
Photoinduced SET to access olefin-acrylate copolymers†
John B.
Garrison
,
Rhys W.
Hughes
,
James B.
Young
and
Brent S.
Sumerlin
 *
*
George & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, PO Box 117200, Gainesville, Florida 32611, USA. E-mail: sumerlin@chem.ufl.edu
First published on 13th January 2022
Abstract
The advantageous material properties that arise from combining non-polar olefin monomers with activated vinyl monomers have led to considerable progress in the development of viable copolymerization strategies. However, unfavorable reactivity ratios during radical copolymerization of the two result in low levels of olefin incorporation, and an abundance of deleterious side reactions arise when attempting to incorporate many polar vinyl monomers via the coordination–insertion pathway typically applied to olefins. We reasoned that design of an activated monomer that is not only well-suited for radical copolymerization with polar vinyl monomers (e.g., acrylates) but is also capable of undergoing post-polymerization modification to unveil an olefin repeat unit would allow for the preparation of statistical olefin-acrylate copolymers. Herein, we report monomers fitting these criteria and introduce a post-polymerization modification strategy based on single-electron transfer (SET)-induced decarboxylative radical generation directly on the polymer backbone. Specifically, SET from an organic photocatalyst (eosin Y) to a polymer containing redox-active phthalimide ester units under green light leads to the generation of reactive carbon-centered radicals on the polymer backbone. We utilized this approach to generate statistical olefin-acrylate copolymers by performing the decarboxylation in the presence of a hydrogen atom donor such that the backbone radical is capped by a hydrogen atom to yield an ethylene or propylene repeat unit. This method allows for the preparation of copolymers with previously inaccessible comonomer distributions and demonstrates the promise of applying SET-based transformations to address long-standing challenges in polymer chemistry.
Introduction
Due to their being significantly less activated, olefins are typically polymerized by methods that are distinct from those applied to common classes of more activated monomers, such as (meth)acrylates.1 The unactivated alkenyl groups present in olefins are best polymerized via transition metal-catalyzed coordination–insertion polymerization. The requisite transition metal catalysts, however, have comparatively more difficulty polymerizing polar monomers due to deleterious interactions between the metal and heteroatoms.1,2 On the other hand, radical polymerization is preferred for activated vinyl monomers due to radical stabilization imparted by their substituents. However, radical polymerization of olefins typically requires high temperature and pressure due to lack of radical stabilization and an increased contribution of undesirable side reactions.3 As neither radical nor coordination–insertion mechanisms are ideal for both monomer classes, tailoring monomer unit distribution and overall chain composition in copolymers of activated and unactivated alkenes is particularly challenging. Despite these complicating factors, copolymer materials comprised of both activated and unactivated monomer units remain desirable due to improvements in many material properties when compared to homopolymers of either monomer.2,4–12Radical copolymerizations of olefins with activated monomers face a substantial barrier that precludes facile copolymerization. The enchainment of a given monomer in a radical copolymerization is governed by its feed ratio and the reactivity ratios of the two monomers. The reactivity ratios of ethylene with methyl acrylate, for example, have been reported to be 0.045 and 5.3, respectively at 220 °C and 2000 bar.3 Despite the rather extreme conditions necessitated by the poor reactivity of ethylene, copolymers with methyl acrylate produced in this manner are limited to low olefin incorporation. A possible strategy to overcome the inherent differences in the reactivity of two monomers in radical copolymerization is to exploit a post-polymerization modification reaction that is specific for one type of monomer unit.13–19 In this manner, copolymerization of two monomers with similar polymerizability can lead to polymeric precursors for copolymers with compositions and microstructures that are inaccessible by direct copolymerization. Indeed, post-polymerization methods20–24 to produce polymers and copolymers containing olefins have been recently reported via either deoxygenation of poly(methyl acrylate)25 or post-functionalization and subsequent grafting-from polymerization from polyethylene or polypropylene.26,27
In this work, we report a post-polymerization modification strategy that enables synthesis of statistical copolymers of olefins and acrylates. Single electron transfer (SET) decarboxylative alkyl radical generation has been heavily represented in recent literature as a facile means to exploit the high reactivity of alkyl radicals.28–38 A common class of SET acceptors capable of decarboxylative radical generation are N-(acyloxy)phthalimide derivatives. Upon receipt of an electron from a SET donor, the N-(acyloxy)phthalimide undergoes a decarboxylation cascade, releasing CO2 and phthalimide to produce an alkyl radical. Radicals generated through SET-induced decarboxylation have been utilized in many radical cross-coupling reactions, such as alkylation, alkenylation, borylation, and more.31,35,38–40 Chapman et al. first reported on SET induced post-polymerization modification utilizing a Ni/Zn system for C–C bond formation to create polymers containing α-olefin repeat units ranging from three-carbon (propylene) repeat units and up.41 Redox-active repeat units were either formed in situ via uronium-based coupling with poly(acrylic acid) using (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo pyridinium 3-oxid hexafluorophosphate (HATU) and N-hydroxytetrachlorophthalimide (TCNHPI) or via preparation of TCNHPI-containing monomer units. We envisaged that inclusion of a phthalimide ester as the pendent group of an acrylate or methacrylate monomer would allow for not only tailorable incorporation in copolymerizations with other activated monomers but also subsequent polymer-based SET-induced reductive decarboxylation akin to the classic Barton decarboxylation (Fig. 1A).42 We specifically investigated non-chlorinated N-hydroxyphthalimide (NHPI) derivatives as a way to significantly improve the economics of these transformations as TCHNPI is substantially more expensive than NHPI as well as the lower molar mass of NHPI compared to TCNHPI providing atom-economic benefits as the phthalimide moiety is ultimately a byproduct. Replacement of the polymer-bound phthalimide ester with a single hydrogen atom reveals either an ethylene or propylene repeat unit depending on the structure of the starting phthalimide-containing monomer (Fig. 1B). Within this approach, the nature of the phthalimide ester monomer has direct implications on the result of the post-polymerization modification, with the acrylate and methacrylate variants serving as pre-ethylene and pre-propylene, respectively. In principle, this allows for a perhaps more versatile system in which a copolymer containing both ethylene and propylene is accessible. Overall, it is shown that unprecedented control over polymer architecture and comonomer distribution is made possible for ethylene-containing radical copolymers. These findings corroborate those of Theato and coworkers that were reported during the preparation of this manuscript, detailing decarboxylation being achieved with a Ru(bpy)3 catalyst system.43,56 The results reported here allow expansion to other olefin-containing comonomer units (i.e., propylene units) and rely on an alternative organocatalytic approach.
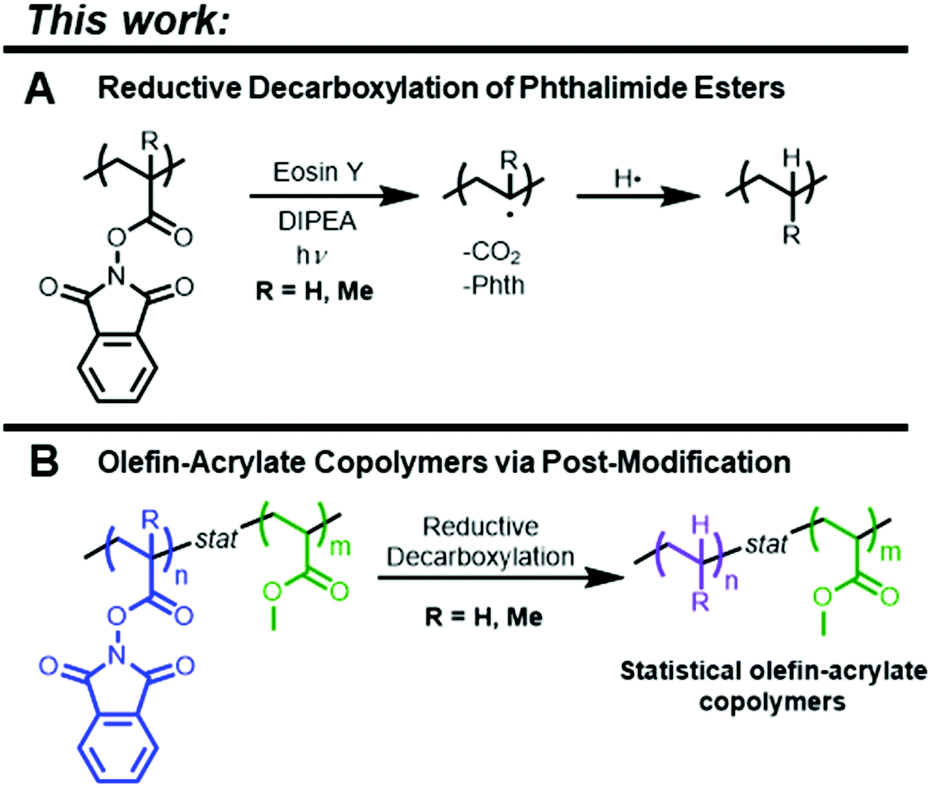 | ||
| Fig. 1 (A) General scheme for polymer-bound reductive decarboxylation of N-(acyloxy)phthalimides. (B) A novel route to previously inaccessible compositions of olefin-acrylate copolymers. | ||
Results and discussion
Small-molecule model studies
We hypothesized that direct attachment of phthalimide ester groups to a polymer backbone would permit light-induced reductive decarboxylation, removing the entire phthalimide ester functionality and replacing it with a single hydrogen atom to yield polyolefin repeat units (Fig. 1). Conditions for photo-induced decarboxylation have been investigated previously and served as a starting point for this reductive variant.28,31 Our conditions here closely follow those presented by Schwarz and coworkers in a recent report which highlights an effective light-responsive system for small-molecule decarboxylative alkylation.31 Eosin Y (EY) is an inexpensive, organic photocatalyst and SET donor that absorbs light in the visible range, allowing for SET under mild, green light.44 Diisopropylethylamine (DIPEA) serves as an electron donor that reduces excited-state EY in a reductive photo-electron transfer process.31,45,46 Decarboxylation of phthalimide esters proceeds upon receipt of an electron from the EY SET donor. The reduced phthalimide ester undergoes decarboxylation, releasing phthalimide and CO2 to yield a reactive carbon-centered radical. SET-induced decarboxylation of N-(acyloxy)phthalimides has been performed using a variety of SET donors in many bond-forming processes.31,35,39,47,48 Carbon–hydrogen bond formation between the carbon-centered radical and a hydrogen atom source has been only lightly explored with derivatives of N-(acyloxy)phthalimides.28,34 Thus, we first sought to investigate reductive decarboxylation of a model compound to determine if conditions for quantitative reaction were attainable.Phthalimidyl cyclohexanoate (PhthCy) was synthesized to serve as a model compound for reductive decarboxylation (Fig. 2A and S1†). We expected that the singular product of reductive decarboxylation of PhthCy would be cyclohexane which would allow for facile monitoring of reaction completion via1H NMR spectroscopy. We selected reaction conditions similar to those previously established for decarboxylative alkylation, with the addition of tributyltin hydride (Bu3SnH) as an H atom donor.31 PhthCy (1 equiv.), EY (0.1 equiv.), DIPEA (2 equiv.), dry DCM (5 mL), and Bu3SnH (5 equiv.) were irradiated with green light for 4 h. 1H NMR spectra of aliquots taken during and after the reaction revealed a gradual disappearance of all proton signals associated with substituted cyclohexane and the evolution of a single proton resonance at 1.43 ppm, relating nicely to reference values for cyclohexane in CDCl3 (Fig. 2B).49 Additionally, a distinct upfield shift had occurred for the aromatic phthalimide protons consistent with a change in their electronic environment upon detachment from the electron-withdrawing ester (Fig. 2C). Quantifying reaction completion via1H NMR spectral integrations revealed quantitative (>95%) conversion from phthalimide ester to free phthalimide. Distillation was performed on the crude mixture to isolate the volatile cyclohexane product (Fig. 2B). Confirmation of quantitative reductive decarboxylation allowed us to move forward and shift our attention to polymer-based systems.
 | ||
Fig. 2 Decarboxylation kinetics of PhthCy in DCM under green light with conditions PhthCy![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EY:DIPEA:Bu3SnH = 1:0.1:2:5. (A) Scheme for reductive decarboxylation of PhthCy. (B) 1H NMR spectra for PhthCy (top), the crude decarboxylation product containing all reaction components at 4 h (middle) and the distillate showing only cyclohexane (bottom). (C) 1H NMR spectra showing shift of phthalimide proton resonances every hour from 0 h to 4 h. :EY:DIPEA:Bu3SnH = 1:0.1:2:5. (A) Scheme for reductive decarboxylation of PhthCy. (B) 1H NMR spectra for PhthCy (top), the crude decarboxylation product containing all reaction components at 4 h (middle) and the distillate showing only cyclohexane (bottom). (C) 1H NMR spectra showing shift of phthalimide proton resonances every hour from 0 h to 4 h. | ||
Polymer model studies
N-(Acyloxy)phthalimides seemed to offer an immediate benefit over other decarboxylating esters such as thiohydroxamates,50,51 including potential amenability to a radical-rich environment such as an active polymerization medium. We sought to verify their stability in a polymerization and to adapt the established reductive decarboxylation conditions to a polymer-based system. Synthesis of a homopolymer with a lone phthalimide ester functional group would allow for both 1H NMR spectroscopy and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) characterization of photocatalytic reductive decarboxylation. Supplemental activator and reducing agent atom transfer radical polymerization (SARA ATRP)52–54 was chosen to prepare the model polymer. A phthalimide ester-containing ATRP initiator (PhthBr) was prepared via acyl substitution of 2-bromo-2-methylpropionyl bromide with N-hydroxyphthalimide (conditions in ESI,† Fig. S2). Poly(methyl acrylate) (PMA) was prepared via SARA ATRP using PhthBr as the initiator and was characterized via1H NMR spectroscopy, gel permeation chromatography (GPC), and MALDI-TOF-MS (Fig. 3 and S5†). The polymerization was well-controlled, yielding low-dispersity PMA with the phthalimide ester still attached to the α-end of the polymer (Phth-PMA-Br). | ||
| Fig. 3 MALDI-TOF-MS analysis of Phth-PMA-Br before (top) and after (bottom) SET-induced reductive decarboxylation, confirming replacement of both the phthalimide ester and chain-end bromine with hydrogen atoms (H-PMA-H). | ||
Inclusion of the N-(acyloxy)phthalimide only on the polymer endgroup allowed for assessment of the polymer before and after decarboxylation via MALDI-TOF-MS and GPC (Fig. 3 and S16†). Phth-PMA-Br was exposed to the same decarboxylating conditions as in the PhthCy model studies (conditions in ESI†). MALDI-TOF-MS analysis of the decarboxylated polymer revealed complete replacement of both the phthalimide ester and chain-end bromine with single hydrogen atoms (H-PMA-H). ω-Modification was observed as expected as it is known that reaction of alkyl bromide-terminated polymers with Bu3SnH leads to dehalogenation.55
Quantitative reductive decarboxylation was once again observed in the polymer model study, corroborating the findings from the small-molecule model study and providing further evidence that H atom replacement of the phthalimide ester is the exclusive product using these reaction conditions.
Moreover, phthalimide esters were benign to the polymerization conditions, opening the door for more expansive post-polymerization modifications via inclusion in polymer pendent groups.
Polymer synthesis
Both acrylate (N-(acryloxy)phthalimide, PhthA) and methacrylate (N-(methacryloxy)phthalimide, PhthMA) phthalimide ester derivatives were synthesized to investigate post-polymerization olefin unit incorporation via reductive decarboxylation (Fig. S3 and S4†).Copolymers of both PhthA and PhthMA with MA were prepared via conventional radical polymerization under argon in dimethyl sulfoxide (DMSO), using azobisisobutyronitrile (AIBN) as a thermal initiator at 70 °C. These copolymerizations were successful, yielding copolymers containing MA and either PhthA or PhthMA. Copolymer CA was prepared with PhthA and MA and copolymer CM was prepared with PhthMA and MA (Table S1†). The polymers were characterized using 1H NMR spectroscopy and GPC prior to further investigation into reductive decarboxylation (Fig. S6, S7, S12 and S13†).
Reversible addition–fragmentation chain transfer (RAFT) polymerization was likewise used to prepare copolymers containing MA and either PhthA or PhthMA by using 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDP) as the chain-transfer agent and AIBN as the thermal initiator at 70 °C. Copolymers RA and RM were prepared by RAFT with PhthA and PhthMA, respectively (Table S1, Fig. S8, S9, S10, S11 S14 and S15†). Copolymers RA2 and RM2 were also synthesized with slightly higher phthalimide molar incorporation, 30% PhthA for RA2 and 36% PhthMA for RM2, to allow for more facile assignment of peaks in 13C NMR spectroscopy and heteronuclear single quantum coherence (HSQC) spectroscopy. One advantage of this reversible deactivation radical polymerization (RDRP) route over the conventional radical polymerization approach discussed above is the ability to pre-determine polymer architecture across a wide array of morphologies. Thus, incorporation of phthalimide ester monomers via RDRP opens the door for facile production of olefin-containing polymers with higher order architecture. Additionally, the pseudo-livingness imparted by RDRP would, in principle, allow for chain extension to yield olefin-containing block copolymers. Another advantage of RDRP is molecular weight and dispersity control. For our purposes, copolymers with lower dispersities would offer a simplified and more striking way to assess the reductive decarboxylation process.
Reductive decarboxylation
Following the success of our model studies, we sought to leverage SET-induced reductive decarboxylation to prepare olefin-acrylate copolymers. Having established that H atom replacement of phthalimide esters is the exclusive product produced when using our standard decarboxylation conditions, reductive decarboxylation of PhthA or PhthMA repeat units within a copolymer would result in ethylene or propylene repeat units, respectively. Accordingly, copolymer RA was irradiated with green light in the presence of EY, DIPEA, and Bu3SnH (full conditions in ESI†). Kinetic aliquots were taken every half-hour and the time-points were analyzed via1H NMR spectroscopy and GPC (Fig. 4). Excitingly, the broad peaks in the 1H NMR spectra corresponding to polymer-bound phthalimide shifted consistently and completely in the upfield direction while also becoming sharper (Fig. 4C and S19†). The change in phthalimide proton shielding is evidence of disassembly of the phthalimide ester and detachment of phthalimide from the polymer. By quantifying the disappearance of the broad, polymer-bound phthalimide peak and the appearance of the sharp, small-molecule phthalimide peak using 1H NMR spectral integration, it was determined that quantitative decarboxylation was achieved in 2.5 h (Fig. 4B). Additionally, an increase in elution volume across timepoints in GPC confirmed a decrease in polymer molecular weight which was expected due to loss of both CO2 and phthalimide and the addition of one single H atom per PhthA repeat unit. Furthermore, lack of molecular weight increase suggests that radical capping with hydrogen occurs quickly enough to prevent radical–radical cross-coupling, which would lead to polymer branching or cross-linking. The decrease in polymer molecular weight halted upon reaching complete decarboxylation with the 2.5 and 3.0 h GPC timepoints overlapping (Fig. 4D). Furthermore, positive identification of the new ethylene repeat units was provided by 1H NMR spectroscopy following purification of the polymer. A new proton resonance was revealed that was not associated with the starting polymer or any reaction components. The broad peak at ∼1.20 ppm is in the region where polyethylene peaks would be expected (Fig. S21 and S22†). Additional 13C NMR spectroscopy of polymer RA2 revealed the presence of a characteristic polyethylene peak as well as disappearance of peaks associated with PhthA repeat units (Fig. S25†). Furthermore, HSQC spectroscopy of RA2 was utilized to verify the identity of the peaks relating to ethylene repeat units (Fig. S28†). Overall, photocatalytic reductive decarboxylation of polymer-bound phthalimide esters led to the release of phthalimide and evolution of CO2 while generating a backbone centered radical which was capped with a hydrogen atom to yield an ethylene-methyl acrylate statistical copolymer (Fig. 4A).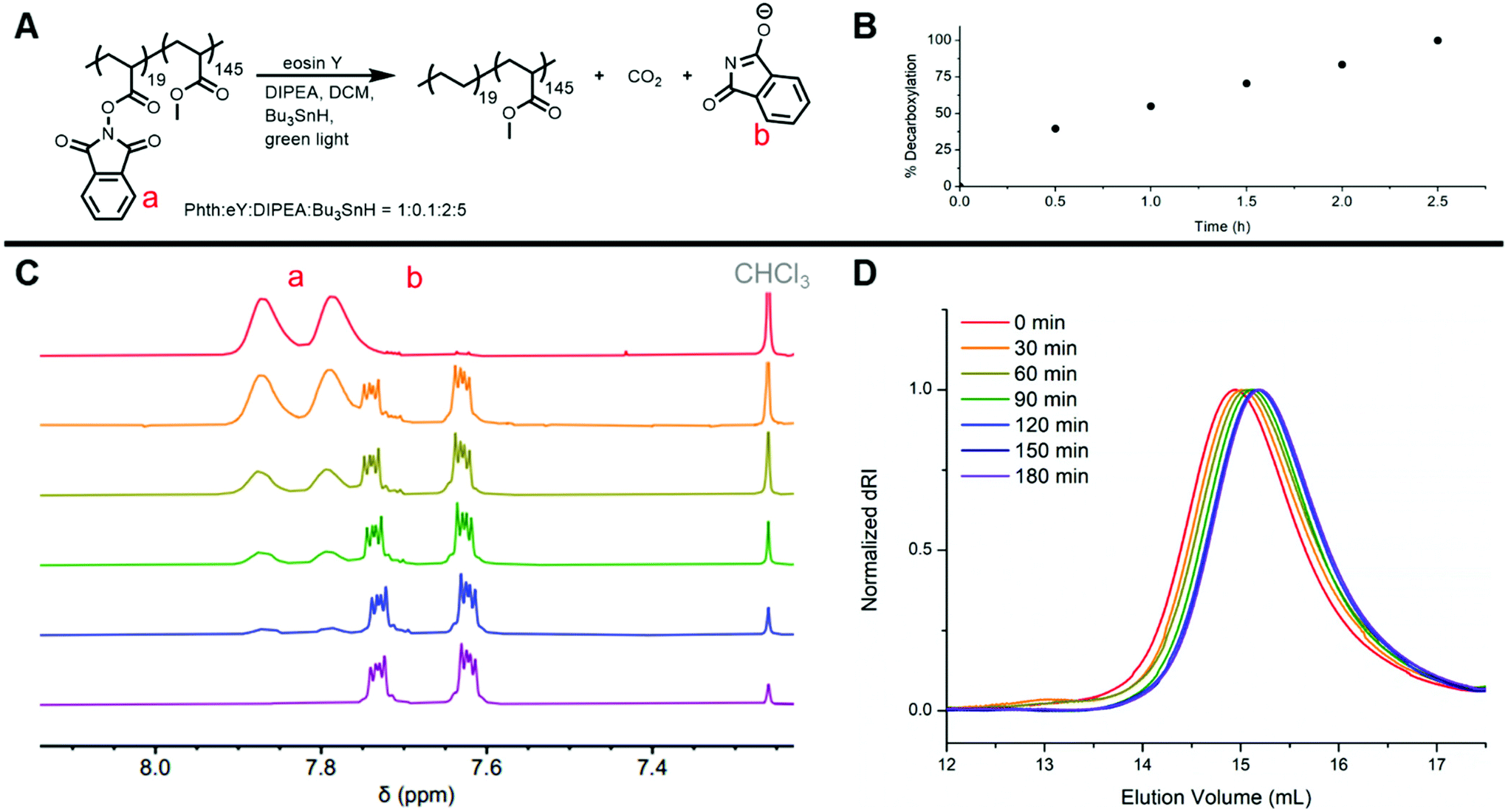 | ||
| Fig. 4 (A) Abbreviated scheme for reductive decarboxylation of RA (complete proposed reductive decarboxylation scheme in Fig. 2). (B) Decarboxylation percentage vs. time showing quantitative conversion at 2.5 h. (C) 1H NMR spectra showing the change in resonance frequency for the phthalimide protons as the reaction proceeded. (D) GPC elugrams showing decrease in polymer molecular weight due to decarboxylation. | ||
Reductive decarboxylation of RM produced similar results to those of RA. When RM was subjected to the standard decarboxylating conditions, a complete, upfield shift of the phthalimide proton resonances was observed in the resulting 1H NMR spectrum (Fig. S20†). Likewise, GPC provided evidence of decreasing polymer molecular weight that ended upon complete decarboxylation (Fig. S20†). 1H NMR analysis of the purified polymer revealed a new proton resonance at 0.9 ppm representing the methyl pendent of a propylene repeat unit (Fig. S23 and S24†). This peak was shown to couple with another peak in the polymer backbone via correlation spectroscopy (COSY) which revealed the methine proton of a propylene repeat unit (Fig. S27†). 13C NMR spectroscopy of RM2 also showed both the disappearance of phthalimide carbonyl peaks, indicating complete decarboxylation and the presence of characteristic polypropylene peaks (Fig. S26†). Again, HSQC spectroscopy was utilized to confirm the assignment of the methine and methyl protons of the new propylene repeat units (Fig. S29†).
Additionally, reductive decarboxylation of the polymers synthesized via conventional radical polymerization (CA, CM) produced a complete shift in phthalimide peaks in 1H NMR spectroscopy corresponding to successful decarboxylation as well as a decrease in polymer molecular weight as shown on GPC (Fig. S17 and S18†). All polymers prepared with phthalimide ester pendants were successfully decarboxylated. These polymers were prepared by either conventional free radical polymerization, which is potentially more scalable, or by RDRP such as RAFT, which affords better control over molecular weight distribution and allows for more advanced architectures.
Conclusions
These results clearly indicate that inclusion of pendent phthalimide ester groups leads to copolymers with latent backbone radical functionality. Activation of the SET-accepting ester unveils a backbone-centered radical which allows for a plethora of radical modification strategies. These phthalimide esters are benign to polymerization conditions and have been included in conventional radical polymers as well as polymers produced by both RAFT and ATRP. Specifically, this technique has been leveraged here to provide a new pathway for the synthesis of both ethylene-acrylate and propylene-acrylate statistical copolymers. Current approaches for copolymerizing these typically incompatible monomer classes rely on extreme conditions and lead to copolymers with uneven monomer distribution within the polymer chain due to the disparate reactivities of acrylates and olefins. Copolymerizing an acrylate with a phthalimide ester-containing monomer, however, allows for eventual olefin unit inclusion through post-polymerization modification. Single electron transfer to the phthalimide ester from the organic photocatalyst under low-energy green light begins a cascade leading to free phthalimide, CO2, and a carbon-centered radical which is capped by a hydrogen atom from a hydrogen atom donor. This reaction has been shown to be quantitative both via small-molecule model studies and polymer modification. Statistical ethylene-acrylate copolymers represent one example of previously inaccessible copolymer materials made possible through this post-polymerization modification pathway. Due to significant recent research focused in decarboxylative cross-coupling chemistry and the efficiency of this reported system, we believe this radical-based post-polymerization modification strategy will enable the creation of many novel polymeric materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation (NSF DMR-1904631) and with partial support under and awarded by DoD through the ARO (W911NF-17-1-0326). MALDI-TOF was supported by funding from NIH S10 OD021758-01A1.Notes and references
- A. Keyes, H. E. Basbug Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y. S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 58, 12370–12391 CrossRef CAS PubMed.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- M. Buback and H. Dietzsch, Macromol. Chem. Phys., 2001, 202, 1173–1181 CrossRef CAS.
- S. Mecking, J. Am. Chem. Soc., 1998, 120, 888–899 CrossRef CAS.
- S. Xiong, M. M. Shoshani, X. Zhang, H. A. Spinney, A. J. Nett, B. S. Henderson, T. F. Miller and T. Agapie, J. Am. Chem. Soc., 2021, 143, 6516–6527 CrossRef CAS PubMed.
- A. Sen, S. Elyashiv, N. Greinerl and S. Liu, J. Am. Chem. Soc., 2002, 43, 53–54 CAS.
- R. Venkatesh, S. Harrisson, D. M. Haddleton and B. Klumperman, Macromolecules, 2004, 37, 4406–4416 CrossRef CAS.
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., 2014, 126, 6801–6804 CrossRef.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed.
- J. Merna, P. Vlček, V. Volkis and J. Michl, Chem. Rev., 2016, 116, 771–785 CrossRef CAS PubMed.
- S. Zhong, Y. Tan, L. Zhong, J. Gao, H. Liao, L. Jiang, H. Gao and Q. Wu, Macromolecules, 2017, 50, 5661–5669 CrossRef CAS.
- Z. Wu, Z. Wang, B. W. Wang, C. H. Peng and X. Fu, Macromolecules, 2020, 53, 212–222 CrossRef CAS.
- H. L. Van De Wouw, E. C. Awuyah, J. I. Baris and R. S. Klausen, Macromolecules, 2018, 51, 6359–6368 CrossRef CAS.
- H. L. van de Wouw, J. Y. Lee, E. C. Awuyah and R. S. Klausen, Angew. Chem., Int. Ed., 2018, 57, 1673–1677 CrossRef CAS PubMed.
- H. L. Van De Wouw, J. Y. Lee and R. S. Klausen, Chem. Commun., 2017, 53, 7262–7265 RSC.
- Y. Ji, T. Zhou, H. L. Van De Wouw and R. S. Klausen, Macromolecules, 2020, 53, 249–255 CrossRef CAS.
- S. N. Mendis, T. Zhou and R. S. Klausen, Macromolecules, 2018, 51, 6859–6864 CrossRef CAS.
- Y. Xia, G. M. Scheutz, C. P. Easterling, J. Zhao and B. S. Sumerlin, Angew. Chem., Int. Ed., 2021, 60, 18537–18541 CrossRef CAS PubMed.
- C. P. Easterling, C. P. Easterling, G. Coste, J. E. Sanchez, G. E. Fanucci and B. S. Sumerlin, Polym. Chem., 2020, 11, 2955–2958 RSC.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS.
- C. P. Easterling, T. Kubo, Z. M. Orr, G. E. Fanucci and B. S. Sumerlin, Chem. Sci., 2017, 8, 7705–7709 RSC.
- W. Xue, H. Mutlu and P. Theato, Eur. Polym. J., 2020, 130, 109660 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- C. Jeon, D. W. Kim, S. Chang, J. G. Kim and M. Seo, ACS Macro Lett., 2019, 8, 1172–1178 CrossRef CAS.
- T. Yan and D. Guironnet, Macromolecules, 2020, 53, 4338–4344 CrossRef CAS.
- T. Yan and D. Guironnet, Angew. Chem., Int. Ed., 2020, 59, 22983–22988 CrossRef CAS PubMed.
- K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736–8738 CrossRef CAS.
- K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed.
- J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 RSC.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. E. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 260–265 CrossRef CAS PubMed.
- J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D. H. Bao, F. L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed.
- M. C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed.
- S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- R. Chapman, D. Melodia, J. B. Qu and M. H. Stenzel, Polym. Chem., 2017, 8, 6636–6643 RSC.
- D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
- S. Frech, E. Molle, A. J. Butzelaar and P. Theato, Macromolecules, 2021, 54, 9937–9946 CrossRef CAS.
- N. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., 2019, 131, 5224–5243 CrossRef.
- C. A. Figg, J. D. Hickman, G. M. Scheutz, S. Shanmugam, R. N. Carmean, B. S. Tucker, C. Boyer and B. S. Sumerlin, Macromolecules, 2018, 51, 1370–1376 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C. M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CrossRef CAS PubMed.
- D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770–2773 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- A. Chimiak, W. Przychodzen and J. Rachon, Heteroat. Chem., 2002, 13, 169–194 CrossRef CAS.
- D. H. R. Barton, D. Bridon, I. Fernandez-Picot and S. Z. Zard, Tetrahedron, 1987, 43, 2733–2740 CrossRef CAS.
- J. Xia, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1998, 31, 5958–5959 CrossRef CAS.
- K. Matyjaszewski, N. V. Tsarevsky, W. A. Braunecker, H. Dong, J. Huang, W. Jakubowski, Y. Kwak, R. Nicolay, W. Tang and J. A. Yoon, Macromolecules, 2007, 40, 7795–7806 CrossRef CAS.
- Y. Zhang, Y. Wang, C. H. Peng, M. Zhong, W. Zhu, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 78–86 CrossRef CAS.
- V. Coessens and K. Matyjaszewski, Macromol. Rapid Commun., 1999, 20, 66–70 CrossRef CAS.
- S. Frech and P. Theato, ACS Macro Lett., 2022, 11, 161–165 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01643a |
| This journal is © The Royal Society of Chemistry 2022 |