
Facile synthesis of polypeptoids bearing bulky sidechains via urea accelerated ring-opening polymerization of α-amino acid N-substituted N-carboxyanhydrides†
Kang
Chen‡
 a,
Yueming
Wu‡
a,
Xue
Wu
b,
Min
Zhou
b,
Ruiyi
Zhou
b,
Jiangzhou
Wang
b,
Ximian
Xiao
b,
Yuan
Yuan
b and
Runhui
Liu
*ab
a,
Yueming
Wu‡
a,
Xue
Wu
b,
Min
Zhou
b,
Ruiyi
Zhou
b,
Jiangzhou
Wang
b,
Ximian
Xiao
b,
Yuan
Yuan
b and
Runhui
Liu
*ab
aState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: rliu@ecust.edu.cn
bKey Laboratory for Ultrafine Materials of Ministry of Education, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, Research Center for Biomedical Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, China
First published on 7th December 2021
Abstract
Polypeptoids, as synthetic mimics of polypeptides, exhibit a variety of biological functions and excellent proteolytic stability. Polypeptoids can be synthesized by the ring-opening polymerization of α-amino acid N-substituted N-carboxyanhydrides (NNCAs); however, they suffer from the generally slow reactivity and poor stability of NNCAs, especially those with bulky substitutes. This long-standing challenge greatly limits the synthesis of polypeptoids with diverse structures. Herein, we found that commercially available 1,3-bis[3,5-bis(trifluoromethyl)phenyl]urea can greatly accelerate the primary amine-initiated ring-opening polymerization of NNCAs by activating the NNCA carbonyl via hydrogen bonding interactions. Urea-catalyzed NNCA polymerization is compatible with diverse NNCAs in preparing polypeptoids with variable polymer lengths and narrow dispersity and is especially suitable for inactive NNCAs bearing bulky N-substitutes, such as cyclohexyl-NNCA. This urea-catalyzed NNCA polymerization strategy will substantially increase the structural diversity and functional study of polypeptoids, implying wider and diverse applications of these polypeptide mimics.
Introduction
Polypeptoids represent a type of polypeptide mimics with a backbone composed of N-substituted glycine, which overcomes peptides’ shortcoming of poor enzyme stability.1 They have been actively studied in the fields of antibacterial agents,2–4 drug and gene delivery,5–7 tissue engineering,8 antifouling,9–11 and heat-responsive materials,12,13 which exhibit their great potential in applications. Polypeptoids were prepared by the ring-opening polymerization of α-amino acid N-substituted N-carboxyanhydrides (NNCAs).14–18 The functional study of polypeptoids involves the diverse functional groups of NNCAs.19,20 In exploring host defense peptide mimicking antimicrobial polypeptides,21–25 it was found that hydrophobic residues, such as the bulky cyclohexyl group, are critical in defining the antimicrobial activity and cytotoxicity of antimicrobial polypeptides.26,27 However, currently only N-methyl glycine N-carboxyanhydride (sarcosine-NCA) is widely used to prepare polypeptoids using various polymerization strategies28–31 due to the poor reactivity especially for NNCAs bearing bulky side chains like cyclohexyl.32 To address this long-standing challenge, we recently reported the fast polymerization of NNCAs to prepare polypeptoids using Li/Na/KHMDS as the initiator.32 Related advances have been used to initiate superfast and moisture-insensitive polymerization of α-amino acid N-carboxyanhydrides (NCAs) to prepare polypeptides.33–35Among all strategies for polymerization of NNCAs, primary amines are the dominantly used initiators for ring-opening polymerization of NNCAs to prepare polypeptoids.36–42 The merit of primary amine initiators lies in the easy introduction of diverse functional groups into the C-termini of polypeptoids via the initiators. Thus, we turn our attention to accelerating primary amine-initiated polymerization of NNCAs, especially for NNCAs bearing bulky side chains and with poor reactivity for polymerization. It has been reported that organocatalysts can accelerate polymerization reactions through hydrogen bonding interactions, thus enabling the preparation of polymers with controllable molecular weight and narrow distribution.43–47 Specifically, this strategy has been used to activate NCAs to achieve fast polymerization using organocatalysts.48,49 Inspired by these precedent studies, herein we explored commercially available 1,3-bis(3,5-bis(trifluoro-ethyl)phenyl)thiourea (TU–S) and 1,3-bis[3,5-bis(trifluoromethyl)phenyl]urea (U–O) as organocatalysts for primary amine-initiated NNCA polymerization and found that this strategy greatly accelerates the reaction via hydrogen bonding with the NNCA carbonyl and results in rapid ring-opening polymerization of NNCAs, even those bearing bulky side chains, to prepare polypeptoids efficiently (Scheme 1).
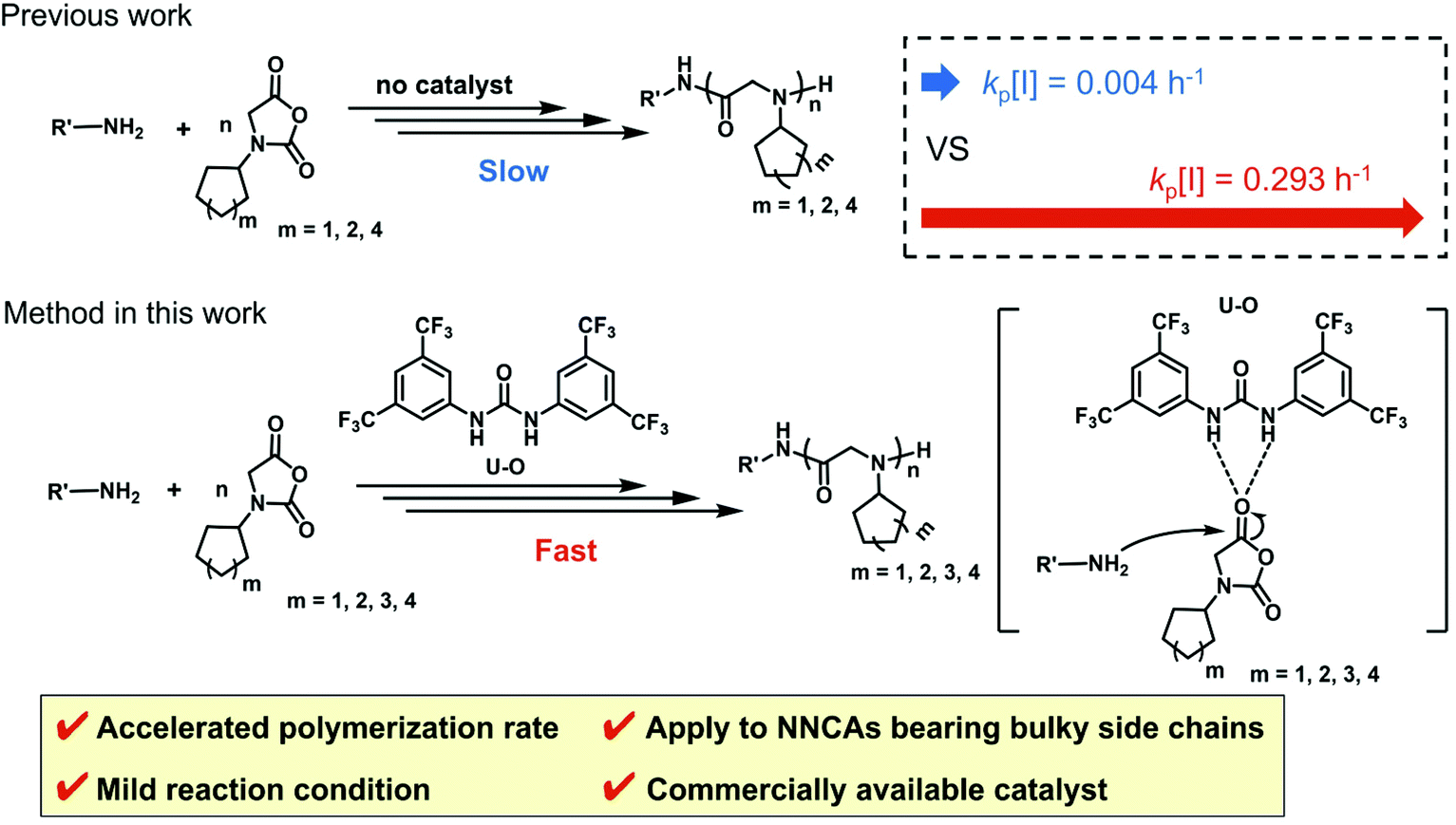 | ||
| Scheme 1 Primary amine-initiated polymerization of NNCAs bearing bulky side chains, with and without using U–O as the catalyst. | ||
Results and discussion
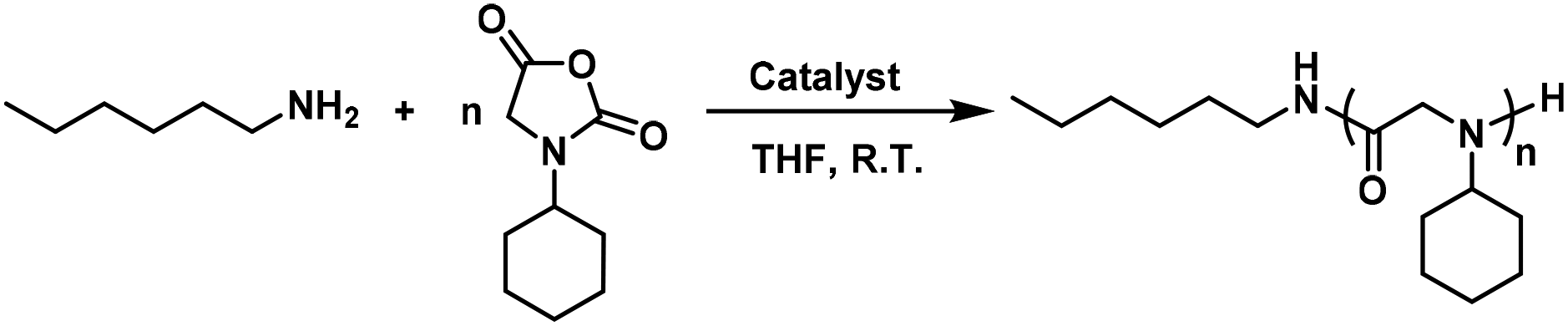
Using N-cyclohexyl glycine N-carboxyanhydride (cyclohexyl-NNCA) as a model, we found that the ring-opening polymerization of NNCA was very slow using a primary amine, n-hexylamine, as the initiator. The polymerization took 7 days to prepare a polymer with a chain length of 20, and took a much longer time to prepare polypeptoids with longer chain lengths (Table 1, entry 1). Using TU–S as the organocatalyst at a concentration of 1/5 that of the NNCA monomer, the ring-opening polymerization of cyclohexyl-NNCA was greatly accelerated to complete within 22 hours and gave polypeptoids at about 20-mer in tetrahydrofuran (THF). However, the GPC trace of the resulting polypeptoid showed a shoulder (Table 1, entry 2, GPC trace in Fig. S1†). So, we continued to examine the urea catalyst U–O for its ability to accelerate the polymerization of NNCAs. We found that U–O, at a concentration of 1/5 that of the NNCA monomer, has superior performance in accelerating the polymerization of NNCA, and reduces the polymerization time from 7 days to 5 hours (Table 1, entry 3). In addition, using U–O as the catalyst, we obtained polypeptoids with expected polymer lengths, a narrow dispersity (Đ = 1.20), and a monomodal distribution without shoulder (Fig. S2†). The observation that U–O showed better catalytic performance in NNCA polymerization than did TU–S is probably owing to the stronger electrophilicity of the oxygen atom in U–O than that of the sulfur atom in TU–S, resulting in a stronger hydrogen bonding interaction between U–O and the carbonyl in the NNCA ring.50|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[Cat] :[I]:[Cat] |
Time | M n,calcd (g mol−1) | M n,SEC (g mol−1) | Đ |
| a The n-hexylamine-initiated ring-opening polymerization of cyclohexyl-NNCA in THF catalyzed by TU–S or U–O ([M]0 = 1 M). b All polypeptoids were characterized by gel permeation chromatography (GPC) using 0.01 M LiBr in DMF as the mobile phase at a flow rate of 1 mL min−1 at 50 °C. Mn,SEC is the number-average molecular weight. Đ is the dispersity index. c GPC curve indicates a shoulder. | ||||||
| 1 | None | 20/1/0 | 7 days | 2900 | 2900 | 1.24 |
| 2c | TU–S | 20/1/4 | 22 h | 2900 | 3000 | 1.20 |
| 3 | U–O | 20/1/4 | 5 h | 2900 | 3400 | 1.20 |
| 4 | U–O | 10/1/1 | 5 h | 1500 | 1900 | 1.13 |
| 5 | U–O | 20/1/1 | 7 h | 2900 | 3400 | 1.20 |
| 6 | U–O | 50/1/2.5 | 12 h | 7100 | 6500 | 1.14 |
| 7 | U–O | 100/1/5 | 16 h | 14000 |
9700 | 1.22 |
| 8 | U–O | 200/1/10 | 21 h | 27900 |
14000 |
1.20 |
The catalytic performance of U–O in accelerating the ring-opening polymerization of cyclohexyl-NNCA was evaluated using polymerization kinetics obtained from HPLC analysis. The polymerization kinetics indicated a very slow ring-opening polymerization of cyclohexyl-NNCA initiated by n-hexylamine with M/I = 50 at an NNCA concentration of 1 M in THF, having the kp[I] value of 0.004 h−1 (Fig. 1). Slow polymerization was due to the weak nucleophilicity of the reactive center, an N-cyclohexyl substituted secondary amine having steric hindrance for continuous nucleophilic addition. We also investigated how the equivalent of U–O relative to the NNCA monomer affects the polymerization rate. The addition of 5% U–O into the polymerization of cyclohexyl-NNCA accelerated the reaction substantially, with the kp[I] value increasing from 0.004 h−1 (without catalyst) to 0.293 h−1. Increasing the amount of U–O catalyst to 10% and 20% relative to the cyclohexyl-NNCA moderately increased the polymerization rate further to have a kp[I] value of 0.362 h−1 and 0.403 h−1, respectively (Fig. 1). These studies demonstrated that U–O has superior performance in catalyzing primary amine-initiated NNCA polymerization to greatly accelerate the polymerization rate of inactive NNCAs up to 100 fold.
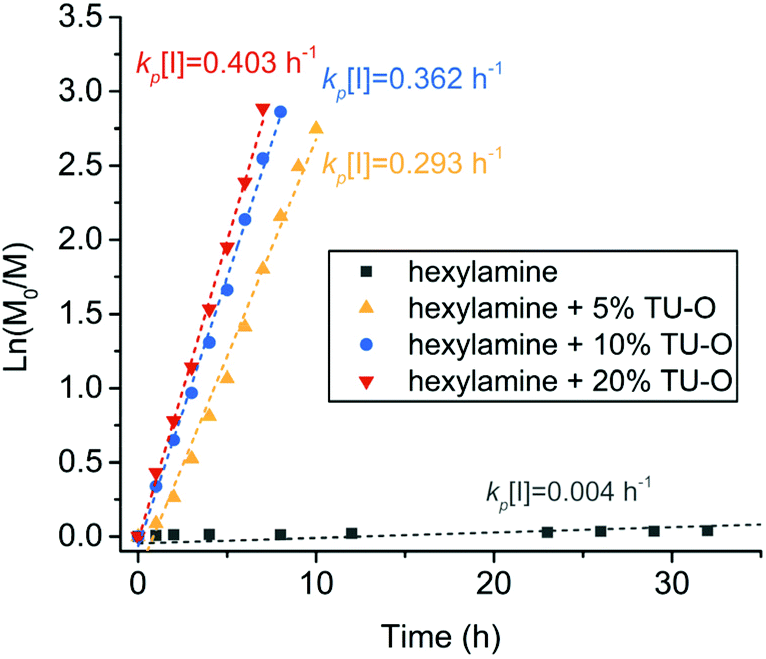 | ||
| Fig. 1 Polymerization kinetics of n-hexylamine-initiated polymerization of cyclohexyl-NNCA catalyzed by different amounts of U–O in THF at room temperature ([M]:[I] = 50:1, [M]0 = 1 M). | ||
U–O catalyzed ring-opening polymerization of cyclohexyl-NNCA using n-hexylamine as the initiator can prepare polypeptoids with variable chain lengths. With an NNCA-to-initiator ratio of 10, the cyclohexyl-NNCA monomers were completely converted to polypeptoids in 5 hours at room temperature with the desired molecular weight (Mn = 1900 g mol−1) and narrow dispersity at Đ = 1.13 (Table 1, entry 4). When the monomer-to-initiator ratio was gradually increased from 20 to 200, the obtained polypeptoids have incrementally increased molecular weight (Mn = 3400–14000 g mol−1) and narrow dispersities (Đ = 1.14–1.22, Table 1, entries 5–8). All the obtained polypeptoids exhibited sharp and monomodal GPC traces (GPC traces in Fig. S3–S7†). This observation echoes the importance of exploring a new synthetic strategy to greatly accelerate the polymerization of NNCAs in preparing polypeptoids with variable molecular weights.32 When monomer-to-initiator ratios of 100 and 200 were used, we observed a lower Mn than the theoretical value, possibly due to intramolecular transamidation in the polymerization process as reported in the precedent literature as a side reaction in the polymerization of NNCA.14,51
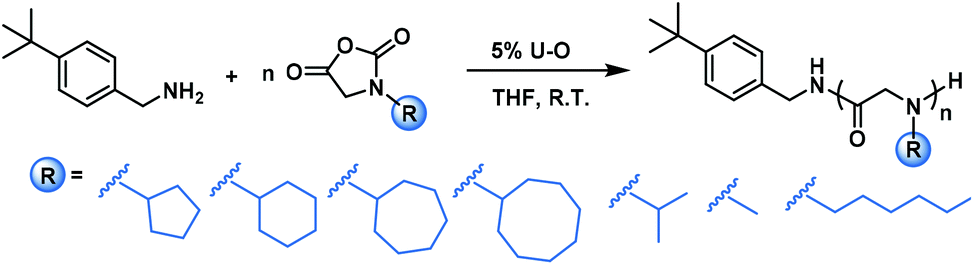
To test the substrate compatibility of this U–O catalyzed polymerization on NNCA, we further examined NNCAs bearing various side chain groups, such as N-cyclopentyl glycine N-carboxyanhydride (cyclopentyl-NNCA), N-cycloheptyl glycine N-carboxyanhydride (cycloheptyl-NNCA), N-cyclooctyl glycine N-carboxyanhydride (cyclooctyl-NNCA) and N-isopropyl glycine N-carboxyanhydride (isopropyl-NNCA). U–O substantially accelerated the polymerization of these NNCAs to complete within 9–12 hours (Table 2, entries 1–5, GPC traces in Fig. S8–S12†). For the polymerization of even more bulky N-tert-butyl glycine N-carboxyanhydride (tert-butyl-NNCA), only oligomers were obtained, which was likely because the great steric hindrance of the tert-butyl group substantially reduces the nucleophilicity of the secondary amine reactive center (Table S2 and Fig. S17†). We also tested U–O catalyzed polymerization of NNCAs bearing less bulky side chains, sarcosine-NCA and N-hexyl glycine N-carboxyanhydride (hexyl-NNCA), and observed very quick completion of polymerization within 10 min and 30 min for sarcosine-NCA and hexyl-NNCA, respectively (Table 2, entries 6 and 7, GPC traces in Fig. S13 and S14†). These studies indicated that U–O is compatible with NNCAs in preparing polypeptoids with diverse side chains.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Monomer | [M]:[I]:[Cat] |
Time | M n,calcd (g mol−1) | M n,SEC (g mol−1) | Đ |
| a The ring-opening tert-butylbenzylamine-initiated polymerization of NNCAs bearing various N-substituted groups catalyzed by 5% U–O (molar ratio, [M]0 = 1 M) in THF at room temperature. b All polypeptoids were characterized by GPC using 0.01 M LiBr in DMF as the mobile phase at a flow rate of 1 mL min−1 at 50 °C. Mn,SEC is the number-average molecular weight. Đ is the dispersity index. | ||||||
| 1 | Cyclopentyl-NNCA | 20/1/1 | 10 h | 2700 | 2500 | 1.27 |
| 2 | Cyclohexyl-NNCA | 20/1/1 | 10 h | 2900 | 3400 | 1.21 |
| 3 | Cycloheptyl-NNCA | 20/1/1 | 11 h | 3200 | 3400 | 1.21 |
| 4 | Cyclooctyl-NNCA | 20/1/1 | 12 h | 3500 | 2700 | 1.25 |
| 5 | Isopropyl-NNCA | 20/1/1 | 9 h | 2100 | 2700 | 1.14 |
| 6 | Sarcosine-NCA | 20/1/1 | 10 min | 1600 | 1400 | 1.20 |
| 7 | Hexyl-NNCA | 20/1/1 | 30 min | 3000 | 2900 | 1.21 |
The superior performance of U–O catalyzed NNCA polymerization to prepare polypeptoids encouraged us to explore the mechanism. We proposed that U–O activated the carbonyl group on the 5 position of the NNCA monomer via hydrogen bonding, which made NNCA more susceptible to be attacked by nucleophilic amines and realized the rapid ring-opening polymerization of NNCA (Scheme 2). In the chain initiation step, primary amine initiators nucleophilically attack the NNCA C5 carbonyl and generate the intermediate which is a secondary amine and act as the reactive center for continuous propagation. In the further chain propagation step, a secondary amine bearing a bulky cyclohexyl substituent acts as the reactive center and has significantly lower nucleophilicity to NNCAs than does the primary amine initiator (Scheme 2).
 | ||
| Scheme 2 Proposed mechanism of primary amine-initiated ring-opening polymerization of NNCA catalyzed by U–O. | ||
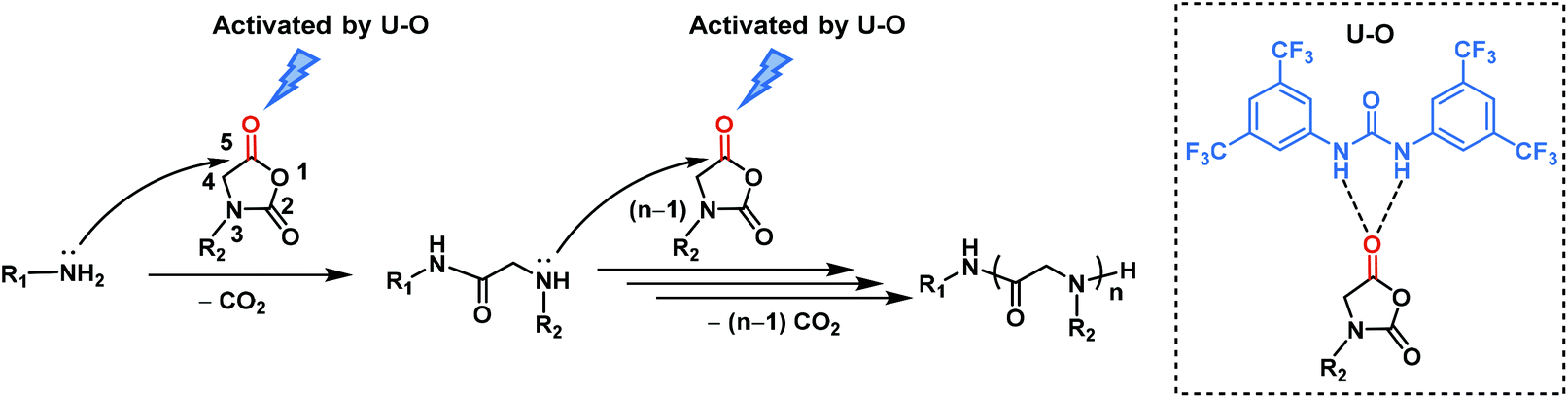
To explore the initiation step, we closely examined the 1:1 molar ratio mixture of an initiator tert-butylbenzylamine and cyclohexyl-NNCA. High resolution electrospray ionization mass spectroscopy (HRESI-MS) showed a characteristic peak at m/z 303.2432, referring to the active species in the initiation step, which supported our proposed mechanism in the chain initiation step (Fig. 2a). We also characterized the final polypeptoid obtained from the polymerization of cyclohexyl-NNCA using 1H NMR spectroscopy, and found the characteristic peaks correlating to the tert-butylbenzyl group, which echoes the above conclusion that the polymerization is initiated via the nucleophilic addition of a primary amine initiator to the NNCA ring and incorporation of a C-terminal functional group by the primary amine (Fig. 2b). Moreover, the existence of tert-butylbenzyl terminal groups and cyclohexyl-NNCA residue repeating units was clearly identified in the characterization using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectroscopy (Fig. 3). All these studies indicated that U–O catalyzed NNCA polymerization follows the normal amine mechanism (NAM).52 We hypothesize that adding U–O into the NNCA polymerization reaction will not change the mechanism, but could activate NNCAs to facilitate the nucleophilic attack step and achieve rapid polymerization.
 | ||
| Fig. 2 (a) HRESI-MS analysis of cyclohexyl-NNCA and tert-butylbenzylamine at the molar ratio of 1:1 with 5% U–O (molar ratio) in THF ([M]0 = 0.5 M). The peak of m/z = 304.2462 belongs to the isotope peak. (b) 1H NMR spectrum of polypeptoid synthesized from tert-butylbenzylamine-initiated polymerization of cyclohexyl-NNCA catalyzed by 5% U–O in THF at room temperature ([M]/[I] = 20, [M]0 = 1 M). | ||
 | ||
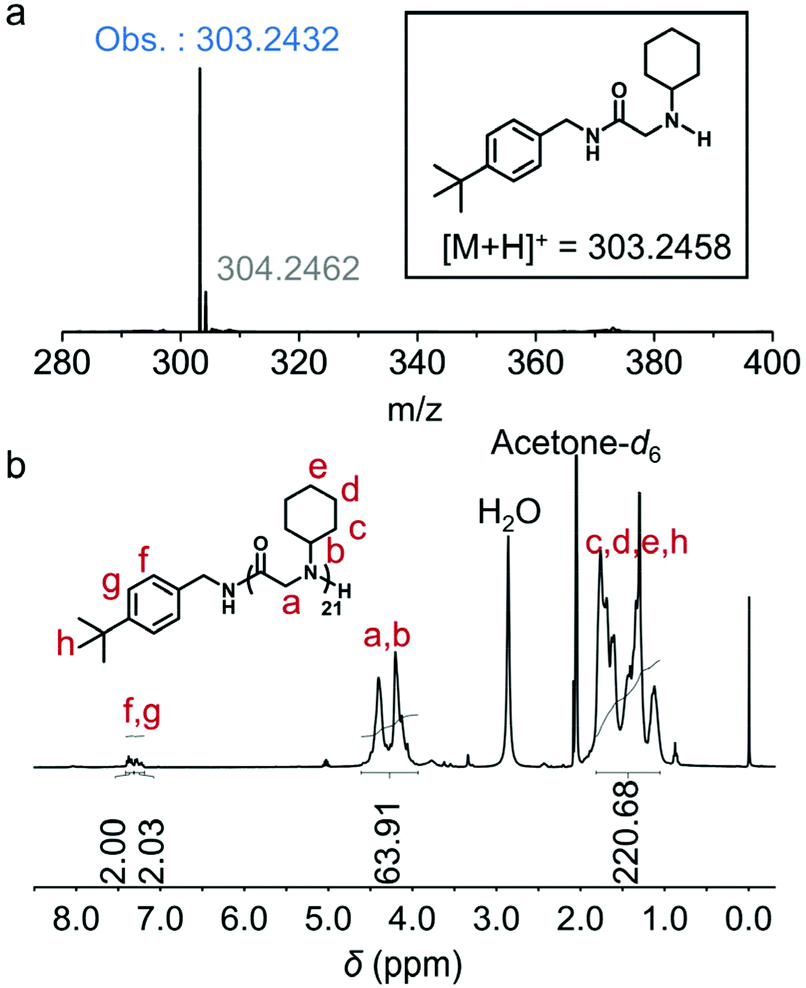
| Fig. 3 MALDI-TOF-MS characterization of polypeptoid prepared from tert-butylbenzylamine-initiated polymerization of cyclohexyl-NNCA catalyzed by 5% U–O (molar ratio) in THF at room temperature ([M]/[I] = 20, [M]0 = 1 M). Detailed analysis is shown in Fig. S15.† | ||
As aforementioned, the addition of U–O significantly increased the polymerization rate of NNCAs bearing bulky substituents, using primary amine as the initiator. This observation encouraged us to determine the catalytic mechanism of U–O using NMR spectroscopy and density functional theory (DFT) calculation. A close comparison of the 1H NMR spectra of U–O and U–O/cyclohexyl-NNCA mixture revealed that the N–H on U–O shifts downfield from 7.67 ppm to 7.73 ppm due to the hydrogen bonding interaction between U–O and cyclohexyl-NNCA, as we proposed (Fig. 4). The 13C NMR spectroscopy revealed that both carbon atoms on the NNCA C5 carbonyl and C2 carbonyl shift downfield slightly after adding U–O as the catalyst (Fig. S16†). This result indicates that U–O could activate both carbonyl groups on the NNCA monomer. Nevertheless, the NNCA C5 carbonyl is more easily attacked by nucleophiles than is the C2 carbonyl due to the higher electrophilicity of the C5 carbonyl. Furthermore, the geometry-optimized structure of U–O and cyclohexyl-NNCA in THF was obtained using DFT calculation, which suggested that U–O served as a hydrogen bond donor to form two hydrogen bonds with one cyclohexyl-NNCA C5 carbonyl with an average hydrogen bond length of 3.10 Å (Fig. 5). The structure also featured a prolongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in cyclohexyl-NNCA induced by the hydrogen bonds. Furthermore, the binding energy of U–O and cyclohexyl-NNCA (ΔE = −7.26 kcal mol−1) in THF suggested that the hydrogen bonds are medium strength hydrogen bonds.53 These studies revealed that U–O activates the cyclohexyl-NNCA monomer through hydrogen bonding interaction to increase its reactivity as an electrophile and accelerate the polymerization of NNCA. DFT calculation also revealed that the hydrogen bonding interaction of TU–S and cyclohexyl-NNCA (ΔE = −7.19 kcal mol−1) was slightly lower than that of U–O and cyclohexyl-NNCA (ΔE = −7.26 kcal mol−1), which is probably caused by the oxygen atom in U–O being more electrophilic than the sulfur atom in TU–S. This result is consistent with the experiment wherein both TU–S and U–O exhibited excellent catalytic performance in speeding up NNCA polymerization and could reduce the polymerization time of cyclohexyl-NNCA ([M]/[I] = 20) from 7 days to 22 hours and 5 hours, respectively (Table 1, entries 2 and 3).
O bond in cyclohexyl-NNCA induced by the hydrogen bonds. Furthermore, the binding energy of U–O and cyclohexyl-NNCA (ΔE = −7.26 kcal mol−1) in THF suggested that the hydrogen bonds are medium strength hydrogen bonds.53 These studies revealed that U–O activates the cyclohexyl-NNCA monomer through hydrogen bonding interaction to increase its reactivity as an electrophile and accelerate the polymerization of NNCA. DFT calculation also revealed that the hydrogen bonding interaction of TU–S and cyclohexyl-NNCA (ΔE = −7.19 kcal mol−1) was slightly lower than that of U–O and cyclohexyl-NNCA (ΔE = −7.26 kcal mol−1), which is probably caused by the oxygen atom in U–O being more electrophilic than the sulfur atom in TU–S. This result is consistent with the experiment wherein both TU–S and U–O exhibited excellent catalytic performance in speeding up NNCA polymerization and could reduce the polymerization time of cyclohexyl-NNCA ([M]/[I] = 20) from 7 days to 22 hours and 5 hours, respectively (Table 1, entries 2 and 3).
 | ||
| Fig. 4 Comparison of 1H NMR spectra for U–O only and cyclohexyl-NNCA mixed with U–O (1:1, molar ratio), in CDCl3/MeCN (CDCl3:MeCN = 85:15, v/v). | ||
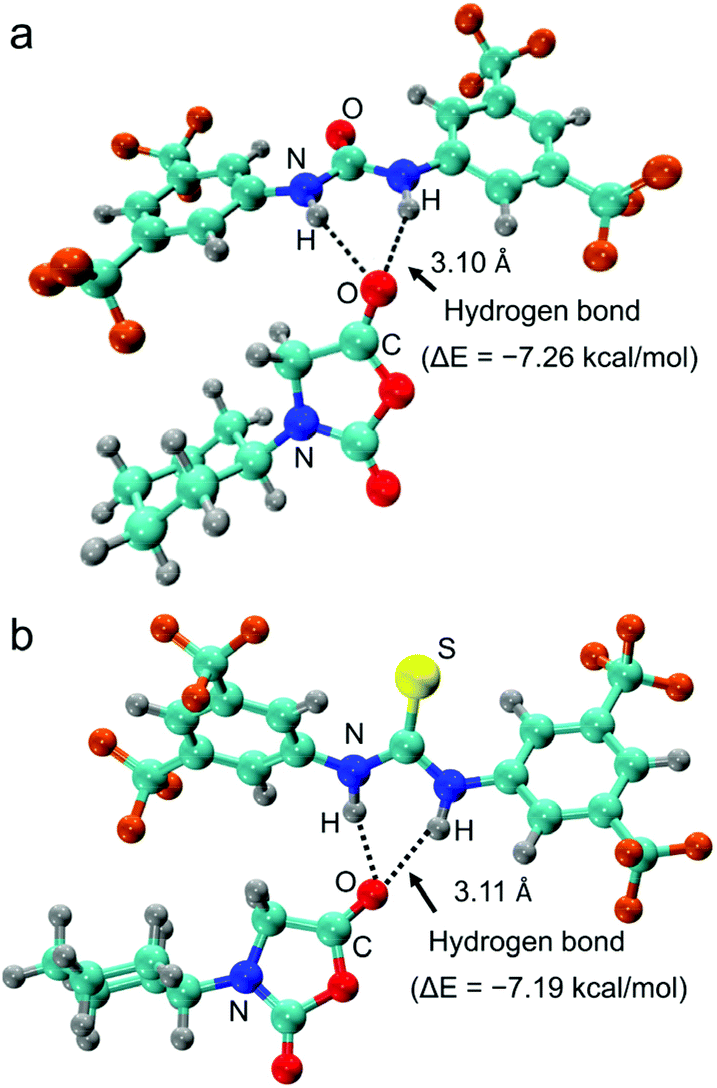 | ||
| Fig. 5 The DFT-optimized geometry structure showing the hydrogen bonding interaction between (a) U–O and cyclohexyl-NNCA in THF (silver: H, cyan: C, blue: N, red: O, and brown: F), (b) TU–S and cyclohexyl-NNCA in THF (silver: H, cyan: C, blue: N, red: O, yellow: S and brown: F). | ||
Conclusions
Polypeptoids have attracted more and more attention as a class of polypeptide mimics for diverse applications and are prepared from the ring-opening polymerization of NNCAs. The generally slow reactivity and poor stability of NNCAs greatly limit the synthesis of polypeptoids with diverse structures and applications. Currently, the N-methyl glycine N-carboxyanhydride (sarcosine-NCA) is dominantly used because sarcosine-NCA is the least bulky N-substitute NNCA to undergo easy polymerization. However, it is a long-standing challenge to prepare polypeptoids with diverse structures from NNCAs with bulky substitutes. In this study, we report a fast NNCA polymerization strategy using primary amine as the initiator and 1,3-bis[3,5-bis(trifluoromethyl)phenyl]urea as the catalyst. The use of urea catalyst greatly accelerates the reaction rate of the polymerization by activating the carbonyl in the NNCA ring via hydrogen bonding interactions, without changing the NAM mechanism for NNCA polymerization. This fast polymerization strategy is compatible with diverse NNCAs, including bulky N-substituted NNCAs, to prepare polypeptoids that are otherwise inaccessible without using the catalyst. The use of a commercially available urea catalyst and compatibility with diverse NNCAs together imply the great potential of this strategy in fast NNCA polymerization to prepare polypeptoids for a variety of applications.Author contributions
K. C., Y. W. and R. L. conceived the idea, proposed the strategy, designed the experiments, evaluated the data, and wrote the manuscript together. K. C. performed the majority of the experiments. X. W. performed DFT calculation and evaluated the data. M. Z. participated in NNCA synthesis and initial polymerization tests. R. Z. participated in HPLC analysis. J. W. participated in NNCA synthesis. X. X. conducted the MALDI-TOF assay. Y. Y. participated in data analysis. All the authors proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 22075078, 21774031, 21861162010, 31800801), Program of Shanghai Academic/Technology Research Leader (20XD1421400), Research Program of State Key Laboratory of Bioreactor Engineering, and the Fundamental Research Funds for the Central Universities (JKD01211520). We also thank the staff members of the Mass Spectrometry System at the National Facility for Protein Science in Shanghai (NFPS), Zhangjiang Lab, China for providing technical support and assistance in data collection and analysis.References
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Bioorg. Med. Chem. Lett., 1994, 4, 2657–2662 CrossRef CAS.
- B. Mojsoska and H. Jenssen, Pharmaceuticals, 2015, 8, 366–415 CrossRef CAS PubMed.
- K. Andreev, M. W. Martynowycz, M. L. Huang, I. Kuzmenko, W. Bu, K. Kirshenbaum and D. Gidalevitz, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1414–1423 CrossRef CAS PubMed.
- J. Xie, M. Zhou, Y. Qian, Z. Cong, S. Chen, W. Zhang, W. Jiang, C. Dai, N. Shao, Z. Ji, J. Zou, X. Xiao, L. Liu, M. Chen, J. Li and R. Liu, Nat. Commun., 2021, 12, 5898 CrossRef CAS PubMed.
- A. Li and D. Zhang, Biomacromolecules, 2016, 17, 852–861 CrossRef CAS PubMed.
- L. Zhu, J. M. Simpson, X. Xu, H. He, D. Zhang and L. Yin, ACS Appl. Mater. Interfaces, 2017, 9, 23476–23486 CrossRef CAS PubMed.
- Y. Zhang, Z. Heidari, Y. Su, T. Yu, S. Xuan, M. Omarova, Y. Aydin, S. Dash, D. Zhang and V. John, Langmuir, 2019, 35, 15335–15343 CrossRef CAS PubMed.
- E. Hara, M. Ueda, C. J. Kim, A. Makino, I. Hara, E. Ozeki and S. Kimura, J. Pept. Sci., 2014, 20, 570–577 CrossRef CAS PubMed.
- K. H. Lau, T. S. Sileika, S. H. Park, A. M. Sousa, P. Burch, I. Szleifer and P. B. Messersmith, Adv. Mater. Interfaces, 2015, 2, 1400225 CrossRef PubMed.
- Y. Hu, Y. Hou, H. Wang and H. Lu, Bioconjugate Chem., 2018, 29, 2232–2238 CrossRef CAS PubMed.
- F. Pan, K. H. Aaron Lau, P. B. Messersmith, J. R. Lu and X. Zhao, Langmuir, 2020, 36, 12309–12318 CrossRef CAS PubMed.
- X. Fu, C. Xing and J. Sun, Biomacromolecules, 2020, 21, 4980–4988 CrossRef CAS PubMed.
- D. Liu and J. Sun, Polymers, 2020, 12, 2973 CrossRef CAS PubMed.
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS PubMed.
- A. Li, L. Lu, X. Li, L. He, C. Do, J. C. Garno and D. Zhang, Macromolecules, 2016, 49, 1163–1171 CrossRef CAS.
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002–2012 CrossRef CAS.
- S. Varlas, P. G. Georgiou, P. Bilalis, J. R. Jones, N. Hadjichristidis and R. K. O'Reilly, Biomacromolecules, 2018, 19, 4453–4462 CrossRef CAS PubMed.
- P. Salas-Ambrosio, A. Tronnet, M. Since, S. Bourgeade-Delmas, J.-L. Stigliani, A. Vax, S. Lecommandoux, B. Dupuy, P. Verhaeghe and C. Bonduelle, J. Am. Chem. Soc., 2021, 143, 3697–3702 CrossRef CAS PubMed.
- X. Fu, Z. Li, J. Wei, J. Sun and Z. Li, Polym. Chem., 2018, 9, 4617–4624 RSC.
- J. Sun, M. Li, M. Lin, B. Zhang and X. Chen, High Antibacterial Activity and Selectivity of the Versatile Polysulfoniums that Combat Drug Resistance, Adv. Mater., 2021, 33, 2104402 CrossRef CAS PubMed.
- C. Ghosh, G. B. Manjunath, P. Akkapeddi, V. Yarlagadda, J. Hoque, D. S. Uppu, M. M. Konai and J. Haldar, J. Med. Chem., 2014, 57, 1428–1436 CrossRef CAS PubMed.
- L. Richter, M. Hijazi, F. Arfeen, C. Krumm and J. C. Tiller, Macromol. Biosci., 2018, 18, 1700389 CrossRef PubMed.
- M. Zhou, Y. Qian, J. Xie, W. Zhang, W. Jiang, X. Xiao, S. Chen, C. Dai, Z. Cong, Z. Ji, N. Shao, L. Liu, Y. Wu and R. Liu, Angew. Chem., Int. Ed., 2020, 59, 6412–6419 CrossRef CAS PubMed.
- L. Wei, R. Gao, M. Wang, Y. Wang, Y. Shi, M. Gu and J. Cai, Biomater. Sci., 2021, 9, 3410–3424 RSC.
- Z. Si, H. W. Lim, M. Y. F. Tay, Y. Du, L. Ruan, H. Qiu, R. Zamudio-Vazquez, S. Reghu, Y. Chen, W. S. Tiong, K. Marimuthu, P. P. De, O. T. Ng, Y. Zhu, Y.-H. Gan, Y. R. Chi, H. Duan, G. C. Bazan, E. P. Greenberg, M. B. Chan-Park and K. Pethe, Angew. Chem., Int. Ed., 2020, 59, 6819–6826 CrossRef CAS PubMed.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. P. Falk, B. P. Mowery, A. J. Karlsson, B. Weisblum, S. P. Palecek, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4333–4342 CrossRef CAS PubMed.
- H. R. Kricheldorf, C. v. Lossow, G. Schwarz and D. Fritsch, Macromol. Chem. Phys., 2005, 206, 1165–1170 CrossRef CAS.
- C. Fetsch and R. Luxenhofer, Macromol. Rapid Commun., 2012, 33, 1708–1713 CrossRef CAS PubMed.
- A. Doriti, S. M. Brosnan, S. M. Weidner and H. Schlaad, Polym. Chem., 2016, 7, 3067–3070 RSC.
- R. M. England, J. I. Moss, A. Gunnarsson, J. S. Parker and M. B. Ashford, Biomacromolecules, 2020, 21, 3332–3341 CrossRef CAS PubMed.
- Y. Wu, M. Zhou, K. Chen, S. Chen, X. Xiao, Z. Ji, J. Zou and R. Liu, Chin. Chem. Lett., 2021, 32, 1675–1678 CrossRef CAS.
- Y. Wu, D. Zhang, P. Ma, R. Zhou, L. Hua and R. Liu, Nat. Commun., 2018, 9, 5297 CrossRef CAS PubMed.
- Y. Wu, W. Zhang, R. Zhou, Q. Chen, C. Xie, H. Xiang, B. Sun, M. Zhu and R. Liu, Chin. J. Polym. Sci., 2020, 38, 1131–1140 CrossRef CAS.
- Y. Wu, K. Chen, X. Wu, L. Liu, W. Zhang, Y. Ding, S. Liu, M. Zhou, N. Shao, Z. Ji, J. Chen, M. Zhu and R. Liu, Angew. Chem., Int. Ed., 2021, 60, 26063–26071 CrossRef CAS PubMed.
- H. R. Kricheldorf, C. von Lossow and G. Schwarz, Macromol. Chem. Phys., 2004, 205, 918–924 CrossRef CAS.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS.
- J. Sun and R. N. Zuckermann, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS PubMed.
- J. Liu and J. Ling, J. Phys. Chem. A, 2015, 119, 7070–7074 CrossRef CAS PubMed.
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Chem. Rev., 2016, 116, 1753–1802 CrossRef CAS PubMed.
- A. Birke, J. Ling and M. Barz, Prog. Polym. Sci., 2018, 81, 163–208 CrossRef CAS.
- Y. Liu, D. Li, J. Ding and X. Chen, Chin. Chem. Lett., 2020, 31, 3001–3014 CrossRef CAS.
- B. Lin and R. M. Waymouth, Macromolecules, 2018, 51, 2932–2938 CrossRef CAS.
- M. Li, Y. Tao, J. Tang, Y. Wang, X. Zhang, Y. Tao and X. Wang, J. Am. Chem. Soc., 2019, 141, 281–289 CrossRef CAS PubMed.
- R. S. Hewawasam, U. L. D. I. Kalana, N. U. Dharmaratne, T. J. Wright, T. J. Bannin, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2019, 52, 9232–9237 CrossRef CAS.
- F. Du, Y. Zheng, W. Yuan, G. Shan, Y. Bao, S. Jie and P. Pan, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 90–100 CrossRef CAS.
- L. Sun, X. Wu, D.-C. Xiong and X.-S. Ye, Angew. Chem., Int. Ed., 2016, 55, 8041–8044 CrossRef CAS PubMed.
- W. Zhao, Y. Lv, J. Li, Z. Feng, Y. Ni and N. Hadjichristidis, Nat. Commun., 2019, 10, 3590 CrossRef PubMed.
- W. Zhao, Y. Lv, J. Li, Z. Feng, Y. Ni and N. Hadjichristidis, Angew. Chem., Int. Ed., 2021, 60, 889–895 CrossRef CAS PubMed.
- N. U. Dharmaratne, J. U. Pothupitiya, T. J. Bannin, O. I. Kazakov and M. K. Kiesewetter, ACS Macro Lett., 2017, 6, 421–425 CrossRef CAS.
- L. Guo, J. Li, Z. Brown, K. Ghale and D. Zhang, Biopolymers, 2011, 96, 596–603 CrossRef CAS PubMed.
- S. G. Waley, J. Watson and A. H. Wilson, Proc. R. Soc. London, Ser. A, 1949, 199, 499–517 CAS.
- N. V. Belkova, M. Besora, L. M. Epstein, A. Lledós, F. Maseras and E. S. Shubina, J. Am. Chem. Soc., 2003, 125, 7715–7725 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01324f |
| ‡ Kang Chen and Yueming Wu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |