
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA waste-minimized protocol for electrochemical reductive amination and its environmental assessment†
Simone
Trastulli Colangeli‡
,
Filippo
Campana‡
,
Francesco
Ferlin
 * and
Luigi
Vaccaro
*
* and
Luigi
Vaccaro
*
Laboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123 – Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; francesco.ferlin@unipg.it; Web: https://greensoc.chm.unipg.it
First published on 3rd December 2024
Abstract
The environmental impact linked to the use of solvents is a recurring criticism of the development of sustainable chemical processes. The possibility of recovering the reaction medium is a key aspect to simplify the isolation of the desired target material. At the same time, it may also allow the minimization of additional solvents necessary for the purification and ultimately minimize all the contributions to the waste generated. Electrochemical processes are of high interest in modern organic synthesis. Anyway, most electrochemical processes nowadays feature the use of unrecoverable solvents or solvent mixtures in high amounts (dilution). The consequent high environmental impact of such procedures is easily understood. Aiming at the definition of effective waste-minimized synthetic protocols, herein, we report our study towards the development of an electrochemical reductive amination protocol employing an acetonitrile–water azeotrope as the recoverable reaction medium. Through the use of gram-scale synthesis and comprehensive sustainability and environmental assessments, we present an efficient management of solvents and electrolytes in an electrochemical methodology for a widely useful chemical transformation.
Introduction
In the realm of modern synthetic chemistry, the search for green and sustainable processes is of fundamental importance.1 Driven by growing environmental concerns and increasingly stringent regulations, research dedicated to unconventional synthetic pathways is gaining ever-increasing attention from the scientific community.2In this context, organic electrosynthesis has been recognized as a powerful tool that effectively fulfills many of the principles of green chemistry. By exploiting electrochemical conditions, it is possible to limit the use of stoichiometric redox reagents and sometimes severe reactive conditions, thus minimizing both waste production and energy consumption.3 This aspect becomes even more attractive when considering the production of fine chemicals and APIs (active pharmaceutical ingredients) – areas featuring high E-factor values among all the fields of chemical manufacturing.4
Given the fundamental presence of the substituted amine moiety in many areas of chemical production, the procedures that allow the installation of C–N bonds are of particular interest.5 Reductive amination is arguably among the most used synthetic tools for the formation of C–N bonds. It allows the production of primary, secondary, or tertiary amines with high yields and selectivity and is easy to operate.6 It is worth noting that this synthetic tool is largely used by the pharmaceutical industry, with at least 25% of carbon–nitrogen bond formation procedures performed via reductive amination.7
In the context of reductive procedures, electrochemistry has proven to be very efficient offering a viable alternative to the use of common strategies that employ stoichiometric hydrides or gaseous hydrogen.8
Early reports on electrochemical reductive amination (ERA) exploited lead cathodes to minimize the hydrogen evolution reaction (HER),9 while recent studies have demonstrated the effectiveness of less toxic and more available electrode materials such as copper, silver, graphite, etc.10 Nevertheless, the role of the solvents in this widely general transformation has not been optimized in view of the definition of a waste minimized and green process.
Solvents are extensively used across all synthetic steps, both as reaction media and during purification. They arguably represent one of the most critical aspects of research in green chemistry.11 In fact, in the preparation of APIs or fine chemicals, solvents account for 85% of the mass of waste.12 The US EPA (Environmental Protection Agency) has estimated that solvent emissions resulting from chemical manufacturing accounted for up to 62% of the total emissions in 2017.13 For this reason, the development of a generally useful procedure in organic synthesis should always consider the efforts to recover the reaction medium. Aqueous azeotropes can be a viable alternative to the use of traditional organic solvents, being able to improve the solubility of reagents of different nature but at the same time being recoverable and reusable.14 By using minimum boiling point azeotropes, it is possible to improve the energy efficiency of a distillation process, facilitating the removal and reuse of the medium.
We believe that this aspect can be very useful and appreciated in the case of organic electrosynthesis, where the use of aqueous mixtures increases the conductivity, improving the scalability and the energy-saving of a protocol.15
For this reason, we have dedicated this study to the widely useful electrochemical reductive amination for which, to the best of our knowledge, the potential utility of azeotropic mixtures has not been investigated.
In this contribution, we report the results that we have obtained in the development of a protocol using inexpensive electrode materials and an acetonitrile–water azeotrope under highly concentrated conditions. Under these conditions, we have optimized the protocol to simplify the isolation of the final products, and the efficiency of our procedure was further demonstrated by expanding the protocol to a larger scale. The greenness of our results was quantified in comparison with the methods known in the literature using multiple assessment levels by evaluating mass metrics, Ecoscale,16 safety/hazard and environmental indicators17 and a Life Cycle Assessment analysis (Fig. 1).
 | ||
| Fig. 1 Features of this work. | ||
Results and discussion
By using 4-fluorobenzaldehyde (1a) and butylamine (2a) as representative reagents, in combination with tetrabutylammonium hexafluorophosphate as the supporting electrolyte and graphite and aluminium, respectively, as the cathode and the anode, the effect of various solvent mixtures was investigated. The results are summarised in Table 1.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Reaction mediumb | Time (min) |
3a′c (%) |
3ac (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), tetrabutylammonium hexafluorophosphate (0.5 mmol), aluminium anode and graphite cathode, charge passed: 3 F mol−1, solvent: 0.1 M, 20 mA. b Percentages reported are % v/v. c Determined by GLC analysis, the remaining materials were unreacted 1a and 2a. d Low miscibility of the mixture. e 4 F mol−1 charge passed. | ||||
| 1 | THF (2% H2O) | 120 | 1 | 69 |
| 2 | THF (9% MeOH) | 120 | 54 | 35 |
| 3d | H2O | 120 | 0 | 0 |
| 4d | TPGS-750 | 120 | 0 | 0 |
| 5 | MeOH | 120 | 95 | 0 |
| 6 | EtOH | 120 | 95 | 0 |
| 7d | 2-MeTHF (2% H2O) | 120 | 95 | 0 |
| 8 | 2-MeTHF (5% MeOH, 2% H2O) | 120 | 87 | 11 |
| 9e | 2-MeTHF (10% MeCN, 2% H2O) | 160 | 8 | 81 |
| 10 | MeCN:H2O (az.) |
120 | 4 | 76 |

On using THF and 2% (v/v) of water, the product was obtained in a modest yield by passing a charge of 3 F mol−1 (Table 1, entry 1). However, on replacing water with methanol (9% v/v), the reaction was slower (Table 1, entry 2). Pure water proved to be ineffective, probably due to the poor solubility of the organic reagents (Table 1, entry 3), and also with the use of TPGS-750 2% aq., the reaction led to no formation of the product (Table 1, entry 4). Both methanol and ethanol were inefficient, leading to no conversion to product 3a (Table 1, entries 5 and 6). Replacing THF with 2-MeTHF–water mixtures led to a biphasic system (Table 1, entry 7), and no reduction of imine occurred. Also, in the case of 2-MeTHF, the presence of methanol decreased the reaction performances, even when the solvent mixture was completely homogeneous (Table 1, entry 8). Interestingly, on using a mixture of 2-MeTHF, acetonitrile, and water, the amine 3a could be obtained in a high yield (Table 1, entry 9). With the use of a simpler acetonitrile–water azeotrope (16% w/w of water), the reaction proceeded with comparable excellent results (Table 1, entry 10). Encouraged by this last result, we decided to employ MeCN:H2O (az.), being easily recoverable and showing excellent conductive ability.
As shown in Table 2, we report a further optimization dedicated to the selection of the supporting electrolyte. As can be seen, the use of inorganic salts does not lead to product formation (Table 2, entries 1–3). Furthermore, when using acidic and basic electrolytes, the imine remained stable but could not be reduced at all (Table 2, entries 4 and 5). However, on changing the cationic counterion of the previously tested salts to tetraalkylammonium moieties, the reaction efficiently formed product 3a. As can be seen, the results did not change when different anionic counterions were used and the tetrabutylammonium cation was kept constant, indicating that the anion does not play a relevant role in the process.
a
|
|
||||
|---|---|---|---|---|
| Entry | Supporting electrolyte | Reaction medium |
3a′b (%) |
3ab (%)b |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), supporting electrolyte (0.5 mmol), aluminium anode and graphite cathode, solvent: 0.1 M, 20 mA, 120 minutes, 3 F mol−1. b Determined by GLC analysis, the remaining materials were unreacted 1a and 2a. c Low solubility or miscibility of the mixture. | ||||
| 1 | NaBF4 | MeCN:H2O (az.) |
95 | 0 |
| 2 | KPF6 | MeCN:H2O (az.) |
95 | 0 |
| 3 | KBr | MeCN:H2O (az.) |
95 | 0 |
| 4 | NaOH | MeCN:H2O (az.) |
95 | 0 |
| 5 | HCl | MeCN:H2O (az.) |
95 | 0 |
| 6 | Bu4NPF6 | MeCN:H2O (az.) |
4 | 76 |
| 7 | Bu4NBr | MeCN:H2O (az.) |
7 | 75 |
| 8 | Bu4NHSO4 | MeCN:H2O (az.) |
3 | 78 |
| 9 | Et4NBr | MeCN:H2O (az.) |
5 | 72 |
| 10c | Et4NBr | 2-MeTHF (10% MeCN, 2% H2O) | 95 | 0 |
In contrast, the tetraalkylammonium cation plays a crucial role (Table 2, entries 6–8). This effect can be ascribed to the stabilization of the anionic intermediate, which is formed following the cathodic reduction of the imine 3a′.10e On replacing the supporting electrolyte with tetraethylammonium bromide, the performance did not change (Table 2, entry 9). Nevertheless, this salt is less soluble in organic solvents, and upon replacing MeCN:H2O (az.) with a 2-MeTHF, acetonitrile and water mixture, the reaction did not occur (Table 2, entry 10).
To develop a simple and wasteless work-up procedure, the tetraethylammonium salt was chosen as the ideal supporting electrolyte. The latter is simpler to separate through a precipitation/filtration sequence compared to the other tetraalkylammonium salts.
Ultimately, the conditions were further optimized to increase the selectivity and yield of product 3a. The results are reported in Table 3.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Anode | Cathode | Current mol−1 (mA mmol−1) | 3a′ (%)b |
3ab (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), supporting electrolyte (0.5 mmol), 3 F mol−1. b Determined by GLC analysis, the remaining materials were unreacted 1a and 2a. c At 0.5 M concentration. d At 1 M concentration. | |||||
| 1 | Aluminium | Graphite | 40 | 5 | 72 |
| 2 | Zinc | Graphite | 40 | 95 | 0 |
| 3 | Graphite | Graphite | 40 | 95 | 0 |
| 4 | Aluminium | Copper | 40 | 95 | 0 |
| 5 | Aluminium | Stainless steel | 40 | 95 | 0 |
| 6 | Aluminium | Nickel | 40 | 95 | 0 |
| 7 | Aluminium | Graphite | 100 | 1 | 99 |
| 8c | Aluminium | Graphite | 100 | 1 | 99 |
| 9d | Aluminium | Graphite | 100 | 1 | 99 |

On replacing the aluminium anode with a zinc or graphite anode, no product formation was observed (Table 3, entries 1–3). Moreover, cathode replacement also led to no product, suggesting that graphite is necessary for imine reduction (Table 3, entries 4–6).
Interestingly, after increasing the current intensity to 100 mA per 1 mmol of 1a, the conversion to 3a increased dramatically (Table 3, entry 7). To minimise waste, the concentration was increased from 0.1 M to 0.5 M and then to 1 M; the performance of the reaction did not change (Table 3, entries 8 and 9). To gather information on the role of dilution,18 the reaction kinetics was determined at various concentrations (Fig. 2). The kinetic analysis demonstrated that reagent 1a is about 5% after 8 min of reaction, indicating that the formation of imine is faster than the cathodic reduction.
 | ||
| Fig. 2 Reaction kinetics. Reaction conditions: 1a (3 mmol), 2a (3 mmol), tetraethylammonium bromide (3 mmol), 300 mA. Blue line:1 M: MeCN:H2O (az.) (3 mL), samples taken every 8 minutes for up to 48 minutes of reaction. Green line:0.5 M: MeCN:H2O (az.) (6 mL), samples taken every 8 minutes for up to 48 minutes. Red line:0.1 M: MeCN:H2O (az.) (30 mL), samples taken every 8 minutes for up to 48 minutes. | ||
The plot depicted in Fig. 2, therefore, shows the percentage of amine 3a in relation to that of imine 3a′. As can be seen, upon changing the concentration, the speed of the reaction does not change. Unlike other electrochemical hydrogenations, the side hydrogen formation (HER) is not relevant at the concentration considered. The system is therefore able to selectively reduce the imine; otherwise, at lower concentrations, there would be a decrease in the reaction kinetics and current efficiency due to the HER.19 From the kinetic analysis, it can also be seen that the reaction is complete after the passage of a charge of 2.75 F mol−1, which results in a current efficiency close to 73%. At this stage, it was decided to test the suitability of the optimized reaction conditions by expanding the substrate scope (Scheme 1).
 | ||
| Scheme 1 Substrate scope of ERA. Reaction conditions: 1a–l (3 mmol), 2a–i (3 mmol), tetraethylammonium bromide (3 mmol), MeCN:H2O (az.) (3 mL), 300 mA until a 2.75 F mol−1 charge is passed (ca. 45 minutes). aPre-step of imine formation is required. Products are isolated as hydrochloride salts. | ||
Using amine 2a, various functionalized aldehydes were tested (1a–h). In all cases, the products 3 were obtained with high purity and their isolation was carried out through a simple filtration procedure using cotton wool, giving generally high yields. Subsequently, using aldehyde 1c, we tested different primary amines. Gratifyingly, the performance of the process did not change, and products 3i–p were isolated in high yields with a very simple work-up procedure.
However, the results were different upon using ketones. The reactions of amine 2a and different acetophenones did not lead to the products. In this case, the formation of the imine occurs much more slowly at room temperature, and for this reason, pre-formation of the imine was obtained by mixing the reagents under solvent-free conditions at 90 °C. In this manner, products 3q–t were isolated as hydrochloride salts.
Expanding the scope of our ERA protocol, biomass-derived furfural was also used with satisfactory results (Fig. 3).
 | ||
| Fig. 3 Electrochemical reductive amination on biomass-derived furfural. Reaction conditions: 1m (3 mmol), 2a (3 mmol), tetraethylammonium bromide (3 mmol), MeCN:H2O (az.) (3 mL), aluminium anode and graphite cathode, 300 mA until a 3 F mol−1 charge is passed (ca. 50 minutes). | ||
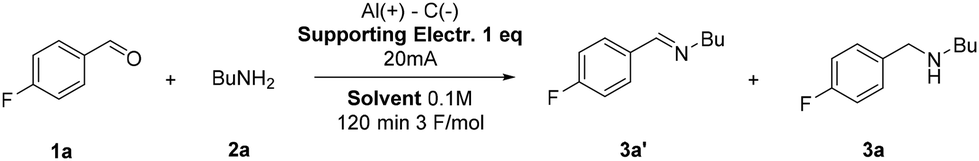
Our waste-minimized protocol was carried out on a larger scale that allowed solvent recovery by simple distillation (Scheme 2). On mixing 30 mmol of reagents 1c and 2a, the 100 mA mmol−1 current used for the 1 mmol scale was not necessary to obtain high selectivity (see optimization in Table 3). In fact, by using a current of 1.5 A (50 mA mmol−1 of substrate), it was possible to decrease the amount of the supporting electrolyte to 0.5 equivalents to achieve sufficient conductivity. At the end of the reaction, 82% of the reaction solvent was recovered. The crude product was dissolved in 100 mL of ethyl acetate and filtered through cotton wool. This step serves to eliminate the supporting electrolyte, which is insoluble in the extraction medium. Through a second distillation process, ethyl acetate was recovered in 85% yield. The product could be obtained as a yellow oil with an overall 78% yield. Furthermore, the reaction solvent was used and recovered for three consecutive runs, by adding the unrecovered portion, without any changes in yield and selectivity.
 | ||
| Scheme 2 Gram-scale ERA with solvent reclaims. Reaction conditions: 1c (30 mmol; 4.217 g), 2a (30 mmol; 2.194 g), tetraethylammonium bromide (15 mmol; 3.152 g), aluminium anode and graphite cathode, MeCN:H2O (az.) (30 mL), 1.5 A, 100 minutes. | ||
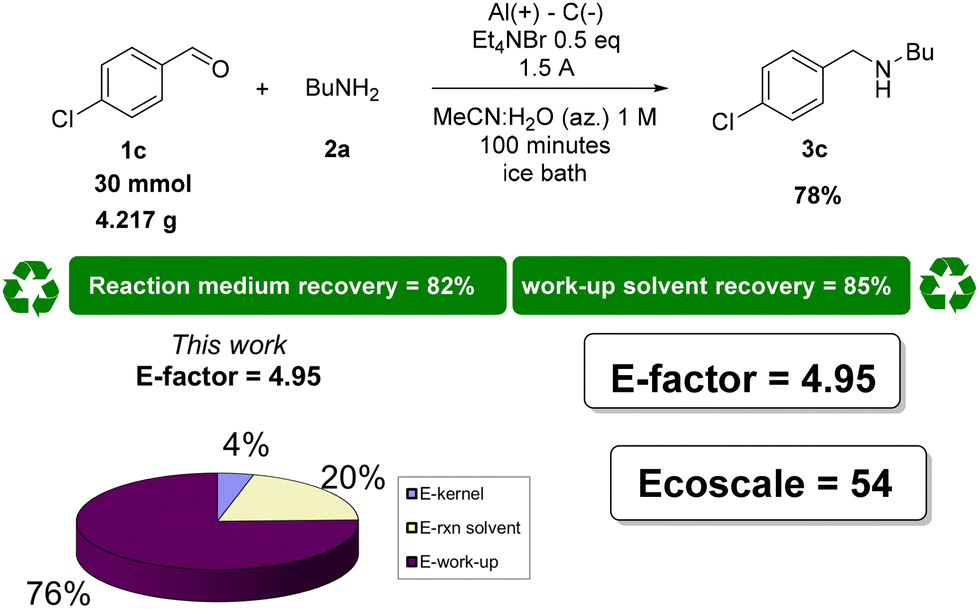
To fully assess our ERA process in terms of waste minimization and overall sustainability, an E-factor value of 4.95 was obtained (see the ESI† for the detailed calculation).
E-Factor distribution analysis highlighted that the significant improvement is ascribable to the highly concentrated conditions and the consequent recovery and recycling of the solvents. The analysis of the E-factor under non-optimized reaction conditions shows that performing the reaction at a concentration of 0.1 M without solvent recovery leads to an E-factor of 73.35. However, at a concentration of 1 M, the E-factor decreases to 25.83 under the same conditions (see the ESI† for analytical calculations). This highlights the critical role of selecting easily recoverable solvents to minimize waste production, reducing the E-factor to as low as 4.95 through recycling.
A further assessment was performed using Ecoscale analysis, which allowed us to include the benefit derived from not needing chromatographic separation.
We therefore compared the sustainability of our methodology with that of selected protocols used for reductive amination.
Our selection includes, to the best of our knowledge, the only two examples of preparative electrochemical reductive amination to date,10c,d as well as a standard procedure that uses NaBH4 in methanol (benchmark route that uses a chemical reductant)20 and a flow methodology reported by Kappe that uses hydrosilane as a hydride source21 that features an excellent E-factor value.
As expected, on analysing the E-factor distribution, it is possible to assign a major contribution to the E-factor to the reaction medium and the work-up solvents, for the ERA procedures and for the protocol that uses NaBH4. It is worth noting that the excessive use of work-up solvents in the ERA procedures is mainly due to the washing procedures needed to remove the supporting electrolytes and their by-products.
In the flow procedure using Et3SiH, given that the authors considered a 100% recovery of the solvents, the E-factor value is only constituted by the excess of the amine and the hydrosilane (Fig. 4). The Ecoscale analysis revealed that, besides the price/availability and safety scores that are almost equal for all the procedures selected and for our protocol, chromatographic separation is the most crucial process that worsens all the values. Additional penalty points are also slightly significant for the chemical routes due to the heating or cooling of the reaction mixture.
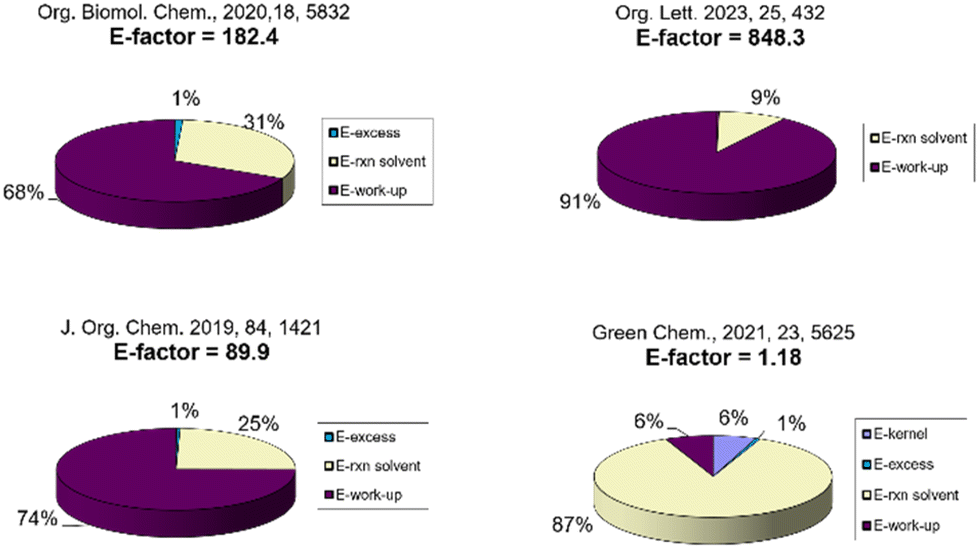 | ||
| Fig. 4 E-Factor and Ecoscale assessment of the literature processes for reductive amination. | ||
An additional level of evaluation was performed by considering the reducing agent safety and environmental index. Chemical reductants, NaBH4 and triethylsilane (Et3SiH) were compared with the supporting electrolytes needed to allow the passage of the electron current in the electrochemical protocols (Scheme 3). The comparisons were based on the Ω values calculated using Andraos algorithms.17 Four categories have been considered for the safety/hazard evaluation: Risk Phrase Potential (RPP), Explosive Vapour Potential (XVP), Flammability Potential (FP) and Hydrogen Generation Potential (HGP), and four categories have been considered for the environmental evaluation: Global Warming Potential (GWP), Bioaccumulation Potential (BAP), Bioconcentration Potential (BCP) and Abiotic Resource Depletion Potential (ARDP).
 | ||
| Scheme 3 Safety hazard and environmental index of the reductant used in the reductive amination process. | ||
As expected, NaBH4 and Et3SiH are associated with higher values in terms of safety and hazard parameters. The magnitude of these values, besides the flammability potential and GSH risk potential, is primarily due to their explosive potential and hydrogen generation potential, which are not associated with the electrolytes used in electrochemical procedures. However, in a stark contrast, NaBH4 has the lowest environmental Ω value, indicating the best environmental profile. On the other hand, triethylsilane has the worst environmental scores, largely due to its tendency to bioaccumulate and bioconcentrate. In general, the cumulative safety/hazard and environmental data for electrolytic substances show lower values compared to chemical reductants. To complete this evaluation, we included the Life Cycle Assessment (LCA) analysis, which provides a cradle-to-gate analysis (Table 4) of the electrolytes and reducing agent, focusing on the distribution of electricity utilization impact on the damage areas.
| Substance | mPts | Electricity impact (mPts) | mPts per damage areas | |
|---|---|---|---|---|
| a Electricity impact cannot be separated as the inventory is taken directly from Ecoinvent. b Data refer to the substances and not to the electricity component. | ||||
| TBAHSO4 | 4.86 | 4.08 (83.9%) | Human health | 3.89 (95.34%) |
| Ecosystems | 0.137 (3.36%) | |||
| Resources | 0.0501 (1.23%) | |||
| TBABF4 | 5.31 | 4.69 (88.4%) | Human health | 4.48 (95.52%) |
| Ecosystems | 0.158 (3.37%) | |||
| Resources | 0.0577 (1.23%) | |||
| NaBH4 | 1.36 | —a | Human healthb | 1.3 (95.6%) |
| Ecosystemsb | 0.0417 (3.07%) | |||
| Resourcesb | 0.0175 (1.29%) | |||
| Et3SiH | 30.4 | 22.26 (73.1%) | Human health | 21.16 (95.10%) |
| Ecosystems | 0.749 (3.37%) | |||
| Resources | 0.273 (1.24%) | |||
| TEABr (this work) | 2.89 | 2.4 (82.9%) | Human health | 2.29 (95.42%) |
| Ecosystems | 0.0808 (3.37%) | |||
| Resources | 0.0295 (1.23%) | |||
| Aluminium (primary) | 0.386 | —a | Human health | 2.29 (95.42%) |
| Ecosystems | 0.0808 (3.37%) | |||
| Resources | 0.0295 (1.23%) | |||
| Aluminium (secondary) | 0.0014 | —a | Human healthb | 0.0013 (93.5%) |
| Ecosystemsb | 0.00056 (4%) | |||
| Resourcesb | 0.00035 (2.54%) | |||
At first instance, we noted that the endpoint single score values of the LCA analysis (Table 4) perfectly confirm the environmental score (environment Ω) trend calculated using Andraos algorithms with Et3SiH and NaBH4, respectively, as the highest and lowest impacting substances. Gratifyingly, among the electrolytes, TEABr possesses a lower impact compared to TBABF4 and TBAHSO4. As a common behaviour, the electricity accounts for the majority of all the LCA endpoint values calculated (ranging from 73% to 88%). Through the analysis of the endpoint characterization (see the ESI† for details) of the non-electricity portion (Scheme 4), it is also possible to highlight that the major influence on the LCA endpoint score value of Et3SiH is ascribable to the usage of ethyl magnesium bromide in its production with minor contribution of the trichlorosilane. Regarding the electrolytes, in the cases where the alkylammonium source has a dominant contribution in the total value (TBABF4 and TBAHSO4 with 90% and 97% contribution, respectively), the LCA scores worsen, while when the alkylammonium source is less relevant, as for TEABr (40% of contribution), the LCA value is better. To provide a complete picture of the LCA analysis, we finally included the data relating to aluminium (the anode material used in our procedure).
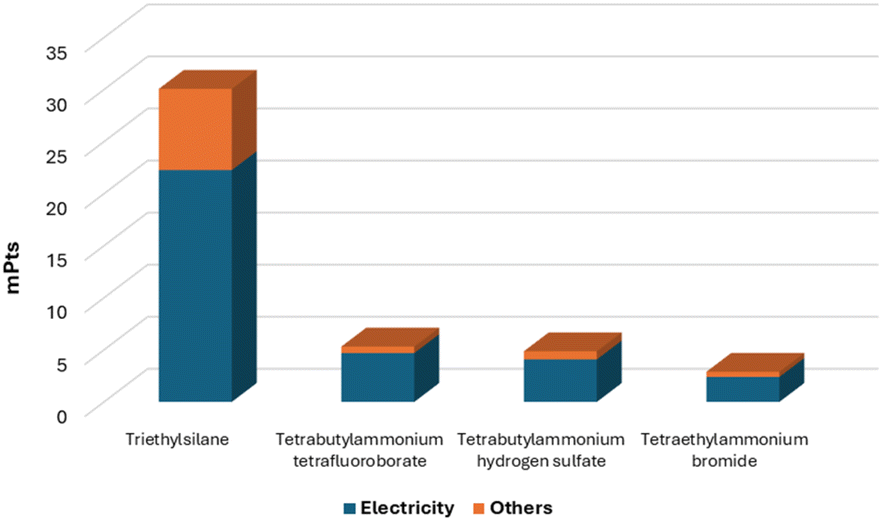 | ||
| Scheme 4 Distribution of the endpoint single score values (mPts) into electricity and non-electricity contributions. | ||
It is worth noting that the cumulative LCA score values (given by the sum of the two endpoint values), currently lower than those reported for Et3SiH or electrolytes, could be further reduced. This reduction could be achieved by using secondary aluminium as the anode material, indicating a promising trajectory for further improvements.22
To summarize the four-level sustainability assessment, Table 5 shows that better E-factor values are associated with the procedure developed by Kappe and our newly developed electrochemical reductive amination.
In terms of Ecoscale, our procedure features the best score, while the Kappe flow protocol and the ERA that uses TBAHSO4 also have good scores.
The safety hazard and environmental cumulative score shows that the TEABr used in our electrochemical protocol is by far associated with the lowest environmental impact and hazard.
The LCA analysis performed shows that NaBH4 possesses the best environmental profile among the substances examined, owing to the lower Endpoint single score value, while Et3SiH clearly possesses the worst.
The TEABr used in our electrochemical reductive amination instead obtained the lowest endpoint value among the electrolytes which is by far much lower compared to Et3SiH.
To further demonstrate the utility of our synthetic protocol, the synthesis of a pharmaceutically relevant product clobenzorex was performed (Scheme 5).
 | ||
| Scheme 5 Clobenzorex synthetic approach and our approach via ERA. Reaction conditions: 1m (3 mmol), 2j (3 mmol), tetraethylammonium bromide (3 mmol), aluminium cathode and graphite cathode, MeCN:H2O (az.) (3 mL), 300 mA until a 2.75 F mol−1 charge is passed (ca. 45 minutes). The product has been isolated as a hydrochloride salt. | ||
This compound is an anorexigenic drug that is widely used for the treatment of obesity since it helps to reduce body weight.23 Although a few examples are reported in the literature, the main procedures for obtaining clobenzorex via traditional chemistry are through reductive amination,24 using sodium borohydride as a reducing agent, or through nucleophilic substitution using benzyl chloride25 (Scheme 5).
With the use of our electrochemical procedure, clobenzorex can be obtained with a yield of 78% and an E-factor associated with the overall process of 3.69.
Experimental section: recovery procedure
In a 50 mL flask equipped with a magnetic stirrer, an ice-bath and a two-electrode system (aluminium anode and graphite cathode), tetraethylammonium bromide (15 mmol), aldehyde 1c (30 mmol), amine 2a (30 mmol) and 30 mL of an azeotropic mixture of acetonitrile:water (84% w/w acetonitrile) were consecutively added and the resulting mixture was electrolyzed in CCE at 1.5 A under stirring at room temperature until a 3 F mol−1 charge was passed (ca. 100 minutes). After reaction completion, the mixture was distilled using a simple distillation apparatus. The acetonitrile:water azeotrope was recovered in 82% yield. The residue was then diluted in 100 mL of ethyl acetate, and then it was filtered through cotton using compressed air to speed up the process. The obtained mixture was then distilled, and ethyl acetate was recovered in 85% yield, giving the product 3c as a yellow oil (78% yield).
Conclusions
In conclusion, this work proves that, with proper optimization, electrochemistry can be efficient from the green chemistry point of view in the reductive amination reaction. The acetonitrile–water azeotrope was seen to be a useful medium for this process, allowing its recovery and enhancing the mass efficiency of the protocol to an excellent level. In line with our recent interest in the development of easily removable and eventually recoverable26 electrolytes, herein we demonstrated that TEABr not only allows its smooth removal from the reaction mixture, minimizing the waste associated with the process, but is also a good environmentally friendly and safer choice compared to other electrolytes previously used in ERA procedures.Moreover, the adoption of highly concentrated conditions allowed us to obtain different substituted amines with low E-factor values. To show the overall significance of our protocol, a comprehensive four-level sustainability assessment of hazard, environmental and mass metrics has been performed, together with LCA analysis. This thorough assessment provides reassurance about the safety and efficiency of our research. The applicability of our protocol was also proven by the one-step synthesis of clobenzorex API in a high yield and with a very low E-factor.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. S. T. C.: investigation, methodology, data analysis, and manuscript preparation–review; F. C.: LCA analysis and manuscript review; F. F. and L. V.: conceptualization, project administration and manuscript review/editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge Università degli Studi di Perugia and MUR for support within the project Vitality. The University of Perugia is acknowledged for financial support to the university project “Fondo Ricerca di Ateneo, edizione 2022”. MUR is also thanked for the PRIN-PNRR 2022 project P2022XKWH7 – CircularWaste.References
- (a) T.-L. Chen, H. Kim, S.-Y. Pan, P.-C. Tseng, Y.-P. Lin and P.-C. Chiang, Sci. Total Environ., 2020, 716, 136998 CrossRef CAS PubMed; (b) A. Ncube, S. Mtetwa, M. Bukhari, G. Fiorentino and R. Passaro, Energies, 2023, 16, 1752 CrossRef CAS.
- (a) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS; (b) T. Hardwick, A. Qurashi, B. Shirinfar and N. Ahmed, ChemSusChem, 2020, 13, 1967–1973 CrossRef PubMed; (c) N. E. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2021, 122, 2487–2649 CrossRef; (d) K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed; (e) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- (a) C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC; (b) G. M. Martins, G. C. Zimmer, S. R. Mendes and N. Ahmed, Green Chem., 2020, 22, 4849–4870 RSC; (c) Y. H. Budnikova, E. L. Dolengovski, M. V. Tarasov and T. V. Gryaznova, J. Solid State Electrochem., 2023, 28, 659–676 CrossRef; (d) N. Sbei, T. Hardwick and N. Ahmed, ACS Sustainable Chem. Eng., 2021, 9, 6148–6169 CrossRef CAS; (e) C. Zhu, N. W. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS.
- (a) R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC; (b) F. Ferlin, G. Brufani, G. Rossini and L. Vaccaro, Green Chem., 2023, 25, 7916–7933 RSC; (c) W. J. Watson, Green Chem., 2012, 14, 251–259 RSC; (d) R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- (a) S. Van Praet, G. Preegel, F. Rammal and B. F. Sels, ACS Sustainable Chem. Eng., 2022, 10, 6503–6508 CrossRef CAS; (b) T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS; (c) V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed; (d) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed; (e) D. Campagna and R. Göstl, Angew. Chem., Int. Ed., 2022, 61, e202207557 CrossRef CAS PubMed.
- (a) O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS; (b) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS; (c) Z. Li, H. Zhang, T. Tan and M. Lei, Catal. Sci. Technol., 2022, 12, 5679–5686 RSC; (d) T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- (a) A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed; (b) E. Podyacheva, O. I. Afanasyev, A. A. Tsygankov, M. Makarova and D. Chusov, Synthesis, 2019, 2667–2677 CAS; (c) K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC; (d) M. Elfinger, T. Schönauer, S. L. Thomä, R. Stäglich, M. Drechsler, M. Zobel, J. Senker and R. Kempe, ChemSusChem, 2021, 14, 2360–2366 CrossRef CAS PubMed; (e) D. Wei, A. Bruneau-Voisine, D. A. Valyaev, N. Lugan and J.-B. Sortais, Chem. Commun., 2018, 54, 4302–4305 RSC; (f) M. Elfinger, C. Bauer, J. Schmauch, M. Moritz, C. Wichmann, C. Papp and R. Kempe, Adv. Synth. Catal., 2023, 365, 4654–4661 CrossRef CAS; (g) Z. Liu, F. Huang, M. Peng, Y. Chen, X. Cai, L. Wang, Z. Hu, X. Wen, N. Wang, D. Xiao, H. Jiang, H. Sun, H. Liu and D. Ma, Nat. Commun., 2021, 12, 6194 CrossRef.
- (a) T. Pienemann and H.-J. Schäfer, Synthesis, 1987, 1005–1007 CrossRef CAS; (b) R. J. Cvetovich, J. S. Amato, L. DiMichele, L. Weinstock and G. Hazen, J. Org. Chem., 1997, 62, 6697–6698 CrossRef CAS; (c) D. K. Root and W. H. Smith, J. Electrochem. Soc., 1982, 129, 1231–1236 CrossRef CAS.
- (a) J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC; (b) Z. J. Schiffer, M. Chung, K. Steinberg and K. Manthiram, Chem. Catal., 2023, 3, 100500 CrossRef CAS; (c) H. Hong, Z. Zou, G. Liang, S. Pu, J. Hu, L. Chen, Z. Zhu, Y. Li and Y. Huang, Org. Biomol. Chem., 2020, 18, 5832–5837 RSC; (d) Y. Fan, W. Ou, M. Chen, Y. Liu, B. Zhang, W. Ruan and C. Su, Org. Lett., 2023, 25, 432–437 CrossRef CAS PubMed; (e) T. Kim, D. I. Park, S. Kim, D. Yadav, S. Hong, S. H. Kim, H. J. Yoon and K. Jin, Chem. Commun., 2023, 59, 4818–4821 RSC.
- (a) V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC; (b) J. D. Chea, A. L. Lehr, J. P. Stengel, M. J. Savelski, C. S. Slater and K. M. Yenkie, Ind. Eng. Chem. Res., 2020, 59, 5931–5944 CrossRef CAS; (c) R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC; (d) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS; (e) B. Nanda, M. Sailaja, P. Mohapatra, R. K. Pradhan and B. B. Nanda, Mater. Today: Proc., 2021, 47, 1234–1240 CAS.
- R. K. Henderson, C. Jiménez-González, D. J. Constable, S. R. Alston, G. G. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC.
- https://19january2017snapshot.epa.gov/global-mitigation-non-co2-greenhouse-gases/global-mitigation-non-co2-greenhouse-gases-solvents_.html .
- (a) F. Valentini and L. Vaccaro, Molecules, 2020, 25, 5264 CrossRef CAS; (b) F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS; (c) F. Valentini, F. Ferlin, E. Tomarelli, H. Mahmoudi, M. Bagherzadeh, M. Calamante and L. Vaccaro, ChemSusChem, 2021, 14, 3359–3366 CrossRef CAS; (d) N. Salameh, F. Ferlin, F. Valentini, I. Anastasiou and L. Vaccaro, ACS Sustainable Chem. Eng., 2022, 10, 3766–3776 CrossRef CAS; (e) F. Ferlin, V. Trombettoni, L. Luciani, S. Fusi, O. Piermatti, S. Santoro and L. Vaccaro, Green Chem., 2018, 20, 1634–1639 RSC; (f) U. Azzena, M. Carraro, A. D. Mamuye, I. Murgia, L. Pisano and G. Zedde, Green Chem., 2015, 17, 3281–3284 RSC; (g) J. Li and T. Wang, Chem. Eng. Process., 2010, 49, 530–535 CrossRef CAS; (h) E. Haaz, B. Szilagyi, D. Fozer and A. J. Toth, Front. Chem. Sci. Eng., 2020, 14, 913–927 CrossRef CAS.
- (a) A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843–3876 RSC; (b) T. S. Zhao and C. Xu, Encyclopedia of Electrochemical Power Sources, 2009, pp. 381–389 Search PubMed.
- (a) R. A. Sheldon, ACS Sustainable Chem. Eng., 2017, 6, 32–48 CrossRef; (b) K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 1–7 CrossRef; (c) M. Tobiszewski, M. Marć, A. Gałuszka and J. Namieśnik, Molecules, 2015, 20, 10928–10946 CrossRef CAS PubMed.
- (a) J. Andraos, Org. Process Res. Dev., 2012, 16, 1482–1506 CrossRef CAS; (b) J. Andraos, Org. Process Res. Dev., 2013, 17, 175–192 CrossRef CAS.
- S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2020, 121, 485–505 CrossRef.
- F. W. Lucas, Y. Fishler and A. Holewinski, Green Chem., 2021, 23, 9154–9164 RSC.
- L.-Y. Fu, J. Ying, X. Qi, J.-B. Peng and X.-F. Wu, J. Org. Chem., 2019, 84, 1421–1429 CrossRef CAS.
- M. Wernik, G. Sipos, B. Buchholcz, F. Darvas, Z. Novák, S. B. Ötvös and C. O. Kappe, Green Chem., 2021, 23, 5625–5632 RSC.
- S. Trastulli Colangeli, F. Ferlin and L. Vaccaro, Green Chem., 2024, 26, 8030–8036 RSC.
- (a) G. D. Apóstol del Rosal, I. D. Limón, I. Martínez and A. Patricio-Martínez, Neurotoxic. Res., 2021, 39, 1405–1417 CrossRef; (b) F. Argüelles-Tello, M. del Carrasco-Portugal, N. A. Carrasco-Portugal, J. C. Aguilar-Carrasco, S. I. Patiño-Camacho, C. F. Valle, G. Reyes-Garcia and F. J. Flores-Murrieta, Pharmacol. Pharm., 2013, 04, 218–221 CrossRef.
- T. Noggle, C. R. Clark, S. V. Andurkar and J. de Ruiter, J. Liq. Chromatogr., 1990, 13, 763–777 CrossRef.
- R. Young, Pharmacol., Biochem. Behav., 1997, 56, 311–316 CrossRef CAS.
- F. Ferlin, F. Valentini, F. Campana and L. Vaccaro, Green Chem., 2024, 26, 6625–6633 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures, full characterization of the synthesized compounds and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4gc04847d |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |