
Sustainable near UV-curable acrylates based on natural phenolics for stereolithography 3D printing†
Rui
Ding
a,
Yuyang
Du
b,
Rebecca B.
Goncalves
c,
Lorraine F.
Francis
 b and
Theresa M.
Reineke
*a
b and
Theresa M.
Reineke
*a
aDepartment of Chemistry and Center for Sustainable Polymers, University of Minnesota, Minneapolis, USA. E-mail: treineke@umn.edu
bDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, USA
cDepartment of Chemistry, The College of New Jersey, New Jersey, USA
First published on 5th February 2019
Abstract
Photocured polymers have recently gained tremendous interest for a wide range of applications, such as industrial prototyping/additive manufacturing, electronics, medical/dental devices, and tissue engineering. However, current development of photoinitiated thermosetting formulations is mostly centered on commercial monomers/oligomers that are petroleum-derived and not environmentally friendly. This work aims to develop natural phenolic-based (meth)acrylates to expand the use of sustainable and mechanically robust 3D printable formulations. Utilizing thiol–ene chemistry, bifunctional 3,6-dioxa-1,8-octanedithiol eugenol acrylate (E) was synthesized through a highly efficient, scalable method. Real-time infrared spectra and photorheology studies revealed that E exhibits rapid photocuring kinetics and that the viscosity, glass transition temperature (Tg) and thermal properties of this material can be tuned by adding a sustainable reactive diluent, guaiacyl methacrylate (G). The effect of adding a crosslinker to binary GE monomers was further investigated by incorporating vanillyl alcohol dimethacrylate (V) or trimethylolpropane trimethacrylate (T). At 20 mol%, V showed a moderate improvement in curing rate and a lower degree of cross-linking than T due to the bifunctionality of V. However, the aromaticity of V provided more resistance to chain deformation and breakage within the network, demonstrating storage moduli and tensile strengths up to 3.4 GPa and 62 MPa, respectively. The distinct impact of the crosslinkers on the tensile behaviors of glassy terpolymers was correlated to the cohesive energy density. Ternary formulations GEV 60–20–20 by mol% with 2 wt% TPO photoinitiator were successfully printed using a commercial desktop stereolithographic 3D printer with 405 nm violet laser source. This work demonstrates a versatile, sustainable, and scalable synthetic strategy to design a class of natural phenolic acrylates for sustainable photocured formulations with potential translation to high performance 3D printing.
Introduction
Photopolymers are widely used in numerous formulations used in UV curable industrial coatings, adhesives, photolithography, and light-based additive manufacturing techniques.1,2 Stereolithography (SLA) 3D printing is one of the most popular additive manufacturing techniques as it utilizes a light source capable of rapid polymerization and crosslinking of photoreactive resins to pattern and fabricate materials in a layer-by-layer manner.1 Acrylates are the most common category of monomers and oligomers used in SLA 3D printing due to their fast reaction rate to radical polymerization, broad available variety in chemical structure and properties, and low cost. Methacrylate monomers offer similar benefits and higher performance but generally have slower reaction kinetics.3 One significant issue for designing new 3D printable photopolymers is the major reliance on the petrochemical feedstocks, which are of significant global concern due to lack of sustainability.4 Rigid building blocks are required to impart sufficient mechanical strength and stiffness to acrylates and many contain petroleum-based aromatic or cyclic aliphatic components. For example, bisphenol A glycol dimethacrylate (Bis-GMA) and ethoxylated bisphenol A dimethacrylate (Bis-EMA) are currently used as the key ingredients for dental sealants and tissue engineering scaffolds, which have been reported to cause toxicity from both a biological and environmental perspective.5 Therefore, sustainable and economical acrylic and methacrylate surrogates derived from renewable biomass that offer competitive mechanical properties and improved biocompatibility would offer significant benefit for many applications in SLA 3D printing.Among the bio-based feedstocks available for rigid building blocks, softwood lignin-derived model compounds are a family of 2-methoxyphenols bearing a single hydroxyl functionality and a spectrum of substituent functional groups (R = –H, –CH3, –CH2CH2CH3, –CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, –CHO, –CHCH–COOH, etc.) at the para-position of the aromatic ring.6 Because of the reactivity for chemical functionalization of –OH and/or –R groups, these natural phenolic compounds are promising alternatives to bisphenol A formulations for high-performance polymers.7,8 Homo- or statistic linear polymethacrylates of these 2-methoxyphenols have been shown to display high Tg (>90 °C) and viscoelastic properties similar to petroleum-derived materials such as polystyrene and polymethyl methacrylate.6,9,10 However, the photoreactivity of the natural phenolic (meth)acrylates and their renewable thermosetting formulations11–14 has not been studied for UV curing or lithographic applications. Several studies on the UV initiated thiol–ene resins based on step-growth polymerization of vinyl ether functionalized derivatives of natural phenolics have been found to exhibit glass transition temperatures below ambient temperatures and elastomeric behavior.15–19 In order to imitate the diphenolic structure of bisphenol A, many strategies on dimerizing functional natural phenolics including etherification,7 esterification,20–23 acetalization, cross metathesis,24 electrophilic condensation,25,26 or enzyme routes,27 have been explored, but to date, liquid diols have not been created (which represent the monomer precursors of (meth)acrylates that are commonly required for resin formulation). Moreover, many previous methods do not enable efficient, affordable, and green synthetic routes for industrial scale manufacturing.
CH2, –CHO, –CHCH–COOH, etc.) at the para-position of the aromatic ring.6 Because of the reactivity for chemical functionalization of –OH and/or –R groups, these natural phenolic compounds are promising alternatives to bisphenol A formulations for high-performance polymers.7,8 Homo- or statistic linear polymethacrylates of these 2-methoxyphenols have been shown to display high Tg (>90 °C) and viscoelastic properties similar to petroleum-derived materials such as polystyrene and polymethyl methacrylate.6,9,10 However, the photoreactivity of the natural phenolic (meth)acrylates and their renewable thermosetting formulations11–14 has not been studied for UV curing or lithographic applications. Several studies on the UV initiated thiol–ene resins based on step-growth polymerization of vinyl ether functionalized derivatives of natural phenolics have been found to exhibit glass transition temperatures below ambient temperatures and elastomeric behavior.15–19 In order to imitate the diphenolic structure of bisphenol A, many strategies on dimerizing functional natural phenolics including etherification,7 esterification,20–23 acetalization, cross metathesis,24 electrophilic condensation,25,26 or enzyme routes,27 have been explored, but to date, liquid diols have not been created (which represent the monomer precursors of (meth)acrylates that are commonly required for resin formulation). Moreover, many previous methods do not enable efficient, affordable, and green synthetic routes for industrial scale manufacturing.
In addition to improving the printing speed, precision, and versatility of SLA technology,28 expanding the scope of photocurable resin formulations is important to accessing a variety of mechanical properties and functions that resemble or even surpass traditional processing methods and feedstocks. For example, Long29,30 and others31 have reported photo-printable aromatic polyimides based on acrylate-modified precursors, which exhibit high Young's modulus, tensile strength, and high thermal stability similar to commercial engineering thermoplastic Kapton. Due to the “click chemistry” characteristics and the benefits of step-growth radical addition polymerization, thiol–ene/–yne monomers have also been explored for light-based 3D printing.3,32 Tough printed photopolymers with good fidelity were obtained in those formulations, which also demonstrated tunable glass transition temperatures (Tg), impact strength, toughness, and semi-crystalline structures via modulating the building block chemistry and thiol–ene/–yne stoichiometry. Dual-curing strategies utilizing the formation of interpenetrating polymer networks in either sequential or concurrent approaches have also been explored to control the heterogeneous structure and properties of stereolithographic polymers.33,34 Hybrid polymers such as acrylate–epoxide, acrylate–thiol, that are capable of multi-photopolymerization mechanisms (i.e. radical, cationic, click) were explored to achieve desirable mechanical performance including shape memory35 and crack resistance.36 Again, these photoinitiated resins were developed merely on the basis of petroleum-derived feedstocks, revealing a gap between high-performance photo 3D printing and sustainability. It is worthy to note that some efforts on addressing the recyclability of 3D printed thermosets still used BPA-based monomers for model studies.37,38
Herein, we report the first development of resin formulations based on natural phenolic acrylates that possess fast photo-curing rates and high thermal and mechanical properties, which are competitive to commercial prototype resins for SLA 3D printing. The formulations contain a structural diacrylate used to provide physical properties similar to bisphenol A-based acrylates, a mono-methacrylate diluent, and a methacrylate crosslinker with a radical photoinitiator. The structural diacrylate was synthesized by a facile dimerization of eugenol (4-allyl-2-methoxyphenol) with a dithiol through the radical thiol–ene “click” reaction. This solvent-free step promoted full conversion and a yield that does not require further purification for the subsequent acrylation reaction. The flexible thioether linkage between a rigid aromatic moiety served to reduce the Tg and viscosity of the monomer to provide a pure liquid able to be 3D printed. The well-defined “hard–soft–hard” structure of the bifunctional monomer proves sufficient Tg and flexibility for a chain-growth polymerized network.39,40 Guaiacol (2-methoxyphenol) methacrylate was explored as a low-cost, low viscous reactive diluent to formulate with the diacrylate. The crosslinker, vanillyl alcohol (4-(hydroxymethyl)-2-methoxyphenol) dimethacrylate or trimethylolpropane trimethacrylate was added in a fixed ratio to further understand the effect on the critical properties for SLA 3D printing. Real-time FTIR and photorheology were used as complementary techniques to investigate the photocuring kinetics of our multi-component natural phenolic (meth)acrylates resin formulations. The tunability of the Tg, high crosslink density, and tensile properties of the photopolymers after curing were achieved by modulating the monomer ratio in the formulations. Indeed, preliminary 3D printed objects were created with these sustainable phenolic acrylate resins demonstrating the potential application of these renewable formulations.
Experimental section
Materials
Eugenol (99%), vanillyl alcohol (≥98%, FG), methacrylic anhydride (94%, contains 2000 ppm topanol A as inhibitor,), acryloyl chloride (≥97%, contains ∼400 ppm phenothiazine as stabilizer), trimethylolpropane trimethacrylate (T, technical grade, contains 250 ppm monomethyl ether hydroquinone as inhibitor), 2,2-dimethoxy-2-phenylacetophenone (99%), diphenyl(2,4,6-trimethylbenzoyl) phosphine oxide (TPO, 97%) were purchased from Sigma-Aldrich (St Louis, MO, USA). Guaiacol (>98.0%), 3,6-dioxa-1,8-octanedithiol (>97.0%), methyl eugenol (>98.0%), isobornyl acrylate (IBA, >90.0%, stabilized with MEHQ), isobornyl methacrylate (IBMA, >85.0%, stabilized with MEHQ) were obtained from TCI America (Portland, OR, USA). 4-Dimethylaminopyridine (98%) was obtained from Oakwood Chemical (West Columbia, SC). All solvents were supplied by Fisher Scientific (Fair Lawn, NJ). All reagents are used as received without further purification unless otherwise noted. (Meth)acrylate monomers were passed through the alumina plug before formulating all photoresins.Synthesis of guaiacol methacrylate (G) and vanillyl alcohol methacrylate (V)
Guaiacol (62 g, 0.50 mol, 1 equiv.) and a catalytic amount of 4-dimethylaminopyridine (3.7 g, 0.030 mol, 0.06 equiv.) were added into a 500 mL round-bottom flask with a magnetic stir bar, which was oven-dried and kept under constant purging with nitrogen gas for at least 1 hour. Methacrylic anhydride (98 g, 0.60 mol, 1.2 equiv.) was then added into the mixture and allowed for stirring at room temperature for 3 hours. The flask was heated to 50 °C for at least 24 hours until the reaction was complete. The reaction mixture was then cooled down, transferred into an Erlenmeyer flask, and followed by adding 1 L of saturated sodium bicarbonate (NaHCO3) aqueous solution. After vigorous stirring for 1 hour with no evident discharge of CO2, 500 mL of dichloromethane was added to extract the product. The organic phase was washed with saturated NaHCO3, cold 1.0 M NaOH aqueous solution, 1.0 M of HCl aqueous solution, and saturated brine in order to completely remove the byproduct of methacrylic acid, unreacted anhydride, and 4-dimethylaminopyridine. The resulting mixture was dried over anhydrous sodium sulfate and concentrated en vacuo. G was obtained as a colorless, low viscosity liquid. (85%) 1H NMR (400 MHz, Chloroform-d) δ ppm: 7.21 (ddd, J = 9.2, 6.5, 1.7 Hz, 1H), 7.07 (dd, J = 7.8, 1.7 Hz, 1H), 7.02–6.91 (m, 2H), 6.36 (t, J = 1.2 Hz, 1H), 5.75 (t, J = 1.6 Hz, 1H), 3.82 (s, 3H), 2.07 (t, J = 1.3 Hz, 3H). ESI-MS (m/z) C11H12NaO3 (M + Na+; G): 215.07.Vanillyl alcohol (38 g, 0.25 mol, 1 equiv.), 4-dimethylaminopyridine (1.5 g, 0.012 mol, 0.05 equiv.) and methacrylic anhydride (98 g, 0.60 mol, 2.4 equiv.) were added into the flask and followed the above synthetic steps to prepare V. After basic, acidic and neutral washes, further purification was performed by dissolving the crude product into ethanol at 50 °C as a saturated solution (V/ethanol ratio: 1 g/2 ml). The solution was then left at −20 °C for 24 h for recrystallization. Highly pure V was obtained as a white, crystalline solid (m.p. = 47.3 °C) (69%).
1H NMR (400 MHz, Chloroform-d) δ 7.05 (d, J = 7.9 Hz, 1H), 7.01–6.95 (m, 2H), 6.36 (t, J = 1.2 Hz, 1H), 6.16 (dd, J = 1.6, 1.0 Hz, 1H), 5.75 (s, 1H), 5.60 (s, 1H), 5.17 (s, 2H), 3.83 (s, 3H), 2.07 (t, J = 1.2 Hz, 3H), 1.97 (t, J = 1.2 Hz, 3H). ESI-MS (m/z) C15H18NaO5 (M + Na+; V): 301.09.
Synthesis of 3,6-dioxa-1,8-octanedithiol eugenol
Eugenol (9.9 g, 0.060 mol, 2 equiv.), 3,6-dioxa-1,8-octanedithiol (5.5 g, 0.030 mol, 1 equiv.) and 4-dimethylaminopyridine (77 mg, 0.5 wt% of the total weight of mixture) were added into a 20 mL nitrogen-filled scintillation vial with a magnetic stir bar. The mixture was stirred constantly and irradiated in a 36 W UV nail dryer curing lamp with 320–400 nm wavelength at room temperature for 8 h. The kinetics of reaction was monitored by removing aliquots at different time points (0.5 h, 1 h, 2 h, 4 h, and 8 h) for immediate 1H-NMR analysis. 3,6-Dioxa-1,8-octanedithiol eugenol was obtained as a viscous liquid (15.4 g, 100%).1H NMR (400 MHz, Chloroform-d) δ ppm: 6.82 (d, J = 7.7 Hz, 2H), 6.65–6.68 (m, 4H), 5.49 (s, 2H), 3.87 (s, 6H), 3.68–3.58 (m, 8H), 2.67 (dt, J = 28.4, 7.3 Hz, 8H), 2.55 (t, J = 7.3 Hz, 4H), 1.87 (ddd, J = 14.8, 8.2, 6.8 Hz, 4H). ESI-MS (m/z) C26H38NaO6S2 (M + Na+; E): 533.27.
Synthesis of 3,6-dioxa-1,8-octanedithiol eugenol acrylate (E)
3,6-Dioxa-1,8-octanedithiol eugenol (56 g, 0.11 mol, 1 equiv.) and triethylamine (24 g, 0.24 mol, 2.2 equiv.) were dissolved in 200 mL of anhydrous dichloromethane and cooled to 0 °C in an ice bath. A solution of acryloyl chloride (21 g, 0.23 mol, 2.1 equiv.) in 100 mL of anhydrous dichloromethane was added dropwise with stirring. The reaction was allowed to warm to room temperature and after 24 h the triethylamine hydrochloride salts were filtered off. The filtrate was concentrated en vacuo and then passed through a silica gel column with a solution of hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate (1:1) as the eluent. BHT (0.05 wt%) was added as a free radical inhibitor and the solvent was removed to afford E as a light yellow, viscous liquid (53 g, 78%).
:ethyl acetate (1:1) as the eluent. BHT (0.05 wt%) was added as a free radical inhibitor and the solvent was removed to afford E as a light yellow, viscous liquid (53 g, 78%).
1H NMR (400 MHz, Chloroform-d) δ ppm: 6.96 (d, J = 8.0 Hz, 2H), 6.82–6.72 (m, 4H), 6.59 (dd, J = 17.3, 1.4 Hz, 2H), 6.34 (dd, J = 17.3, 10.4 Hz, 2H), 5.99 (dd, J = 10.4, 1.4 Hz, 2H), 3.80 (s, 6H), 3.68–3.58 (m, 8H), 2.71 (td, J = 8.1, 7.5, 5.5 Hz, 8H), 2.58 (t, J = 7.2 Hz, 4H), 1.97–1.85 (m, 4H). ESI-MS (m/z) C32H42NaO8S2 (M + Na+; E): 641.22.
Formulation of resins and preparation of photo-cured specimens
All resin formulations were prepared in scintillation vials at a scale of 2 g. For binary formulations, E were blended with G at different molar ratios of G:E (25:75, 50:50, 67:33 and 75:25). For ternary formulations, T or V were added to the GE compositions at 20 mol% of total, yielding ternary formulation with molar ratios of G:E:T (or G:E:V) (20:60:20, 40:40:20, 53:27:20, 60:20:20). The abbreviation protocol here is described using ternary formulation containing G, E, and T with ratio of 20:60:20 as an example: GET 20–60–20 for the uncured resin and pGET 20–60–20 for the resulting polymers (Fig. 1). V was heated to a low viscous liquid at 50 °C to promote ease of mixing. The photoinitiator TPO (400 mg, 2 wt%) was then added to the resin and sonicated for 3 hours until it was completely dissolved into the liquid resin mixture. Sonication was then also performed to degas the viscous resins. The formulated resins were then were kept in the dark to prevent unwanted photo-polymerization. To prepare thin films of the photo-cured specimens, 0.2 g of liquid acrylate resins was transferred by a 1 ml syringe on to a clean glass slide with a pair of spacers with 0.17 mm thickness on both short ends. The second glass slide was covered on the top and fixed by a pair of binder clips, in order to reduce air exposure during photocuring. The acrylate resins were then subjected to photo-curing for 3 min under the 36 W UV curing lamp with 320–400 nm wavelength and irradiation intensity of 2.6 ± 0.4 mW cm−2 and then a thermal post-cure at 120 °C was performed for 12 h. To prepare dog-bone shaped bars, a silicone mold (0.86 mmm thickness) with a hollow dog-bone shape (typical gauge dimensions of 11 mm (L) × 2.8 mm (W) × 0.85 mm (T)) was used instead of the spacers in the same way as described. The acrylate resins were allowed to photo-cure for 6 min under the same UV lamp and followed by thermal post-curing at 120 °C for 12 h.
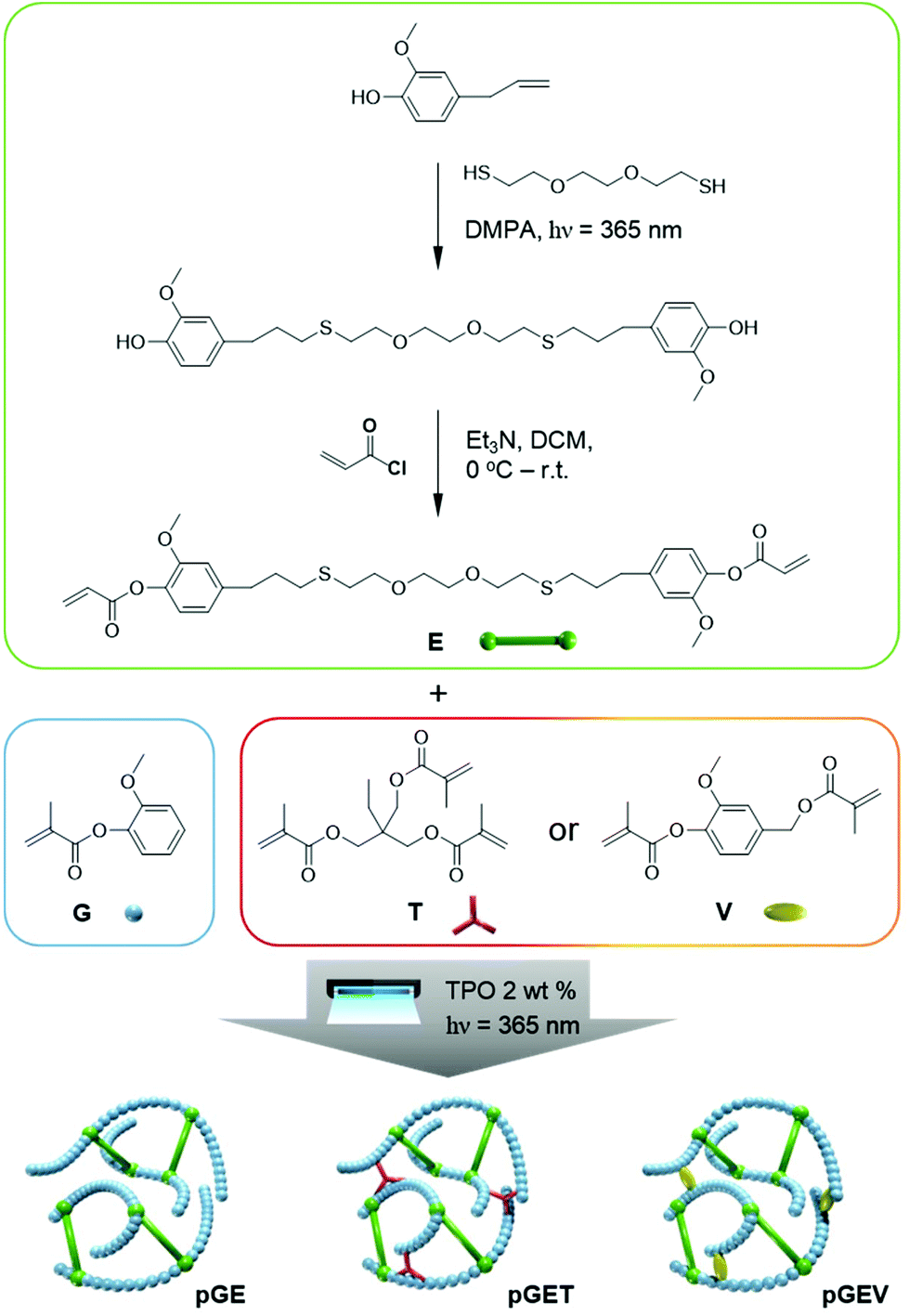 | ||
| Fig. 1 The synthetic route of the E monomer and its binary formulation GE, ternary formulations GET and GEV that are viable for photopolymerization with 2 wt% of TPO to form different cross-linking networks. | ||
SLA 3D printing
The GEV 60–20–20 (with 2 wt% TPO) formulation was selected for a 3D printing test using Formlabs Form 1+ stereolithography desktop printer. The printer is equipped with a violet laser source at 405 nm wavelength and spot size of 300 μm. The formulated resins (about 50 g) were poured into the vat with the dimension of 150 mm by 150 mm. The 3D models of University of Minnesota logo “M” (20.0 mm × 11.8 mm × 2.9 mm) and a standard dogbone specimen (31.8 mm × 4.8 mm × 1.6 mm) were created using Autodesk software and saved as a STL file. In the Preform software that interfacing the CAD models with the Formlabs 3D printer, the Z resolution was set at 100 μm. The printing parameters were set using the default FL clear resin version for testing our resins, and the laser scan speed was 1550 mm s−1. Supporting base and joints were added in the design template as a protocol for printing reliability. The 3D models were laid horizontally with length and width as X–Y plane. The total printing time for the “M” logo was about 15 min and the dogbone specimens was about 20 min. After the layer-by-layer photocuring process was finished, the “green” printed objects were removed from the platform with a scraper, followed by soaking in isopropanol for 10 min to wash off uncured resin liquid. Post-photocuring was conducted in the UV chamber for 30 min.Characterization
C double bond absorption peak. The (meth)acrylate CC double bond absorption peak at 1636 cm−1 was monitored for monomer conversion as a function of irradiation time. The aromatic absorption peak at 1604 cm−1 was used as the reference peak. The double bond conversion was calculated with the ratio of monitored peak areas at irradiation time (ACC,t) to the peak area prior to polymerization (ACC,∅), normalized by the ratio of reference beak area (Aref,t/Aref,∅), shown in eqn (1). All reactions were performed under ambient conditions.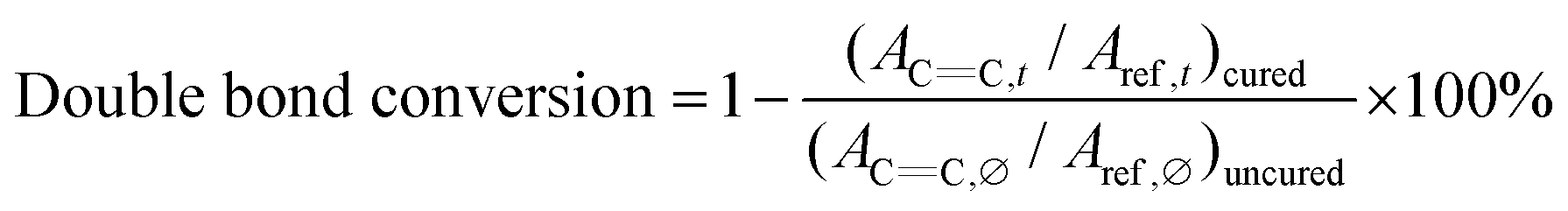 | (1) |
The morphology of 3D printed model was examined using a field emission gun – scanning electron microscope (FEG-SEM) (JEOL 6500) operating at 5 kV under secondary electron imaging mode. The sample was prepared by sputtering a 5 nm layer of platinum coating.
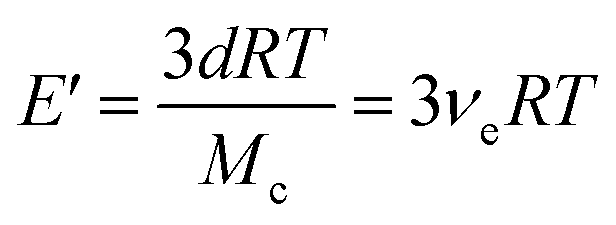 | (2) |
In eqn (2), E is the storage modulus in tension mode, d is the density of the polymer, R is the gas constant, T is the absolute temperature at Tg + 50 °C, and Mc is the average molecular weight of elastically active network chains between cross-links, which is inversely proportional to the crosslink density, νe. Tensile testing was performed using a Shimadzu Autograph AGS-X Series tensile tester on specimens with a dogbone geometry (typical gauge dimensions of 11 mm (L) × 2.8 mm (W) × 0.85 mm (T)) at a uniaxial extension rate of 5 mm min−1. Reported data were the average of at least three replicates. Young's modulus (E) values were calculated by taking the slope of stress–strain curve from 0 to 1% strain.
Results and discussion
To evaluate the efficiency of the first reaction step to synthesize the E monomer, a smaller scale reaction (about 2.5 g) of the thiol–ene reaction between eugenol and 3,6-dioxa-1,8-octanedithiol was conducted in the bulk without the presence of 2,2-dimethoxy-2-phenylacetophenone initiator. The reaction was monitored via NMR and the eugenol allyl protons (–CH2–CHCH2, at 3.30–3.33 ppm, 5.89–5.99 ppm, and 5.03–5.09 ppm, respectively) and dithiol protons (–SH, at 1.56–1.65 ppm) were found to diminish as the proton signals of –CH2–CH2–S–(CH2)2–O– (at 1.85–1.90 ppm) groups were found to increase in a stoichiometric manner over the irradiation time (Fig. S11†). As the reaction proceeded, an apparent increase in viscosity was also observed in the bulk mixture. An addition of a small amount of the 0.5 wt% 2,2-dimethoxy-2-phenylacetophenone initiator was necessary to accelerate the coupling reaction performed in larger scales (about 15 g), reaching full conversion in 8 h according to 1H-NMR. Compared to other thiol–ene click radical reactions,17,19,32,41–44 the relatively slow kinetics of the 3,6-dioxa-1,8-octanedithiol eugenol was attributed to the presence of phenol hydroxyl group in the eugenol, which scavenge radicals to retard the thiol–ene click reaction through additional routes proposed in Fig. S12.† This hypothesis was supported by the fact that methyl protected eugenol showed an initial reaction rate 1.5 times as fast as eugenol reacting with dithiol (Fig. S11†). Nevertheless, the final conversion of this solvent-free thiol–ene reaction was not affected and still proceeded to nearly 100%. 3,6-Dioxa-1,8-octanedithiol eugenol was thus directly used for the next acrylation step without the need for further purification. The resulting E monomer was obtained in 78% yield and offered a viscosity of 5.2 ± 0.5 Pa s at ambient and a Tg of −36.7 °C (Fig. S9†) amenable to 3D printing.
Next, the G monomer was synthesized using a reported solvent-free procedure using methacrylic anhydride. Among the 2-methoxyphenol derivatives found in softwood lignin-based bio-oils, G was selected as the monofunctional reactive diluent due to the optimal balance of low cost, low viscosity, low volatility, and desirable Tg and thermomechanical properties.6,9 While others have reported the synthesis and chromatographic techniques used for isolation and purification,12 it should be noted that this is the first report to create the V crosslinker in high purity and large-scale (50 g) using a modified purification method via a simple recrystallization from ethanol. Though pure V is a crystalline solid at room temperature, the low melting point of 47.3 °C (Fig. S10†) renders it facile mixing with other monomers by mild heating into a low viscosity liquid. The high reactivity of the vinyl group on V towards free-radical polymerization was discovered from the observation that its crude product forms a gel automatically over the course of a few days at room temperature (or in hours at elevated temperature). The potential of V for use as a reactive bifunctional photo-crosslinker from a bio source has been further detailed in the following sections.
Free radical-based photopolymerization of E monomer and G–E monomers
The intrinsic reactivity of the phenolic (meth)acrylates towards photo-initiated polymerization is crucial to the successful application of SLA or other light-based 3D printing processes. To understand this, real time FTIR was performed comparing monomers synthesized in this study and their blends with commercial monomers and 3D printable resins. In Fig. 2, the maximum polymerization rate (Rp) of the G monomer was 0.044 s−1, about 2.1 times higher than that of IBA, although it is still half of the rate of IBA, for all formulations containing 2 wt% TPO and irradiated in the same conditions. In agreement with the structural effects study on the (meth)acrylate reactivity, the methoxyphenyl side groups contribute a higher dipole moment than the isobornyl substituent to the (meth)acrylate, resulting in an enhancement of the inherent reactivity. The bifunctional monomer, E, shows the maximum Rp as 0.084 s−1, similar to 0.083 s−1 of IBMA and slightly less than 0.101 s−1 of Formlabs clear resin. The CC conversion of the E monomer after 60 s irradiation was found to be 88.5% and reached 99.4% (almost full conversion after 10 min irradiation). The fully cured E polymer showed a decomposition temperature (Td at less than 5% weight loss) of 323 °C under nitrogen (Fig. S15†). The pendant phenolic moieties on the acrylic backbones are thermally stable enough to prevent early decomposition before random backbone chain scission normally occurring at 300–350 °C for polyacrylates. In comparison to the commercially available acrylated epoxidized soybean oil (Tg at 38.0 °C determined by tanδ peak and elastic moduli of 0.30 GPa),45 our E polymer shows comparable Tg (45.0 °C) and superior elastic moduli (0.96 GPa at 25 °C). This is likely attributed to the presence of a bulky and rigid 4-methoxy phenyl moiety, present as side-chain groups on the acrylates.
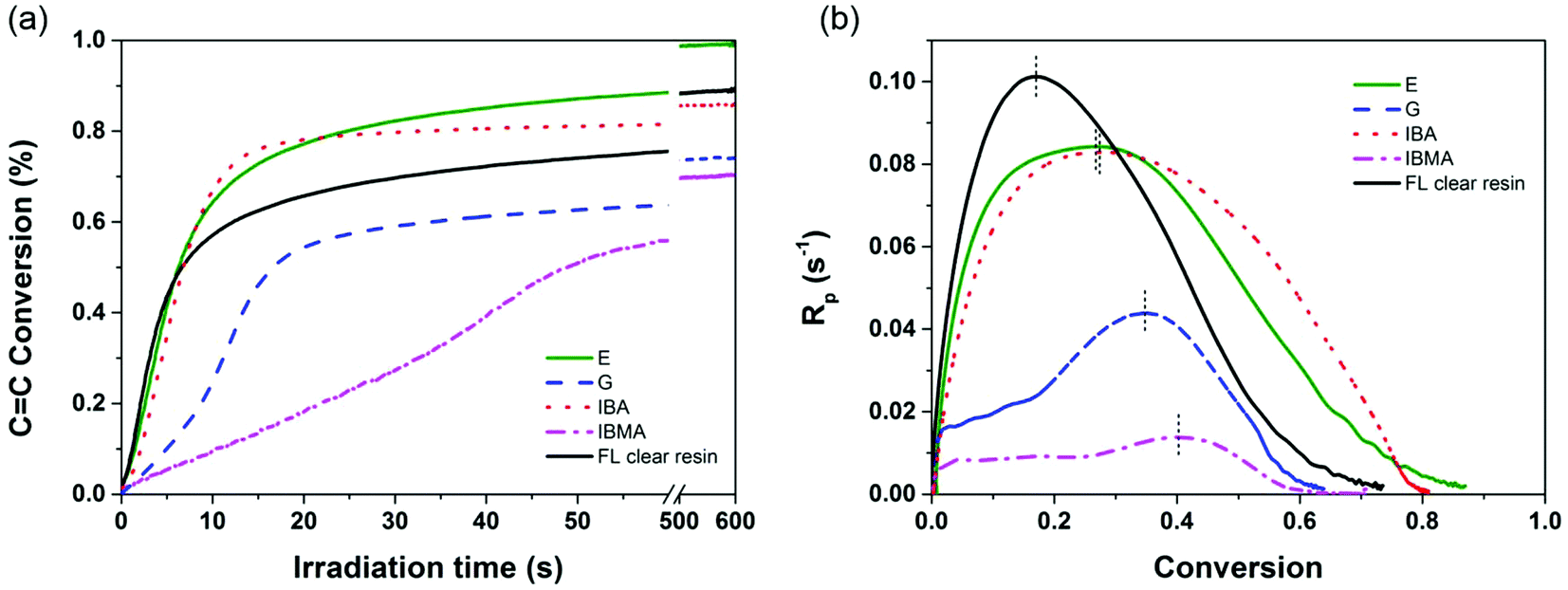 | ||
| Fig. 2 Photopolymerization kinetic profiles of E and G compared to commercially available monomers (IBA and IBMA) and 3D printable prototype resins determined by real time-FTIR. (a) Plot of the double bond conversion as a function of UV irradiation time. (b) Plot of the polymerization rate as a function of monomer conversion at irradiation intensity of 2 mW cm−2. The UV irradiation condition was 365 nm at 2 mW cm−2. | ||
The GE monomer blend of varied ratios demonstrated an unexpected shift of curing kinetics. As shown in Fig. 3, a drastic drop (instead of a ratio-dependent decrease) in the reaction rate with increasing G loading was observed, which is in agreement with the shift of gel time. The retardance in photo-copolymerization rate of the methacrylate–acrylate monomer mixture could be attributed to the dominance of a more stable methacrylate radical during the chain propagation step even at a fraction as low as 20%.46 The addition of G into E led to a slight decrease of CC conversion from ∼90% to below 80%, likely due to the intrinsic low conversion of G itself. The Td of GE binary polymers decreases rapidly from 320 °C to 267 °C with G:E molar ratio, attributed to the higher content of methacrylate backbone with less thermal stability. The Tg and Young's modulus (E′) of the GE binary polymers was raised up to 80 °C and 2.7 GPa respectively with an increase to 75 mol% of G (Table S2†). After photocuring, the incorporation of G exhibited acceptable improvement in thermo-mechanical properties as a compatible monomer with E, as shown by the single tanδ peak with high intensity (Fig. S21†). Nevertheless, the limited reaction rate with varying the G:E ratio leads to the necessity of incorporating a third component, a photoreactive crosslinker, to achieve the high photo-reactivity need to for commercial SLA printing.
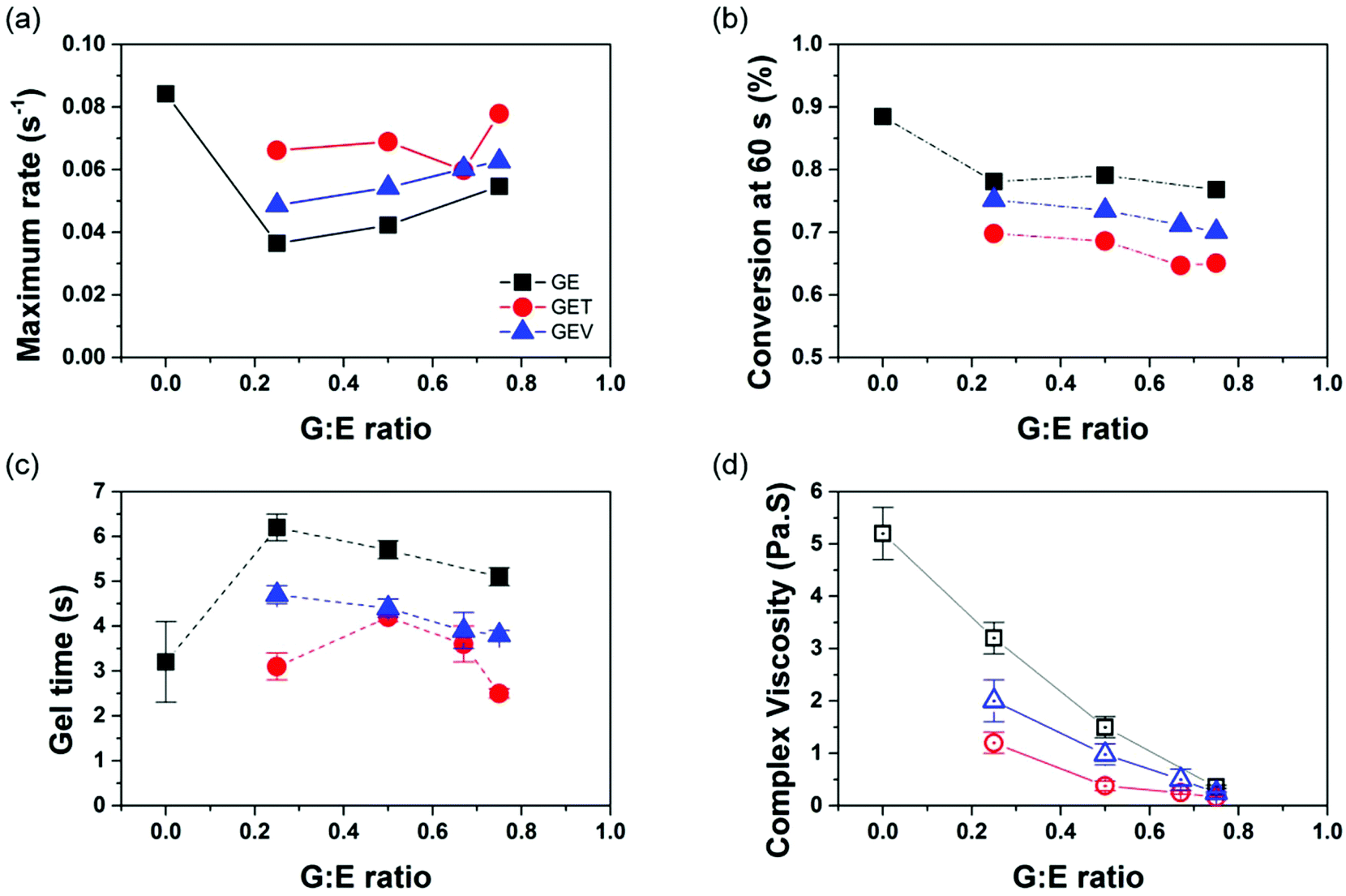 | ||
| Fig. 3 Dependence of the maximum polymerization rate (a), reaction conversion at 60 s from IR (b), gel time (c) and initial complex viscosity of photoresins from photorheology (d) on the ratio of G to E in different formulations. | ||
The influence of cross-linkers (T and V) on GE monomers: photo-curing behaviors
Two low-Mw crosslinkers, T (Mw = 338.4) and V (Mw = 290.3) were thus added to the formulation at 20 mol% and their effects on the properties were investigated and compared. T is a trifunctional, low viscous, highly reactive and readily available aliphatic methacrylate that is widely used in UV curable materials. V is a bifunctional, biorenewable (meth)acrylate that contains natural phenolic structure based on the natural product vanillin. By varying the G:E ratio, the average (meth)acrylate functionality of the ternary system GET ranges between 1.6 and 2 while that of GEV ranges between 1.4 and 1.8 (Table S1†). The dependence of photocuring behavior on the different crosslinkers is illustrated in Fig. 3. As expected, both T and V boost the maximum reaction rate and shorten the gel time of binary GE polymers. T shows a higher efficiency than V on reaction acceleration because of the higher number of methacrylates in the T monomer. For the GET formulation at the highest G:E ratio, it exhibits similar photoreactivity to the E monomer but meanwhile, the viscosity of GET drops to as low as 0.16 Pa s, which is comparable to 0.74 Pa s of Formlabs clear resin. The ability to maintain good flow is important for liquid 3D printable resins to uniformly recoat the fabricated object on each printed layer (to enable repeating the printing process). Due to the higher functionality, T causes more of a conversion drop than V, so that extra caution of T loading need to be taken for the complex resin formulation.
The influence of cross-linkers (T and V) on GE monomers: glass transition, cross-link density and network structure
As expected, the incorporation of T into the binary GE polymers increases the Tg significantly by 50 °C for all G:E ratios. However, a smaller increase in Tg was found for the formulations incorporating V (Table S2†). The origin of further Tg increase is likely attributed to increasing the number of covalent cross-linking sites rather than the side group bulkiness or rigidity of crosslinkers, which restricts the cooperative segmental motions in the network. Compared to the pGET formulation that offers the highest Tg (up to 130.9 °C), the Tg of the pGEV terpolymers reaches 107.5 °C at 60–20–20 mol%. While lower, this increase is sufficient for various practical applications (Table 1). As for the ambient E′ value, it is noteworthy that V outperformed T as a crosslinker; indeed a modulus of 3.40 GPa was found for the pGEV 60–20–20 formulations, which is much higher than 2.51 GPa for the pGET formulation (as shown in Fig. 4a). Here, the higher fraction of natural phenolic moieties in the GEV formulations that are either pendant or crosslinked to the mainchain appeared to play the main role in the higher stiffness of the photocured polymers in the glassy state to resist elastic deformation.
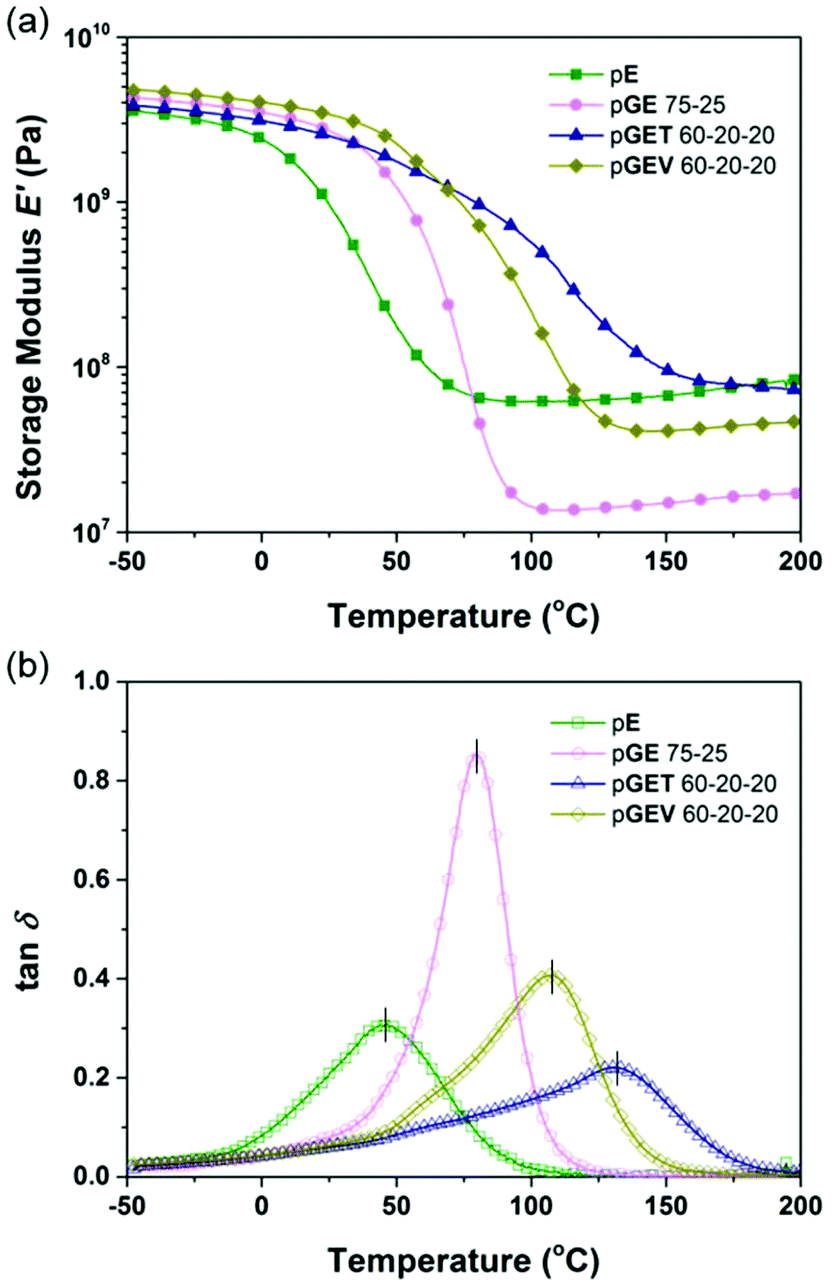 | ||
| Fig. 4 Dynamic mechanical behavior of the formulated resins: (a) storage modulus and (b) tan delta of the photocured E polymer, binary pGE 75–25, and ternary pGET 60–20–20 and pGEV 60–20–20. | ||
| Polymer | T g (°C) | tanδmax |
E′ at 25 °C (GPa) | E′ at Tg + 50 °C (MPa) | ν e (×103 mol m−3) | M c (kg mol−1) |
|---|---|---|---|---|---|---|
| pE | 45.0 | 0.31 | 0.96 | 62 | 3.9 | 0.15 |
| pGE 75–25 | 79.7 | 0.85 | 2.70 | 14 | 2.8 | 0.72 |
| pGET 60–20–20 | 130.9 | 0.22 | 2.51 | 77 | 6.8 | 0.15 |
| pGEV 60–20–20 | 107.5 | 0.41 | 3.40 | 42 | 3.9 | 0.26 |
| FL clear resin | 114.5 | 0.64 | 2.23 | 33 | 3.0 | 0.33 |
The rubbery E′ plateau observed at 50 °C above the Tg is modulated higher than 30 MPa for both pGET and pGEV terpolymers, indicating a highly cross-linked network. The lower calculated cross-link density of the pGEV terpolymers is directly associated with the lower average functionality (less cross-linking sites) of its resin formulation compared to pGET terpolymers (Table S2†). As shown in Fig. 4(b), pGET 60–20–20 exhibits a tanδ peak representing the glass transition regime much broader than the other polymers with same G:E ratio, as well as the neat E polymer with the same rubbery E′ plateau. The other GET formulations show similar results, indicating that pGET terpolymers form a less homogeneous network than pGEV counterparts. In summary, pGEV 60–20–20 demonstrates the most competitive dynamic mechanical properties to the Formlabs clear resin among all the formulations listed in Table 1 and Table S2.†
The influence of cross-linkers (T and V) on G–E thermal and mechanical behavior
With the incorporation of one of the methacrylate crosslinkers, T or V, the thermal stability of the GE binary polymers improved (Fig. S22 and 23†). The overall Td of the pGET terpolymers was found to be above 320 °C, similar to or even higher than that found for the E homopolymer. For the pGEV polymers, the Td remained around 300 °C with less than a 20 °C shift by the G:E ratio variation. For the acrylate–methacrylate hybrid systems, the appropriate degree of cross-linking between polymer backbones appears to be beneficial for increasing methacrylate fraction without deteriorating the thermal stabilities.
Uniaxial tensile tests were performed to investigate the effect of crosslinkers with distinct functionality and molecular structure on the mechanical performance of the photocuring-enhanced terpolymers. In general, all the polymers exhibited stiff and brittle behaviors characteristic of unmodified acrylic materials as shown in Fig. 5. Further comparison between two ternary systems reveals interesting stress–strain behaviors. Using T as the trifunctional crosslinker, pGET terpolymers showed a continuous increase of Young's modulus along with the decrease of strain at break with increasing G:E ratio (Table 2). As a result, pGET 60–20–20 at the highest G:E ratio exhibited the highest tensile modulus yet the lowest tensile strength. In contrast, pGEV terpolymers comprising the bifunctional V crosslinker demonstrated that both tensile modulus and strength increase continuously without compromising the strain at break when increasing G:E ratio. The highest tensile strength (up to 62 MPa) was obtained for the pGEV 60–20–20 formulation, which yielded similar results to the commercial Formlabs clear resin (83 MPa as measured for photocured specimens) and conventional acrylic polymers. In order to understand how the crosslinkers affect the impact resistance of the photocured ternary materials, toughness was found by integrating the area under stress versus strain curve for the two terpolymers and plotted in relation to the rubbery storage modulus (an indicator for crosslink density) in Fig. 6. As the G:E ratio was increased, the toughness values decreased for the pGET terpolymers, which is completely opposite to the case for pGEV terpolymers. These results indicated that for the glassy crosslinked polymers, toughness is not solely dictated by the cross-link density but also related to the chemical structure of crosslinkers that affects intermolecular interaction in the multicomponent network.
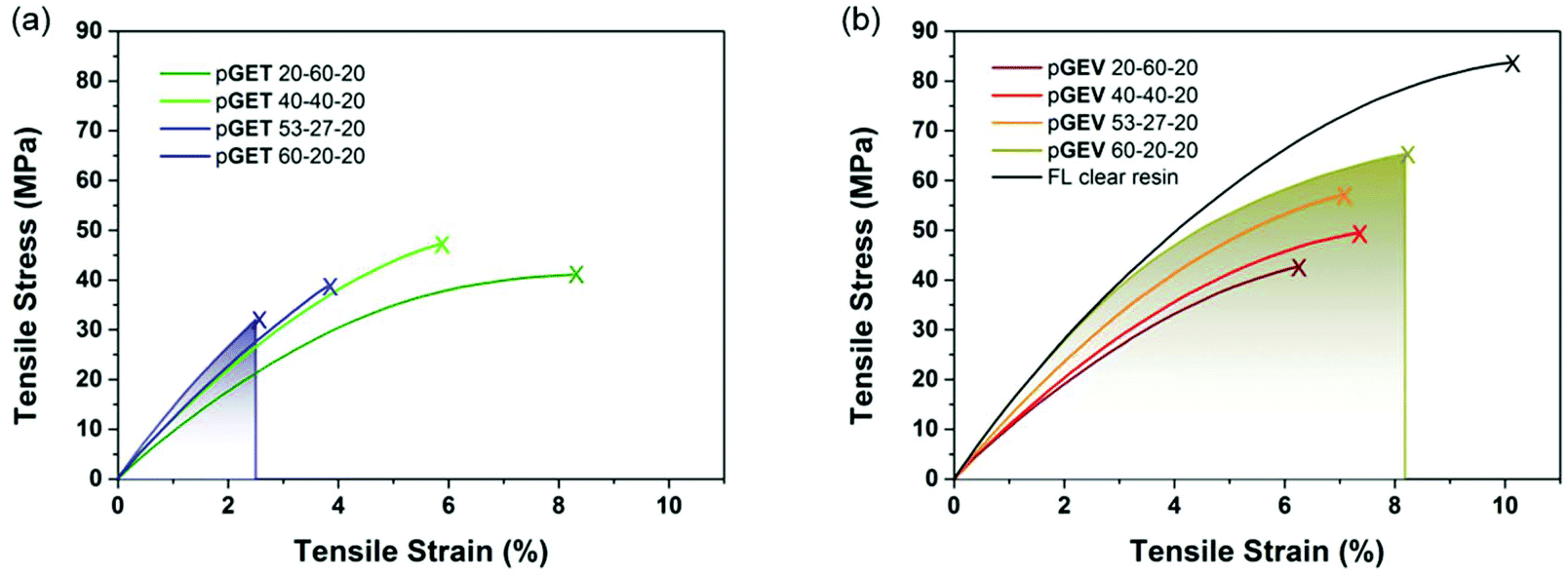 | ||
| Fig. 5 Tensile stress–strain curves of the photocured polymers from ternary formulations (a) GET and (b) GEV as a function of varying the ratios of G to E. The shaded regions denote the integrated areas of stress–strain curves determining toughness. | ||
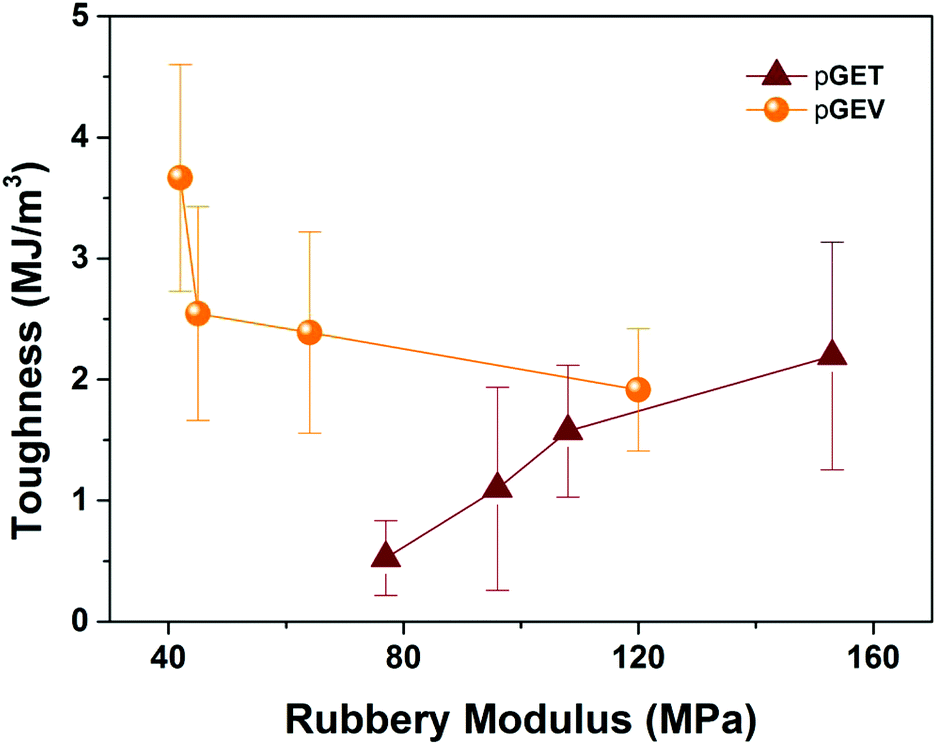 | ||
| Fig. 6 Toughness versus rubbery modulus for the pGET and pGEV terpolymer formulations. | ||
| Polymers | T d5 (°C) | Young's modulus (GPa) | Tensile strength (MPa) | Strain at break (%) | Toughness (MJ m−3) |
|---|---|---|---|---|---|
| pGET 20–60–20 | 338 | 0.83 ± 0.07 | 38.8 ± 4.1 | 8.2 ± 2.7 | 2.2 ± 0.9 |
| pGET 40–40–20 | 329 | 1.04 ± 0.04 | 46.3 ± 6.2 | 6.0 ± 0.9 | 1.6 ± 0.5 |
| pGET 53–27–20 | 321 | 1.18 ± 0.06 | 41.6 ± 9.4 | 3.9 ± 0.8 | 1.1 ± 0.8 |
| pGET 60–20–20 | 324 | 1.35 ± 0.06 | 33.1 ± 7.4 | 2.8 ± 0.9 | 0.5 ± 0.3 |
| pGEV 20–60–20 | 306 | 1.02 ± 0.02 | 44.6 ± 1.8 | 6.9 ± 1.1 | 1.9 ± 0.5 |
| pGEV 40–40–20 | 319 | 1.09 ± 0.02 | 49.7 ± 2.8 | 7.6 ± 1.6 | 2.4 ± 0.8 |
| pGEV 53–27–20 | 309 | 1.19 ± 0.01 | 57.4 ± 4.6 | 7.0 ± 1.6 | 2.5 ± 0.9 |
| pGEV 60–20–20 | 300 | 1.23 ± 0.07 | 61.7 ± 5.1 | 8.9 ± 1.6 | 3.7 ± 0.9 |
| FL clear resin | 240 | 1.38 ± 0.07 | 83.4 ± 2.0 | 10.1± 1.8 | 5.6 ± 1.5 |
By comparing the cohesive energy density (CED), a molecular parameter that is correlated to the interactions between polymer chains and their chemical structure, we were able to rationalize the distinct crosslinker effect on the tensile behaviors. Herein, the method from Fedors et al. was used to calculate and compare the cohesive energy density for the samples (CED = Ecoh/V).47 In this equation, the Ecoh is the cohesive energy (J mol−1) and V is the molar volume (cm3 mol−1). The calculated CED values of G, E, T, V moieties incorporated in the polymers were calculated to be 500, 483, 458 and 509 MPa, respectively. As the T crosslinker has a lower CED value than the G and E monomers, the results imply: (i) the average CED value for pGET terpolymers is lower than the pGE binary polymers and (ii) the increase of G:E ratio increases the CED discrepancy between T and GE binary components, and consequently causes lower homogeneity in network structure. On the contrary, the V crosslinker has a similar and slightly higher CED value than G and E monomer and thus the CED discrepancy between V and GE is diminished in its case. To this end, the pGEV terpolymers are found to have a higher average CED value (and thus stronger intermolecular forces in a more homogeneous network) compared to pGET terpolymers, which appears to influence its tensile behavior.
3D printing performance
Among the ternary formulations discussed in the previous section, GEV 60–20–20 outperforms the others in the following aspects: high bio-renewable content, high curing rate and low viscosity of the monomers, and the photocured polymer product offered high Tg, tensile modulus, strength and good toughness. Therefore, the 3D printing capability was investigated using this formulation in a 50 g scale on a desktop Form 1+ SLA printer. As shown in Fig. 7a, the designed CAD models were successfully printed with reasonable fidelity. The topography of the printed objects was also examined by SEM in Fig. 7b, showing that the actual layer thickness measured by Image J varied from 60 μm to 90 μm, which is lower than the Z resolution (set as 100 μm). A few defects observed on the printed surface is indicative of the existence of certain amount of sol fraction after laser curing. Optimization of the printing parameters such as increasing the laser exposure time or intensity is expected to improve the monomer conversion in the gel state. For a specific photo-initiated resin formulation, the laser exposure necessary to achieve a prescribed cure depth is defined by the penetration depth and the critical exposure of the resin. Factors like photoinitiator and UV absorber could significantly affect both of the resin parameters that are key to determine printability and should be further understood. Selecting a UV absorber compatible with the photo-initiated resin at an appropriate loading48 could increase the UV absorption and therefore allow for improved printing precision. Indeed, those parameters are currently being optimized systematically to further improve the accuracy and precision of SLA 3D printing, which is beyond the scope of this current work.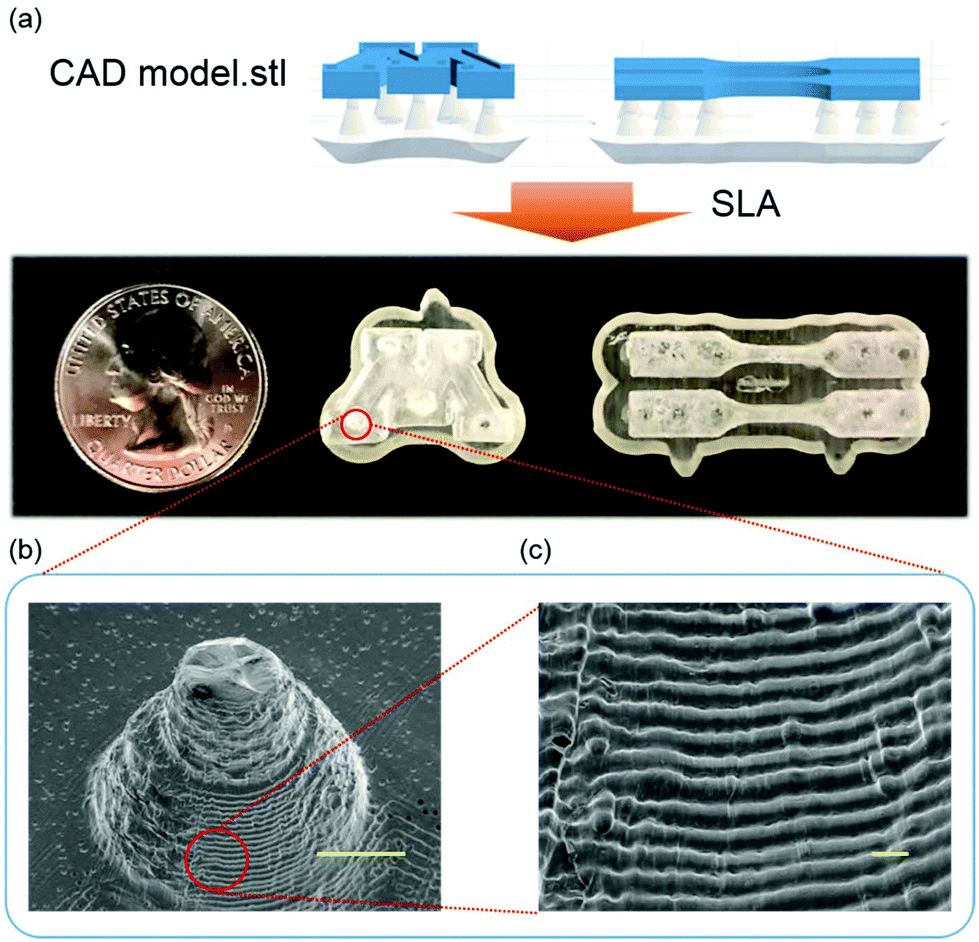 | ||
| Fig. 7 SLA printed object examples from the GEV 60–20–20 photoresin with 2% TPO photoinitiator. (a) A printed “M” logo and dogbone tensile bars on the substrate. (b) Scanning electron microscopy image of a support cone was taken to observe the print resolution of objects, exhibiting the stair-casing feature. Scale bar is 100 mm. (c) Magnified image with scale bar of 100 μm. | ||
Conclusion
This work demonstrates that natural phenolic (meth)acrylates derived from softwood lignin provide promising thermomechanical behavior as sustainable thermosetting resins. Combining the convenience of thiol–ene click chemistry and the inherent unsaturation of natural phenolics, novel acrylate monomers, (E) can be created through efficient, green, and scalable synthetic routes to incorporate aromatic biobased building blocks. Photokinetic studies of G showed its superior photoreactivity to isobornyl methacrylate. The third incorporated natural phenolic photo-crosslinker, V was further blended with E and G to achieve the formulation GEV, which offered a promising combination of fast reactivity for SLA 3D printing and high Tg, modulus, tensile strength and thermal stability. All together, this work shows that functionalized natural phenolics from biomass yield excellent photocuring kinetics to form covalent crosslinked networks that enable light-based 3D printing of mechanically robust and high bio-content objects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the funding source from the Center for Sustainable Polymers supported by the National Science Foundation (CHE-1413862). We would like to thank Prof. Christopher J. Ellison and Dr. Heonjoo Ha from the Department of Chemical Engineering and Materials Science for the access to FTIR and UV-Vis spectrometer and kind guidance on ATR-IR and real-time IR kinetic tests. We would also like to thank Dr. David Giles for the assistance in photorheology and dynamic mechanical analysis experiment. We are grateful to Dr. Leon Lillie and Kunwei Liu for their valuable thoughts to this project. YD and LFF acknowledge support from the industrial partners of the Coating Process Fundamentals Program and advice from Prof. Alon McCormick.References
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349–1352 CrossRef CAS PubMed.
- A. Oesterreicher, J. Wiener, M. Roth, A. Moser, R. Gmeiner, M. Edler, G. Pinter and T. Griesser, Polym. Chem., 2016, 7, 5169–5180 RSC.
- J. T. Sutton, K. Rajan, D. P. Harper and S. C. Chmely, ACS Appl. Mater. Interfaces, 2018, 10, 36456–36463 CrossRef CAS PubMed.
- N. B. Cramer, J. W. Stansbury and C. N. Bowman, J. Dent. Res., 2011, 90, 402–416 CrossRef CAS PubMed.
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- J. Wan, J. Zhao, B. Gan, C. Li, J. Molina-Aldareguia, Y. Zhao, Y.-T. Pan and D.-Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 2869–2880 CrossRef CAS.
- J. F. Stanzione, J. M. Sadler, J. J. La Scala and R. P. Wool, ChemSusChem, 2012, 5, 1291–1297 CrossRef CAS PubMed.
- J. F. Stanzione, P. A. Giangiulio, J. M. Sadler, J. J. La Scala and R. P. Wool, ACS Sustainable Chem. Eng., 2013, 1, 419–426 CrossRef CAS.
- K. Liu, S. A. Madbouly and M. R. Kessler, Eur. Polym. J., 2015, 69, 16–28 CrossRef CAS.
- Y. Zhang, V. K. Thakur, Y. Li, T. F. Garrison, Z. Gao, J. Gu and M. R. Kessler, Macromol. Mater. Eng., 2018, 303, 1700278 CrossRef.
- Y. Zhang, Y. Li, L. Wang, Z. Gao and M. R. Kessler, ACS Sustainable Chem. Eng., 2017, 5, 8876–8883 CrossRef CAS.
- Y. Zhang, Y. Li, V. K. Thakur, L. Wang, J. Gu, Z. Gao, B. Fan, Q. Wu and M. R. Kessler, RSC Adv., 2018, 8, 13780–13788 RSC.
- J. Dai, S. Ma, L. Zhu, S. Wang, L. Yang, Z. Song, X. Liu and J. Zhu, Polymer, 2017, 108, 215–222 CrossRef CAS.
- T. Yoshimura, T. Shimasaki, N. Teramoto and M. Shibata, Eur. Polym. J., 2015, 67, 397–408 CrossRef CAS.
- T. Modjinou, H. Rodriguez-Tobias, G. Morales, D.-L. Versace, V. Langlois, D. Grande and E. Renard, RSC Adv., 2016, 6, 88135–88142 RSC.
- G. Yang, S. L. Kristufek, L. A. Link, K. L. Wooley and M. L. Robertson, Macromolecules, 2015, 48, 8418–8427 CrossRef CAS.
- G. Yang, S. L. Kristufek, L. A. Link, K. L. Wooley and M. L. Robertson, Macromolecules, 2016, 49, 7737–7748 CrossRef CAS.
- M. Firdaus and M. A. R. Meier, Eur. Polym. J., 2013, 49, 156–166 CrossRef CAS.
- I. Barbara, A. L. Flourat and F. Allais, Eur. Polym. J., 2015, 62, 236–243 CrossRef CAS.
- M. A. Ouimet, J. Griffin, A. L. Carbone-Howell, W.-H. Wu, N. D. Stebbins, R. Di and K. E. Uhrich, Biomacromolecules, 2013, 14, 854–861 CrossRef CAS PubMed.
- M. Z. Oulame, F. Pion, S. Allauddin, K. V. S. N. Raju, P. H. Ducrot and F. Allais, Eur. Polym. J., 2015, 63, 186–193 CrossRef CAS.
- B. G. Harvey, C. M. Sahagun, A. J. Guenthner, T. J. Groshens, L. R. Cambrea, J. T. Reams and J. M. Mabry, ChemSusChem, 2014, 7, 1964–1969 CrossRef CAS PubMed.
- E. D. Hernandez, A. W. Bassett, J. M. Sadler, J. J. La Scala and J. F. Stanzione, ACS Sustainable Chem. Eng., 2016, 4, 4328–4339 CrossRef CAS.
- Q. Chen, W. Huang, P. Chen, C. Peng, H. Xie, Z. K. Zhao, M. Sohail and M. Bao, ChemCatChem, 2015, 7, 1083–1089 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 7693–7700 RSC.
- R. L. Truby and J. A. Lewis, Nature, 2016, 540, 371–378 CrossRef CAS.
- M. Hegde, V. Meenakshisundaram, N. Chartrain, S. Sekhar, D. Tafti, C. B. Williams and T. E. Long, Adv. Mater., 2017, 29, 1701240 CrossRef PubMed.
- J. Herzberger, V. Meenakshisundaram, C. B. Williams and T. E. Long, ACS Macro Lett., 2018, 7, 493–497 CrossRef CAS.
- Y. Guo, Z. Ji, Y. Zhang, X. Wang and F. Zhou, J. Mater. Chem. A, 2017, 5, 16307–16314 RSC.
- D. G. Sycks, T. Wu, H. S. Park and K. Gall, J. Appl. Polym. Sci., 2018, 135, 46259 CrossRef.
- G. J. Berg, T. Gong, C. R. Fenoli and C. N. Bowman, Macromolecules, 2014, 47, 3473–3482 CrossRef CAS.
- O. Konuray, X. Fernández-Francos, X. Ramis and À. Serra, Polymer, 2018, 10, 178 Search PubMed.
- R. Yu, X. Yang, Y. Zhang, X. Zhao, X. Wu, T. Zhao, Y. Zhao and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 1820–1829 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 CrossRef.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- S.-H. Pyo, P. Wang, H. H. Hwang, W. Zhu, J. Warner and S. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 836–844 CrossRef CAS PubMed.
- R. Ding, Y. Xia, T. C. Mauldin and M. R. Kessler, Polymer, 2014, 55, 5718–5726 CrossRef CAS.
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Polym. Chem., 2018, 9, 3272–3278 RSC.
- Q. Zou, L. Ba, X. Tan, M. Tu, J. Cheng and J. Zhang, J. Mater. Sci., 2016, 51, 10596–10607 CrossRef CAS.
- J. Heo, T. Kang, S. G. Jang, D. S. Hwang, J. M. Spruell, K. L. Killops, J. H. Waite and C. J. Hawker, J. Am. Chem. Soc., 2012, 134, 20139–20145 CrossRef CAS PubMed.
- B. R. Donovan, J. S. Cobb, E. F. T. Hoff and D. L. Patton, RSC Adv., 2014, 4, 61927–61935 RSC.
- S. N. Khot, J. J. Lascala, E. Can, S. S. Morye, G. I. Williams, G. R. Palmese, S. H. Kusefoglu and R. P. Wool, J. Appl. Polym. Sci., 2001, 82, 703–723 CrossRef CAS.
- M. Buback and E. Müller, Macromol. Chem. Phys., 2007, 208, 581–593 CrossRef CAS.
- R. F. Fedors, Polym. Eng. Sci., 1974, 14, 147–154 CrossRef CAS.
- H. Gong, B. P. Bickham, A. T. Woolley and G. P. Nordin, Lab Chip, 2017, 17, 2899–2909 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py01652f |
| This journal is © The Royal Society of Chemistry 2019 |