
Excited-state dynamics and fluorescence lifetime of cryogenically cooled green fluorescent protein chromophore anions†
Anne P.
Rasmusssen
 ,
Henrik B.
Pedersen
and
Lars H.
Andersen
*
,
Henrik B.
Pedersen
and
Lars H.
Andersen
*
Department of Physics and Astronomy, Aarhus University, Aarhus 8000, Denmark. E-mail: lha@phys.au.dk
First published on 29th November 2023
Abstract
Time-resolved action spectroscopy together with a fs-pump probe scheme is used in an electrostatic ion-storage ring to address lifetimes of specific vibrational levels in electronically excited states. Here we specifically consider the excited-state lifetime of cryogenically cooled green fluorescent protein (GFP) chromophore anions which is systematically measured across the S0–S1 spectral region (450–482 nm). A long lifetime of 5.2 ± 0.3 ns is measured at the S0–S1 band origin. When exciting higher vibrational levels in S1, the lifetime changes dramatically. It decreases by more than two orders of magnitude in a narrow energy region ∼250 cm−1 (31 meV) above the 0–0 transition. This is attributed to the opening of internal conversion over an excited-state energy barrier. The applied experimental technique provides a new way to uncover even small energy barriers, which are crucial for excited-state dynamics.
The green fluorescent protein (GFP), originally found in the jellyfish Aequorea victoria,1,2 is widely used as a biomarker within biological and life sciences due to its characteristic green bioluminescence.3–6 The protein has two strong absorption bands, one at 398 nm pertaining to a neutral chromophore, and one at 478 nm which is ascribed to a deprotonated (hence anionic) form.7 The green emission is solely linked to fluorescence from the deprotonated chromophore in the protein. The GFP chromophore is burried in an 11 stranded β-barrel structure2,8 which protects it from surroundings. It enables efficient fluorescence, with a high quantum yield.3 The fluorescence is quenched when the chromophore is removed from the protein environment, emphasizing an important protection by the protein structure.
To understand the properties of the heart of GFP, its chromophore, p-hydroxybenzylidene-2,3-dimethylimidazolinone (HBDI) is frequently used as a model chromophore, in gas-phase experiments9–31 and theoretically.20,22,29,32–50 Deprotonated HBDI− is shown in Fig. 1c. It has a π conjugated structure with a phenol- and an imidazolinone-ring, held together by a methine bridge. In GFP, the two methyl groups on the imidazolinone-ring are replaced by covalent bonds to the protein structure without extending the electronic conjugation. Indeed, in terms of spectroscopy, the HBDI−-model chromophore absorbs in gas phase essentially at the same wavelength as the entire protein when holding a deprotonated chromophore.9 The overall spectral shift has been determined to be only 221 cm−1 (27 meV),31 emphasising the relevance of the small HBDI-model chromophore.
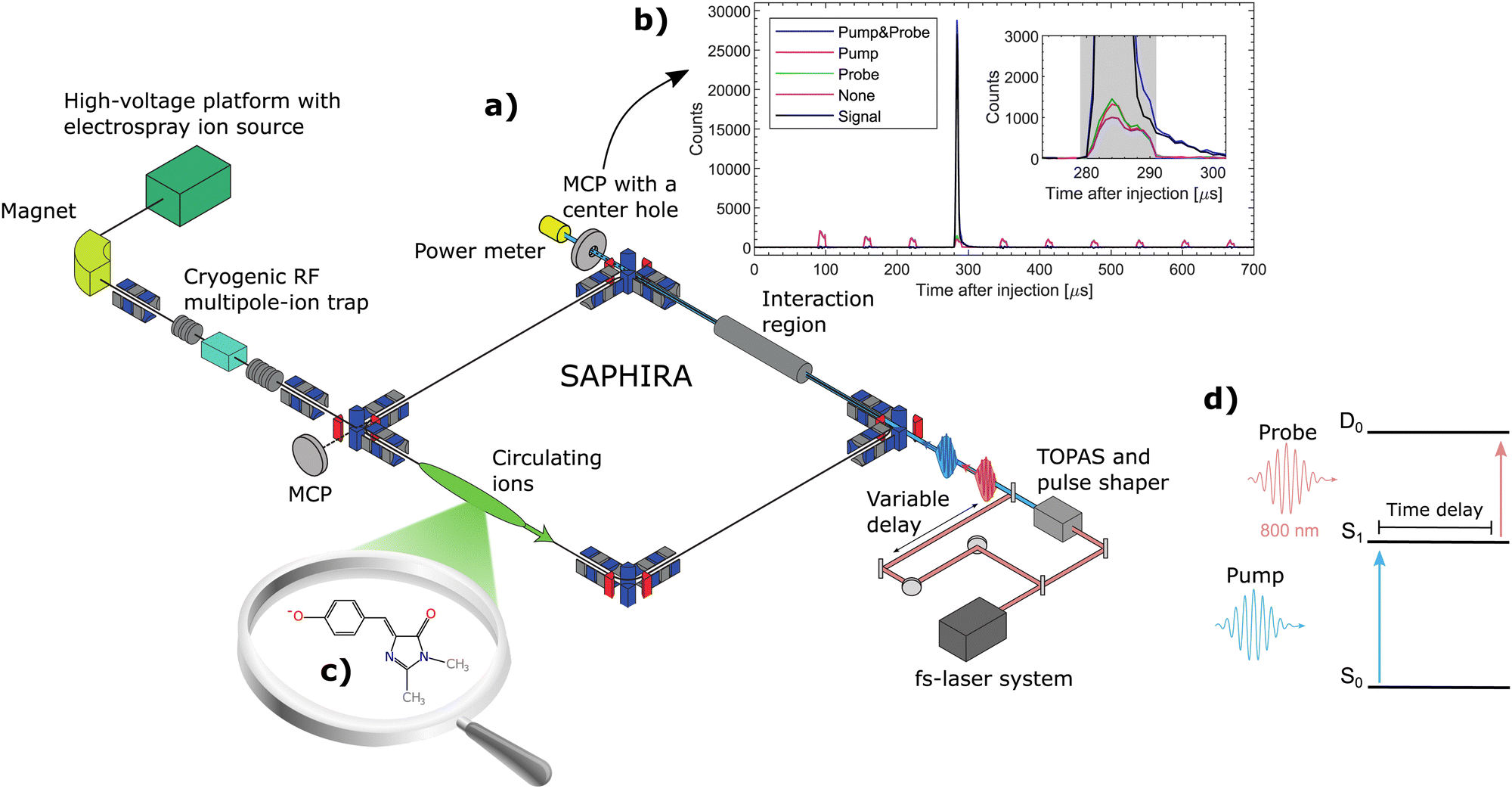 | ||
| Fig. 1 (a) The SAPHIRA ion-storage ring. The ion-source, detectors and laser system are shown schematically. The ions are irradiated in the interaction region. (b) Histogram of neutral counts as a function of time after injection measured on the MCP detector placed directly after the interaction region. The measurements are run with 4 cycles, where either both the pump and probe pulse are on (blue), the pump alone is on(red), the probe alone is on (green) or both are off (purple). The final signal (black) is calculated as the pump–probe signal with the background from the individual and the no laser is subtracted. (c) Molecular structure of the deprotonated GFP chromophore model HBDI− (mass 215 amu). (d) Illustration of the pump–probe scheme with anion electronic states S0 and S1. The ground state of the radical neutral, D0, is produced by two-photon pump–probe excitation within the S1 excited-state lifetime. | ||
In solution51–54 as well as in gas-phase55 the chromophore may undergo ultrafast radiationless decay by twisting the imidazolinone ring with respect to the bridge moiety, thereby opening a fast excited-state decay channel back to the electronic ground state by internal conversion (IC) through conical intersections (CI),20,56 thus preventing fluorescence.57,58 Internal conversion from the S1 electronically excited state may however be hindered by energy barriers for the rotation of the imidazolinone ring.55,59 The effect of such barriers is clearly most pronounced when the internal energy of the chromophore in S1 is low, i.e. at low temperature and for excitation near the S0–S1 band origin.
At room temperature, the first electronically excited state of gas-phase HBDI− decays bi-exponentially with lifetimes of 1.3 ± 0.2 ps and 11.5 ± 0.5 ps. At 100 K, the lifetimes are increased to 4.6 ± 2.1 ps, 27 ± 2 ps and an additional 1.2 ± 0.1 ns long component is appearing.55 These times reflect the nuclear dynamics in S1, in particular related to rotations of the two rings of HBDI.55,56 The longest time observed so far (1.2 ns)55 is long enough that it may be associated with radiative stabilization, potentially in combination with the nuclear dynamics.
Recently, action-absorption spectroscopy of cryogenically cooled HBDI− chromophores in gas phase has revealed immense structures in the action-absorption band pertaining to the vibrational structure and excited-state dynamics of S1.31 From spectral analysis, it was found that the S0 to S1 absorption band could be divided into four spectral regions, defined by the presence (or lack of) IC and vibrational autodetachment (VAD). The lowest energy region was defined as the region closed to internal conversion by barriers in the excited state. This region manifested itself experimentally by the appearance of resonantly enhanced multiphoton-electron detachment, but little internal conversion followed by photo fragmentation out of a hot electronic ground state. This below-the-barrier region was estimated to have a spectral width of only 31 meV (∼250 cm−1)31 meaning that it has little consequences for the dynamics at room temperature where the average internal energy is around 300 meV.10,55
The HBDI− chromophore has been studied under cold conditions in a few previous cases involving absorption- and photo-electron spectroscopy.30,31,60 In the present work, we report on state specific excited-state dynamics of cryogenically cooled HBDI− in gas phase in spectral regions (i) below barriers where the decay is closed for IC, (ii) in a region where IC is operative, and (iii) in a region where, in addition, non-adiabatic VAD is energetically allowed (see later discussion of Fig. 2). We determine both the intrinsic fluorescence lifetime of HBDI− and the height of the energy barriers in S1 that gives rise to fluorescence.
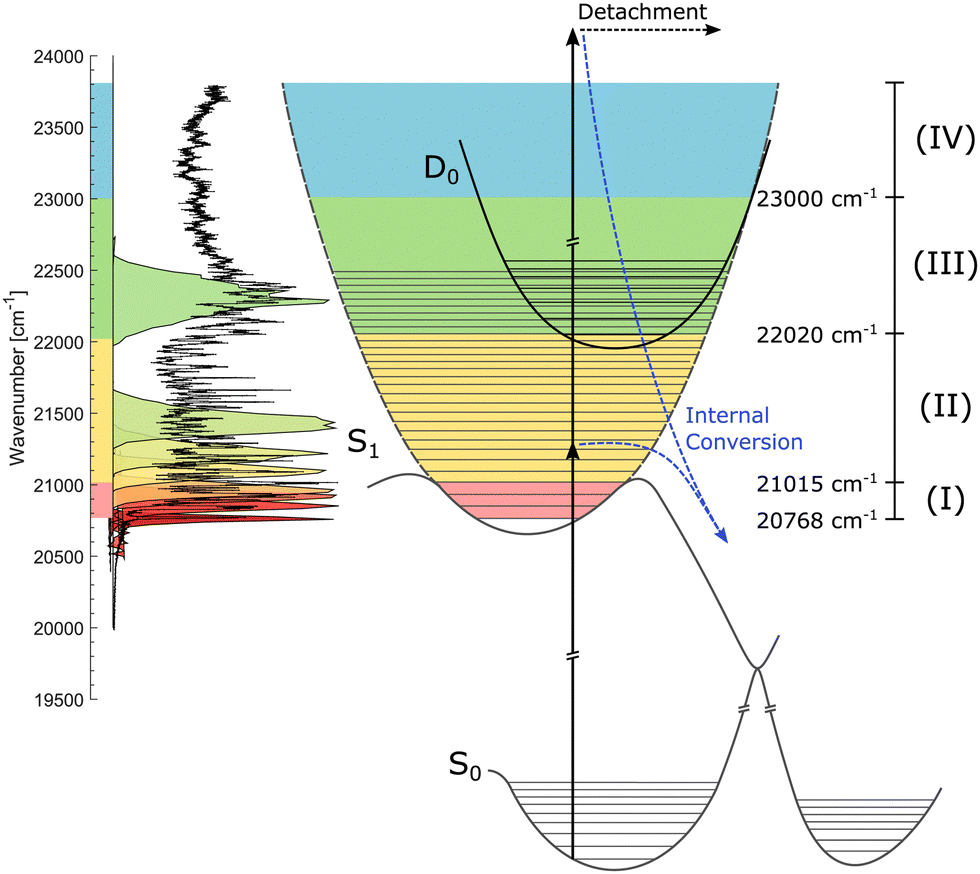 | ||
| Fig. 2 Illustration of the S0, S1, and D0 potential-energy curves. The action-absorption spectrum of HBDI− at cryogenic temperature is shown on the vertical axis to the left.31 Four spectral regions are shown in colors. Red: near the band-origin region below the barrier for internal conversion, yellow: below the detachment-threshold region, where internal conversion is operative, green: the region open for vibrational autodetachment (VAD) as well as IC, and blue: the region where S1 is electronically unbound. The measured spectral shapes of the fs-pump pulses are shown as colored areas on top of the spectrum. More details are found in Fig. 3. | ||
The measurements were performed at the ion-storage ring SAPHIRA61,62,63 (see Fig. 1a). An electrospray ionization-ion source was used to bring HBDI− anions into gas-phase. The ions were first accumulated in a 16-pole radio-frequency (RF) ion trap and then accelerated to 4 keV. After mass-to-charge selection by an electromagnet, the ions were decelerated and trapped in another 16-pole RF-ion trap held at 6 K.61 Here the ions were cooled for 150 ms by cold He-buffer gas. After cooling, the ions were once again accelerated to 4 keV and guided into the ring. The ring was held at room-temperature with a pressure between 10−9 to 10−8 mbar. The ions circulated only a few revolutions in the ring (revolution time of 60 μs – see Fig. 1b) before being irradiated in a straight section of the ring. No significant heating occured before irradiation.31 Neutral photofragments were detected on an MCP detector right after the interaction region. The detector had a center hole to allow the laser beams to exit onto a power meter for pulse-energy reading.
A pump–probe scheme was used to follow the time-evolution of the excited state of HBDI− (see Fig. 1d).62–66 800 nm (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1) fs-pulses were generated by a Coherent Libra UFS-HE femto-second laser. The laser beam was split into two separate beams. One beam, designated the pump beam, was guided through a tunable optical parametric amplifier system (Light Conversion TOPAS) to produce specific photon energies for the S0 to S1 excitation. The other 800-nm beam was guided through a variable delay stage to produce probe pulses. The life time of S1 was directly monitored by detecting the number of prompt two-photon absorption events on the MCP detector as a function of pump–probe delay (see Fig. 1d). The pump–probe signal was corrected for a small number of single-pulse events and for collision-induced fragmentation (see Fig. 1b). Since the probe-pulse wavelength was always 800 nm, ‘photon energy’ means in the following ‘pump-photon energy’.
500 cm−1) fs-pulses were generated by a Coherent Libra UFS-HE femto-second laser. The laser beam was split into two separate beams. One beam, designated the pump beam, was guided through a tunable optical parametric amplifier system (Light Conversion TOPAS) to produce specific photon energies for the S0 to S1 excitation. The other 800-nm beam was guided through a variable delay stage to produce probe pulses. The life time of S1 was directly monitored by detecting the number of prompt two-photon absorption events on the MCP detector as a function of pump–probe delay (see Fig. 1d). The pump–probe signal was corrected for a small number of single-pulse events and for collision-induced fragmentation (see Fig. 1b). Since the probe-pulse wavelength was always 800 nm, ‘photon energy’ means in the following ‘pump-photon energy’.
To follow the dynamics of individual (or small groups of) vibrational levels in S1, it was necessary to narrow the bandwidth of the pump-laser beam. The pump-laser pulses produced by the TOPAS had a temporal width of about 100 fs and a spectral width of about 300 cm−1. A pulse shaper67,68 was used to bring the spectral width down to 50 cm−1 at a cost of increasing the temporal width to ∼700 fs, which is still short compared to the lifetimes considered in the present study. The pulse shaper is described in detail in the ESI.†
We address three spectral regions (I) to (III) where S1 is electronically bound with respect to D0. The situation is depicted in Fig. 2 showing schematically the potential-energy curves of S0, S1, and D0. The vibrationally-resolved action-absorption spectrum is shown as well, together with the applied laser-energy profiles by which we address the regions of interest.
The signal as a function of the pump–probe delay for several photon energies of the pump laser is shown in Fig. 3. Panel (a) shows data with long decays obtained in the spectral region below and slightly above the excited-state energy barrier (discussed further below). The middle panel (b) shows the pump–probe signal up to a delay of 70 ps for all considered photon energies, while the right panel (c) displays the spectral shape of the fs-pump pulses as colored areas overlaid on the prompt action-absorption spectrum measured previously by nanosecond-laser pulses.31 To determine the lifetime, a single-exponential function folded with a Gaussian with the width of the cross-correlation between the pump and probe pulses was fitted to the data.
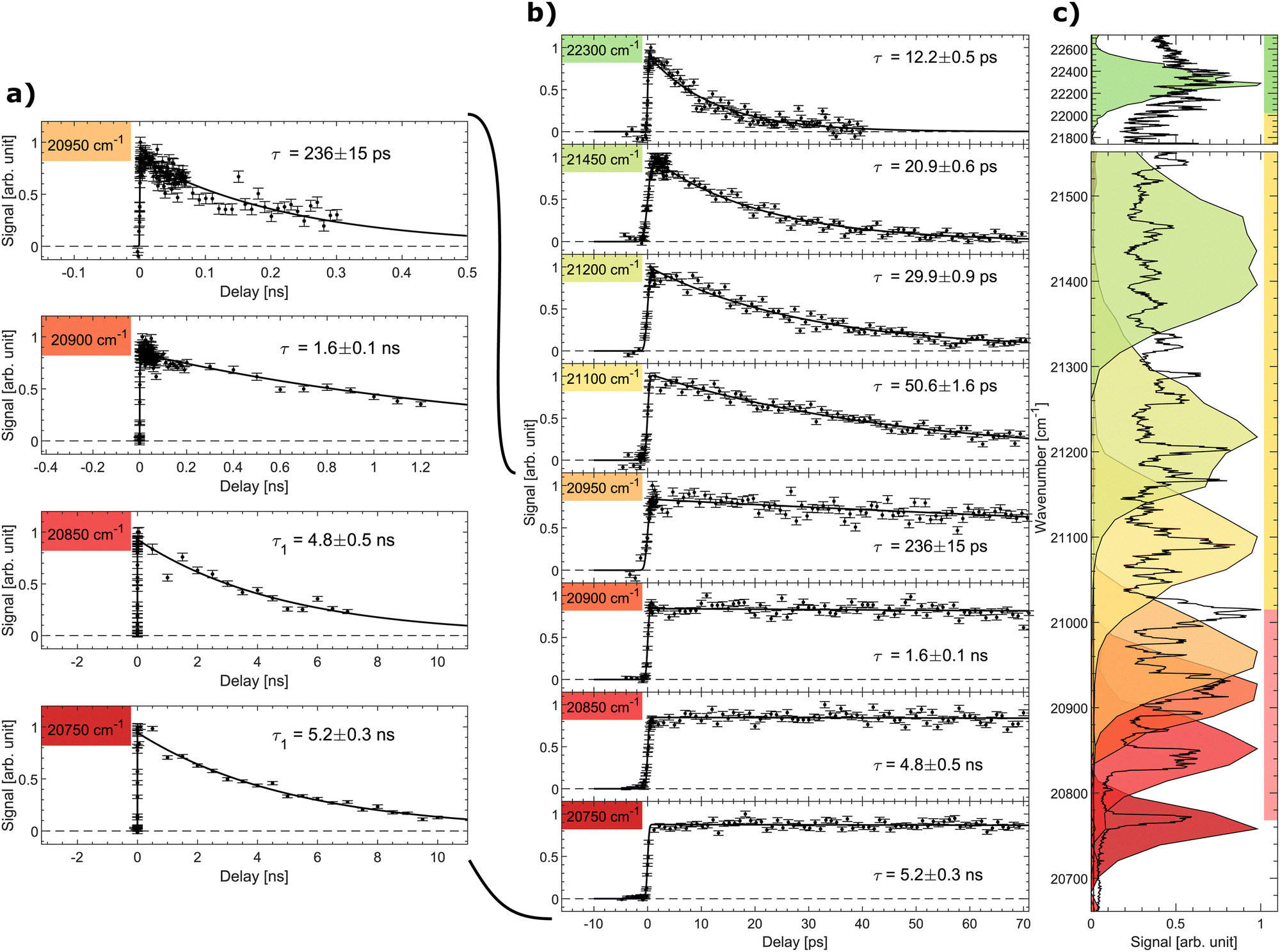 | ||
| Fig. 3 Signal as a function of pump–probe delay recorded at different photon energies (given in cm−1). (a) Decays with long ns lifetimes, (b) all data (pump–probe delays up to 70 ps). (c) The recorded pulse shape of the pump pulses are shown as colored areas overlaid on the vibrationally resolved prompt action-absorption spectrum of HBDI−.31 See Fig. 2 for the color code applied on the vertival axis of panel (c). | ||
In region (III), above the detachment threshold but below the S1 and D0 crossing point, VAD as well as IC are operative. The lifetime is found to be 12.2 ± 0.5 ps, which is comparable to the lifetime of 11.5 ± 0.5 ps measured previously with a photon energy of 20800 cm−1 at room temperature.55 The lifetime is determined by the rate for vibrational energy in S1 to couple to electronic energy (D0) by VAD and the rate for IC (S1 to S0). In region III, the ∼1500–1600 cm−1 VAD-active modes (stretching vibrations) may directly be photo excited, but due to the broad energy profile of the fs laser, we cannot specifically address individual VAD-active modes, although they are clearly seen by ns-laser pulses.31 The low-energy out-of-plane twisting modes promoting IC, on the other hand, are not active upon S0 to S1 excitation.31 The IC rate thus depends on IVR mediated energy transfer to these modes.
As the photon energy is decreased to the middle of spectral region (II) below the detachment threshold, the lifetime increases to 20.9 ± 0.6 ps. Vibrational autodetachment is here no longer possible and the lifetime is a direct measure of the IC-decay rate. When the photon energy is further decreased the lifetime increases gradually to 50.6 ps reflecting that transfer of vibrational energy into relevant IC-active modes31 becomes slower with less energy available.20
The lifetime increases drastically when the photon energy approaches the top of the S1 barrier. Data with a ∼ns lifetime (region I) are shown in Fig. 3a. At a photon energy of around 20950 cm−1 (477.3 nm) the lifetime is 236 ± 15 ps. Close to but below the top of the energy barrier (20900 cm−1) we measure a lifetime of 1.6 ± 0.1 ns, which is shorter than longest detected lifetime of 5 ns. This implies that tunnelling is likely operational close to the top of the barrier.
At a photon energy of 20850 cm−1 the ions are fully trapped behind the S1-energy barrier. A strong reduction of two-photon induced statistical (delayed) fragmentation out of S0 by ns-pulses was observed in this region.31 This shows that internal conversion as a result of direct coupling between S1 and highly vibrationally excited levels in S0 is almost negligible. The 4.8 ± 0.5 ns lifetime is hence ascribed to fluorescence. Lowering the photon energy further to 20750 cm−1 ensures that only the lowest vibrational state in S1 is populated (see the dark red marked area in Fig. 3c). Here a lifetime of 5.2 ± 0.3 ns is measured. Although fluorescence was not directly measured in this experiment, we conclude that 5.2 ± 0.3 ns is the intrinsic fluorescence lifetime of the GFP model chromophore HBDI−.
Fig. 4a shows both prompt (blue) and fragment mass 200 amu (red) action-absorption spectra.31 The black vertical line shows where the top the S1-energy barrier was estimated to be, based on the spectroscopy. Fig. 4b shows the measured lifetimes as a function of photon energy. There is a steep drop in the lifetime at around 21000 cm−1 which we ascribe to the top of the energy barrier. Based on a 0–0 transition energy of 20768 cm−131 a barrier height of about 232 cm−1 is derived. Thus, both spectroscopy and the dynamics agree well on a ∼ 250 cm−1 energy-barrier height. This barrier height does not account for any possible vibrational ground-state energy of the ions. The exact temperature of the ions is not known. The cold trap is cooled to 6 K and a rotational temperature of OH− has previously been found to be 25 ± 2 K with the same setup.61 We therefore expect the ions to have a temperature in that regime. At 10, 20 and 30 K the ions have on average 0.06, 2.82, and 12.3 cm−1 of vibrational energy respectively.31 Although small, this vibrational energy may be added to get the full barrier height.
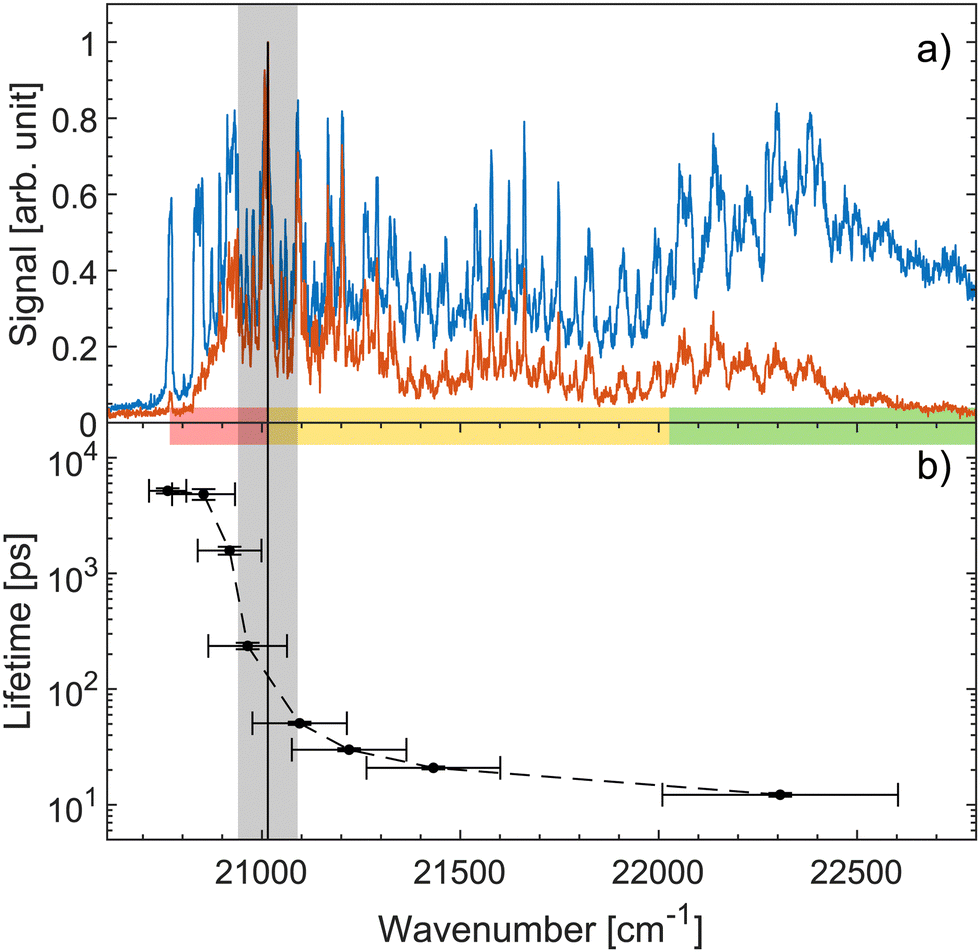 | ||
| Fig. 4 (a) Action-absorption spectra of HBDI− reported recently.31 The blue curve is the prompt-detachment signal and the red curve is the signal corresponding to detection of mass 200 amu photo-fragmentation signal (loss of methyl). (b) The lifetime as a function of photon energy. The horizontal errorbars are the FWHM of the pump pulses, while the vertical errorbars are from the exponential fit to the data. The vertical black line is the energy barrier-top position proposed based on the spectroscopy31 and the grey band indicates the energy-barrier top position estimated from the sudden change in lifetime. Notice the logarithmic scale showing over two orders of magnitude span in lifetimes. The energy-barrier height is determined as the difference between the band origin at 20768 cm−1 and the energy-barrier top position. The horizontal colored bar indicates the different spectral regions, also shown in Fig. 2 and 3. | ||
The gas-phase chromophore HBDI− will only fluoresce outside of the protein environment if trapping occurs behind this small barrier, hindering rotation of the imidazolinone ring.56 Here, this is achieved by reducing the internal energy and thereby the nuclear motion of the system. In the protein the chromophore is kept in a preferred configuration through a hydrogen-bond network3 resulting in a fluorescence lifetime of 2.8 ns.69
In summary, a fs-pump probe scheme in an ion-storage ring with ions prepared at cryogenic temperatures was applied to achieve the intrinsic excited-state dynamics of the GFP-model chromophore, HBDI−. We specifically address different spectral regions, including a region with only the vibrational ground state in S1 populated. Here the lifetime is found to be 5.2 ± 0.3 ns. We find the height of the S1-energy barrier for internal conversion to be 250 cm−1 by establishing when the decay channel for internal conversion opens and causes a drastic decrease in the excited-state lifetime by more than two orders of magnitude. The lack of direct detection of fluorescence from HBDI− seems to be a result of non radiative decay by internal conversion above this small energy barrier. The barrier supresses internal conversion through imidazolinone-ring rotation about the central methine bridge in S1 only when the chromophore is cold or held in a protein environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Peter Balling for providing details about the pulse shaping technique. This publication is based upon work from COST Action CA18212–Molecular Dynamics in the GAS phase (MD-GAS), supported by COST (European Cooperation in Science and Technology).References
- O. Shimomura, F. H. Johnson and Y. Saiga, Extraction, Purification and Properties of Aequorin, a Bioluminescent Protein from the Luminous Hydromedusan, Aequorea, J. Cell. Physiol., 1962, 59(3), 223–239 CrossRef CAS PubMed.
- M. Ormö, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Crystal Structure of the Aequorea victoria Green Fluorescent, Protein Sci., 1996, 273(5280), 1392–1395 Search PubMed.
- R. Y. Tsien, The Green Fluorescent Protein, Annu. Rev. Biochem., 1998, 67(1), 509–544 CrossRef CAS PubMed.
- M. Zimmer, Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior, Chem. Rev., 2002, 102(3), 759–782 CrossRef CAS PubMed.
- R. N. Day and M. W. Davidson, The fluorescent protein palette: tools for cellular imaging, Chem. Soc. Rev., 2009, 38(10), 2887 RSC.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues, Physiol. Rev., 2010, 90(3), 1103–1163 CrossRef CAS PubMed.
- M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(16), 8362–8367 CrossRef CAS PubMed.
- H. Niwa, S. Inouye, T. Hirano, T. Matsuno, S. Kojima and M. Kubota, et al., Chemical nature of the light emitter of the Aequorea green fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(24), 13617–13622 CrossRef CAS PubMed.
- S. B. Nielsen, A. Lapierre, J. U. Andersen, U. V. Pedersen, S. Tomita and L. H. Andersen, Absorption Spectrum of the Green Fluorescent Protein Chromophore Anion In Vacuo, Phys. Rev. Lett., 2001, 87, 228102 CrossRef CAS PubMed.
- L. H. Andersen, H. Bluhme, S. Boye, T. J. D. Jørgensen, H. Krogh and I. B. Nielsen, et al., Experimental studies of the photophysics of gas-phase fluorescent protein chromophores, Phys. Chem. Chem. Phys., 2004, 6(10), 2617–2627 RSC.
- L. Lammich, M. A. Petersen, M. B. Nielsen and L. H. Andersen, The gas-phase absorption spectrum of a neutral GFP model chromophore, Biophys. J., 2007, 92(1), 201–207 CrossRef CAS PubMed.
- J. Rajput, D. B. Rahbek, L. H. Andersen, T. Rocha-Rinza, O. Christiansen and K. B. Bravaya, et al., Photoabsorption studies of neutral green fluorescent protein model chromophores in vacuo, Phys. Chem. Chem. Phys., 2009, 11(43), 9996–10002 RSC.
- M. W. Forbes and R. A. Jockusch, Deactivation Pathways of an Isolated Green Fluorescent Protein Model Chromophore Studied by Electronic Action Spectroscopy, J. Am. Chem. Soc., 2009, 131(47), 17038–17039 CrossRef CAS PubMed.
- M. W. Forbes, A. M. Nagy and R. A. Jockusch, Photofragmentation of and electron photodetachment from a GFP model chromophore in a quadrupole ion trap, Int. J. Mass Spectrom., 2011, 308(2–3), 155–166 CrossRef CAS.
- M. B. Nielsen, L. H. Andersen and T. Rocha-Rinza, Absorption tuning of the green fluorescent protein chromophore: synthesis and studies of model compounds, Monatsh. Chem., 2011, 142(7), 709–715 CrossRef CAS; 24th European Colloquium on Heterocyclic Chemistry (ECHC), Vienna, Austria, 2010.
- D. A. Horke and J. R. R. Verlet, Photoelectron spectroscopy of the model GFP chromophore anion, Phys. Chem. Chem. Phys., 2012, 14(24), 8511–8515 RSC.
- C. R. S. Mooney, M. E. Sanz, A. R. McKay, R. J. Fitzmaurice, A. E. Aliev and S. Caddick, et al., Photodetachment Spectra of Deprotonated Fluorescent Protein Chromophore Anions, J. Phys. Chem. A, 2012, 116(30), 7943–7949 CrossRef CAS PubMed.
- C. W. West, A. S. Hudson, S. L. Cobb and J. R. R. Verlet, Communication: Autodetachment versus internal conversion from the S1 state of the isolated GFP chromophore anion, J. Chem. Phys., 2013, 139(7), 071104 CrossRef PubMed.
- C. R. S. Mooney, D. A. Horke, A. S. Chatterley, A. Simperler, H. H. Fielding and J. R. R. Verlet, Taking the green fluorescence out of the protein: dynamics of the isolated GFP chromophore anion, Chem. Sci., 2013, 4(3), 921–927 RSC.
- A. V. Bochenkova and L. H. Andersen, Ultrafast dual photoresponse of isolated biological chromophores: link to the photoinduced mode-specific non-adiabatic dynamics in proteins, Faraday Discuss., 2013, 163, 297–319 RSC.
- J. B. Greenwood, J. Miles, S. De Camillis, P. Mulholland, L. Zhang and M. A. Parkes, et al., Resonantly Enhanced Multiphoton Ionization Spectrum of the Neutral Green Fluorescent Protein Chromophore, J. Phys. Chem. Lett., 2014, 5(20), 3588–3592 CrossRef CAS PubMed.
- A. V. Bochenkova, B. Klaerke, D. B. Rahbek, J. Rajput, Y. Toker and L. H. Andersen, UV Excited-State Photoresponse of Biochromophore Negative Ions, Angew. Chem., Int. Ed., 2014, 53(37), 9797–9801 CrossRef CAS PubMed.
- C. W. West, J. N. Bull, A. S. Hudson, S. L. Cobb and J. R. R. Verlet, Excited State Dynamics of the Isolated Green Fluorescent Protein Chromophore Anion Following UV Excitation, J. Phys. Chem. B, 2015, 119(10), 3982–3987 CrossRef CAS PubMed.
- H. V. Kiefer, E. Lattouf, N. W. Persen, A. V. Bochenkova and L. H. Andersen, How far can a single hydrogen bond tune the spectral properties of the GFP chromophore?, Phys. Chem. Chem. Phys., 2015, 17(31), 20056–20060 RSC.
- A. V. Bochenkova, C. R. S. Mooney, M. A. Parkes, J. L. Woodhouse, L. Zhang and R. Lewin, et al., Mechanism of resonant electron emission from the deprotonated GFP chromophore and its biomimetics, Chem. Sci., 2017, 8(4), 3154–3163 RSC.
- J. Langeland, C. Kjær, L. H. Andersen and S. B. Nielsen, The Effect of an Electric Field on the Spectroscopic Properties of the Isolated Green Fluorescent Protein Chromophore Anion, Chem. Phys. Chem., 2018, 19(14), 1686–1690 CrossRef CAS PubMed.
- E. Carrascosa, J. N. Bull, M. S. Scholz, N. J. A. Coughlan, S. Olsen and U. Wille, et al., Reversible Photoisomerization of the Isolated Green Fluorescent Protein Chromophore, J. Phys. Chem. Lett., 2018, 9(10), 2647–2651 CrossRef CAS PubMed.
- J. Langeland, N. W. Persen, E. Gruber, H. V. Kiefer, L. H. Andersen and A. M. Kabylda, et al., Controlling Light-Induced Proton Transfer from the GFP Chromophore, Chem. Phys. Chem., 2021, 22(9), 807 CrossRef CAS PubMed.
- A. V. Bochenkova and L. H. Andersen, Action-Absorption Spectroscopy at the Band Origin of the Deprotonated Green Fluorescent Protein Chromophore In Vacuo, J. Phys. Chem. Lett., 2022, 13, 6683–6685 CrossRef CAS PubMed.
- W. Zagorec-Marks, M. M. Foreman, J. R. R. Verlet and J. M. Weber, Cryogenic Ion Spectroscopy of the Green Fluorescent Protein Chromophore in Vacuo, J. Phys. Chem. Lett., 2019, 10(24), 7817–7822 CrossRef CAS PubMed.
- L. H. Andersen, P. R. Rasmussen, H. B. Pedersen, O. B. Belatsan and A. V. Bochenkova, High-resolution spectroscopy and selective photoresponse of cryogenically cooled Green Fluorescent Protein chromophore anions, J. Phys. Chem. Lett., 2023, 14, 6395–6401 CrossRef CAS PubMed.
- V. Helms, C. Winstead and P. Langhoff, World Assoc Theoretical Organ Chem. Low-lying electronic excitations of the green fluorescent protein chromophore, J. Mol. Struct.: THEOCHEM., 2000, 506, 179–189 CrossRef CAS ; 5th World Congress of Theoretically Oriented Chemists (WATOC), Imperical Coll, London, England, 1999.
- M. Marques, X. Lopez, D. Varsano, A. Castro and A. Rubio, Time-dependent density-functional approach for biological chromophores: The case of the green fluorescent protein, Phys. Rev. Lett., 2003, 90, 25 CrossRef PubMed.
- M. E. Martin, F. Negri and M. Olivucci, Origin, Nature, and Fate of the Fluorescent State of the Green Fluorescent Protein Chromophore at the CASPT2//CASSCF Resolution, J. Am. Chem. Soc., 2004, 126(17), 5452–5464 CrossRef CAS PubMed.
- A. Toniolo, S. Olsen, L. Manohar and T. J. Martinez, Conical intersection dynamics in solution: the chromophore of Green Fluorescent Protein, Faraday Discuss., 2004, 127, 149–163 RSC; General Meeting on Non-Adiabiatic Effects in Chemical Dynamics, Univ Oxford, Oxford, England, 2004 Search PubMed.
- K. B. Bravaya, A. V. Bochenkova, A. A. Granovskii and A. V. Nemukhin, Modeling of the Structure and Electronic Spectra of Green Fluorescent Protein Chromophore, Russ. J. Phys. Chem., 2008, 2(5), 671–675 CrossRef.
- S. Olsen and R. H. McKenzie, A diabatic three-state representation of photoisomerization in the green fluorescent protein chromophore, J. Chem. Phys., 2009, 130, 18 CrossRef PubMed.
- C. Filippi, M. Ziccheddu and F. Buda, Absorption Spectrum of the Green Fluorescent Protein Chromophore: A Difficult Case for ab Initio Methods?, J. Chem. Theory Comput., 2009, 5(8), 2074–2087 CrossRef CAS PubMed.
- I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Nemukhin, Potential Energy Landscape of the Electronic States of the GFP Chromophore in Different Protonation Forms: Electronic Transition Energies and Conical Intersections, J. Chem. Theory Comput., 2010, 6(8), 2377–2387 CrossRef CAS PubMed.
- E. Kamarchik and A. I. Krylov, Non-Condon Effects in the One- and Two-Photon Absorption Spectra of the Green Fluorescent Protein, J. Phys. Chem. Lett., 2011, 2(5), 488–492 CrossRef CAS.
- S. Rafiq, B. K. Rajbongshi, N. N. Nair, P. Sen and G. Ramanathan, Excited State Relaxation Dynamics of Model Green Fluorescent Protein Chromophore Analogs: Evidence for Cis-Trans Isomerism, J. Phys. Chem. A, 2011, 115(47), 13733–13742 CrossRef CAS PubMed.
- D. Ghosh, A. Acharya, S. C. Tiwari and A. I. Krylov, Toward Understanding the Redox Properties of Model Chromophores from the Green Fluorescent Protein Family: An Interplay between Conjugation, Resonance Stabilization, and Solvent Effects, J. Phys. Chem. B, 2012, 116(41), 12398–12405 CrossRef CAS PubMed.
- A. H. Steindal, J. M. H. Olsen, K. Ruud, L. Frediani and J. Kongsted, A combined quantum mechanics/molecular mechanics study of the one- and two-photon absorption in the green fluorescent protein, Phys. Chem. Chem. Phys., 2012, 14(16), 5440–5451 RSC.
- B. K. Paul and N. Guchhait, Looking at the Green Fluorescent Protein (GFP) chromophore from a different perspective: A computational insight, Spectrochim. Acta, Part A, 2013, 103, 295–303 CrossRef CAS PubMed.
- K. B. Bravaya and A. I. Krylov, On the Photodetachment from the Green Fluorescent Protein Chrornophore, J. Phys. Chem. A, 2013, 117(46), 11815–11822 CrossRef CAS PubMed.
- L. Zhao, P. W. Zhou, B. Li, A. H. Gao and K. L. Han, Non-adiabatic dynamics of isolated green fluorescent protein chromophore anion, J. Chem. Phys., 2014, 141, 235101 CrossRef PubMed.
- R. Send, C. M. Suomivuori, V. R. I. Kaila and D. Sundholm, Coupled-Cluster Studies of Extensive Green Fluorescent Protein Models Using the Reduced Virtual Space Approach, J. Phys. Chem. B, 2015, 119(7), 2933–2945 CrossRef CAS PubMed.
- F. Zutterman, V. Liegeois and B. Champagne, Simulation of the UV/Visible Absorption Spectra of Fluorescent Protein Chromophore Models, ChemPhotoChem, 2017, 1(6), 281–296 CrossRef CAS.
- M. G. Khrenova, A. V. Nemukhin and V. G. Tsirelson, Origin of the pi-stacking induced shifts in absorption spectral bands of the green fluorescent protein chromophore, Chem. Phys., 2019, 522, 32–38 CrossRef CAS.
- X. Jin, W. J. Glover and X. He, Fragment Quantum Mechanical Method for Excited States of Proteins: Development and Application to the Green Fluorescent Protein, J. Chem. Theory Comput., 2020, 16(8), 5174–5188 CrossRef CAS PubMed.
- N. M. Webber, K. L. Litvinenko and S. R. Meech, Radiationless Relaxation in a Synthetic Analogue of the Green Fluorescent Protein Chromophore, J. Phys. Chem. B, 2001, 105(33), 8036–8039 CrossRef CAS.
- P. Altoe', F. Bernardi, M. Garavelli, G. Orlandi and F. Negri, Solvent Effects on the Vibrational Activity and Photodynamics of the Green Fluorescent Protein Chromophore: A Quantum-Chemical Study, J. Am. Chem. Soc., 2005, 127(11), 3952–3963 CrossRef PubMed.
- K. M. Solntsev, O. Poizat, J. Dong, J. Rehault, Y. Lou and C. Burda, et al., Meta and Para Effects in the Ultrafast Excited-State Dynamics of the Green Fluorescent Protein Chromophores, J. Phys. Chem. B, 2008, 112(9), 2700–2711 CrossRef CAS PubMed.
- K. Addison, I. A. Heisler, J. Conyard, T. Dixon, P. C. Bulman Page and S. R. Meech, Ultrafast excited state dynamics of the green fluorescent protein chromophore and its kindling fluorescent protein analogue, Faraday Discuss., 2013, 163, 277 RSC.
- A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, Origin of the Intrinsic Fluorescence of the Green Fluorescent Protein, J. Am. Chem. Soc., 2017, 139(25), 8766–8771 CrossRef CAS PubMed.
- N. H. List, C. M. Jones and T. J. Martínez, Internal conversion of the anionic GFP chromophore: in and out of the I-twisted S1/S0 conical intersection seam, Chem. Sci., 2022, 13(2), 373–385 RSC.
- A. Baldridge, K. M. Solntsev, C. Song, T. Tanioka, J. Kowalik and K. Hardcastle, et al., Inhibition of twisting of a green fluorescent protein-like chromophore by metal complexation, Chem. Commun., 2010, 46(31), 5686 RSC.
- L. M. Tolbert, A. Baldridge, J. Kowalik and K. M. Solntsev, Collapse and Recovery of Green Fluorescent Protein Chromophore Emission through Topological Effects, Acc. Chem. Res., 2012, 45(2), 171–181 CrossRef CAS PubMed.
- K. L. Litvinenko, N. M. Webber and S. R. Meech, Internal Conversion in the Chromophore of the Green Fluorescent Protein: Temperature Dependence and Isoviscosity Analysis, J. Phys. Chem. A, 2003, 107(15), 2616–2623 CrossRef CAS.
- S. H. M. Deng, X. Y. Kong, G. Zhang, Y. Yang, W. J. Zheng and Z. R. Sun, et al., Vibrationally Resolved Photoelectron Spectroscopy of the Model GFP Chromophore Anion Revealing the Photoexcited S1 State Being Both Vertically and Adiabatically Bound against the Photodetached D0 Continuum, J. Phys. Chem. Lett., 2014, 5(12), 2155–2159 CrossRef CAS PubMed.
- H. B. Pedersen, H. Juul, F. K. Mikkelsen, A. P. Rasmussen and L. H. Andersen, Inline cryogenically cooled radio-frequency ion trap as a universal injector for cold ions into an electrostatic ion-beam storage ring: Probing and modeling the dynamics of rotational cooling of OH−, Phys. Rev. A, 2022, 106, 053111 CrossRef CAS.
- H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, Decoupling Electronic versus Nuclear Photoresponse of Isolated Green Fluorescent Protein Chromophores Using Short Laser Pulses, Phys. Rev. Lett., 2016, 117, 243004 CrossRef PubMed.
- H. V. Kiefer, E. Gruber, J. Langeland, P. A. Kusochek, A. V. Bochenkova and L. H. Andersen, Intrinsic photoisomerization dynamics of protonated Schiff-base retinal, Nat. Commun., 2019, 10, 1210, DOI:10.1038/s41467-019-09225-7.
- E. Gruber, R. Teiwes, C. Kjær, S. B. Nielsen and L. H. Andersen, Tuning fast excited-state decay by ligand attachment in isolated chlorophyll a, Phys. Chem. Chem. Phys., 2021, 24(1), 149–155 RSC.
- A. P. Rasmussen, E. Gruber, R. Teiwes, M. Sheves and L. H. Andersen, Spectroscopy and photoisomerization of protonated Schiff-base retinal derivatives in vacuo, Phys. Chem. Chem. Phys., 2021, 23(48), 27227–27233 RSC.
- E. Gruber, A. M. Kabylda, M. B. Nielsen, A. P. Rasmussen, R. Teiwes and P. A. Kusochek, et al., Light Driven Ultrafast Bioinspired Molecular Motors: Steering and Accelerating Photoisomerization Dynamics of Retinal, J. Am. Chem. Soc., 2022, 144(1), 69–73 CrossRef CAS PubMed.
- P. Balling, D. J. Maas and L. D. Noordam, Interference in climbing a quantum ladder system with frequency-chirped laser pulses, Phys. Rev. A, 1994, 50(5), 4276–4285 CrossRef CAS PubMed.
- A. M. Weiner, J. P. Heritage and E. M. Kirschner, High-resolution femtosecond pulse shaping, J. Opt. Soc. Am. B, 1988, 5(8), 1563 CrossRef CAS.
- M. Kneen, J. Farinas, Y. Li and A. S. Verkman, Green Fluorescent Protein as a Noninvasive Intracellular pH Indicator, Biophys. J., 1998, 74(3), 1591–1599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04696f |
| This journal is © the Owner Societies 2023 |