DOI:
10.1039/C2RA20253K
(Paper)
RSC Adv., 2012,
2, 5585-5590
Simultaneous sensitization and hole activation in carbon nitride polymer sensitized TiO2†
Received
13th February 2012
, Accepted 4th April 2012
First published on 11th April 2012
Abstract
Experimental and theoretical methods have combined to explore the role of carbon nitride polymer (CNP) in the photocatalytic performance of CNP-sensitized TiO2 (P25). In this work, CNP rather than its precursors has been used to directly sensitize TiO2 at 170 °C based on acid–base interaction and the photocatalytic performance of CNP-sensitized TiO2 is evaluated by degradation of methyl orange under visible light irradiation. The obtained results show the photocatalytic performance of CNP-sensitized TiO2 is obviously stronger than TiO2 and CNP. The high performance results not only from transferring the photoinduced electrons from CNP to the conductivity band (CB) of TiO2 and the increased separation efficiency of photoinduced electrons and holes, but also possibly from hole activation. Hole activation may result from the interaction between TiO2 and CNP, which increases the oxidation ability of the photoinduced hole that leaves the CNP skeleton after light irradiation.
Introduction
Global energy shortages and environmental pollution stimulate scientists around the world to look for new energies that may replace fossil energy. The energy from sun to earth in one hour is 4.3 × 1020 J, being more than all of the energy consumed by human being in one year (2001, 4.1 × 1020 J),1 which makes solar energy one of the attractive new energies. Until now, lots of photocatalysts have been synthesized to harvest solar energy, mainly including inorganic materials (e.g. metal oxides, nitrides, sulfides, phosphides),2 organic metal complex (e.g. Ru-complex, Pt-complex) 3 and polymeric semiconductor.4 Titanium oxide (TiO2) is regarded as the most potential photocatalysts since it is cheap, stable and non-toxic.5 However, the band gaps of TiO2 are relatively broad (3.2 eV), which limit its application. Many methods have been developed to allow TiO2 to have visible light response, such as doping and sensitization.1,2,6 Sensitization is an efficient method to extend the absorption of TiO2.7 It is well accepted that the role of sensitizer is to transfer its excited electrons from visible light irradiation to the CB of TiO2 and increase the separation efficiency of photoinduced electrons and holes.1,2,6 Since the discovery of carbon nitride polymer (CNP) as a metal-free photocatalyst,4,8 it has also been used as sensitizer to expand the absorption edge of metal oxide photocatalysts, including TiO2.9–11 In addition, many endeavours have been devoted to clarify the nature of nitrogen species in “N-doped” TiO2 and it has proposed that polymeric carbon nitride may also make a contribution for the increased photocatalytic performance of “N-doped” TiO2.9–12 Until now, CNP-sensitized photocatalysts are generally synthesized by using some precursors, such as urea, to mix with the wide band gap photocatalysts, followed by the treatment at high temperature.
The formation of CNP on a photocatalyst surface and N-doping into the photocatalyst most likely occurs simultaneously when the precursors of CNP are used to sensitize TiO2, which make it difficult to judge of the contribution of CNP to the photocatalytic performance of CNP-sensitized TiO2. For deep understanding of the performance of CNP as a sensitizer, in the current work we used CNP rather than its precursors to directly sensitize TiO2 (P25) at 170 °C based on acid–base interactions (Scheme 1),13–15 and investigated the photocatalytic performance of CNP-sensitized P25 by experimental and theoretical methods. The obtained results show that the MO photodegradation performance of CNP-sensitized P25 is nearly four times faster than P25 and three times faster than CNP within two hours under visible light irradiation. Based on experimental and theoretical results, the high MO photodegradation performance results not only from transferring the photoinduced electrons from CNP to the CB of P25 and the increased separation efficiency of photoinduced electrons and holes, but also possibly from hole activation, which may result from the interaction between P25 and CNP. In this way, the holes leaving the CNP skeleton participate in the photodegradation of MO as well. This finding provides a new insight for the role of the sensitizer in the photocatalytic performance of the sensitized composite.
 |
| | Scheme 1 A synthesis scheme of the CNP-sensitized P25 by the mixing of P25 and CNP at 170 °C in N,N-dimethylacetamide for 12 h. | |
Results and discussion
Characterization of CNP material
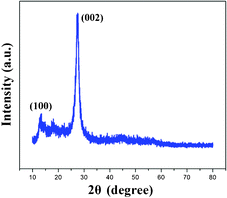
From the (002) and (100) peaks of the X-ray diffraction (XRD) result (Fig. 1), the as-synthesized CNP has graphite-like structure.4 Several strong bands in the 1200–1650 cm−1 region are found in Fourier transform infrared (FT-IR) spectra of the CNP material (Fig. 2), corresponding to the typical stretching modes of CN heterocycles, while the broad band at around 3000 cm−1 is assigned to these NH and NH2 groups.16
Characterization of P25CNP composites
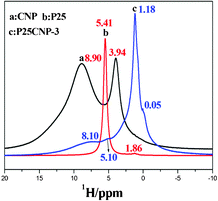
Based on the ratio between P25 and CNP, these P25CNP composites are named as P25CNP-0.7 (100 g P25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7 g CNP), P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7. According to Fig. 3, 1H NMR signals at 8.90 ppm and 3.94 ppm in CNP are assigned to the amine proton and adsorbed water,8 while those at 5.41 ppm and 1.86 ppm in P25 correspond to the terminal and bridging hydroxyl groups.17 Comparing the 1H NMR signals of CNP and P25, those of the amine proton, the terminal and bridging hydroxyl groups in the P25CNP-3 sample all shift to high field, while that of the adsorbed water moves to low field. This may be due to the surface of P25 becoming negative after the proton from the hydroxyl group in P25 transfers to the nitrogen atoms in CNP and the N-bound protons in CNP are shielded by the negative P25 surface (Scheme 1).14 The proton that transfers to the nitrogen atoms of CNP may further migrate to the oxygen atom of the adsorbed water, which leads to its 1H NMR signal shifting to low field.
:0.7 g CNP), P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7. According to Fig. 3, 1H NMR signals at 8.90 ppm and 3.94 ppm in CNP are assigned to the amine proton and adsorbed water,8 while those at 5.41 ppm and 1.86 ppm in P25 correspond to the terminal and bridging hydroxyl groups.17 Comparing the 1H NMR signals of CNP and P25, those of the amine proton, the terminal and bridging hydroxyl groups in the P25CNP-3 sample all shift to high field, while that of the adsorbed water moves to low field. This may be due to the surface of P25 becoming negative after the proton from the hydroxyl group in P25 transfers to the nitrogen atoms in CNP and the N-bound protons in CNP are shielded by the negative P25 surface (Scheme 1).14 The proton that transfers to the nitrogen atoms of CNP may further migrate to the oxygen atom of the adsorbed water, which leads to its 1H NMR signal shifting to low field.
From X-ray Photoelectron Spectroscopy (XPS) results shown in Fig. 4, the peak of the N signals in P25CNP-3 becomes a little broader (398.8, 399.4, 401.1 and 402.3) with respect to that in CNP (398.2, 400-401), indicating that there is an interaction between the Lewis base center N in CNP and the Lewis acid center Ti in P25.15 Since the amount of CNP in P25CNP-3 is very little, the intensity of the N signals in P25CNP-3 is obviously weaker than those in CNP. Compared with CNP, the N signals in P25CNP-3 become diverse, which may be due to the fact that the interactions between N and Ti are versatile. For deep understanding of this phenomenon, we constructed CNP and P25 models and investigated the interaction types between them by first principle calculations.18 A model containing three melem units was constructed to simulate the structure of CNP. From Fig. S1†, the HOMO mainly derives from the combination of nitrogen pz orbitals, while the LUMO localizes predominantly in C–N bond orbitals. This is the same as the previous report.4,8 The structure of P25 is simulated by a (TiO2)10H2O cluster, in which H2O has been split into OH and H between Ti and O (Fig. S2†). TiO2 cluster has been widely used to explore the band structure and the photocatalytic reaction.19,20 The CB of TiO2 is formed by Ti3d states and O2p, which is consistent with previous reports (Fig. S2†).19–21 According to Fig. 5, the type and strength of Ti–N interaction in P25CNP composite are varied. Both N atoms of the amino groups and CN heterocycle are involved in the interaction with Ti of P25. And the interaction strength is different since the distance between N and Ti varies in different cases (Fig. 5). The interaction between Ti and N may make the electrons of the N atoms more difficult to excite than in CNP, which account for why the N signals in P25CNP-3 are a little larger than those in CNP.
 |
| | Fig. 4 XPS spectra of CNP (left) and P25CNP-3 samples (right). Shown are the 1s binding energies of nitrogen. | |
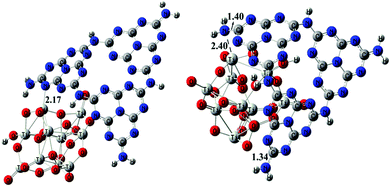 |
| | Fig. 5 The optimized geometries of P25CNP models with different Ti–N interactions. | |
MO photodegradation by P25CNP-3 composites synthesized at different temperature has been performed to explore the effect of reaction temperature on the photocatalytic performance of the P25CNP composite. According to Fig. 6, MO is stable under visible light irradiation. The photodegradation performance of P25CNP-3 increased with the enhancing reaction temperature. So, high temperature favors the formation of acid–base interactions between P25 and CNP. This also further supports the successful preparation of P25CNP composites at 170 °C.
 |
| | Fig. 6 The photodegradation of methyl orange (4 mg L−1) under visible light (λ > 420 nm) irradiation by P25CNP-3 composites synthesized at 30 °C, 80 °C and 170 °C, respectively. | |
The TEM result provided solid evidence for the formation of the P25CNP composite. From Fig. 7, it is clearly to see that very fine CNP flakes have been well attached to the P25 surface. The XRD patterns of all P25CNP samples are nearly identical to that of P25 (Fig. S3†), owing to the fact that the characterization diffraction peak of CNP overlaps with that of P25 at a 2θ value of 28°. The typical stretching modes of a CN heterocycle at 1409 and 1240 cm−1 are found in the FT-IR spectra of P25CNP-3 (Fig. S4†). According to the UV results (Fig. 8), the absorption edges of the P25CNP samples are longer in wavelength than the corresponding P25. The sensitization of P25 by CNP is not obvious since the absorption edge did not shift significantly to the lower energy side, which may be due to just a small amount of CNP being used in our work to sensitize P25. But in previous reports,9,10 the amounts of precursors used to produce CNP for sensitizing wide gap photocatalysts are very large.
 |
| | Fig. 8 UV-Vis absorption spectra of P25, P25CNP-0.7, P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7 samples. | |
Photocatalytic performance of P25CNP composites
The photocatalytic activities of P25, CNP, P25CNP-3-mix, P25CNP-0.7, P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7 samples have been evaluated by 4 mg L−1 MO photodegradation under visible light (λ > 420 nm) irradiation, as shown in Fig. 9. The P25CNP-3-mix sample is obtained by directly mixing P25 and CNP in the ratio of 100 g P25:3 g CNP at room temperature. From Fig. 9, the photocatalytic activity of the P25CNP-3-mix is higher than that of P25, and lower than that of CNP, implying that CNP cannot effectively sensitize P25 simply by mixing. The reactivity of CNP is a little lower than that reported,22 due to the different synthesis conditions. All P25CNP samples have a higher MO photodegradation performance than P25 and CNP. The P25CNP-3 sample can completely photodegrade MO in two hours, which is nearly four times faster than P25 and three times faster than CNP. Thus, using CNP to sensitize P25 can effectively increase the photocatalytic performance.
 |
| | Fig. 9 The photodegradation of methyl orange (4 mg L-1) under visible light (λ > 420 nm) irradiation by P25, CNP, P25CNP-3-mix, P25CNP-0.7, P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7 samples, respectively. | |
To confirm that methyl orange was really photodegraded by the P25CNP samples under light irradiation, we investigated the chemical oxygen demand (COD) of the system before and after reaction. It is well known that COD is an important evaluation criterion for the mineralization of organic pollutants. After photodegradation, the COD of the system decreased by 19%, implying that some of the MO has been decomposed into CO2. In addition, some of the MO molecules are degraded into species containing aromatic rings since a new absorption peak appeared at 230 nm with the decrease of the absorption peak at 460 nm (Fig. 10). The decrease of the absorption peak at 460 nm implies the breaking of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in MO, while the new peak at 230 nm indicates the formation of a species containing an aromatic ring. So, MO is really photodegraded in this reaction.
N bond in MO, while the new peak at 230 nm indicates the formation of a species containing an aromatic ring. So, MO is really photodegraded in this reaction.
MO photodegradation mechanism of P25CNP composites
From Fig. 11, the fluorescence intensity of P25CNP-3 is weaker than that of P25 and P25CNP-3-mix, implying that the interaction between P25 and CNP is favourable for the separation of the photoinduced electron and hole. So, the high photocatalytic performance of P25CNP samples is from the contribution of sensitization and the increased separation efficiency of the photoinduced electron and hole. This is consistent with the general sensitization mechanism.7,9–11
It is well known that photoinduced electron generally takes part in the photocatalytic reaction by combing with O2 to form a superoxide radical or hydroxyl radical. So undergoing the reaction under a N2 atmosphere will prevent the combination of a photoinduced electron and O2 and annihilate the activity of photoinduced electron. For photoinduced holes, methanol can be used to annihilate its activity. For example, Yan et al.22 have found that only photoinduced electrons are responsible for the CNP-catalyzed MO degradation since the rate of MO degradation did not change after methanol was added into the system, but decreases greatly under N2 atmosphere. To explore the contribution of the photoinduced electrons and holes to the photocatalytic performance of P25CNP, three comparison experiments have been performed. From Fig. 12, the reaction rate decreases to nearly 50% from the original 100% when the reaction proceeds under N2 atmosphere, implying that photoinduced electron is involved in P25CNP-catalyzed MO photodegradation. Interestingly, the rate also nearly decreases to 50% from the original 100% after methanol is added into the system. This suggests that the photoinduced hole participates in the P25CNP-catalyzed MO photodegradation as well. Thus, photoinduced electrons and holes both take part in the MO photodegradation by the P25CNP composite.
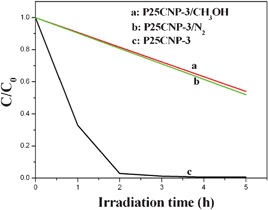 |
| | Fig. 12 Photodegradation of methyl orange (4 mg L-1) by P25CNP-3/N2, P25CNP-3/CH3OH and P25CNP-3 samples under visible light (λ > 420 nm) irradiation. | |
Why is it that the photoinduced hole in P25CNP can take part in MO photodegradation, but that in CNP cannot take part in MO photodegradation? According to the VBXPS results for CNP and P25CNP, shown in Fig. 13, the energy of the valence band in CNP is 1.38 V, while that of P25CNP is 1.90 V, implying that the oxidation ability of the photoinduced hole in P25CNP is stronger than that in CNP. This accounts well for the experimental observation.
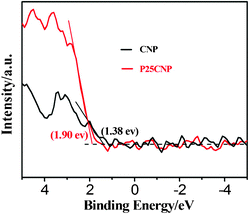 |
| | Fig. 13 VBXPS spectra of the CNP and P25CNP-3 samples. | |
For deep understanding of this point, first principle calculations have been performed. The calculated band gap for the P25 model is 4.7 eV (Fig. 14), which is larger than the experimental result for TiO2,21 due to the fact that we use a cluster model. The calculated band gap of 3.6eV for CNP model is also larger than the experimental result for CNP.8 However, the gap in the CNP model is narrower than that in the P25 model and the LUMO energy of the CNP model is higher than that of the P25 model. Both of these two points are consistent with the following experimental results: 9,10 (1) the band gap of CNP is narrower than that of TiO2; (2) the CB energy of CNP is higher than that of TiO2, which is the reason why CNP can sensitize TiO2. Furthermore, the calculated energy order of HOMO in CNP model and P25CNP are consistent with that measured with VBXPS method: the energy of the HOMO in P25CNP is larger than that in CNP (Fig. 13 and 14). This means that the P25 and CNP models used can provide reliable information for this system. From Fig. 14, the LUMO of P25CNP is distributed in both the CNP and P25 moieties, while the HOMO is located in the CNP moieties. The gap for the P25CNP model is 3.5 eV, which is smaller than that (4.8 eV) of the P25 model. This is consistent with the traditional CNP sensitization mechanism: electron transfers from the CB of CNP to the CB of P25 under light irradiation.10 In addition, the gap for the P25CNP model is just a little smaller than that of the CNP model (3.5 eV versus 3.6 eV), implying that using CNP to sensitize P25 cannot lead to an obvious red shift in the UV-Vis spectra (Fig. 8). The HOMO energy (−6.6 eV) of the P25CNP model is lower than that (−5.8 eV) of the CNP model, while the energy of the LUMO (−3.2 eV) in P25CNP is lower than that of CNP (−2.3 eV) (Fig. 14). It is well known that the HOMO and LUMO orbitals in a cluster are the counterparts of the valence band (VB) and CB in the material.23,24 This suggests that the oxidation ability of the photoinduced hole in P25CNP is stronger than that in CNP and the reduction ability of the photoinduced electron in P25CNP is weaker than that in CNP. Thus, forming P25CNP by the interaction between P25 and CNP will increase the oxidation ability of the photoinduced hole and decrease the reduction ability of the photoinduced electron. Thus, the photocatalytic performance of P25CNP will have a maximum with the ratio increase of CNP in P25CNP. This accounts well for why P25CNP-3 showed the highest photocatalytic activity among all samples (Fig. 9). It has been reported that doping S into CNP can change the energy of the CB in CNP.25 In a word, the high MO photodegradation of P25CNP results not only from transferring the photoinduced electrons from CNP to the CB of P25 and the increased separation efficiency of photoinduced electrons and holes, but also from hole activation.
 |
| | Fig. 14 Orbitals illustration for the formation of the P25CNP model from the P25 and CNP models. | |
A possible photodegradation mechanism has been proposed based on the results mentioned above (see Scheme 2). Under visible light irradiation, the electrons in the HOMO orbital located in the CNP moiety of the P25CNP composite are excited into its LUMO orbital and holes stay in the CNP moiety. Then the active species, such as superoxide radical and hydroxyl radicals, are generated by photoinduced electron reduction and hole oxidation, respectively.26 Subsequently, MO is degraded into small molecules by these active species in both the CNP and P25 moieties.
 |
| | Scheme 2 The possible MO photodegradation mechanism of the P25CNP composite under visible light irradiation. | |
Conclusion
In summary, detailed experimental and theoretical investigations have been performed to investigate the role of CNP in the photocatalytic performance of CNP-sensitized P25. The obtained results show that the high MO photodegradation performance results not only from transferring the photoinduced electrons of CNP to the CB of P25 and the high separation efficiency of photoinduced electrons and holes, but also possibly from hole activation. The hole activation is due to the interaction between P25 and CNP, which increases the oxidation ability of the photoinduced holes that stay in the CNP skeleton. This finding provides a new insight for the role of the sensitizer in the photocatalytic performance of sensitized photocatalysts and is helpful to develop high efficiency composite photocatalysts.
Experimental Section
I. Preparation of carbon nitride polymer (CNP).
CNP was prepared by the following way: 10 g melamine powder was put into an alumina crucible with a cover, and then this system was heated to 520 °C in a muffle furnace. Heating rate was 7 °C min−1 and the temperature maintained for 4 h.
II. Preparation of P25CNP samples.
Then, CNP-sensitized TiO2 (Degussa, P25) composites were synthesized by mixing CNP and TiO2 in a glass bottle with dimethylacetamide as solvent, followed by increasing the temperature to 170 °C in oil bath and keeping the temperature for 12 h. Based on the ratio between TiO2 and CNP, these composites are named as P25CNP-0.7 (100 g P25:0.7 g CNP), P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7.
III. Characterization of CNP and P25CNP samples.
The samples were characterized by X-ray diffraction (XRD) for phase identification on the Rigaku RINT2000 diffractometer. UV-Vis diffuse reflection spectra were measured using a UV-Vis spectrophotometer (Varian CARY 100, USA) and converted from reflection to absorbance by the Kubelka–Munk method. All solid-state NMR experiments were performed on a Bruker Aduvance III 400WB spectrometer equipped with a 9.4 T magnet. For 1H NMR, the relaxation delay was 10–60 s, the 90 pulse width was 4 μs, and a three-pulse sequence pulse program was applied to suppress deading and background signal. The chemical shift was referenced to trimethylsilane (TMS). Chemical oxygen demand (COD) measurements were carried out on Hach DR1010 COD analyzer to evaluate the mineralization of MO.
The photocatalytic activities of P25, CNP, P25CNP-3-mix, P25CNP-0.7, P25CNP-1, P25CNP-3, P25CNP-5 and P25CNP-7 samples were evaluated by photodegrading methyl orange (MO) in a Pyrex reactor. 0.2 g sample was dispersed in MO aqueous solution (100 mL, 4 mg L−1). The light irradiation system contains a 300 W Xe lamp with a filter (420 nm) for visible light and a water filter to eliminate the temperature effect.
IV. Computational details.
All calculations are performed with Gaussian 03 program.
B3LYP/3-21g method was used to optimize P25, CNP and P25CNP models. HOMO and LUMO orbitals of P25, CNP, P25CNP models were constructed with Gview program based the B3LYP/3-21g-optimized results.
Acknowledgements
We are grateful the financially support from NSFC (20871067 and 20873059), Analysis Center of Nanjing University and high performance computing center station of Nanjing University.
References
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercawand and C. Creutz, Chem. Rev., 2001, 101, 953 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- D. Ravelli, D. Dondi, M. Fagnoniand and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC and references therein.
- C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef.
- M. Nolan, Chem. Commun., 2011, 47, 8617 RSC.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS.
- L. Pan, J. J. Zou, X. W. Zhang and L. Wang, J. Am. Chem. Soc., 2011, 133, 10000 CrossRef CAS.
- Y. Guo, S. Chu, S. C. Yan, Y. Wang and Z. G. Zou, Chem. Commun., 2010, 46, 7325 RSC.
- H. Kisch and D. Mitoraj, Chem.–Eur. J., 2010, 16, 261 CrossRef.
- F. Peng, X. S. Zhou, H. J. Wang, H. Yu and Y. P. Fang, Chem. Commun., 2011, 47, 10323 RSC.
- T. Kako, Q. Y. Li, B. Yue, H. Iwai and J. H. Ye, J. Phys. Chem. C, 2010, 114, 4100 Search PubMed.
- D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 9975 CrossRef CAS.
- B. Bonelli, M. Cozzolino, R. Tesser, M. Di Serio, M. Piumetti, E. Garrone and E. Santacesaria, J. Catal., 2007, 246, 293 CrossRef CAS.
- R. H. Goncalves, W. H. Schreiner and E. R. Leite, Langmuir, 2010, 26, 11657 CrossRef CAS.
- N. Nakayama and T. Hayashi, Colloids Surf., A, 2008, 317, 543 CrossRef CAS.
- M. J. Bojdys, J. O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177 CrossRef CAS.
- M. Crocker, R. H. M. Herold, A. E. Wilson, M. Mackay, C. A. Emeis and A. M. Hoogendoorn, J. Chem. Soc., Faraday Trans., 1996, 92, 2791 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. Valdes and G. J. Kroes, J. Phys. Chem. C, 2010, 114, 1701 CAS.
- J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 3022 CrossRef.
- Y. C. Nah, I. Paramasivam and P. Schmuki, ChemPhysChem, 2010, 11, 2698 CrossRef CAS.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397 CrossRef CAS.
- Z. W. Qu and G. J. Kroes, J. Phys. Chem. B, 2006, 110, 8998 CrossRef CAS.
- S. S. Sun, J. Mater. Sci.: Mater. Electron., 2007, 18, 1143 CrossRef CAS.
- J. S. Zhang, J. H. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Z. Fu and X. C. Wang, Energy Environ. Sci., 2011, 4, 675 CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20253k |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.