
Diels–Alder dynamic crosslinked polyurethane/polydopamine composites with NIR triggered self-healing function†
Li
Yang
,
Xili
Lu
,
Zhanhua
Wang
 and
Hesheng
Xia
*
and
Hesheng
Xia
*
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute, Sichuan University, Chengdu, 610065, China. E-mail: xiahs@scu.edu.cn
First published on 23rd March 2018
Abstract
A new kind of ultrafast near-infrared light responsive shape memory assisted self-healing polymer composite was prepared by introducing polydopamine particles (PDAPs) into polyurethane containing Diels–Alder bonds. The mechanical properties of the polyurethane composites are significantly enhanced with the addition of PDAPs due to the strong interface interaction between PDAPs and polyurethane segments. Owing to the outstanding photothermal effect of PDAPs and excellent dynamic properties of Diels–Alder bonds, the composites possess rapid light responsive shape memory and self-healing properties. The shape memory ability of the materials also plays an important role in improving the self-healing efficiency. Due to the excellent dispersion and easy preparation method, PDAPs have great potential to be used as high-efficiency and environmentally friendly fillers to obtain novel photoactive functional polymer composites.
Introduction
With the demand for improving the reliability and resource utilization efficiency, and extending the lifetime of materials, self-healing polymer materials that mimic the ability of living systems have become an important research field in recent years.1–4 Two main strategies have been used to obtain self-healing polymers. First, the microcapsules or micro-fluid channel networks capable of storing and releasing healing agents are dispersed into polymer materials to repair the damaged crack.5–7 Second, reversible covalent or non-covalent bonds8,9 such as dynamic disulfide metathesis,10 Diels–Alder reaction,11–13 imine bond exchange,14 hydrogen bonding,15,16 metal–ligand interactions,17,18 electrostatic interactions19 and so on, are introduced into polymer materials to form intrinsic self-healing polymers. Of the two approaches, intrinsic self-healing polymers have received increasing attention as no external materials were introduced and also multiple repairing can be easily attained. Generally, a self-repair procedure includes molecular diffusion, the contact of failure surfaces, and the rupture and formation of dynamic bonds or interactions. In practical applications, the destruction of the material is often accompanied by a large deformation or crack inside the material which is very difficult to repair. In order to address this challenge, the shape memory function was introduced into self-healing materials to recover the shape and close the crack. Previously shape memory alloy wires or shape memory polymer fibres were embedded into a polymer matrix to improve the self-healing performance.20–24 Mather et al. reported another strategy in which they blended thermoplastic linear poly(3-caprolactone) (PCL) with a crosslinked PCL network or distributed PCL fibres randomly in a shape memory epoxy matrix.25,26 Lu et al. synthesized a new kind of ultrasound healable shape memory dynamic PCL-based polyurethane bearing Diels–Alder bonds, which not only possesses high mechanical strength and toughness, but also exhibits shape memory assisted crack-closure and healing upon exposure to ultrasound.27Light is the most favourite power supply mode which can spatially and temporally heat the materials, and the light-induced temperature increase can be tuned by adjusting the light intensity.28–31 The long-wavelength near-infrared (NIR) light can efficiently penetrate through materials which makes NIR light a perfect candidate for remote stimulus control, especially for biomaterials.32–34 The most common strategy for preparing NIR-responsive materials is to introduce NIR-responsive fillers into the polymer matrix. A variety of photothermal conversion reagents such as carbon nanotubes,35 graphene oxide,36 organic dyes37 and conjugated polymers38 were used to trigger self-healing and shape-memory by absorbing NIR light and converting it into thermal energy.39 One drawback of these materials is the poor interface interaction between the filler and polymer matrix, which will seriously affect the performance and stability of the composites. Developing novel NIR rapid responsive fillers possessing good compatibility with the polymer matrix is still highly deserved.
Mussel-inspired polydopamine has attracted great attention since it was reported as a smart coating material in 2007.40 As a derivative of natural melanin (eumelanin), polydopamine was rapidly applied in a wide range of fields in chemical, materials and biological science.41 Furthermore, polydopamine has been used as a photothermal agent with strong NIR absorption and high photothermal conversion efficiency, which is much higher than those of previously reported photothermal agents, such as 100 times higher than those of carbon nanotubes.42 Li et al. reported a polydopamine coated shape memory polymer which enables the light triggered shape recovery, light controlled shape reprogramming and surface functionalization of polymers.43 Considering the excellent photothermal effect, multifunctional groups and availability of polydopamine, it is of great interest to fabricate smart polymer composites by using polydopamine particles as fillers.
Herein, we present a universal approach to prepare photoactive shape memory assisted self-healing polymer composites by utilizing PDAPs as fillers in dynamic crosslinked polyurethane containing DA bonds.27 DA bonds were introduced into the polymer networks due to their dynamic properties, endowing the composites with self-healing properties. PDAPs were selected because of their excellent photothermal effect, which can induce the temperature increase of the composites to achieve the shape recovery and self-healing. Moreover, compared with other fillers, PDAPs are much easier to be evenly dispersed in the matrix materials, and have the strong interface interaction with the DAPU segment, and thus can improve the mechanical properties dramatically.
Experimental
Materials
Unless otherwise stated, all reagents and solvents were commercially supplied and used without further purification. Polycaprolactone diol (Perstorp, Capa 2402, Mn = 3000 g mol−1) was dried under vacuum at 110 °C for 12 h prior to use. PDAPs with various grain diameters were obtained in a weak alkaline aqueous solution according to the literature.44Synthesis of crosslinked PU containing Diels–Alder bonds (DAPU)
The DAPU was synthesized following the route shown in Scheme S1.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 Typically, a dual hydroxyl group functionalized small molecule containing DA bonds was firstly synthesized. Then the isocyanated terminated PCL prepolymer was synthesized by the reaction between 4,4′-methylene diphenyl diisocyanate (MDI) and polycaprolactone diol (Mn = 3000 g mol−1) with a molar ratio of 2:1 at 80 °C. Finally, the as-synthesized DA compound, prepolymer and the hexamethylene diisocyanate trimer (tri-HDI) were mixed and reacted to obtain a pale yellow and non-transparent solid.
27 Typically, a dual hydroxyl group functionalized small molecule containing DA bonds was firstly synthesized. Then the isocyanated terminated PCL prepolymer was synthesized by the reaction between 4,4′-methylene diphenyl diisocyanate (MDI) and polycaprolactone diol (Mn = 3000 g mol−1) with a molar ratio of 2:1 at 80 °C. Finally, the as-synthesized DA compound, prepolymer and the hexamethylene diisocyanate trimer (tri-HDI) were mixed and reacted to obtain a pale yellow and non-transparent solid.
Preparation of polydopamine particles (PDAPs)
For a typical synthesis of PDAPs with an average size of 340 nm, the aqueous ammonia solution (NH4OH, 5 mL, 28–30%) was mixed with ethanol (80 mL) and deionized water (180 mL) under stirring at 30 °C for 30 min. Dopamine hydrochloride (1 g) was dissolved in deionized water (20 mL) and then added into the above mixture. The color of this solution immediately turned pale brown and gradually changed to dark brown. The reaction was allowed to proceed for 24 h. The PDAPs were obtained by freeze drying after centrifugation and washed with water three times. The control over the size of PDAPs was achieved by adjusting the amount of aqueous ammonia solution (3, 4, 6 mL for 730, 540, 220 nm, respectively).Preparation of DAPU–PDAP composites
The dried DAPU powders were pre-mixed with PDAPs (0.01–1 wt%) by using a high speed mixer and the mixture was extruded using a twin-screw extruder (L/D: 36, model: SHJ-25, Nanjing Chengmeng Plastics Machinery Industry Company, Ltd, China) at 120 °C, and cut into pellets and then dried in a vacuum oven for 6 h. These dried pellets were then injection molded by using a Haake Minijet to obtain the DAPU–PDAP composite samples. Typical injection parameters are listed in Table S1.† In order to express more clearly, we named the sample with X wt% PDAPs as DAPU–PDAP-X. Unless otherwise stated, the size of the particles used in DAPU–PDAP composites was 340 nm.The biocompatibility of DAPU and DAPU–PDAP-1
The fibroblasts were used for the cytotoxicity evaluation experiment. The DAPU and DAPU–PDAP-1 sample were cut into discs (diameter: 0.4 cm) by punching. 100 μL of fibroblast suspension (1 × 104 cells per mL) was seeded in a 96-well plate, with five replicates for each group, then samples were added to the well and the cells cultured in the well without sample served as control. All the samples and wells were washed with phosphate buffered saline (PBS, pH: 7.4) and then 100 μL fresh Dulbecco's Modified Eagle's Medium (DMEM) was added to each well. The cell numbers of the samples that were incubated at 37 °C were counted with CCK8 at the time points of 1, 3, 5 and 7 days. 10 μL of CCK8 were added to each well. The absorbance was measured at 450 nm by using a Thermofisher Scientific Varioskan Flash.Characterization
FTIR spectra were recorded on a Nicolet 560 FTIR spectrometer. The fracture surface was observed by using an optical microscope (VHX-1000, KEYENCE, Japan). The morphology of PDAPs and the dispersion in polymer matrixes were observed on an FEI-inspect F SEM with an acceleration voltage of 15 kV. The DAPU–PDAP composites were cut into films with a thickness of about 80 nm using a Leica EMUC6/FC6 ultra-thin slicer, and then the film was placed in a copper mesh for transmission electron microscopy (TEM) observation by using a FEC Tecnai F20 S-TWIN (the acceleration voltage was 200 kV). Mechanical properties were measured with an Instron Machine (Model 5567) at room temperature, and thermal response shape memory behavior was also tested on the same Instron Machine at Ttrans of DAPU, by stretching the sample to 50% and 100%, respectively (sample size: length ∼35 mm, width ∼5 mm, and thickness ∼3 mm). The elongation rate was 20 mm min−1.The dynamic thermomechanical properties of the samples (thickness: ∼0.5 mm) were tested by using a TA Q800 dynamic mechanical analyzer in the stretching mode, the test temperature was from −110 °C to 120 °C, the heating rate was 3 °C min−1, and the stretching frequency was 1 Hz. The photothermal effect of the composite was recorded in real time by using an Infrared Thermal Imager (Testo 875-1i). In this work, we studied the photothermal effect with an 808 nm near-infrared (NIR) light.
The NIR triggered self-healing test was conducted as follows: the rectangular sample was first cut into two pieces and were then brought into contact immediately at room temperature, and exposed to NIR light with diverse irradiation intensities and times. As to the thermal healing test, the damaged and contacted samples were thermally treated successively at 110 °C for 1 h and at 80 °C for different times. Additionally, the self-healing properties were evaluated by tensile-stress curves and the healing efficiencies were calculated from the ratio of tensile strength of healed and original samples.
Results and discussion
Preparation and characterization of PDAP and DAPU–PDAP composites
PDAPs with various diameters could be obtained by oxidation polymerization of dopamine in the weak alkaline aqueous solution at room temperature (Fig. 1a).44 The dopamine was spontaneously oxidized and polymerized into spherical PDAPs via intra/intermolecular cross-linking. SEM and TEM were employed to study the morphology and size distribution of the particles. As shown in Fig. 1(d–g) the obtained PDAPs were uniformly spherical and the average particle size from 220 to 730 nm could be obtained by tuning the ratio of ammonia to dopamine. The particle size distributions were statistically analyzed from the SEM images by Nano Measurer 1.2, and it can be seen that the particle size distribution is nearly monodisperse (Fig. 1b). The typical TEM image in Fig. 1c showed that the particles are solid and the surface is a little bit rough.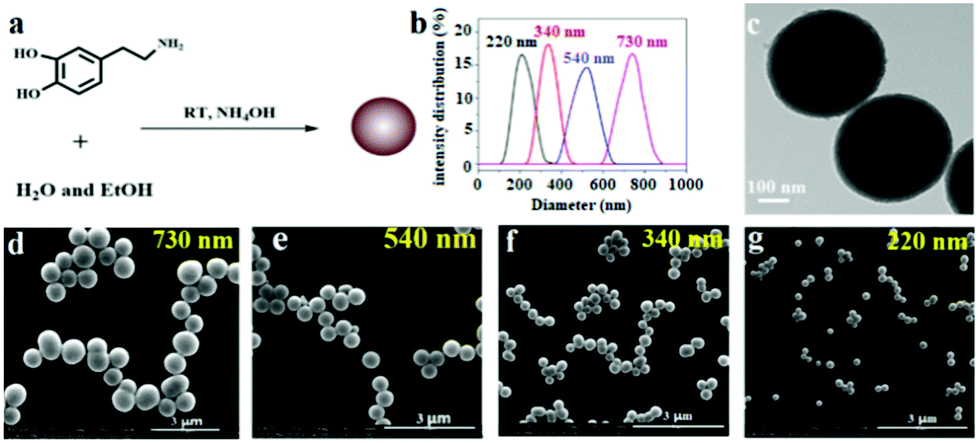 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of PDAPs. (b) PDAP diameter distribution statistical analyzed from SEM images by Nano Measurer 1.2. (c) Typical TEM image of PDAPs with an average diameter of 340 nm. (d–g) SEM images of PDAPs with different diameters prepared at different ratios of ammonia to dopamine. | ||
With the increase of the PDAP concentration, the samples become darker (Fig. S1†). The SEM images of blank DAPU and DAPU–PDAP composites are shown in Fig. 2a and b, respectively. It can be observed that polydopamine particles are uniformly distributed in the DAPU matrix. It seems that the morphology of the particles changes possibly due to the external force by melt extrusion. The TEM image in Fig. 2c also confirms this. The light-colored areas at the particle edge may be due to the mutual wetting of the particle and the matrix. Unlike inorganic particles, such as metal oxide and silica particles, which usually have a large phase contrast and clear interface between the polymer matrixes, the observed interface between PDAPs and DAPU is vague, indicating a good compatibilization between DAPU and PDAPs. PDAPs have a large amount of amino and hydroxyl groups on the surface, which can interact with the polyurethane to form strong bonding.
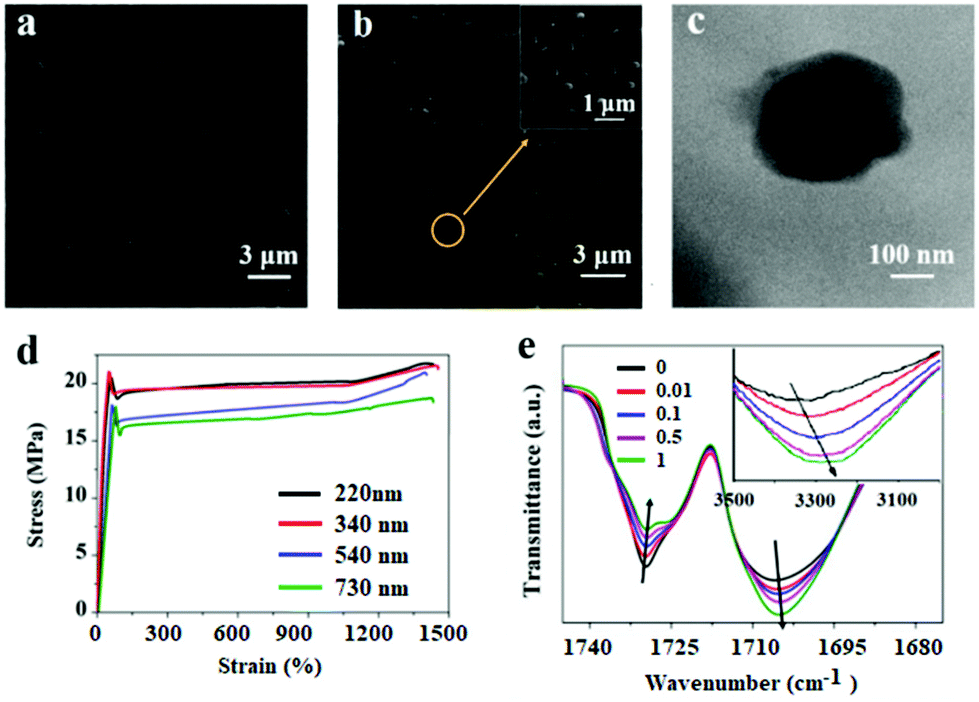 | ||
| Fig. 2 SEM images of (a) blank DAPU and (b) DAPU–PDAP composites (PDAP content: 1 wt%); (c) typical TEM image of PDAPs in DAPU–PDAP composites (PDAP content: 1 wt%); (d) effect of PDAP particle size on the tensile properties of DAPU–PDAP-1 with 1 wt% PDAP content; (e) FTIR spectra of DAPU–PDAP composites with different contents of PDAPs (PDAPs size: ∼340 nm). | ||
The mechanical properties of DAPU–PDAP composites are listed in Table 1. A significant increase in mechanical properties was observed with increasing PDAP content. Surprisingly, at only 1 wt% PDAP content the strength was dramatically enhanced from 16.3 to 22.6 MPa, while the strain at break was almost unchanged. Also the modulus was enhanced from 216 to 247 MPa. This is different from the inorganic filler reinforced composite system in which usually the increase in the tensile strength and modulus will sacrifice the flexibility. The effect of PDAP particle size on the mechanical properties was also studied. From Fig. 2d, the tensile strength of DAPU–PDAPs at 1 wt% PDAP content increases from 18.3 to 22.6 MPa when PDAP particle size decreases from 730 nm to 220 nm. This may be because at the same mass fraction of PDAPs, for smaller size particles a larger number of particles were required, and thus strong bonding can be formed to enhance the mechanical properties of the polymer.
| PDAP content [wt%] | Stress at break [MPa] | Strain at break [%] | Young's modulus (MPa) | Yield strength (MPa) |
|---|---|---|---|---|
| 0 | 16.3 ± 1.3 | 1386 ± 117 | 216 ± 3 | 15.6 ± 0.4 |
| 0.01 | 17.8 ± 1.2 | 1373 ± 132 | 224 ± 4 | 16.2 ± 0.3 |
| 0.1 | 19.3 ± 1.6 | 1336 ± 133 | 228 ± 3 | 18.3 ± 0.4 |
| 0.5 | 20.4 ± 1.4 | 1320 ± 142 | 236 ± 3 | 19.5 ± 0.4 |
| 1 | 22.6 ± 2.2 | 1356 ± 109 | 247 ± 4 | 21.7 ± 0.3 |
FTIR spectra in Fig. 2e recorded the transmittance intensity variations of free carbonyl at 1732 cm−1 and hydrogen-bonded carbonyl groups at 1706 cm−1 of DAPU–PDAP composites.45 With the increase of the PDAP content, the peak intensity of free carbonyl decreases while that of the hydrogen-bonded carbonyl groups increases significantly. In addition, the –OH stretching band in the spectra shifts from 3322 cm−1 of neat DAPU to lower wavenumbers 3285 cm−1 of DAPU–PDAP-1 with the increase of the PDAP content. These results demonstrate that PDAPs can form strong hydrogen bonding with the polymer matrix, and thus further enhance the mechanical properties of the composites by improving the interfacial interaction with the DAPU matrix. An analogous hydrogen bonding interaction was also found in melanin particle filled polyurethane composites reported by Wang et al.46 There are also a lot of amino and hydroxyl groups on the melanin particles, which can form hydrogen bonds with PU molecular chains. Xiong et al. prepared PDAP/poly(vinyl alcohol) (PVA) composites in which the hydrogen bonds were formed between PDAPs and PVA.47
Photothermal effect of DAPU–PDAP composites
The photothermal effect is the prerequisite for the light active shape memory and self-healing behavior. UV-vis spectra in Fig. S4† show that the PDAPs have an excellent absorption over the whole light region. Considering the mild non-invasiveness and the penetrability of NIR light, the photothermal effect for the disk-shape DAPU–PDAP samples with a diameter of 20 mm and a thickness of 4 mm was investigated under 808 nm NIR light. The temperature increase as a function of irradiation time under NIR light was recorded with a thermal infrared camera. Fig. 3b shows that for the DAPU–PDAP-0.1 samples, under NIR irradiation with a light intensity of 0.75 W cm−2, the temperature increases rapidly to 122 °C in 20 s, and then remains constant. Fig. 3b shows that with increasing PDAP content, the heating rate increases significantly and the equilibrium temperature is further increased. At a PDAP content of 1 wt%, the temperature can reach 155 °C. Increasing the NIR light intensity will lead to a higher equilibrium temperature (Fig. 3c). For example, at a PDAP content of ∼0.01 wt%, the equilibrium temperature changes from 38 to 121 °C when increasing the light intensity from 0.25 to 2 W cm−2, suggesting a good light controlling behavior.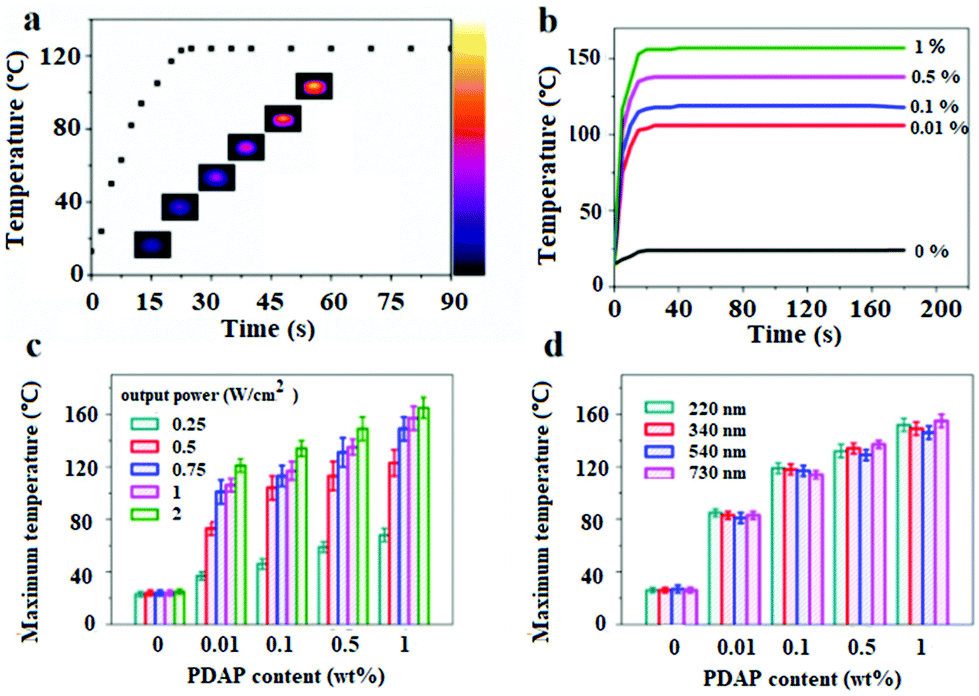 | ||
| Fig. 3 (a) Temperature increase as a function of NIR exposure time for DAPU–PDAP-0.1, PDAP size: 340 nm; (b) temperature increase of DAPU–PDAP composites with different PDAP contents (wt%) exposed to a 808 nm NIR light with a light intensity of 0.75 W cm−2, PDAP size: 340 nm; (c) the equilibrium temperature of DAPU–PDAP composites containing PDAPs over NIR light intensity, PDAP size: 340 nm; (d) the equilibrium temperature of DAPU–PDAP composites with different PDAP particle sizes exposed to a 808 nm NIR light with a light intensity of 0.75 W cm−2. | ||
Fig. 3d shows that the effect of the particle size on the temperature increase is not so obvious within our investigated size range from 220–730 nm. Compared with the conventionally used photothermal Au nanoparticles (AuNPs), both PDAPs and AuNPs displayed an efficient photothermal effect at very low filler content and irradiation intensity, however, PDAPs reported a higher photothermal conversion efficiency.30,31,48 Liu et al. reported that except for the high photothermal conversion efficiency, PDAPs exhibit much better photostability than AuNPs allowing PDAPs to absorb more light and convert it into heat during NIR irradiation.48
Photo-stimulated shape memory assisted self-healing PDAP/DAPU composites
The DAPU–PDAP composite samples with different PDAP contents were deformed at 65 °C and cut as shown in Fig. 4e and f. When the sample was irradiated with NIR light (0.75 W cm−2), the crack immediately closed due to its shape memory function and healed after the crack closure due to the dynamic Diels–Alder reaction.27Fig. 4a shows the stress–strain curves for the original, damaged and healed samples at different healing times (samples were cut at the middle). The original ultimate strain at break and tensile strength of the materials is ∼1385% and ∼22 MPa and decreases to ∼80% and ∼6 MPa respectively after the sample is damaged. When the sample is subjected to NIR irradiation (0.75 W cm−2), the healing starts and the mechanical strength recovers with time. After NIR treatment for 60 s, the sample exhibits almost the same stress and strain at break as the original material, showing a self-healing efficiency of ∼96%. The thermal healing test was also conducted as shown in Fig. 4a. After the two fractured surfaces were brought into contact and stored at 110 °C for 1 hour, they were healed at 80 °C for 60 s, 12 h and 24 h respectively. The samples can't be repaired by heating for 60 s; when the samples were heated for 24 h, the repair efficiency is ∼76%, much lower than that of samples subjected to 60 s NIR irradiation. Compared to the conventional heating, the spatially light controlled heating and cooling process allows the material to be ultra-fast repaired. The repairing time was shortened from few hours or even days to several minutes. The self-repairing behavior of the DAPU–PDAP composites can also be observed by using the optical microscope. The 3D images in Fig. 4g confirmed the NIR-triggered shape memory assisted repairing process. The damaged area by mechanical scratch was repaired immediately after exposure to NIR light.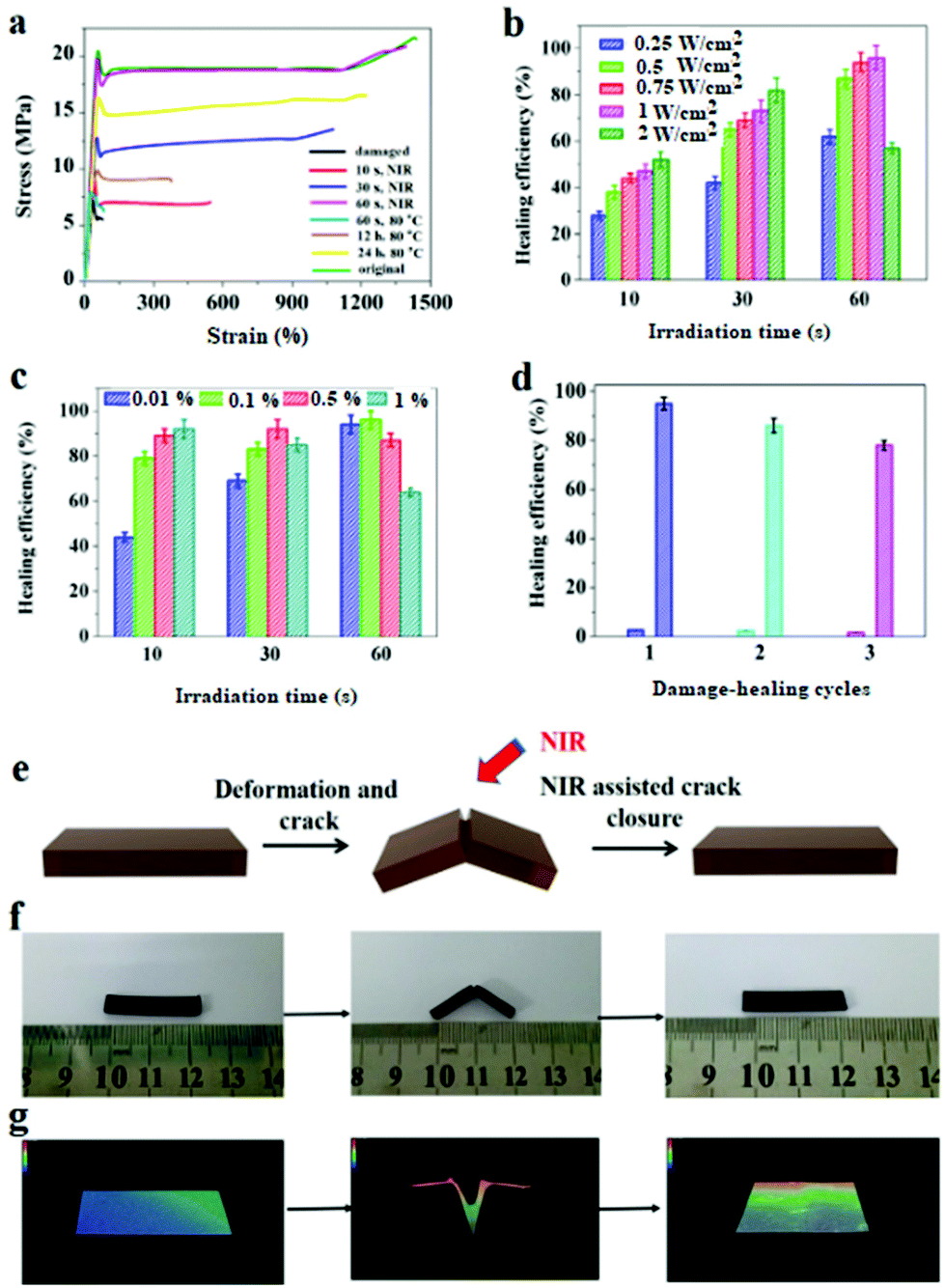 | ||
| Fig. 4 (a) Stress–strain behavior of the damaged and healed DAPU–PDAP-0.01 (NIR output power: 0.75 W cm−2), (b) effect of the NIR output power on the self-healing efficiency of DAPU–PDAP-0.01, (c) effect of the PDAP content on the self-healing efficiency of DAPU–PDAP composites under 0.75 W cm−2 NIR irradiation, (d) self-healing efficiency of DAPU–PDAP-0.01 after multiple “damage–healing” cycles (NIR output power: 1 W cm−2, irradiation time: 60 s), schematic illustration (e), photos (f) and 3D images (g) of the NIR-triggered self-healing process of DAPU–PDAP-0.01 (PDAP particle size: 340 nm). | ||
The NIR triggered self-healing behaviors of DAPU–PDAP composites were further investigated by varying the NIR light intensity and PDAP concentration. As shown in Fig. 4b, when the light intensity was below 2 W cm−2, the healing efficiency can be improved with the increase of NIR light intensity as long as the irradiation time was appropriate. However, long time NIR irradiation will damage the materials and decrease the healing efficiency. After 30 s NIR exposure at a light intensity of 2 W cm−2, the healing efficiency is ∼81%, and decreases to ∼57% after NIR treatment for 60 s. The decrease should be attributed to the high temperature induced damage on the sample when the light intensity is too high or the irradiation time is too long. With increasing PDAP content, the photo-thermal effect is significantly enhanced. However, the healing efficiency of the sample at a high PDAP content decreases with the prolonged irradiation time. For 1 wt% PDAP composite, the healing efficiency can be ∼93% after 10 s irradiation, but drops to 63% when the irradiation time extends to 60 s (Fig. 4c). Multiple damage–repair cycles for the sample DAPU–PDAP-0.01 were also conducted. The healing efficiency after the first, second and third damage–repair cycles is ∼96%, ∼87% and ∼78%, respectively (Fig. 4d), suggesting that the materials can be repetitively healed. All of these observations illustrated that the DAPU–PDAP composites possess excellent and repeatable NIR triggered self-healing properties.
The thermally induced self-healing mechanism of DAPU has been studied based on temperature-dependent reversible covalent cross-linking of the DA bond in our previous work.27 A retro-DA (rDA) reaction for DAPU will proceed at a high temperature of above 120 °C; the disconnected DAPU chains with furan and maleimide moieties resulted from the rDA reaction have enough mobility and will reform the DA bond through network reconnection when cooling down to room temperature, consequently, the fractured crack of the sample can be healed. In our systems, under NIR irradiation, PDAPs absorb the light and convert it into heat energy, which results in the temperature increase of composites for the shape recovery and rDA reaction. Since NIR light can spatially and temporally heat the crack area without causing the overall temperature increase of the sample, once NIR light is removed, the irradiated parts can quickly cool down and the broken DA bonds will be reformed to achieve a shape memory assisted self-healing function (Fig. 5a). Replacing heating with NIR light has not only improved the controllability, but also greatly improved energy efficiency. It is worth mentioning that in our self-healing polyurethane system DA bonds were distributed at both the cross-linking points and the main chains.27 When DA bonds are broken, the cross-linked polymer will be degraded into small molecular weight chains to improve the molecular motility, this is also the one of the reasons for the ultra-fast self-healing.
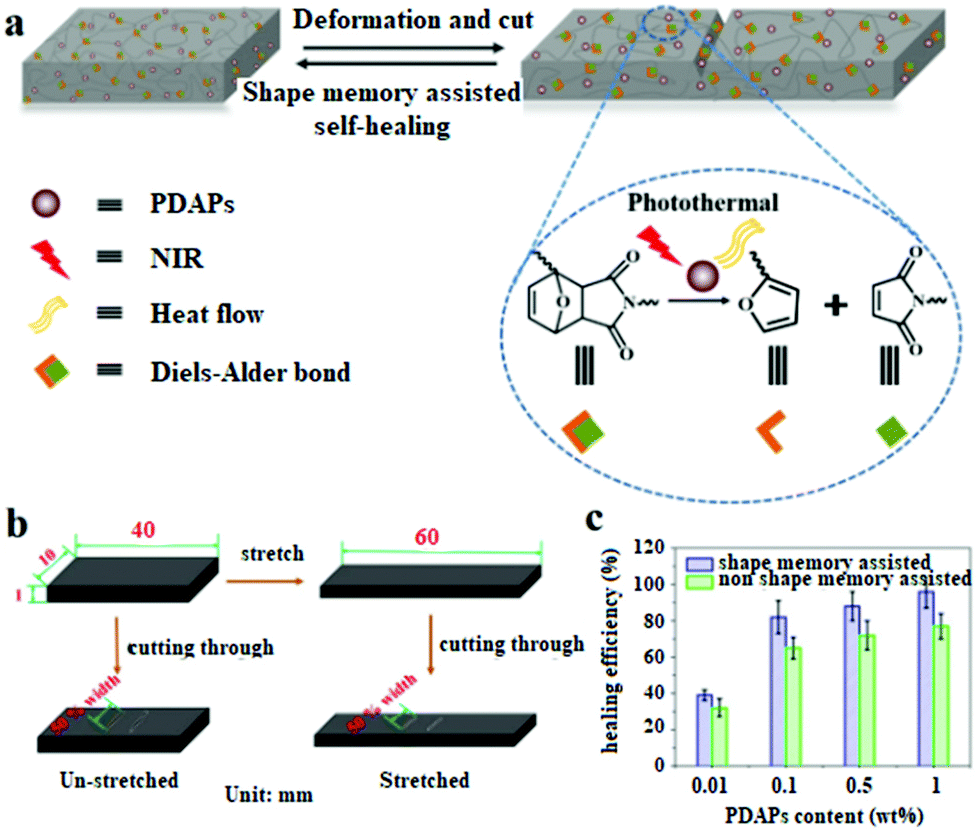 | ||
| Fig. 5 (a) Schematic illustration of NIR triggered shape memory assisted self-healing process of DAPU–PDAP composites; (b) in order to confirm the shape memory assisting the self-healing, the samples were stretched at 65 °C to 50% strain, and fixed at 10 °C under stress. The stress was removed after the temporary shape was fixed to obtain the stretched samples. Then the stretched samples were cut through in the middle of samples to obtain a 50% width crack. The un-stretched samples were cut in the same way as a contrast; (c) the healing efficiency of DAPU–PDAP composites cut with stretching and non-stretching obtained from the method of (b) under 30 s NIR irradiation (0.75 W cm−2) (PDAP particle size: 340 nm). | ||
In order to confirm the role of the shape memory effect in the self-healing process, the stretched DAPU–PDAP samples with cracks were prepared. As shown in Fig. 5b, the DAPU–PDAP samples were stretched slowly at 65 °C to 50% strain, and fixed at 10 °C, and then the stretched samples were cut to obtain two cracks with ∼50% of the sheet width at the middle of the sample. The un-stretched samples were also cut in the same manner as a contrast. The two groups of samples were repaired under the same conditions (0.75 W cm−2, 30 s NIR irradiation). The results in Fig. 5c showed that the stretched samples possess a higher self-healing efficiency. At a PDAP content of 1 wt%, the healing efficiency of the stretched samples is ∼96%, while only ∼76% for the un-stretched sample. Fig. S3† shows that all the PDAP–DAPU composites have a good shape recovery ratio (Rr, ∼99%), and can recover to the original shape at ∼55 °C. As a result, when the NIR light is irradiated on the cracks, the stretched samples will shrink and help cracks close before the temperature increase up to the self-healing temperature. These observations suggest that the shape memory function plays a very important role in promoting the self-healing process.
Polyurethane is one of the most common biomedical polymer materials with good biocompatibility. Polydopamine has also been reported as a biomedical material since it is polymerized by dopamine, which is a kind of naturopathic neurotransmitter secreted by the human body. The biocompatibility for the DAPU and DAPU–PDAP composites was characterized by the cytotoxicity evaluation experiments. The cell numbers on the samples were counted by means of CCK8 at the time points: 1, 3, 5 and 7 days after the fibroblasts were seeded. The cell numbers on the DAPU–PDAP composites increased with time, and there was no significant difference between the DAPU–PDAP composites and blank control (Fig. 6), indicating that both DAPU and DAPU–PDAP composites do not inhibit the cell growth and DAPU–PDAP composites have great potential to be used as biomaterials.
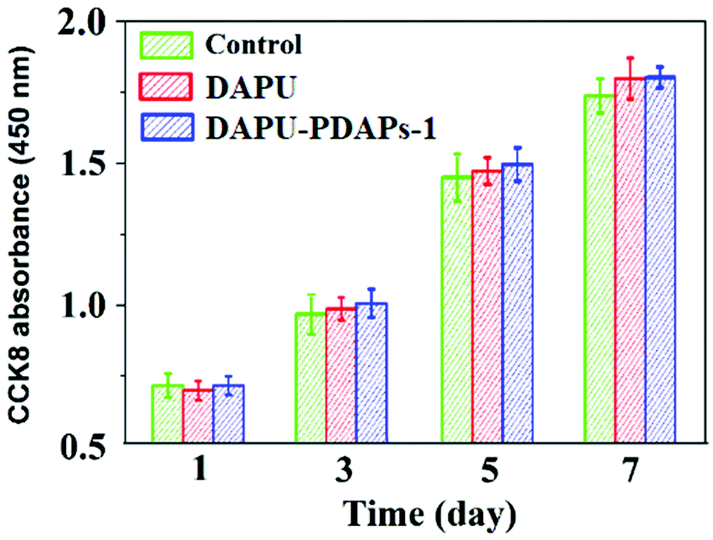 | ||
| Fig. 6 Cell numbers on DAPU and DAPU–PDAP-1 composite samples in blank culture medium (control) at different times (PDAP particle size: 340 nm). | ||
Conclusions
Based on the strong NIR absorption, high photothermal conversion efficiency and photostability of PDAPs, we developed a new kind of DAPU–PDAP composite which possess NIR remote-controlled shape memory assisted self-healing properties. Upon NIR exposure, the temperature of the irradiated polymer parts can increase up to the phase transition temperature rapidly. The shape memory and self-healing can be achieved at a very low PDAP content (0.01 wt%) in a very short time (60 s). The shape memory function plays an important role in improving the self-healing efficiency by promoting crack closure. The mechanical properties of DAPU–PDAP composites are enhanced significantly with increasing PDAP content, because the large number of active groups on the surface of PDAPs can form strong hydrogen bonding with PU segments and the interface interactions between PDAPs and DAPU were improved. Due to the excellent dispersion and easy preparation method, PDAPs have great potential to be used as high-efficiency and environmentally friendly fillers to obtain novel photoactive functional polymer composite materials. We believe that the obtained self-healing composites have potential applications in intelligent devices, bionic self-repairable coatings, etc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 51433006) and the Young Talent Team Science and Technology Innovation Project of Sichuan Province (Grant No. 2016TD0010).Notes and references
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363 CrossRef CAS PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49, 34 CrossRef.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446 RSC.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973 RSC.
- A. Lavrenova, J. Farkas, C. Weder and Y. C. Simon, ACS Appl. Mater. Interfaces, 2015, 7, 21828 CAS.
- J. D. Rule, E. N. Brown, N. R. Sottos, S. R. White and J. S. Moore, Adv. Mater., 2005, 17, 205 CrossRef CAS.
- J. H. Huang, J. Kim, N. Agrawal, A. P. Sudarsan, J. E. Maxim, A. Jayaraman and V. M. Ugaz, Adv. Mater., 2009, 21, 3567 CrossRef CAS.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- N. Roy, B. Bruchmann and J. M. Lehn, Chem. Soc. Rev., 2015, 44, 3786 RSC.
- A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237 RSC.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS PubMed.
- A. Fuhrmann, R. Göstl, R. Wendt, J. Kötteritzsch, M. D. Hager, U. S. Schubert, K. Brademann-Jock, A. F. Thünemann, U. Nöchel, M. Behl and S. Hecht, Nat. Commun., 2016, 7, 13623 CrossRef PubMed.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561 CrossRef CAS PubMed.
- A. Chao, I. Negulescu and D. Zhang, Macromolecules, 2016, 49, 6277 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
- Z. Wang, Y. Yang, R. Burtovyy, I. Luzinov and M. W. Urban, J. Mater. Chem. A, 2014, 2, 15527 CAS.
- Z. Wang and M. W. Urban, Polym. Chem., 2013, 4, 4897 RSC.
- Z. Wang, E. van Andel, S. P. Pujari, H. Feng, J. A. Dijksman, M. M. Smulders and H. Zuilhof, J. Mater. Chem. B, 2017, 5, 6728 RSC.
- E. L. Kirkby, J. D. Rule, V. J. Michaud, N. R. Sottos, S. R. White and J. A. E. Månson, Adv. Funct. Mater., 2008, 18, 2253 CrossRef CAS.
- E. L. Kirkby, V. J. Michaud, J. A. Månson, N. R. Sottos and S. R. White, Polymer, 2009, 50, 5533 CrossRef CAS.
- G. Li, O. Ajisafe and H. Meng, Polymer, 2013, 54, 920 CrossRef CAS.
- N. S. euser, V. Michaud and S. R. White, Polymer, 2012, 53, 370 CrossRef.
- G. Li, H. Meng and J. Hu, J. R. Soc., Interface, 2012, rsif20120409 Search PubMed.
- E. D. Rodriguez, X. Luo and P. T. Mather, ACS Appl. Mater. Interfaces, 2011, 3, 152 CAS.
- X. Luo and P. T. Mather, ACS Macro Lett., 2013, 2, 152 CrossRef CAS.
- X. L. Lu, G. X. Fei, H. S. Xia and Y. Zhao, J. Mater. Chem. A, 2014, 2, 16051 CAS.
- D. Habault, H. J. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244 RSC.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278 RSC.
- H. J. Zhang, D. Han, Q. Yan, D. Fortin, H. S. Xia and Y. Zhao, J. Mater. Chem. A, 2014, 2, 13373 CAS.
- H. J. Zhang, H. S. Xia and Y. Zhao, ACS Macro Lett., 2014, 3, 940 CrossRef CAS.
- G. D. Moon, S. W. Choi, X. Cai, W. Li, E. C. Cho, U. Jeong and Y. Xia, J. Am. Chem. Soc., 2011, 133, 4762 CrossRef CAS PubMed.
- C. Y. Wang, C. H. Yang, Y. S. Lin, C. H. Chen and K. S. Huang, Biomaterials, 2012, 33, 1547 CrossRef CAS PubMed.
- H. Y. Huang, S. H. Hu, S. Y. Hung, C. S. Chiang, H. L. Liu, T. L. Chiu and S. Y. Chen, J. Controlled Release, 2013, 172, 118 CrossRef CAS PubMed.
- Y. Ji, Y. Y. Huang, R. Rungsawang and E. M. Terentjev, Adv. Mater., 2010, 22, 3436 CrossRef CAS PubMed.
- J. Huang, L. Zhao, T. Wang, W. Sun and Z. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 12384 CAS.
- L. T. de Haan, V. Gimenez-Pinto, A. Konya, T. S. Nguyen, J. Verjans, C. Sánchez-Somolinos, J. V. Selinger, R. L. Selinger, D. J. Broer and A. P. Schenning, Adv. Funct. Mater., 2014, 24, 1251 CrossRef CAS.
- W. Liu, L. X. Guo, B. P. Lin, X. Q. Zhang, Y. Sun and H. Yang, Macromolecules, 2016, 49, 4023 CrossRef CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1 CrossRef CAS.
- B. E. Stranger, M. S. Forrest, M. Dunning, C. E. Ingle, C. Beazley, N. Thorne and C. Tyler-Smith, Science, 2007, 315, 848 CrossRef CAS PubMed.
- Y. L. Liu, K. L. Ai and L. H. Lu, Chem. Rev., 2014, 114, 5057 CrossRef CAS PubMed.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353 CrossRef CAS PubMed.
- Z. Li, X. Zhang, S. Wang, Y. Yang, B. Qin, K. Wang, T. Xie, Y. Wei and Y. Ji, Chem. Sci., 2016, 7, 4741 RSC.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. M. Lu, Adv. Mater., 2013, 25, 998 CrossRef CAS PubMed.
- L. Zhang, S. S. Shams, Y. Wei, X. Liu, S. Ma, R. Zhang and J. Zhu, J. Mater. Chem. A, 2014, 2, 20010 CAS.
- Y. Wang, T. Li, X. Wang, P. Ma, H. Bai, W. Dong, Y. Xie and M. Chen, Biomacromolecule, 2016, 17, 3782 CrossRef CAS PubMed.
- S. Xiong, Y. Wang, J. Yu, L. Chen, J. Zhu and Z. Hu, J. Mater. Chem. A, 2014, 2, 7578 CAS.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py00162f |
| This journal is © The Royal Society of Chemistry 2018 |