
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenated non-innocent vanadium(V) Schiff base complexes: chemical and anti-proliferative properties†‡
Allison A.
Haase
 a,
Skyler A.
Markham
a,
Heide A.
Murakami
a,
John
Hagan
a,
Kateryna
Kostenkova
a,
Jordan T.
Koehn
a,
Canan
Uslan
bc,
Cheryle N.
Beuning
a,
Lee
Brandenburg
a,
Joseph M.
Zadrozny
a,
Aviva
Levina
bc,
Peter A.
Lay
*bc and
Debbie C.
Crans
*ad
a,
Skyler A.
Markham
a,
Heide A.
Murakami
a,
John
Hagan
a,
Kateryna
Kostenkova
a,
Jordan T.
Koehn
a,
Canan
Uslan
bc,
Cheryle N.
Beuning
a,
Lee
Brandenburg
a,
Joseph M.
Zadrozny
a,
Aviva
Levina
bc,
Peter A.
Lay
*bc and
Debbie C.
Crans
*ad
aDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA
bSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: peter.lay@sydney.edu.au
cSydney Analytical, The University of Sydney, Sydney, NSW 2006, Australia
dThe Cell and Molecular Biology Program, Colorado State University, Fort Collins, CO 80523, USA. E-mail: Debbie.Crans@colostate.edu
First published on 30th May 2024
Abstract
A series of non-innocent halogen substituted Schiff base vanadium catecholates were added with different electron donating and withdrawing substituents on the catecholates to investigate the electronic effects on the properties of this class of compounds. We hypothesized that the electronic changes would be reflected in their redox properties and stabilities, which should lead to differences in their biological properties. Using UV-spectroscopy, we measured their hydrolytic stability, and using electrochemistry, we characterized their redox properties. Adding one substituent on the catecholate group on the complexes changed the redox potentials of the complexes; however, less impact on the hydrolytic stability was observed. We discovered that hydrolytic stability was crucial to the anti-proliferative effects on glioblastoma cells, and most of these compounds had effects similar to vanadates regardless of their different redox properties. Hence, we could not determine the importance of changing the electronic properties and redox potential on the anti-proliferative effects of mono-substituted catecholates. However, the studies did show that the pKa of the substituted catecholate showed a linear correlation with the redox potential of the non-innocent Schiff base vanadium complexes, which will be important in future investigations into this class of complexes.
Introduction
Metal complexes incorporating Schiff bases have shown and continue to be developed for a wide variety of applications, from catalysis1,2 to bioactive compounds with typical metal ions used for the bioactive complexes spanning Cu(II), Co(II), Fe(II), and Ni(II).3–6 Non-innocent Schiff base vanadium-catecholates (V-catecholates) have interesting electronic properties with the potential for redox taking place not only on the metal center but also on the ligand.3,7–9 The subclass of non-innocent Schiff base V-catecholates have recently been discovered to have desirable pharmaceutical properties3–6,8–16 which has expanded the knowledge on anti-diabetic and anti-cancer properties of vanadium complexes.3,4,6,8–18 The reported antiproliferative properties of the class of non-innocent Schiff base V-catecholates were reported to surpass the efficacy of cisplatin in human chondrosarcoma (SW1353) bone cells, T98G glioblastoma cells, aggressive mesenchymal-like MDA-MB-231 cells, A549 (lung) and PANC-1 (pancreas) cancer cell lines, and HFF-1 (human foreskin fibroblasts) normal cells. The efficacy of these molecules elicited proposals for intratumoral injections19 as a viable treatment because their potential properties enable intact complexes for cellular uptake which then hydrolyze over time into much less toxic components.8,9,20 The key properties of the most potent anti-cancer complexes from this series are complexes that contain a sterically hindered catechol and high hydrophobicity.8,9,20–22 However, little is known about the effects of electronics on the biological properties of the compounds. Considering the reported effects of electronics on the HOMO–LUMO gap,7 we set out to investigate the effects of a range of substituents on complex stability, biological activity, and electrochemistry as the electronic substituents were changed.The electronic properties of vanadium coordination complexes have been investigated using solid and solution 51V NMR spectroscopy, X-ray absorption spectroscopy, and DFT calculations.7,23–25 Studies were undertaken with non-innocent Schiff base vanadium catecholate complexes when the Schiff base was (N-(salicylideneaminato)-N′-(2-hydroxyethyl)-1,2-ethanediamine), abbreviated Hshed.7 This class of complexes were first described as [VVO(Hshed)(catecholate)] where catecholate changes moved 51V NMR chemical shifts from −200 to 400 ppm.26 The NMR parameters in these vanadium complexes are highly sensitive to the local environment because 51V is a half-integer (I = 7/2) quadrupolar nucleus.7 The studies demonstrate that substituting the catechol ligand with electron-donating groups results in an increase in the HOMO–LUMO gap and can be directly followed by an upfield shift for the 51V signal. In contrast, the substitution of the catechol ligand with electron-withdrawing groups results in a decrease in the HOMO–LUMO gap and can directly be followed by a downfield shift for the 51V NMR peak.
We designed and synthesized a series of non-innocent Schiff base vanadium catecholates with different electron donating and withdrawing substituents shown in Fig. 1. We hypothesized that the electronic changes would be reflected in their redox properties and their stabilities, which should lead to differences in their biological properties. Previously, we discovered that chloro-substitution of the Schiff base on the vanadium complexes increased stability, hydrophobicity, and reduction of the complex compared to the parent Schiff base vanadium(V) complexes9 and chose to investigate chloro-substituted Schiff base complexes because of their increased stability. Herein, we describe the synthesis and characterization of the chemical and biological properties of a series of new complexes to determine if changes in electronic properties of the catecholato ligand in the non-innocent vanadium catecholate complex are being reflected in their effects on glioblastoma cells.
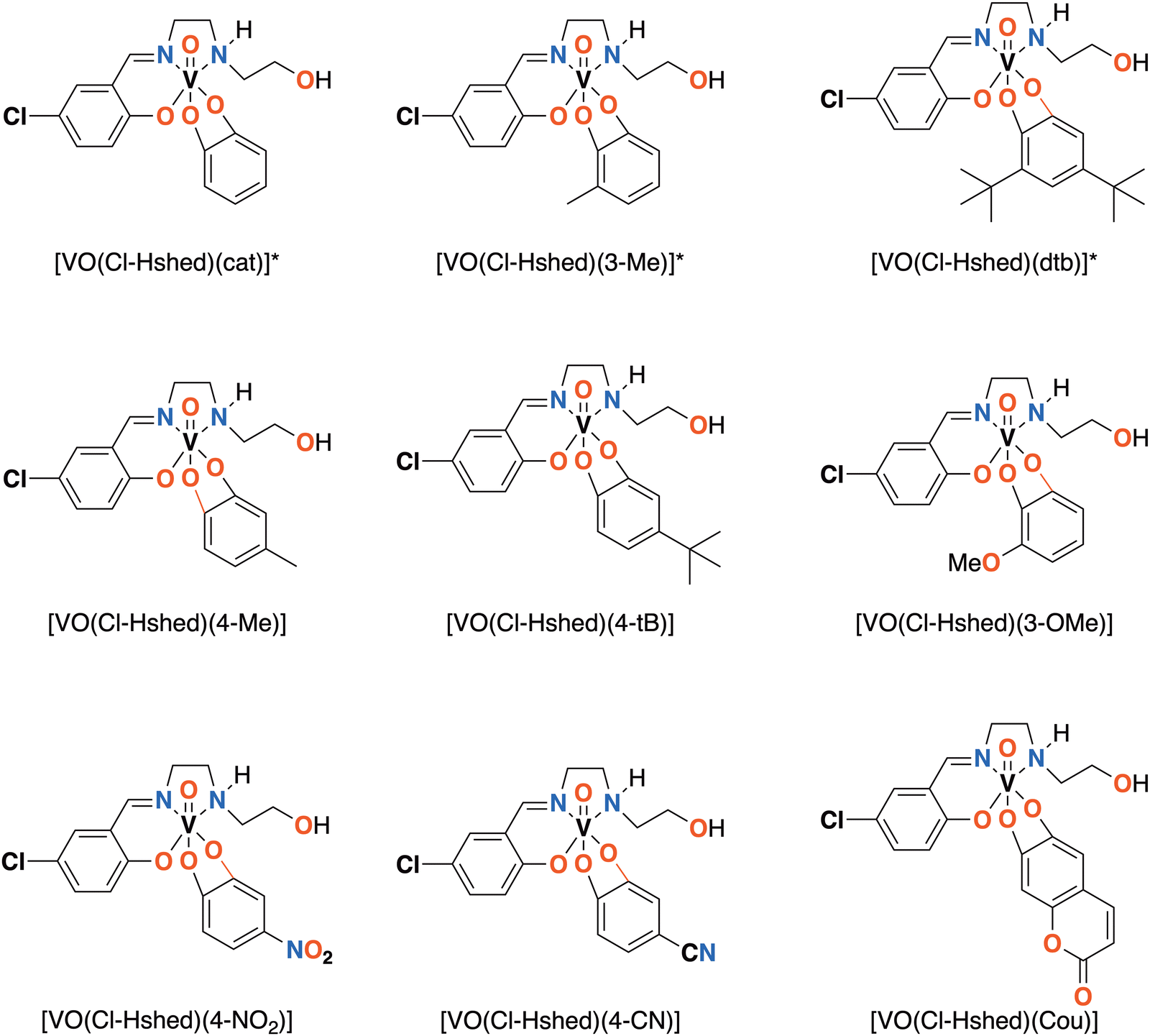 | ||
| Fig. 1 Novel [VO(Cl-Hshed)] structures presented in this paper. *Compounds previously synthesized and reported.9 | ||
Experimental
General materials
Vanadyl sulfate, 4-nitrilecatechol, 4-methylcatechol, coumarincatechol, 3-methoxycatechol, 4-nitrocatechol, 4-tertbutylcatechol were purchased from Sigma Aldrich and used without further purification (all greater than 99% purity at purchase). ACS-reagent-grade ethyl acetate, acetone, and n-hexane were also purchased from Sigma-Aldrich. Argon gas used for the Schlenk line work was of ultra-high purity and was purchased from Airgas. The [VO2(Cl-Hshed)] precursor was synthesized, as described previously using Schlenk techniques9,19,26 and the syntheses of known derivates were optimized and their characterization compared to those reported previously.9 Six new compounds are described in detail in this work. In the ESI,‡ figures and tables detailing the properties of these compounds are summarized.Dimethyl sulfoxide (DMSO) was purchased from Millipore-Sigma. Salts for the phosphate buffer solution (PBS) were purchased from Fischer Scientific, and included sodium phosphate dibasic, potassium phosphate monobasic, sodium chloride, potassium chloride, calcium chloride dihydrate, and magnesium sulfate. Electrochemical grade tetra-n-butylammonium perchlorate (TBAP) and silver nitrate (AgNO3) were purchased from Millipore-Sigma. Ferrocene was purchased from Alfa Aesar. Ultrapure argon (AR UHP300) from Airgas was used to degas solutions. All chemicals were used as provided without further purification. Dry acetonitrile (CH3CN) was used as the solvent for non-aqueous electrochemistry experiments because of its large potential window and the compound solubility and relative stability.9 All synthesized compounds (both solids and solutions) were stored in a −20 °C freezer or a desiccator before being used in electrochemical experiments. TBAP was used as a supporting electrolyte at 100 mM in CH3CN.
General methods and instrumentation
All compounds were characterized by routine methods including multi-nuclear NMR spectroscopy, IR spectroscopy, UV-vis-NIR spectrophotometry, and mass spectrometry or elemental analysis (EAI, Lexington, KY).NMR spectroscopy
NMR spectra were recorded at ambient temperature using a Bruker NEO400 NMR spectrometer equipped with a Prodigy™ BBFO probe and SampleCase™. The instrument was located at the Analytical Resources Core – Materials and Molecular Analysis Center at Colorado State University. The NMR spectrometer was set at 400.13 MHz for 1H and 105.2 MHz for 51V spectra. The 1H NMR spectroscopic experiments were performed using standard parameters at ambient temperature (26 °C) by running 32 scans and were locked onto the deuterated solvent (DMSO-d6). The 1D 1H NMR spectra in DMSO-d6 were referenced to DMSO's internal residual solvent peak at 2.50 ppm. The parameters used for the 51V NMR spectra were as follows: 4096 scans in the f1 domain, spectral window from −320 to −780 ppm, 0.01 s relaxation delay, 45° pulse angle, and 16 µs pulse. The 51V NMR spectra were reported relative to a neat VOCl3 solution at 0.00 ppm and referenced against an external reference of an aqueous NaVO4 solution at pH 12 that produced two signals at −535.7 ppm (vanadate monomer, HVO42−) ppm and −560.4 ppm (vanadate dimer, HV2O73−) relative to VOCl3.27,28 The data were processed using MestreNova NMR processing software (version 14.0.1).UV-vis-NIR spectrophotometry
The AvaLight UV/Vis/NIR light source and AvaSpex-UL S2048 fiber-optic spectrometer were used for characterization. AfterMath v 1.5.9807 was the software used during data acquisition, and Microsoft Excel, OriginPro 2024 (64-bit) SR1 10.1.0.178 (student version) were used for analysis.Stability studies
A 10× PBS stock solution was made using double deionized water (DDI) water with 1 mM CaCl2·2H2O, 1 mM MgSO4, 100 mM Na2HPO4, 17.6 mM KH2PO4, 1.37 M NaCl, and 26.8 mM KCl. PBS stocks were diluted to 1× and pH adjusted to 7.4 before mixing with DMSO. The spectra were recorded at 0.10 mM and prepared from a concentrated stock solution made from the compound dissolved in DMSO. Spectra were taken once every minute for the first hour then at the 2-, 4-, and 24-hour marks for the DMSO, and at every minute for 1 hour for the mixed solutions.Electrochemistry
The potentiostat used was a WaveDriver 40 DC Bipotentiostat/Galvanostat purchased from Pine Research. A typical three electrode setup was used for all electrochemical experiments. All electrodes were purchased from BASi Research products. The working electrode was a glassy carbon electrode (GC) with a 3.0 mm diameter. The counter electrode was a 7.5-cm platinum wire electrode. A non-aqueous silver/silver ion (Ag+/Ag) reference electrode was used, which consisted of a 30 cm long Ag wire with a 0.5 mm diameter, contained in a double-junction reference electrode chamber with 1/8″ long Porous CoralPor™ Tips. All electrochemical data were collected using Aftermath (ver. 1.5.9807) without iR compensation. Microsoft Excel, and OriginPro 2024 (64-bit) SR1 10.1.0.178 (Student Version) were used for further data processing and analysis.Samples of each complex (10 mL, 2 mM) were prepared in triplicate. The external standard ferrocene and precursor complex [VO2(Cl-Hshed)] was also at a 2 mM concentration, while the concentration of free catechol ligand was approximately 5 mM. Each cyclic voltammetry (CV) sample was bubbled with argon for 10 min to remove ambient water and oxygen. The refill solution for the reference electrode was made using dry CH3CN with TBAP (100 mM) and AgNO3 (10 mM). All electrodes were cleaned between each collection. Most CV experiments were run where the initial voltage was 1.0 V, the vertex was −1.5 V, and the final voltage was 1.0 V with a 100 mV s−1 sweep rate. Sometimes this voltage range was shortened, mainly when hysteresis experiments were run after the half-wave potential for the compound had already been identified.
Mass spectrometry
The high-resolution positive-ion ESI-MS was recorded on a Thermo Velos Pro Orbitrap mass spectrometer. The resolving power used was 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at 200 m/z. Before any analysis, the instrument was externally calibrated and internally calibrated by locking on dioctyl phthalate. Analysis solutions (∼10 µM V in acetonitrile) were injected using a syringe pump at a flow rate of 8 µL per min. The spectra obtained were averages of 100–200 scans (scan time, 100 ms). The isotopic patterns were simulated using Bruker Compass Data Analysis 5.0 software.
000 at 200 m/z. Before any analysis, the instrument was externally calibrated and internally calibrated by locking on dioctyl phthalate. Analysis solutions (∼10 µM V in acetonitrile) were injected using a syringe pump at a flow rate of 8 µL per min. The spectra obtained were averages of 100–200 scans (scan time, 100 ms). The isotopic patterns were simulated using Bruker Compass Data Analysis 5.0 software.
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P, polarizability and pKa calculations
P, polarizability and pKa calculations
Chemicalize (https://chemicalize.com/, developed by ChemAxon, used in November 2023) was used for the prediction of structural properties, and to estimate the pKa, lipophilicity, logP, solubility, and molecular geometry for the free catechol ligands and the vanadium complexes. Since the program is designed for organic structures, the calculations are best for the catechol ligands and less reliable for the metal complexes. Most estimated are provided in the ESI‡ (Fig. S6a–d).
Cell culture studies
The human glioblastoma multiforme cell line (T98G) was obtained from the American Type Culture Collection (ATCC, cat. no. CRL-1690). The cells were grown in Advanced DMEM (Thermo Fisher Scientific cat. no. 12491-015), supplemented with 1% vol. L-Glutamine (2.0 mM stock), 1% vol. antibiotic–antimycotic (a mixture of 100 U mL−1 penicillin, 100 mg mL−1 streptomycin, and 0.25 mg mL−1 amphotericin B), and heat-inactivated fetal calf serum (FCS; 2% vol). For proliferation experiments, cells were seeded in 96-well plates at an initial density of 1.5 Å ∼103 viable cells per well in 100 µL medium; they were left to attach to the plates overnight. Freshly prepared 10 mM stock solutions in DMSO of V(V) complexes were further diluted with DMSO so that all the cell treatments, including controls, contained 1.0% (vol) of DMSO. Previous studies9,21,29 have demonstrated that 1% DMSO did not affect the cell growth during the assays. Solutions of the treatment complexes in DMSO were diluted 100-fold with fully supplemented cell culture media. The resultant media were either added to the cells within 30 s (fresh solutions) or left in cell culture incubator (310 K, 5% CO2) for 24 h prior to the cell treatments (aged solutions).9,21,29 Generally, the V(V) complexes treating the cells were added in a series of nine two-fold dilutions, starting at 100 µM V.Treatments consisted of six replicate wells with cells and two background wells without cells that contained the same assay components. After the treatment by the V(V) complexes, the plates were incubated for 72 h at 310 K and 5% CO2. To visualize the response the MTT reagent [1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan] from Sigma M5655 was added (50 µL per well of freshly prepared 2.0 mg mL−1 solution in assay solution), and the plate was incubated for another 4–6 h at which point the medium was removed. The blue formazan crystals were dissolved in 0.10 mL per well of DMSO, and the absorbance at 600 nm was measured using a Victor V3 plate reader. The calculation and fitting of the experimental data to determine the IC50 values were performed using Origin 6.1 software (Microcal Origin, 1999). For the data to be of publishable quality, consistent results were obtained in at least two independent experiments that employed different passages of cells and varying stock solutions of the treatment complexes.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching: 943 cm−1, UV-vis-NIR, λmax: 546 nm (ε 5.7 × 103), 868 nm (ε 7.2 × 103). 1H (DMSO-d6 of major isomer): 8.75 ppm (s, 1H), 7.61 ppm (s, 1H), 7.41 ppm, (d, 1H), 6.74 ppm (d, 1H), 6.37 ppm (m, 1H), 6.17 ppm (s, 1H), 4.68–4.74 ppm (m, 2H), 4.15 ppm (d, 1H), 3.98 ppm (d, 1H), 3.66–3.72 ppm (m, 1H), 3.53–3.58 ppm (m, 1H), 3.44–3.50 ppm (m, 2H), 3.27 ppm (m, 3H), 2.83–2.86 ppm (d, 1H). 51V (DMSO-d6): 419 ppm, 401 ppm, 365 ppm, 346 ppm (major). Mass spec expected: +430.05 m/z. Obs.: +431.06 m/z.
O stretching, UV-vis-NIR λmax 539 nm (ε 6.8 × 103), 882 nm (ε 1.0 × 104). 1H (DMSO-d6 of major isomer): 8.76 ppm (s, 1H), 7.62 ppm (s, 1H), 7.42 ppm (d, 1H), 6.76 ppm (d, 1H), 6.63–6.70 ppm (m, 1H), 6.01 ppm (d, 1H), 5.94 ppm (d, 1H), 4.76 ppm (s, 1H), 4.13–4.21 ppm (m, 1H), 3.95–4.00 ppm (m, 1H), 3.80 (s, 3H), 3.64–3.73 ppm (s, 3H), 3.44–3.55 ppm (m, 3H), 3.33 ppm (s, 6H), 3.27 ppm (s, 3H), 2.80–2.86 ppm (m, 1H), 2.37 ppm (s, 1H). 51V (DMSO-d6): 400 ppm (major), 386 ppm, 315 ppm, 299 ppm. Mass spec expected: 472.10 m/z. Obs.: 473.11 m/z.
O stretching. UV-vis-NIR λmax 562 nm (ε 5.8 × 103), 831 nm (ε 5.0 × 103).1H (DMSO-d6 of major isomer): 8.75 ppm (s, 1H), 7.61 ppm (s, 1H), 7.41 ppm (d, 1H), 6.75 ppm (d, 1H), 6.40 ppm (s, 1H), 6.32 ppm (s, 1H), 4.77 ppm (m, 2H), 4.15 ppm (d, 1H), 3.97–4.01 ppm (m,1H), 3.70–3.74 ppm (m, 1H), 3.48–3.52 ppm (m, 1H), 3.27 ppm (m, 1H) 2.79–288 ppm (m, 1H). 51V (DMSO-d6): 410 ppm, 390 ppm, 363 ppm (major). Mass spec expected 446.04 m/z obs.: 447.05 m/z.
O stretching. UV-vis-NIR λmax 411 nm (ε 1.4 × 104), 770 nm (ε 4.1 × 103). 1H (DMSO-d6 of major isomers): 9.00 ppm (s, 1H), 7.81 ppm (m), 7.55–7.63 ppm (m), 7.36–7.44 ppm (m), 6.86–7.02 ppm (s, 1H), 6.91 ppm (m), 6.42 ppm (d, 1H), 6.22 ppm (d, 1H), 5.43 ppm (s, 1H), 4.30 ppm (m, 1H), 4.00–4.06 ppm (m, 2H), 3.75–3.80 ppm (m, 1H), 3.67–3.72 ppm (m, 2H), 3.54–3.60 ppm (m), 3.06 ppm (s, 1H), 2.86–3.00 ppm (m, 2H). 51V (DMSO-d6): −98 ppm, −174 ppm (major), −285 ppm, −525 ppm. Mass spec expected: 461.02 m/z. Obs. 462.03 m/z.
O stretching. 1H (MeCN-d3 of major isomer): 8.58 ppm (s, 1H), 7.53 ppm (m, 1H), 7.41 ppm (m, 1H), 7.05 ppm (m, 1H), 6.81 ppm, (m, 1H), 6.72 ppm (d, 1H), 4.45 ppm (s, 1H), 4.10–4.13 ppm (m, 1H), 3.96–4.01 ppm (m, 1H), 3.67 ppm (m, 1H), 3.56–3.59 ppm (m, 1H), 3.41 ppm (m, 1H), 3.27 ppm (m, 1H), 2.84–2.90 ppm (m, 1H), 2.71 (s, 1H). 51V (MeCN-d3) −73 ppm, −98 ppm (major). Mass spec expected: 441.03 m/z obs.: 429.15 m/z.
O stretching. UV-vis-NIR λmax 359 nm (ε 1.9 × 104), 524 nm (ε 6.4 × 103), 881 nm (ε 1.1 × 104). 1H (MeCN-d3 of the major isomer): 8.55 ppm (s, 1H), 7.60 ppm (d, 2H), 7.49 ppm (s, 1H), 7.38 ppm (d, 1H), 6.89 ppm (s, 1H), 6.72 ppm (s, 1H), 6.56 ppm (s, 1H), 6.16 ppm (s, 1H), 6.07 ppm (s, 1H), 5.88 ppm (d, 1H), 4.39 ppm (s, 1H), 4.08–4.11 ppm (m, 1H), 3.96–4.02 ppm (m, 1H), 3.68 ppm (m, 1H), 3.56–3.59 ppm (m, 1H), 3.48 ppm (m, 1H), 3.4 1 ppm (m, 1H), 3.27 ppm (m, 1H), 2.88–2.94 ppm (m, 2H), 2.72 ppm (s, 1H). 51V (MeCN-d3): 439 ppm, 410 ppm (major). Mass spec. expected: 484.02. Obs.: 485.03 m/z.
O stretching: 943 cm−1, UV-vis-NIR, λmax: 546 nm (ε 5.7 × 103), 868 nm (ε 7.2 × 103). 1H (DMSO-d6 of major isomer): 8.75 ppm (s, 1H), 7.61 ppm (s, 1H), 7.41 ppm, (d, 1H), 6.74 ppm (d, 1H), 6.37 ppm (m, 1H), 6.17 ppm (s, 1H), 4.68–4.74 ppm (m, 2H), 4.15 ppm (d, 1H), 3.98 ppm (d, 1H), 3.66–3.72 ppm (m, 1H), 3.53–3.58 ppm (m, 1H), 3.44–3.50 ppm (m, 2H), 3.27 ppm (m, 3H), 2.83–2.86 ppm (d, 1H). 51V (DMSO-d6): 419 ppm, 401 ppm, 365 ppm, 346 ppm (major). Mass spec expected: +430.05 m/z. Obs.: +431.06 m/z.
O stretching, UV-vis-NIR λmax 539 nm (ε 6.8 × 103), 882 nm (ε 1.0 × 104). 1H (DMSO-d6 of major isomer): 8.76 ppm (s, 1H), 7.62 ppm (s, 1H), 7.42 ppm (d, 1H), 6.76 ppm (d, 1H), 6.63–6.70 ppm (m, 1H), 6.01 ppm (d, 1H), 5.94 ppm (d, 1H), 4.76 ppm (s, 1H), 4.13–4.21 ppm (m, 1H), 3.95–4.00 ppm (m, 1H), 3.80 (s, 3H), 3.64–3.73 ppm (s, 3H), 3.44–3.55 ppm (m, 3H), 3.33 ppm (s, 6H), 3.27 ppm (s, 3H), 2.80–2.86 ppm (m, 1H), 2.37 ppm (s, 1H). 51V (DMSO-d6): 400 ppm (major), 386 ppm, 315 ppm, 299 ppm. Mass spec expected: 472.10 m/z. Obs.: 473.11 m/z.
O stretching. UV-vis-NIR λmax 562 nm (ε 5.8 × 103), 831 nm (ε 5.0 × 103).1H (DMSO-d6 of major isomer): 8.75 ppm (s, 1H), 7.61 ppm (s, 1H), 7.41 ppm (d, 1H), 6.75 ppm (d, 1H), 6.40 ppm (s, 1H), 6.32 ppm (s, 1H), 4.77 ppm (m, 2H), 4.15 ppm (d, 1H), 3.97–4.01 ppm (m,1H), 3.70–3.74 ppm (m, 1H), 3.48–3.52 ppm (m, 1H), 3.27 ppm (m, 1H) 2.79–288 ppm (m, 1H). 51V (DMSO-d6): 410 ppm, 390 ppm, 363 ppm (major). Mass spec expected 446.04 m/z obs.: 447.05 m/z.
O stretching. UV-vis-NIR λmax 411 nm (ε 1.4 × 104), 770 nm (ε 4.1 × 103). 1H (DMSO-d6 of major isomers): 9.00 ppm (s, 1H), 7.81 ppm (m), 7.55–7.63 ppm (m), 7.36–7.44 ppm (m), 6.86–7.02 ppm (s, 1H), 6.91 ppm (m), 6.42 ppm (d, 1H), 6.22 ppm (d, 1H), 5.43 ppm (s, 1H), 4.30 ppm (m, 1H), 4.00–4.06 ppm (m, 2H), 3.75–3.80 ppm (m, 1H), 3.67–3.72 ppm (m, 2H), 3.54–3.60 ppm (m), 3.06 ppm (s, 1H), 2.86–3.00 ppm (m, 2H). 51V (DMSO-d6): −98 ppm, −174 ppm (major), −285 ppm, −525 ppm. Mass spec expected: 461.02 m/z. Obs. 462.03 m/z.
O stretching. 1H (MeCN-d3 of major isomer): 8.58 ppm (s, 1H), 7.53 ppm (m, 1H), 7.41 ppm (m, 1H), 7.05 ppm (m, 1H), 6.81 ppm, (m, 1H), 6.72 ppm (d, 1H), 4.45 ppm (s, 1H), 4.10–4.13 ppm (m, 1H), 3.96–4.01 ppm (m, 1H), 3.67 ppm (m, 1H), 3.56–3.59 ppm (m, 1H), 3.41 ppm (m, 1H), 3.27 ppm (m, 1H), 2.84–2.90 ppm (m, 1H), 2.71 (s, 1H). 51V (MeCN-d3) −73 ppm, −98 ppm (major). Mass spec expected: 441.03 m/z obs.: 429.15 m/z.
O stretching. UV-vis-NIR λmax 359 nm (ε 1.9 × 104), 524 nm (ε 6.4 × 103), 881 nm (ε 1.1 × 104). 1H (MeCN-d3 of the major isomer): 8.55 ppm (s, 1H), 7.60 ppm (d, 2H), 7.49 ppm (s, 1H), 7.38 ppm (d, 1H), 6.89 ppm (s, 1H), 6.72 ppm (s, 1H), 6.56 ppm (s, 1H), 6.16 ppm (s, 1H), 6.07 ppm (s, 1H), 5.88 ppm (d, 1H), 4.39 ppm (s, 1H), 4.08–4.11 ppm (m, 1H), 3.96–4.02 ppm (m, 1H), 3.68 ppm (m, 1H), 3.56–3.59 ppm (m, 1H), 3.48 ppm (m, 1H), 3.4 1 ppm (m, 1H), 3.27 ppm (m, 1H), 2.88–2.94 ppm (m, 2H), 2.72 ppm (s, 1H). 51V (MeCN-d3): 439 ppm, 410 ppm (major). Mass spec. expected: 484.02. Obs.: 485.03 m/z.
Results and discussion
Syntheses of mono-substituted chloro Schiff base V(V) catecholate complexes
The syntheses of a series of chloro-Schiff base V(V) complexes with mono-substituted catecholates allowed the correlation of structures, spectroscopic properties, stabilities in organic solvents and PBS medium and their anti-proliferative activities against T98G glioblastoma cancer cells.9Methods previously reported for several different vanadium non-innocent Schiff base complexes range from 20 to 80%9,20,21,26 (for the three compounds reported previously [VO(Cl-Hshed)(cat)] – 80%, [VO(Cl-Hshed)(3-MeCat)] – 79%, [VO(Cl-Hshed)(dtb)] – 70%9 and for the parent complex [VO(Hshed)(cat)] – 40%26). In this work, we report a series of novel chloro-substituted complexes with mono-substituted catechols. Catecholates with a bulky aliphatic group such as t-butyl generated chloro substituted Schiff base complexes with yields around 70% in agreement with the literature report on corresponding parent complexes.9,26 However, initial syntheses of other substituted complexes generated significantly lower yields, on the order of 20–40%.26 We, therefore, optimized the procedures for these complexes by changing solvents, reagent ratios, reaction times, and temperature. As a result, the procedures reported in this manuscript have yields of these novel compounds in the range of 50 to 80%. Syntheses optimization showed that the parameters most important to obtain increased yields for most complexes were the nature of the reaction solvent and the reaction time.
Optimizations of the syntheses were initially tested on [VO(Cl-Hshed)(dtb)] by changing the reagent ratio, temperature, reaction time, concentration, and solvent. Changes in the reagent ratio from 1:1 to 1:1.2 did not increase yields and, since product could precipitate during the reaction, the increase in catechol only decreased product purity. Increases in the reaction temperature even from ambient temperature (23 °C) to 30 °C caused a dramatic decrease in yields. Reactions run at 0 °C and −78 °C led to pure products with slightly increased yields, about 5% yield increase 0 °C and no further increase occurred upon running the reaction at −78 °C. The major improvement at the lower temperature is attributed to product precipitation. However, the modest yield increase in our hands did not overcome the inconvenience and speed of reaction set-up. The overall effect of increasing reaction time led to no yield change at 6 h, but it was increased by 5% at 24 h and 48 h. This modest yield increase was appreciable and used going forward. Optimization of the nature of the solvent led to the most significant yield increase. Testing precursor solubility in various solvents found 11 solvents, predominantly polar solvents, that showed appreciable solubility that were suitable for running the reaction, shown in the ESI‡ (S1c). Most reactions led to similar results compared to acetone with a notable exception that the use of ethyl acetate increased the yield by 15% increase with no effect on purity.
Characterization by 51V and 1H-NMR spectroscopies
51V-NMR spectroscopy was used to verify the purity of the compounds. As shown in Fig. 2 at 10 mM solutions of the Cl-Hshed/catecholato complexes in DMSO-d6 and CD3CN, we observe more than one isomer for these complexes. As shown in Fig. 2, the major peaks of all Cl-Hshed/catecholato complexes in DMSO-d6 and CD3CN shifted 50 ppm on average and there is no precursor signal at −531 ppm in CD3CN, [VO2(Cl-Hshed)]. Since more than one signal was observed it was first necessary to confirm that the different signals were isomers and not impurities or hydrolysis products. Since different numbers of isomers were observed in varying solvents this is consistent with interconversion of signals. This isomer hypothesis was confirmed by monitoring the linewidth changes in a series of concentration and time dependent studies. The linewidth changes of the different isomers align with previous literature reports stating that the signals are interchanging consistently with kinetic isomeric exchange in the different solvents.9,21 These data suggest that the signals observed probably are different isomers and not the result of impurities in solution. Our studies also showed that most of these complexes decompose rapidly in DMSO-d6 and decompose slower in CD3CN. The 1D NMR spectra were recorded to determine the spectra for the complexes in DMSO-d6, CD3CN, and CD3OD. [VO(Cl-Hshed)(4-tB)] was chosen to illustrate the spectroscopic signature because of its high stability in comparison to the rest of the mono-substituted catechol compounds described in this paper. Because the complexes are dissolved in DMSO for biological studies, this solvent was our first solvent tried. However, the low stability of the complex in DMSO-d6 precluded further investigation in DMSO. 2D 1H–1H NMR spectra were then attempted in CD3CN but due to the poor solubility (less than 4 mM) led to spectra with poor signal to noise ratio. However, the [VO(Cl-Hshed)(4-tB)] complex was found to be soluble at 10 mM concentration in CD3OD and the decomposition rate was found to be low in solution (Fig. S2e and f, ESI‡). The 1H–1H COSY 2D and 1H–1H NOESY 2D spectra were therefore recorded in CD3OD and the subsequent analysis allowed us to determine the solution structure based on the spectra shown for [VO(Cl-Hshed)(4-tB)] in Fig. 3 and in the ESI‡ (Fig. S2c and d). Due to multiple isomers observed by 51V NMR in solution and possible candidates shown in Fig. 4, we used integration of the 1H NMR spectrum to identify the signals associated with the large isomer in the 1H NMR spectrum. Then, we identified the three major spin systems (X, Y, and Z, Fig. 3) associated with this compound and were able to make the following assignments as detailed below.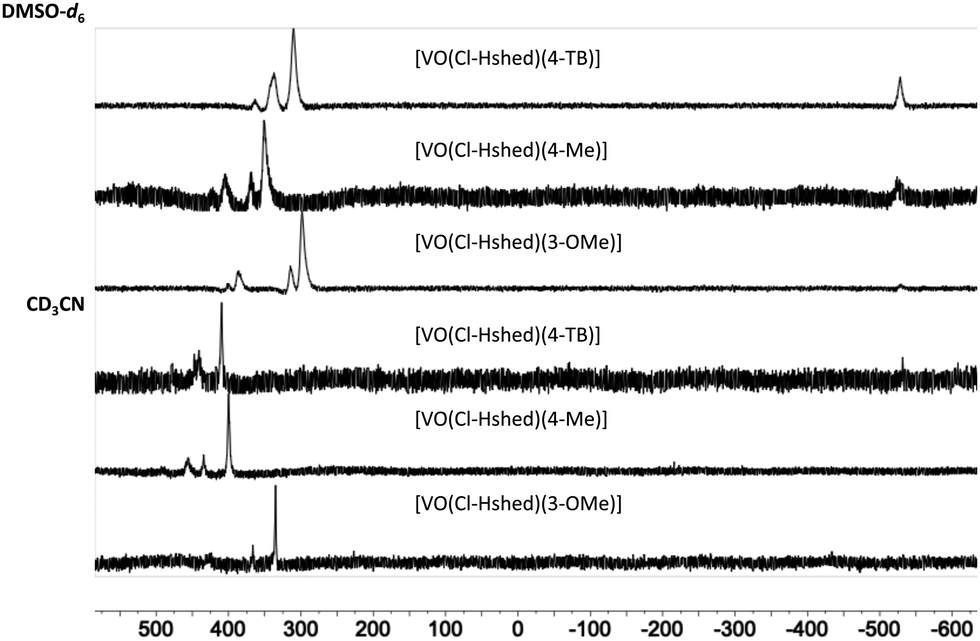 | ||
| Fig. 2 51V NMR (400 MHz) of [VO(Cl-Hshed)(4-tB)], [VO(Cl-Hshed)(3-OMe)], and [VO(Cl-Hshed)(4-Me)] in DMSO-d6 and CD3CN. | ||
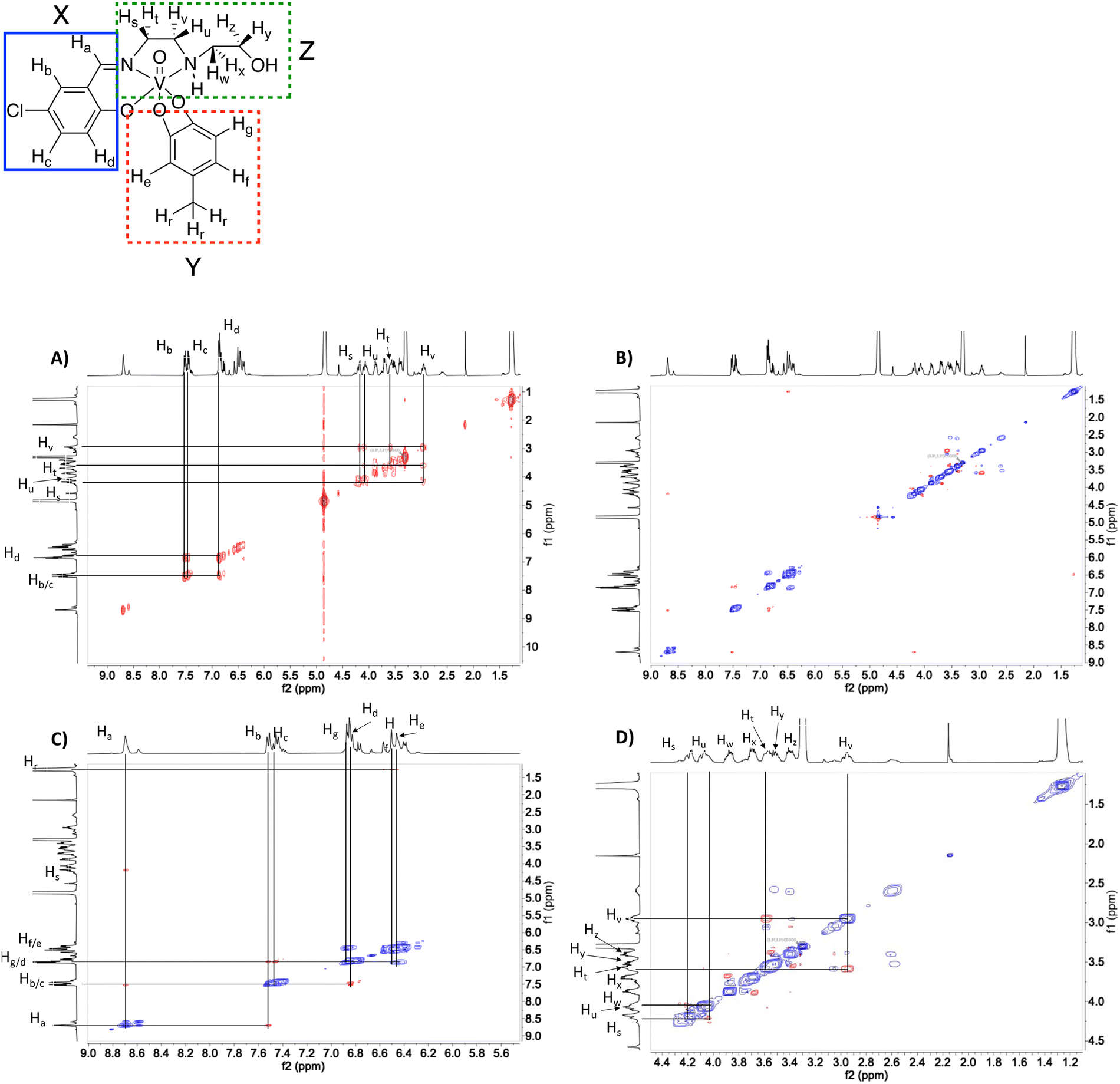 | ||
| Fig. 3 Structure of [VO(Cl-Hshed)(4-tB)] shown with three distinct spin systems labelled X, Y, and Z. (A) 1H–1H COSY and (B) 1H–1H NOESY spectrum were run in 10.0 mM CD3OD at room temperature. (C) Zoom in view of the aromatic region of the NOESY spectrum. (D) Zoom in view of the alkyl region of the NOESY spectrum. Red intensity contours represent positive NOEs, and blue intensity contours represent negative NOEs. A standard NOESY sequence was used, which consisted of 200–256 transients with 16 scans in the f1 domain using an X mixing time and X relaxation delay. The structure of [VO(Cl-Hshed)(4-tB)] is shown with a proton-labelling scheme at the top. | ||
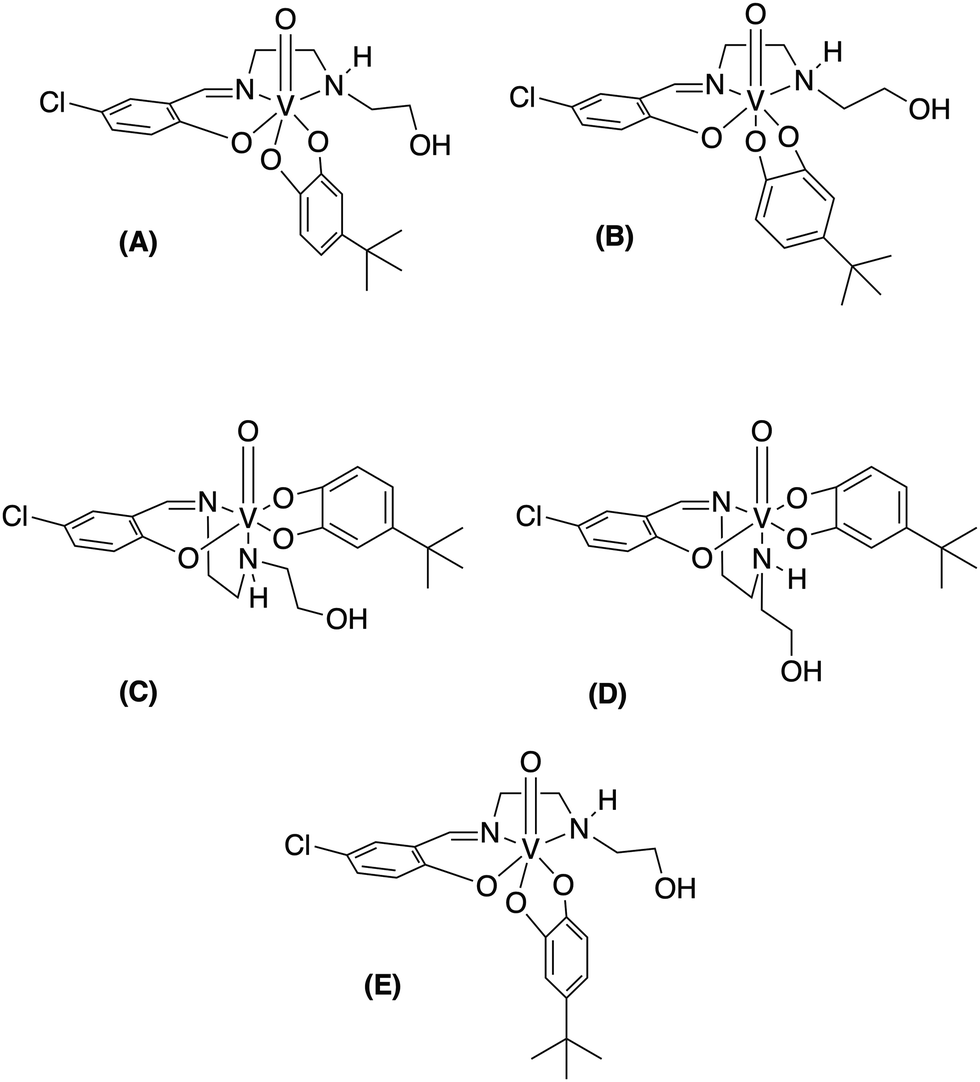 | ||
| Fig. 4 Potential isomers of [VO(Cl-Hshed)(4-tB)] in solution with the major isomer identified as (A) and other observed as potential isomers labeled as (B)–(E). The structures vary by how the catechol and Schiff base are coordinated to the vanadium center. Note that each isomer shown in this figure is chiral and forms racemic mixtures in solution as opposed to the single isomer shown. | ||
Based on integrations in the 1H-NMR and cross peaks in the COSY spectrum, the spin system X consisting of the imine proton (8.70 ppm) labelled as Ha, and aromatic protons Hb (7.52 ppm), Hc (7.45 ppm), and Hd (6.85 ppm) assigned as the protons on the Schiff base. In spin system Z, peaks Hs (4.21 ppm) and Hu (4.18 ppm) were found to have a cross peak. Ht (3.56 ppm) and Hv (2.95 ppm) are also shown to have a cross peak. Peak Hz is the only multiplet in the alkyl region with a splitting pattern of triplet of doublets connecting this proton with Ht and Hv. We also find that Hs and Hu are correlated with Hv showing the correlations for the 1,2-ethanediamine arm. This observation demonstrate that the additional four protons belong to the 2-hydroxyethylamine chain.
1H–1H NOESY spectra indicated through-space interactions as off-diagonal cross-peaks between signals at a distance up to 5 Å.30 In the NOESY spectrum of [VO(Cl-Hshed)(4-tB)] cross-peaks between the imine proton Ha and the aromatic proton Hb are observed and confirmed the assignment of Ha and Hb from the COSY spectrum Fig. 3(A). The imine proton Ha was correlated with one of the protons on the 1,2-ethanediamine arm, labelled Hs (4.18 ppm). He (6.50 ppm) and Hf (6.46 ppm) were assigned by the cross-peaks with the tert-butyl protons on the catechol, labelled Hr (1.27 ppm) Fig. 3(B) and (C). The peaks Hg (6.87 ppm) and Hd overlapped in the spectrum, and their identity was determined by Hd, which had cross-peaks with both Hb and Hc, while Hg showed a negative NOE with both He and Hf. To determine the proximity of the aromatic rings in spin systems X and Y, cross-peaks with proton Hd did not have any NOE with either He or Hg and consistent with both protons were located at a distance greater than 5 Å, Fig. 3(B) and (C). The predicted 1H spectrum had Hg as the least deshielded aromatic proton, which is not observed in this spectrum. We assign Hg to the ethanolamine arm folding over the catecholate group to the H-bond, providing a shielding of the Hg that is shifted downfield. The spin system of the N-(2-hydroxyethyl)-1,2-ethanediamine arm was also determined based on the 1H–1H NOESY spectrum and cross-peaks observed in Fig. 3(D). The 2D 1H–1H NMR spectrum shown here is assigned to the structure shown in Fig. 4(A) which is different than the conformation of [VO(Cl-Hshed)(dtb)] identified in our previous work.9 The change in the major isomer orientation of the catechol is likely due to the lack of a second sterically hindered group on the catechol, which locked [VO(Cl-Hshed)(dtb)] in a structure determined by the two tert-butyl groups.
UV-vis-NIR studies of the electronic structure and hydrolytic stabilities
To understand the anti-proliferative effects, we sought to determine the hydrolytic stability and speciation of the V(V) complexes. Hence, UV-vis-NIR spectra were recorded on all complexes to assess changes in the electronic structure with ligand and their hydrolytic stability.Spectra were collected in DMSO and are shown in Fig. 5. All complexes show intense absorption peaks consistent with the overall deep colors of the complexes. For most complexes, there are two primary absorption peaks at—approximately 550 and 850 nm. The exceptions are [VO(Cl-Hshed)(4-NO2)], which showed peaks at 411 and 770 nm, and [VO(Cl-Hshed)(4-CN)], which was too chemically unstable under these conditions to record reliable spectra. The exact wavelengths for these peaks (λmax) are listed in Table 1 alongside three additional, previously reported compounds: [VO(Cl-Hshed)(dtb)], [VO(Cl-Hshed)(cat)] and [VO(Cl-Hshed)(3-Me)]. The molar absorption coefficients (ε) for these two lowest energy peaks are all large and range from 5.4 × 103 M−1 cm−1 for the highest-energy peaks of [VO(Cl-Hshed)(cat)] and [VO(Cl-Hshed)(3-Me)] up to 14 × 103 M−1 cm−1 for the lowest-energy band of [VO(Cl-Hshed)(4-NO2)]. Ligand-based π–π* transitions are nearly always in the UV,31 and not the visible range like the peaks here. Furthermore, the V(V) ions in all these species are d0, ruling out d–d transitions as the source of the intense colors.32 Finally, the large ε values observed are more commonly associated with charge-transfer transitions (metal-to-ligand or ligand-to-metal). Owing to these three points and the lack of such peaks in the precursor complex that lacks catecholato ligands, we assign the observed electronic transitions to ligand-to-metal-charge transfer (LMCT) transitions, which is consistent with literature interpretations for complexes like those studied here,21,33,34 and their presence in homoleptic tris(catecholato)vanadium complexes.35 Note that this assignment is also consistent with the fact that changes in the electronic structure of the ligand affect the peak positions. For example, adding the Cl functional group shifted peaks relative to the non-chlorinated “parent” versions: [VO(Cl-Hshed)(dtb)] 552/863 nm compared to parent absorbance signals at (551/861 nm), [VO(Cl-Hshed)(cat)] (531/873 nm compared to parent absorbance signals at 528/873 nm) and [VO(Cl-Hshed)(3-Me)] 549/873 nm compared to parent absorbance signals at (546/875 nm).
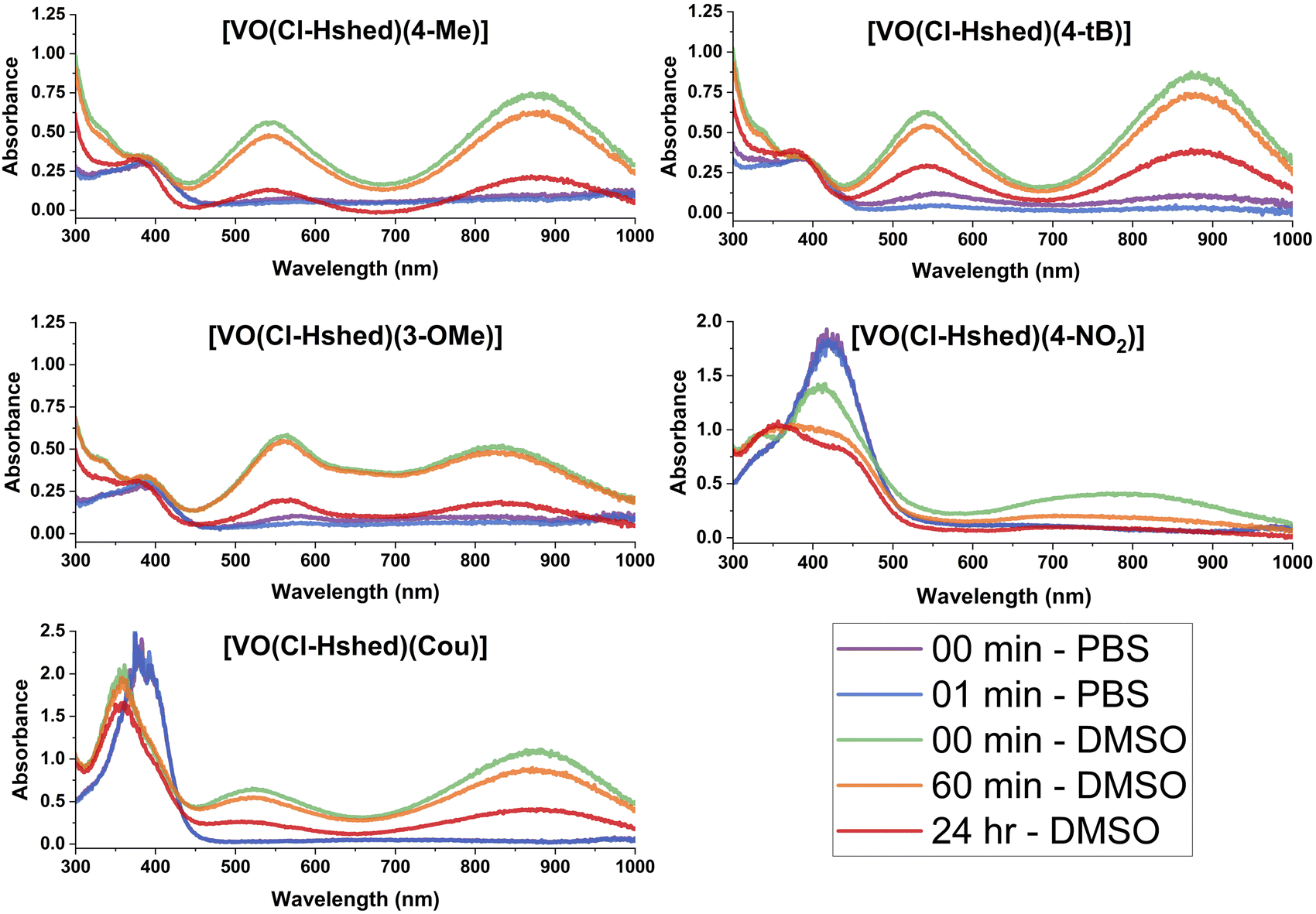 | ||
| Fig. 5 UV-vis-NIR spectra of 0.10 mM catecholate complexes, recorded in both DMSO and PBS as a function of time. The complex formed with 4-CN was excluded due to low stability. | ||
| Compound | Peak 1, λmax (εmax) | Peak 2, λmax (εmax) | t 1/2 (min) |
|---|---|---|---|
|
a Data were collected in 8:1 DMSO:PBS solutions immediately after formation for λmax and εmax data.
b Units for λmax is nm, units for εmax is M−1 cm−1.
c Previously reported compounds were measured for proper comparison.
d This compound was only measured in DMSO, not DMSO:PBS due to stability issues.
|
|||
| [VO(Cl-Hshed)(4-Me)] | 546 (5.7 × 103) | 868 (7.2 × 103) | 13.5 |
| [VO(Cl-Hshed)(4-tB)] | 539 (6.8 × 103) | 882 (1.0 × 104) | 9 |
| [VO(Cl-Hshed)(3-OMe)] | 562 (5.8 × 103) | 831 (5.0 × 103) | 11 |
| [VO(Cl-Hshed)(4-NO2)] | 411 (1.4 × 104) | 770 (4.1 × 103) | 33 DMSOd |
| [VO(Cl-Hshed)(4-CN)] | Unable to measure | Unable to measure | Unable to measure |
| [VO(Cl-Hshed)(Cou)] | 524 (6.4 × 103) | 881 (1.1 × 104) | 1 |
| [VO(Cl-Hshed)(cat)] | 529 (5.4 × 103) | 869 (6.8 × 103) | 2.5 |
| [VO(Cl-Hshed)(3-Me)] | 551 (5.4 × 103) | 864 (5.6 × 103) | 3.5 |
| [VO(Cl-Hshed)(dtb)] | 529 (3.8 × 103) | 871 (4.2 × 103) | 42 |
All complexes were also evaluated for stability in DMSO, a phosphate buffer solution (PBS), and a mixture of the two to evaluate compatibility with biological studies. The chemical stability here was evaluated by analysis of the UV-vis-NIR spectrum as a function of time after dissolution (see Fig. 5). Spectra are shown for [VO(Cl-Hshed)(4-Me)], [VO(Cl-Hshed)(4-tBu)], [VO(Cl-Hshed)(3-MeO)], [VO(Cl-Hshed)(Cou)] and [VO(Cl-Hshed)(4-NO2)] in DMSO immediately upon dissolution and after 60 min and 24 h in solution, which illustrates the stability for a few hours in DMSO but near complete decomposition over the course of 24 h.
The stability of these compounds in phosphate-buffered saline (PBS) alone was also measured to determine if the compounds were stable for any length of time in aqueous media, which was used for cell studies. The 4-CN complex was not evaluated, as it degraded upon exposure to PBS or water and degraded quickly in DMSO, which was observed as an immediate visible colour change to a yellow on dissolution. Since many anti-proliferative cell assays used a minimum of 1 h for brief exposure studies, and up to 72 h for longer term studies,22 it is important to determine how quickly the compounds decompose to ascertain whether activity was due to the intact complexes or their decomposition products.
Comparison of both the pure PBS and DMSO stability studies (Fig. 5 and 6) demonstrated that the only compounds that are slightly stable in buffered saline solution are [VO(Cl-Hshed)(4-tBu)] and possibly [VO(Cl-Hshed)(3-OMe)], as indicated by the presence of the characteristic peaks at 550 nm and 900 nm in the PBS spectra at timepoints 0 and 1 min. The rest of these compounds decomposed before being measured even for the 0 and 1 min timepoints.
Because we were interested in comparing the effects of substituents with different electronic properties, and the hydrolysis in pure water was too rapid to study compound degradation accurately but solutions of DMSO more stable, we opted to study the stabilities in DMSO–PBS mixtures (Fig. 6). Specifically, we evaluated the stability in a solution consisting of a ratio of 8 parts DMSO to 1 part PBS to compare the stability of the compounds with different substituents in solutions containing some aqueous component. The UV-vis-NIR spectra were measured for [VO(Cl-Hshed)(R)] (R = Cat, 3-Me, dtb, 4-Me, 4-tB, 3-OMe, 4-NO2 and Cou) in a ratio of 8 parts DMSO to 1 part PBS. All samples still produced the two characteristic peaks in this solvent mixture, with the first peak at about 525 nm observed between 480 and 620 nm, while the second peak was at about 850 nm, observed between 750 and 950 nm. Note that a slight shift in the peak location with solvent is expected given the LMCT assignment.36 The exceptions to the general spectra similarities were observed for R = 4-NO2, which had the first peak at 411 nm, and R = Cou, which had a third peak at 359 nm. The compounds with the R = 4-NO2 and 4-CN substituents were by far the most unstable, as the R = 4-NO2 could only be measured in DMSO, and R = 4-CN was not even stable in that solvent and is thus not shown. All samples showed decomposition in water, except for R = dtb, which was by far the most stable. Based on the data shown in Fig. 6, we were able to plot the absorbances as a function of time and determine the half-lives (t1/2) in the mixed DMSO:PBS solution, which are listed in Table 1. The increasing order of stability, with the least-stable compound first, is [VO(Cl-Hshed)(4-NO2)] < [VO(Cl-Hshed)(Cou)] < [VO(Cl-Hshed)(cat)] < [VO(Cl-Hshed)(3-Me)] < [VO(Cl-Hshed)(4-tB)] < [VO(Cl-Hshed)(3-OMe)] < [VO(Cl-Hshed)(4-Me)] < [VO(Cl-Hshed)(dtb)]. As stability is vital to the biological activity of these compounds, a similar order in PBS is anticipated, which was tested for the biological activities of these compounds.
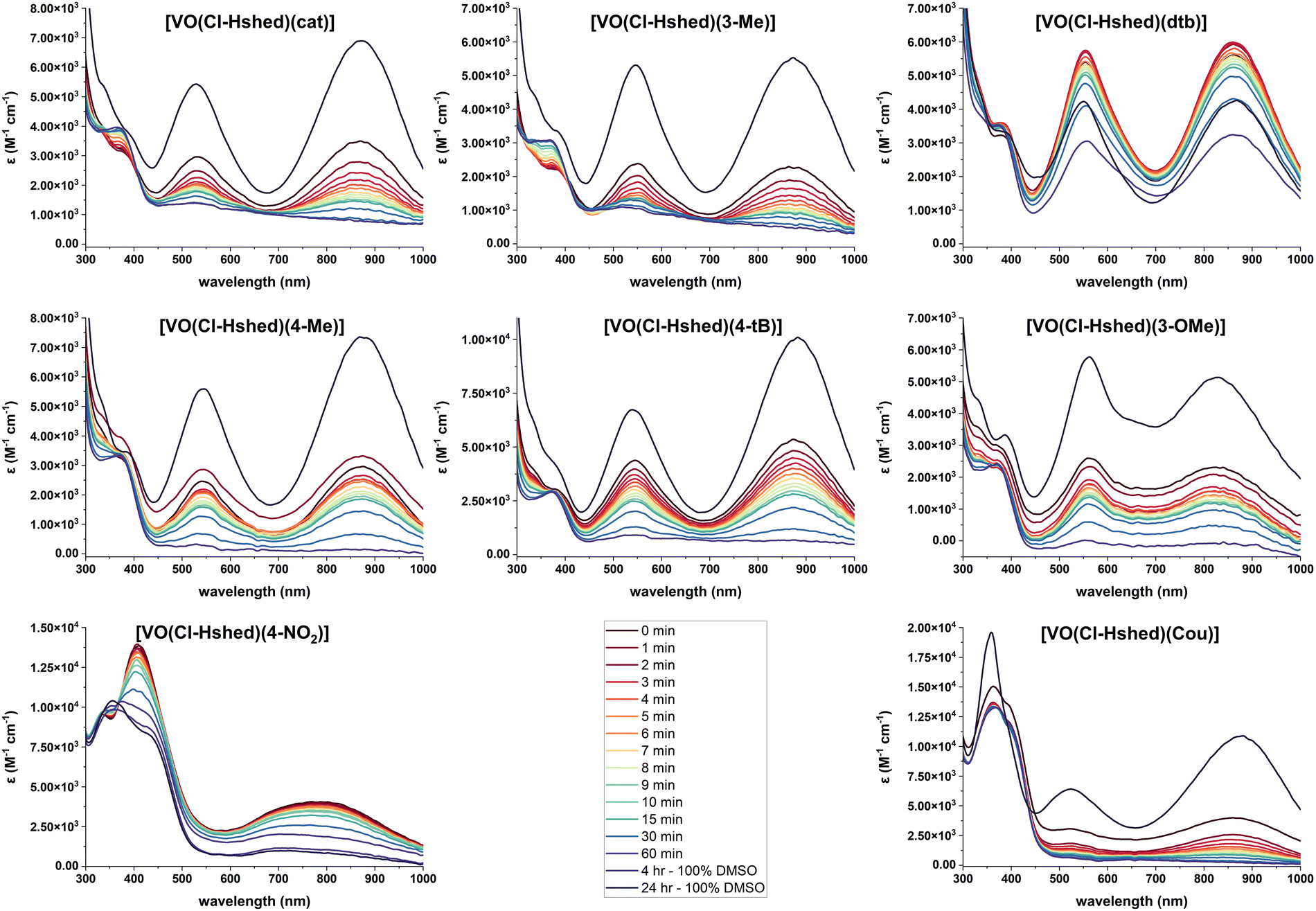 | ||
| Fig. 6 UV-vis-NIR spectra of 0.10 mM catecholate complexes, recorded in 8:1 DMSO to PBS as a function of time and compared to the 24 h DMSO sample. | ||
Electrochemistry of non-innocent vanadium complexes in CH3CN
Metal-based therapeutics are prone to undergo redox reactions and the applications of many promising examples of such compounds have been reported.37–42 Direct injection can reduce undesirable redox chemistry and stabilize the drug in a reducing tumor environment.37–46 The generation of reactive oxygen species (ROS),37–42 changes with the type of cancer,43 the state of the cancer's adaptability and ROS generation within the cells.44 Oxidative and antioxidative treatments can suppress cancer,47 and while oxidative stress is known to be a cancer promoter, some therapeutics use ROS as their primary mode of action.42,47,48 Vanadium forms in the oxidation states V, IV and III under physiological conditions and the redox cycle with the oxidation state V(IV) is most prevalent under reducing environments.49 However, V in oxidation state III might also be present in biological systems.50–53 We therefore explore here the redox properties, the compound stabilities and the biological activities of electronically varied complexes to investigate if any relationships emerge. Specifically, we plan to compare the compounds’ half wave potentials with their chemical and biological properties, considering that the more negative potentials indicate the relative electrochemical stability of the compounds within a reducing environment.CVs of 2 mM complexes were recorded at 100 mV s−1 at a glassy carbon working electrode in CH3CN in the presence of 0.1 M TBAP as the supporting electrolyte. Ferrocene was run as an external reference and E1/2 values are reported versus the Fc+/0 redox couple. CH3CN was chosen as the preferred solvent because it provided a large solvent window that included redox processes from both the vanadium redox couple and the catechol ligands. The CVs shown in Fig. 7 are only displayed over the potential window of the V(V/IV) redox couple (+0.5 V to −1.25 V vs. Fc+/0). The complete CVs are shown in the ESI‡ (Fig. S4a-1 and a-2) and show the oxidation peak of the catechol to the semiquinone, along with the CVs for the catechol ligands, and the [VO2(Cl-Hshed)] precursor complex. Previous studies have established that the redox reaction for these non-innocent vanadium complexes occurs at the vanadium center and not on the non-innocent ligands.9 While oxidation of the catechol to the semiquinone and quinone does occur, these processes are present at much more positive potentials.
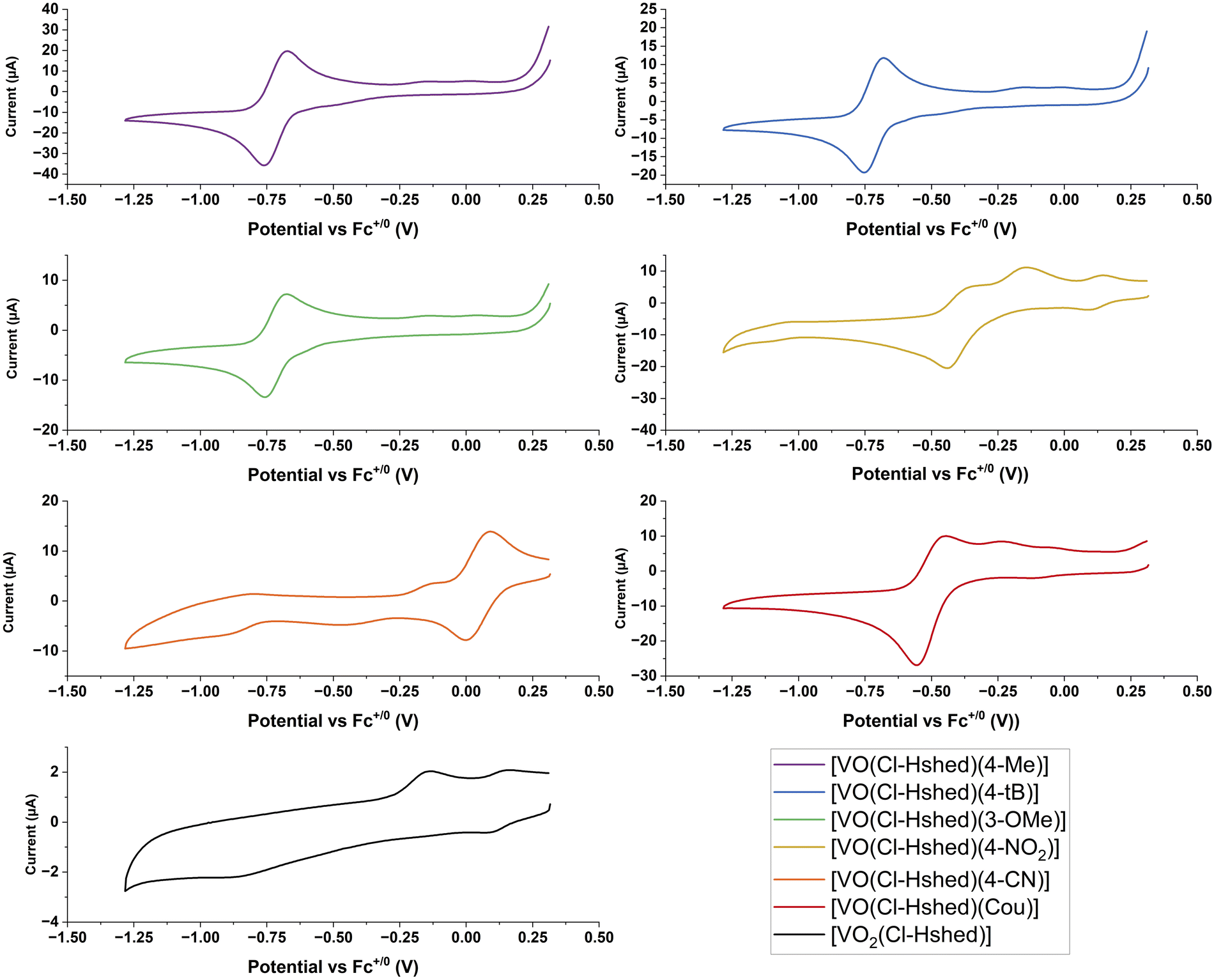 | ||
| Fig. 7 Cyclic voltammograms of tested compounds at 2 mM in CH3CN in the presence of 0.1 M TBAP as the supporting electrolyte. Scans were cycled at 100 mV s−1 from approx. 0.5 V vs. Fc+/0 to −1.1 V using a glassy carbon working electrode. | ||
For the CVs for the catechol complexes, the one-oxidation peak of the catechol to the semiquinone and quinone occurs in the region +0.25 to +1.0 V vs. Fc+/0 similar to those of the corresponding free catechol ligands (Fig. S 4a-2–c-2). Two redox processes that form one quasi-reversible V(V/IV) redox couple are observed between −1.0 and 0.0 V vs. Fc+/0 apart from the 4-CN which has a redox peak at approx. +0.05 V vs. Fc+/0 (Fig. 7 and Table 2). From the ESI‡ data, the E1/2 of the free catechol ligand occurs around +0.25 to +0.50 V vs. Fc+/0 with the cathodic peak between +0.0 and +0.25 V vs. Fc+/0 and the anodic peak at +0.5 to +0.75 V vs. Fc+/0, as observed when run in the presence of free ligand. These CVs can be seen clearly in the 4-tB, 4-Me, and 3-OMe spectra, as well as ones reported previously.9,21 For a few complexes, such as the Cou and 4-NO2 complexes, a small, secondary peak can be seen in the −0.5 V to 0 V region, and this might be due to degradation of products, as these complexes are not very stable. The presence of these secondary peaks further away from the primary redox peak, and the fact that the intensity of the major peak is so small, indicates that the complex has already slightly degraded by the time the CV is recorded. 4-CN is the outlier in this trend of negative V(V/IV) redox couples, which may be due to its increased instability when dissolved and may have entirely degraded by the time the scan has run, as the sample needs to degas for 10 minutes under argon to remove oxygen from the sample.
| Parameter | cat* | 3-Me* | dtb* | 4-Me | 4-tB |
|---|---|---|---|---|---|
| E 1/2 vs. Fc+/0 (V) | −0.678 ± 0.002 | −0.709 ± 0.005 | −0.782 ± 0.004 | −0.720 ± 0.001 | −0.719 ± 0.001 |
| I pa (µA) | 31 ± 4 | 29 ± 4 | 26 ± 2 | 27.6 ± 0.7 | 14.6 ± 0.01 |
| I pc (µA) | −31 ± 5 | −28 ± 5 | −27 ± 4 | −25 ± 1 | −12.3 ± 0.2 |
| Average Ipa/Ipc (µA) | 1.06 ± 0.09 | 1.02 ± 0.03 | 0.98 ± 0.08 | 1.7 ± 0.04 | 1.19 ± 0.02 |
| ΔEp (V) | 0.17 ± 0.01 | 0.145 ± 0.003 | 0.14 ± 0.03 | 0.084 ± 0.002 | 0.074 ± 0.002 |
| Parameter | 3-OMe | 4-NO2 | 4-CN | Cou |
|---|---|---|---|---|
| E 1/2 vs. Fc+/0 (V) | −0.719 ± 0.002 | −0.386 ± 0.006 | 0.042 ± 0.006 | −0.503 ± 0.001 |
| I pa (µA) | 8.8 ± 0.2 | 8.4 ± 0.4 | 12.7 ± 0.2 | 15.1 ± 0.2 |
| I pc (µA) | −8.1 ± 0.6 | −10.8 ± 0.3 | −14 ± 1 | −16.8 ± 0.5 |
| Average Ipa/Ipc (µA) | 1.1 ± 0.1 | 0.78 ± 0.2 | 0.91 ± 0.08 | 0.90 ± 0.02 |
| ΔEp (V) | 0.074 ± 0.002 | 0.11 ± 0.01 | 0.09 ± 0.02 | 0.093 ± 0.005 |
The catechol complex with the 4-CN substituent was the easiest to reduce, followed by those with 4-NO2 and Cou ligands, and the following substituents on the catechol ligand, 3-OMe, 4-Me, and 4-t-Bu were the hardest to reduce. The anodic and cathodic peak ratios (Ipa and Ipc) were recorded and are listed in Table 2. The Ipa/Ipc ratios of ∼1 indicate chemically reversible electrochemical redox reactions, and all the CVs were consistent for over five cycles except for [VO(Cl-Hshed)(Cou)], which showed a slight decrease in the cathodic peak. A slight deviation from 1 (between 0.9 and 1.1) in the Ipa/Ipc ratio indicated a quasi-reversible couple, which includes most of the complexes tested, except for 4-NO2. This electrochemical quasi-reversibly was similar to that observed previously,9 even though most of the V(V) complexes had limited stability in acetonitrile and showed that their V(IV) counterparts were stable on the electrochemical timescale.
As was observed previously for the catechol and dtb complexes,9 quasi-reversibility was confirmed by the large ΔEp values when compared to that of the Fc+/0 couple under the same conditions which averaged 0.095 ± 0.003 V which showed that the large ΔEp values for 4-NO2 were not due to lack of iR compensation. It is also consistent with the presence of multiple geometric isomers observed in the NMR results, which would be in rapid equilibrium on the electrochemical timescale when only one redox couple was observed in most instances. Where a single reduction and a single oxidation peak was observed for the V(V/IV) couple, the position of these peaks will correspond to a weighted average of the reduction of the V(V) isomers observed in the NMR spectra, and the corresponding oxidation, the weighted average of oxidation of the V(IV) isomers. Since the ratio of isomers would be different in the two oxidation states given changes in steric and electronic interactions, this will lead to larger values of ΔEp than those observed for the V(V/IV) couple of individual isomers if the CVs are undertaken at sufficiently high scan rates so that the electrochemical timescale is faster than the isomerization timescale.
Fast exchange between individuals isomers was observed for the sterically hindered Schiff base dtb complexes,9 and the acetone linkage isomerization in the Ru(III/II) couple of the [Ru(NH3)5(acetone)]3+/2+ couple.34–36 In particular, the 51V NMR chemical shifts are very sensitive to the electron density on the V(V) center,7,20,21,23 hence, the spread of chemical shifts. In contrast, the diffusion coefficients are much more sensitive to the degradation of the compounds in solution since their computation relies on the Ipa value and the concentration of the species that creates the redox peak. Decomposition prevents an exact concentration measurement because the concentration changes as the solution is measured, and these values are thus not reported, especially since the NMR data showed degradation over a short time frame even in CH3CN.
Anti-proliferative studies with the mono-substituted catecholato V(V) complexes
New anticancerous complexes lacking systemic toxicity will relieve side effects include targeted treatment and intertumoral administration, which delivers a payload of chemotherapeutic agents directly into the tumor.19 While these methods are not effective against cancers that have metastasized, they allow for a greater variety of suitable compounds that are reactive and appropriate for intravenous applications.54,55 Previous work from our group has shown the efficacy of the [VO(Hshed)(dtb)] complex in in vitro anti-proliferative assays against the brain cancer cell lines, and T98G (glioma multiforme), as well as A549 (lung), PANC-1 (pancreatic), SW1353 (bone chondrosarcoma), and triple-negative breast cancer (being more cytotoxic than cisplatin, while lacking cisplatin's characteristic toxicity when tested against both normal human foreskin fibroblasts (HFF-1) and mice).9,20 Furthermore, the previous studies showed that the presence of the chloro substituent on the Schiff base increased the hydrolytic stability of the V(V) complexes compared with the complexes with the Hshed ligand.9 Hence, we chose to study the series of V(V) complexes with the Cl-Hshed ligand in the anticipation that some of the mono-substituted catecholato complexes in this series would have sufficient stability to enter the cells intact before decomposition to exert their anti-proliferative activity.9The anti-proliferative studies with the series of compounds prepared in this work were carried out and IC50 values were determined (Table 3) both for the compounds that were added to the cells immediately after the addition of stock solutions in DMSO to cell culture medium (designated ‘fresh’) and for the same compounds that were pre-incubated with the cell culture medium for 24 h at 310 K and 5% CO2 before the addition to cells (designated ‘aged’). Previous studies have demonstrated significant differences in anti-proliferative activities between the fresh and aged solutions of some of the V(V)-Schiff base-catecholato complexes.8,21
| Compound | IC50 (fresh),a μM | IC50 (aged),b μM |
|---|---|---|
| a Freshly prepared dilutions of compounds (0.040–10 mM) in DMSO were diluted 100-fold with fully supplemented cell culture medium, and the resulting solutions were added to the cells within ∼30 s and incubated for 72 h before the determination of cell viability by MTT assays. b Dilutions of the compounds in cell culture medium were kept for 24 h at 310 K and 5% CO2, then added to the cells and incubated for 72 h. c Previously reported data.9 | ||
| [VO(Cl-Hshed)(4-Me)] | 60 ± 7 | 29 ± 2 |
| [VO(Cl-Hshed)(4-tB)] | 46 ± 9 | 38 ± 3 |
| [VO(Cl-Hshed)(3-OMe)] | 61 ± 10 | 71 ± 12 |
| [VO(Cl-Hshed)(4-NO2)] | 64 ± 7 | 25 ± 2 |
| [VO(Cl-Hshed)(4-CN)] | 34 ± 4 | 13 ± 2 |
| [VO(Cl-Hshed)(Cou)] | 67 ± 12 | 81 ± 6 |
| [VO(Cl-Hshed)(cat)] | 19 ± 2 | 8.1 ± 0.6 |
| [VO(Cl-Hshed)(3-Me)] | 34 ± 7 | 18 ± 3 |
| [VO(Cl-Hshed)(dtb)] | 4.1 ± 0.5 | 9 ± 1 |
| [VO2(Cl-Hshed)] | 34 ± 7 | 67 ± 7 |
| Na3VO4 | 26 ± 4 | 24 ± 4 |
Four compounds had IC50 (fresh) values between 60 to 70 μM; two of these compounds showed the same activity as vanadate when the solutions were aged and these two complexes were [VO(Cl-Hshed)(4-NO2)] and [VO(Cl-Hshed)(4-Me)], which decomposed immediately upon dissolution in the DMSO/PBS media and the other after 13 min. The other two complexes [VO(Cl-Hshed)(3-OMe)] and [VO(Cl-Hshed)(Cou)] remained weakly inhibitory after 72 hours, akin to the Schiff base framework [VO2(Cl-Hshed)]. Three complexes, [VO(Cl-Hshed)(4-tB)], [VO(Cl-Hshed)(3-Ome)] and [VO2(Cl-Hshed)], had IC50 (fresh) values similar to vanadate, around 35–45 μM; the [VO(Cl-Hshed)(4-tB)] complex upon aging stayed at the same activity as vanadate, whereas [VO(Cl-Hshed)(3-Me)] became more inhibitory and the scaffold less inhibitory. Finally, two complexes [VO(Cl-Hshed)(cat)] and [VO(Cl-Hshed)(dtb)], were more inhibitory than vanadate both for the IC50 (fresh) and for the IC50 (aged) samples. However, these activities are expected to arise from completely different mechanisms, as reported previously for the anti-proliferative activities of the analogous parent HSHED complexes in breast cancer cells.22
Based on the UV-vis-NIR results, the complexes lifetimes in the DMSO–PBS mixtures are divided into four groups. The first group immediately decomposed and included complexes [VO(Cl-Hshed)(4-CN)], [VO(Cl-Hshed)(4-NO2)] and [VO(Cl-Hshed)(Cou)]. Two complexes, [VO(Cl-Hshed)(cat)] and [VO(Cl-Hshed)(3-Me)] survived for about 3 min. Another three complexes, [VO(Cl-Hshed)(4-tB)], [VO(Cl-Hshed)(3-OMe)] and [VO(Cl-Hshed)(4-Me)] survived for 9–13 min, and finally [VO(Cl-Hshed)(dtb)] survived for 44 min. However, the lifetimes in PBS are expected to be much shorter than DMSO/PBS mixtures. Hence, they likely have insufficient stabilities to enable the less stable and less hydrophobic mono-substituted catecholato complexes to enter the cells intact.9,25 In particular, from the organization of the IC50 (fresh) and the IC50 (aged) values listed above, it was clear that these IC50-values did not follow the life-times of the compounds. However, as listed in Table 3, the IC50 (fresh) and the IC50 (aged) values, except for those of [VO(Cl-Hshed)(dtb)], [VO(Cl-Hshed)(4-CN)] and [VO(Cl-Hshed)(cat)] are all close to the values observed with vanadate. This is consistent with the interpretation that all these complexes hydrolyze to vanadate before there is sufficient time to be taken up by the cells, and that the observed anti-proliferative effects could be explained by the formation of vanadate.
However, cell culture media are complex mixtures and alternative explanations exist including the possibility that some proportion of the cytotoxicity of these complexes could be explained by other species forming or limited permeability of intact complexes into cells. Although improved activity may be possible by modified procedures,9,25,56 the weak activities of these complexes do not warrant additional studies. Hence, within the series, the effects of [VO(Cl-Hshed)(dtb)] and [VO(Cl-Hshed)(cat)] were different and of most interest. Recently the mechanism of the corresponding parent complexes [VO(Hshed)(dtb)] and [VO(Hshed)(cat)] were investigated in detail and both complexes showed high antiproliferative activities in triple-negative human breast cancer (MDA-MB-231) cells.22 However, the mechanisms of their activities were radically different. [VO(Hshed)(dtb)] formed noncovalent adducts with human serum albumin, and rapidly entered cells via passive diffusion. This conclusion was made because the complex had an IC50 (fresh) value 20-fold more potent than the IC50 (aged) values. In the case of the [VO(Cl-Hshed)(dtb)] complex the activity of [VO(Cl-Hshed)(dtb)] decreased two-fold after its decomposition. In contrast the [VO(Hshed)(cat)] showed lower activity in both fresh and aged solution (around 12 μM) which was interpreted as the complex hydrolyzing immediately. The anti-proliferative activity of [VO(Hshed)(cat)] complex was rationalized to be due to V(V) decomposition products, free catechol, and O2 in the cell culture medium. In the case of the [VO(Cl-Hshed)(cat)] complex the activities of fresh and aged solutions were within a factor of two that of the [VO(Hshed)(cat)] complex and more potent than vanadate alone.
In summary, for most complexes, except for [VO(Cl-Hshed)(dtb)] and [VO(Cl-Hshed)(cat)], the anti-proliferative activities were relatively low (IC50 ∼ 30 μM) and close to those of vanadate and the fresh solution of the precursor complex, [VO2(Cl-Hshed)]. This observation is consistent with the rapid loss of catechol ligands from these complexes under cell culture conditions. The aged solutions of [VO(Cl-Hshed)(3-OMe)], [VO(Cl-Hshed)(Cou)], and [VO2(Cl-Hshed)] showed lower activities. However, generally, the EC50 values of aged solutions of V(V) complexes in cell culture media were higher and less efficacious than those of the fresh solutions. A similar pattern was also observed for the sterically hindered and hydrophobic [VO(Cl-Hshed)(dtb)] complex,22 however the aged compound was much more efficacious presumably because of the reaction of the V(V/IV) decomposition products with released ligands in the presence of molecular oxygen, which led to some toxic V(V)-peroxido complexes.
Investigation of potential relationships between the complexes chemical and physical properties
Since we have determined in detail the properties of the complexes with different catechol ligands, the potential relationships between the electronic properties and various other properties were investigated. The study of several sulfa drugs showed correlations between their antibacterial potency with both their E1/2 values and their pKa values.57 This linear trend has shown consistency for phenolic compounds and sulfa drugs and provided a rationale for this study's observations. We know that hydrophobicity, which is related to the logP and steric hindrance is important for anti-proliferative properties.9,21 Other properties that we were interested in examining included compound stability, complex or ligand acidity and pKa values, and isomer content, as well as anti-proliferative effects. However, the anti-proliferative properties studied for the series of complexes with mono-substituted catecholato ligands in this work were similar and resembled both vanadate and the precursor scaffold.
Interpretation of this observation suggests that although the series of compounds prepared in this work are very different with varying redox properties, the pattern of their anti-proliferative effects are mainly similar to vanadate. However, the similar anti-proliferative effects of all the mono-substituted catecholate complexes to vanadate are consistent with decomposition to form vanadate. Although we cannot discount the possibility that a proportion of the activity is due to the intact complexes for the more stable complexes in the series. The only exception to this observation is the [VO(Cl-Hshed)(dtb)] complex that was significantly more stable and showed a much higher anti-proliferative effect. In summary, this supports the interpretation that stability is important for anti-proliferative properties, and we suggest that this is related to the fact that the complex needs time to be absorbed into the cell. Furthermore, the observation that compound hydrophobicity is important to anti-proliferative properties is demonstrated.9,20
The studies presented in this work have expanded the understanding of the properties of the classes of non-innocent Schiff base vanadium(V) catecholate complexes. Effects of electronic substituents have been extensively studied for a wide range of compounds using Hammett parameters and have provided information on the general properties of each class of materials. Such studies have been most successful for aromatic compounds and since the catechol functionality is the moiety substituted with different groups, this approach is appropriate to obtain more information on these non-innocent Schiff base vanadium(V) catecholate complexes. The pure Hammett correlations would involve modified Hammett relationships since the relevant unit in our system is a catechol.58 Indeed substituted catechol derivatives have been investigated but in place of known Hammett parameter values the polarizability and pKa values were used due to the second OH group on the catechol in addition to the added substituents, which made it challenging to define pure Hammett parameters.59
To characterize the electronic effects of catechol substituents on the non-innocent vanadium-Schiff base complexes, we systematically measured the properties of the catechol as the substituent changed. Substituted catechols with an electron withdrawing substituent have pKa values less than the parent catechol and the catechol with electron donating substituents, using plots of redox potential or stability of the complex on the x-axis and various ligand properties on the y-axis. Several potential relationships were investigated, shown in the ESI‡ (Fig. S4d). Here, we show one successful relationship correlating the catechol ligand's protonation state with the corresponding complex's redox potential.
The Brønsted acidity of the catechol ligand is expected to correlate with the Lewis basicity of the catecholato ligands and, hence, the electron density at the V(V) center upon ligand binding. Previous studies showed linear correlations between oxidative half-wave potentials and pKa values for para-substituted phenols as a suitable solvent–electrolyte system.60 Jovanovic and coworkers also showed that Hammett constants and electron potentials for para-substituted phenoxy radicals are correlated.61 These correlations have also been observed for other systems including biological processes.57 Similar correlations have been shown between Co(III/II) redox potentials of cage complexes with substituent parameters, NMR chemical shifts, and pKa values of related molecules, including the use of these techniques for obtaining substituent parameters.62,63 Therefore, it is not surprising that we observe a linear relationship between the electronic properties of the complexes introduced in this work and the catechol pKa values as described in the following.
The catechol coordinated to the vanadium is deprotonated and these ligand catechol pKa values are sensitive to the substitution. By estimating these pKa values using Chemicalize, a series of pKa values was obtained for the mono-substituted catechols. There are two OH-groups on the catechol, and both deprotonate forming charged phenoxy groups that coordinate directly to the vanadium. However, the most acidic OH-group deprotonates first (the first pKa value) forming a charged phenoxy group, resulting in the V-atom becoming more susceptible to reduction. Indeed, it has previously been reported that a lower pKa value for the catechols will result in a more readily reduced complex.64 As a result, we plotted the first pKa values of the most acidic OH-group on the catechol as a function of the complex's redox potential (Fig. 8). We observed a linear correlation between the first pKa value of the catechol and the half wave potential of the Schiff base vanadium(V) catecholate complex as shown in Fig. 8. The one system that does not adhere to this relationship is that of the 4-CN substituted catechol ligand that forms the [VO(Cl-Hshed)(4-CN)] complex. This complex decomposes very rapidly in the solvents investigated and hence the observed E½vs. Fc+/0 (V) may not be very representative for the intact complex.
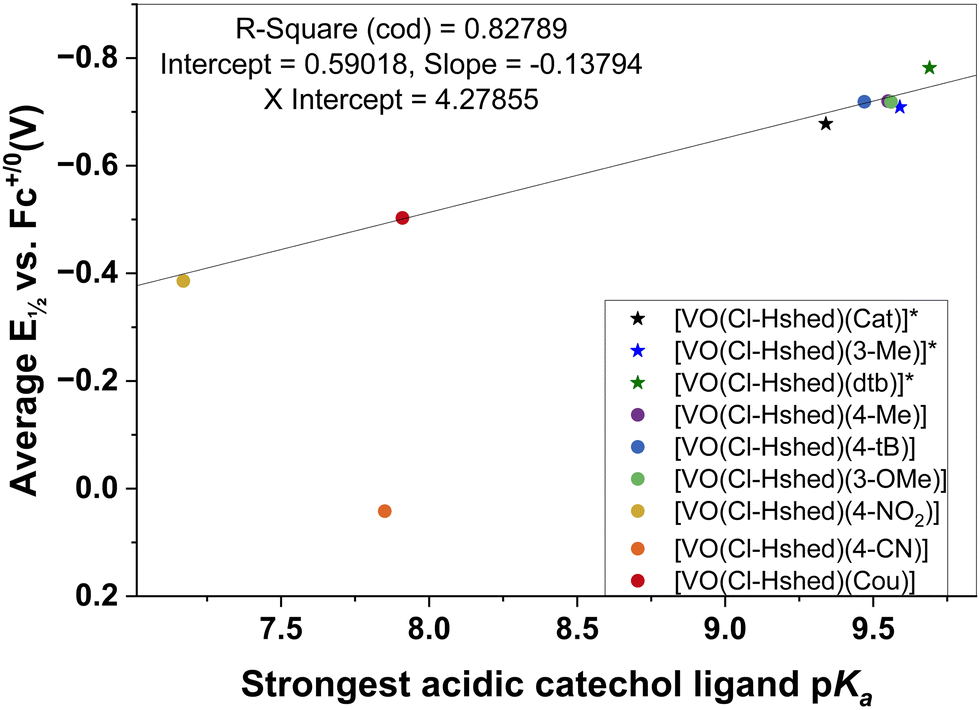 | ||
| Fig. 8 Average half wave potentials of tested [VO(Cl-Hshed)(R)] complexes referenced to the ferrocene redox couple compared to the first pKa value calculated via chemicalize for the free catechol ligand. | ||
Conclusions
We have synthesized and characterized a series of chloro-substituted Schiff base vanadium mono-substituted catecholate complexes with the catechol having a variety of electronically different substituents. The effects against T98G glioblastoma cells and the resulting chemical, physical and biological properties of the parent series and their chlorinated daughter compounds lead us to conclude that some aqueous compound stability is an essential factor for the design of an anti-proliferative compound. As such, we have expanded the knowledge of the structure activity relationships developed in our previous studies to provide important insights into future compound development in the series.Specifically, several observations can be made. First, while the chloro-substituted Schiff base complexes exhibited the expected increased stability in aqueous media, it was insufficient to probe the effect of V(V/IV) redox potential on anti-proliferative activities. Such a study would require more sterically hindered systems. Second, the range of redox potentials that these substituent effects could obtain was −0.5 to −0.8 V versus the Fc+/0 couple in acetonitrile. While the complexes are too unstable to measure these potentials in aqueous media due to their hydrolytic instability, we can estimate their range by first converting the potentials to the less solvent-dependent decamethylferrocenium/decamethyferrocence redox standard (0 to −0.3 V vs. Me10Fc+/0 in acetonitrile) and then estimating the value in aqueous media to be −0.1 to −0.4 V versus the Ag/AgCl/KCl(sat) electrode,65 which corresponds to −0.3 to −0.6 V vs. the standard hydrogen electrode. For comparison, the redox potential of the cellular glutathione/glutathione disulfide (GSH/GSSG) couple was estimated to be −0.26 to −0.15 V and that of NADPH/NADP+ −0.4 V.66 Since both these biological redox couples are important in apoptotic and other fundamental processes of cancer cells,67 the design of these complexes met its goal to span a biologically relevant range. Despite showing the lack of hydrolytic stability, these results demonstrated the ability to span a biologically relevant range of V(V/IV) redox potentials to probe the effects of redox potential on anti-proliferative activities by changing substituents on the catecholato ligand and act as a blueprint for designing more hydrolytically stable systems as described above. In summary, the series of electronically different chlorinated Schiff base vanadium mono-substituted catecholate complexes designed to explore the role of redox properties of the anti-proliferative effects on glioblastoma T98G cells all hydrolyzed very rapidly and hence, had similar anti-proliferative effects on the T98G cells as free vanadates. These studies demonstrate that although electronic effects may play a role, systems to demonstrate such effects must also include catecholates with sterically hindered groups to assure a greater stability of the complexes.
Finally, the studies of this series of compounds did demonstrate that changes in electronic properties in the catechols lead to a linear correlation of the half wave redox potentials of the Schiff base vanadium catecholato complexes in a modified Hammett relationship with the most acidic proton of the catechol. The latter relationship will allow us to design compounds with differing redox properties in the future and determine the importance of this property on the anti-proliferative effects on glioblastoma T98G cells.
Author contributions
D. C. C. and P. A. L. conceived the project. A. A. H., H. A. M., J. T. K., S. A. M. designed and performed with the help of J. H., K. K., C. N. B., L. E. the chemical studies. J. M. Z. helped with the UV-vis-NIR studies. A. l. and C. U. designed and performed the biological studies. A. A. H., S. A. M. and D. C. C. wrote the initial draft. D. C. C. and P. A. L. supervised the project. All authors contributed to and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Tribute to George M. Whitesides on his 85th birthday. Happy B-day George! Debbie C. Crans was in your laboratory in 1981–1985 and not only learned to appreciate your larger-than-life vision in chemistry and science but also adopted the interdisciplinary approach to science that has served her and her students so well. In addition, you made Chris R. Roberts and Debbie C. Crans’ lab-partners, and Chris has been and is still her soulmate and husband of 33 years and with whom she has three daughters Patricia, Gerri, and Mia. Thank you for everything. D. C. C. thanks the Arthur Cope Foundation, Colorado State University, and a private donor for support. P. A. L. thanks the Australian Research Council (ARC) grants for an ARC Senior Research Associate position for A. L. (DP160104172). D. C. C. and P. A. L. thank the University of Sydney for an International Scholar Award to D. C. C. The authors wish to thank the Colorado State University Analytical Resources Core Facility, RRID:SCR_021758 for instrumental access and a specific thanks to Dr Michele Mailhot for assistance with the NMR studies. J. M. Z. acknowledged the National Science foundation (CAREER award 2047325) for support. The authors acknowledge the facilities at the Australian Centre for Microscopy & Microanalysis at The University of Sydney (Drs Minh Huynh and Ellie Kable) for the use of the cell culture laboratory. We thank Dr Nicholas Proschogo (Chemistry Mass Spectrometry Facility, The University of Sydney) for performing ICP-MS measurements.References
- M. Sutradhar, L. M. Martins, M. F. C. G. da Silva and A. J. Pombeiro, Vanadium complexes: Recent progress in oxidation catalysis, Coord. Chem. Rev., 2015, 301, 200–239 CrossRef.
- A. Paul, R. A. Khan, G. M. Shaik, J. P. Shaik, D. S. Nesterov, M. F. C. G. da Silva and A. J. Pombeiro, Synthesis, characterization, and anti-cancer potential of pyrene-appended Schiff base tin(IV) complexes: experimental and computational insights, New J. Chem., 2024, 48(7), 2907–2919 RSC.
- P. Nunes, Y. Yildizhan, Z. Adiguzel, F. Marques, J. C. Pessoa, C. Acilan and I. Correia, Copper(II) and oxidovanadium(IV) complexes of chromone Schiff bases as potential anti-cancer agents, J. Biol. Inorg. Chem., 2022, 27(89), 89–109 CrossRef CAS PubMed.
- N. Ribeiro, P. F. Farinha, J. O. Pinho, H. Luiz, J. P. Mészáros, A. M. Galvão, J. C. Pessoa, É. A. Enyedy, C. P. Reis, I. Correia and M. M. Gaspar, Metal Coordination and Biological Screening of a Schiff Base Derived from 8-Hydroxyquinoline and Benzothiazole, Pharmaceutics, 2022, 14, 2583 CrossRef CAS PubMed.
- H. Kargar, A. A. Ardakani, M. N. Tahir, M. Ashfaq and K. S. Munawar, Synthesis, spectral characterization, crystal structure and antibacterial activity of nickel(II), copper(II) and zinc(II) complexes containing ONNO donor Schiff base ligands, J. Mol. Struct., 2021, 1255, 130112 CrossRef.
- M. Woźniczka, M. Sutradhar, M. Chmiela, W. Gonciarz and M. Pająk, Equilibria in the aqueous system of cobalt(II) based on 2-picolinehydroxamic acid and N-(2-hydroxybenzyl) phenylalanine and its ability to inhibit the propagation of cancer cells, J. Inorg. Biochem., 2023, 249, 112389 CrossRef PubMed.
- P. B. Chatterjee, O. Goncharov-Zapata, L. L. Quinn, G. Hou, H. Hamaed, R. W. Schurko, T. Polenova and D. C. Crans, Characterization of noninnocent metal complexes using solid-state NMR spectroscopy: o-dioxolene vanadium complexes, Inorg. Chem., 2011, 50, 9794–9803 CrossRef CAS PubMed.
- A. Levina, A. Pires Vieira, A. Wijetunga, R. Kaur, J. T. Koehn, D. C. Crans, P. A. Lay and A. Short-Lived, but Highly Cytotoxic Vanadium(V) Complex as a Potential Drug Lead for Brain Cancer Treatment by Intratumoral Injections, Angew. Chem., Int. Ed., 2020, 59, 15834 CrossRef CAS PubMed.
- H. A. Murakami, C. Uslan, A. A. Haase, J. T. Koehn, A. P. Vieira, D. J. Gaebler, J. Hagan, C. N. Beuning, N. Proschogo, A. Levina, P. A. Lay and D. C. Crans, Vanadium Chloro-Substituted Schiff Base Catecholate Complexes Are Reducible, Lipophilic, Water Stable, and Have Anticancer Activities, Inorg. Chem., 2022, 61(51), 20757–20773 CrossRef CAS PubMed.
- S. A. Patra, A. Banerjee, G. Sahu, M. Mohanty, S. Lima, D. Mohapatra, H. G. Görls, W. Plass and R. Dinda, Evaluation of DNA/BSA interaction and in vitro cell cytotoxicity of μ2-oxido bridged divanadium(V) complexes containing ONO donor ligands, J. Inorg. Biochem., 2022, 233, 111852 CrossRef CAS PubMed.
- M. F. A. Santos, G. Sciortino, I. Correia, A. C. P. Fernandes, T. Santos-Silva, F. Pisanu, E. Garribba and J. C. Pessoa, Binding of VIVO2+, VIVOL, VIVOL2 and VVO2L Moieties to Proteins: X-ray/Theoretical Characterization and Biological Implications, Chem. – Eur. J., 2022, 28, e202200105 CrossRef CAS PubMed.
- A. Banerjee, S. A. Patra, G. Sahu, G. Sciortino, F. Pisanu, E. Garribba, M. F. N. N. Carvalho, I. Correia, J. C. Pessoa, H. Reuter, R. Dinda and A. Series, of Non-Oxido VIV Complexes of Dibasic ONS Donor Ligands: Solution Stability, Chemical Transformations, Protein Interactions, and Antiproliferative Activity, Inorg. Chem., 2023, 62, 7932–7953 CrossRef CAS PubMed.
- M. F. Mosquillo, P. Smircich, A. Lima, S. A. Gehrke, G. Scalese, I. Machado, D. Gambino, B. Garat and L. Pérez-Díaz, High Throughput Approaches to Unravel the Mechanism of Action of a New Vanadium-Based Compound against Trypanosoma cruzi, Bioinorg. Chem. Appl., 2020, 2020, 1–10 CrossRef PubMed.
- G. Sahu, S. A. Patra, P. D. Pattanayak, W. Kaminsky and R. Dinda, LVVO-Ethyl Maltol-Based Metallodrugs (L2− = Tridentate ONO Ligands): Hydrophobicity, Hydrolytic Stability, and Cytotoxicity via ROS-Mediated Apoptosis, Inorg. Chem., 2023, 62, 6722–6739 CrossRef CAS PubMed.
- G. Ferraro, M. Paolillo, G. Sciortino, F. Pisanu, E. Garribba and A. Melrino, Implications of Protein Interaction in the Speciation of Potential VIVO−Pyridinone Drugs, Inorg. Chem., 2023, 62, 8407–8417 CrossRef CAS PubMed.
- K. Dankhoff, A. Ahmad, B. Weber, B. Biersack and R. Schobert, Anticancer properties of a new non-oxido vanadium(IV) complex with a catechol-modified 3,3′-diindolylmethane ligand, J. Inorg. Biochem., 2019, 194, 1–6 CrossRef CAS PubMed.
- L. Ni, H. Zhao, L. Tao, X. Li, Z. Zhou, Y. Sun, C. Chen, D. Wei, Y. Liu and G. Diao, Synthesis, in vitro cytotoxicity, and structure–activity relationships (SAR) of multidentate oxidovanadium(IV) complexes as anti-cancer agents, Dalton Trans., 2018, 47, 10035–10045 RSC.
- A. Levina and P. A. Lay, Metal-Based Anti-diabetic Drugs: Advances and Challenges, Dalton Trans., 2011, 40(44), 11675–11686 RSC.
- A. Levina, D. C. Crans and P. A. Lay, Advantageous Reactivity of Unstable Metal Complexes: Potential Applications of Metal-Based Anti-cancer Drugs for Intratumoral Injections, Pharmaceutics, 2022, 14(4), 790 CrossRef CAS PubMed.
- D. C. Crans, J. T. Koehn, S. M. Petry, C. M. Glover, A. Wijetunga, R. Kaur, A. Levina and P. A. Lay, Hydrophobicity May Enhance Membrane Affinity and Anti-Cancer Effects of Schiff Base Vanadium(V) Catecholate Complexes, Dalton Trans., 2019, 48, 6383–6395 RSC.
- K. Kostenkova, A. Levina, D. A. Walters, H. A. Murakami, P. A. Lay and D. C. Crans, Vanadium(V) pyridine-containing Schiff-Base catecholate complexes are novel lipophilic, redox-active, and selectively cytotoxic in glioblastoma (T98g) cells, Chem. – Eur. J., 2023, 29(68), e202302271 CrossRef CAS PubMed.
- A. Levina, C. Uslan, H. A. Murakami, D. C. Crans and P. A. Lay, Substitution Kinetics, Albumin and Transferrin Affinities, and Hypoxia All Affect the Biological Activities of Anti-cancer Vanadium(V) Complexes, Inorg. Chem., 2023, 62(43), 17804–17817 CrossRef CAS PubMed.
- K. J. Ooms, S. E. Bolte, J. Smee, B. Baruah, D. C. Crans and T. Polenova, Investigating the vanadium environments in hydroxylamido V(V) dipicolinate complexes using 51 V NMR spectroscopy and density functional theory, Inorg. Chem., 2007, 46, 9286–9293 CrossRef PubMed.
- K. J. Ooms, S. E. Bolte, B. Baruah, M. A. Choudhary, D. C. Crans and T. Polenova, 51V solid-state NMR and density functional theory studies of eight-coordinate non-oxo vanadium complexes: Oxidized amavadin, Dalton Trans., 2009, 3262–3269 CAS.
- J. J. Smee, J. A. Epps, K. Ooms, S. Bolte, T. Polenova, B. Baruah, L. Yang, W. Ding, M. Li, G. R. Willsky, A. L. Cour, O. P. Anderson and D. C. Crans, Chloro-substituted dipicolinate vanadium complexes: Synthesis, solution, solid-state and insulin enhancing properties, J. Inorg. Biochem., 2009, 103, 575–584 CrossRef CAS PubMed.
- C. R. Cornman, G. J. Colpas, J. D. Hoeschele, J. Kampf and V. L. Pecoraro, Implications for the Spectroscopic Assignment of Vanadium Biomolecules: Structural and Spectroscopic Characterization of Monooxovanadium(V) Complexes Containing Catecholate and Hydroximate Based Noninnocent Ligands, J. Am. Chem. Soc., 1992, 114, 9925–9933 CrossRef CAS.
- M. Žižić, Z. Miladinović, M. Stanić, M. Hadžibrahimović, M. Živić and J. Zakrzewska, 51V NMR Investigation of Cell-Associated Vanadate Species in Phycomyces Blakesleeanus Mycelium, Res. Microbiol., 2016, 167(6), 521–528 CrossRef PubMed.
- D. C. Crans, B. J. Peters, X. Wu and C. C. McLauchlan, Does anion-cation organization in Na+-containing X-ray crystal structures relate to solution interactions in inhomogeneous nanoscale environments: Sodium-decavanadate in solid state materials, minerals, and microemulsions, Coord. Chem. Rev., 2017, 344, 115–130 CrossRef CAS.
- Y. Ilieva, L. Dimitrova, M. M. Zaharieva, M. Kaleva, P. Alov, I. Tsakovska, T. Pencheva, I. Pencheva-El Tibi, H. Najdenski and I. T. Pajeva, Cytotoxicity and Microbicidal Activity of Commonly Used Organic Solvents: A Comparative Study and Application to a Standardized Extract from Vaccinium macrocarpon, Toxics, 2021, 9(5), 92 CrossRef CAS PubMed.
- C. R. Jones, C. P. Butts and J. N. Harvey, Accuracy in determining interproton distances using Nuclear Overhauser Effect data from a flexible molecule, Beilstein J. Org. Chem., 2011, 7, 145–150 CrossRef CAS PubMed.
- N. Turro, J. Scaiano and V. Ramamurthy, Modern Molecular Photochemistry, University Science Books, Herndon, VA, 2010 Search PubMed.
- B. Figgis and M. Hitchman, Ligand field theory and its applications, Wiley-VCH, New York, 2000 Search PubMed.
- S. N. Choing, A. J. Francis, G. Clendenning, M. S. Schuurman, R. D. Sommer, I. Tamblyn, W. W. Weare and T. Cuk, Long-Lived LMCT in a d0 Vanadium(V) Complex by Internal Conversion to a State of 3dxy Character, J. Phys. Chem. C, 2015, 119(30), 17029–17038 CrossRef CAS.
- J. A. Bonadies and C. J. Carrano, Vanadium phenolates as models for vanadium in biological systems. 1. Synthesis, spectroscopy, and electrochemistry of vanadium complexes of ethylenebis[(o-hydroxyphenyl)glycine] and its derivatives, J. Am. Chem. Soc., 1986, 108(14), 4088–4095 CrossRef CAS.
- C. Milsmann, A. Levina, H. H. Harris, G. J. Foran, P. Turner and P. A. Lay, Charge Distribution in Chromium and Vanadium Catecholato Complexes: X-ray Absorption Spectroscopic and Computational Studies, Inorg. Chem., 2006, 45, 4743–4754 CrossRef CAS PubMed.
- A. A. Saleh and R. J. Crutchley, Solvent dependence of ligand to metal charge-transfer oscillator strength: outer-sphere perturbation of the ruthenium(III)-cyanamide bond, Inorg. Chem., 1990, 29(11), 2132–2135 CrossRef CAS.
- D. I. Ugwu and J. Conradiea, Metal complexes derived from bidentate ligands: Synthesis, catalytic and biological applications, Inorg. Chim. Acta, 2023, 553, 121518 CrossRef CAS.
- N. Ribeiro, I. Bulut, B. Cevatemre, C. Teixeira, Y. Yildizhan, V. André, P. Adão, J. C. Pessoa, C. Acilan and I. Correia, Cu(II) and V(IV)O complexes with tri- or tetradentate ligands based on (2-hydroxybenzyl)-l-alanines reveal promising anti-cancer therapeutic potential, Dalton Trans., 2021, 50, 157–169 RSC.
- S. Hazra, A. Paul, G. Sharma, B. Koch, M. F. C. G. da Silva and A. J. Pombeiro, Sulfonated Schiff base Sn(IV) complexes as potential anti-cancer agents, J. Inorg. Biochem., 2016, 162, 83–95 CrossRef CAS PubMed.
- M. Woźniczka, M. Lichawska, M. Sutradhar, M. Chmiela, W. Gonciarz and M. Pająk, Chemical characterization and biological evaluation of new cobalt(II) complexes with bioactive ligands, 2-picolinehydroxamic acid and reduced Schiff base N-(2-hydroxybenzyl) alanine, in terms of DNA binding and antimicrobial activity, Pharmaceuticals, 2021, 14(12), 1254 CrossRef PubMed.
- S. Petanidis, E. Kioseoglou, M. Hadzopoulou-Cladaras and A. Salifoglou, Novel ternary vanadium-betaine-peroxido species suppresses H-ras and matrix metalloproteinase-2 expression by increasing reactive oxygen species-mediated apoptosis in cancer cells, Cancer Lett., 2013, 335(2), 387–396 CrossRef CAS PubMed.
- S. Petanidis, E. Kioseoglou and A. Salifoglou, Metallodrugs in targeted cancer therapeutics: aiming at chemoresistance-related patterns and immunosuppressive tumor networks, Curr. Med. Chem., 2019, 26(4), 607–623 CrossRef CAS PubMed.
- S. Rodic and M. D. Vincent, Reactive Oxygen Species (ROS) Are a Key Determinant of Cancer's Metabolic Phenotype, Int. J. Cancer, 2018, 142(3), 440–448 CrossRef CAS PubMed.
- J. E. Klaunig and L. M. Kamendulis, The Role of Oxidative Stress in Carcinogenesis, Annu. Rev. Pharmacol. Toxicol., 2004, 44, 239–267 CrossRef CAS PubMed.
- D. Shi, Cancer Cell Surface Negative Charges: A Bio-Physical Manifestation of the Warburg Effect, Nano Life, 2017, 7(03n04), 1771001 CrossRef CAS.
- V. V. Khramtsov and R. J. Gillies, Janus-Faced Tumor Microenvironment and Redox, Antioxid. Redox Signaling, 2014, 21(5), 723–729 CrossRef CAS PubMed.
- S. J. Kim, H. S. Kim and Y. R. Seo, Understanding of ROS-Inducing Strategy in Anti-cancer Therapy, Oxid. Med. Cell. Longevity, 2019, 2019, 5381692 Search PubMed.
- T. Zaidieh, J. R. Smith, K. E. Ball and Q. An, ROS as a Novel Indicator to Predict Anti-cancer Drug Efficacy, BMC Cancer, 2019, 19(1), 1–14 CrossRef PubMed.
- D. Rehder, Biological and Medicinal Aspects of Vanadium, Inorg. Chem. Commun., 2003, 6, 604–617 CrossRef CAS.
- A. Levina, A. I. McLeod and P. A. Lay, Vanadium Speciation by XANES Spectroscopy: A Three-Dimensional Approach, Chem. – Eur. J., 2014, 20(38), 12056–12060 CrossRef CAS PubMed.
- D. C. Horton, D. VanDerveer, J. Krzystek, J. Telser, T. Pittman, D. C. Crans and A. A. Holder, Spectroscopic characterization of L-ascorbic acid-induced reduction of vanadium(V) dipicolinates: Forming V(III) and V(IV) complexes from V(V) dipicolinate derivatives, Inorg. Chem., 2014, 420, 112–119 CAS.
- J. Krzystek, A. Ozarowski, J. Telser and D. C. Crans, High-frequency and -field electron paramagnetic resonance of vanadium(IV, III, and II) complexes, Coord. Chem. Rev., 2015, 301–302, 123–133 CrossRef CAS.
- A. Levina, A. I. McLeod, L. E. Kremer, J. B. Aitken, C. J. Glover, B. Johannessen and P. A. Lay, Reactivity-Activity Relationships of Oral Anti-Diabetic Vanadium Complexes in Gastrointestinal Media: An X-ray Absorption Spectroscopic Study, Metallomics, 2014, 6, 1880–1888 CrossRef CAS PubMed.
- A. Huang, M. M. Pressnall, R. Lu, S. G. Huayamares, J. D. Griffin, C. Groer, B. J. DeKosky, M. L. Forrest and C. J. Berkland, Human Intratumoral Therapy: Linking Drug Properties and Tumor Transport of Drugs in Clinical Trials, J. Controlled Release, 2020, 326, 203–221 CrossRef CAS PubMed.
- A. Tasdogan, J. M. Ubellacker and S. J. Morrison, Redox Regulation in Cancer Cells during Metastasis, Cancer Discovery, 2021, 11(11), 2682–2692 CrossRef CAS PubMed.
- A. Levina and P. A. Lay, Vanadium(V/IV)−Transferrin Binding Disrupts the Transferrin Cycle and Reduces Vanadium Uptake and Antiproliferative Activity in Human Lung Cancer Cells, Inorg. Chem., 2020, 59(22), 16143–16153 CrossRef CAS PubMed.
- R. Andreoli, G. Battistuzzi Gavioli, G. Grandi, L. Benedetti and A. Rastelli, Half-wave potential of sulfa drugs as structural index, J. Electroanal. Chem., 1980, 108, 77–86 CrossRef CAS.
- K. C. Gross and P. G. Seybold, Substituent Effects on the Physical Properties and pKa of Phenol, Int. J. Quantum Chem., 2001, 85(4–5), 569–579 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, A Survey of Hammett Substituent Constants and Resonance and Field Parameters, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- H. N. Simpson, C. K. Hancock and E. A. Meyers, The Correlation of the Electronic Spectra, Acidities, and Polarographic Oxidation Half-Wave Potentials of 4-Substituted 2-Chlorophenols with Substituent Constants1, J. Org. Chem., 1965, 30, 2678–2683 CrossRef CAS.
- S. V. Jovanovic, M. Tosic and M. G. Simic, Use of the Hammett Correlation and σ+ for Calculation of One-Electron Redox Potentials of Antioxidants, J. Phys. Chem., 1991, 95, 10824–10827 CrossRef CAS.
- A. M. Bond, G. A. Lawrance, P. A. Lay and A. M. Sargeson, Electrochemistry of Macrobicyclic (Hexaamine)cobalt(III) Complexes. Metal-Centered and Substituent Reductions, Inorg. Chem., 1983, 22, 2010–2021 CrossRef CAS.
- G. A. Lawrance, P. A. Lay and A. M. Sargeson, Organic Substituent Effects in Macrobicyclic Hexaamine Cobalt(III/II) Complexes: A New Method for Obtaining Polar Substituent Constants, Inorg. Chem., 1990, 29, 4808–4816 CrossRef CAS.
- R. Pinnataip and B. P. Lee, Oxidation Chemistry of Catechol Utilized in Designing Stimuli-Responsive Adhesives and Antipathogenic Biomaterials, ACS Omega, 2021, 6(8), 5113–5118 CrossRef CAS PubMed.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS.
- D. P. Jones, Redox Potential of the GSH/GSSG Couple: Assay and Biological Significance, Methods Enzymol., 2002, 348, 93–112 CAS.
- F. Xing, Q. Hu, Y. Qin, J. Xu, B. Zhang, X. Yu and W. Wang, The relationship of redox with hallmarks of Cancer: the importance of homeostasis and context, Front. Oncol., 2022,(12), 862743 CrossRef CAS PubMed.
Footnotes |
| † This publication is also part of the special issue for the International Vanadium Symposium in Lisbon, Portugal, November 22–24, 2023 and apart of the collection of papers celebrating the 85th birthday of George M. Whitesides. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj01223b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |