
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLinking structural and rheological memory in disordered soft materials†
Krutarth M.
Kamani
*a,
Yul Hui
Shim
b,
James
Griebler
a,
Suresh
Narayanan
c,
Qingteng
Zhang
 c,
Robert L.
Leheny
d,
James L.
Harden
e,
Alexander
Deptula
f,
Rosa M.
Espinosa-Marzal
g and
Simon A.
Rogers
a
c,
Robert L.
Leheny
d,
James L.
Harden
e,
Alexander
Deptula
f,
Rosa M.
Espinosa-Marzal
g and
Simon A.
Rogers
a
aDepartment of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Champaign, IL 61801, USA. E-mail: sarogers@illinois.edu
bSchool of Chemical and Materials Engineering, The University of Suwon, Hwaseong-si, Gyeonggi-do, 18323, Republic of Korea
cAdvanced Photon Source, Argonne National Laboratory, Argonne, IL 60439, USA
dDepartment of Physics and Astronomy, Johns Hopkins University, Baltimore, MD 21218, USA
eDepartment of Physics, University of Ottawa, Ottawa, ON K1N 6N5, Canada
fDepartment of Materials Science and Engineering, University of Illinois at Urbana-Champaign, Illinois, USA 61801
gDepartment of Civil and Environmental Engineering, University of Illinois at Urbana-Champaign, Illinois, USA 61801
First published on 6th January 2025
Abstract
Linking the macroscopic flow properties and nanoscopic structure is a fundamental challenge to understanding, predicting, and designing disordered soft materials. Under small stresses, these materials are soft solids, while larger loads can lead to yielding and the acquisition of plastic strain, which adds complexity to the task. In this work, we connect the transient structure and rheological memory of a colloidal gel under cyclic shearing across a range of amplitudes via a generalized memory function using rheo-X-ray photon correlation spectroscopy (rheo-XPCS). Our rheo-XPCS data show that the nanometer scale aggregate-level structure recorrelates whenever the change in recoverable strain over some interval is zero. The macroscopic recoverable strain is therefore a measure of the nano-scale structural memory. We further show that yielding in disordered colloidal materials is strongly heterogeneous and that memories of prior deformation can exist even after the material has been subjected to flow.
1 Introduction
When many soft materials are placed under mechanical loading above some threshold, they change from being viscoelastic solids, where deformations are recoverable, to deforming plastically, where a part of the deformation cannot be recovered. This yielding behavior is of fundamental significance for a wide range of applications including biomaterials important for healthcare,1–9 industrial materials such as battery slurries,10,11 foods and cosmetics,12,13 and environmental flows.14,15 A long-standing challenge in designing these materials is to link microstructural characteristics to the macroscopic flow properties above and below the yielding condition.Systems that have been taken out of equilibrium often have memories of the process that got them to the state. Mechanical memory formation has been studied in materials with various microstructures and inter-particle forces with several themes such as ability of disordered materials to remember a direction16–27 and cyclic driving in jammed and glassy systems.18,28–34
Structural memory has been studied using various techniques including interfacial stress rheometry and light scattering.33,35–44 Recent studies of non-Brownian suspensions45,46 and jammed systems18,28–31,47 have demonstrated that memories are stored during oscillatory shearing at some long-applied ‘training’ amplitude. When viewed stroboscopically, it is possible to read out the training amplitude at some later time. The addition of noise provides such systems with the ability to store multiple memories from different training amplitudes.48 In these studies, the memories and the mechanism of memory retention are explained in terms of reversible and irreversible, but still periodic, particle trajectories.
Studies that use light scattering have explored the mechanism of structural memory through time-resolved measurements of the changes occurring within the microstructure during large amplitude oscillatory shear (LAOS). Many studies that combine LAOS with diffusing wave spectroscopy (DWS) have provided significant insight into the shear-driven structural dynamics of colloidal glasses, foams, and other soft materials.18,28,49,50 Similar approaches that combine LAOS and X-ray photon correlation spectroscopy (XPCS) have studied memory loss through irreversible microscopic rearrangements in a stroboscopic manner.37
XPCS provides information at small length scales and long time scales, which makes it well-matched for the study of nanoscale structural dynamics.35,36 At any given time, the speckle pattern that results from the interference effects in the coherent X-ray beam scattered by the complex fluid contains information regarding all the scatterers within the scattering volume. Therefore, the speckle is an instantaneous representation of the nanoscale structure, and its temporal evolution contains information about sample dynamics. An understanding of the nanoscale dynamics through variation of speckle patterns at each moment during an experiment remains elusive when a material is dynamically deformed, such as by oscillatory shearing or startup of flow. This knowledge gap is a critical hindrance for fully understanding and ultimately designing material behaviors for a wide range of applications. Rather, existing studies adopt a stroboscopic approach where the structural information derived from the speckle patterns is compared between cycles at discrete points of the oscillation.36,37
Prior studies18,28,33,37,43,45,47,49–51 have provided only limited discrete structural measures of remembered deformations. However, the correlation of these structural measures to a continuous rheological quantity, which would facilitate the formation of dynamic structure–property–processing relations, is a long-standing and currently unsolved problem. Additionally, the discussions of memory retention have been limited to discrete points during the experiment, an approach referred to as the stroboscopic view, leaving a continuous study of memory formation and loss unexplored.
Numerous microscopic models have been proposed to link microstructure to rheology, including mode coupling theory and shear transformation zone theory.52–54 Despite their usefulness, these theories face challenges in experimental applications due to the need for fitting parameters and the reliance on difficult-to-measure empirical relations.55,56 In addition, given that the experimental structural measurements have been restricted to a stroboscopic approach, these theories share the same limitation. This has resulted in a lack of continuous structure–property relations, which are crucial for processing applications. A number of phenomenological models have been proposed to explain the macroscopic rheological behaviour of materials that yield,57–60 but studies that form structure–property–processing relationships by relating model predictions to structural measurements remain lacking.
Rheological memory in soft materials has been studied extensively in the guise of viscoelasticity, thixotropy, and aging, as well as particular studies that focus on the memory materials have of the direction of an applied shear deformation.61,62 In the simplest cases, those of Newtonian fluids and Hookean elastic solids, memory effects are conceptually simple. A Newtonian fluid has no memory of prior deformations and will remain in its current state when loads are removed, while a Hookean elastic solid has a perfect memory and will always return to its initial configuration when any applied loads are removed.
Viscoelastic memory is intermediate between the two simplest cases, and is often introduced in terms of the Boltzmann superposition principle and the conceptually simple step-strain experiment in which a material is subjected to a small step increase in strain of magnitude γ. Once the small strain has been imposed, the stress, σ(t), relaxes over some timescale. From this observation, it is possible to define the relaxation modulus, G(t), as
 | (1) |
The rheological memory function, m(t), is then defined in terms of the rate at which the relaxation modulus decays
 | (2) |
The negative sign accounts for the fact that the relaxation modulus is a decaying function of time, allowing the rheological memory function to be interpreted as the rate at which memory is lost. In studies of viscoelasticity, memory is therefore discussed as a property that fades over timescales given by the spectrum of relaxation times. Fading memory encompasses the phenomena of stress relaxation and creep retardation.63
The Boltzmann superposition principle for linear viscoelasticity relates the present stress to the memory function via an integral over all past times,64,65
 | (3) |
It is difficult to reconcile the traditional rheological memory function with stroboscopically-observed structural studies, however, given that it originates from consideration of the step-strain test through the relaxation modulus. Built into the concepts surrounding its definition, shown in eqn (2), is that the cause of the stress relaxation was an instantaneous, or very rapid, imposition of strain immediately prior to the measurement. The formulation of the memory function is therefore predicated on an assumption of when the memory was formed. It's only able to quantify the rate at which memory is lost, and is unable to provide any information about when the memory was formed. This makes it difficult to apply to long-lasting transient nonlinear rheological protocols where strains are large and vary significantly in time. Examples of such circumstances include steady shear start-up, creep, and LAOS, as well as realistic flows where deformation may be acquired in multiple different processing steps that may even encompass different kinematics.
The limitation of defining rheological memory in the traditional way includes its applicability to small deformations only. The assumption that the relaxation modulus is independent of the strain is not valid for large deformations. There are ways that nonlinear deformations can be accounted for, but these still rely on the linear viscoelastic memory function with a modification through the introduction of the damping function.66–71
In this work, we form a continuous structure–property–processing relationship by making the direct comparison between nanoscale structural memory determined using two-time plots from rheo-X-ray photon correlation spectroscopy (rheo-XPCS)72 and a proposed two-time macroscale rheological memory function, defined in terms of the recoverable strain acquired over an interval. We determine the macroscale rheological memory using bulk rheometry and add predictions from the Kamani–Donley–Rogers (KDR) model to show correspondence to the nanoscale memory measured in the rheo-XPCS experiments. We use a model fumed silica colloidal suspension to demonstrate the proposed connection.
2 Theory, materials, and methods
2.1 A revised memory function
Before introducing our proposed memory function, it is necessary to make a few fundamental observations. The first is that memories are formed and lost over intervals of time. While this may be obvious to state, it makes clear that any generalized rheological memory ought also to be defined over an interval, rather than at a particular instant, as is the case with the traditional memory function presented in eqn (2). As discussed earlier, the traditional memory function makes an assumption about when the memory was imparted and only describes the rate at which such memories are forgotten subsequent to that formation instant. Because we wish to use a function that can inform us about when memories are formed and when they are lost, the assumption of when a memory is formed must be relaxed. The second observation is that there are two ways to describe a rheological memory. It's possible to refer to an absolute memory as how much is remembered? or a relative memory as how much of what was applied is remembered? Here, we present the definition of absolute memory and its merits, while a discussion of relative memory can be found in the ESI.†We define the absolute rheological memory of the material over the time interval [t1,t2], Mabs(t1,t2), as the change in the ultimate recoverable strain, γrec, acquired over that interval,
 | (4) |
As shown in the ESI,† we also define the relative rheological memory of the material over the time interval [t1,t2], Mrel(t1,t2), as the change in the ultimate recoverable strain relative to the change in the total strain over the same interval.
Our proposal to relate the recoverable strain to the rheological memory is consistent with White's interpretation of the Weissenberg number, Wi, which is intended to describe the nonlinearity of the rheological response73,74 and is typically defined today for steady-shear applications by the product of the applied shear rate, ![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) , and the linear viscoelastic relaxation time, λ, Wi = λ. White originally defined the Weissenberg number as the product of the steady-state shear compliance, Je = γrec/σ, the viscosity, η = σ/, and a velocity gradient U/L = , so that JeηU/L = γrec, making clear his interpretation of the Weissenberg number as the amount of recoverable strain in the fluid. This interpretation is echoed in a comment by Dealy and Wissbrun, who stated that, “This group is a measure of the extent to which elastic or memory effects will play a role”.65 Pipkin also commented on this group, interpreting it as, “the amount of shear in one relaxation time,… the part of the deformation that the fluid can remember…”.75 In this respect, we draw a direct equivalence between the interpretations of the Weissenberg number as the recoverable strain and the memory.
, and the linear viscoelastic relaxation time, λ, Wi = λ. White originally defined the Weissenberg number as the product of the steady-state shear compliance, Je = γrec/σ, the viscosity, η = σ/, and a velocity gradient U/L = , so that JeηU/L = γrec, making clear his interpretation of the Weissenberg number as the amount of recoverable strain in the fluid. This interpretation is echoed in a comment by Dealy and Wissbrun, who stated that, “This group is a measure of the extent to which elastic or memory effects will play a role”.65 Pipkin also commented on this group, interpreting it as, “the amount of shear in one relaxation time,… the part of the deformation that the fluid can remember…”.75 In this respect, we draw a direct equivalence between the interpretations of the Weissenberg number as the recoverable strain and the memory.
Our proposed definition of memory, which we use to connect the structure to the rheology, is unitless, in contrast to the traditional definition shown in eqn (2), which has units of stress per unit time. From the definition, it can be seen that the absolute memory is antisymmetric with respect to the times over which it is defined, Mabs(t1,t2) = −Mabs(t2,t1).
It can also be seen from the definition of the absolute memory given in eqn (4) that for simple step strain applied to a Hookean solid, in which all of the strain is acquired recoverably, γ = γrec, its absolute memory is equal to the applied deformation, Mabs = γ. That is, the absolute memory says it remembers a total of γ amount of strain. For a Newtonian fluid that only ever acquires strain unrecoverably, both its absolute and relative memories are zero.
For cyclic experiments on an arbitrary material, a special case can arise where the change in recoverable strain over some interval is equal to zero. This situation will always happen in the stroboscopic case at steady state, where the interval is a full period. When the system is returned to a state of the same recoverable strain, the absolute memory is zero.
In this work, we preferentially focus on oscillatory experiments with the aim of making connections to prior studies that have also used cyclic shearing. Oscillatory shearing is advantageous as it allows the material to be brought back to the same macroscopic strain and observe any structural similarities and differences. We present a limited set of results for the case of a steady shear startup in the ESI.†
Our proposed definition of absolute memory given by eqn (4) is generally applicable to any flow or deformation protocol for any material. Further, this measure provides information about the rheological memory over an interval between times t1 and t2, in contrast to the traditional definition that presumes that time t1 is immediately after the strain has been applied and a memory has been imparted.
Since the proposed memory function is defined as an integral between two times t1 and t2, it is a multivariable function that requires surface or contour plots to display effectively. In this work, we make direct analogies to two-time plots from XPCS.72 We focus in this work only on the steady alternating state, which requires a specific choice for where to call ‘the beginning’ of the oscillation. We define the beginning of the period to be at the point where the imposed strain is zero and increasing. Since the XPCS correlation function can only provide information about how much the microstructure is correlated between any two times t1 and t2, we compare the contour plots of the two-time correlation function and the magnitude of the absolute rheological memory function i.e.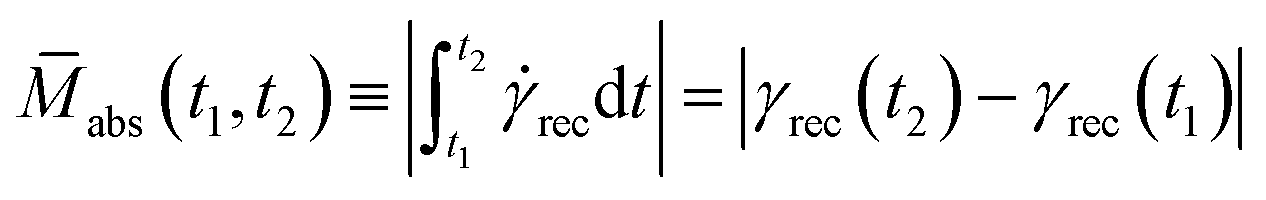 .
.
2.2 Material
The fumed silica colloidal suspension used in this study was selected as a representative disordered soft material that exhibits a yield stress that has good contrast for X-rays. It was prepared as noted elsewhere,76–79 by dispersing 2.9 vol% of hydrophobic fumed silica (R972, Evonik) in highly refined paraffin oil (18512, Sigma-Aldrich) and large molecular weight polyisobutylene (H25, Indopol), at a ratio of 69 wt%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31 wt%. Mixing was achieved using a Thinky mixer (ARE-310) running at 2000 rpm for 1 h. The frequency dependence of the dynamic moduli in the linear viscoelastic regime and the amplitude dependence of the dynamic moduli at ω = 1 rad s−1 are shown in Fig. 1. The material is a soft solid at small deformations, as evidenced by the material storing more energy than it dissipates in the linear viscoelastic regime shown in Fig. 1(a). However, the suspension can be made to yield via the imposition of larger deformations, as shown in Fig. 1(b), where larger amplitudes lead to more energy being dissipated than stored. The amplitude dependence of the dynamic moduli include an overshoot in G′′, a behavior known to arise from acquisition of unrecoverable strain.59,80
:31 wt%. Mixing was achieved using a Thinky mixer (ARE-310) running at 2000 rpm for 1 h. The frequency dependence of the dynamic moduli in the linear viscoelastic regime and the amplitude dependence of the dynamic moduli at ω = 1 rad s−1 are shown in Fig. 1. The material is a soft solid at small deformations, as evidenced by the material storing more energy than it dissipates in the linear viscoelastic regime shown in Fig. 1(a). However, the suspension can be made to yield via the imposition of larger deformations, as shown in Fig. 1(b), where larger amplitudes lead to more energy being dissipated than stored. The amplitude dependence of the dynamic moduli include an overshoot in G′′, a behavior known to arise from acquisition of unrecoverable strain.59,80
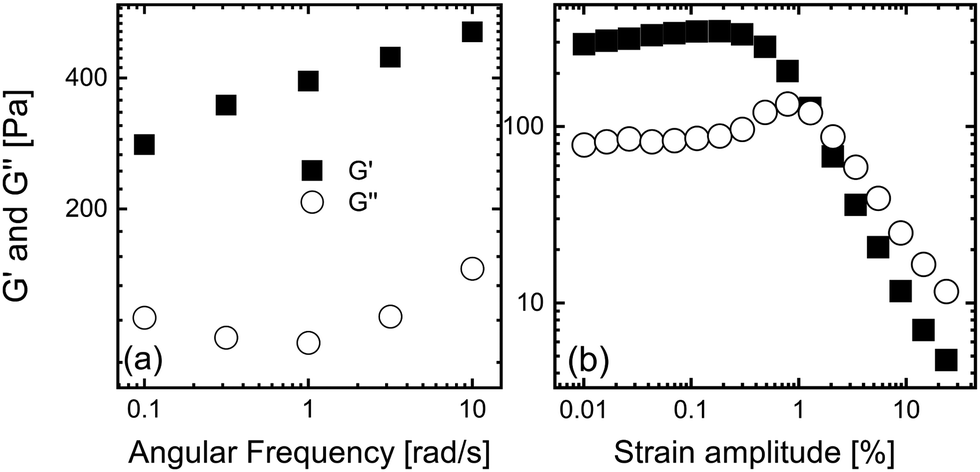 | ||
| Fig. 1 The storage and loss moduli, G′ and G′′, for the fumed silica suspension as functions of (a) the frequency in the linear viscoelastic regime and (b) the strain amplitude at a frequency of 1 rad s−1. | ||
2.3 Kamani–Donley–Rogers (KDR) model
To provide a theoretical prediction of rheological memory, we use the recently proposed KDR model,59,60 which unifies the physics of yielding below and above the yield stress. The 1D version of the model is, | (5) |
) is the rate-dependent relaxation time that is a consequence of combining the recoverable and unrecoverable components, | (6) |
The model can be completely parameterized experimentally. Two parameters govern the way the model acquires strain recoverably,  and
and  are determined from the linear viscoelastic response at an angular frequency of ω = 1 rad s−1, as shown in Fig. 1(a). Three parameters govern the acquisition of unrecoverable strain, σy = 2 Pa, k = 8 Pa sn, and n = 0.05. These parameters can be determined from the steady-state flow curve or, as we have done here, from the large amplitude response of an amplitude sweep shown in Fig. 1(b), as it is known that this behavior is predominantly unrecoverable.59,80 Once all five parameters have been determined from these two tests, they are kept fixed. We then present the model's predictions of the rheological memory during LAOS in the main manuscript and steady shear startup in the ESI.†
are determined from the linear viscoelastic response at an angular frequency of ω = 1 rad s−1, as shown in Fig. 1(a). Three parameters govern the acquisition of unrecoverable strain, σy = 2 Pa, k = 8 Pa sn, and n = 0.05. These parameters can be determined from the steady-state flow curve or, as we have done here, from the large amplitude response of an amplitude sweep shown in Fig. 1(b), as it is known that this behavior is predominantly unrecoverable.59,80 Once all five parameters have been determined from these two tests, they are kept fixed. We then present the model's predictions of the rheological memory during LAOS in the main manuscript and steady shear startup in the ESI.†
2.4 Rheo-XPCS
We display the schematic representation of our XPCS setup in Fig. 2(a). When a coherent X-ray beam is incident on the fumed silica sample (details can be found in the Materials and methods section), the interference generates a highly irregular scattering intensity pattern of “speckles”. The speckle pattern at any one time contains information regarding all the scatterers within the scattering volume. Therefore, the speckle pattern is an instantaneous representation of the nano-scale structure, and its temporal evolution contains information about sample dynamics. In the case of X-ray scattering, which probes density–density correlations, the coherent intensity of the scattered X-rays varies over time as the density correlations within the material fluctuate.35,36,81 Analyzing these temporal variations in intensity allows us to unveil the time correlations in the density. | ||
| Fig. 2 (a) Schematic diagram of rheo-XPCS setup: the incident coherent X-ray beam is incident on a complex fluid, and interference results in a highly irregular speckle pattern. Rheo-XPCS results: (b) two-time correlation plots from oscillatory shear were obtained for various strain amplitudes at ω = 1 rps. As an example, we show results for strain amplitude of 0.1%. Before each oscillatory shear, an interval of zero shear rates was included. The black lines parallel to t1 = t2 diagonal show the average of correlation function C(t1,t1 − n2π) over multiple oscillation periods, indicated by n. (c) Two-time correlation plot corresponding to a steady state oscillatory shear. The starting time for t1 and t2 corresponds to zero imposed strain and increasing. | ||
For coherent scattering intensity measurements, area detectors are commonly used, enabling simultaneous monitoring of multiple speckles. This approach determines the intensity over a range of scattering wave vectors in a single measurement at a time interval Δt, which is set by the detector frame rate, as illustrated in Fig. 2(a).
To determine the relationship between our absolute rheological memory and structural memory, we show the XPCS result of one example oscillatory shear test at a strain amplitude of 0.1% in Fig. 2(b) and (c). These plots are reminiscent of the full 2D distribution of correlation p(s,t|s,t(0)) shown in Teich et al.82 The time-resolved scattering data displayed as the two-time correlation plot allows one to observe the time dependence of the microstructural dynamics characterized by the two-time correlation function of the scattering intensity83
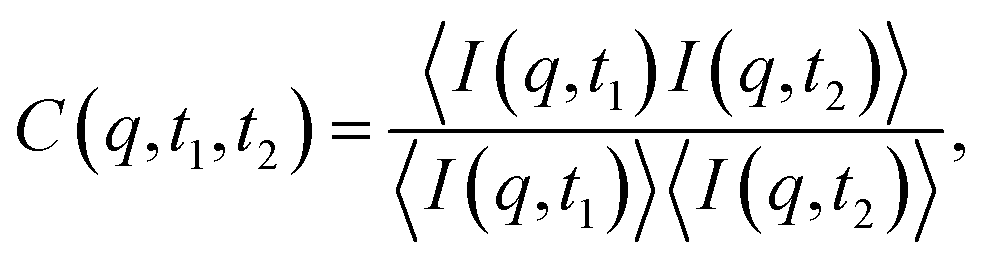 | (7) |
= 0 while the second interval corresponds to our desired oscillatory shear conditions. Recording the correlation prior to deformation allows us to correlate the microstructure at any time during the oscillatory test to the initially quiescent microstructure.
Previous studies focusing on structural memory have been limited to stroboscopic viewing of the structure, where the correlation functions are investigated at discrete points during the period of oscillation with a time period of T, such as C(q,nT,nT − T), when the strain is zero and the rate is maximum, or at C(q,n(T + T/4),n(T − T + T/4)), when the strain is maximum, and the rate is zero.28,29,32,33,38,42 In this study, as shown in Fig. 2(c), we extend this view significantly, focusing on understanding the memory formation and retention transiently throughout an oscillation. Therefore, in contrast to previous studies that limit the comparison of structure at discrete points during an oscillation, our definition of rheological memory naturally allows for its comparison to the structural memory obtained from XPCS at every point of the oscillation in a continuous manner.
3 Results and discussion
3.1 Hookean solid and Maxwell model
To situate our new proposal within known behaviors, in Fig. 3, we compare the color-maps of the absolute memory for Hookean solids and Maxwellian viscoelastic materials, under an applied sinusoidal strain of amplitude γ0 = 1 over a single period. We also show the recoverable strain as a function of time. For clarity and ease of comparison, the times have been normalized by the period of oscillation, T. Regions of zero memory, where the system has the same recoverable strain at t2 and t1 are shown as dark blue, while white regions correspond to highest memory. By definition, the absolute memory along the t1 = t2 line is always zero. | ||
| Fig. 3 The absolute rheological memory function M(t1,t2) of the Hookean solid (a) and Maxwell model (b) at De = 1, for oscillatory shear. The color map representing the change in unrecoverable strain between t1 and t2 is shown along with the recoverable strain as a function of the fraction of the period, outside the color map. Three specific points in (a) are shown as an example to understand how the color map of the absolute memory function is generated. | ||
For the Hookean solid, the sinusoidal form of the recoverable strain indicates that it is perfectly in phase with the applied sinusoidal strain. A line perpendicular to the t1 = t2 line traces a line of constant age, defined as tage = (t1 + t2)/2. Moving along a line of constant age away from the t1 = t2 point, the delay time, |t1 − t2|, increases. As shown in Fig. 3(a), the absolute memory is zero at ages of tage/T = 0.25 and tage/T = 0.75 because of the symmetry of the recoverable strain. The memory across any interval centered on these times will always be zero.
We show in Fig. 3(b) the variation of the absolute memory for a Maxwellian viscoelastic material at a Deborah number De = λω = 1, where λ is the relaxation time and ω is the angular frequency. A lower value of recoverable strain is observed for the Maxwell model, as compared to a Hookean solid, because part of the total applied strain is acquired unrecoverably. At this frequency, the stress and the recoverable strain are out of phase with the applied strain.84 As compared to the Hookean solid, the age diagonal at which the absolute memory is zero is shifted to a lower age value of tage/T = 0.125, indicating the symmetry of the recoverable strain around that point. From this, we conclude that whenever there is a measurable or significant unrecoverable strain, which signifies flow, the points of zero absolute memory will shift to earlier times, along age diagonals tage/T < 0.25.
3.2 Yield stress fluid – fumed silica
In the previous section we concentrated on understanding the absolute rheological memory for the ideal cases of a Hookean elastic solid and a Maxwellian viscoelastic fluid, for which the underlying physics is well understood. In this section, we focus on determining the relation between the absolute rheological memory and the structural memory for the fumed silica colloidal suspension, which has been selected as a representative yield stress fluid. Since our proposed rheological memory function provides information about the memory over an interval between any times t1 and t2, it can be directly compared with the two-time correlation function (C(q,t1,t2)) determined from rheo-XPCS. Because of the symmetries present in the expected results, we focus from now on only on the correlations obtained during half of an oscillation period.We show in Fig. 4 how the absolute rheological memory compares with the two-time correlation function C(q,t1,t2) measured using rheo-XPCS at various strain amplitudes. We choose strain amplitudes such that the storage modulus is larger than the loss modulus, G′ > G′′, where they are equal, G′ = G′′, and where the loss modulus is larger than the storage modulus, G′ < G′′ to account for responses with different degrees of yielding.59,80,85 The minimum resolution for our recoverable strain measurements is 5 × 10−4 [−]. Anything smaller than this is treated as being zero. For a fair comparison, the same approach was taken for determining the recoverable strain using the KDR model. The regions with zero absolute rheological memory, ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) abs = 0, and hence the same recoverable strain at times t1 and t2, are shown in blue in Fig. 4(b), (f) and (j) for the experiments and in Fig. 4(c), (g) and (k) for the model predictions. The white regions indicate intervals where the absolute rheological memory is greater than zero, abs > 0, and hence the system has different recoverable strains at times t1 and t2. We show in the rheo-XPCS plots of Fig. 4(d), (h) and (l) the intervals of high structural correlation, C(q,t1,t2) > 1.09, as purple, while white regions indicate lower correlation. In all the three cases, the two-time correlation functions obtained from XPCS, rheological measurements, and model predictions show that within the uncertainties of our measurements, the nanoscopic structure is the same whenever the change in the bulk recoverable strain over the same interval is zero. That is, the macroscopic measurement and the theoretical prediction from the KDR model of the recoverable strain are equivalent to a measurement of structural similarity at the nanometer scale.
abs = 0, and hence the same recoverable strain at times t1 and t2, are shown in blue in Fig. 4(b), (f) and (j) for the experiments and in Fig. 4(c), (g) and (k) for the model predictions. The white regions indicate intervals where the absolute rheological memory is greater than zero, abs > 0, and hence the system has different recoverable strains at times t1 and t2. We show in the rheo-XPCS plots of Fig. 4(d), (h) and (l) the intervals of high structural correlation, C(q,t1,t2) > 1.09, as purple, while white regions indicate lower correlation. In all the three cases, the two-time correlation functions obtained from XPCS, rheological measurements, and model predictions show that within the uncertainties of our measurements, the nanoscopic structure is the same whenever the change in the bulk recoverable strain over the same interval is zero. That is, the macroscopic measurement and the theoretical prediction from the KDR model of the recoverable strain are equivalent to a measurement of structural similarity at the nanometer scale.
 | ||
| Fig. 4 Comparison of the absolute rheological memory and structural memory: (a), (e), (i) the dynamic moduli G′(γ0) and G′′(γ0) as functions of strain amplitude. The rheological memory obtained from recovery measurements (b), (f), (j) and the KDR model (c), (g), (k) is shown as blue color. The corresponding two-time correlation plot (d), (h), (l) obtained from rheo-XPCS measurements is shown as purple color. The reference diagonal lines t1 = t2 and t2/T = 0.5 − t1/T are shown as dashed lines. The vertical line in (a), (e), (i) shows the strain amplitude to which the rheological and structural memory correspond to. | ||
At a small strain amplitude of γ0 = 0.251%, which is too small to induce yielding, more energy is stored than dissipated as indicated by the dynamic storage modulus being larger than the loss modulus, G′ ≫ G′′. Under such circumstances the material is a viscoelastic solid. From the perspective of the model, this behavior occurs because the rate-dependent relaxation time λ() is always longer than the period of oscillation. The absolute rheological memory M(t1,t2 = 0.5 − t1) is zero at an age of tage/T = 0.25 for both the rheological measurements and the KDR model, as is expected for a viscoelastic solid. The corresponding XPCS color-map, shown in Fig. 4(d) shows high correlation C(q,t1,t2 = 0.5 − t1) at the same age of tage/T = 0.25.
At an intermediate strain amplitude of γ0 = 1.58%, as shown in Fig. 4(e), the dynamic moduli are approximately equal, G′ = G′′, indicating that equal amounts of energy are being stored and dissipated, a condition typically associated with yielding. Under such conditions, the absolute rheological memory is zero and the structure recorrelates at a normalized age of tage/T = 0.22 as shown in Fig. 4(f), (g) and (h). Under these conditions, the relaxation time of the KDR model decreases because of the higher applied shear rate compared to smaller strain amplitudes. This results in a more fluid-like behavior, where a larger portion of the total strain is acquired unrecoverably. Similar behavior is observed at large strain amplitudes, including the case of γ0 = 6.31%, where G′ < G′′, as shown in Fig. 4(i). At the larger amplitudes, the normalized age at which recorrelation takes place shifts to earlier in the period, reflecting the great acquisition of unrecoverable strain.
The benefit of our protocol, which begins measurement of structural correlation before the application of any deformation, is that we can clearly see that some of the structural recorrelation that takes place during testing is not just the same within a period, but is the same as before any deformation began. Even at the largest strain amplitudes we investigated, where the loss modulus is larger than the storage modulus, indicating that more energy is dissipated than is stored, the rheo-XPCS measurements still show some structural correlation with the initial microstructure present during the quiescent state, as indicated by correlation values C(q,t1,t2= −0.5 s) shown in the ESI.† This clearly indicates that even beyond the yielding transition, part of the microstructure that was present before applying any oscillations remains unchanged and still recorrelates each period. Yielding therefore happens in a spatially heterogeneous manner, and memories of prior deformation can exist in disordered colloidal materials even when they are made to flow.
With the data of Fig. 4, we have established a qualitative equivalence between the absolute rheological memory and the structural measures in the form of the XPCS two-time correlation during an oscillation. This qualitative equivalence allows for a continuous description of memory across multiple length scales. It's difficult to make a quantitative comparison due to contrast and shear flow moving scatterers from the scattering window, so we now make use of the average normalized values. For the rheology, we normalize the maximum recoverable strain during the cycle i.e. the recoverable strain amplitude γrec,0 by the total strain amplitude γ0 to look at the relative memory. For the XPCS, we consider the average correlation at time differences equal to n periods by taking an average from the two-time correlation along a path parallel to t1 = t2 but shifted by n periods. This is expressed as
 | (8) |
 | ||
| Fig. 5 (a) Comparison of the normalized rheological metric and the XPCS line average correlation at various strain amplitudes, for both, model and experiments. (b) Direct comparison of the rheological normalized amplitude with the XPCS normalized amplitude. The dashed line is y = x. | ||
The relation between the XPCS and rheological measurements is clear when both are plotted against each other, as shown in Fig. 5(b). All of the data follow the y = x diagonal, showing that a one-to-one relation exists between the experimentally measured and theoretically predicted macroscopic rheological quantity γrec,0/γ0 and the measured nanoscale structural quantity (Clineavg − 1)/(Clineavg − 1)LR. Having already established a qualitative equivalence in Fig. 4, we provide in Fig. 5 a quantitative relation between the macroscopic and nanoscale memories.
When similar comparisons are made for n > 1, they reveal important insights about the nature of yielding at the microstructural level. The normalized line average from the XPCS correlation across multiple cycles can be calculated from eqn (8). We choose to look across two and four periods as examples, taking values of n = 2 and 4 in eqn (8). We compare this to the normalized rheological amplitude for n > 1, (γrec,0/γ0)n, raising it to the power of n, which assumes that the structure responsible for recoverable strain acquired within a period is independent of the responsible structure in another period. The results are shown in Fig. 6. At large strain amplitudes, we observe that the measured XPCS correlations are significantly greater than the bulk rheological measures, indicating that the microscopic deformation associated with the recoverable strain is heterogeneous. The reversible dynamics are therefore spatially confined to long-lived portions of the sample, and the recoverable strain in the non-linear regime is a manifestation of constrained positional fluctuations of the fumed silica particles.
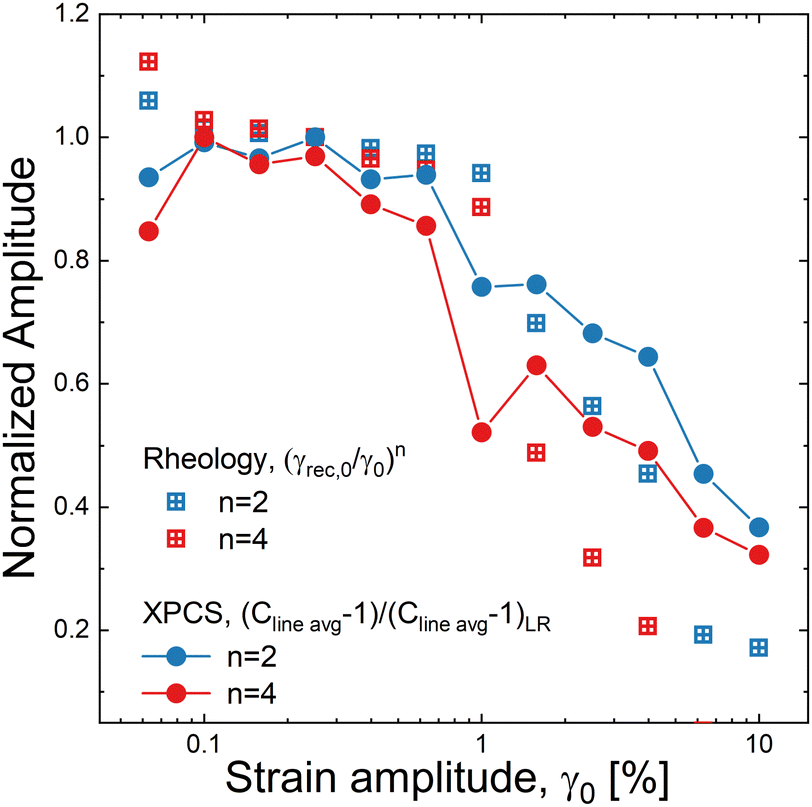 | ||
| Fig. 6 Comparison of the normalized rheological metric and the XPCS line average correlation over n cycles, as a function of strain amplitude. | ||
4 Conclusions
We have formed a continuous structure–property–processing relation by linking transient structural memory at the nanometer scale to macroscopic deformational memory in a model disordered soft material. The rheological memory function is defined over an interval [t1,t2] and provides qualitative and quantitative connection to structural measures across a range of deformations that span the yielding transition. The function provides insights into the rheological memory formation in soft materials in a clear, unambiguous, and protocol-agnostic way. Our new memory function extends the conventional rheological concept by accounting for when memories are formed as well as when they are lost during complex transient protocols such as cyclic deformation and startup of steady flow. A direct comparison is made between the two-time rheological memory function obtained from bulk measurements and the KDR model and structural memory obtained from rheo-XPCS measurements of two-time intensity correlation functions. Our rheo-XPCS data show that structural recorrelation at the nanometer scale is maximized whenever the bulk recoverable strain returns to the same value over an interval. The macroscopic recoverable strain is therefore a measure of the nano-structural memory, suitably averaged over the scattering volume. We show this to be true for oscillatory shear and steady shear startup tests (shown in the ESI†), indicating the generality of our approach in making connection between bulk rheological and structural measures.The connections that have been made between structural and rheological memory in this study also indicate that yielding in disordered colloidal materials is strongly heterogeneous and that memories of prior deformation can exist even when they are made to flow.
Author contributions
KMK designed and performed the research, analyzed data, and wrote the paper. YS performed research and analyzed data. JG performed research and analyzed data. SN designed and performed the research, analyzed data, and wrote the paper. QZ performed research and analyzed data. RLL analyzed data and wrote the paper. JLH analyzed data and wrote the paper. AD performed research and analyzed data. RME analyzed data. SAR designed the research, analyzed data, and wrote the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by NSF Grant 1847389. We thank Anton Paar for the use of the TwinDrive MCR 702 through their academic program. This research was performed at beamline 8-ID-I of the Advanced Photon Source and the Center for Nanoscale Materials, US Department of Energy (DOE) Office of Science User Facilities operated for the DOE Office of Science by ANL (contract no. DE-AC02-06CH11357). Y. H. S. acknowledges support from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A6A3A03039172). R. L. L. acknowledges funding from NSF grant no. CBET-1804721. J. L. H. acknowledges the support of the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant Program.References
- T. Bhattacharjee, S. M. Zehnder, K. G. Rowe, S. Jain, R. M. Nixon, W. G. Sawyer and T. E. Angelini, Sci. Adv., 2015, 1, e1500655 CrossRef PubMed.
- C. Yu, F. Yao and J. Li, Acta Biomater., 2022, 139, 4–21 CrossRef CAS PubMed.
- T. J. Hinton, Q. Jallerat, R. N. Palchesko, J. H. Park, M. S. Grodzicki, H.-J. Shue, M. H. Ramadan, A. R. Hudson and A. W. Feinberg, Sci. Adv., 2015, 1, e1500758 CrossRef PubMed.
- T. H. Qazi, V. G. Muir and J. A. Burdick, ACS Biomater. Sci. Eng., 2022, 8, 1427–1442 CrossRef CAS PubMed.
- M. Schaffner, P. A. Rühs, F. Coulter, S. Kilcher and A. R. Studart, Sci. Adv., 2017, 3, eaao6804 CrossRef PubMed.
- Y. Cao, Y. Zhou, Z. Chen, Z. Zhang, X. Chen and C. He, Adv. Healthcare Mater., 2021, 10, 2100814 CrossRef CAS PubMed.
- S. Xin, K. A. Deo, J. Dai, N. K. R. Pandian, D. Chimene, R. M. Moebius, A. Jain, A. Han, A. K. Gaharwar and D. L. Alge, Sci. Adv., 2021, 7, eabk3087 CrossRef CAS PubMed.
- A. K. Grosskopf, L. Labanieh, D. D. Klysz, G. A. Roth, P. Xu, O. Adebowale, E. C. Gale, C. K. Jons, J. H. Klich and J. Yan, et al. , Sci. Adv., 2022, 8, eabn8264 CrossRef CAS PubMed.
- G. Negro, L. N. Carenza, G. Gonnella, F. Mackay, A. Morozov and D. Marenduzzo, Sci. Adv., 2023, 9, eadf8106 CrossRef PubMed.
- B. G. Compton and J. A. Lewis, Adv. Mater., 2014, 26, 5930–5935 CrossRef CAS PubMed.
- K. Fu, Y. Wang, C. Yan, Y. Yao, Y. Chen, J. Dai, S. Lacey, Y. Wang, J. Wan and T. Li, et al. , Adv. Mater., 2016, 28, 2587–2594 CrossRef CAS PubMed.
- J. J. Griebler and S. A. Rogers, Phys. Fluids, 2022, 34, 023107 CrossRef CAS.
- H. Melito, C. Daubert and E. Foegeding, J. Texture Stud., 2013, 44, 253–288 CrossRef.
- N. J. Balmforth, I. A. Frigaard and G. Ovarlez, Annu. Rev. Fluid Mech., 2014, 46, 121–146 CrossRef.
- H. O’Ghaffari, M. Peč, T. Mittal, U. Mok, H. Chang and B. Evans, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305667120 CrossRef PubMed.
- A. Bot, I. A. van Amerongen, R. D. Groot, N. L. Hoekstra and W. G. Agterof, Polym. Gels Networks, 1996, 4, 189–227 CrossRef CAS.
- S. Kitade, A. Ichikawa, N. Imura, Y. Takahashi and I. Noda, J. Rheol., 1997, 41, 1039–1060 CrossRef CAS.
- G. Petekidis, A. Moussad and P. N. Pusey, Phys. Rev. E, 2002, 66, 051402 CrossRef CAS PubMed.
- P. C. Møller, J. Mewis and D. Bonn, Soft Matter, 2006, 2, 274–283 RSC.
- R. H. Ewoldt, C. Clasen, A. E. Hosoi and G. H. McKinley, Soft Matter, 2007, 3, 634–643 RSC.
- J.-C. Baudez, Appl. Rheol., 2008, 18, 13495 Search PubMed.
- A. S. Negi and C. O. Osuji, J. Rheol., 2010, 54, 943–958 CrossRef CAS.
- W. Sun, Y. Yang, T. Wang, H. Huang, X. Liu and Z. Tong, J. Colloid Interface Sci., 2012, 376, 76–82 CrossRef CAS PubMed.
- R. R. Fernandes, D. E. Andrade, A. T. Franco and C. O. Negrão, Rheol. Acta, 2017, 56, 743–752 CrossRef CAS.
- S. A. Rogers, D. Vlassopoulos and P. Callaghan, Phys. Rev. Lett., 2008, 100, 128304 CrossRef CAS PubMed.
- S. Rogers, P. Callaghan, G. Petekidis and D. Vlassopoulos, J. Rheol., 2010, 54, 133–158 CrossRef CAS.
- S. A. Rogers, B. M. Erwin, D. Vlassopoulos and M. Cloitre, J. Rheol., 2011, 55, 435–458 CrossRef CAS.
- P. Hébraud, F. Lequeux, J. P. Munch and D. J. Pine, Phys. Rev. Lett., 1997, 78, 4657–4660 CrossRef.
- N. C. Keim and P. E. Arratia, Phys. Rev. Lett., 2014, 112, 028302 CrossRef PubMed.
- K. Hima Nagamanasa, S. Gokhale, A. K. Sood and R. Ganapathy, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 062308 CrossRef CAS PubMed.
- S. Slotterback, M. Mailman, K. Ronaszegi, M. van Hecke, M. Girvan and W. Losert, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 021309 CrossRef.
- K. Galloway, E. Teich, X. Ma, C. Kammer, I. Graham, N. Keim, C. Reina, D. Jerolmack, A. Yodh and P. Arratia, Nat. Phys., 2022, 18, 565–570 Search PubMed.
- S. Aime, D. Truzzolillo, D. J. Pine, L. Ramos and L. Cipelletti, Nat. Phys., 2023, 1–7 Search PubMed.
- R. Angelini, E. Zaccarelli, F. A. de Melo Marques, M. Sztucki, A. Fluerasu, G. Ruocco and B. Ruzicka, Nat. Commun., 2014, 5, 4049 Search PubMed.
- R. L. Leheny, Curr. Opin. Colloid Interface Sci., 2012, 17, 3–12 CrossRef CAS.
- R. L. Leheny, M. C. Rogers, K. Chen, S. Narayanan and J. L. Harden, Curr. Opin. Colloid Interface Sci., 2015, 20, 261–271 CrossRef CAS.
- M. C. Rogers, K. Chen, L. Andrzejewski, S. Narayanan, S. Ramakrishnan, R. L. Leheny and J. L. Harden, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 90, 062310 CrossRef PubMed.
- M. C. Rogers, K. Chen, M. J. Pagenkopp, T. G. Mason, S. Narayanan, J. L. Harden and R. L. Leheny, Phys. Rev. Mater., 2018, 2, 095601 CrossRef CAS.
- Z. Evenson, B. Ruta, S. Hechler, M. Stolpe, E. Pineda, I. Gallino and R. Busch, Phys. Rev. Lett., 2015, 115, 175701 CrossRef PubMed.
- A. Das, P. M. Derlet, C. Liu, E. M. Dufresne and R. Maaß, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- M. Lüttich, V. M. Giordano, S. Le Floch, E. Pineda, F. Zontone, Y. Luo, K. Samwer and B. Ruta, Phys. Rev. Lett., 2018, 120, 135504 CrossRef.
- S. Küchemann, C. Liu, E. M. Dufresne, J. Shin and R. Maaß, Phys. Rev. B, 2018, 97, 014204 CrossRef.
- A. Pommella, A.-M. Philippe, T. Phou, L. Ramos and L. Cipelletti, Phys. Rev. Appl., 2019, 11, 034073 CrossRef CAS.
- G. J. Donley, S. Narayanan, M. A. Wade, J. D. Park, R. L. Leheny, J. L. Harden and S. A. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2215517120 CrossRef CAS PubMed.
- N. C. Keim and S. R. Nagel, Phys. Rev. Lett., 2011, 107, 010603 CrossRef PubMed.
- J. D. Paulsen, N. C. Keim and S. R. Nagel, Phys. Rev. Lett., 2014, 113, 068301 CrossRef CAS PubMed.
- N. C. Keim and P. E. Arratia, Soft Matter, 2013, 9, 6222–6225 RSC.
- N. C. Keim, J. D. Paulsen, Z. Zeravcic, S. Sastry and S. R. Nagel, Rev. Mod. Phys., 2019, 91, 035002 CrossRef CAS.
- R. Höhler, S. Cohen-Addad and H. Hoballah, Phys. Rev. Lett., 1997, 79, 1154 CrossRef.
- M. Laurati, S. Egelhaaf and G. Petekidis, J. Rheol., 2014, 58, 1395–1417 CrossRef CAS.
- K. L. Galloway, X. Ma, N. C. Keim, D. J. Jerolmack, A. G. Yodh and P. E. Arratia, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 11887–11893 CrossRef CAS PubMed.
- M. L. Falk and J. S. Langer, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 7192 CrossRef CAS.
- A. Argon, Acta Metall., 1979, 27, 47–58 CrossRef CAS.
- S. Slotterback, M. Mailman, K. Ronaszegi, M. Van Hecke, M. Girvan and W. Losert, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 85, 021309 CrossRef PubMed.
- E. Bouchbinder and J. Langer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 061503 CrossRef PubMed.
- M. Siebenbürger, M. Fuchs, H. Winter and M. Ballauff, J. Rheol., 2009, 53, 707–726 CrossRef.
- P. Saramito, J. Non-Newtonian Fluid Mech., 2009, 158, 154–161 CrossRef CAS.
- C. J. Dimitriou and G. H. McKinley, J. Non-Newtonian Fluid Mech., 2019, 265, 116–132 CrossRef CAS.
- K. Kamani, G. J. Donley and S. A. Rogers, Phys. Rev. Lett., 2021, 126, 218002 CrossRef CAS PubMed.
- K. M. Kamani and S. A. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2401409121 CrossRef CAS PubMed.
- F. Gadala-Maria and A. Acrivos, J. Rheol., 1980, 24, 799–814 CrossRef CAS.
- M. Toiya, J. Stambaugh and W. Losert, Phys. Rev. Lett., 2004, 93, 088001 CrossRef PubMed.
- A. Y. Malkin and A. I. Isayev, Rheology Concepts, Methods, And Applications, Chem Tec Publishing, Ontario, 2012, pp. 1–8 Search PubMed.
- C. Macosko, Rheology: Principles, Measurements, and Applications, Wiley, New York, 1994 Search PubMed.
- J. M. Dealy and K. F. Wissbrun, Melt rheology and its role in plastics processing: theory and applications, Springer, New York, 1999 Search PubMed.
- B. Bernstein, E. Kearsley and L. Zapas, Trans. Soc. Rheol., 1963, 7, 391–410 Search PubMed.
- A. Kaye, College of Aeronautics Note 134 & 149, 1962.
- R. B. Bird, R. C. Armstrong and O. Hassager, Dynamics of Polymeric Liquids, Volume 1: Fluid Mechanics, Wiley, New York, 1987 Search PubMed.
- R. G. Larson, Constitutive equations for polymer melts and solutions: Butterworths series in chemical engineering, Butterworth-Heinemann, 2013 Search PubMed.
- M. Wagner, et al. , Rheol. Acta, 2009, 48, 245–284 CrossRef.
- J. L. White and N. Tokita, J. Phys. Soc. Jpn., 1967, 22, 719–724 CrossRef CAS.
- O. Bikondoa, J. Appl. Crystallogr., 2017, 50, 357–368 CrossRef CAS PubMed.
- J. L. White, J. Appl. Polym. Sci., 1964, 8, 2339–2357 CrossRef CAS.
- J. Dealy, Rheol. Bull., 2010, 79, 14–18 Search PubMed.
- A. C. Pipkin, Lectures on viscoelasticity theory, ed. A. C. Pipkin, Springer-Verlag, New York, 1972, vol. viii, p. 180 Search PubMed.
- K. Dullaert and J. Mewis, Rheol. Acta, 2005, 45, 23–32 CrossRef CAS.
- Y. Wei, M. J. Solomon and R. G. Larson, J. Rheol., 2016, 60, 1301 CrossRef CAS.
- J. Choi and S. A. Rogers, Rheol. Acta, 2020, 59, 921–934 CrossRef CAS.
- M. Mahmoudabadbozchelou, K. M. Kamani, S. A. Rogers and S. Jamali, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202234119 CrossRef CAS PubMed.
- G. J. Donley, P. K. Singh, A. Shetty and S. A. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21945–21952 CrossRef CAS PubMed.
- M. Sutton, C. R. Phys., 2008, 9, 657–667 CrossRef CAS.
- E. G. Teich, K. L. Galloway, P. E. Arratia and D. S. Bassett, Sci. Adv., 2021, 7, eabe3392 CrossRef PubMed.
- A. Madsen, R. L. Leheny, H. Guo, M. Sprung and O. Czakkel, New J. Phys., 2010, 12, 055001 CrossRef.
- N. W. Tschoegl, Representation of linear viscoelastic behavior by mathematical models, Springer, 1989, pp. 314–364 Search PubMed.
- K. M. Kamani, G. J. Donley, R. Rao, A. M. Grillet, C. Roberts, A. Shetty and S. A. Rogers, J. Rheol., 2023, 67, 331–352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00953c |
| This journal is © The Royal Society of Chemistry 2025 |