Recent advances in RAFT dispersion polymerization for preparation of block copolymer aggregates
Jiao-Tong
Sun
,
Chun-Yan
Hong
* and
Cai-Yuan
Pan
*
CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, People's Republic of China. E-mail: pcy@ustc.edu.cn; hongcy@ustc.edu.cn
First published on 3rd September 2012
Abstract
Differently from bulk, solution, suspension, emulsion, and miniemulsion polymerizations, the controlled radical dispersion polymerization (CRDP) demonstrates self-assembly of the block copolymers formed in the homogeneous system, forming various kinds of micelles or vesicles. Thus, this technology can prepare both the block copolymers and the polymeric aggregates directly. Among CRDP, the reversible addition-fragmentation chain transfer (RAFT) dispersion polymerization has been studied in relative detail and has been successfully developed to prepare a diverse range of assemblies. Several typical systems for RAFT dispersion polymerization are presented in detail and the factors influencing the polymerization and the in situ self-assembly are also highlighted in this minireview.
 Jiao-Tong Sun | Jiao-Tong Sun was born in 1984 in Jiangsu, China. He received his Bachelor in polymer science and engineering in 2007 from Nanjing University of Science and Technology. He is currently pursuing his PhD at University of Science and Technology of China under the supervision of Professors Chunyan Hong and Caiyuan Pan, and works on synthesis of responsive polymers by controlled radical polymerization. |
 Chun-Yan Hong | Chun-Yan Hong is a professor of University of Science and Technology of China (USTC). She obtained a PhD in chemistry from USTC in 2002. Her research interests include controlled radical polymerization, synthesis of stimuli-responsive polymers and biodegradable polymers, functionalization of nanomaterials, and their applications in drug or gene delivery. |
 Cai-Yuan Pan | Professor Cai-Yuan Pan joined the University of Science and Technology of China as a lecturer in 1977, and became a full professor in 1989. His interests include the synthesis and characterization of polymers, especially nonlinear polymers, and the preparation and properties of nanomaterials. |
Introduction
Initially, controlled radical polymerization (CRP) techniques, including stable free radical polymerization (SFRP), atom transfer radical polymerization (ATRP) and reversible addition-fragmentation chain transfer (RAFT) polymerization were homogeneously performed in organic solvents or in water, and later heterogeneous CRP methods, namely suspension, emulsion, miniemulsion and dispersion polymerization have been developed.1,2 Controlled radical dispersion polymerization has become interesting only in recent years. Strictly speaking, dispersion polymerization should be considered as a semi-homogeneous polymerization, since the initial polymerization system is homogeneous while the resultant polymer is not soluble in the polymerization medium. The produced polymer chains precipitate to form spherical particles stabilized by a stabilizer, as polymerization proceeds in the presence of any stabilizers.3 Later on, a soluble macromolecular initiator or macromolecular chain transfer agent (macro-CTA) was used as a stabilizer in the dispersion polymerization system, and then the polymerization process makes a difference. In the homogeneous polymerization stage, with the growth of the insoluble block, the resultant amphiphilic block copolymer chains become more and more unstable unimolecularly. When the chain length of the insoluble block exceeds the critical micelle degree of polymerization (CMDP), the block copolymer aggregates to form spherical micelles and the phase separation process is called polymerization-induced self-assembly. After the formation of core–shell nanoparticles, the polymerization proceeds mainly in the micelles, and the polymerization rate is controlled by monomer concentration in the cores and the diffusion rate of the monomer. The difference in polymerization rates before and after phase separation was therefore often observed. If the polymerization rate is preserved and the chain length ratio of insoluble block to soluble block increases to a certain critical value, transition of the formed morphology might take place, which is called polymerization-induced reorganization. The in situ formed aggregates also affect the RAFT polymerization, and polymers with ultrahigh molecular weights, which are usually hard to prepare by homogeneous polymerization, can be obtained. The above viewpoints are emphasized in this minireview.The SFRP, ATRP and RAFT polymerizations have all been conducted in the form of dispersion polymerization. When atom transfer radical dispersion polymerization (ATRDP) is applied to prepare the block copolymer aggregates, it is difficult to remove the copper salt from the assemblies. Thus, the studies of ATRDP are relatively few.4–6 Although there is no need to remove any additives in the stable free radical dispersion polymerization, the versatile nitroxide radical initiator is not easy to obtain.7,8 Therefore, RAFT dispersion polymerization is the most widely studied technique in CRDP. In this minireview, the discussion is limited to recent progress in RAFT dispersion polymerization. If the readers are interested in the other controlled radical dispersion polymerizations, several detailed reviews supply further information.3,9,10 Since the dispersion polymerization requires that a monomer should be soluble in the solvent but its polymer should not, there are only several successful examples reported so far. These typical polymerization systems are presented as follows.
Dispersion polymerization of 4-vinylpyridine in cyclohexane
The first trial of RAFT dispersion polymerization dates back to the polymerization of 4-vinylpyridine (4VP) in cyclohexane in 2006.11 Because 4VP and polystyrene (PSt) are both soluble in hot cyclohexane but poly(4-vinylpyridine) (P4VP) is insoluble in cyclohexane, 4VP was chosen to extend the dithiobenzoate-terminated polystyrene (PSt–SC(S)Ph) chains in cyclohexane. Divinylbenzene (DVB) was used to cross-link the core of the formed micelles. Multi-angle laser light scattering (MALLS) and transmission electron microscopy (TEM) measurements confirmed the formation of micelles with PSt as the shell and poly(4VP-co-DVB) as the core (Fig. 1).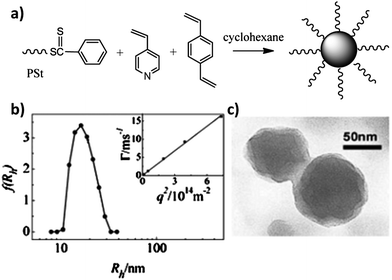 | ||
| Fig. 1 (a) Schematic representation for the preparation of core-crosslinked micelles. (b) Hydrodynamic radius distributions of the micelles with cross-linked cores in THF measured by the dynamic light scattering method. The inset shows that Γ/q2 is independent of the angle of detection. (c) TEM photo of the core cross-linked micelles.11 | ||
The kinetics of the dispersion polymerization was further investigated to disclose the possible formation mechanism of the micelles. As a control experiment, the solution polymerization of 4VP in tetrahydrofuran (THF) was conducted with the same feed ratio as the dispersion polymerization of 4VP in cyclohexane. However, their kinetic behaviours as shown in Fig. 2 are different. Fig. 2A shows a linear relationship of 4VP conversion with polymerization time, but in Fig. 2B the polymerization in cyclohexane levelled off at about 5 h. Therefore, it could be concluded that the RAFT polymerization process of 4VP in cyclohexane differed from the regular one in THF. The turning point in Fig. 2B was supposed to indicate the aggregation of block copolymers. The active end groups of the macro-RAFT agent were trapped inside the hard core of the micelles, where it was difficult for the monomer to penetrate. Subsequently, the polymerization in the cores of micelles was retarded. Thus, the micellization was induced by the polymerization and the formed micelles slowed down the polymerization dramatically.
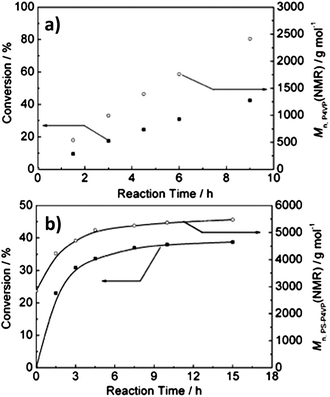 | ||
| Fig. 2 Evolution of 4VP conversion and the number-average molecular weights of the P4VP block and the PSt–P4VP with polymerization time in THF (a) and in cyclohexane (b), respectively. There was no cross-linker DVB in either polymerization system.11 | ||
Dispersion copolymerization of styrene and maleic anhydride in chloroform
Another successful RAFT-mediated dispersion polymerization system was reported by Chen and co-workers.12 The poly(ethylene oxide) (PEO) macro-CTA was used in the copolymerization of styrene (St) and maleic anhydride (MAn) in chloroform (Fig. 3a). Since the growing P(St-alt-MAn) block is insoluble in chloroform while the PEO block is soluble, the polymerization-induced self-assembly occurs in this dispersion polymerization system. The time-resolved laser light scattering was employed to monitor the polymerization-induced micellization. From the result shown in Fig. 3b, three stages including the induction period, the formation of loose aggregates and the formation of micelles could be identified. When the DP of the P(St-alt-MAn) block was far below the CMDP, the PEO-b-P(St-alt-MAn) remained as individual polymer chains, as the PEO macro-CTAs (phase I). With the DP increasing to a certain value, close to CMDP, some of the PEO-b-P(St-alt-MAn) chains were associated together to form loose aggregates (phase II), which was indicated by the slight increase in the scattered intensity. Once the DP of the P(St-alt-MAn) block exceeded the CMDP, the loose aggregates and the other single polymer chains were gradually transformed into micelles, as shown in phase III. In addition, the value of CMDP could be calculated by measuring the conversion of monomers at the turning point for transition from loose aggregates to micelles. | ||
| Fig. 3 (a) RAFT dispersion copolymerization of styrene and maleic anhydride in chloroform with a PEO macro-CTA. (b) Time dependence of the scattering intensity of the polymerization system at scattering angles of 30° and 90°. The inset shows three respective stages during the polymerization-induced self-assembly process.12 | ||
The critical micelle concentrations (cmc) of amphiphilic block copolymers in water decrease with the increase in hydrophobic block chain length when the hydrophilic block is fixed. With the growth of the core-forming block, the cmc value decreases gradually. When the cmc is equal to the concentration of the existing block copolymer, the chain length of the core-forming block reaches the CMDP. Hence, the CMDP depends on the concentration of the macro-RAFT agent. The scattering intensities of the polymerization systems with different concentrations of PEO macro-CTA at 1.38, 2.50, and 5.00 mg mL−1 were monitored, as shown in Fig. 4. The intensity data was normalized for direct comparison. The polymerization-induced micellization of PEO-b-P(St-alt-MAn) at all PEO macro-CTA concentrations exhibited a similar profile for scattering intensity. The major difference was that the polymerization carried out at higher concentrations of PEO macro-CTA showed a shorter induction period and a faster increase in the scattered intensity, because the high concentration of macro-CTA accelerated the polymerization rate and increased the concentration of the amphiphilic block copolymer. Therefore, the concentration of macro-CTA is also a crucial parameter in the RAFT-mediated polymerization-induced micellization process.
 | ||
| Fig. 4 Time dependence of the scattering intensity of the polymerization-induced self-assembly process at 90° at different PEO macro-CTA concentrations.12 | ||
Dispersion polymerization of styrene in methanol
The most reported RAFT dispersion polymerization system in organic solvents is the polymerization of styrene in methanol.13–20 Our group first carried out the RAFT dispersion polymerization of St in methanol with trithiocarbonate-terminated poly(4-vinylpyridine) (P4VP–TTC) as a macro-RAFT agent (Fig. 5A). Methanol is a solvent for P4VP and St, but not for PSt, so the polymerization-induced self-assembly is also expected to happen in this system. However, differing from those systems reported before, spherical micelles are not the only assembly during the whole polymerization process. It is well-known that different aggregates could be formed by changing the chain length ratio of the two blocks when the block copolymers self-assemble in a selective solvent. Nevertheless, the kinetics studies have revealed that in situ micellization of the prepared block copolymer usually hinders further polymerization and postpones the morphological transition. Hence, the key is how to maintain the rate of polymerization after microphase separation. One easy way is to swell the PSt cores by addition of more St in the polymerization system since the monomer St is a solvent for the PSt block. When different feed molar ratios of St/P4VP–TTC/AIBN/CH3OH were adopted, different morphological transitions for block polymer aggregates were observed. Under the conditions of St/P4VP–TTC = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/1 and St/CH3OH = 1/0.7, the block copolymer aggregates prepared by RAFT dispersion polymerization are recorded at different times in Fig. 5B. When St/P4VP–TTC = 100000/1 and St/CH3OH = 2/1 are applied, the TEM images are shown in Fig. 5C. The critical compositions for rod-like micelles in Fig. 5B and vesicles in Fig. 5C are P4VP99-b-PSt770 and P4VP99-b-PSt2040, respectively. Rod-like micelles as an intermediate assembly were not observed in Fig. 5C, probably because the almost two-fold feed of St caused chain propagation to proceed smoothly in the formed loose micelles. The faster polymerization made the hydrophobic block propagate rapidly and the spherical micelles were reorganized to form vesicles directly. In addition, this CRP made sure that the resultant block copolymers had low polydispersity (PDI < 1.25) and uniform assemblies were eventually obtained.
000/1 and St/CH3OH = 1/0.7, the block copolymer aggregates prepared by RAFT dispersion polymerization are recorded at different times in Fig. 5B. When St/P4VP–TTC = 100000/1 and St/CH3OH = 2/1 are applied, the TEM images are shown in Fig. 5C. The critical compositions for rod-like micelles in Fig. 5B and vesicles in Fig. 5C are P4VP99-b-PSt770 and P4VP99-b-PSt2040, respectively. Rod-like micelles as an intermediate assembly were not observed in Fig. 5C, probably because the almost two-fold feed of St caused chain propagation to proceed smoothly in the formed loose micelles. The faster polymerization made the hydrophobic block propagate rapidly and the spherical micelles were reorganized to form vesicles directly. In addition, this CRP made sure that the resultant block copolymers had low polydispersity (PDI < 1.25) and uniform assemblies were eventually obtained.
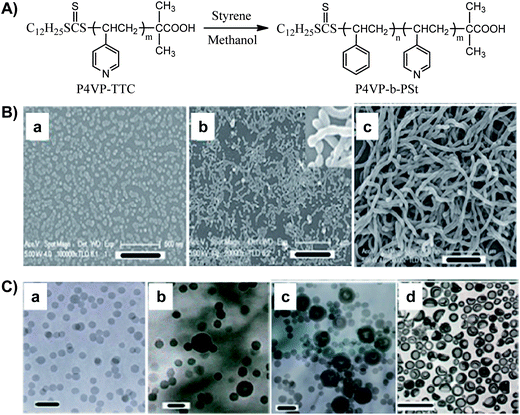 | ||
| Fig. 5 (A) RAFT polymerization of styrene in methanol using P4VP–TTC as macro-CTA. (B) FESEM images of the polymeric aggregates prepared by RAFT polymerization for 3 h (a), 4 h (b), and 24 h (c). Scale bar: (a) 500, (b) 2000, (c) 1000 nm. (C) TEM images of the polymeric aggregates with another different feed ratio at different polymerization times: 2 h (a), 4 h (b), 6 h (c), and 24 h (d). Scale bars: (a) 100 nm, (b and c) 200 nm, (d) 1000 nm.13,15 | ||
Apart from P4VP–TTC and PEO–TTC used as macro-CTA,17 the dithiobenzoate-terminated poly(2-dimethylaminoethyl methacrylate) (PDMAEMA–DTB)18 and the trithiocarbonate-terminated poly(acrylic acid) (PAA–TTC) were also used as macro-CTAs in the RAFT dispersion polymerization of St in methanol, and various morphologies were obtained due to the polymerization-induced self-assembly and reorganization. Under the condition that the feed ratio of St/CH3OH was fixed, when the ratio of St/macro-CTA was higher, the more complex assemblies would finally be obtained (Fig. 6).19 When the molar ratio of macro-CTA/AIBN was adjusted to 1/1 from the general ratio of 10/1, the aggregate with an interesting morphology called yolk–shell was observed.20 Hence, the morphological transitions greatly depend on the feed ratios of all contents.
 | ||
| Fig. 6 TEM images of PAA-b-PSt assemblies formed at different molar ratios after 20 h of polymerization. St/PAA–TTC = 250 (a), 500 (b), 750 (c), 1000 (d), and 1250 (e).19 | ||
The formed aggregate morphologies also affect the dispersion polymerization. One of the interesting polymerization behaviours is the formation of high molecular weight polymers, which is never obtained in the homogeneous RAFT polymerization, even in the RAFT dispersion polymerization without transition of morphology. When the polymerization with feed molar ratio of P4VP–TTC/St/AIBN = 10/100000/1 was carried out in methanol, the block copolymer, P4VP-b-PSt with Mn = 3.9 × 105 g mol−1 was formed while the transition of spherical micelles to vesicles occurred (Fig. 7).16
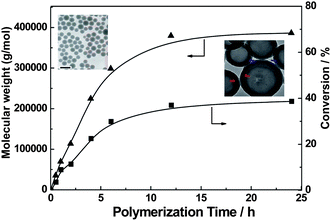 | ||
| Fig. 7 The relationship of molecular weights and conversion to the polymerization time. Feed molar ratio: P4VP–TTC/St/AIBN = 10/100000/1, methanol: 0.9 g, St: 2 g, 80 °C.16 | ||
Aqueous dispersion polymerization of N-isopropylacrylamide, N,N-diethylacrylamide or other thermoresponsive (co)monomers
Hawker et al. reported the first example of aqueous RAFT dispersion polymerization in 2007.21 Two types of trithiocarbonate-terminated poly(dimethylacrylamide)s (PDMA–TTC) with different end groups, macro-CTA-1 with hydrophobic dodecyl tails and fully hydrophilic macro-CTA-2 without surface activity, were used as macro-CTAs to control the RAFT polymerization of N-isopropylacrylamide (NIPAAm) at 70 °C (Fig. 8). This system undergoes dispersion polymerization because PDMA and NIPAAm are soluble, but PNIPAAm is insoluble in water at the polymerization temperature of 70 °C. In the presence of N,N′-methylenebisacrylamide (MBA), the cross-linked nanoparticles produced using the amphiphilic macro-CTA-1 were less uniform than those produced using the fully hydrophilic macro-CTA-2 because there are two competing particle nucleation mechanisms in the former case, resulting in higher polydispersity of the particles. Therefore, as steric stabilizers, it is not necessary for the macro-CTAs to have a surfactant-like structure for the stabilization of the formed particles. After cooling to room temperature, the resulting fully hydrophilic nanoparticles are swollen in the water, while maintaining the integrity of the nanoparticles. While no cross-linker is used, the well-defined double hydrophilic block copolymers are obtained after cooling to room temperature, due to the transition of the PNIPAAm block to being soluble below its lower critical solution temperature (LCST).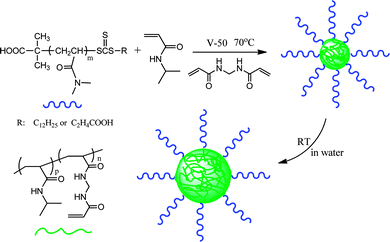 | ||
| Fig. 8 RAFT dispersion polymerization of N-isopropylacrylamide for the preparation of nanostructured hydrogels.21 | ||
Later on, the pegylated thermoresponsive core–shell nanoparticles were also synthesized by RAFT-mediated aqueous dispersion polymerization.22 The PEO–TTC was used as a macro-CTA in the copolymerization of N,N-diethylacrylamide (DEAAm) and crosslinker MBA. Similar to NIPAAm, DEAAm is also water-soluble and poly(N,N-diethylacrylamide) (PDEAAm) exhibits a LCST at 32 °C. The thermoresponsive nanogel particles were therefore obtained after the phase separation at the reaction temperature of 70 °C. Very recently, Cao and An obtained biocompatible and thermosensitive nanogels by RAFT dispersion polymerization of di(ethylene glycol) methyl ether methacrylate (MEO2MA) or copolymerization of MEO2MA and oligo(ethylene glycol) methyl ether methacrylate (OEGMA) using linear PEG and brush-like PEG CTAs to mediate the polymerizations.23 The uniform nanogels with tunable size were produced by changing the feed ratios. The nanogels containing brush-like PEG as the shell had enhanced stability during the freeze–thawing process and in biologically relevant solutions. They are therefore possibly applicable in drug delivery.
Aqueous dispersion polymerization of 2-hydroxypropyl methacrylate
Armes and Li expanded the scope of aqueous RAFT dispersion polymerization by introducing the commercial monomer 2-hydroxypropyl methacrylate (HPMA).24,25 In contrast to the thermoresponsive PNIPAAm, poly(2-hydroxypropyl methacrylate) (PHPMA) is always insoluble in water. The sterically stabilized methacrylic nanolatexes were synthesized in situ by aqueous RAFT dispersion polymerization of HPMA using the dithiobenzoate-terminated poly(glycerol monomethacrylate) (PGMA–DTB) as a macro-CTA. When the polymerizations were conducted at a total solid content of 10% and 20%, the spherical micelles and the vesicles were obtained, respectively. This result agrees with the conclusion obtained in the aqueous RAFT dispersion polymerization of HPMA when the biomimetic poly(2-(methacryloyloxy)ethylphosphoryl choline) (PMPC) was used as the macro-CTA.26 As shown in Fig. 9, the final particle morphology obtained at full monomer conversion depends on the target degree of polymerization of the PHPMA block and the total solids concentration. A detailed phase diagram has also been drawn for this aqueous dispersion polymerization formulation. Moreover, the morphological transformations from spheres to worms to vesicles during the polymerization of HPMA were also recorded. Later, Blanazs et al. investigated the transition of block copolymer morphologies by carefully monitoring the in situ polymerization with TEM measurement.27 Various novel intermediate structures were seized and provided important information for understanding the mechanism of the evolution of the particle morphology. Herein, the glass transition temperature (Tg) of the core-forming PHPMA block is the key factor. Due to the fact that the Tg is lower than the polymerization temperature, the growing PHPMA block chains are highly mobile and the HPMA molecules can diffuse easily into the formed loose aggregates to extend the chains. Almost complete conversion (>99%) of HPMA was therefore achieved in 2 h. | ||
| Fig. 9 Trend relationships of in situ formed assemblies with total solids concentration and chain length of the PHPMA block during RAFT aqueous dispersion polymerization of HPMA.26 | ||
Very recently, a reversible transition between wormlike micelles and spherical micelles was reported by Blanazs et al.28 As shown in Fig. 10, three well-defined PGMA54–PHPMAn diblock copolymers (where n = 90, 140 and 220) with narrowly distributed molecular weights (PDI = 1.10, 1.10 and 1.12) were synthesized with the same polymerization formulation mentioned before. At the same copolymer concentration of 10.0 wt%, a self-supporting gel is formed by the wormlike micelles at 21 °C, as opposed to the free-flowing low-viscosity fluids formed by either spherical micelles or vesicles (Fig. 10b). The free-standing physical hydrogel was formed, probably due to the interworm entanglements at 21 °C. The more interesting phenomenon is that gel dissolution occurs on cooling to 4 °C and the gelation could be repeated on heating to 21 °C. The instrumental measurements confirmed that this process was an unusual worm-to-sphere order–order transition (Fig. 11). This thermo-reversible behavior allows the facile preparation of sterile gels by conducting the ultrafiltration at 4 °C to remove micrometer-sized bacteria.
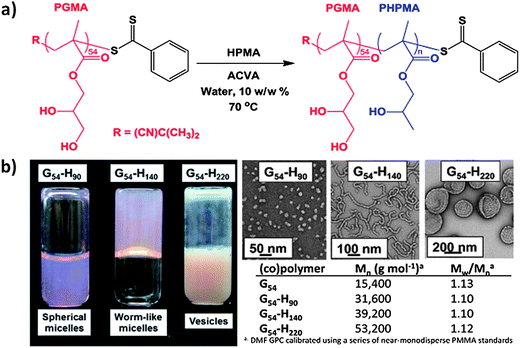 | ||
| Fig. 10 (a) RAFT aqueous dispersion polymerization of HPMA monomer using a PGMA54 macro-CTA at 10 wt% and 70 °C. (b) Digital photographs and TEM images of three G54-Hn copolymer dispersions with the corresponding molecular weight data.28 | ||
 | ||
| Fig. 11 Thermoresponsive aqueous solution behavior of a 10 wt% aqueous dispersion of G54-H140 diblock copolymer particles.28 | ||
Aqueous dispersion polymerization of 2-methoxyethyl acrylate
Recently, Liu and co-workers successfully developed another system for aqueous RAFT dispersion polymerization.29,30 The monomer/polymer pair is 2-methoxyethyl acrylate (MEA)/poly(MEA) (PMEA). The MEA is highly water-soluble but PMEA is not; the nanoparticles can therefore be formed when the trithiocarbonate-terminated poly[oligo(ethylene glycol) methyl ether methacrylate] (POEGMA–TTC) chains are extended by MEA in water (Fig. 12A). The RAFT dispersion polymerization of MEA in water is highly efficient using a redox initiator at low temperature. The size of the nanoparticles increases by increasing the molar ratio of MEA/macro-CTA. While increasing the total solids content but fixing the molar ratio of MEA/macro-CTA, the size of the nanoparticles in most cases does not change obviously. Fig. 12B illustrates the DLS size distribution (Dh = 54 nm, PDI = 0.06) and the AFM image of the cross-linked nanoparticles, which indeed demonstrate that the formed nanoparticles are highly monodisperse. | ||
| Fig. 12 (A) Dispersion polymerization of MEA using POEGMA macro-CTA in water. (B) Dynamic light scattering (DLS) result (a) and atomic force microscopy (AFM) graph (b) of the produced cross-linked nanoparticles.29 | ||
Dispersion polymerization of methacrylates, acrylamides, and styrenics in supercritical carbon dioxide
In the polymer industries, the use of environmentally friendly media with low cost is always expected. In addition to water, there has recently been increasing interest in supercritical carbon dioxide (scCO2). The scCO2 is non-flammable, nontoxic, and inexpensive. Besides, the solubility and viscosity of scCO2 are easily adjustable by varying the temperature and pressure in the supercritical region. Since most monomers are soluble but their polymers are insoluble in scCO2, it is therefore a suitable solvent for dispersion polymerization. The dispersion polymerizations of vinyl monomers such as styrene and methyl methacrylate (MMA) and so on in scCO2 with steric stabilizers, like poly(dimethylsiloxane monomethyl methacrylate) (PDMS–MA), were all studied before.31–33 However, one-pot synthesis of block copolymers in scCO2 by sequential RAFT dispersion polymerization has been reported by Howdle and co-workers for the first time.34 The well-known PDMS–MA was also added and acted as a grafting steric stabilizer in the polymerization of MMA in scCO2. Then a wide range of monomers, including methacrylates, acrylamides, and styrenics were subsequently utilized to extend the resulting trithiocarbonate-terminated PMMA-RAFT precursor for preparation of the block copolymers (Fig. 13A). The low viscosity and high diffusivity of scCO2 ensure efficient plasticization of PMMA by scCO2, and hence it allows excellent access of the growing chain ends and the RAFT agent to the incoming monomer. Consequently, the second block grew in the PMMA spherical particles, resulting in block copolymers with the same external morphology (Fig. 13B), that is, no transition in morphology occurred, even during growth of the second blocks, which is different from the dispersion polymerization discussed above. However, the internal morphology of the block copolymer microparticles reveals a diverse range of nanostructures formed via phase segregation of the incompatible blocks. Since the RAFT dispersion polymerization in scCO2 can produce diblock copolymer microparticles with various nanostructures, so this work is reviewed in this article. | ||
| Fig. 13 (A) The block copolymers synthesized by RAFT dispersion polymerization in scCO2 from PMMA-RAFT precursors (a): PMMA-b-PSt (b), PMMA-b-P4VP (c), PMMA-b-PBzMA (d), PMMA-b-PDMA (e), PMMA-b-PDMAEMA (f). (B) SEM of the block copolymer particles. Scale bar in each image is 5 μm.34 | ||
Summary and outlook
Controlled free radical dispersion polymerizations have made significant progress in recent years, and one of the most important examples is RAFT dispersion polymerization using a macro-RAFT agent. This technology can prepare not only the block copolymers with a narrow molecular weight distribution, but also directly fabricate polymeric assemblies including spherical micelles, rod-like micelles, vesicles, nanotubes and other aggregates with various complex morphologies on a large scale. The media used in this polymerization technique could be organic solvents, water and supercritical carbon dioxide. Compared to bulk and solution polymerizations, this polymerization system has low viscosity after phase separation. While in contrast to emulsion and miniemulsion polymerizations, there is no need to add additional surfactants, which usually reduce the quality of polymeric materials. More interestingly, by selecting appropriate feed ratios, block copolymers with narrowly-distributed and ultrahigh molecular weights, even over one million Da, can be obtained. In addition, the polymeric assemblies are expected to have potential applications in coatings, drug delivery, biomedical materials, temperature sensors and so on.Although the monomer/polymer pairs studied in RAFT dispersion polymerization are limited, combination of them with different macro-CTAs could produce many different block copolymers. Copolymerization of two different monomers and applying mixed solvents in the dispersion polymerization have also been studied for the preparation of functional and interesting polymeric assemblies.35–38 Further exploring more monomer/polymer pairs suitable for RAFT dispersion polymerization may be helpful in creating new morphologies, and should also benefit further understanding of the polymerization-induced self-assembly and reorganization. In addition, the investigation of the properties of these aggregates and their stimuli-responsive transitions are also urgent. It is promising for RAFT dispersion polymerization to find its place in industrial production.
Acknowledgements
This project was supported by National Natural Science Foundation of China (no. 20974103, 21074121 and 21090354).References
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS.
- J. Qiu, B. Charleux and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 2083–2134 CrossRef CAS.
- S. Kawaguchi and K. Ito, Adv. Polym. Sci., 2005, 175, 299–328 CAS.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS.
- H. Minami, A. Tanaka, Y. Kagawa and M. Okubo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2578–2584 CrossRef CAS.
- J. Jiang, Y. Zhang, X. Guo and H. Zhang, RSC Adv., 2012, 2, 5651–5662 RSC.
- G. Delaittre, J. Nicolas, C. Lefay, M. Save and B. Charleux, Chem. Commun., 2005, 614–616 RSC.
- S. Brusseau, J. Belleney, S. Magnet, L. Couvreur and B. Charleux, Polym. Chem., 2010, 1, 720–729 RSC.
- G. Riess and C. Labbe, Macromol. Rapid Commun., 2004, 25, 401–435 CrossRef CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 3753–3767 Search PubMed.
- G.-H. Zheng and C.-Y. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- W. Ji, J. Yan, E. Chen, Z. Li and D. Liang, Macromolecules, 2008, 41, 4914–4919 CrossRef CAS.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883–5885 RSC.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- W.-M. Wan, X.-L. Sun and C.-Y. Pan, Macromolecules, 2009, 42, 4950–4952 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W.-M. Cai, W.-M. Wan, C.-Y. Hong, C.-Q. Huang and C.-Y. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2010, 43, 2672–2675 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C. K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. J. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- Y. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Ian Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–563 CrossRef CAS.
- W. X. Wang, M. R. Giles, D. Bratton, D. J. Irvine, S. P. Armes, J. V. W. Weaver and S. M. Howdle, Polymer, 2003, 44, 3803–3809 CrossRef CAS.
- K. J. Thurecht, A. M. Gregory, W. X. Wang and S. M. Howdle, Macromolecules, 2007, 40, 2965–2967 CrossRef CAS.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS.
- C.-Q. Huang, Y. Wang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2011, 32, 1174–1179 CrossRef CAS.
- X. Zhang, S. Boisse, C. Bui, P.-A. Albouy, A. Brulet, M.-H. Li, J. Rieger and B. Charleux, Soft Matter, 2012, 8, 1130–1141 RSC.
- X. Wang, J. Xu, Y. Zhang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2452–2462 CrossRef CAS.
- X. Zhang, J. Rieger and B. Charleux, Polym. Chem., 2012, 3, 1502–1509 RSC.
| This journal is © The Royal Society of Chemistry 2013 |