DOI:
10.1039/D4TA09149C
(Paper)
J. Mater. Chem. A, 2025,
13, 13440-13456
Nitrogen-induced deep reconstruction and formation of a high-valent nickel species γ-NiOOH surface layer on NiFealloy/NiFeN pre-catalysts for efficient water oxidation†
Received
24th December 2024
, Accepted 19th March 2025
First published on 21st March 2025
Abstract
Transition metal-based electrocatalysts undergo electrochemical surface reconstruction to generate metal oxy-hydroxide-based hybrids, which are regarded as the actual active sites for the oxygen evolution reaction (OER). Many efforts have been devoted to understanding the electrochemical surface reconstruction, but there are not many studies that have identified the origin of the improved OER performance derived from the substrate. Herein, we report the electrochemical synthesis of an amorphous γ-NiOOH surface layer on NiFealloy/NiFeN pre-catalysts for efficient water oxidation. However, the conversion of β-NiOOH to γ-NiOOH was a thermodynamically unfavorable process, which demanded a much higher applied potential to drive the reaction and subsequently catalyze the OER. We identified that the NiFe-bimetallic active sites can promote the OER catalytic activity more than the Ni-monometallic active sites. Moreover, nitrogen could reduce the potential required to generate γ-NiOOH OER-active sites from β-NiOOH by generating the NO3− anion, which promoted the formation of γ-NiOOH. The electrochemical analysis and in situ spectroscopic approaches, including cyclic voltammetry (CV), linear sweep voltammetry (LSV), and Raman spectroscopy, revealed that the Ni species in NiFealloy/NiFeN thermodynamically favored the formation of γ-NiOOH more than the Nialloy/NiN and NiFeLDH. Furthermore, X-ray photoelectron spectroscopy (XPS), high-resolution transmission electron microscopy (HRTEM), and Bode plots demonstrated that Fe doping and nitrogen significantly increased the electrochemically active sites. Additionally, DFT calculation results showed that the electronic structure of the catalysts was modulated by Fe doping, and the surface reconstruction optimized the adsorption energy of the oxygen-containing species and enhanced the OER catalytic activity. This work provides a new design for constructing transition metal-based electrocatalysts for water oxidation.
1. Introduction
Electrochemical water splitting (EC-WS) is considered an attractive pathway to produce hydrogen via electrical energy conversion.1,2 EC-WS has two half-reactions, namely, the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode, which generate hydrogen (H2) and oxygen (O2) gases, respectively.3,4 The sluggish kinetics of the four-proton coupled electron transfer in the OER process compared with the HER process limits the application of water electrolysis, and hence, highly effective electrocatalysts are urgently needed.5,6 In recent studies, NiFe-based catalysts have emerged as one of the best catalysts for water oxidation.7,8 A variety of NiFe-based catalysts (alloys, oxides, hydroxides, sulfides, phosphides, and nitrides) have been reported as electrocatalysts that exhibit extraordinary catalytic behavior for the OER under alkaline conditions.9,10 Recently, NiFe-based catalysts are known as pre-catalysts which undergo electrochemical surface reconstruction and form oxyhydroxide-based hybrids as the true catalysts for OER.11,12 The reconstructed active metal oxyhydroxide hybrids usually have much higher OER catalytic performance than their bulk counterparts prepared via conventional methods.13,14 However, the mechanism between the reconstructed metal oxyhydroxide and pre-catalysts is still unclear, especially with regards to understanding the OER performance and the structural correlation.15,16 Therefore, identifying the origin of the enhanced OER performance and providing design principles for these pre-catalysts are highly desirable for constructing highly active electrocatalysts. For the NiFe-based catalysts, many efforts have been made to achieve a better OER performance, such as by increasing specific surface areas to increase the number of active sites via a synthesis strategy and enhancing the intrinsic activity of active sites via multi-element doping or construction of a heterostructure.17,18 Indeed, the amorphous NiFe-based catalysts revealed excellent potential in OER because the lattice jumbling and distortion can form more effective catalytic active sites.19,20 Recently, operando spectroscopies have revealed that β-Ni(OH)2 deprotonated to generate high-valent species β-NiOOH can further transform to γ-NiOOH, which is regarded as the actual active species for the OER.21,22 However, the conversion of β-NiOOH to γ-NiOOH is a thermodynamically unfavorable process, which demands a much higher applied potential to drive the reaction and subsequently catalyze the OER.23,24 Hence, promoting the formation of the γ-NiOOH active species under operando conditions to boost the OER performance of NiFe-based electrocatalysts has been rarely investigated.
Herein, we report a novel heterostructure of the Fe-doped Ni alloy coupled with Ni nitride (NiFealloy/NiFeN) for efficient water oxidation and provide insight into its surface reconstruction phenomenon, which promotes the formation of the γ-NiOOH active species during OER under alkaline conditions. Fe-doped Ni(OH)2 (NiFeLDH) nanosheets with extremely high specific surface area were first fabricated via a novel hydrothermal modification method. Subsequently, NiFeLDH undergoes a transformation into the NiFealloy/NiFeN heterostructure via a vapor phase nitration process in one step using LiNH2 in a nitrogen (N2) atmosphere. As a pre-catalyst, NiFealloy/NiFeN undergoes electrochemical activation using cyclic voltammetry (CV) to form the amorphous high-valent nickel species γ-NiOOH, which exhibits superior catalytic activity and stability. Due to the high electronegativity of nitrogen and the high stability of the NiFe alloy, CV activation lead to the formation of a very thin layer of γ-NiOOH on the surface of the NiFealloy/NiFeN heterostructure, which is the actual catalyst for efficient water oxidation. To overcome this problem, chronopotentiometry (CP) at different current densities was used to achieve superior surface activation, which revealed that the surface was highly activated upon increasing the current density by 40 mA cm−2. In situ Raman spectroscopy and electrochemical measurements reveal that NiFealloy/NiFeN more readily forms γ-NiOOH. Furthermore, the catalytic system γ-NiOOH/NiFealloy/NiFeN exhibited excellent electrochemical activity compared to γ-NiOOH/Nialloy/NiN, and γ-NiOOH/NiFeLDH, which led to superior OER catalytic performance. DFT calculation results also show that Fe doping can modulate the electronic structure of the catalysts.
2. Experimental section
2.1. Materials and chemicals
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O; 99%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O; 98%), and potassium hydroxide (KOH; ≥95%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Trimesic acid (H3BTC, C9H6O6; 98%) was purchased from Energy Chemical Co., Ltd. Polyvinylpyrrolidone (PVP, (C6H9NO)n; 99%), lithium amide (LiNH2; 99.9%), oleylamine (OAM, C18H37N; 99%), N,N-dimethylformamide (DMF, HCON(CH3)2; 99.9%), and ethanol (C2H5OH; 99.9%) were purchased from Aladdin Reagent Co., Ltd. The ultrapure water was obtained from a Millipore Autopure system with a resistivity of 18.25 MΩ cm. All other materials in this study were used as received without any further purification.
2.2. Synthesis of Ni MOF
The one-step hydrothermal reaction was used to prepare Ni MOF. First, a piece of nickel foam with an area of 2 cm × 3 cm was sonicated in HCl (3 M), deionized water, and ethanol for 15 min each before drying under vacuum to remove any surface contaminants, such as oils, dust, or other impurities before use. After this, 0.216 g of Ni(NO3)2·6H2O, 0.75 g of PVP, and 0.075 g of H3BTC were dissolved in a mixture of 10 mL of ethanol, 10 mL of water, and 10 mL of DMF with stirring. Then, stirring was maintained with the solution for 30 minutes to ensure homogeneity. The solution was transferred to a Teflon-lined stainless-steel autoclave with a capacity of 50 mL containing a piece of NF, and sealed at 150 °C for 10 h with an increasing rate of 2 °C min−1. After cooling down to room temperature, Ni MOF/NF was collected and cleaned several times with ethanol and UP water. Finally, it was dried under vacuum condition overnight.
2.3. Synthesis of the iron-doped nickel-layered double hydroxide nanosheets (NiFeLDH)
The one-step hydrothermal reaction was used to prepare all samples. A solution with x g of Fe(NO3)3·9H2O (x = 0, 25, 50, 75, and 100) was dissolved in 20 mL UP water. Then, 0.5 mL of OAM was dissolved in 10 mL of ethanol, and added dropwise to the above solution with stirring. Stirring was then maintained for 30 minutes to ensure homogeneity. Then, the solution was transferred to a Teflon-lined stainless-steel autoclave with a capacity of 50 mL containing a piece of Ni MOF/NF, and sealed at 140 °C for 10 h with an increasing rate of 2 °C min−1. After cooling down to room temperature, the NiFeLDH/NF samples were collected and cleaned several times with ethanol and UP water. Finally, it was dried under vacuum condition overnight.
2.4. Synthesis of the iron-doped nickel alloy/nickel nitride heterostructure (NiFealloy/NiFeN)
NiFealloy/NiFeN samples were prepared via the one-step vapor phase nitration method. In a typical procedure, NiFeLDH/NF samples were placed downstream of a ceramic boat, and LiNH2 was placed upstream at the other side with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The system was then covered by another ceramic boat, and placed in a tube furnace filled with nitrogen gas (N2) with a flow rate of 5 sccm at 400 °C for 2 h (heating rate of 2 °C min−1). After cooling down to room temperature, the NiFealloy/NiFeN/NF samples were collected.
:5. The system was then covered by another ceramic boat, and placed in a tube furnace filled with nitrogen gas (N2) with a flow rate of 5 sccm at 400 °C for 2 h (heating rate of 2 °C min−1). After cooling down to room temperature, the NiFealloy/NiFeN/NF samples were collected.
2.5. Synthesis of iron-doped nickel oxide (NiFeO)
A NiFeO sample was prepared via calcination of NiFeLDH/NF in a tube furnace at 400 °C for 2 h in air (heating rate 2 °C min−1). After cooling down to room temperature, NiFeO/NF was collected.
2.6. Electrochemical surface reconstruction via rapid CV activation to form the γ-NiOOH active surface layer
For NiFeLDH, NiFeO, and NiFealloy/NiFeN, cyclic voltammetry (CV) in the potential range of 0.92–1.82 (V vs. RHE) with a sweep rate of 500 mV s−1 was carried out to activate and clear up the surface to form an amorphous layer of high-valent β-NiOOH, which converts to γ-NiOOH before the OER.
2.7. Electrochemical surface reconstruction via slow rate CV until steady-state to form the γ-NiOOH active surface layer
Cyclic voltammetry (CV) with a sweep rate of 5 mV s−1 was carried out to activate the surface until reaching the steady-state performance to reveal the formation of high-valent β, γ-NiOOH before the OER.
2.8. Deep electrochemical surface reconstruction via chronopotentiometry (CP) at different current densities
Chronopotentiometry at a different current density range of 10–100 mA was carried out for deep electrochemical reconstruction of NiFealloy/NiFeN to optimize the number of γ-NiOOH active sites on the surface for superior OER catalytic performance.
2.9. Physical characterization
Field emission scanning electron microscope (FESEM) graphs were obtained by Zeiss SIGMA to investigate the surface morphology and fine structure of the catalysts. Transmission electron microscopy (TEM) coupled with high-resolution transmission electron microscopy (HRTEM), energy dispersive X-ray spectroscopy (EDX), and selected area electron diffraction (SAED) was performed by FEI Tecnai G2 F30 S-TWIN. The crystal structure was obtained by a Bruker D8 Advance X-ray diffractometer (XRD) with Cu Kα (λ = 1.54056 Å) radiation. X-ray photoelectron spectroscopy (XPS) was performed by Thermo Fisher's ESCALAB Xi+ instrument, where the C 1s binding energy was calibrated at 284.6 eV, and the data were analyzed using Casa XPS software version 2.3.19. The surface area was analyzed by the Brunauer–Emmett–Teller (BET) method with the N2 adsorption–desorption isotherm obtained by TriStar II 3020. Inductively coupled plasma-atomic emission spectroscopy (ICP, Agilent 5110) was used to determine the quantitative elemental analyses. Raman spectra were collected using a confocal Raman system (InVia, Renishaw) equipped with laser excitation (λ = 532 nm) to investigate the change in the catalysts's structure and investigate the possible vibration modes of oxides/hydroxides before and during the OER. X-ray absorption near structure (XANES) coupled with extended X-ray absorption fine structure (EXAFS) spectroscopy was carried out at room temperature on a (Table-XAFS-500A) to obtain the electronic structures and coordination environments of the heterostructure pre-catalysts at the atomic level at Specreation Instrument Co., Ltd, Hefei, China. The spectra were analyzed (background subtraction, normalization, and Fourier transform) using Athena software.
2.10. Electrochemical measurements
The electrochemical measurements were conducted at room temperature (30 °C) on a CHI-760E electrochemical workstation (CH Instruments, Inc.) in a standard three-electrode system. The three-electrode system comprised a catalyst loaded on a nickel foam working electrode, a platinum counter electrode, and a Hg/HgO reference electrode filled with 1 M KOH. After the surface activation, linear sweep voltammetry (LSV) was investigated at a sweep rate of 5 mV s−1 in 1 M KOH to investigate the electrochemical performance of the catalysts. Unless otherwise noted, all recorded potentials in this study were converted to a relative reversible hydrogen electrode (RHE) by comparing them to the corresponding reference electrode, according to the following equation:
| ERHE = EHg/HgO + 0.059 × pH + 0.098 |
The overpotential (η) was obtained from the following equation:
where
ERHE,
EHg/HgO, and
η are potentials for the reversible hydrogen electrode, Hg/HgO reference electrode, and the overpotential, respectively, and all curves were recorded with 90%
iR correction. The charge transfer resistance was utilized using electrochemical impedance spectroscopy (EIS) in a frequency range of 0.01–100 kHz from 1.52 to 1.67 (V
vs. RHE). The logarithmic relationship between the current density and voltage was utilized to obtain the Tafel slope of the catalysts, depending on the following equation:
| η = a + b × logj |
where
j represents the current density normalized to the geometric area of the working electrode,
b is the Tafel slope, and
a is the equation constant. The electrical double layer capacitance (
Cdl) was investigated by CV curves in the non-Faraday region in the range of 1.17–1.27 (V
vs. RHE) at scan rates ranging from 10 to 50 mV s
−1. The
Cdl values were estimated by determining the line slope obtained from plotting Δ
J/2 (Δ
J =
Ja −
Jc) at 1.22 V (
vs. RHE) against the scan rate, where
Ja is the anodic current density and
Jc is the cathodic current density. The long-term stability was determined by the chronopotentiometry (CP) test at a current density of 100 mA cm
−2 without
iR correction.
3. Results and discussion
3.1. Construction and structure characterization of the pre-catalysts
As shown in Fig. 1a, the schematic diagram illustrates the construction of the NiFealloy/NiFeN heterostructure as a pre-catalyst. The NiFealloy/NiFeN heterostructure was synthesized starting with the synthesized Ni MOF as a Ni precursor via hydrothermal reaction, followed by Fe doping to prepare the NiFeLDH precursors via a hydrothermal reaction. Finally, a nitridation process at 400 °C for 2 h in a nitrogen (N2) environment to obtain NiFealloy/NiFeN heterostructure pre-catalysts. Powder X-ray diffraction (XRD) patterns were obtained to characterize the crystalline structure and chemical composition. All XRD patterns were measured for powder catalysts, which were obtained by ultrasonic treatment on the nickel foam in ethanol, followed by collecting the powder catalysts to dry overnight under vacuum before measurement. The XRD pattern of Ni MOF (Fig. 1b) confirms the good formation of the crystalline structure of Ni MOF. Furthermore, Ni MOF reveals diffraction peaks at 2θ = 9.49°, 11.17°, 12.79°, 15.61°, 18.75°, 22.16°, 25.58°, and 28.61°, corresponding to the (111), (2−10), (2−11), (002), (300), (4−10), (2−13), and (5−10) crystal planes, respectively, which is in line with those reported by S. N. Shreyanka et al.25,26Fig. 1c shows the XRD pattern of NiFeLDH with clear peaks at 2θ = 19.25°, 33.06°, 38.54°, 52.1°, 59.05°, and 62.72°, corresponding to the (001), (100), (101), (102), (110), and (111) plane phases, respectively, which matched well with Ni(OH)2 (PDF#14-0117).27 The XRD pattern of NiFeO (Fig. 1d) showed clear peaks at 2θ = 37.24°, 43.27°, 62.87°, 75.41°, and 79.40° that correspond to the (111), (200), (220), (311), and (222) plane phases, respectively, and matched well with NiO (PDF#47-1049).28 The XRD pattern of the NiFealloy/NiFeN heterostructure pre-catalyst (Fig. 1e and S1, ESI†) showed certain peaks at 2θ = 44.5°, 51.84°, and 76.37° that correspond to the (111), (200), and (220) plane phases, respectively, and matched well with Ni (PDF#04-0850). Finally, the peaks at 2θ = 38.94°, 42.11°, 44.48°, 58.51°, 70.6°, and 78.38° corresponding to the (110), (002), (111), (112), (300), and (113) plane phases, respectively, matched well with Ni3N (PDF#10-0280).29,30 We notice that all characterization peaks of Ni and Ni3N were shifted to negative values, which confirms the successful Fe doping for both materials to form the NiFealloy/NiFeN heterostructure pre-catalyst. In order to assess the impact of the Ni foam substrate on the catalyst, the XRD pattern of the NiFealloy/NiFeN heterostructure was produced using the same experimental procedure without using Ni foam (Fig. S1, ESI†), and showed no statistically significant change. This indicates that the Ni alloy obtained in the catalyst is influenced by the experimental procedure, not by the Ni foam. Furthermore, the hysteresis loop in the BET curve indicated that the NiFealloy/NiFeN heterostructure pre-catalysts are a separate type IV isotherm (H3), as seen in their nitrogen adsorption and desorption isotherms, suggesting the presence of many mesoporous structures with a BET surface area of about 98 m2 g−1 and small pore sizes of 7.4 nm (Fig. 1f and g).31
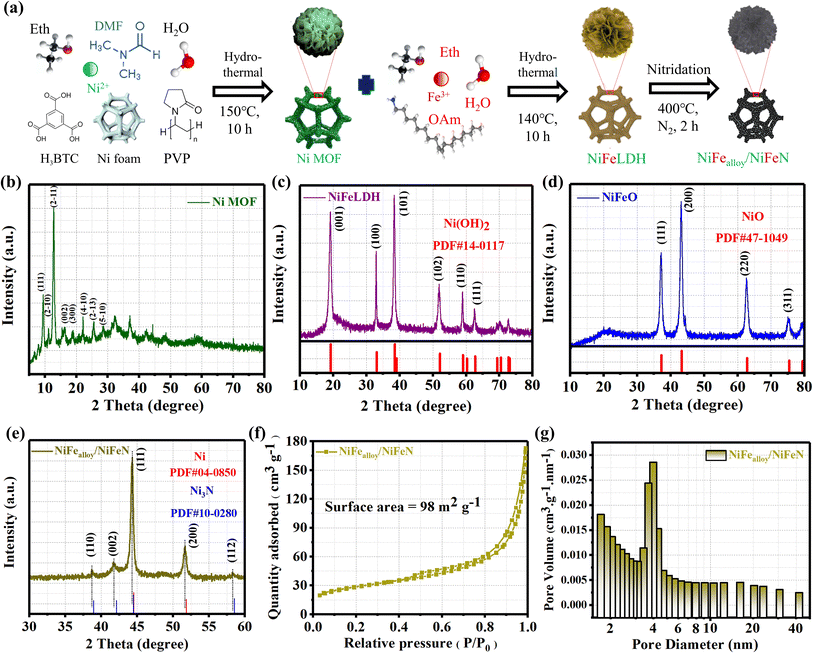 |
| | Fig. 1 (a) Schematic illustrating the construction of NiFealloy/NiFeN as a pre-catalyst; phase and bonding structure of the materials under study; XRD patterns of (b) Ni MOF; (c) NiFeLDH; (d) NiFeO; and (e) NiFealloy/NiFeN; (f) N2 adsorption–desorption isotherms of NiFealloy/NiFeN; and (g) pore size distribution curves of NiFealloy/NiFeN. | |
SEM and TEM were implemented to investigate the morphologies and microstructure of the NiFealloy/NiFeN heterostructure pre-catalyst and the other prepared catalysts. First, the SEM images (Fig. S2, ESI†) of the Ni MOF revealed a hollow nanosphere morphology.32,33 Moreover, the SEM images (Fig. S3 and S4, ESI†) of the NiFeLDH and NiFeO, respectively, revealed a two-dimensional (2D) morphology with nanosheets, indicating a layer double structure with a very high surface area.34 The SEM (Fig. S5, ESI†) and TEM (Fig. 2a and b) of the NiFealloy/NiFeN heterostructure pre-catalyst revealed a 2D morphology with nanosheets. The HRTEM imaging of the NiFealloy/NiFeN heterostructure (Fig. 2c) coupled with the FFT image (Fig. 2d) of the selected area in Fig. 2b revealed the presence of lattice fringes in certain areas with an interplanar spacing of 0.221 nm, corresponding to the (111) crystal plane of Ni (PDF#10-0280) (Fig. 2e). This is in line with the yellow dashed marked point on the FFT image, and 0.214 and 0.231 nm corresponding to the (002) and (110) crystal planes of Ni3N (PDF#10-0280), respectively (Fig. 2f). This is also in line with the red dashed marked points on the FFT image, and with its XRD patterns. The atomic lattice image (Fig. S6, ESI†) revealed the presence of numerous stacking faults and dislocations in the NiFealloy/NiFeN heterostructure, which clearly indicated that the successful Fe doping caused lattice distortion.35,36 The SAED graph (Fig. 2g) provided additional proof of the creation of the NiFealloy/NiFeN heterostructure, with discrete diffraction rings corresponding to a polycrystalline structure with Ni crystal planes (220) and (111) and Ni3N crystal planes (004) and (112). The overall distribution of the Ni, Fe, N, and C elements is established by HAADF STEM in conjunction with elemental EDX mapping (Fig. 2h), demonstrating the homogeneous distribution of the elements over the complete surface of the NiFealloy/NiFeN heterostructure. The elemental composition of the NiFe bimetallic-based catalysts was ascertained using ICP-OES (Table S1, ESI†). Based on the ICP-OES results, the metal ion ratio of the NiFe bimetallic-based catalysts can be written as Ni0.975Fe0.025N and Ni0.96Fe0.04LDH, which is fairly close to their feeding ratio. Moreover, as Fig. S7, ESI,† and the inset table Fig. 2h, illustrate, the EDX elements ratio demonstrates that the metal ion ratio of the NiFealloy/NiFeN heterostructure is quite close to the ICP-metal ions ratio. In conclusion, a one-step hydrothermal process and a vapor phase nitridation reaction were both successful in producing the NiFe bimetallic-based nitride (NiFealloy/NiFeN) heterostructure.
 |
| | Fig. 2 Morphology and structure characterization of NiFealloy/NiFeN: (a and b) TEM images; (c) HRTEM image; and (d) FFT image of the surface; (e) atomic lattice image of Ni alloy of the yellow dashed area in (c); (f) atomic lattice image of Ni–N of the red dashed area in (c); (g) SAED pattern; (h) HADAAF image and EDX spectra with the main metal element mappings. | |
3.2. Electronic structure and valence state analysis
XPS analysis was used to determine the electronic structure of the NiFealloy/NiFeN heterostructure pre-catalysts and NiFeLDH precursor in order to identify the reasons for the more efficient surface reconstruction. The XPS survey spectra (Fig. 3a) confirm the presence of Ni, Fe, O, and C in the NiFealloy/NiFeN heterostructure and NiFeLDH precursor. Moreover, an additional peak of N 1s was detected for the NiFealloy/NiFeN heterostructure, confirming the successful nitridation process, which also matched well with the EDS results. Furthermore, the peak at 110.5 eV corresponds to Ni 3s, and the peaks at 640.4 eV and 777.7 eV correspond to Ni Auger. The absence of any unexpected peaks indicates that the constructed catalysts are pure and free of impurities or contaminants. The high-resolution Ni 2p XPS profile (Fig. 3b) shows a spin–orbit doublet of Ni 2p3/2 and Ni 2p1/2, with partially oxidized Ni2+ and Ni3+ coupled with two satellite humps, which refer to the formation of Ni–NOx. The two peaks at 852.6 eV and 869.9 eV are indexed to Ni0, which confirms the formation of the Ni alloy (Ni–M, where M = Ni, Fe).37,38 The observation of two small peaks at 853.49 eV and 871.35 eV, which are very close to nickel metal, indicates the existence of Ni–N.39 The formation of a lower oxidation state of Ni0 and Ni–N leads to a slightly lower Ni2+ binding energy of the NiFealloy/NiFeN heterostructure compared to NiFeLDH, which is estimated to be 0.3 eV. The high-resolution Fe 2p XPS spectrum (Fig. 3c) shows a two-spin orbit doublet of Fe 2p3/2 and Fe 2p1/2 with partially oxidized Fe2+ and Fe3+, along with two satellite humps.40 Same as Ni, Fe exhibited slightly lower Fe3+ binding energy of the NiFealloy/NiFeN heterostructure compared to NiFeLDH, which was estimated to be 0.5 eV. Additionally, the NiFealloy/NiFeN heterostructure and NiFeLDH showed an Auger peak of Ni2+ near the Fe2+ peak at ≈706 eV.41 The high-resolution N 1s XPS spectrum (Fig. 3d) of the NiFealloy/NiFeN heterostructure displays two peaks at binding energies of 397.91 eV and 399.68 eV, which can be assigned to M–N and N–H, respectively, suggesting the chemical bonding between NiFe-bimetallic and nitrogen atoms during the nitridation process.42,43Fig. 3e represents the high-resolution XPS profile of the C 1s spectra of the NiFealloy/NiFeN heterostructure and NiFeLDH, which exhibited three main fitted peaks at 284.6, 285.8, and 288.01 eV, corresponding to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, and C–O groups, respectively. However, the peak of CN was absent from the spectra of NiFeLDH.44,45 The high-resolution XPS profile of the O 1s spectra of the NiFealloy/NiFeN heterostructure and NiFeLDH (Fig. S8, ESI†) exhibited three main fitted peaks at 529.22, 531, and 532.55 eV, corresponding to the O−2, OH−, and C–O groups, respectively.46,47
C, CN, and C–O groups, respectively. However, the peak of CN was absent from the spectra of NiFeLDH.44,45 The high-resolution XPS profile of the O 1s spectra of the NiFealloy/NiFeN heterostructure and NiFeLDH (Fig. S8, ESI†) exhibited three main fitted peaks at 529.22, 531, and 532.55 eV, corresponding to the O−2, OH−, and C–O groups, respectively.46,47
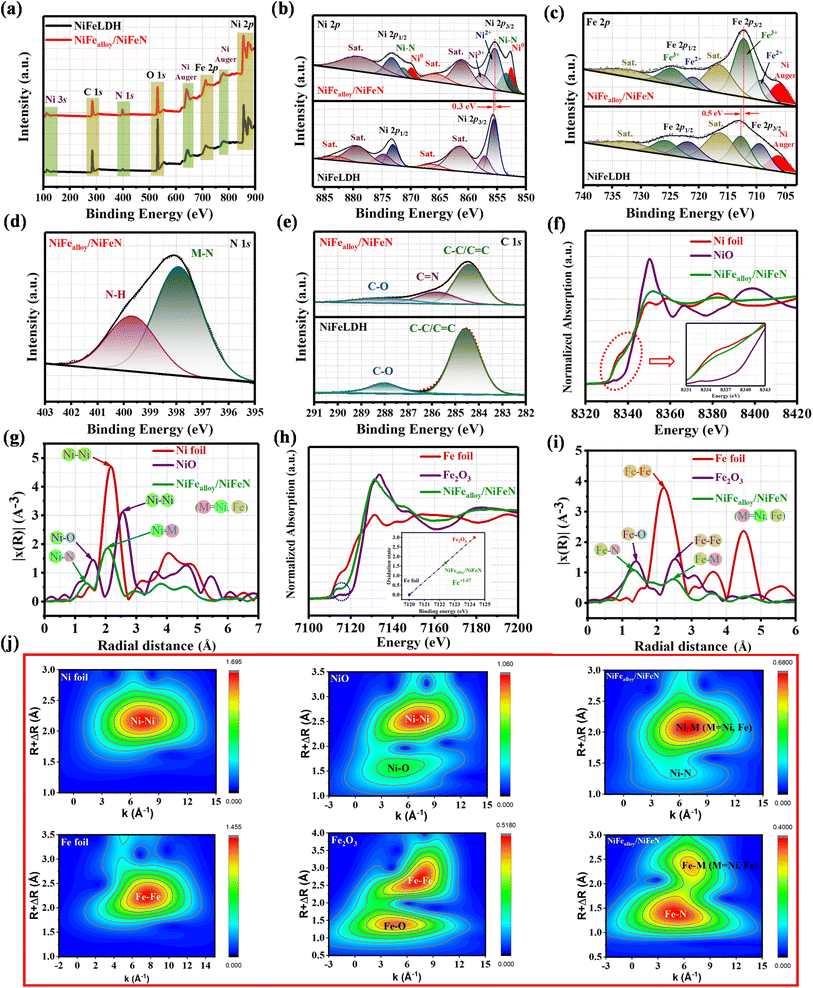 |
| | Fig. 3 XPS fine spectra of NiFealloy/NiFeN and NiFeLDH precursors: (a) XPS survey spectra; (b) high-resolution Ni 2p XPS; (c) Fe 2p XPS; (d) N 1s XPS; and (e) C 1s XPS. XANES coupled with EXAFS spectroscopy of NiFealloy/NiFeN and other compared samples: (f) normalized Ni K-edge XANES spectra of NiFealloy/NiFeN, NiO, and Ni foil; (g) corresponding Ni R-space FT-EXAFS spectra of NiFealloy/NiFeN, NiO, and Ni foil; (h) normalized Fe K-edge XANES spectra of NiFealloy/NiFeN, Fe2O3, and Fe foil; (i) corresponding Fe R-space FT-EXAFS spectra of NiFealloy/NiFeN, Fe2O3, and Fe foil. (j) Corresponding Ni and Fe K-edge WT-EXAFS spectra of NiFealloy/NiFeN and other compared samples. | |
X-ray absorption near structure (XANES) coupled with extended X-ray absorption fine structure (EXAFS) spectroscopy was employed to obtain the electronic structures and coordination environments of the NiFealloy/NiFeN heterostructure pre-catalysts at the atomic level. To avoid the effect of nickel foam, the catalyst was prepared by the same procedure without Ni foam for characterization. XANES spectra at the Ni K-edge (Fig. 3f) show a spectral profile very close to that of Ni foil, further proving that the average oxidation state of Ni is +δ (+δ less than 1). The overlapped Ni K-edge XANES spectra of the NiFealloy/NiFeN heterostructure also verify that the electronic environments of the Ni sites correspond to the average oxidation state of the Ni metal and Ni–N, which is consistent with the results of the Ni 2p XPS.48,49 The Fourier transform of EXAFS (FT-EXAFS) of the NiFealloy/NiFeN heterostructure at the Ni K-edge consists of two major peaks (Fig. 3g). A peak located at 1.3 Å is related to the typical Ni–N or Ni–O bond. It is worth noting that O and N have similar scattering capabilities in FT-EXAFS, as the Ni–O and Ni–N peaks are very close. Depending on the XPS results, which reveal clear peaks for Ni–N, we can designate it as Ni–N. Another peak related to the Ni–M scattering path at 2.1 Å in the R space, which is very close to the Ni–Ni peak of the Ni foil, suggests the existence of metallic Ni.49–51 To further understand the electronic effect on Fe after the nitridation process, the XANES spectra at the Fe K-edge were measured. As shown in Fig. 3h, the position of the absorption edge for the NiFealloy/NiFeN heterostructure is located between the Fe foil and Fe2O3. This indicates that the chemical valence state of Fe in the NiFealloy/NiFeN heterostructure is between 0 and +3, which is calculated to be +1.67 (inset Fig. 3h).52 As exhibited in Fig. 3h, the higher intensity of the pre-edge peak of the NiFealloy/NiFeN heterostructure and the pre-edge peak of the NiFealloy/NiFeN heterostructure shift negatively to lower photon energy (inset circle) compared with Fe2O3, suggesting the lower oxidation state and coordination numbers of Fe after the nitridation reaction.53 The FT-EXAFS spectrum of the NiFealloy/NiFeN heterostructure at the Fe K-edge (Fig. 3i) revealed a peak related to the Fe–Fe scattering path at 2.5 Å in R space at the same position as the Fe–Fe peak of Fe2O3. Furthermore, the peak located at 1.3 Å is related to the typical Fe–N bond, which is very close to the Fe–O peak of Fe2O3, and attributed to the first coordination shell of the Fe–N/O scattering.54,55 It is worth noting that O and N have similar scattering capabilities in FT-EXAFS. Furthermore, the peaks of Fe–O and Fe–N are very close. Therefore, the form written as Fe–N/O indicates the existence of Fe–N and Fe–O signals in Fe.49,50 Moreover, the wavelet transforms (WT) contour plots for the Ni K-edge EXAFS of the NiFealloy/NiFeN heterostructure (Fig. 3j) show one intensity maximum at ≈2.1 Å−1 corresponding to the Ni–Ni coordination and another intensity maximum at ≈1.3 Å−1 corresponding to the Ni–N scattering.56 Furthermore, the corresponding WT images for the Fe K-edge EXAFS are shown in Fig. 3j, and show one intensity maximum at ≈2.4 Å−1 that corresponds to the Fe–Fe coordination and another intensity maximum at ≈1.4 Å−1 corresponding to the Fe–N scattering.53 The WT plots are coupled with the FT-EXAFS results, further demonstrating the existence of the NiFe alloy and NiFe nitride moieties in the NiFealloy/NiFeN heterostructure, which is in line with the XPS results.
3.3. Electrochemical performance of NiFealloy/NiFeN and other comparison samples
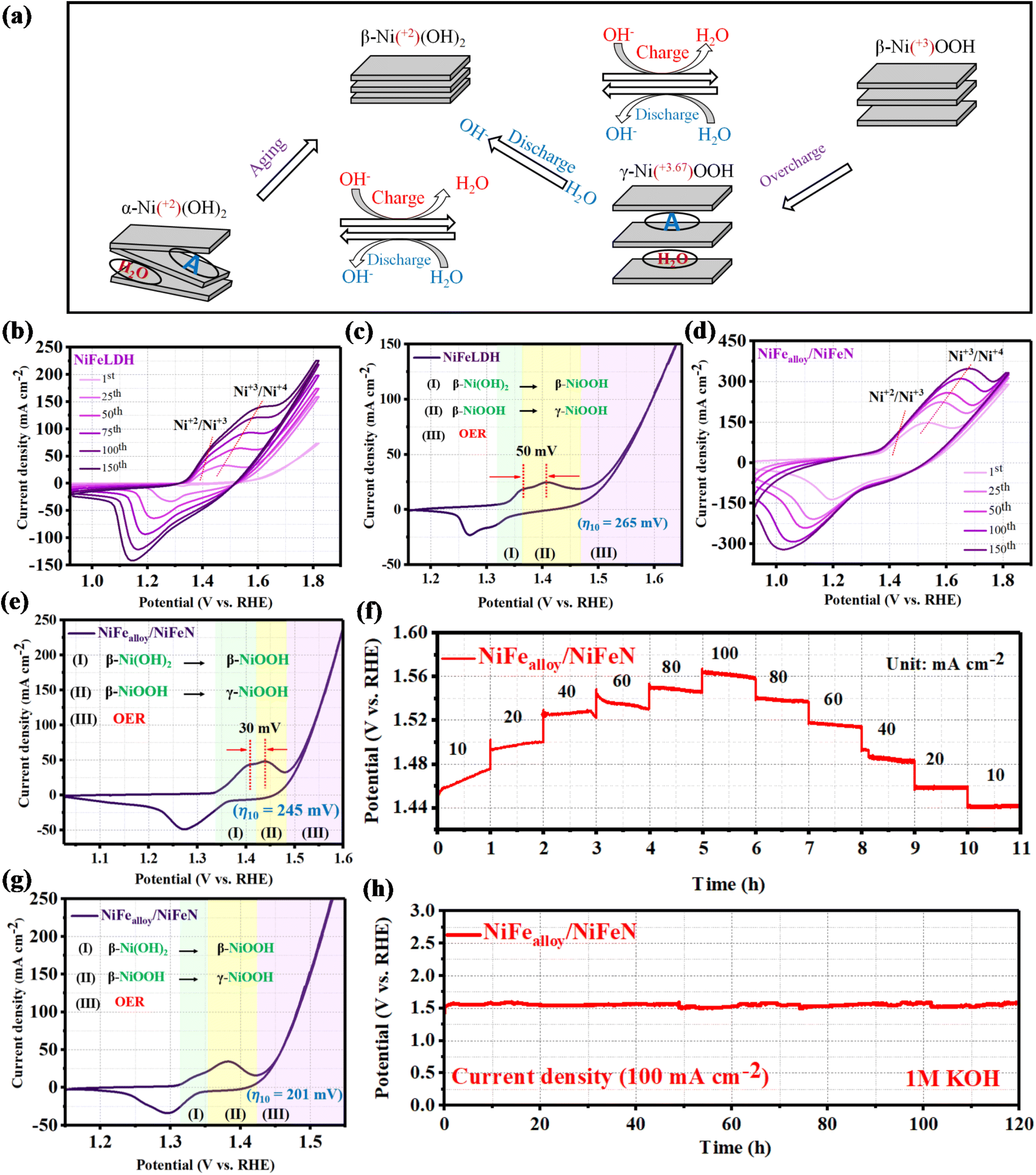
The electrocatalytic OER performances of NiFealloy/NiFeN, NiFeLDH, and other comparison samples were investigated in a 1 M KOH aqueous solution using a typical three-electrode system. As shown in Fig. 4a, after reaching the steady-state performance with 90% iR, the LSV curves reveal that NiFealloy/NiFeN exhibited the best OER electrocatalytic activity with a lower overpotential of 245 mV at 10 mA cm−2 (η10 = 245 mV). This is in line with its steady-state performance (Fig. S9a, ESI†). This overpotential is lower than that for NiFeLDH (η10 = 265 mV), Nialloy/NiN (η10 = 285 mV), and NiLDH (η10 = 341 mV). This result shows that the high OER catalytic activity is associated with the synergistic effect of Ni and Fe sites. Therefore, the heterostructure of metal alloys and metal nitrides has a huge effect on the OER electrocatalytic activity of NiFealloy/NiFeN. Thus, Fe could improve the electronic properties of Ni, which was proven by XPS. Furthermore, N could lead to the generation of more active sites, which would decrease the potential required to obtain γ-NiOOH actual active sites and promote the OER performance. Fig. 4b exhibits the overpotential plot at current densities 10, 50, and 100 mA cm−2, where NiFealloy/NiFeN shows significantly better electrocatalytic behavior than other catalysts. The Tafel slope was derived from LSV curves, which represent the OER kinetics of the catalysts and are shown in Fig. 4c. NiFealloy/NiFeN showed the lowest Tafel slope of 20.08 mV dec−1, demonstrating excellent electrochemical OER kinetics, which is smaller than Nialloy/NiN (37.4 mV dec−1), NiLDH (66.85 mV dec−1), and NiFeLDH (45.47 mV dec−1). The results confirm that NiFealloy/NiFeN could accelerate the OER kinetics for the formation of oxygen-containing intermediates more than other prepared catalysts. The intrinsic properties of the catalyst's surface were investigated using Cdl obtained from the CV curve with a non-faradaic region at different scan rates. Fig. 4d shows that NiFealloy/NiFeN has the higher Cdl value of 126 mF cm−2 than Nialloy/NiN (111 mF cm−2), NiLDH (22.55 mF cm−2), and NiFeLDH (87.5 mF cm−2). The Cdl of the catalysts was measured to reveal the ECSA of the prepared catalysts, where the ECSA is directly proportional to Cdl. The findings show that compared to other catalysts, NiFealloy/NiFeN has the highest ECSA, which suggests its largest number of active sites, indicating its advanced performance in the OER test. Electrochemical impedance spectroscopy (EIS) was employed to investigate the kinetics of the electrocatalytic reactions, and the charge transfer dynamics between the working electrode and electrolyte at a potential of 1.52 V (vs. RHE). By fitting the Nyquist plots in Fig. 4e with the equivalent circuit (Fig. S10, ESI†), it is revealed that NiFealloy/NiFeN has the smallest charge transfer resistance (Rct) value (0.5 Ω), compared to Nialloy/NiN (3.3 Ω), NiLDH (16 Ω), and NiFeLDH (1.1 Ω), which exhibit the fastest charge transfer kinetics and improved OH− transfer at the surface of the catalyst during OER due to the high ECSA and the abundance of active sites on the surface. The CV durability test yielded an intriguing finding: the area under peaks grew as the number of cycles increased. This suggests adding more active sites, which enhances the catalytic activity of NiFealloy/NiFeN as the number of cycles increases and stabilizes after 1000 cycles (Fig. 4f). The results exhibited that something happened on the catalyst's surface during the electrochemical reaction. The investigation into this will be covered in the following section. NiFealloy/NiFeN also demonstrated exceptional stability as it maintained its stable state for 5000 cycles of CV activation after reaching it.
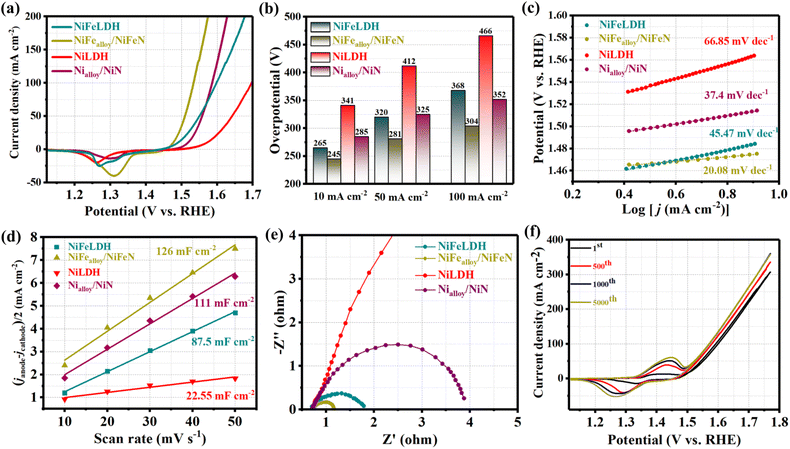 |
| | Fig. 4 Electrochemical measurement results of different samples in 1 M KOH: (a) LSV curves of OER; (b) overpotential histogram at 10, 50, and 100 mA cm−2; (c) Tafel plots; (d) capacitive current at 1.22 V vs. RHE as a function of the scan rate; (e) Nyquist plots measured at 0.6 V versus Hg/HgO; and (f) LSV curves of NiFealloy/NiFeN before and after multiple CV cycles. | |
3.4. Insights into electrochemical surface reconstruction based on CV and CP
In water-based alkaline solutions, the Bode scheme (Fig. 5a) illustrates that α-Ni(OH)2 and β-Ni(OH)2 undergo oxidation to provide β-NiOOH and γ-NiOOH, respectively. The crystallographic structure of β-NiOOH is more well defined than that of γ-NiOOH because it results from only proton insertion during the manufacturing of β-Ni(OH)2. In addition, because γ-NiOOH contains water molecules, acidic anions, and alkali cations in its crystallographic structure, it has disordered layer stacking and broad interlayer spacing. Inserting water molecules, acidic anions, and alkali cations increases the charge capacity of α-Ni(OH)2 and γ-NiOOH, allowing for quicker H+ movement inside the material. Although α-Ni(OH)2 and γ-NiOOH have similar properties, β-Ni(OH)2 is more commonly employed in practical applications due to its superior stability. Chemical aging in an aqueous alkaline solution transforms α-Ni(OH)2 into β-Ni(OH)2, while γ-NiOOH is reduced to β-Ni(OH)2.57–59 As seen in Fig. 5b, the rapid CV activation curves of the NiFeLDH precursor exhibit a noticeable uplift (expansion). Increases in the current density of OER, a continuous increase in peak area, and more pronounced redox peaks all accompany this expansion. Two oxidation peaks were observed in the CV, which corresponds to the change from Ni(OH)2 to β-NiOOH and then to γ-NiOOH. The steady-state performance curve shown in Fig. 5c of the NiFeLDH precursor was obtained using CV at a scan rate of 5 mV s−1, and the steady-state current response was recorded after several cycles. Moreover, the steady-state performance curve showed two oxidation peaks prior to OER that line up with the CV curves.23 The findings demonstrate that the actual active sites for the electrochemical water oxidation process are γ-NiOOH, and an overpotential of 265 mV is required for the system γ-NiOOH/NiFeLDH in order to achieve the OER at a current density of 10 mA cm−2. As shown in Fig. 5d, the NiFealloy/NiFeN CV curves exhibit a significant uplift (expansion) that is correlated with more pronounced redox peaks, and an almost constant current density of OER. As the number of CV cycles increases, the curve extending below zero, coupled with the increasing reduction peak area, suggests the successful activation of the catalyst surface, leading to the formation of more active sites and enhanced electrochemical activity. The NiFealloy/NiFeN steady-state performance curve (Fig. 5e) was obtained using CV at a scan rate of 5 mV s−1, and the steady-state current response was recorded after several cycles. The findings also examine two oxidation peaks, corresponding to the formation of β-NiOOH and γ-NiOOH. The LSV curves revealed that the potential required for transformation from β-NiOOH to γ-NiOOH of NiFealloy/NiFeN is lower than NiFeLDH, which is estimated at 20 mV. At a current density of 10 mA cm−2, the produced system γ-NiOOH/NiFealloy/NiFeN requires an overpotential of 245 mV to achieve the OER. The results indicate that N can modulate the electronic structure and the energy required to obtain γ-NiOOH actual active sites, which could promote the OER catalytic activity. Chronopotentiometry was used for deep activation of the surface at various current densities in order to overcome the electronegativity of nitride. We can comprehend what happens to the overpotential as the current density increases, owing to Fig. 5f. We observed that the overpotential increased with increasing current density, reaching a steady-state point at 40 mA cm−2 (Fig. S9b, ESI†). After that, increasing the current density results in a gradual drop in the NiFealloy/NiFeN overpotential. This indicates that some surface modifications occurred during the electrochemical reaction. To ascertain what transpired on the surface following CP activation, the CV curve of the steady-state performance (Fig. 5g) was measured after the degradation of the potential and reaching the steady state. As observed during the initial current density ramp-up phase, the potential gradually increased over the 1 hour holding period. Meanwhile, in the subsequent current density ramp-down phase, the potential remained nearly stable and revealed the steady state performance. The CP activation resulted in more pronounced redox peaks and a consistent increase in peak area. This lowers the peak intensity of β-NiOOH, while increasing the peak intensity of γ-NiOOH, which increases the number of actual active sites. At a current density of 10 mA cm−2, the produced system γ-NiOOH/NiFealloy/NiFeN requires an overpotential of 201 mV to achieve the OER. Moreover, throughout a variety of current densities, γ-NiOOH/NiFealloy/NiFeN showed dynamic stability and a flexible response to diverse electrochemical conditions. In order to confirm the durability, a chronopotentiometric test was used to assess the long-term electrochemical durability of the as-prepared NiFealloy/NiFeN catalyst, as illustrated in Fig. 5h. Remarkably, the data showed good long-term stability, with no discernible decline during the 120 hours at 100 mA. The long-term durability at 10 mA (Fig. S11, ESI†) was also investigated. The brown ring test was performed (Fig. S12, ESI†) for the electrolyte before and after the durability. The results reveal that the brown ring appeared after the durability, and none appeared for the electrolyte before the durability. The presence of NO3− in the electrolyte after durability could be introducing between the layers of NiOOH and promoting the formation of γ-NiOOH on the surface.
 |
| | Fig. 5 (a) Overview of the transformations between distinct phases of Ni(OH)2 and NiOOH. Note that A represents the possible anion of the acid (e.g., NO3−2; SO4−2; PO4−3) that can be incorporated between the layer of hydroxide. Electrochemical surface reconstruction of NiFeLDH and NiFealloy/NiFeN: (b) cyclic voltammetry curves of NiFeLDH in the potential range from 0.9 to 1.8 (V vs. RHE). (c) Steady-state performance curve of NiFeLDH after 150 cycles of CV activation. (d) Cyclic voltammetry curves of NiFealloy/NiFeN in the potential range from 0.9 to 1.8 (V vs. RHE). (e) Steady-state performance curve of NiFealloy/NiFeN after 150 cycles of CV activation. (f) Chronopotentiometry activation of NiFealloy/NiFeN at different current densities ranging from 10 to 100 mA cm−2. (g) Steady-state performance curve of NiFealloy/NiFeN after chronopotentiometry activation. (h) Long-term stability test of NiFealloy/NiFeN at a current density of 100 mA cm−2. | |
3.5.
In situ Raman spectroscopy of NiFealloy/NiFeN and NiFeLDH
While the prior electrochemical analysis distinctly confirmed the exceptional OER activity of NiFealloy/NiFeN due to electrochemical surface reconstruction and the formation of γ-NiOOH active sites, additional in situ Raman spectroscopy studies on the evolution of the active site and surface structure during OER are essential for elucidating its mechanism. According to reports, during OER measurements, a coating of metal hydroxides, oxides, and/or oxyhydroxides would build on the surface.60,61 Using a specially made electrochemical cell, in situ Raman spectra were acquired in the potential range from the open-circuit potential (OCP) to 1.53 V (vs. RHE) in order to elucidate the structure reconstruction during the OER process. The in situ Raman spectra (Fig. 6a and b) reveal a prominent Raman scattering signal at approximately 437 cm−1 and 447 cm−1 due to NiII–OH of α-Ni(OH)2 or β-Ni(OH)2 of NiFealloy/NiFeN and NiFeLDH, respectively. Another Raman scattering signal at approximately 487 cm−1 related to NiII–O was found at OCP using NiFealloy/NiFeN and NiFeLDH.62 After submerging the samples in the alkaline electrolyte, hydroxide and oxide are formed at the surface, which explains this. When the applied external potential for the NiFealloy/NiFeN catalyst is higher than 1.33 V, two distinct peaks can be seen at approximately 475 and 551 cm−1, which correspond to the Eg bending and A1g stretching vibration modes of NiIII–O in γ-NiOOH, respectively.62,63 The emergence of these two unique peaks suggests that the active sites for NiFealloy/NiFeN in the OER process are γ-NiOOH. The strength of the peaks for NiII–OH and NiII–O decreased concurrently with the potential rise, and almost vanished at 1.53 V vs. RHE. The findings allude to the buildup of the γ-NiOOH active sites on the outside. At applied potentials greater than 1.38 V vs. (RHE), the Raman spectra of NiFeLDH showed phase transition processes that were similar to those of NiFealloy/NiFeN. Furthermore, because of the extremely low Fe, it can be challenging to identify Raman peaks for Fe species. The higher I475/I551 ratio of NiFealloy/NiFeN than NiFeLDH at 1.53 (V vs. RHE) should be attributed to the presence of more γ-NiOOH active sites, and an approximate oxidation state of +3.67 is revealed.63,64 As a result, for the as-activated NiFealloy/NiFeN, the formation of the active species is simpler, and more active Ni+3/+4 accumulates, which facilitates the acquisition of an optimal electronic structure for OER.64 Consistent with the earlier electrochemical cyclic voltammetry results, the above results show that the substitution of nitrogen hinders the phase change and enhances the oxidation of NiII–OH and NiII–O to NiIII–O in γ-NiOOH. For this reason, the chronopotentiometric curve at different current densities for NiFealloy/NiFeN demonstrates a progressive stabilization of the catalytic performance during the initial few hours. Subsequent to the electrochemical process and surface reconstruction, a slow decrease in the NiFealloy/NiFeN overpotential resulting from surface changes ultimately led to the creation of the γ-NiOOH active sites.
 |
| | Fig. 6
In situ Raman spectra of (a) NiFealloy/NiFeN and (b) NiFeLDH under various applied potentials (vs. RHE). Morphology and structure characterization of NiFealloy/NiFeN after OER: (c) TEM image; (d) HRTEM image. (e–g) Atomic lattice image of the three selected dashed areas in (d). (h) SAED pattern and (i) HADAAF image and EDX spectra with the main metal element mappings. | |
3.6. Insights into the electrochemical surface reconstruction based on HRTEM, XRD, XPS, and Bode plot
TEM (Fig. 6c) and SEM (Fig. S13, ESI†) were used to study the surface morphology of the post-OER NiFealloy/NiFeN catalyst. The results showed that the surface morphology of the nanosheets remained unchanged after 5000 CV cycles. Additionally, HRTEM imaging was used to examine the structure of the post-OER NiFealloy/NiFeN catalyst (Fig. 6d). This analysis showed a heterostructure made up of amorphous regions (Fig. 6e and f) and crystalline region (Fig. 6g). The (011) crystal plane of NiOOH (JCPDS No. 03-0953) corresponds to the lattice fringes in the crystalline area, which have an interplanar spacing of 0.239 nm. HRTEM verified the creation of an amorphous and crystalline NiOOH layer on the surface, as well as the excellent preservation of the interior structure. Polycrystalline materials with a low crystalline structure, as depicted in the SAED image (Fig. 6h), have unique diffraction rings that correlate to the (200), (111), and (220) crystal planes of Ni metal, matching well with (PDF#04-0850). Furthermore, the (400) and (113) crystal planes of Ni3N matched well with (PDF#10-0280), and the (002) crystal plane of NiOOH matched well with (PDF#27-0956). The post-OER HAADF STEM and the homogenous distribution of Ni, Fe, N, O, and C elements along the complete surface of NiFealloy/NiFeN were demonstrated by the elemental EDX mapping (Fig. 6i and S13a, ESI†). The results reveal that the Fe atomic percentage is considerably lower than that of Ni, as Fe is used for element doping. Additionally, XRD was used to examine the chemical makeup of the post-OER of the NiFealloy/NiFeN catalyst (Fig. S14b, ESI†). The findings revealed no discernible changes in any of the peak positions, with the exception of a little drop in peak strength and the absence of any new peaks that would have indicated the creation of a very thin amorphous surface layer on the NiFealloy/NiFeN surface.
Based on the examination of the electrochemical segment, it can be inferred that the surface reconstruction evolved as the electrocatalytic reaction progressed. Furthermore, the change in the electronic structure and the electrochemical surface reconstruction were investigated based on the XPS spectrum comparison of initial and post-OER after 5000 CVs of NiFealloy/NiFeN heterostructure catalysts. The XPS survey spectra (Fig. 7a) confirm the distribution of Ni, Fe, O, N, and C in the NiFealloy/NiFeN heterostructure after OER with an increase in the intensity of the O peak, which refers to some oxidation process that occurred on the surface during the OER process. The high-resolution Ni 2p XPS profile (Fig. 7b) reveals a slightly positive shift of Ni2+ binding energy, estimated to be 0.1 eV, and the peaks of Ni0 (Ni–Ni and Ni–Fe) disappeared after OER. However, the peak of Ni–N still appeared, which may be due to the large electronegativity of nitride.37,65 The observed increase in the Ni2+ intensity after the OER is primarily due to the disappearance of the Ni0 peaks, which suggests a complete oxidation of the Ni metal on the surface, resulting in the formation of the Ni2+ species. Additionally, the slight decrease in the peak intensity of Ni–N reveals that the partial oxidation of Ni–N is likely responsible for the formation of Ni–NOx, which further explains the increasing of the Ni2+ peak intensity after OER. The high-resolution Fe 2p XPS profile (Fig. 7c) reveals no obvious shift of the peak position. This could be caused by the large electronegativity of nitrogen. This leads to the binding energy of the Fe 2p peaks in nitrates being located in the higher binding energy position, which was very close to the Fe-based (oxy)hydroxide.66,67 The high-resolution N 1s XPS profile (Fig. 7d) reveals a decrease in the M–N peak intensity with the appearance of a new peak corresponding to N–O; the results refer to the partially oxidized NiFe-based nitride. The high-resolution C 1s XPS profile (Fig. 5e) is additional evidence of the electrochemical surface reconstruction. The appearance of two new peaks could be attributed to the surface chemical changes that occurred on the catalyst surface during the reaction. The peak at around 288.9 eV with π–π* shakeup satellite at 292.5 eV typically corresponds to O–CO bonds, reflecting the presence of the carbon–oxygen species such as hydroxyls on the catalyst surface during the reaction.68 Shedding light on the charge transfer pathways between the NiFealloy/NiFeN and surface species, the disappearance of Ni0 and decreasing intensity of the M–N peaks during the activation process refer to the fact that NiFe-based catalysts are usually electrochemically oxidized into N-containing species (nitrates NO3−, NO2−, or NO−). This is also the main mode of N–O binding, which could dissolve in alkali to facilitate the electrochemical surface reconstruction, generating MOOH active sites and accelerating the electron transfer during the OER.69 The charge transfers between NiFealloy/NiFeN and MOOH are very important for enhancing the catalytic activity, electron transfer, and long-term stability. Moreover, Fig. 7f and g exhibits the Bode plots of NiFealloy/NiFeN and its precursor, and was coupled with other compared samples (Fig. S15, ESI†). Due to the heterogeneous charge distribution of the surface oxide species, which correlates to the OER process, the catalysts produced notable response signals in the low-frequency range.70 In particular, the NiFealloy/NiFeN response signal was skewed more toward positive than NiFeLDH at all voltages. This is due to the large number of oxidations from Ni2+ to Ni3+, which lead to an increase in the number of active sites, matching well with the higher Cdl and ECSA.71,72Fig. 7h shows that at all voltages, NiFealloy/NiFeN has the shortest phase angle, indicating that more electrons are participating in the OER reaction.70Fig. 7i illustrates a near-volcano-type relationship between the Fe dopant amount and overpotential. The results reveal that bimetallic sites have lower overpotential and better catalytic activity than monometallic sites. By including 50 mg of Fe during the experiment, the optimal Ni:Fe ratio with the optimum electrical structure was achieved. The valence states and electronic structure analysis discussed above not only demonstrated the significance of the right ratio of the NiFe bimetallic centers for controlling the electronic structure, but also provided more insight into the increase in the electrochemical active site.
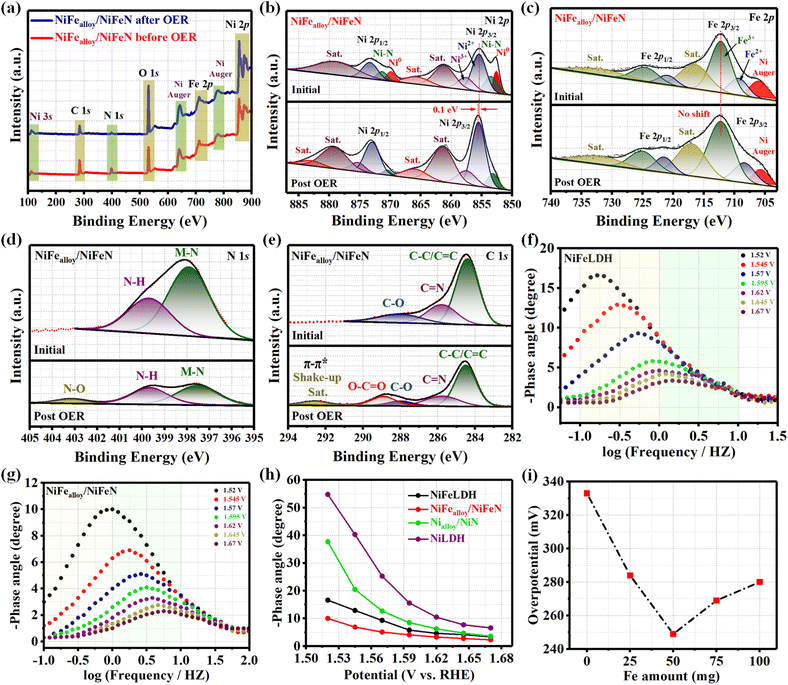 |
| | Fig. 7 XPS fine spectra of NiFealloy/NiFeN before and after OER: (a) XPS survey spectra; (b) high-resolution Ni 2p XPS; (c) Fe 2p XPS; (d) N 1s XPS; and (e) C 1s XPS. Bode plots: (f) NiFeLDH and (g) NiFealloy/NiFeN at the potential range from 1.52 to 1.67 V (vs. RHE). (h) Potential dependence of the phase angle of NiFealloy/NiFeN and other compared samples; and (i) overpotential at a current density of 10 mA cm−2 for NiFealloy/NiFeN with different Fe atomic ratios. | |
For comparison, other reactions were used to fabricate nitrides. NiN was prepared based on Ni MOF as a precursor, and the XRD pattern of NiN (Fig. S16, ESI†) confirms the good preparation of nickel nitride. NiFeN was obtained based on NiFeO as a precursor, and the XRD pattern (Fig. S17, ESI†) reveals the formation of the heterostructure of nickel nitride and nickel alloy. The SEM image of NiN (Fig. S18, ESI†) reveals nanosphere surface morphology, which is in line with its precursor. Moreover, the SEM image of NiFeN (Fig. S19, ESI†) reveals the wormlike surface morphology. The electrocatalytic OER performances of NiN, NiFeN, and other samples were also investigated in a 1 M KOH aqueous solution. As shown in Fig. S20a,† the LSV curves reveal that NiFeN exhibited the best OER electrocatalytic activity with a lower overpotential of 284 mV at 10 mA cm−2 (η10 = 284 mV). This is lower than NiN (η10 = 333 mV), NiFeO (η10 = 320 mV), and NiO (η10 = 357 mV). Fig. S20b† exhibits the overpotential plot at current densities of 10, 50, and 100 mA cm−2, where NiFeN shows significantly better electrocatalytic behavior than the other catalysts. The Tafel slope (Fig. S20c†) reveals that NiFeN showed the lowest Tafel slope of 33.42 mV dec−1, which is smaller than NiN (61.08 mV dec−1), NiFeO (35.32 mV dec−1), and NiO (64.83 mV dec−1). The intrinsic properties of the catalyst's surface were investigated using Cdl obtained from the CV curve with a non-faradaic region at different scan rates. Fig. S20d† shows that NiFeN has the higher Cdl value of 102.8 mF cm−2 than NiN (56.2 mF cm−2), NiFeO (26 mF cm−2), and NiO (17.5 mF cm−2). By fitting the Nyquist plots in Fig. S20e,† it is revealed that NiFeN has the smallest charge transfer resistance (Rct) value (2.2 Ω), compared to NiN (9 Ω), NiFeO (6 Ω), and NiO (17 Ω). The findings show that compared to other nitride catalysts, NiFealloy/NiFeN has the best catalytic activity with a low overpotential toward OER. The highest ECSA suggests its largest number of active sites, indicating its advanced performance in OER tests. All of the above conclusions point to the extraordinary electrochemical activity of γ-NiOOH/NiFealloy/NiFeN, which is better than most of the NiFe-based electrocatalysts that have been reported previously (Fig. S20f and Table S2, ESI†).
3.7. Density function theory calculations (DFT)
DFT simulations were performed in order to gain a profound understanding of the electrochemical surface reconstruction process during OER, the OER catalytic mechanism of γ-NiFeOOH, and the effect of Fe doping on the OER catalytic process (detailed calculations are shown in ESI†). Fig. 8a reveals the d-orbital density of states (d-DOS) of the Ni 3d orbital in γ-NiFeOOH and γ-NiOOH to predict the adsorption strength between the catalyst surface and the OER intermediates. Each of them shows unique Ni metallic properties. As is well known, the bonding and antibonding orbitals are created when the metal's 3d orbital overlaps with the O's 2p orbital. A higher energy of the d-band center indicates that the stronger the metal's adsorption (interaction) to the oxygen-containing intermediate, the fewer electrons are filled in the antibonding orbital. A strong adsorption between the metal sites and oxygenated species results from the Ni site in γ-NiFeOOH having the greatest d-band center (−1.57 eV), which reveals the strongest adsorption capacity of the oxygen intermediates. Notably, a stronger rate-determining step (RDS) energy barrier results from such intense adsorption, which facilitates the adsorption and evolution of intermediates containing oxygen. Moreover, the calculated d-band center of γ-NiOOH is −1.78 eV. The results reveal that the incorporation amount of Fe can increase the d-band center of Ni and be close to the Fermi level, which modulates the electronic properties of the Ni active sites. The Gibbs free energy fluctuation for several catalyst intermediates in the basic OER phases is shown in Fig. 8b. It is evident that the RDS of OER is *O formation (*OH to *O). Notably, the γ-NiFeOOH reaction energy barrier is 1.901 eV, which is lower than that of γ-NiOOH (2.047 eV). The results reveal that an incorruption amount of Fe can modulate the electronic properties, which is thermodynamically favorable. The results agree with the DOS results. The aforementioned results demonstrate the validity of the theoretical calculations by showing good agreement with the overpotential trends discovered experimentally. To further evaluate the interaction between the catalyst surface and the intermediates, the typical 4 e− mechanism of OER for the γ-NiFeOOH electrocatalysts is illustrated in Fig. 8c. In the OER process, four intermediate adsorbed species, *, *OH, *O, and *OOH, are present in the adsorbate evolution mechanism (AEM). Moreover, the structure of the four intermediates of γ-NiOOH is also revealed in Fig. S21, ESI.† The results revealed that the NiFealloy/NiFeN pre-catalyst underwent electrochemical surface reconstruction after the OER activation process to form β-NiOOH, which underwent a higher oxidation process to form γ-NiOOH on the surface as actual active sites. Afterward, the electrocatalytic system underwent a four-electron coupled four-proton reaction process to generate O2 molecules.
 |
| | Fig. 8 (a) DOS of γ-NiFeOOH and γ-NiOOH with respect to the Ni 3d orbital. (b) Calculated Gibbs free energy diagram of γ-NiFeOOH and γ-NiOOH over the four steps of the OER process. (c) Adsorbate evolution mechanism (AEM) of the γ-NiFeOOH OER catalytic process. | |
4. Conclusion
In summary, three easy processes were used to produce the NiFealloy/NiFeN heterostructure. First, Ni MOF was created as a Ni precursor via a one-step hydrothermal procedure. Second, NiFeLDH was created by combining Ni MOF with a variable quantity of Fe in a one-step hydrothermal process. Ultimately, nitridation via chemical vapor deposition was used to create the NiFealloy/NiFeN heterostructure. Concurrently, the synthesized NiFealloy/NiFeN heterostructure nanosheets of NiFealloy/NiFeN exhibited remarkable surface area and good crystallinity, together with exceptional electrocatalytic activity towards OER and remarkable electrochemical durability under alkaline conditions. Based on CV and operando spectroscopy, the enhanced electrocatalytic activity of the NiFealloy/NiFeN heterostructure nanosheets can be ascribed to the bimetallic center (Ni and Fe doping) and N active sites, which expedite the electrochemical surface reconstruction and modify the electronic structure during the electrochemical process by forming γ-NiOOH active sites. According to DFT calculations, OER is facilitated, and the energy barrier of the rate-determining step is lowered when electrons are redistributed as a result of surface reconfiguration during electrochemical processes. Moreover, the Fe doping could modulate the electronic structure and decrease the Gibbs free energy. Future electrochemical surface reconstruction electrocatalyst design and preparation will benefit from the current work as well.
Data availability
The supplemental materials that are attached to this article contain the ESI data† of this study.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The Large-scale Instrument and Equipment Sharing Foundation of Wuhan University and the Natural Science Foundation of Hubei Province (2021CFB144) provided financial support for this work.
References
- J. Gautam, S. Y. Lee and S. J. Park, Adv. Compos. Hybrid Mater., 2024, 7, 1–15 CrossRef
 .
.
- I. Slobodkin, E. Davydova, M. Sananis, A. Breytus and A. Rothschild, Nat. Mater., 2024, 23, 398–405 CrossRef CAS PubMed .
- W. Li, C. Wang and X. Lu, J. Mater. Chem. A, 2021, 9, 3786–3827 RSC .
- C. Li, B. Kim, Z. Li, R. Thapa, Y. Zhang, J. M. Seo, R. Guan, F. Tang, J. H. Baek, Y. H. Kim, J. P. Jeon, N. Park and J. B. Baek, Adv. Mater., 2024, 36, 2403151 CrossRef CAS .
- Q. Huang, G. J. Xia, B. Huang, D. Xie, J. Wang, D. Wen, D. Lin, C. Xu, L. Gao, Z. Wu, J. Wu, F. Xie, W. Guo and R. Zou, Energy Environ. Sci., 2024, 17, 5260–5272 RSC .
- G. Helal, Z. Xu, W. Zuo, Y. Yu, J. Liu, H. Su, J. Xu, H. Li, G. Cheng and P. Zhao, RSC Adv., 2024, 14, 10182–10190 RSC .
- J. Liu, Y. Liu, X. Mu, H. Jang, Z. Lei, S. Jiao, P. Yan, M. G. Kim and R. Cao, Adv. Funct. Mater., 2022, 32, 2204086 CrossRef CAS .
- X. Liu, S. Jing, C. Ban, K. Wang, Y. Feng, C. Wang, J. Ding, B. Zhang, K. Zhou, L. Gan and X. Zhou, Nano Energy, 2022, 98, 107328 CrossRef CAS .
- Z. Xu, Y. Ying, G. Zhang, K. Li, Y. Liu, N. Fu, X. Guo, F. Yu and H. Huang, J. Mater. Chem. A, 2020, 8, 26130–26138 RSC .
- Z. Zheng, D. Wu, G. Chen, N. Zhang, H. Wan, X. Liu and R. Ma, Carbon Energy, 2022, 4, 901–913 CrossRef CAS .
- H. Lei, L. Ma, Q. Wan, S. Tan, B. Yang, Z. Wang, W. Mai and H. J. Fan, Adv. Energy Mater., 2022, 12, 2202522 CrossRef CAS .
- J. Nie, J. Shi, T. Huang, M. Y. Xie, Z. Y. Ouyang, M. H. Xian, G. F. Huang, H. Wan, W. Hu and W. Q. Huang, Adv. Funct. Mater., 2024, 34, 2314172 CrossRef CAS .
- Z. Xu, W. Zuo, Y. Yu, J. Liu, G. Cheng and P. Zhao, Adv. Sci., 2024, 11, 2306758 CrossRef CAS PubMed .
- N. Yao, G. Wang, H. Jia, J. Yin, H. Cong, S. Chen and W. Luo, Angew. Chem., Int. Ed., 2022, 61, e202117178 CrossRef CAS PubMed .
- J. Ferreira de Araújo, F. Dionigi, T. Merzdorf, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14981–14988 CrossRef PubMed .
- S. Tao, G. Zhang, B. Qian, J. Yang, S. Chu, C. Sun, D. Wu, W. Chu and L. Song, Appl. Catal. B., 2023, 330, 122600 CrossRef CAS .
- S. Javaid, X. Xu, W. Chen, J. Chen, H. Y. Hsu, S. Wang, X. Yang, Y. Li, Z. Shao, F. Jones and G. Jia, Nano Energy, 2021, 89, 106463 CrossRef CAS .
- P. Xiong, X. Zhang, H. Wan, S. Wang, Y. Zhao, J. Zhang, D. Zhou, W. Gao, R. Ma, T. Sasaki and G. Wang, Nano Lett., 2019, 19, 4518–4526 CrossRef CAS PubMed .
- Z. Yin, R. He, Y. Zhang, L. Feng, X. Wu, T. Wågberg and G. Hu, J. Energy Chem., 2022, 69, 585–592 CrossRef CAS .
- J. Kwon, H. Han, S. Jo, S. Choi, K. Y. Chung, G. Ali, K. Park, U. Paik and T. Song, Adv. Energy Mater., 2021, 11, 2100624 CAS .
- Y. Yao, G. Zhao, X. Guo, P. Xiong, Z. Xu, L. Zhang, C. Chen, C. Xu, T. S. Wu, Y. L. Soo, Z. Cui, M. M. J. Li and Y. Zhu, J. Am. Chem. Soc., 2024, 146, 15219–15229 CrossRef CAS PubMed .
- H. Wang, X. Liu, G. Liu, Y. Wang, X. Du and J. Li, Chem Catal., 2023, 3, 100552 CAS .
- R. Zhao, S. Xu, D. Liu, L. Wei, S. Yang, X. Yan, Y. Chen, Z. Zhou, J. Su, L. Guo and C. Burda, Appl. Catal. B., 2023, 338, 123027 CAS .
- Z. Qiu, Y. Ma and T. Edvinsson, Nano Energy, 2019, 66, 104118 CAS .
- S. N. Shreyanka, J. Theerthagiri, S. J. Lee, Y. Yu and M. Y. Choi, Chem. Eng. J., 2022, 446, 137045 CrossRef .
- F. Israr, D. Chun, Y. Kim and D. K. Kim, Ultrason. Sonochem., 2016, 31, 93–101 CrossRef CAS PubMed .
- L. Chen, X. Yang, Y. Gao, Y. Tian, Y. Wang, X. Zhao, X. Lei and F. Zhang, Int. J. Hydrogen Energy, 2023, 48, 26148–26161 CrossRef CAS .
- H. Yan, D. Zhang, J. Xu, Y. Lu, Y. Liu, K. Qiu, Y. Zhang and Y. Luo, Nanoscale Res. Lett., 2014, 9, 1–7 CrossRef CAS PubMed .
- T. Wu, E. Song, S. Zhang, M. Luo, C. Zhao, W. Zhao, J. Liu and F. Huang, Adv. Mater., 2022, 34, 2108505 CrossRef CAS PubMed .
- P. Zhou, G. Zhai, X. Lv, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Appl. Catal. B., 2021, 283, 119590 CrossRef CAS .
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC .
- Y. Qiu, Y. Lin, H. Yang, L. Wang, M. Wang and B. Wen, Chem. Eng. J., 2020, 383, 123207 CrossRef CAS .
- X. Zhang, J. Qiao, Y. Jiang, F. Wang, X. Tian, Z. Wang, L. Wu, W. Liu and J. Liu, Nano-Micro Lett., 2021, 13, 1–31 CrossRef CAS PubMed .
- H. Gu, G. Shi, H. C. Chen, S. Xie, Y. Li, H. Tong, C. Yang, C. Zhu, J. T. Mefford, H. Xia, W. C. Chueh, H. M. Chen and L. Zhang, ACS Energy Lett., 2020, 5, 3185–3194 CrossRef CAS .
- K. Huang, D. Peng, Z. Yao, J. Xia, B. Zhang, H. Liu, Z. Chen, F. Wu, J. Wu and Y. Huang, Chem. Eng. J., 2021, 425, 131533 CAS .
- Y. Wang, T. Wang, H. Arandiyan, G. Song, H. Sun, Y. Sabri, C. Zhao, Z. Shao and S. Kawi, Adv. Mater., 2024, 36, 2313378 CrossRef CAS PubMed .
- J. Tang, J. L. Xu, Z. G. Ye, X. B. Li and J. M. Luo, J. Mater. Sci. Technol., 2021, 79, 171–177 CrossRef CAS .
- J. Li, K. Zheng, C. Zhang, L. Jiao, Z. Dong, X. Tao, R. Su, H. Xie and C. Xu, Chem. Eng. J., 2023, 462, 142267 CAS .
- X. Lin, L. Chen, X. Zhong, A. BaQais, W. Dang, M. A. Amin, H. Huang, H. Li, G. Liang, G. Liu and Z. Yang, Adv. Compos. Hybrid Mater., 2023, 6, 79 CAS .
- H. Liao, T. Luo, P. Tan, K. Chen, L. Lu, Y. Liu, M. Liu and J. Pan, Adv. Funct. Mater., 2021, 31, 2102772 CrossRef CAS .
- W. Zuo, Z. Xu, M. Hu, Y. Yu, J. Liu, G. Cheng and P. Zhao, Inorg. Chem. Front., 2023, 10, 2697–2707 RSC .
- Y. Men, S. Jia, P. Li, Y. Tan, J. Wang, P. Zhao, G. Cheng, S. Chen and W. Luo, Chem. Eng. J., 2022, 433, 133831 CrossRef CAS .
- S. Zhang, L. Wang, T. Xie, Q. Chen, W. Peng, Y. Li, F. Zhanga and X. Fan, J. Mater. Chem. A, 2022, 10, 21523–21530 RSC .
- C. Wang, H. Yang, Y. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2019, 58, 6099–6103 CrossRef CAS PubMed .
- H. Xu, K. Ye, J. Yin, K. Zhu, J. Yan, G. Wang and D. Cao, J. Power Sources, 2021, 491, 229592 CrossRef CAS .
- K. Shen, Y. Tang, Q. Zhou, Y. Zhang, W. Ge, X. Shai, S. Deng, P. Yang, S. Deng and J. Wang, Chem. Eng. J., 2023, 471, 144827 CrossRef CAS .
- J. Cai, H. Zhang, L. Zhang, Y. Xiong, T. Ouyang and Z. Q. Liu, Adv. Mater., 2023, 35, 2303488 CrossRef CAS PubMed .
- S. Guo, Y. Wu, C. Wang, Y. Gao, M. Li, B. Zhang and C. Liu, Nat. Commun., 2022, 13, 5297 CrossRef CAS PubMed .
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, 1901666 CrossRef PubMed .
- H. Li, G. Yan, H. Zhao, P. C. Howlett, X. Wang and J. Fang, Adv. Mater., 2024, 36, 2311272 CrossRef CAS PubMed .
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed .
- S. Yin, L. Chen, J. Yang, X. Cheng, H. Zeng, Y. Hong, H. Huang, X. Kuai, Y. Lin, R. Huang, Y. Jiang and S. Sun, Nat. Commun., 2024, 15, 7489 CrossRef CAS PubMed .
- H. Liao, G. Ni, P. Tan, Y. Liu, K. Chen, G. Wang, M. Liu and J. Pan, Appl. Catal. B., 2022, 317, 121713 CrossRef CAS .
- X. Wan, Q. Liu, J. Liu, S. Liu, X. Liu, L. Zheng, J. Shang, R. Yu and J. Shui, Nat. Commun., 2022, 13, 2963 CrossRef CAS PubMed .
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., 2017, 129, 625–629 CrossRef .
- P. Zhang, H. Chen, L. Chen, Y. Xiong, Z. Sun, H. Yang, Y. Fu, Y. Zhang, T. Liao and F. Li, Chin. J. Catal., 2023, 45, 152–161 CrossRef CAS .
- S. Fujita, S. Baranton, C. Coutanceau and G. Jerkiewicz, Energy Technol., 2024, 12, 2301268 CrossRef CAS .
- A. Van der Ven, D. Morgan, Y. S. Meng and G. Ceder, J. Electrochem. Soc., 2005, 153, A210 CrossRef .
- P. Oliva, J. Leonardi, J. F. Laurent, C. Delmas, J. J. Braconnier, M. Figlarz, F. Fievet and A. De Guibert, J. Power Sources, 1982, 8, 229–255 CrossRef CAS .
- P. Yan, Q. Liu, H. Zhang, L. C. Qiu, H. B. Wu and X. Y. Yu, J. Mater. Chem. A, 2021, 9, 15586–15594 RSC .
- X. Bo, R. K. Hocking, S. Zhou, Y. B. Li, X. J. Chen, J. C. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13, 4225–4237 RSC .
- L. Wei, K. Zhang, R. Zhao, L. Zhang, Y. Zhang, S. Yang and J. Su, Nano Res., 2024, 17, 4720–4728 CrossRef CAS .
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. Von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed .
- P. Zhai, C. Wang, Y. Zhao, Y. Zhang, J. Gao, L. Sun and J. Hou, Nat. Commun., 2023, 14, 1873 CrossRef CAS PubMed .
- Y. Kang, Y. Guo, J. Zhao, B. Jiang, J. Guo, Y. Tang, H. Li, V. Malgras, M. A. Amin, H. Nara, Y. Sugahara, Y. Yamauchi and T. Asahi, Small, 2022, 18, 2203411 CrossRef CAS PubMed .
- W. Tang, G. Zhang and Y. Qiu, Int. J. Hydrogen Energy, 2020, 45, 28566–28575 CrossRef CAS .
- P. Zhou, L. Wu, Z. Ji, C. Fan, X. Shen, G. Zhu and L. Xu, J. Colloid Interface Sci., 2023, 646, 98–106 CrossRef CAS PubMed .
- G. Helal, Z. Xu, W. Zuo, Y. Yu, J. Liu, H. Su, J. Xu, H. Li, G. Cheng and P. Zhao, RSC Adv., 2024, 14, 17202–17212 RSC .
- Y. Zeng, M. Zhao, Z. Huang, W. Zhu, J. Zheng, Q. Jiang, Z. Wang and H. Liang, Adv. Energy Mater., 2022, 12, 2201713 CrossRef CAS .
- Y. Qi, Y. Zhang, L. Yang, Y. Zhao, Y. Zhu, H. Jiang and C. Li, Nat. Commun., 2022, 13, 4602 CrossRef CAS PubMed .
- Y. Wang, B. Liu, X. Shen, H. Arandiyan, T. Zhao, Y. Li, M. Garbrecht, Z. Su, L. Han, A. Tricoli and C. Zhao, Adv. Energy Mater., 2021, 11, 2003759 CrossRef CAS .
- D. Zhou, S. Wang, Y. Jia, X. Xiong, H. Yang, S. Liu, J. Tang, J. Zhang, D. Liu, L. Zheng, Y. Kuang, X. Sun and B. Liu, Angew Chem. Int. Ed. Engl., 2019, 58, 736–740 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Pingping
Zhao
*b and
Pingping
Zhao