
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
X-Ray absorption spectroscopy of H3O+
Julius
Schwarz
 a,
Fridtjof
Kielgast
a,
Ivan
Baev
a,
Simon
Reinwardt
a,
Florian
Trinter
bc,
Stephan
Klumpp
b,
Alexander
Perry-Sassmannshausen
d,
Ticia
Buhr
d,
Stefan
Schippers
d,
Alfred
Müller
d,
Sadia
Bari
be,
Valerie
Mondes
f,
Roman
Flesch
f,
Eckart
Rühl
f and
Michael
Martins
*a
a,
Fridtjof
Kielgast
a,
Ivan
Baev
a,
Simon
Reinwardt
a,
Florian
Trinter
bc,
Stephan
Klumpp
b,
Alexander
Perry-Sassmannshausen
d,
Ticia
Buhr
d,
Stefan
Schippers
d,
Alfred
Müller
d,
Sadia
Bari
be,
Valerie
Mondes
f,
Roman
Flesch
f,
Eckart
Rühl
f and
Michael
Martins
*a
aInstitut für Experimentalphysik, Universität Hamburg, Luruper Chaussee 149, Hamburg, Germany. E-mail: michael.martins@uni-hamburg.de
bDeutsches Elektronen-Synchrotron DESY, Notkestraße 85, Hamburg, Germany
cMolecular Physics, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
dI. Physikalisches Institut, Justus-Liebig-Universität Gießen, Leihgesterner Weg 217, 35292 Gießen, Germany
eZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
fInstitut für Chemie und Biochemie, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 24th August 2022
Abstract
We report the X-ray absorption of isolated H3O+ cations at the O 1s edge. The molecular ions were prepared in a flowing afterglow ion source which was designed for the production of small water clusters, protonated water clusters, and hydrated ions. Isolated H2O+ cations have been analyzed for comparison. The spectra show significant differences in resonance energies and widths compared to neutral H2O with resonances shifting to higher energies by as much as 10 eV and resonance widths increasing by as much as a factor of 5. The experimental results are supported by time-dependent density functional theory calculations performed for both molecular cations, showing a good agreement with the experimental data. The spectra reported here could enable the identification of the individual molecules in charged small water clusters or liquid water using X-ray absorption spectroscopy.
1 Introduction
Water is arguably the most important substance on the planet, due to its prominent role in all living organisms.1 A plethora of critical chemical reactions take place in aqueous solution, making detailed knowledge of water at the molecular level of high interest not only for itself, but also for its applications in chemistry and pharmaceutics.2The effects that shape the solvation process arise from microscopic-scale solvation states. The number of water molecules in the first hydration shell of the form (H2O)nX involved in the solvation process is as small as n = 1 to n = 4, depending on the solution.3,4 Special cases of this ionic solution process are found in protolysis and proton transfer. Facilitated by the hydrogen-bond network of water, protons in liquid water diffuse at a rapid rate, a much debated phenomenon, with several possible explanations invoking (H2O)2H+ and H3O+.5 Understanding water at the molecular level, by this reasoning, also involves the study of the isolated, small cationic species, such as H3O+, that facilitate these solution and transfer processes. Considerable difficulty is associated with the selective investigation of these small cationic species in the solid and liquid phase. In the gas phase, mass-resolved studies of the appearance energies and valence photoelectron spectra in water clusters and protonated water clusters of the form (H2O)nH+ were conducted using supersonic molecular beams.6–9 Identification of contributions from specific species usually requires either their isolation from the bulk of the beam or a comparison to electronic-structure calculations.
Recently, on an even smaller scale than water clusters, the H2O+ cation has been investigated in a liquid-jet experiment, aiming at the time scales of the radiolysis of liquid water.10 In this study, proton transfer to H3O+ determining the H2O+ cation lifetime in the elementary proton transfer following radiolysis in liquid water was reported. As species associated with elementary proton transfer, this study also presented theoretical gas-phase absorption spectra of H3O+ and H2O+.10 Despite their evident importance, both for elementary proton transfer, as well as the aqueous solvation process as a whole, experimental data for X-ray absorption are not available in the literature to the best of our knowledge.
Among the many ways of gaining knowledge about the electronic-state configurations of H3O+ and H2O+, X-ray absorption spectroscopy has been established as a valuable and reliable analysis approach for neutral as well as ionic molecules and even ionic clusters.11–13 In particular, due to its element- and site-specificity, X-ray absorption spectroscopy enables probing of specific atoms in solution as well as in the gas phase. It was previously used to probe the proton transfer facilitating hydrogen-bond networks in liquid water at the O 1s site.14
Characterization of charged atoms, molecules, and clusters using X-ray absorption spectroscopy is one of the goals of the photon-ion spectrometer at PETRA III (PIPE) at DESY (Hamburg, Germany).15,16 The PIPE setup allows for mass selection and detection of charged atomic, molecular, and cluster beams as well as for investigating their interaction with soft X-ray beams in a merged-beam section.11 It is a permanent end-station at branch 2 of PETRA III's P04 beamline which operates in a photon-energy range from 250 eV to 2600 eV. PIPE was previously used to measure ion-yield spectra over a large dynamic range with cross-sections ranging from several 10 Mb down to the 1 b regime.17
The challenge in studying cationic species such as H3O+ and H2O+ at the PIPE setup lies in the construction of an optimized and reliable ion source for their production. Recently, a flowing afterglow ion source (FLAGS), for the production of small protonated water clusters and solvated ions, has been constructed for this purpose.
In this study, we present mass spectra recorded using the new flowing afterglow ion source designed to produce protonated water clusters of the form (H2O)nH+ up to n = 10 and X-ray absorption spectra of H3O+ and H2O+ following excitation of an O 1s electron using the photon-ion merged-beams technique as implemented in the PIPE experiment. This article is structured as follows: Section 2 details the design, discusses its operating parameters, and shows the mass spectra of the ion source. In Section 3, the X-ray absorption spectra of H2O+ and H3O+ upon core-hole excitation are discussed with regards to neutral H2O spectra and contrasted with the results of corresponding time-dependent density functional theory (TD-DFT) calculations. Lastly, Section 4 highlights the vast differences in electronic structure between H2O, H2O+, and H3O+ and gives an outlook on the implications of these results for the identification of H3O+ in spectroscopy experiments. Our findings can serve as a reference point for any arguments in the ongoing debate about the facilitator species in protolysis and, in particular, the role of its electronic structure.5,18,19
2 Experiments
Fig. 1 shows a schematic of the FLAGS, which can be used to produce protonated water clusters and even solvated ions (ionic species surrounded by a solvation shell). The flowing afterglow technique extracts ionic species from the afterglow of a plasma that is propagating through a drift tube. It has been used in ion chemistry for decades and is a well-established tool for cation and cationic cluster production.20,21 For the production of protonated water clusters, the main challenge in the operation of the FLAGS lies in a precise control of the plasma ignition parameters in order to extend the clustering within the flowing afterglow to (H2O)nH+ to numbers n beyond 1, while simultaneously extracting a strong and focused ion beam. In order to fulfill these requirements, the source is divided into three regions, as shown in Fig. 1.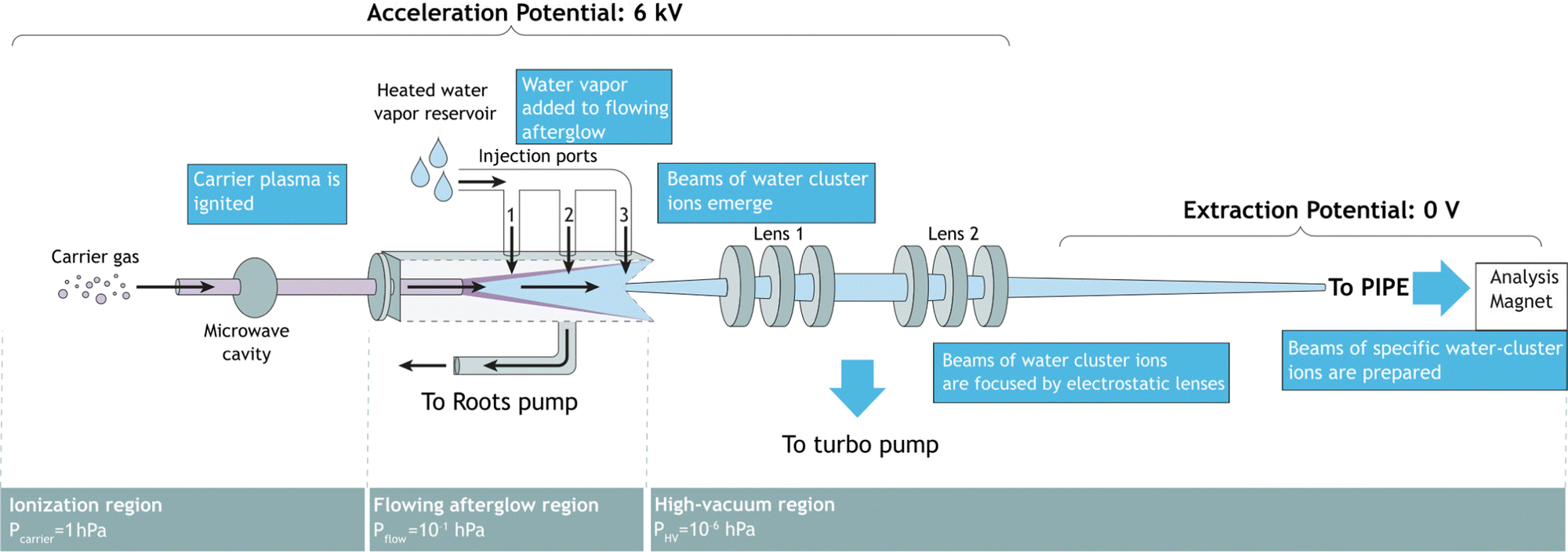 | ||
| Fig. 1 Flowing afterglow ion source schematic. Three different conceptual regions are shown: the ionization region, the flowing afterglow region, and the high-vacuum region. | ||
2.1 Ionization region
The ionization region serves to ignite and sustain a carrier plasma, typically a noble gas. Argon was chosen for the present experiments, because of its atomic mass being close to that of (H2O)2H+. In the ionization region of the ion source, carrier gas enters into an evacuated glass capillary. Influx pressures were set between 1.0 hPa and 2.0 hPa for ignition and maintained at 1.0 hPa during operation. The carrier gas was ionized by microwave heating within the confinement of an Evenson cavity. The Evenson cavity was highlighted by Fehsenfeld et al. as a suitable choice for microwave-ionization applications.22 Microwave power to the cavity was supplied by a solid-state microwave generator (SAIREM GMS450WSM 450 W, 2450 MHz). It was found that the carrier-gas inlet pressure, the microwave-generator settings, and the microwave-cavity tuning all serve to control the state of the carrier-gas plasma, the flowing afterglow and, thus, the ion-production rate.2.2 Flowing afterglow region
In the flowing afterglow region, the ionized carrier gas interacts with an added reactant gas, while flowing through a tube under the action of a Roots pump, which ensures a fast throughput of carrier gas and reactant gas. The target ionic clusters are produced by an ion–molecule reaction coupled with a three-body clustering reaction.23,24 A water reservoir supplies continuously water vapor to act as the reactant gas. Three injection ports allow for the injection of reactant gas in front of, directly above, or right behind the flowing afterglow maximum. Setting the water temperature and water pressure as well as choosing the injection port allows for control of the reaction conditions in the flowing afterglow region.2.3 Ion extraction region
In this region, ions are extracted from the flowing afterglow plasma by an electrically insulated skimmer with an inner diameter of 1 mm and an outer diameter of 50 mm over a length of 50 mm. The skimmer is able to maintain potentials of up to +400 V relative to the acceleration potential. Fig. 1 shows ions that are extracted into a high-vacuum region of 1 × 10−6 hPa. The ion beam is focused inside the chamber by an electrostatic lens system. The potentials applied to the focusing lenses determine the focal point of the beam. The ions are then extracted to the PIPE setup by applying a high voltage of 6 kV to the entire FLAGS chamber, mounted on the ion-source high-voltage platform of the PIPE experiment. The first ion-optical element of the electrically grounded PIPE ion beamline is an extraction lens which is electrically separated from the FLAGS chamber by an isolation flange.2.4 Operation
The production of certain ionic species, particularly ones involving a large hydration shell, using the flowing afterglow technique, depends largely on the plasma parameters of the flowing afterglow, most notably the flow pressure and the sample injection pressure. This is in turn dependent on the list of parameters contained in Table 1. The two settings displayed here use different pressure balances and varying microwave ignition power. These experimental variables affect which ionic species can be extracted. In its current technical design, the components of each region were designed for the production of small protonated water clusters of the form (H2O)nH+ with sizes up to n ≤ 10. This region could likely be modified by adjusting the skimmer size and the FLAGS parameters for the extraction of larger ionic clusters which goes beyond the scope of this work.| Parameter | H3O+ | Cluster ((H2O)2,3,4H+) |
|---|---|---|
| Inlet pressure (hPa) | 5.76 × 10−1 | 3.17 × 10−1 |
| Flow pressure (hPa) | 1.3 | 4.58 × 10−1 |
| Water pressure (hPa) | 25 | 15 |
| Microwave power (W) | 130 | 100 |
| Cavity position (cm) | 7.0 | 7.5 |
| Skimmer potential (V) | 244 | 248 |
| Lens 1 potential (V) | 214 | 198 |
| Lens 2 potential (V) | 188 | 200 |
During operation of the flowing afterglow ion source at PIPE, mass spectra of the ion beams have been taken depicted in Fig. 2. They showcase the ion source's capability of providing strong ion currents approaching the 1 nA regime of small, protonated water clusters. The hydronium-dominant setting shown in Table 1 has been chosen for XAS measurements.
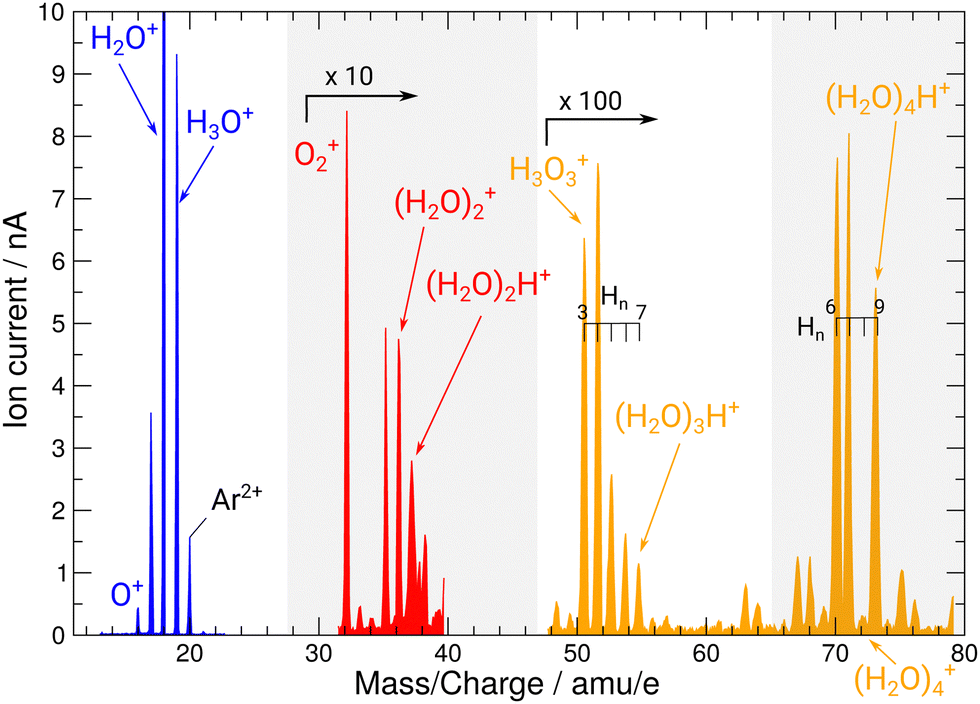 | ||
| Fig. 2 Mass spectrum of ion beams from the FLAGS in the regions featuring the relevant cationic species and protonated water clusters. The mass spectrum was produced using the cluster setting of the ion source (third column in Table 1). For clusters containing three and four oxygen atoms, a variety of clusters with a different number of H atoms are produced. Mass ranges with no water cluster signal (22–30 u e−1 and 38–48 u e−1) are omitted. | ||
A pure H3O+ beam was selected by setting the analyzing magnet accordingly and then transported to the 1.7 m long interaction region, where it was collimated and merged with the P04 photon beam. The product ions were separated from the parent ion beam by a second magnet, the demerger magnet, inside of which the primary ion beam was collected in a Faraday cup. After having exited the demerger magnet, the photoionized ions passed through a spherical 180-deg out-of-plane electrical deflector to suppress background from stray electrons or photons, and the fragment ions were then counted by a single-particle detector with near-100% detection efficiency.25 The photon flux was measured with a calibrated photodiode. During the H3O+ parent ion measurements, the average photon flux was 7 × 1013 photons per s. Photon-energy scans were conducted in the photon-energy range of 520 eV to 570 eV, with a grating of 400 lines per mm. The exit slit was opened to 1500 μm, resulting in an estimated energy resolution of 0.5 eV.
Using a measurement of the O+ → O2+ channel at the O 1s ionization edge, photon energies for the H2O+ → O2+ and H2O+→ O+ channels were photon-energy-calibrated using the results reported by Bizau et al.26 They reported an absolute energy uncertainty of 240 meV for the relevant findings.
3 Results and discussion
During commissioning of the new FLAGS, the H3O+-dominant setting (values provided in the second column of Table 1) resulted in ion peaks for H3O+, H2O+, OH+, and O+ at the masses 19 u, 18 u, 17 u, and 16 u, respectively. The spectral indicators for (H2O)3,4H+ expected as peaks in the cluster regime at 55 u, 73 u, or higher mass-to-charge ratios, were not present in the mass spectra obtained from this setting. This could be changed by adjusting injection pressures and microwave power to a cluster-dominant setting of the flowing afterglow ion source (values provided in the third column of Table 1). The mass/charge spectrum depicted in Fig. 2 was obtained from this setting. It shows the ion source's output at 37 u, 55 u, and 73 u, respectively, and underscores the suitability of the FLAGS as a means to produce cluster ions of the (H2O)2,3,4H+ species for X-ray absorption spectroscopy experiments. Beside (H2O)2,3,4H+ species also various other molecules with a smaller number of hydrogen atoms, e.g., H3O3+ could be produced. The comparison of the ion currents of (H2O)2H+ and (H2O)3H+ to the ion currents of the respective non-protonated cluster species (H2O)2+ and (H2O)3+ shows a weaker relative production efficiency for the protonated ions. However, this is different for (H2O)4H+. Fig. 2 shows a strong disparity in the production of (H2O)4H+ as compared to (H2O)4+. The (H2O)4+ signal is almost vanishing, whereas all other clusters with four oxygen atoms have a similar signal strength. As the protonated species can be fragments of larger neutral clusters, this disparity can be attributed to the neutral size distribution within the cluster beam. Its relative strength may make (H2O)4H+ a more accessible target for XAS measurements than (H2O)4+ in future experiments.In Fig. 3, the acquired X-ray ion-yield spectra of H2O+ (panels b and c) and H3O+ (panel d) following an O 1s excitation or ionization are shown. The displayed spectra for H2O+ → O+ (c) and H2O+ → O2+ (b) were measured for comparison purpose. An electron-cyclotron resonance (ECR) ion source, also operated at the PIPE experiment was used to produce the H2O+ ions.28 The total ion yield of parent neutral H2O, measured by Piancastelli et al., following the pioneering work of Wight et al., is also depicted, as it has been corroborated previously and serves as a reference for the XAS spectrum of the H2O O 1s excitation.27,29,30
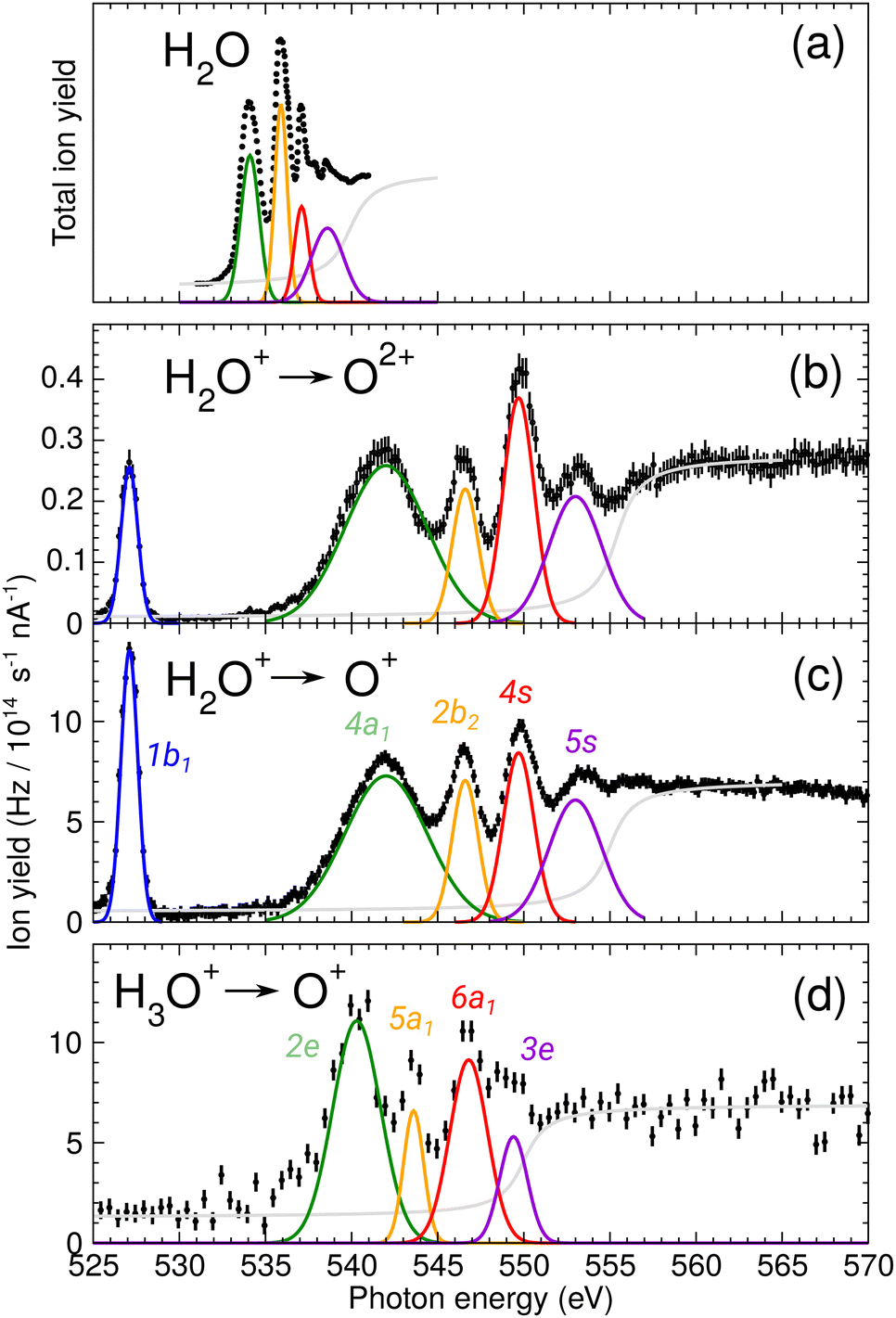 | ||
| Fig. 3 (a) Total ion yield of neutral H2O, digitized from the work of Piancastelli et al.,27 (b) H2O+ → O2+, (c) H2O+ → O+, and (d) H3O+ → O+. The 1b1, 4a1/3s, 2b2/3p, 4s, and 5s resonance peaks have been fitted by Gaussians in blue, green, yellow, red, and purple, respectively. For H3O+ parent ions, the corresponding resonances have been fitted and are indicated in the same colors. An arc tangent function representing the ionization edge is plotted in grey for all channels. In panels (b)–(d), a constant background has been subtracted from the experimental data (see text). | ||
The presented data was analyzed as follows: a constant background of different origin, as detailed below, was subtracted from each cation channel and the rising ionization edge was fitted by an arc tangent function. For the comparison of resonance intensities, Gaussian fits of noticeable resonances were carried out as shown in Fig. 3. For the H2O+ → O2+ channel, the background due to ion-atom collisions was negligible (e.g., less than 0.01 Hz nA−1). For the H2O+ → O+ channel, the background was quite intense, of the order of 50 Hz nA−1. For the H3O+ → O+ channel, the background was found to be of the order of 13 Hz nA−1.
For the H3O+ parent ion, only the fragmentation channel H3O+ → O+ was observed. It should be noted here that the channels H3O+ → H2O+ and H3O+ → OH+ were found to be too weak and burdened by background to result in useful spectra within the limited time available. For the H2O+ product channel, this is likely due to noise produced by the parent ion beam. For the OH+ product channel, this could also be indicative for a fragmentation that is accompanied by a kinetic energy release inhibiting the detection of the fragments.31 This is suggested by the findings of Pedersen et al., insofar as their fragmentation analysis of H3O+ following valence shell ionization showed both fragmentation channels to result in a high kinetic energy release (as compared to other dissociation processes) of 4 eV to 5.7 eV for H3O+ → H2O+ and 3.3 eV for H3O+ → OH+.32
For H2O+ parent ions, two different fragmentation channels could be measured, namely the O+ and the O2+ product channel. The O+ channel is roughly two orders of magnitude stronger in count rate than the O2+ channel and the relative intensities of the resonances in the two background-corrected cation channels vary slightly over the photon-energy scan range. The strong O+ channel is in accordance with the findings of Piancastelli et al. for neutral water.27 Further, the relative variations are similar, in H2O+ the 2b2/3p resonance is stronger as compared to H2O. In comparison to the calculated XAS spectra, the 4a1/3s resonance energy seems to be too low. This is a hint at different fragmentation pathways for these resonances, which could not be resolved in our experiment.
Fragmentation pathways and their associated kinetic energy releases (KER) also contribute to the two orders of magnitude difference in ion yield for the measured H2O+ → O+ and H2O+ → O2+ product channels. While the single-particle detector has an detection efficiency of almost 100%, its 10 mm diameter limits the acceptance to a 2–3 eV KER range for both product channels. The maximum KER due to the Coulomb repulsion of an O+ and a singly charged H+ or H2+ fragment is of the order of 13 eV, i.e., about 20% of the produced O+ ions are detected. Due to the higher product charge of the H2O+→ O2+ channel compared to the O+ channel, the KER in the O2+ channel can be up to twice as large, resulting in a smaller measured intensity of O2+ fragments. This can partially explain the strong difference in overall intensity in the two product channels.
For the H2O parent molecule, the arc tangent ionization threshold was centered at 539.9 eV, following the work of Piancastelli et al.27 The fitting of the ionization edges in the cationic species H2O+ and H3O+ was carried out on the basis of the relative positions of their O 1s binding energies in relation to the neutral H2O molecule. These ionization energy shifts have been estimated from Hartree–Fock calculations with GAMESS, using a cc-pVTZ basis set.33 In Fig. 3, the estimated ionization threshold shifts from 539.9 eV in H2O to 555.3 eV in H2O+ in accordance with the calculated shift in the O 1s binding energy of about 15.6 eV. In H3O+, the estimated O 1s binding energy is shifted by about 12.2 eV as compared to H2O. The fitted ionization threshold of H3O+ at 550.1 eV shows a 2 eV difference from this calculation, exceeding by far 0.24 eV energy uncertainty of the photon-energy calibration. While this energy difference may be attributed to the approximation of the H3O+ O 1s binding energy, the shifts in position of the ionization threshold and their shape in the cationic species H2O+ and H3O+ call for further investigation of ionization thresholds in these species.
The assignments of the experimentally observed features for the two parent ions are presented in Table 2. Due to the similarity of the NEXAFS spectra, a comparison of the results for H2O, H2O+, and H3O+ is possible. For the H2O+ parent ion, the assignment of the spectral features was carried out on the basis of the work by Piancastelli et al.27 For the parent ion H3O+, the assignment of the spectral features was done using the results from time-dependent density functional theory (TD-DFT) calculations as well as results from previous work on ammonia by Schirmer et al.34 This assignment of the spectral features allows for a comparison of the observed excitation energies of the two parent ions. The 1b1 resonance does not occur in the H3O+, due to the closed-shell configuration of the neutral molecule.
| H2O+ excitation | ħω (eV) | Width (eV) |
|
|
|---|---|---|---|---|
| O+ | O2+ | |||
| 1b1 | 527.1 (0.24) | 1.2 (0.5) | 13.6 (0.3) | 0.27 (0.02) |
| 4a1/3s | 541.7 (0.24) | 5.5 (2.3) | 7.4 (0.3) | 0.26 (0.02) |
| 2b2/3p | 546.5 (0.24) | 2.1 (0.9) | 6.8 (0.3) | 0.23 (0.02) |
| 4s | 549.7 (0.24) | 2.0 (0.9) | 7.3 (0.3) | 0.37 (0.03) |
| 5s | 553.3 (0.24) | 3.3 (1.4) | 6.1 (0.3) | 0.21 (0.02) |
| H3O+ excitation | ħω (eV) | Width (eV) |
|
|---|---|---|---|
| 2e/3p | 540.2 (0.24) | 3.3 (1.4) | 9.5 (1.1) |
| 5a1/3p | 543.5 (0.24) | 1.4 (0.6) | 6.5 (1.1) |
| 6a1/4s | 546.7 (0.24) | 2.7 (1.1) | 8.1 (1.1) |
| 3e/3p | 548.8 (0.24) | 1.9 (0.8) | 5.2 (1.1) |
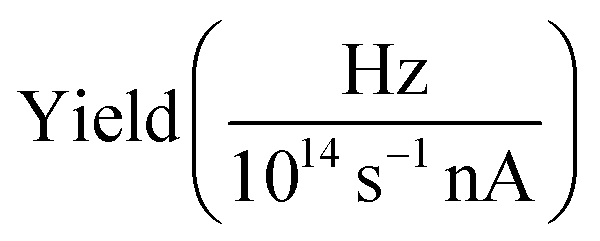
To gain insight into the nature of the observed transitions, time-dependent density functional theory (TD-DFT) calculations have been performed using the quantum-chemical program ORCA.35,36 These TD-DFT calculations were conducted on the isolated H2O+ and H3O+ species with the goal to simulate the XAS spectra of the two molecular cations. We opted to use the range-separated hybrid ωB97X-V functional introduced by Mardirossian et al.37 The admixture of exact exchange in the long range has been shown to improve the description of Rydberg-type states, which are of interest to this work, and it has been shown that in this respect the ωB97X-V functional performs better than others.37 As a basis set, we used def2-TZVPP, which has been shown to perform well close to the basis-set limit for DFT calculations on properties of small molecules.38,39 In order to more accurately describe the excited Rydberg-type states, additional uncontracted Gaussian s- and p-type functions were included, following the work of Kaufmann et al.40 The number of added functions was set to 10 s- and p-type functions for each atom, as this greatly improves the shape of the spectra while keeping the numerical cost reasonable. To further improve agreement between calculation and experiment, the range-separation parameter of the ωB97X-V functional was adjusted, similar to previous reports of optimally tuned range-separated hybrids.41,42 This tuning has been shown to improve XAS simulations for water.42 For H2O+, a range separation of 0.35 a0−1, with a0 being the Bohr radius, was found to perform best, while for H3O+, best agreement was achieved with a range-separation parameter of 0.49 a0−1.
The excitation energies were shifted to achieve maximum overlap with the experiment. For a comparison to the experimental results, spectra were broadened by a Voigt profile by convoluting the computed “stick spectrum” with a Lorentzian with a FWHM of 0.1 eV to simulate lifetime broadening and a Gaussian with a FWHM of 0.5 eV to account for the experimental resolution.
The results of these calculations can be seen in Fig. 4 and 5, for H2O+ and H3O+, respectively. Furthermore, plane cuts through the natural transition orbitals (NTOs) for the strongest of these excited states are shown. The conversion from electron orbitals to NTOs was conducted with the quantum-chemical program ORCA, following the method outlined in the work of Martin et al.43 For clarity, the NTOs in Fig. 4 and 5 are brought to a similar scale. With increasing photon energy the size of the NTOs is increasing which can be seen from the size of the H2O+ or H3O+ molecules shown in all NTOs. Fig. 4 reveals that the low-energy NTOs of H2O+ are similar to the unoccupied molecular orbitals of the ground state.10 The first, single peak is due to a transition into the singly occupied 1b1 orbital. The second, broad peak at around 542 eV consists of an A1 and B2 state in the C2v symmetry character table, which could not be resolved experimentally. It is labeled as the 4a1/3s feature. The next peak comprises one excited state of A1 and B1 symmetry each and is labeled as the 2b2/3p feature. Lastly, the peak at around 549.7 eV stems mostly from a B2, an A1, and a B1 state. It is labeled as the 4s feature.
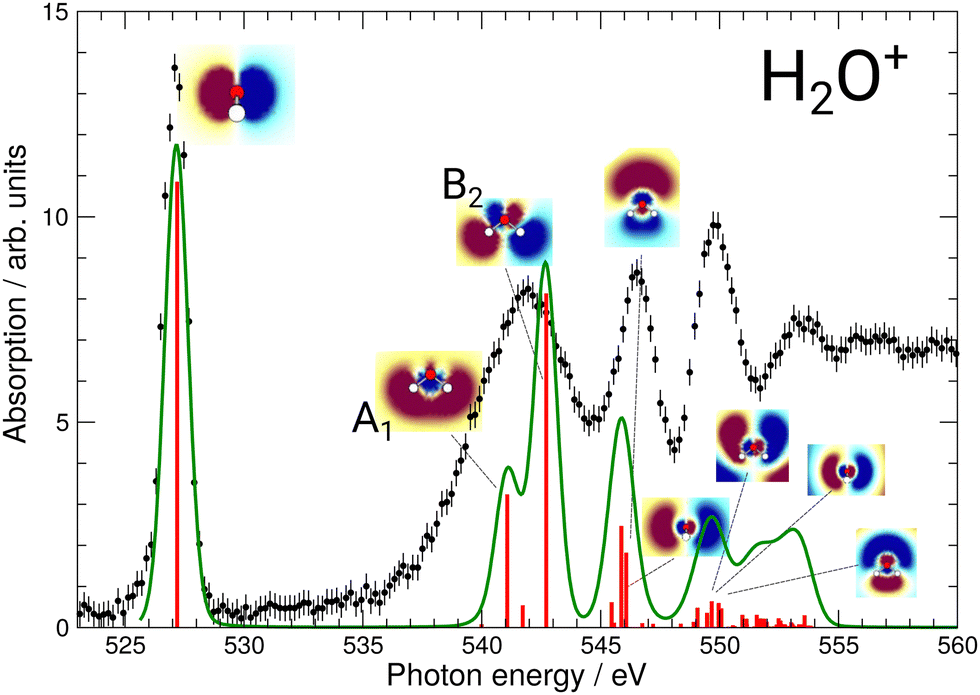 | ||
| Fig. 4 Calculated XAS spectrum of H2O+ compared to the experimental results. The strongest excitations are shown with their respective natural transition orbitals. For the broad 4a1/3s resonance the two natural transition orbitals (NTO) are denoted as A1 and B2. Note that, for clarity, the NTOs are brought to a similar scale as mentioned in the text. | ||
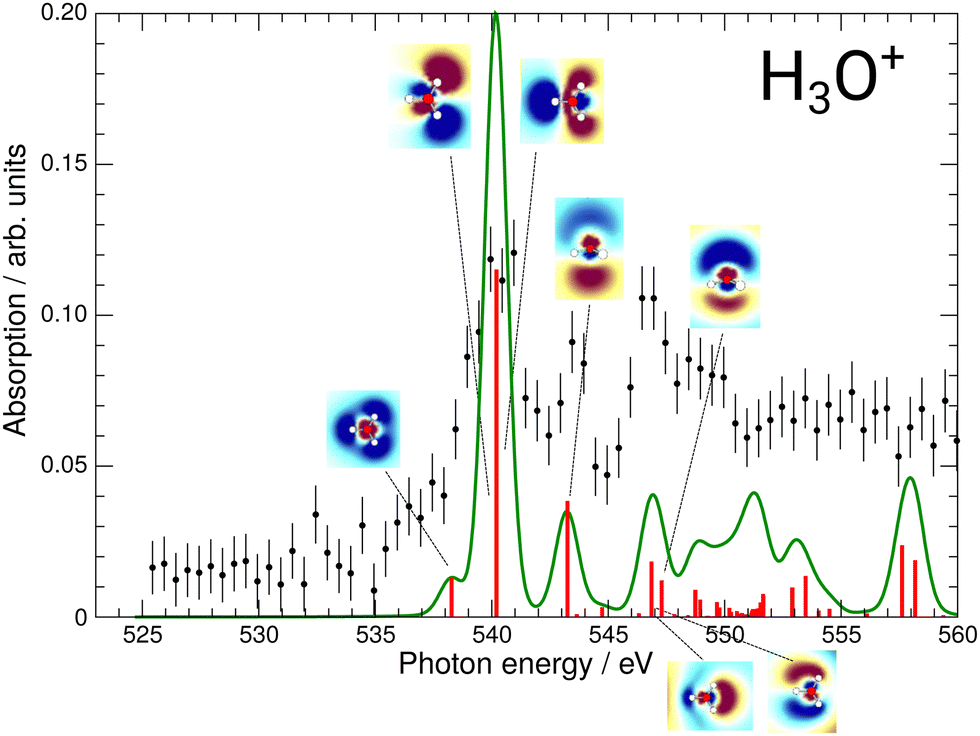 | ||
| Fig. 5 Calculated XAS spectrum of H3O+ compared to the experimental results. The strongest excitations are shown with their respective natural transition orbitals. | ||
The effect of the NTO shapes on the possible fragmentation pathways can be exemplarily discussed for the 4a1/3s resonance. This feature is a result from the A1 and B2 orbitals denoted in Fig. 4 experimentally not resolved due to large widths of the resonances. While the A1 orbital shows a node between the O and both H atoms, the NTO suggests a bonding character for the two H atoms between each other. The B2 state, which is energetically closest, similarly shows a node between the O and both H atoms, yet this state also features a node between each H atom. It is straightforward to assume that the induced fragmentation occurs with distinctly favored product channels at this point in the spectrum, e.g., the A1 state might fragment into O and H2 and the B2 state into O and two hydrogen atoms. While no information about the charge states of these fragmentation pathways is available from this experiment and calculation alone, this could result in a different KER of the charged O fragments for the two channels and, hence, a variation in the measured ion yield. This could lead to the broad 4a1/3s feature detected in both the O+ and the O2+ product channels which might be resolved into their two largest contributing states due to their different fragmentation routes. Hence, the fragmentation pathways for the different resonances will vary and might result in different KER of the charged O fragments. This calls for a KER-resolved study of the H2O+ fragmentation dynamics of the type recently conducted by Jahnke et al.44
For H3O+, the first experimental excitation is once again a combination of two separate excitations, in this case A1 and E, labeled as the 2e/3p feature. This is followed by two A1 excitations, the first of which constitutes the second peak and the second one being quite weak, the 5a1/3p feature. The third peak is once again a combination of an E and an A1 state, the 6a1/4s feature, as is the shoulder which is visible starting at 548.5 eV and labeled 3e/3p.
While the 4a1/3s feature in the H2O+ spectrum gave some insights into the possible fragmentation of the H2O+ molecular ion, the case is less intuitive for the H3O+ 2e/3p feature. The A1 and E excitations overlap and their NTOs do not allow for much reasoning towards their preferential fragmentation channels. A KER-resolved experiment might, however, reveal a separation between the small shoulder and the main feature for some KER ranges.
Revisiting the preliminary assignments presented in Table 2 we conclude that, while accurate for the observed excitations, several contributions to the spectroscopic features predicted by our calculations could not be resolved. Comparing the ion yields of H2O+ and H3O+ to the calculated XAS spectra shown in Fig. 4 and 5, the resolved resonances match well. We can identify distinct resonance energies, so that these features can be unequivocally assigned.
Fig. 3 reveals the shifts in spectral features of the H3O+ → O+ channel relative to the H2O+ parent ion and the H2O spectrum. While the quadruple-peak structure of the H2O parent is preserved throughout all spectra, the resonances shift drastically from species to species. For an overview, these shifts are depicted in Fig. 6 for H2O+ and H3O+ relative to the corresponding resonances in neutral water molecules. The first 4a1/3s peak in H2O, found at 534 eV by Piancastelli et al., shifts to 542 eV for the H2O+ parent ion, then back by 2 eV to 540 eV for the corresponding 2e/3p excitation in the H3O+ parent ion. From H2O to H3O+, the peak shifts by 6 eV.
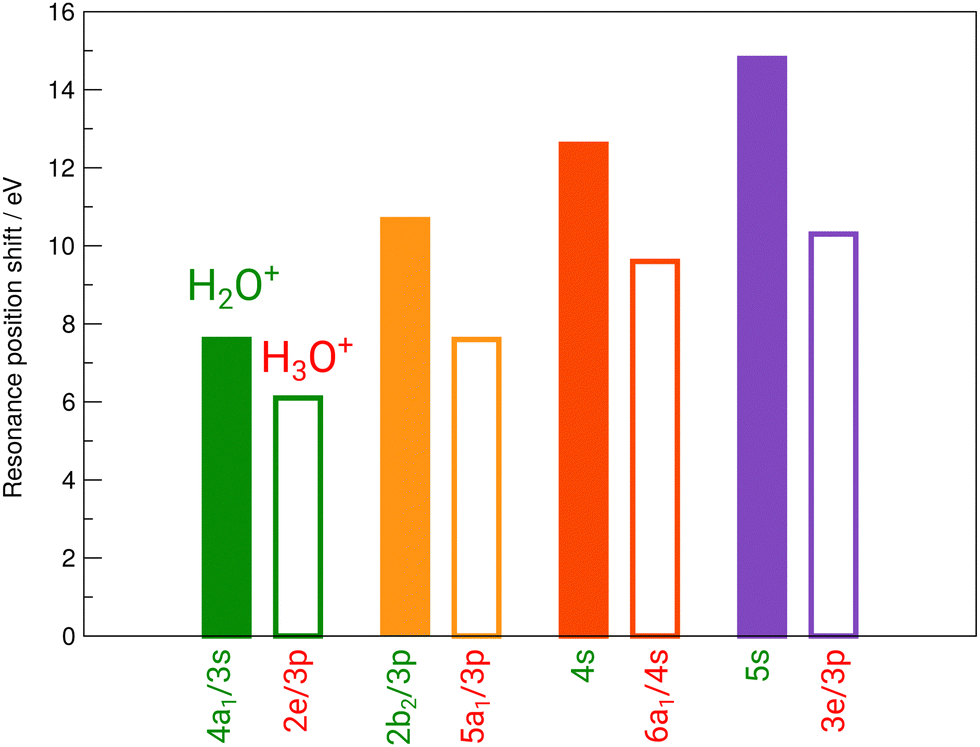 | ||
| Fig. 6 Experimental shifts of the individual resonances of H2O+ (full bars) and H3O+ (open bars) relative to the corresponding resonances in neutral H2O. | ||
The 2b2/3p peak shifts by more than 10 eV from H2O to 546.5 eV in H2O+, compared to a redshift by 3 eV to 543.5 eV in the corresponding 5a1/3p peak in H3O+. From H2O to H3O+, the peak shifts by 7 eV.
The 4s resonance shifts by 12.7 eV from H2O to 549.7 eV in H2O+. The corresponding 6a1/4s peak in H3O+ at 546.7 eV results in a 3 eV shift from H2O+ to H3O+ once more. From H2O to H3O+, the peak shifts by 9.7 eV.
The 5s resonance is close to the ionization edge in the H2O fits, as in the H2O+ fits. This also holds for the corresponding feature in H3O+, the 3e/3p peak. The excitation energy shifts by 14.8 eV to 553.3 eV from neutral H2O to H2O+. The shift to the corresponding 3e/3p peak in H3O+ at 548.8 eV, with 4.5 eV, is the largest observed shift between H2O+ and H3O+. From H2O to H3O+, the peak shifts by 10.3 eV.
A systematic pattern emerges from these results: resonance energies in H2O are drastically shifted to higher photon energies in their cationic form H2O+, i.e., by the removal of one electron. The corresponding shifts to higher photon energies in the H3O+ cation by the addition of a proton are smaller by several electronvolt in comparison. The observed shifts follow the difference in ionization energy for both cations.
The peak structure of the ionic spectra is similar to that of neutral H2O, but the splitting of the four observed features is strongly increased by a factor of about 5. Furthermore, the H2O+ peaks are much broader as compared to neutral H2O, e.g., the width of the 4a1/3s peak in H2O+ is 5.5 eV, i.e., increased by a factor of 5 as compared to H2O. Interestingly, the width of the corresponding 2e/3p peak in H3O+ is again smaller amounting to 3.3 eV. Partially, this large observed width can be explained by the calculated A1 and B2 states. However, with the employed photon-energy resolution these states should be resolved, which is obviously not the case. Such broadening effect has been found also for the σ* resonance in the HF+ cation and the NH series and might be a hint at a more general and different dissociation dynamics related to the repulsion of anti-bonding states in hydrogen-containing molecular cations.15,31
4 Conclusions
X-ray absorption studies of the water H2O+ and hydronium H3O+ cations were conducted at the O 1s site using the photon-ion merged-beams technique. In comparison to the neutral water molecule, strong shifts of photoabsorption resonances are found and the widths of the resonance features are strongly increased. This might be a hint for a steeper potential energy surface which would result in a faster dissociation dynamics in the cation compared to the neutral species. These processes could be studied by kinetic-energy-release-resolved experiments as demonstrated for HF+.31The H3O+ cation has been produced using a transportable flowing afterglow ion source which can be used to provide intense protonated water cluster beams for merged-beam and ion-trap X-ray absorption spectroscopy experiments. This represents a novel approach to produce small-scale protonated water clusters for determining their electronic structure and dynamics.
The resulting spectra for H2O+ and H3O+ mirror the results reported by Loh et al., but the increased energy resolution provided four clearly resolved spectral features, assigned in Table 2 that enable a clear spectroscopic distinction between the H3O+ cation, H2O, H2O+, and other species.10 This distinction is due to the large changes in electronic structure caused by the removal of an electron or the addition of a single proton to the H2O molecule.10
The distinct differences in absorption spectra between neutral H2O and its cationic protonated and unprotonated counterparts facilitate an accurate spectral assignment of the H3O+ species in multi-species absorption experiments. In the future, it should be possible to distinguish between H3O+ and similar species more easily. Due to the nature of this experiment, these results can serve the understanding of future spectroscopy experiments as a reference.
The distinct spectral shifts and changes in line width should also shed more light on experiments and calculations conducted in the liquid phase, akin to the one presented by Loh et al.10 The high-resolution spectra presented here are well-suited to be used for further spectroscopy experiments and calculations aimed at the dynamics of molecular water and small cations, such as H3O+ and H2O+ in protolysis and proton-transfer processes. With some additional difficulty, it could also be possible to use these results for spectroscopic studies on these cations within the region of strong absorption by liquid water.
By providing an X-ray absorption spectrum of isolated H3O+ and H2O+ following O 1s excitation, insights into the drastic differences in electronic structure of the cations compared to the neutral ground state molecule may contribute to the ongoing debate on possible facilitators of the protolysis in liquid water, particularly when arguments call on effects of the electronic structure of H3O+.5,18,19 Our results show for the H2O molecule that both the addition of a proton or the removal of an electron lead to large shifts in its cationic counterparts' ionization energies, resonance energies as well as a broadening of the spectra's features, while leaving the general shape of the recognizable features. There is also a strong difference in the absorption spectra of H2O+ and H3O+ when compared to each other, as for both the blue shift of resonance peaks as well as their broadening, the removal of an electron from the electronic configuration of H2O has a notably larger effect on the resonance features and their shape than the addition of a proton. As the cationic clusters of the form (H2O)nH+ with small n are also considered as possible candidates for facilitating proton transfer, future spectroscopy experiments using the flowing afterglow ion source shall investigate small size-selected water clusters for exploring their electronic structure and dynamics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank the team of beamline P04 for assistance in using P04 and PIPE. Beamtime was allocated for proposal I-20190066. Funding was provided by the German Ministry of Education and Research (BMBF), contracts 05K16GUC, 05K16RG1, O5K19RG3, and 05K19GU3 and the Deutsche Forschungsgemeinschaft (DFG), projects SFB925/A3 and 389115454 (Schi378/12). E. Rühl acknowledges financial support by BMBF (contracts 05KS7KE1 and 05K10KEA). S. Bari acknowledges funding by the Helmholtz Initiative and Networking Fund through the Young Investigator Groups Program. F. Trinter acknowledges support by the MaxWater initiative of the Max-Planck-Gesellschaft. This research was supported by the Centre for Molecular Water Science (CMWS) in an Early Science Project.Notes and references
- L. J. Rothschild and R. L. Mancinelli, Nature, 2001, 409, 1092–1101 CrossRef CAS PubMed.
- E. A. Oprzeska-Zingrebe and J. Smiatek, Biophys. Rev., 2018, 10, 809–824 CrossRef CAS PubMed.
- W. H. Robertson, E. G. Diken, E. A. Price, J.-W. Shin and M. A. Johnson, Science, 2003, 299, 1367–1372 CrossRef CAS PubMed.
- H.-P. Cheng, J. Phys. Chem. A, 1998, 102, 6201–6204 CrossRef CAS.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- S. Barth, M. Ončák, V. Ulrich, M. Mucke, T. Lischke, P. Slavíček and U. Hergenhahn, J. Phys. Chem. A, 2009, 113, 13519–13527 CrossRef CAS PubMed.
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 10075–10083 CrossRef CAS PubMed.
- H. Shiromaru, H. Shinohara, N. Washida, H.-S. Yoo and K. Kimura, Chem. Phys. Lett., 1987, 141, 7–11 CrossRef CAS.
- C. Y. Ng, D. J. Trevor, P. W. Tiedemann, S. T. Ceyer, P. L. Kronebusch, B. H. Mahan and Y. T. Lee, J. Chem. Phys., 1977, 67, 4235 CrossRef CAS.
- Z.-H. Loh, G. Doumy, C. Arnold, L. Kjellsson, S. H. Southworth, A. Al Haddad, Y. Kumagai, M.-F. Tu, P. J. Ho, A. M. March, R. D. Schaller, M. S. Bin Mohd Yusof, T. Debnath, M. Simon, R. Welsch, L. Inhester, K. Khalili, K. Nanda, A. I. Krylov, S. Moeller, G. Coslovich, J. Koralek, M. P. Minitti, W. F. Schlotter, J.-E. Rubensson, R. Santra and L. Young, Science, 2020, 367, 179–182 CrossRef CAS PubMed.
- J. Hellhund, A. Borovik, Jr., K. Holste, S. Klumpp, M. Martins, S. Ricz, S. Schippers and A. Müller, Phys. Rev. A, 2015, 92, 013413 CrossRef.
- S. Bari, L. Inhester, K. Schubert, K. Mertens, J. O. Schunck, S. Dörner, S. Deinert, L. Schwob, S. Schippers, A. Müller, S. Klumpp and M. Martins, Phys. Chem. Chem. Phys., 2019, 21, 16505–16514 RSC.
- J. T. Lau, J. Rittmann, V. Zamudio-Bayer, M. Vogel, K. Hirsch, Ph. Klar, F. Lofink, T. Möller and B. v. Issendorff, Phys. Rev. Lett., 2008, 101, 153401 CrossRef CAS PubMed.
- N. Huse, H. Wen, D. Nordlund, E. Szilagyi, D. Daranciang, T. A. Miller, A. Nilsson, R. W. Schoenlein and A. M. Lindenberg, Phys. Chem. Chem. Phys., 2009, 11, 3951–3957 RSC.
- S. Schippers, S. Ricz, T. Buhr, A. Borovik Jr, J. Hellhund, K. Holste, K. Huber, H.-J. Schäfer, D. Schury, S. Klumpp, K. Mertens, M. Martins, R. Flesch, G. Ulrich, E. Rühl, T. Jahnke, J. Lower, D. Metz, L. P. H. Schmidt, M. Schöffler, J. B. Williams, L. Glaser, F. Scholz, J. Seltmann, J. Viefhaus, A. Dorn, A. Wolf, J. Ullrich and A. Müller, J. Phys. B: At., Mol. Opt. Phys., 2014, 47, 115602 CrossRef.
- S. Schippers, T. Buhr, A. Borovik Jr., K. Holste, A. Perry-Sassmannshausen, K. Mertens, S. Reinwardt, M. Martins, S. Klumpp, K. Schubert, S. Bari, R. Beerwerth, S. Fritzsche, S. Ricz, J. Hellhund and A. Müller, X-Ray Spectrom., 2020, 49, 11–20 CrossRef CAS.
- A. Müller, M. Martins, A. Borovik, Jr., T. Buhr, A. Perry-Sassmannshausen, S. Reinwardt, F. Trinter, S. Schippers, S. Fritzsche and A. S. Kheifets, Phys. Rev. A, 2021, 104, 033105 CrossRef.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601–604 CrossRef CAS.
- Y. Zeng, A. Li and T. Yan, J. Phys. Chem. B, 2020, 124, 1817–1823 CAS.
- V. M. Bierbaum, Int. J. Mass Spectrom., 2015, 377, 456–466 CrossRef CAS.
- D. Smith and N. G. Adams, J. Phys. D: Appl. Phys., 1980, 13, 1267 CrossRef CAS.
- F. C. Fehsenfeld, K. M. Evenson and H. P. Broida, Rev. Sci. Instrum., 1965, 36, 294 CrossRef.
- D. Smith, N. G. Adams and M. J. Henchman, J. Chem. Phys., 1980, 72, 4951 CrossRef CAS.
- R. C. Bolden and N. D. Twiddy, Faraday Discuss. Chem. Soc., 1972, 53, 192–200 RSC.
- A. Müller, D. Bernhardt, A. Borovik Jr., T. Buhr, J. Hellhund, K. Holste, A. L. D. Kilcoyne, S. Klumpp, M. Martins, S. Ricz, J. Seltmann, J. Viefhaus and S. Schippers, Astrophys. J., 2017, 836, 166 CrossRef.
- J. M. Bizau, D. Cubaynes, S. Guilbaud, M. M. Al Shorman, M. F. Gharaibeh, I. Q. Ababneh, C. Blancard and B. M. McLaughlin, Phys. Rev. A, 2015, 92, 023401 CrossRef.
- M. N. Piancastelli, A. Hempelmann, F. Heiser, O. Gessner, A. Rüdel and U. Becker, Phys. Rev. A, 1999, 59, 300 CrossRef CAS.
- M. Schlapp, R. Trassl, E. Salzborn, R. W. McCullough, T. K. McLaughlin and H. B. Gilbody, Nucl. Instrum. Methods Phys. Res. B, 1995, 98, 525–527 CrossRef CAS.
- G. R. Wight and C. E. Brion, J. Electron Spectrosc. Relat. Phenom., 1974, 4, 25–42 CrossRef CAS.
- I. Hjelte, L. Karlsson, S. Svensson, A. De Fanis, V. Carravetta, N. Saito, M. Kitajima, H. Tanaka, H. Yoshida, A. Hiraya, I. Koyano, K. Ueda and M. N. Piancastelli, J. Chem. Phys., 2005, 122, 084306 CrossRef CAS PubMed.
- M. Martins, S. Reinwardt, J. O. Schunck, J. Schwarz, K. Baev, A. Müller, T. Buhr, A. Perry-Sassmannshausen, S. Klumpp and S. Schippers, J. Phys. Chem. Lett., 2021, 12, 1390–1395 CrossRef CAS PubMed.
- H. B. Pedersen, S. Altevogt, B. Jordon-Thaden, O. Heber, L. Lammich, M. L. Rappaport, D. Schwalm, J. Ullrich, D. Zajfman, R. Treusch, N. Guerassimova, M. Martins and A. Wolf, Phys. Rev. A, 2009, 80, 012707 CrossRef.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- J. Schirmer, A. B. Trofimov, K. J. Randall, J. Feldhaus, A. M. Bradshaw, Y. Ma, C. T. Chen and F. Sette, Phys. Rev. A, 1993, 47, 1136 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- N. Mardirossian and M. Head-Gordon, Phys. Chem. Chem. Phys., 2014, 16, 9904–9924 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- K. Kaufmann, W. Baumeister and M. Jungen, J. Phys. B: At., Mol. Opt. Phys., 1989, 22, 2223 CrossRef CAS.
- R. Baer, E. Livshits and U. Salzner, Annu. Rev. Phys. Chem., 2010, 61, 85–109 CrossRef CAS PubMed.
- P. Cabral do Couto, D. Hollas and P. Slavíček, J. Chem. Theory Comput., 2015, 11, 3234–3244 CrossRef CAS PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
- T. Jahnke, R. Guillemin, L. Inhester, S.-K. Son, G. Kastirke, M. Ilchen, J. Rist, D. Trabert, N. Melzer, N. Anders, T. Mazza, R. Boll, A. De Fanis, V. Music, Th. Weber, M. Weller, S. Eckart, K. Fehre, S. Grundmann, A. Hartung, M. Hofmann, C. Janke, M. Kircher, G. Nalin, A. Pier, J. Siebert, N. Strenger, I. Vela-Perez, T. M. Baumann, P. Grychtol, J. Montano, Y. Ovcharenko, N. Rennhack, D. E. Rivas, R. Wagner, P. Ziolkowski, P. Schmidt, T. Marchenko, O. Travnikova, L. Journel, I. Ismail, E. Kukk, J. Niskanen, F. Trinter, C. Vozzi, M. Devetta, S. Stagira, M. Gisselbrecht, A. L. Jäger, X. Li, Y. Malakar, M. Martins, R. Feifel, L. Ph. H. Schmidt, A. Czasch, G. Sansone, D. Rolles, A. Rudenko, R. Moshammer, R. Dörner, M. Meyer, T. Pfeifer, M. S. Schöffler, R. Santra, M. Simon and M. N. Piancastelli, Phys. Rev. X, 2021, 11, 041044 CAS.
| This journal is © the Owner Societies 2022 |