
Reprocessable polyhydroxyurethane networks exhibiting full property recovery and concurrent associative and dissociative dynamic chemistry via transcarbamoylation and reversible cyclic carbonate aminolysis†
Xi
Chen
 a,
Lingqiao
Li
a,
Kailong
Jin
a and
John M.
Torkelson
*ab
a,
Lingqiao
Li
a,
Kailong
Jin
a and
John M.
Torkelson
*ab
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: j-torkelson@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 21st September 2017
Abstract
We discovered that polyhydroxyurethane (PHU) networks synthesized in the presence of a catalyst from five-membered cyclic carbonates are intrinsically reprocessable with full property recovery via transcarbamoylation exchange reactions and reversible cyclic carbonate aminolysis. Through a judicious choice of monomers, we demonstrated that PHU networks can be recycled multiple times with full property retention. The presence of reversible reactions in addition to exchange reactions in PHU network reprocessing should spur reconsideration of the underlying reprocessing chemistries associated with some dynamic covalent polymer networks which have been ascribed solely to exchange reactions. With excellent reprocessability, this synthetic framework also serves as a sustainable non-isocyanate-based alternative to traditional polyurethane (PU) networks.
Traditional covalent polymer networks cannot be recycled for high value applications because permanent cross-links prevent them from being melt-reprocessed.1 Studies focused on reprocessable polymer networks have been ongoing for fifteen years, with goals of producing self-healing networks and recycling of cross-linked polymers with complete (or nearly complete) recovery of properties associated with cross-links.1–7 Reprocessable polymer networks, also called covalent adaptable networks8 (CANs) and dynamic covalent polymer networks4 (DCPNs), are cross-linked polymers that contain sufficient dynamic bonds for network reconfiguration under appropriate conditions. Such networks undergo dynamic chemistry which can be divided into two types:3 (1) dissociative dynamic chemistry, in which dynamic covalent bonds break under a stimulus (e.g., high temperature) and later reform when the stimulus is removed, and (2) associative dynamic exchange chemistry in which there is an exchange of covalent bonds with the number of bonds remaining constant. Examples of dissociative dynamic chemistry used in DCPNs include alkoxyamine dynamic bonds9,10 and Diels–Alder (and hetero-Diels–Alder) chemistry;11–14 examples of associative dynamic chemistry include transamination15 and transesterification.16,17 The name “vitrimer”16 has been given to dynamic covalent cross-linked polymers that undergo exchange reactions leading to the materials relaxing stresses and being malleable at sufficiently high temperature but having a “vitrified” cross-linked topology at sufficiently low temperature.
Notably, although some dynamic functionalities that can be incorporated into DCPNs have the potential to undergo associative exchange reactions as well as dissociative reversible reactions, to the best of our knowledge, experimental studies claiming (complete or nearly complete) success in achieving reprocessable polymer networks with full property recovery have been described as involving one or the other of the dynamic chemistries but not both. Possibly this is because the two types of chemistries exhibit decidedly different temperature dependences, making one essentially fully dominant at the reprocessing temperature of choice. In other cases, deleterious side reactions may accompany dissociative reactions at the high temperatures required for significant levels of dissociation to contribute meaningfully to the dynamic chemistry, making it impossible to achieve complete or nearly complete recovery of the cross-link density. In any case, experimentally studied DCPNs leading to successful reprocessing have been characterized as involving either dissociative or associative dynamic chemistry, but not both. Additionally, in most studies reported to date, full property recovery (within error) associated with the cross-link density has not been achieved in reprocessed DCPNs.11,18–22 Very recently, a small number of studies have reported full recovery of such properties after multiple reprocessing steps,10,15,23–25 which proves that the potential sustainability benefits of recycling spent polymer networks for high-value applications are attainable in selected circumstances.
Unfortunately, conventional polyurethane networks are unable to be reprocessed for such applications. Polyurethane (PU) is the sixth most widely produced polymer, with worldwide annual production estimated to have reached 18 million tons in 2016.26 Recently, a number of studies have been published on PU-based networks with dynamic covalent bonds. With the exception of research by a single research group,27 the studies focused on dynamic PU-based networks have incorporated non-carbamate functionalities into the networks (e.g., Diels–Alder bonds, alkoxyamines or disulfide bonds).15,24,25,28–35 The reformulation of the PU-based networks with other dynamic functionalities comes at the cost of often complex, sophisticated chemistry which may render commercial application impractical. However, the incorporation of other dynamic functionalities was justified because although the carbamate bonds in PU are exchangeable, transcarbamoylation in PU networks is “sluggish”21 and attempts to speed these reactions by going to high temperature exceeding 200 °C also result in carbamate dissociation to alcohols and isocyanates with deleterious side reactions.21 In 2016, Zhao, Xie and coworkers employed PU networks “with carbamate bonds as the only potentially (dynamically) active structural moieties”.27 By adding dibutyltin dilaurate as a catalyst and using a 130 °C reprocessing temperature, they demonstrated that the carbamate exchange reaction was effective at providing thermal plasticity necessary to achieve permanent reshaping of a thermoset shape-memory PU. Nevertheless, they also stated that the carbamate exchange reaction was insufficient for effectively reprocessing PU networks.27 Thus, in the absence of non-carbamate functionalities, conventional isocyanate-based PU networks are not effectively reprocessable.
An alternative to an isocyanate-based PU is a non-isocyanate PU (NIPU),36 the most common being polyhydroxyurethane (PHU) which can be synthesized via reaction of cyclic carbonate functional groups with amine functional groups. As such, PHUs have additional sustainability (via human health) benefits as they eliminate the use of toxic isocyanates involved in conventional PU production. Numerous studies have been published on single-phase PHUs, nanophase-separated PHUs, and cross-linked PHU networks,21,37–60 but only two studies have focused on reprocessable PHU networks.21,60 Dichtel, Hillmyer, and coworkers indicated that PHU thermosets derived from bis(six-membered cyclic carbonates) (6CCs) and amines can achieve network rearrangement through transcarbamoylation between the carbamate group and the pendant hydroxyl group; they classified this PHU network as a new class of vitrimers.21,60 The material recovered 75% of the original tensile properties after reprocessing at 160 °C for 8 h, and they attributed the loss of properties to minor decomposition in the network at elevated temperature over long times.21 They also compared PHU networks synthesized from bis(five-membered cyclic carbonates) (5CCs) and 6CCs and concluded that 5CC-derived PHUs decompose to some extent at elevated temperature due to reversion of the PHU linkage and subsequent side reactions, hence having poor reprocessability.60 In both cases, the PHUs were synthesized and reprocessed in the absence of a catalyst.21,60
Here, we have reconsidered the viability of PHU networks in achieving reprocessable DCPNs with full property recovery related to the cross-link density. We have used two types of five-membered cyclic carbonates and two types of amines, with 4-(dimethylamino)pyridine (DMAP) as the catalyst for network rearrangement, thus allowing for lower reprocessing temperature than employed in ref. 21 and 60. By using a trifunctional cyclic carbonate and a bifunctional amine, we obtained a PHU network that can be reprocessed multiple times with full recovery of the rubbery plateau modulus and ultimate tensile strength. Additionally, at the reprocessing temperature employed in our study and in the presence of DMAP catalyst, we discovered that, in addition to the transcarbamoylation exchange reaction, reversible cyclic carbonate aminolysis also plays an important role in network rearrangement.
We first synthesized a PHU network elastomer using poly(propylene glycol) dicyclocarbonate (PPGDC) and tris(2-aminoethyl)amine (TAEA). The resulting elastomer (PPGDC-TAEA) can be recycled multiple times through reprocessing at 140 °C for 2 h, a lower temperature and a shorter timescale than those used in previous PHU network reprocessability studies.21,60 The reprocessing steps were performed by cutting the network into pieces and remolding them in a compression mold with a 7-ton ram force (generating a pressure of ∼11 MPa). Scheme 1 shows the mechanism of formation and rearrangement of this PPGDC-TAEA network. Intact films were formed after reprocessing the PHU networks, as shown in Fig. 1(a). All samples successfully maintained their intact forms and swelled after being immersed in solvent for 24 h, verifying the existence of cross-links in original and reprocessed networks. These results indicate that the cross-linked chains rearranged at 140 °C under high pressure so that dynamic covalent bonds are formed to bring the broken pieces into a consolidated film.
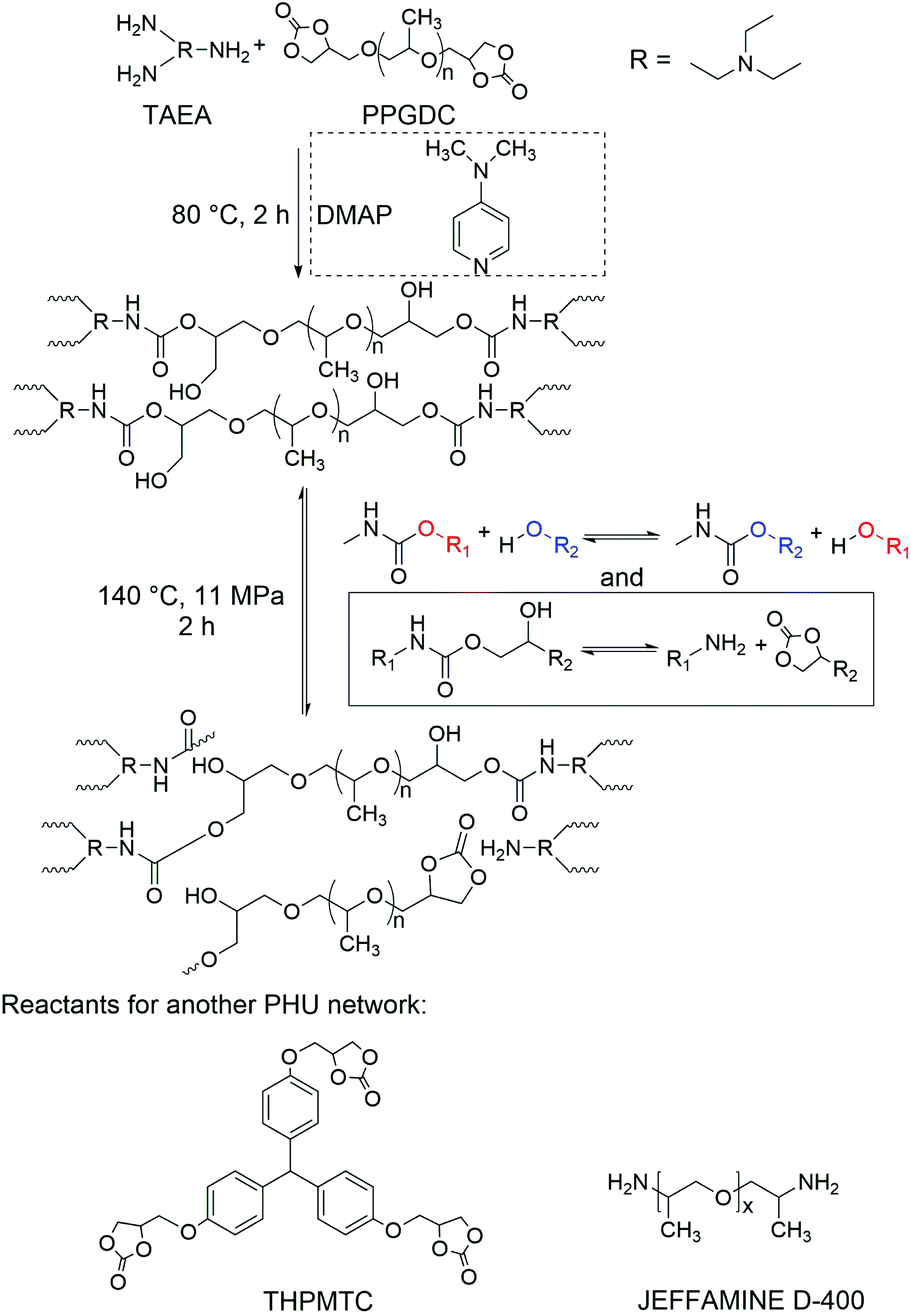 | ||
| Scheme 1 Mechanism of the synthesis and rearrangement of PPGDC-TAEA PHU networks (molecular structures of reactants used in another PHU network are shown at the bottom). | ||
 | ||
Fig. 1 (a) Photograph of a PPGDC-TAEA PHU sample before and after reprocessing. (b) Dynamic mechanical responses of PPGDC-TAEA PHU networks, E′ (half-open symbols) and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ (open symbols) as a function of temperature for the 1st reprocessed (squares), 2nd reprocessed (circles) and 3rd reprocessed (triangles) samples. δ (open symbols) as a function of temperature for the 1st reprocessed (squares), 2nd reprocessed (circles) and 3rd reprocessed (triangles) samples. | ||
As shown in Fig. 1(b), we characterized the PPGDC-TAEA networks by dynamic mechanical analysis (DMA). The rubbery plateaus at high temperature are consistent with the cross-linked nature of the reprocessed PHU samples. However, a 30% decrease in the rubbery plateau value of tensile storage modulus, E′, was observed after each reprocessing step, indicating a loss in the cross-link density during reprocessing. (Recall from ideal rubber elasticity theory that the rubbery plateau modulus is linearly related to the cross-link density.61) The increasing tanδ peak values with the increasing number of recycles also indicate an increasingly decross-linked nature of the material.62 In addition, the decreasing Tgs as indicated by the shifting tanδ peaks (often used as a signature of shifted Tg)62 and by differential scanning calorimetry (Fig. S1(a)†) are also consistent with the loss of cross-link density. Table 1 summarizes the mechanical properties of the 1st, 2nd and 3rd reprocessed PPGDC-TAEA networks characterized at room temperature. The strain-at-break values of these samples were the same within error. However, there was a noticeable decrease in tensile strength from 1.6 MPa to 1.2 MPa upon reprocessing for the third time. This reduction in ultimate tensile strength can be attributed to a decrease in the cross-link density.
| Sample | Tensile strength (MPa) | Strain at break (%) | |
|---|---|---|---|
| PPGDC-TAEA | 1st reprocessed | 1.7 ± 0.1 | 105 ± 9 |
| 2nd reprocessed | 1.6 ± 0.1 | 113 ± 13 | |
| 3rd reprocessed | 1.2 ± 0.1 | 115 ± 14 | |
| THPMTC-JEFFAMINE® D-400 | 1st reprocessed | 9.7 ± 1.1 | 200 ± 18 |
| 2nd reprocessed | 9.4 ± 1.3 | 198 ± 30 | |
| 3rd reprocessed | 10.1 ± 1.6 | 234 ± 24 | |
To reveal the underlying mechanism of network rearrangement and explain the decreasing cross-link density after reprocessing, we performed Fourier transform infrared (FTIR) spectroscopy to investigate the changes of chemical bonds in original and reprocessed cross-linked PPGDC-TAEA PHU samples. Fig. 2(a) shows the normalized spectra of original and recycled samples from 1850 cm−1 to 1750 cm−1, with the cyclic carbonate peak centered at ∼1800 cm−1 (for full spectra, see Fig. S2†). The spectra were normalized with respect to the ether peak at ∼1100 cm−1, which is constant during synthesis and reprocessing steps. In the original PHU sample, the cyclic carbonate peak could not be observed, indicating essentially full conversion of reactants into the network. However, the cyclic carbonate peaks appeared in the reprocessed samples, with an increasing trend in peak height as the network was increasingly reprocessed. The fact that the level of cyclic carbonate in the network increased after each reprocessing is evidence of the reverse reaction of aminolysis under reprocessing conditions. This reverse reaction must decross-link the networks to a certain extent and cause losses in the cross-link density as well as associated properties after reprocessing.
 | ||
| Fig. 2 (a) Normalized FTIR transmittance of the original (solid), 1st reprocessed (dash), 2nd reprocessed (dot) and 3rd reprocessed (dash dot) PPGDC-TAEA samples from 1850 cm−1 to 1750 cm−1. (b) FTIR transmittance of the 1st reprocessed sample (dash), the 1st reprocessed sample after 2 h annealing at 80 °C with the additional TAEA and DMF (solid), and the 1st reprocessed sample after 2 h annealing at 80 °C with DMF only (dot). (c) Normalized FTIR transmittance of the original sample (solid), the original sample after 2 h annealing at 140 °C (dot), and the 1st reprocessed sample (dash). | ||
When we annealed the 1st reprocessed sample for 12 h at reaction temperature (80 °C) in an attempt to recover the cross-links, we found that the cyclic carbonate peak did not change (Fig. S3†). This is because at 140 °C, TAEA is relatively volatile (and possibly unstable due to the tertiary amine structure which is present in many commonly used catalysts including DMAP, triazabicyclodecene and triethylamine). During the 1st reprocessing step at 140 °C, the TAEA component may generate from the reverse reaction and evaporate from the system (and possibly undergo side reactions), further pushing the reversible reaction toward cyclic carbonates and leaving them with no amine to react with. We also performed thermogravimetric analysis on the original PPGDC-TAEA network; the sample showed a weight loss of 2.43% after being heated under a helium flow from 25 °C to 140 °C and held at 140 °C for 150 min (Fig. S4(a)†). The volatile fragments from the furnace were passed through a gas chromatography-mass spectrometry (GC-MS) system. The complex MS spectrum (Fig. S4(b)†) rules out the possibility of solely water desorption during heating and confirms the volatilization (and possible side reactions) of the TAEA component.
To further investigate the appearance of cyclic carbonate after reprocessing, we placed two pieces of the 1st reprocessed samples in different vials, and added an equal weight of TAEA to one of the vials. Anhydrous dimethylformamide (DMF) was then added to both vials, aiming to facilitate the diffusion of amines into the network. Both vials were held at 80 °C for 2 h, and the samples were then characterized by FTIR spectroscopy, as shown in Fig. 2(b). The cyclic carbonate peak at ∼1800 cm−1 was retained in the sample annealed without TAEA, whereas the cyclic carbonate peak disappeared in the sample annealed with TAEA. Thus, the cyclic carbonate that was formed in the network after reprocessing was able to react with amines under appropriate conditions. These results indicate the presence of reversible cyclic carbonate aminolysis in our PHU system during reprocessing, as shown in the boxed region in Scheme 1.
We studied factors influencing the reversibility of cyclic carbonate aminolysis by performing a control experiment. We annealed an original sample at 140 °C for 2 h and compared its normalized FTIR spectrum with the original and 1st reprocessed samples, as shown in Fig. 2(c). In contrast to the original sample, the annealed sample showed a cyclic carbonate peak at ∼1800 cm−1, indicating that high temperature plays an important role in pushing the aminolysis backward during reprocessing. In addition, the fact that the 1st reprocessed sample showed a higher cyclic carbonate peak than the original sample after annealing makes evident the effect of high pressure in accelerating the reverse reaction during reprocessing.
We also designed a small-molecule model system for further validation of the reversibility of cyclic carbonate aminolysis at the reprocessing temperature. The model molecule was synthesized using divinyl benzene dicyclocarbonate (DVBDC) and monofunctional cyclohexylamine (CYCHA) (see Fig. S5† for both structures). The reaction was run at 80 °C for 36 h, and the product was then annealed at 140 °C for 8 h, which allows for evaporation of any free amine. Nuclear magnetic resonance (NMR) spectroscopy was used to monitor the change in the cyclic carbonate level during this process. As shown in Fig. S6,† the NMR peaks associated with cyclic carbonate disappeared completely after the 36 h reaction and reappeared after the 8 h annealing at 140 °C, indicating the reverse reaction of cyclic carbonate aminolysis. This result indicates that the reversibility of cyclic carbonate aminolysis at high temperature exists not only in cross-linked systems, but also in linear systems. This fact may be important in future studies of linear or branched PHUs adopting this chemistry in order to avoid the loss of volatile components resulting from the reverse reaction.
To eliminate the loss resulting from volatilization (and possible side reactions) of TAEA in the cross-link density and associated properties after reprocessing, we synthesized another PHU network with tris(4-hydroxyphenyl)methane tricyclocarbonate (THPMTC) and JEFFAMINE® D-400 (see Scheme 1 for both structures), which are both non-volatile and stable at 140 °C. The mechanisms of synthesis and rearrangement of this network are shown in Fig. S7.† The resulting network (THPMTC-JEFFAMINE® D-400) was reprocessed three times at 140 °C for 1 h, and the cross-linked nature of the reprocessed products was confirmed by their insolubility in good solvents. Fig. 3(a) shows the DMA results for the reprocessed THPMTC-JEFFAMINE® D-400 networks. The three reprocessed samples exhibited identical E′ values (within error) in the rubbery plateau region at 110 °C (0.85 ± 0.02 MPa, 0.82 ± 0.02 MPa, 0.84 ± 0.05 MPa for the 1st, 2nd and 3rd reprocessed samples) and very similar tanδ curves, indicating full retention of the cross-link density after reprocessing. Also, the differential scanning calorimetry curves of the reprocessed samples showed identical Tgs within an experimental error of ±1 °C (Fig. S1(b)†). These results indicate that the decross-linking caused by reverse cyclic carbonate aminolysis can be fully reversed if both reagents remain in the network. We then performed tensile testing on reprocessed THPMTC-JEFFAMINE® D-400 networks at room temperature. Tensile strength and elongation at break results are summarized in Table 1, and representative stress–strain curves are shown in Fig. 3(b). These samples exhibited a tensile strength of ∼10 MPa and elongation at break of ∼200%. Within error, the results were the same for all reprocessed samples. Therefore, our THPMTC-JEFFAMINE® D-400 network possesses excellent reproduction of both mechanical and thermal properties after multiple reprocessing steps.
 | ||
| Fig. 3 (a) Dynamic mechanical responses of THPMTC-JEFFAMINE® D-400 PHU networks, E′ (half-open symbols) and tanδ (open symbols) as a function of temperature for the 1st reprocessed (squares), 2nd reprocessed (circles) and 3rd reprocessed (triangles) samples. (b) Representative stress–strain curves of the 1st reprocessed (solid), 2nd reprocessed (dash) and 3rd reprocessed (dot) THPMTC-JEFFAMINE® D-400 PHU networks. | ||
We performed a control experiment to show the effect of catalyst on network reprocessing. A THPMTC-JEFFAMINE® D-400 network was synthesized in the absence of a catalyst and identical reprocessing conditions (140 °C, 1 h) were applied to this network. Compared to the network with a catalyst, the network without a catalyst cannot be reprocessed under these conditions, showing cracks and disconnected regions (Fig. S8(a)†). Unlike the network with a catalyst which maintained its integral form in good solvents, the network without a catalyst broke into pieces within a few hours when immersed in DMF (Fig. S8(b)†). These results indicate the role of a catalyst in affecting the rate of dynamic chemistry during reprocessing. When the catalyst is eliminated from the system, the network reprocessing requires a longer time and/or a higher temperature. Fortman et al.60 indicated that, compared to 6CCs, 5CCs have greater thermodynamic stability, and hence PHU networks derived from 5CCs are prone to reversion and associated side reactions at 160 °C or a higher temperature needed for reprocessing in the absence of the catalyst. We also observed this reversion in our PPGDC-TAEA networks during reprocessing at 140 °C in the presence of the catalyst, as shown in Fig. 2(a) and (c). However, by switching to precursors that are non-volatile and stable at the reprocessing temperature (THPMTC and JEFFAMINE® D-400), the stability of the PHU network was enhanced significantly. Fig. S9† shows the FTIR spectra of the 1st, 2nd and 3rd reprocessed THPMTC-JEFFAMINE® D-400 networks. The curves overlap much better than those for the reprocessed PPGDC-TAEA samples shown in Fig. S2,† indicating improved stability of the network chemical structure during remolding. Therefore, we regard the instability and poor reprocessability of PPGDC-TAEA networks at 140 °C in the presence of the catalyst as mainly a result of the use of TAEA precursors and not because of deleterious side reactions accompanying the reverse aminolysis. (TAEA was also used in the reprocessable PHU network studies by Fortman et al.,21,60 which may have contributed to the relatively limited reprocessability of their networks.) Also, the relatively short reprocessing time (1 h), relatively low reprocessing temperature (140 °C), and the use of a catalyst associated with our THPMTC-JEFFAMINE® D-400 networks made any possible side reactions of the liberated amine and cyclic carbonate less likely to happen, further benefiting their stability.
To demonstrate the existence of transcarbamoylation exchange reactions during reprocessing, we performed a decross-linking study on the THPMTC-JEFFAMINE® D-400 network. We added equal masses of the 1st reprocessed network into two test tubes. In tube 1, an excess amount of 1,4-butanediol and some dimethyl sulfoxide (DMSO) solvent was added. In tube 2, only DMSO was added (Fig. S10(a)†). The test tubes were placed in an oil bath at the reprocessing temperature (140 °C). After 24 h, the mixture in tube 1 became completely transparent (Fig. S10(b)†), indicating decross-linking of the network. In contrast, network materials remained swelled in tube 2, showing no sign of decross-linking. These results indicate that the carbamate groups in our PHU networks undergo transcarbamoylation exchange reactions with hydroxyl groups at the reprocessing temperature, making the network capable of being completely decross-linked when linear alcohols are added. Therefore, the combined results for the PPGDC-TAEA network and the THPMTC-JEFFAMINE® D-400 network demonstrate that both reversible cyclic carbonate aminolysis (through a dynamic equilibrium between hydroxyurethane and cyclic carbonate/amine groups) and transcarbamoylation exchange reactions (between carbamate groups and hydroxyl groups) contribute significantly to the network rearrangement during reprocessing. Thus, unlike the PHU networks described as vitrimers in ref. 21 and 60, our PHU is not a vitrimer as the reprocessing involves both associative and dissociative chemistry.
Our results on PHU networks suggest that the impact of reversible reactions in addition to exchange reactions on material properties should also be considered for networks made by other step-growth chemistry. For example, Obadia et al. reported a reprocessable ion-conducting network through transalkylation exchanges by cross-linking linear poly(1,2,3-triazolium ionic liquid) with 1,6-dibromohexane.20 The resulting network showed a 50% loss in modulus and tensile strength after each reprocessing step, which was attributed to chain-scission side reactions although such evidence was absent. In fact, their network may have suffered from decross-linking by reverse reaction and loss of volatile 1,6-dibromohexane reactants at the reprocessing temperature of 160 °C. Future work is warranted on the effect of the reprocessing temperature, external mechanical stress, and catalyst load on the rate of network rearrangement, which will give a better understanding of the optimal reprocessing conditions. Additionally, the equilibrium reactant conversion as a function of temperature should be determined for reversible cyclic carbonate aminolysis in order to gain more insight into this chemistry.
Conclusions
We have developed reprocessable PHU networks with full property recovery after multiple reprocessing steps. We also discovered that dissociative reversible cyclic carbonate aminolysis participates with associative transcarbamoylation exchange reactions in the rearrangement of our PHU networks during reprocessing. This discovery indicates that some reprocessable PHU networks do not fall into the vitrimer categorization21 due to the existence and important roles of both associative and dissociative dynamic chemistry during reprocessing. Utilizing transcarbamoylation and reversible cyclic carbonate aminolysis, PHU networks can be designed to be intrinsically reprocessable with full property retention. With excellent reprocessability and more environmentally benign generation and the sustainable use of materials, non-isocyanate-based PHU networks have the potential to serve as effective replacements for conventional PU networks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work made use of central facilities supported by the MRSEC program of the National Science Foundation (DMR-1121262) at the Northwestern University Materials Research Science and Engineering Center as well as facilities supported by Northwestern University at the Integrated Molecular Structure Education and Research Center (IMSERC). We also acknowledge support of Northwestern University via discretionary funds associated with a Walter P. Murphy Professorship (J. M. T.), an ISEN Fellowship (L. L.), and a Terminal Year Fellowship (K. J.).References
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- E. Trovatti, T. M. Lacerda, A. J. F. Carvalho and A. Gandini, Adv. Mater., 2015, 27, 2242–2245 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- A. M. Schenzel, N. Moszner and C. Barner-Kowollik, Polym. Chem., 2017, 8, 414–420 RSC.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 10108–10117 RSC.
- W. Gao, M. Bie, F. Liu, P. Chang and Y. Quan, ACS Appl. Mater. Interfaces, 2017, 9, 15798–15808 CAS.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS PubMed.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- N. Zheng, Z. Fang, W. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- P. Du, M. Wu, X. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, J. Appl. Polym. Sci., 2014, 131, 40234 Search PubMed.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- Y. Zhong, X. Wang, Z. Zheng and P. Du, J. Appl. Polym. Sci., 2015, 132, 41944 Search PubMed.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- Y. Yang and M. W. Urban, Polym. Chem., 2017, 8, 303–309 RSC.
- L. Zhang, L. Chen and S. J. Rowan, Macromol. Chem. Phys., 2017, 218, 1600320 CrossRef.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2012, 113, 80–118 CrossRef PubMed.
- N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2765–2773 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 860–867 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 851–859 CrossRef CAS.
- V. M. Lombardo, E. A. Dhulst, E. K. Leitsch, N. Wilmot, W. H. Heath, A. P. Gies, M. D. Miller, J. M. Torkelson and K. A. Scheidt, Eur. J. Org. Chem., 2015, 2791–2795 CrossRef CAS.
- E. K. Leitsch, G. Beniah, K. Liu, T. Lan, W. H. Heath, K. A. Scheidt and J. M. Torkelson, ACS Macro Lett., 2016, 5, 424–429 CrossRef CAS.
- G. Beniah, K. Liu, W. H. Heath, M. D. Miller, K. A. Scheidt and J. M. Torkelson, Eur. Polym. J., 2016, 84, 770–783 CrossRef CAS.
- G. Beniah, B. E. Uno, T. Lan, J. Jeon, W. H. Heath, K. A. Scheidt and J. M. Torkelson, Polymer, 2017, 110, 218–227 CrossRef CAS.
- G. Beniah, X. Chen, B. E. Uno, K. Liu, E. K. Leitsch, J. Jeon, W. H. Heath, K. A. Scheidt and J. M. Torkelson, Macromolecules, 2017, 50, 3193–3203 CrossRef CAS.
- G. Beniah, W. H. Heath, J. Jeon and J. M. Torkelson, J. Appl. Polym. Sci., 2017, 134, 44942 CrossRef.
- G. Beniah, D. J. Fortman, W. H. Heath, W. R. Dichtel and J. M. Torkelson, Macromolecules, 2017, 50, 4425–4434 CrossRef CAS.
- G. Beniah, W. H. Heath and J. M. Torkelson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3347–3351 CrossRef CAS.
- J. Nanclares, Z. S. Petrović, I. Javni, M. Ionescu and F. Jaramillo, J. Appl. Polym. Sci., 2015, 132, 42492 CrossRef.
- C. Carré, H. Zoccheddu, S. Delalande, P. Pichon and L. Avérous, Eur. Polym. J., 2016, 84, 759–769 CrossRef.
- A. Yuen, A. Bossion, E. Gomez-Bengoa, F. Ruiperez, M. Isik, J. L. Hedrick, D. Mecerreyes, Y. Y. Yang and H. Sardon, Polym. Chem., 2016, 7, 2105–2111 RSC.
- K. Zhang, A. M. Nelson, S. J. Talley, M. Chen, E. Margaretta, A. G. Hudson, R. B. Moore and T. E. Long, Green Chem., 2016, 18, 4667–4681 RSC.
- M. M. Mazurek-Budzyńska, G. Rokicki, M. Drzewicz, P. A. Guńka and J. Zachara, Eur. Polym. J., 2016, 84, 799–811 CrossRef.
- L. Poussard, J. Mariage, B. Grignard, C. Detrembleur, C. Jérôme, C. Calberg, B. Heinrichs, J. De Winter, P. Gerbaux, J. M. Raquez, L. Bonnaud and P. Dubois, Macromolecules, 2016, 49, 2162–2171 CrossRef CAS.
- B. Grignard, J.-M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J.-M. Raquez, P. Dubois, M.-P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- H. Blattmann and R. Mülhaupt, Macromolecules, 2016, 49, 742–751 CrossRef CAS.
- A. Cornille, G. Michaud, F. Simon, S. Fouquay, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 404–420 CrossRef CAS.
- A. Cornille, C. Guillet, S. Benyahya, C. Negrell, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 873–888 CrossRef CAS.
- H. Matsukizono and T. Endo, RSC Adv., 2015, 5, 71360–71369 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, United States, 1953 Search PubMed.
- K. P. Menard, Dynamic mechanical analysis: a practical introduction, CRC Press, Boca Raton, United States, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, experimental details, and additional data. See DOI: 10.1039/c7py01160a |
| This journal is © The Royal Society of Chemistry 2017 |