0.3 | 100:1:1 |
0.15 | 76 | 12.1 | 7.2 | 3.5 [1.17]h |
| 17 | 5 | 1.0 | 50:1:1 |
0.15 | >99 | 8.0 | 7.5 | 4.9 [1.27] |
| 18 | 5 | 1.0 | 100:1:1 |
0.15 | >99 | 15.8 | 10.3 | 5.5 [1.19]h |
| 19 | 5 | 1.0 | 200:1:1 |
0.15 | >99 | 31.4 | 12.2 | 4.3 [1.19]h |
| 20 | 5 | 2.0 | 100:1:1 |
0.15 | >99 | 15.8 | 11.5 | 6.0 [1.23]h |
The ROP of 2 proceeded rapidly, yet reaching a maximum monomer conversion that was dependent on the initial monomer concentration: from 28% (for [2]0 = 1 mol L−1), 54% (2 mol L−1) and 59% (3 mol L−1), to 65% (4 mol L−1) (Table 2, entries 1, 3, 5 and 6; Fig. S68‡). This equilibrium-limited polymerisation is reminiscent of that of unsaturated xanthate 1,21 and typical of ROP of monomers with limited ring-strain.29,51–54 However, monomer concentration at equilibrium should only depend on temperature (Table S1,‡ data for 1), and here we observe an unexpected increase in [2]eq with [2]0 (Fig. S69‡), which we do not have any explanation for and are currently investigating. Next, the temperature dependence of the ROP equilibrium was investigated between 0–60 °C in 1,2-dichloroethane ([2]0 = 2 mol L−1; Table S1‡). As expected, maximum conversion decreased with increased temperature. A plot of ln([2]eq) vs. the reciprocal of the absolute temperature gave an approximation of the polymerisation thermodynamic parameters (Fig. S70‡): ΔHp = −3.41 kJ mol−1 and ΔSp = −15.7 J mol−1 K−1 (normalised to 1 mol L−1).44 For comparison, under similar conditions, we have reported for the ROP of 1 an enthalpic driving force of −25.6 kJ mol−1 and accompanying −80 J mol−1 K−1 entropy decrease.21 The significant increase in both ΔHp and ΔSp with the saturation of the C6 pyranose sugar ring was attributed to the decreased ring strain and rigidity of monomer 2 compared to 1.
The ROP of monothiocarbonate 3 seemed also limited by thermodynamics, reaching for example a maximum monomer conversion of 42% at room temperature for [3]0 of 2 mol L−1 (Table 2, entry 9). However, a rigorous analysis of this equilibrium was complicated by the rapid precipitation of a solid from the reaction mixture. This product was highly insoluble in all common organic solvents and in water. However, solubility in a chloroform–hexafluoroisopropanol (HFIP) mixture confirmed its polymeric nature and enabled an estimation of Mn and ĐM by SEC (vide infra). While precipitation of the polymer may be seen as beneficial to drive the equilibrium towards monomer conversion, during that process, the concentrations of both monomer and initiating chains decrease, impeding further monomer conversion and slowing down the reaction, respectively.
The thermodynamics limitations in the ROP of xanthate and thiocarbonate monomers 1–3 are in stark contrast with the C6-sugar pyranose-based cyclic carbonate monomer 5 for which the ROP equilibrium lies heavily towards polymeric products (over 99% observed conversion) (Table 2, entries 16–20). When exchanging the carbonate functionality for a thionocarbonate, a minor decrease in reactivity is observed (Table 2, entry 11), however, the ROP of 4 still proceeds at a faster rate than both xanthate monomers and to full conversion (>99% in 1 hour; Table 2, entries 12–15).
As evaluated by SEC, polymers of up to 7800 (ĐM 1.23), 2800 (ĐM 1.46), 8200 g mol−1 (ĐM 1.31) and 6000 (ĐM 1.23) could be obtained from the ROP of 2, 3, 4 and 5 respectively. While broadly speaking molar masses increase with decreasing quantities of initiator as expected, some discrepancy with theoretical molar masses currently limits the controlled polymerisation of these monomers. This is attributed to the presence of impurities in the monomers, such as diols and other protic impurities, acting as chain transfer agents and limiting the increase in Mn at high monomer:initiator ratios. Bimodal distributions are also occasionally observed, and this is attributed to undesired inter- or intra-(cyclisation) transesterification reactions during polymer growth. Similar behaviour has been reported by Wooley and coworkers for the unsaturated D-glucal based cyclic carbonate.37 End-group analysis was performed using NMR spectroscopy, and with the exception of polymers produced from 5, good agreement between Mn,NMR and Mn,SEC values suggested the absence of major cyclic polymer species. For poly(5), Mn,NMR values were approximately double those found by SEC, consistent with the major presence of cyclic species, as suspected from bimodal distributions. These conclusions are to be taken carefully without further support from supplementary end-group analysis or determination of absolute molar masses. However, MALDI-ToF mass spectrometry proved unsuccessful to probe further the macromolecular structure of the polymers.
Monomer structural, spectroscopic and computational analysis
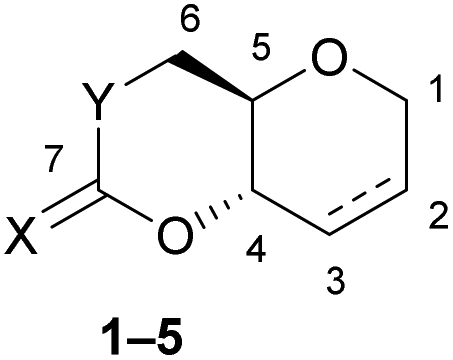
The impact of incorporating sulfur into the monomer structures was further investigated by comparing structural, spectroscopic and computational data with the conversion observed experimentally (Table 3).a
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Monomer | C7![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X (ppm) X (ppm) |
mpb (°C) | C7X (Å) |
C7–Y (Å) | O–C7–Y (°) | O–C4–C5–C6 (°) | Δχc (°) | ΔHROPd |
Max Conv.e (%) |
|
a Structural data collected for monomers 1–5 by NMR, DSC, XRD (with [estimated standard deviations]) and computational analysis, M = monomer.
b mp = melting point, generated by DSC analysis.
c Change in torsion angle, calculated based on the relative change in the O–C4–C5–C6 torsion angle of monomer 3vs. other monomers.
d Enthalpy change for the isodesmic reactions of monomers 1–5, units given in kcal mol−1.
e Maximum monomer conversion to polymer for [M]0 = 2 mol L−1 and [M]0:[TBD]0:[4-MeBnOH]0 = 100:1:1, calculated based on the relative integration of the monomer proton signals and polymer signals, in the 1H NMR spectrum.
|
|||||||||
| 1 | 208 | 122 | 1.646[2] | 1.733[2] | 122.9[2] | 74.3[2] | 5.0 | −6.4 | 90 |
| 2 | 207 | 110 | 1.655[2] | 1.726[2] | 123.9[2] | 67.2[3] | 2.1 | −6.2 | 54 |
| 3 | 165 | 122 | 1.207[2] | 1.761[2] | 122.5[1] | 69.3[2] | 0 | −4.9 | 42 |
| 4 | 189 | 90 | 1.649[2] | 1.327[2] | 120.5[2] | 60.3[2] | 7.1 | −8.6 | 99 |
| 5 | 148 | 92 | 1.204[2] | 1.333[2] | 120.1[2] | 62.2[2] | 9.0 | −8.9 | 99 |
To complement experimental ROP data, the enthalpy of ring-opening of each monomer was evaluated by computational modelling (DFT). Recently, Ramprasad and coworkers55 have developed a first-principles computational scheme to calculate ΔHROP with high precision, by involving conformational sampling and taking into account finite size effect. Here, a more conventional and basic approach, involving the isodesmic ring-opening reaction of the monomers, was used.56 While such method may not yield accurate absolute values, the calculated ΔHROP values were found to reproduce relative experimental ROP trends, with monomers having the most negative ΔHROP values showing the highest monomer conversion at equilibrium, and vice versa (Fig. S115 and S116‡). This suggests that the ROP of these monomers is mainly driven by the release of their ring strain.
Experimental ROP data was therefore evaluated against some of the structural features of each monomer, as determined by X-ray diffraction of single crystals (Fig. 2). In particular, the torsion angle between atoms O–C4–C5–C6 was taken as a good representation of ring strain. Monomer 3, which experimentally show the lowest polymerisability and computationally the least negative ΔHROP, was taken as a reference, from which deviations in torsion angle (Δχ) were calculated for each monomer (Table 3). Strong correlations were observed between Δχ and the maximum monomer conversion observed (Fig. 3a) or ΔHROP (Fig. 3b). Therefore, for this type of cyclic monomers, it appears possible to explain and predict polymerisability from a simple structural parameter.
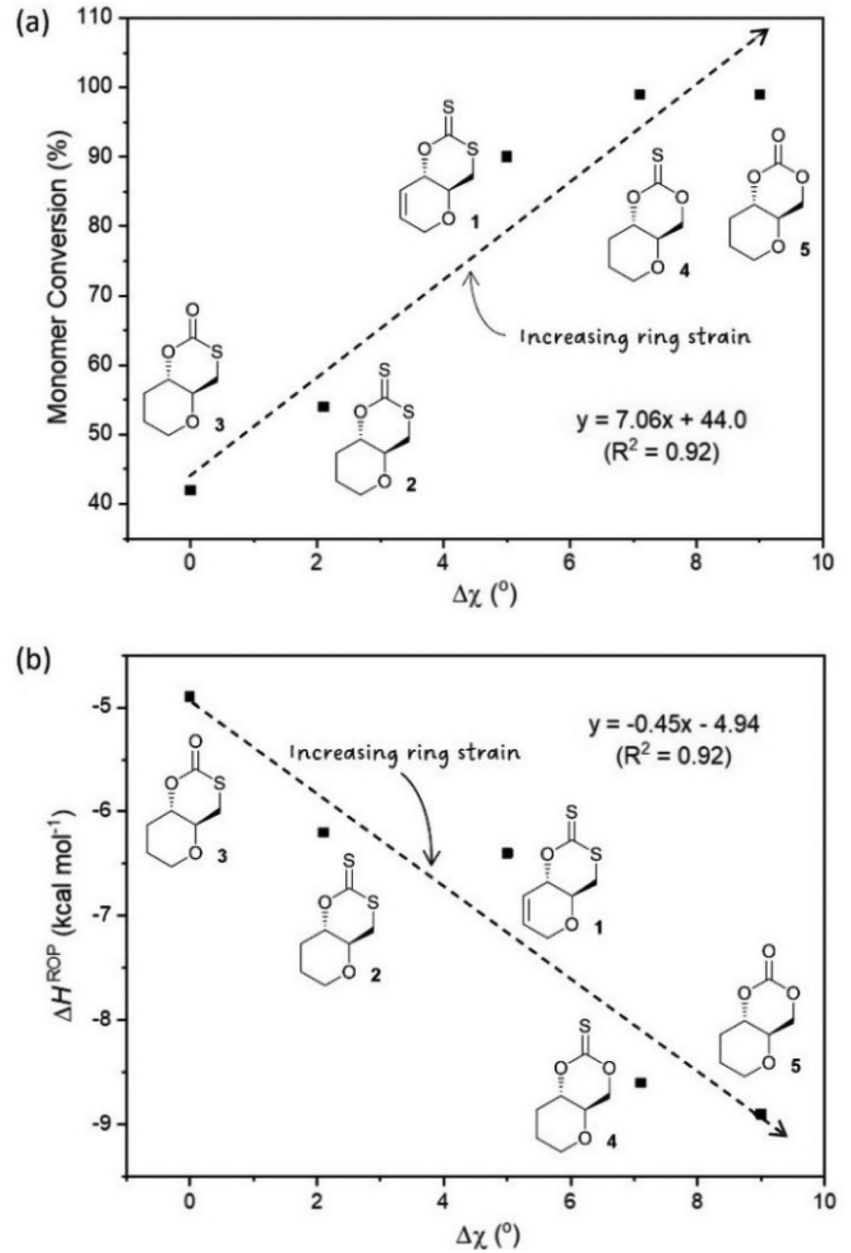 | ||
| Fig. 3 (a) Plot of Δχ vs. monomer conversion (b) relationship between Δχ obtained from XRD data and ΔHROP calculated as a representation of ring strain. | ||
Unlike the influence of the saturation of the C6 pyranose ring on monomer polymerisability (1vs. 2), that of sulfur incorporation within the carbonate functional group was less clear (5vs. 4, 3 and 2). We found that in general, exchanging an oxygen for a sulfur atom decreased ring strain and polymerisability. However, the position of this substitution mattered. As expected, when inserting a sulfur atom into the C6-alkoxy position of the carbonate group and lengthening the C7–Y bond from 1.333[2] (in 5) to 1.761[2] Å (in 3) a large decrease in both ring strain and polymerisability was observed (Δχ = 9.0° and ΔHROP = +3.99 kcal mol−1) and led to a significant decrease to monomer conversion at equilibrium (Table 3). To a larger extent, the same observation was made when inserting a sulfur atom into the C6-alkoxy position of the thionocarbonate group, with a decrease in monomer conversion at equilibrium between 4 and xanthate 2 (Table 3). Replacing the carbonyl oxygen atom for sulfur and lengthening the C7X from 1.204[2] (in 5) to 1.649[2] Å (in 4) had little influence on the ROP equilibrium (both monomers polymerise >99%), even if kinetics were slightly faster for the carbonate (76% conversion after 15 min for 5vs. 38% for 4; Table 2 entries 16 and 11). On the other hand, replacing the carbonyl oxygen atom in monothiocarbonate 3 for sulfur, and lengthening the C7X in xanthate 2 (1.655[2] Å vs. 1.207[2] Å in 3), unexpectedly led to a decrease in the C7–Y bond distance (from 1.761[2] (in 3) to 1.726[2] Å (in 2)), which may explain the positive impact seen on ring strain and polymerisability.
Polymer structures
Initial insight into the structure of the polymers produced from 1–5 was gained by FT-IR spectroscopy (Fig. S67, S88, S101 and S113‡). The xanthate-derived polymer, poly(2), displayed multiple strong absorption bands within the 1290–1150 cm−1 region, which were assigned to the carbon–sulfur double bond (CS) vibrational modes. The thiono-derived polymer, poly(4), displayed a similar broad absorption band at 1200 cm−1. In contrast, strong carbonyl (CO) frequencies were seen for both monothiocarbonate-derived poly(3) and carbonate-derived poly(5), within the 1750–1700 cm−1 region. Notably, the vibrational mode belonging to poly(3) appears at a lower frequency (1710 cm−1) than polycarbonate poly(5) (C(O)O2) (1740 cm−1), likely due to the presence of the more electropositive sulfur atom and corroborating thiocarbonate (C(O)SO) linkages.
13C{1H} NMR spectroscopy proved more decisive in identifying the microstructure of the polymer species (Fig. 4). For poly(2), two major thiocarbonyl resonances at δC ≈ 194 and 223 ppm of similar intensity could be seen. Based on literature precedent,21,27 these resonances were assigned to a mono-thionocarbonate (C(S)O2) environment and a tri-thiocarbonate environment (C(S)S2), respectively. This data suggests that, like its unsaturated counterpart 1, the ROP of 2 proceeds via the alternating opening of the monomer at that both ends of the xanthate linkage, followed by the subsequent propagation of the polymer chain to produce regio-regular polymers with alternating C(S)S2 and C(S)O2 linkages.
 | ||
| Fig. 4 Comparison of the 13C{1H} NMR spectra (500 MHz, chloroform-d) of monomers 2–5 (top) and their respective polymers. (a) Cyclic xanthate monomer 2 (bottom) and polyxanthate poly(2) (top) (Table 2, entry 3), (inset) alternating C(S)S2 and C(S)O2 linkages observed for the respective polymer. (b) Cyclic thiocarbonate monomer 3 (bottom) and polythiocarbonate poly(3) (top, signals due to hexafluoroisopropanol visible at 121.5 (q) and 69.7 (m) ppm (Table 2, entry 9), (inset) C(S)SO linkage observed for the respective polymer. (c) Cyclic thionocarbonate monomer 4 (bottom) and polythionocarbonate poly(4) (top) (Table 2, entry 13), (inset) TT, TH and HH linkages observed for the respective polymer. (d) Cyclic carbonate monomer 5 (bottom) and polycarbonate poly(5) (top) (Table 2, entry 18), (inset) TT, TH and HH linkages observed for the respective polymer. | ||
The 13C{1H} NMR spectrum of poly(5) revealed three carbonate environments centred at 155.2, 154.2 and 153.3 ppm, consistent with tail–tail (TT), tail–head (TH) and head–head (HH) linkages that arise from cleavage at either side of the asymmetric carbonate by either a free secondary or primary propagating alcohol chain. Relative assignments were suggested based upon previous work within and outside of our group.29,30,37 Further splitting observed within the carbon environments is attributed to long range sequence effects. Notably, compared to the 0.5:1:0.5 TT:TH:HH integration ratio that would be expected for a regiorandom polymer, a 0.37:1:0.35 (TT:TH:HH) ratio was observed. Like for its unsaturated analogue,37 this suggests a slight regioregularity, with the acyl–oxygen bond cleavage occurs preferentially on one side of the carbonate carbonyl. It is worth noting that Wooley and coworkers have identified the importance of side-chain functionalities (absent here) to promote regioregular ROP of similar glucose-derived polycarbonates.57
A similar pattern was observed for poly(4), with 13C{1H} NMR analysis revealing three thionocarbonate environments centred at 195.4, 194.1 and 193.1 ppm, consistent with TT, TH and HH linkages respectively.27 Importantly, the absence of other (thio)carbonyl signals ruled out any O–S exchange reactions during ROP. Similar to the polycarbonate, a 0.37:1:0.34 (TT:TH:HH) integration ratio suggested a slight regioregularity.
The 13C{1H} NMR spectrum of poly(3) showed a single thiocarbonate environment at δC ≈ 165 ppm, attributed to monothiocarbonate (C(O)SO) linkages. The absence of other thiocarbonate environments suggests that unlike for 2, 4 and 5, the ROP proceeds via the regioregular opening of the monomer on one side of the thiocarbonate, resulting in only TH-type linkages within the polymer backbone. Based on the stronger acidity of a primary thiol vs. a secondary alcohol, we propose that the ring opening takes place preferentially via acyl–sulfur cleavage favouring the formation of the thiocarbonate linkage.
All polymer structures were further confirmed by 1H NMR spectroscopy, with carbonate and thionocarbonate linkages showing significantly de-shielded C6 proton environments (δH ≈ 4.68–4.14 ppm; CH2–O) compared to trithiocarbonate and monothiocarbonate linkages (δH ≈ 3.80–3.24 ppm; CH2–S).37,48
Thermal properties
The thermal properties of the polymers were investigated by thermogravimetric analysis (TGA) (performed under argon atmosphere) and differential scanning calorimetry (DSC) (Table 4). All polymers were shown to be amorphous, with no observation of endothermic transitions associated with melting.| Polymer | M n,SEC [ĐM] | T g (°C) | T d5% (°C) | T d,max (°C) |
|---|---|---|---|---|
| a Number-average molar mass and dispersity (Mn,SEC, Mw,SEC, Đ), calculated by SEC relative to polystyrene standards in THF eluent, units given in kg mol−1. b Number-average molar mass and dispersity (Mn,SEC, Mw,SEC, Đ), calculated by SEC relative to polystyrene standards in chloroform eluent, units given in kg mol−1. c Glass transition temperatures obtained from the second heating cycle. | ||||
| Poly(1)21 | 6.8 [1.60] | 65 | 188 | 219 |
| Poly(2) | 5.5 [1.54] | 78 | 185 | 202 |
| Poly(2) | 7.7 [1.58] | 92 | 191 | 204 |
| Poly(3) | 2.6 [1.50] | — | 202 | 228 |
| Poly(3) | 2.8 [1.46]b | — | 203 | 241 |
| Poly(4) | 5.7 [1.38] | 86 | 136 | 303 |
| Poly(4) | 7.1 [1.32] | 104 | 140 | 312 |
| Poly(5) | 4.9 [1.27]b | 93 | 215 | 256 |
| Poly(5) | 6.0 [1.23] | 90 | 215 | 262 |
For poly(2), which alternate thionocarbonate and trithiocarbonate linkages, glass transition temperatures (Tgs) of 78 and 92 °C were observed in the second heating cycles, for polymers of Mn 5700 g mol−1 (ĐM 1.54) and 7700 g mol−1 (ĐM 1.58), respectively (Fig. S61 and S63‡). However, changes could be observed from the third heating cycles (Fig. S62 and S64‡) which was attributed to some depolymerisation. This was confirmed by NMR and GPC analysis which showed that heating of poly(2) at 180 °C for 5 minutes resulted in the almost quantitative reformation of 2 (Fig. S71–S74‡). The onset of thermal degradation (Td5%) occurred around 191 °C, with a final mass loss of 97% (for Mn 7700 g mol−1 (Đ 1.54)) (Fig. S65 and S66‡). Direct comparison with unsaturated analogue poly(1) (Mn 6800 g mol−1; ĐM 1.60; Tg 65 °C; Td5% 188 °C),21 reveals non-significant change in thermal stability and a slight decrease in Tg upon hydrogenation of the CC bonds, perhaps due to increased flexibility of the pyranose rings and of the polymer backbone.
For poly(5), Tgs of 93 and 90 °C were observed for polymers of Mn 4900 g mol−1 (Đ 1.27) and 6000 g mol−1 (Đ 1.23) respectively (Fig. S109 and S110‡). Td5% occurred around 215 °C, with a final mass loss of 96% (for Mn 4900 g mol−1 (Đ 1.27)) (Fig. S111 and S112‡). While direct comparison is not possible because of a different substituent in C1, the unsaturated, glucal-derived polycarbonate reported by Wooley and coworkers, with a OiPr group in the anomeric position, displayed lower Tg (≈69 °C) for a polymer of Mn 11800 g mol−1.37 It therefore appears that regardless of the presence of sulfur in the polymer backbone, an unsaturation in the pyranose ring looks to decrease Tg.
Poly(4) displayed a unique glass transition, with Tg of 86 and 104 °C for Mn 5700 (Đ 1.32) and 7900 g mol−1 (Đ 1.38) respectively (Fig. S96–S98‡). However, when heated above 120 °C, a broad exothermic transition was observed, as well as a loss of the Tg transition within the next heating cycle. Therefore, this transition most likely corresponds to the degradation the polymer structure. This theory was further corroborated by TGA data, which revealed Td5% to be around 136 °C, with a final mass loss of 97% (for Mn 5700 g mol−1 (Đ 1.32)) (Fig. S99 and S100‡).
Poly(3) samples did not display any thermal transitions when analysed by DSC (Fig. S82 and S83‡). Dynamic mechanical analysis (DMA) was also performed but remained inconclusive, even if a single broad transition around Tmax = 150 °C may correspond to the softening of the short polymers analysed (Fig. S86 and S87‡). TGA data was however much clearer and revealed an onset of degradation (Td5%) occurring around 203 °C, with a final mass loss of 98% (Fig. S84 and S85‡).
Collectively, even if direct comparisons are not always possible due to concomitant changes in polymer sequence and regioregularity, it appears from the data gathered that replacing oxygen atoms with sulfur atoms leads to a decrease in Tg values for those pyranose-derived poly(thio)carbonates (Fig. 5).
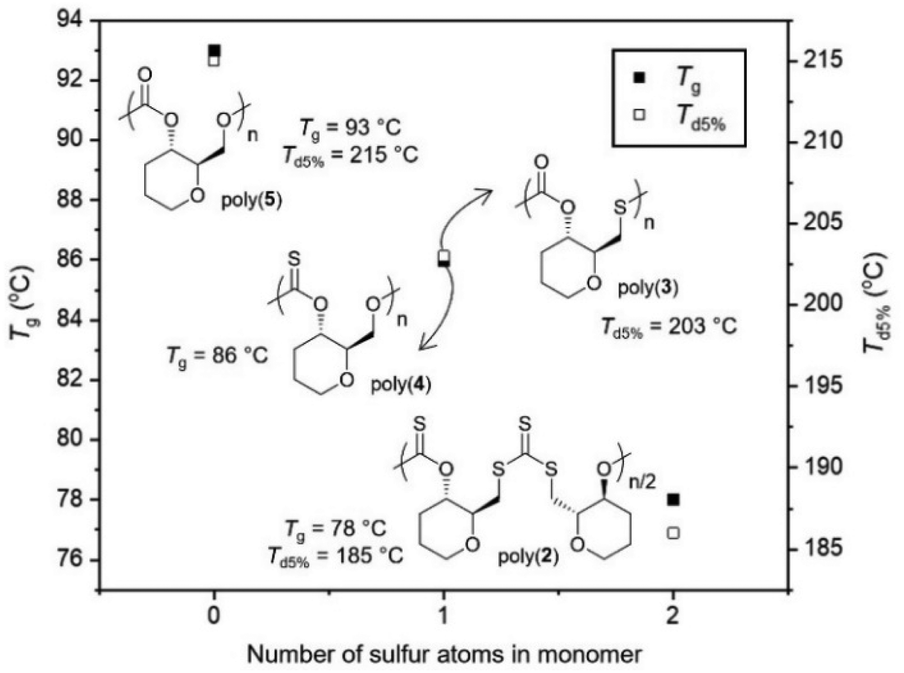 | ||
| Fig. 5 Glass transition and degradation temperatures of poly(2–5) vs. the quantity of sulfur atoms in their monomers. Notable omissions include: poly(3) (no Tg detected) and poly(4) (Td5% = 136 °C). | ||
When within the polymer backbone, this may be due to both the increase in the length of the C–S bond involved in the polymer linkage (1.73 Å vs. 1.33 Å for C–O), and the increase in the van der Waals radius between O (1.52 Å) and S (1.85 Å) atoms. This would increase the free volume within the polymer structure and in turn decrease Tg. While not directly involved in the polymer backbone, changing a carbonyl bond (CO; 1.21 Å) for a thiocarbonyl (CS; 1.65 Å) appears to have a similar influence, that could also be due to an increase in free volume. In general, introducing an element of lesser electronegativity than oxygen may decrease the polarity of the polymer which may facilitate mobility and decrease Tg.
Likewise, a decrease in Td5% was also observed when increasing the quantity of sulfur atoms within the polymer backbone, which is likely due to the reduction in bond strength from C–O to C–S bonds. However, the extreme decrease in thermal stability observed for poly(4) is much harder to explain. The thionocarbonate (C(S)O2) linkages appears to be extremely susceptible to thermal degradation. Based upon the multi-stage degradation profile observed this may also be due to intramolecular reactions (e.g., O/S exchanges) taking place upon heating.
Polymer degradation under UV light
Building upon some of our previous studies, in which poly(1),21 as well as other sulfur containing polymers,25 were readily degraded under UV radiation (λ = 365 nm), the UV-degradability of the polymers was explored in THF at room temperature (Fig. 6). As suspected from its similarity with its unsaturated analogue poly(1), poly(2) was completely degraded within 6 hours into oligomeric products, with 1H NMR analysis revealing the disappearance of trithiocarbonate linkages (loss of the H-6′ environment; Fig. S118‡). Conversely, only 11% reduction in Mn was observed for poly(4) within 6 hours, likely via the degradation of its thiono–carbonate linkages. This was further supported by 1H NMR spectroscopy (Fig. S119 and S120‡), which revealed new proton environments as well as a reduction in the relative integration of the H-6′environments. As expected, poly(5) did not degrade and unfortunately, poly(3) could not be investigated due to solubility issues. Systematic comparisons and definitive conclusions are therefore not possible at this stage, including due to differences in polymer structures. However, the incorporation of sulfur atoms within carbonate linkage appears to enhance UV degradability, with trithiocarbonate linkages degrading faster than thionocarbonate ones.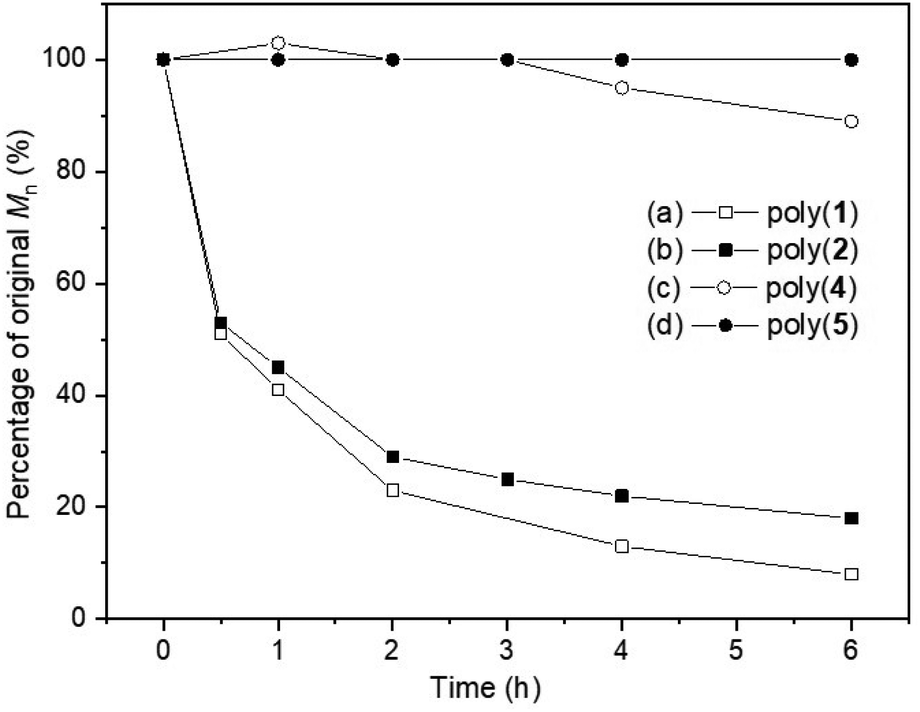 | ||
| Fig. 6 Plot of original Mn percentage vs. time, following the degradation of poly(1) (Mn,SEC = 10000 g mol−1, ĐM = 1.77),21 poly(2) (Mn,SEC = 5500 g mol−1, ĐM = 1.54), poly(4) (Mn,SEC = 6300 g mol−1, ĐM = 1.30) and poly(5) (Mn,SEC = 4900 g mol−1, ĐM = 1.16) when exposed to UV light (λ = 365 nm) for 6 hours, at room temperature, in THF solution. | ||
Conclusions
In summary, we have synthesised a series of novel cyclic carbonate and thiocarbonate monomers derived from carbohydrate derivative D-glucal, in which the number and position of sulfur atoms in the 6-membered polymerisable ring have been varied. All monomers were amenable to organocatalytic ROP but presented various degrees of thermodynamics limitations. Through DFT calculations and the analysis of their experimental solid-state structures, the polymerisability of the monomers was related to their ring strain, indicating that ROP thermodynamics are dominated by enthalpy changes. While the impact of the thiocarbonyl group varied across the series, introducing a sulfur atom inside the polymerisable ring decreased ring strain and maximum monomer conversion at equilibrium. All polymers were characterised, and their sequence identified by NMR spectroscopy. While direct comparisons were not always possible, thermal properties trends could still be identified. Sulfur did not induce crystallinity in the polymers studied, but the onset of thermal degradation and the glass transition temperature were shown to decrease with the amount of sulfur in the polymer linkages, regardless of its position in the polymer linkage. The incorporation of sulfur also promoted degradability under UV light. While the incorporation of sulfur is increasingly seen as a powerful handle to modulate polymerisation thermodynamics and facilitate polymer chemical recycling to monomer, the desired physical properties should be preserved. By exploring the structure–property relationship of sulfur containing polymers and their oxygenated analogues, this work provides fundamental insight for the design of future sulfur-containing renewable polymeric materials, combining adequate properties, recyclability, and degradability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Royal Society (UF\160021, fellowship to AB; RGF\EA\201023; RGF\EA\180028, studentship to CAH) for research funding.References
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed.
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Top. Curr. Chem., 2019, 377, 16 CrossRef PubMed.
- S. Wu, M. Luo, D. J. Darensbourg and X. Zuo, Macromolecules, 2019, 52, 8596–8603 CrossRef.
- J. Bearinger, S. Terrettaz, R. Michel, N. Tirelli, H. Vogel, M. Textor and J. Hubbell, Nat. Mater., 2003, 2, 259–264 CrossRef PubMed.
- E. Marianucci, C. Berti, F. Pilati, P. Manaresi, M. Guaita and O. Chiantore, Polymer, 1994, 35, 1564–1566 CrossRef.
- M. Olshavsky and H. R. Allcock, Macromolecules, 1997, 30, 4179–4183 CrossRef.
- X.-H. Zhang and P. Theato, Sulfur-containing polymers: from synthesis to functional materials, John Wiley & Sons, 2021 Search PubMed.
- T.-J. Yue, L.-Y. Wang and W.-M. Ren, Polym. Chem., 2021, 12, 6650–6666 RSC.
- M. Luo, Y. Li, Y.-Y. Zhang and X.-H. Zhang, Polymer, 2016, 82, 406–431 CrossRef.
- G. Montaudo, C. Puglisi, C. Berti, E. Marianucci and F. Pilati, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2277–2290 CrossRef.
- H. R. Kricheldorf and G. Schwarz, J. Macromol. Sci., Part A: Pure Appl.Chem., 2007, 44, 625–649 CrossRef.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, J. Am. Chem. Soc., 2019, 141, 4928–4935 CrossRef.
- H.-Z. Fan, X. Yang, J.-H. Chen, Y.-M. Tu, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202117639 Search PubMed.
- L.-G. Li, Q.-Y. Wang, Q.-Y. Zheng, F.-S. Du and Z.-C. Li, Macromolecules, 2021, 54, 6745–6752 CrossRef.
- J.-Z. Zhao, T.-J. Yue, B.-H. Ren, Y. Liu, W.-M. Ren and X.-B. Lu, Macromolecules, 2022, 55, 8651–8658 CrossRef.
- M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef PubMed.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef.
- T. J. Bannin and M. K. Kiesewetter, Macromolecules, 2015, 48, 5481–5486 CrossRef PubMed.
- P. P. Datta and M. K. Kiesewetter, Macromolecules, 2016, 49, 774–780 CrossRef.
- C. Hardy, G. Kociok-Köhn and A. Buchard, Chem. Commun., 2022, 58, 5463–5466 RSC.
- M. Piccini, D. J. Leak, C. J. Chuck and A. Buchard, Polym. Chem., 2020, 11, 2681–2691 RSC.
- M. Piccini, J. Lightfoot, B. C. Dominguez and A. Buchard, ACS Appl. Polym. Mater., 2021, 3, 5870–5881 CrossRef.
- T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- T. M. McGuire, E. F. Clark and A. Buchard, Macromolecules, 2021, 54, 5094–5105 CrossRef.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2017, 8, 2093–2104 RSC.
- G. L. Gregory, E. M. Hierons, G. Kociok-Köhn, R. I. Sharma and A. Buchard, Polym. Chem., 2017, 8, 1714–1721 RSC.
- G. L. Gregory, L. M. Jenisch, B. Charles, G. Kociok-Köhn and A. Buchard, Macromolecules, 2016, 49, 7165–7169 CrossRef CAS.
- Y. Wang, M. Li, J. Chen, Y. Tao and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 22547–22553 CrossRef CAS PubMed.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303 CrossRef CAS.
- C. G. Overberger and J. K. Weise, J. Am. Chem. Soc., 1968, 90, 3533–3537 CrossRef CAS.
- K. Hirofumi, T. Norio and E. Takeshi, Chem. Lett., 2005, 34, 376–377 CrossRef.
- G. L. Gregory, E. M. López-Vidal and A. Buchard, Chem. Commun., 2017, 53, 2198–2217 RSC.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- A. T. Lonnecker, Y. H. Lim and K. L. Wooley, ACS Macro Lett., 2017, 6, 748–753 CrossRef CAS.
- M. Langlais, O. Coutelier, S. Moins, J. De Winter, O. Coulembier and M. Destarac, Polym. Chem., 2018, 9, 2769–2774 RSC.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- C. G. Seefried Jr. and J. V. Kodeske, Polym. Eng. Sci., 1976, 16, 526–528 CrossRef.
- U. L. D. I. Kalana, P. P. Datta, R. S. Hewawasam, E. T. Kiesewetter and M. K. Kiesewetter, Polym. Chem., 2021, 12, 1458–1464 RSC.
- N. F. Zaaba and M. Jaafar, Polym. Eng. Sci., 2020, 60, 2061–2075 CrossRef CAS.
- J. P. MacDonald and M. P. Shaver, Polym. Chem., 2016, 7, 553–559 RSC.
- K. A. Stellmach, M. K. Paul, M. Xu, Y.-L. Su, L. Fu, A. R. Toland, H. Tran, L. Chen, R. Ramprasad and W. R. Gutekunst, ACS Macro Lett., 2022, 11, 895–901 CrossRef CAS.
- H. Nishida, M. Yamashita, T. Endo and Y. Tokiwa, Macromolecules, 2000, 33, 6982–6986 CrossRef CAS.
- Y. Kato, T. Futanaga and T. Nomura, Bioorg. Med. Chem. Lett., 2019, 29, 664–667 CrossRef CAS PubMed.
- B. Quiclet-Sire and S. Z. Zard, Bull. Korean Chem. Soc., 2010, 31, 543–544 CrossRef.
- T. M. McGuire, E. M. López-Vidal, G. L. Gregory and A. Buchard, J. CO2 Util., 2018, 27, 283–288 CrossRef.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef.
- P. Olsén, J. Undin, K. Odelius, H. Keul and A.-C. Albertsson, Biomacromolecules, 2016, 17, 3995–4002 CrossRef.
- A. Duda and A. Kowalski, in Handbook of Ring–Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, 2009, ch. 1, pp. 1–51 Search PubMed.
- M. A. F. Delgove, A. A. Wróblewska, J. Stouten, C. A. M. R. van Slagmaat, J. Noordijk, S. M. A. De Wildeman and K. V. Bernaerts, Polym. Chem., 2020, 11, 3573–3584 RSC.
- H. Tran, A. Toland, K. Stellmach, M. K. Paul, W. Gutekunst and R. Ramprasad, J. Phys. Chem. Lett., 2022, 13, 4778–4785 CrossRef CAS.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450–4458 CrossRef CAS.
- Y. Song, X. Yang, Y. Shen, M. Dong, Y.-N. Lin, M. B. Hall and K. L. Wooley, J. Am. Chem. Soc., 2020, 142, 16974–16981 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Dominique Zann, an inspiring teacher, on the occasion of her retirement. |
| ‡ Electronic supplementary information (ESI) available: Experimental and computational details; spectroscopic and crystallographic data for 1–5; SEC traces, spectroscopic and thermal (TGA, DSC, DMA) data for the polymers; DFT calculations data and associated digital repositories. CCDC 2089243, 2095960 and 2177255–2177257. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2py01366e |
| This journal is © The Royal Society of Chemistry 2023 |