
Preparation and nanoaggregate formation ability in water of amphiphilic ladder-like polymers with parallelly linked hydrophilic polyether and hydrophobic polysiloxane chains†
Shiori
Matsuo
a,
Sho
Nonaka
a,
Aki
Mihata
a,
Kazuhiro
Shikinaka
 b and
Yoshiro
Kaneko
*a
b and
Yoshiro
Kaneko
*a
aGraduate School of Science and Engineering, Kagoshima University, 1-21-40 Korimoto, Kagoshima 890-0065, Japan. E-mail: ykaneko@eng.kagoshima-u.ac.jp
bResearch Institute for Chemical Process Technology, National Institute of Advanced Industrial Science and Technology (AIST), Nigatake, 4-2-1, Miyagino-ku, Sendai, Miyagi 983-8551, Japan
First published on 2nd January 2025
Abstract
Aiming to enhance the high-temperature stability of nanoaggregates of amphiphilic polymers in water, an amphiphilic ladder-like (double-chain) polymer (PGE–PAMS) was successfully prepared via the thiol–ene reaction between 3-mercaptopropyldimethoxymethylsilane and poly(allyl glycidyl ether), followed by intramolecular polycondensation (template polymerization) of the dimethoxysilyl groups in the side chains of the resulting polymer. Solubility tests, FT-IR spectroscopy, 1H NMR spectroscopy, 29Si NMR spectroscopy and transmission electron microscopy confirmed that PGE–PAMS possesses a ladder-like structure, where the hydrophilic poly(glycidyl ether) chain and the hydrophobic polyalkylmethylsiloxane chain are linked in parallel by sulfide bonds. PGE–PAMS formed nanoaggregates with a diameter of ca. 60–70 nm in water, which were stable even at high temperatures (80 °C–90 °C). Additionally, in water containing a small amount of N,N-dimethylformamide, the PGE–PAMS nanoaggregates could solubilize tetraphenylporphyrin, a hydrophobic dye, and stably retain it even at a high temperature of 90 °C.
Introduction
Amphiphilic polymers contain both hydrophilic and hydrophobic segments within a single polymer chain, resulting in not only emulsifying, dispersing and solubilizing properties similar to those of low-molecular-weight surfactants but also diverse functionalities through self-assembly, such as the reversible formation of well-defined aggregates depending on the surrounding environment. By virtue of these characteristics, amphiphilic polymers are widely used as functional materials in various fields, including drug delivery systems,1,2 surface modification and functionalization3 and nanoscale patterning.4,5Amphiphilic molecules can be classified into ionic and nonionic types. Nonionic amphiphilic molecules are more commonly used because their self-assembly and interfacial activity functions are maintained even in salt-containing water, and they can also be combined with ionic amphiphilic molecules. Polyethylene glycol (PEG) is frequently used as the hydrophilic segment of nonionic amphiphilic polymers, particularly in biotechnological applications like drug delivery systems, owing to its biocompatibility.6–15 As the hydrophobic segments of nonionic PEG-containing amphiphilic polymers, materials such as polypropylene glycol,6,7 poly(ε-caprolactone),8–10 polylactic acid,11,12 poly(lactic acid-co-glycolic acid)13,14 and polydimethylsiloxane (PDMS)15 are used.
In water, nonionic amphiphilic polymers form nanoaggregates such as micelles and vesicles, many of which exhibit temperature responsiveness because the hydrophilic PEG chain, which is surrounded by water molecules, undergo reversible hydration at low temperatures and dehydration at high temperatures.16–18 As a result, nanoaggregates of nonionic amphiphilic polymers can alter their shape in response to external stimuli such as temperature, which renders them promising candidates for smart materials applications.
However, only a limited number of nonionic amphiphilic polymers can stably maintain the nanoaggregates at high temperatures; instead, large aggregates that cause turbidity in water dispersions are formed. This is because, at high temperatures, water becomes a poor solvent not only for the hydrophobic segments but also for PEG.19 Considering that the hydration/dehydration of hydrophilic polymer chains is related to their conformation,16 suppressing the conformational change of the PEG chain could help maintain its hydrophilicity even at high temperatures.
In fact, in a previous study, Honda et al. reported that a macrocyclic amphiphilic polymer composed of a hydrophilic PEG chain and a hydrophobic poly(butyl acrylate) chain exhibited a cloud point (Tc) of ca. 70 °C in water, which was ca. 40 °C higher than that of a linear amphiphilic polymer having the same components.20 This suggests that the micelles formed by macrocyclic amphiphilic polymers remain stable even at relatively high temperatures (70 °C), which may be attributed to the inhibition of conformational changes due to the unique cyclic shape of the polymers. However, the relationship between the shape of amphiphilic polymers and the stability of their nanoaggregates at high temperatures has not yet been fully clarified. Unveiling this relationship is expected to contribute to the development of amphiphilic polymers with novel structures.
In this study, we were interested in investigating amphiphilic polymers with a ladder-like structure, which are expected to exhibit different properties from single-chain polymers due to the suppression of conformational changes.21,22 For example, Gu et al. reported that a low-defect conjugated ladder-like polymer forms stable aggregates in solution and remains aggregated at high temperatures up to 75 °C in chlorobenzene compared with a conjugated non-ladder-like polymer control.23
Ladder-like polymers can be prepared using various methods. For instance, the hydrolytic polycondensation of limited organotrialkoxysilanes, i.e., the sol–gel reaction, yields ladder-like polysilsesquioxanes,24–35 which exhibit more rigid properties than linear polysiloxanes. Meanwhile, template polymerization is a widely recognized method for preparing ladder-like organic polymers. In this approach, polymerizable groups are introduced into the side chains of a base polymer, which then undergoes intramolecular polymerization. Kämmerer et al. reported the preparation of a ladder-like oligomer via intramolecular free-radical polymerization of a p-cresol oligomer containing acrylic groups.36,37 Similarly, Jantas et al. prepared a ladder-like polymer by the intramolecular free-radical polymerization of multivinyl monomers based on polyvinyl alcohol and poly(meth)acrylate.38–42 Saito et al. prepared a ladder-like polymer through the intramolecular atom transfer radical polymerization of poly(2-methacryloyloxyethyl methacrylate).43,44 Recently, we reported a ladder-like polysiloxane with two siloxane main chains, which was prepared by introducing diethoxysilyl groups as polymerizable groups into the side chains of ammonium-containing polysiloxane,45 followed by intramolecular polycondensation.46
Herein, to investigate the effect of the inhibition of conformational changes due to the unique shape of the double chains on the stability of the self-assembly, we adopted the template polymerization method to prepare a ladder-like nonionic amphiphilic polymer comprising a poly(glycidyl ether) (PGE) chain as the hydrophilic segment and a polyalkylmethylsiloxane (PAMS) chain as the hydrophobic segment, which were linked in parallel. To the best of our knowledge, a ladder-like polymer consisting of two polymer chains with such distinct properties has not been reported to date. The resulting PGE–PAMS polymer formed nanoaggregates with a diameter of ca. 60–70 nm in water, and these nanoaggregates remained stable even at high temperatures (80 °C–90 °C). Furthermore, in water containing a small amount of N,N-dimethylformamide (DMF), the PGE–PAMS nanoaggregates could solubilize tetraphenylporphyrin (TPP), a hydrophobic dye. Even when the solution was heated to 90 °C, TPP did not precipitate, suggesting that the PGE–PAMS nanoaggregates can stably encapsulate TPP at high temperatures.
Results and discussion
Preparation of poly(allyl glycidyl ether) (PAGE)
PAGE was prepared as the precursor polymer via the anionic ring-opening polymerization of allyl glycidyl ether, following a previously reported procedure.47,48 PAGE was soluble in dimethylsulfoxide (DMSO), DMF, methanol (CH3OH), acetone, chloroform, ethyl acetate, tetrahydrofuran (THF) and diethyl ether but insoluble in hexane and water. The number-average molecular weight (Mn) and molecular weight distribution (Mw/Mn) of PAGE were estimated to be 2.5 × 103 and 1.31, respectively, using gel permeation chromatography (GPC). Furthermore, its average degree of polymerization (DP) was calculated to be 46 according to end-group quantification using 1H NMR spectroscopy. This calculation considered the formula weights of both the repeating and initiating units, yielding an Mn of 5.3 × 103 (Fig. S1†).Preparation of PGE–PAMS
PGE–PAMS was prepared by means of the thiol–ene reaction between 3-mercaptopropyldimethoxymethylsilane (MPDMMS) and PAGE, followed by intramolecular polycondensation (template polymerization) of the dimethoxysilyl groups in the side chains of the resulting polymer.First, the thiol–ene reaction of MPDMMS with PAGE was performed using phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO) as a photoradical initiator in dehydrated THF, resulting in poly(glycidyl ether) containing dimethoxysilyl groups as side chains (PGE–DS, Scheme 1a). To confirm the progress of the thiol–ene reaction, we attempted to isolate the solid product via reprecipitation. However, the reprecipitated product was insoluble in any solvent. The cause of insolubility is believed to be the partial hydrolysis of the diethoxysilyl groups, which leads to intermolecular dehydration condensation involving the formed silanol groups, resulting in the formation of intermolecular crosslinking. Therefore, after completing the reaction, a large amount of DMF was added to the reaction solution to prepare a dilute solution (1.0 × 10−3 mol unit per L) without isolating the product. To avoid the intermolecular reaction between the dimethoxysilyl groups in PGE–DS side chains, which should react only intramolecularly, the solution concentration was reduced during the subsequent polycondensation.
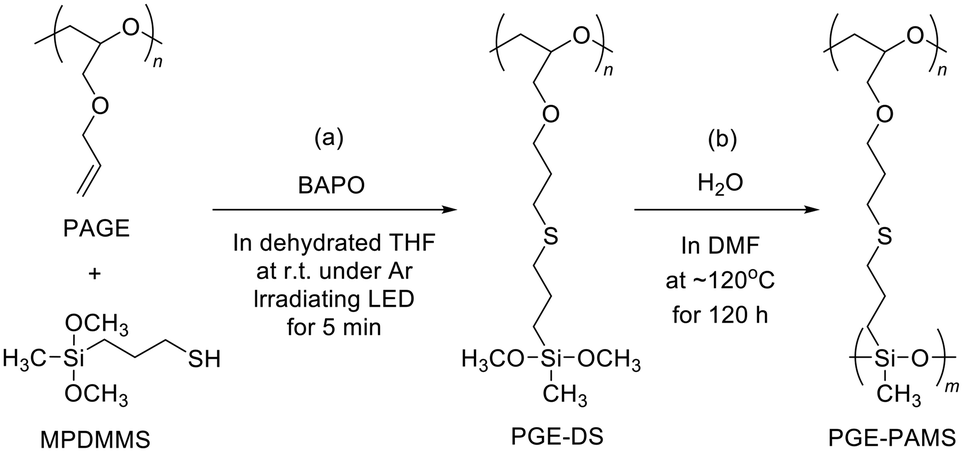 | ||
| Scheme 1 Preparation of (a) poly(glycidyl ether) containing dimethoxysilyl groups (PGE–DS) and (b) a ladder-like polymer with a poly(glycidyl ether) (PGE) chain as the hydrophilic segment and a polyalkylmethylsiloxane (PAMS) chain as the hydrophobic segment (PGE–PAMS). | ||
The intramolecular polycondensation of the dimethoxysilyl groups attached to the polymer chain of PGE–DS was performed by adding purified water, followed by stirring at 120 °C for 120 h (Scheme 1b). To isolate the product, the DMF solution was concentrated and reprecipitated from diethyl ether. The diethyl ether-insoluble fraction was then isolated by decantation and washed with diethyl ether. Finally, the product was dried under reduced pressure to yield PGE–PAMS. PGE–PAMS was soluble in water and in organic solvents such as DMSO, CH3OH, ethanol (C2H5OH), and isopropyl alcohol (iso-C3H7OH) (Fig. 1a–e), demonstrating its amphiphilic nature. Meanwhile, it was insoluble in organic solvents such as DMF, acetone, THF, chloroform, toluene and hexane.
 | ||
| Fig. 1 Photographs of PGE–PAMS solutions in (a) water, (b) DMSO, (c) CH3OH, (d) C2H5OH and (e) iso-C3H7OH (concentration: 0.1 w/v%). | ||
Characterization of PGE–PAMS
PGE–PAMS was characterized via1H NMR, 29Si NMR and transmission electron microscopy (TEM) analyses, which confirmed its ladder-like structure with PGE and PAMS chains linked in parallel via sulfide bonds.In the 1H NMR spectrum of PGE–PAMS in CD3OD, the signals attributed to the allyl groups (5.40–5.10 ppm) almost disappeared, indicating the progress of the thiol–ene reaction between MPDMMS and PAGE. Additionally, the average DP of the PAMS chain was estimated to be ca. 39.1 by calculating the integral ratio of signal k to signal b, which correspond to the phenolate-initiated end group and the methylene proton adjacent to the Si atom, respectively, in the 1H NMR spectrum (Fig. 2). Furthermore, the average DP of the PGE chain was ca. 36.4, of which 33.5 were units linked to PAMS chains and 2.9 were units with unreacted allyl groups, indicating that 8.0% of the PGE chains contained units with unreacted allyl groups. This was determined from the integral ratio of signal k to signals d and i, which are attributed to the phenolate-initiated end group, methylene protons and double bonds, respectively, in the 1H NMR spectrum (Fig. 2).
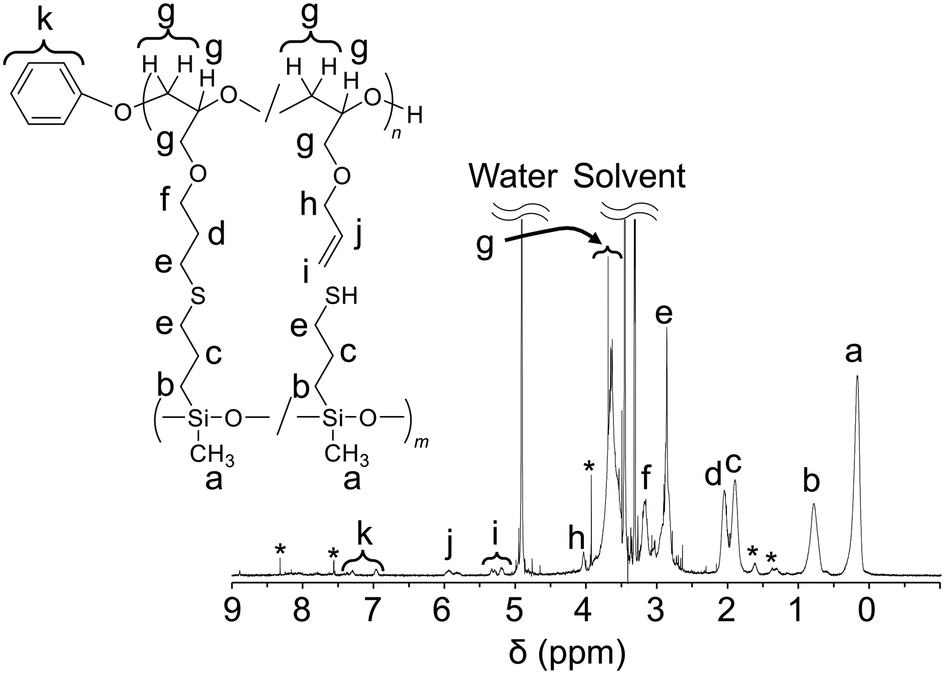 | ||
| Fig. 2 1H NMR spectrum of PGE–PAMS in CD3OD. The chemical shifts are referenced to CD3OD (δ 3.31). *: Unknown signal. | ||
Thus, the 1H NMR end-group quantification revealed a difference between the DP of the PGE chain (36.4) and that of the PAMS chain (39.1). This suggests that the PAMS chains contain units with mercapto groups in addition to units linked to the PGE chains. The integral ratio of signal b to signal d was 2.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.72, and that of signal c to signal d was 2.00:1.61 (Fig. 2), further supporting the higher DP of the PAMS chain compared with that of the PGE chain. According to the integral ratio of signal b to signal d, ca. 14.3% of the PAMS chain contains units with mercapto groups. Assuming an average DP of 39.1 for the PAMS chain, 33.5 units are linked to the PGE chains and 5.6 units contain mercapto groups. Furthermore, from the average DPs of the PGE and PAMS chains, the Mn of PGE–PAMS was estimated to be ca. 1.0 × 104.
:1.72, and that of signal c to signal d was 2.00:1.61 (Fig. 2), further supporting the higher DP of the PAMS chain compared with that of the PGE chain. According to the integral ratio of signal b to signal d, ca. 14.3% of the PAMS chain contains units with mercapto groups. Assuming an average DP of 39.1 for the PAMS chain, 33.5 units are linked to the PGE chains and 5.6 units contain mercapto groups. Furthermore, from the average DPs of the PGE and PAMS chains, the Mn of PGE–PAMS was estimated to be ca. 1.0 × 104.
The 29Si NMR spectrum of PGE–PAMS in CD3OD at 40 °C exhibited two broad signals in the D2 regions at −17 to −21 ppm and −21 to −24 ppm, with an integral ratio of 68:32, whereas no signals attributable to a D1 or D0 structure were detected (Fig. 3a). The predominance of the D2 structure indicates that sufficient polycondensation of the dimethoxysilyl groups in PGE–DS occurred, resulting in the formation of the Si–O–Si bond main chain.
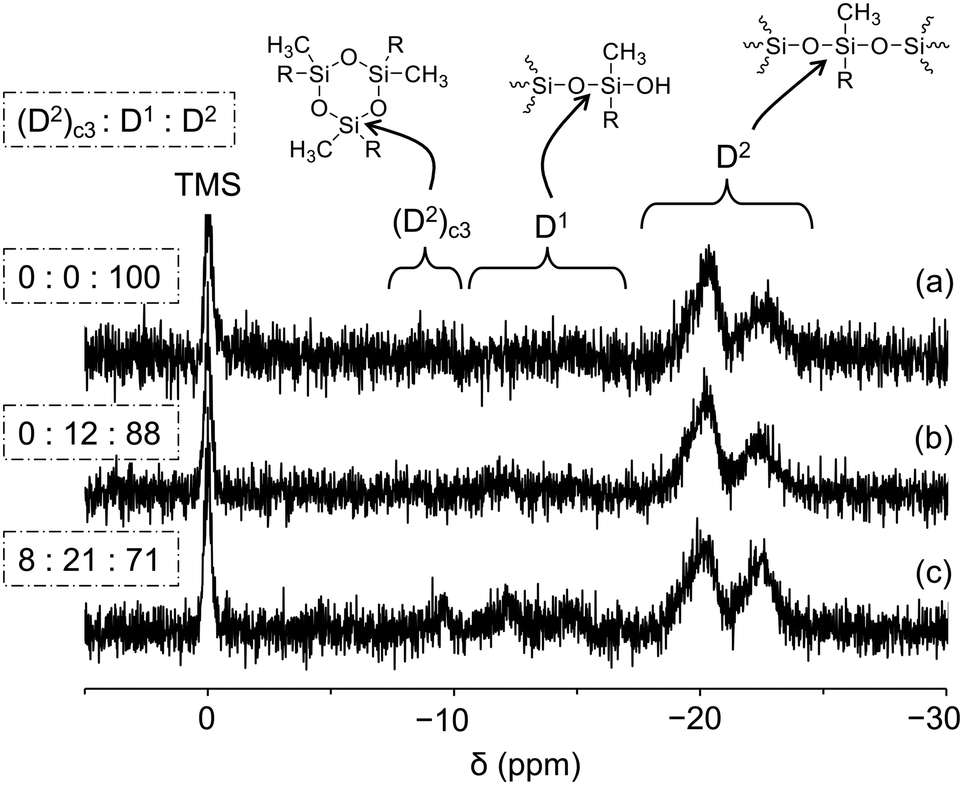 | ||
| Fig. 3 29Si NMR spectra of PGE–PAMS obtained via intramolecular polycondensation of PGE–DS for (a) 120 h, (b) 96 h and (c) 48 h in CD3OD at 40 °C. A small amount of Cr(acac)3 was added as a relaxation agent. The chemical shifts are referenced to tetramethylsilane (TMS, δ 0.0). | ||
As mentioned earlier, the signals attributed to the D2 structure of the PAMS chain were split into two. These signals are presumed to correspond to units linked to the PGE chain via sulfide bonds (−17 to −21 ppm) and units with mercapto groups (−21 to −24 ppm) (Fig. 3a). According to the 1H NMR spectrum (Fig. 2), ca. 14.3% of the repeating units in the PAMS chain contain mercapto group components, supporting this interpretation. Although the estimated proportions of mercapto-containing units in the PAMS chains, as determined by 29Si and 1H NMR measurements, differ (ca. 32% and 14.3%, respectively), this discrepancy is considered to fall within the range of experimental error.
Since the repeating unit length of the PAMS chain, which consists of two main atoms (Si–O), is shorter than that of the PGE chain, which has three main atoms (C–C–O), introduction of mercapto-containing units in the PAMS chain most likely helps balance the chain lengths between PGE and PAMS, thereby alleviating strain. From an alternative perspective, the strain might also be alleviated by the long crosslinking units even without the introduction of MPDMMS. However, in such a case, it is speculated that the molecular chains would curve rather than straighten due to the mismatch in the repeating unit lengths of the PAMS and PGE chains. As evidenced by TEM observations (Fig. 4), described in detail below, the molecular chains are straight, suggesting that the introduction of MPDMMS plausibly explains this outcome.
 | ||
| Fig. 4 TEM image of a sample obtained by drying an aqueous solution of PGE–PAMS. | ||
Performing the intramolecular polycondensation of PGE–DS for shorter times (96 and 48 h) resulted in a decrease in the proportion of D2 signals and an increase in the proportion of D1 (D1: C2Si(OSi)(OR), R = H or CH3) and (D2)c3 signals ((D2)c3 is a cyclic trimer component49) in the 29Si NMR spectra of the resulting products (Fig. 3b and c). These results indicate that a reaction time of 120 h is essential for obtaining PGE–PAMS via the intramolecular polycondensation of PGE–DS. Therefore, subsequent experiments were performed using PGE–PAMS prepared with an intramolecular polycondensation time of 120 h.
To examine the structure of PGE–PAMS, TEM measurements were performed. A droplet of an aqueous solution of PGE–PAMS was placed onto a TEM grid and dried without electron staining. The TEM image of the resulting sample revealed straight polymer chains arranged in parallel stripe patterns (Fig. 4), indicating that PGE–PAMS possesses a one-dimensional structure. The width of a single black-and-white stripe set was estimated to be ca. 3.33 nm. The black-and-white regions can be attributed to the inorganic PAMS components (Si-rich regions) and the organic PGE components (C-rich regions), respectively. On the basis of the molecular dimensions of a model compound representing the repeating unit of PGE–PAMS (Fig. 5a), the molecular width of PGE–PAMS was estimated to be ca. 1.76 nm. Accordingly, the stripe widths of ca. 3.33 nm observed in the TEM image suggest that the polymer chains are arranged in a regular pattern in which identical segments are aligned face-to-face (Fig. 5b).
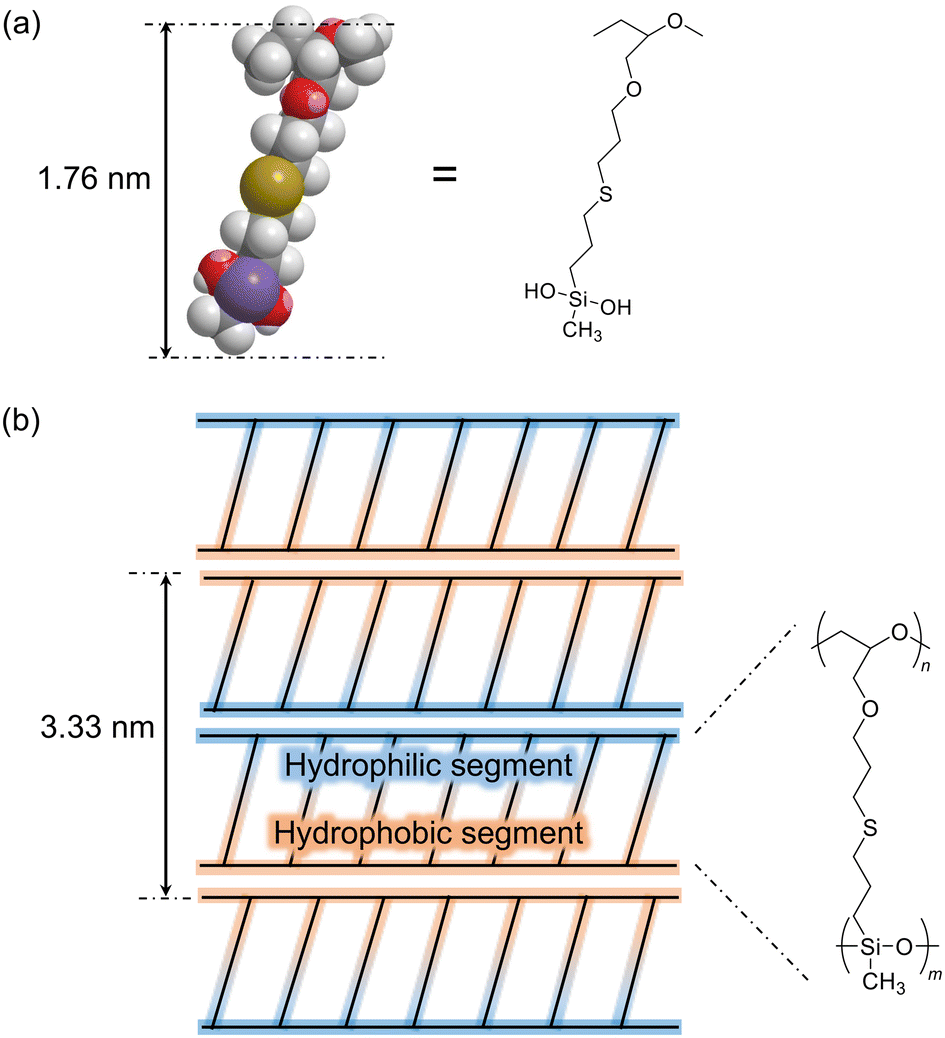 | ||
| Fig. 5 (a) Molecular dimensions of a model compound representing the repeating unit of PGE–PAMS, energy-minimized using MM2 in the CS Chem3D program package. (b) Plausible stacking structure of PGE–PAMS. | ||
The results of the structural analysis of PGE–PAMS can be summarized as follows. Solubility tests in various solvents indicated the absence of intermolecular crosslinking in PGE–PAMS. The 1H NMR spectrum confirmed the successful thiol–ene reaction between MPDMMS and PAGE. The 29Si NMR spectra showed that the dimethyoxysilyl groups of PGE–DS underwent polycondensation to form the PAMS chains, although some units with mercapto group components were also present. The TEM image revealed that PGE–PAMS had a one-dimensional structure. Taken together, these results suggest that PGE–PAMS exhibits a ladder-like structure consisting of PGE and PAMS chains linked in parallel by sulfide bonds, as illustrated in Fig. 6.
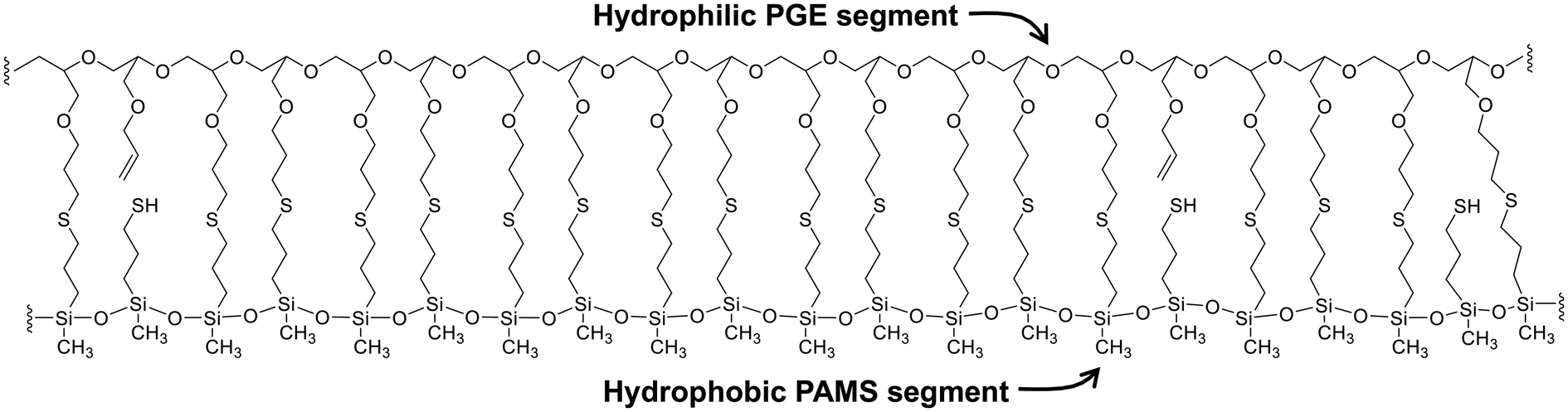 | ||
| Fig. 6 Proposed molecular structure of PGE–PAMS. | ||
Aggregation behaviour of PGE–PAMS in various solvents
The volume-average particle size of PGE–PAMS was determined to be 67.6 ± 24.1 nm using dynamic light scattering (DLS) measurements in water (0.1 w/v%) at 25 °C (Fig. 7a). These particles are considered to be nanoaggregates formed by the self-assembly of PGE–PAMS. Although amphiphilic diblock copolymers with hydrophilic and hydrophobic segments linked in series are known to form micelles around 100 nm in size, the measured size of 67.6 ± 24.1 nm is too large to be classified as micelles, likely due to the parallel arrangement of these segments in PGE–PAMS, as observed in the TEM image. This regular arrangement suggests that PGE–PAMS may form multilayer vesicles in water; a more detailed structural analysis of these nanoaggregates to confirm this hypothesis is still under investigation.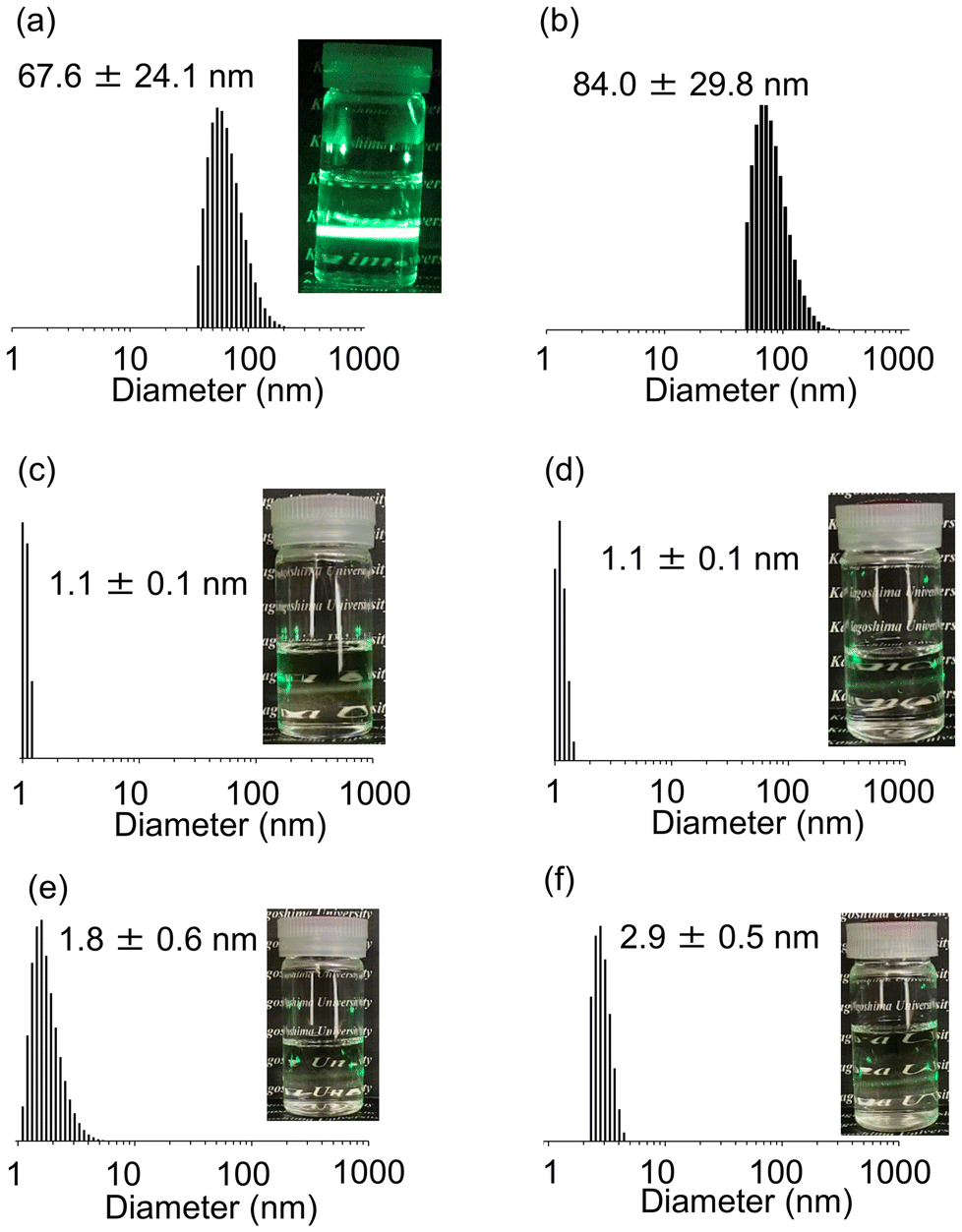 | ||
| Fig. 7 Volume-average particle size distributions of PGE–PAMS measured by DLS in water at (a) 25 °C and (b) 80 °C and in (c) DMSO, (d) CH3OH, (e) C2H5OH and (f) iso-C3H7OH at 25 °C (concentration: 0.1 w/v%). | ||
Meanwhile, the particle size was estimated to be ca. 1–3 nm via DLS measurements in organic solvents such as DMSO, CH3OH, C2H5OH and iso-C3H7OH, suggesting that PGE–PAMS dissolved as unimers (single polymer molecules) without forming nanoaggregates (Fig. 7c–f).
Stability of PGE–PAMS nanoaggregates formed in water at high temperatures
To evaluate the stability of the PGE–PAMS nanoaggregates formed in water at high temperatures, the transmittance (λ = 500 nm) of an aqueous solution of PGE–PAMS was measured while heating from 5 °C to 95 °C. The Tc was defined as the temperature at which the transmittance of the solution reached 90%. For comparison, a triblock copolymer consisting of PDMS segments in the center and PEG segments at both ends (PEG-block-PDMS-block-PEG) as well as a PEG-grafted PDMS copolymer (PDMS-graft-PEG) were evaluated. These polymers are block and graft copolymers containing hydrophobic polysiloxane and hydrophilic polyether chains.The transmittances of the aqueous solutions of PEG-block-PDMS-block-PEG and PDMS-graft-PEG sharply decreased at 61 °C and 81 °C, respectively (Fig. 8a and b). Visual observation also confirmed that these aqueous solutions became cloudy at 90 °C (Fig. 8d and e). These findings suggest the formation of large aggregates above these temperatures, indicating that block and graft copolymers containing PDMS and PEG components have the Tc of 61 °C and 81 °C. In contrast, the transmittance of the PGE–PAMS aqueous solution did not decrease upon heating to 90 °C (Fig. 8c). Visual observation revealed that the PGE–PAMS solution remained transparent at 90 °C, without the formation of turbidity or a precipitate (Fig. 8f), indicating that the nanoaggregates remained stable even at high temperatures.
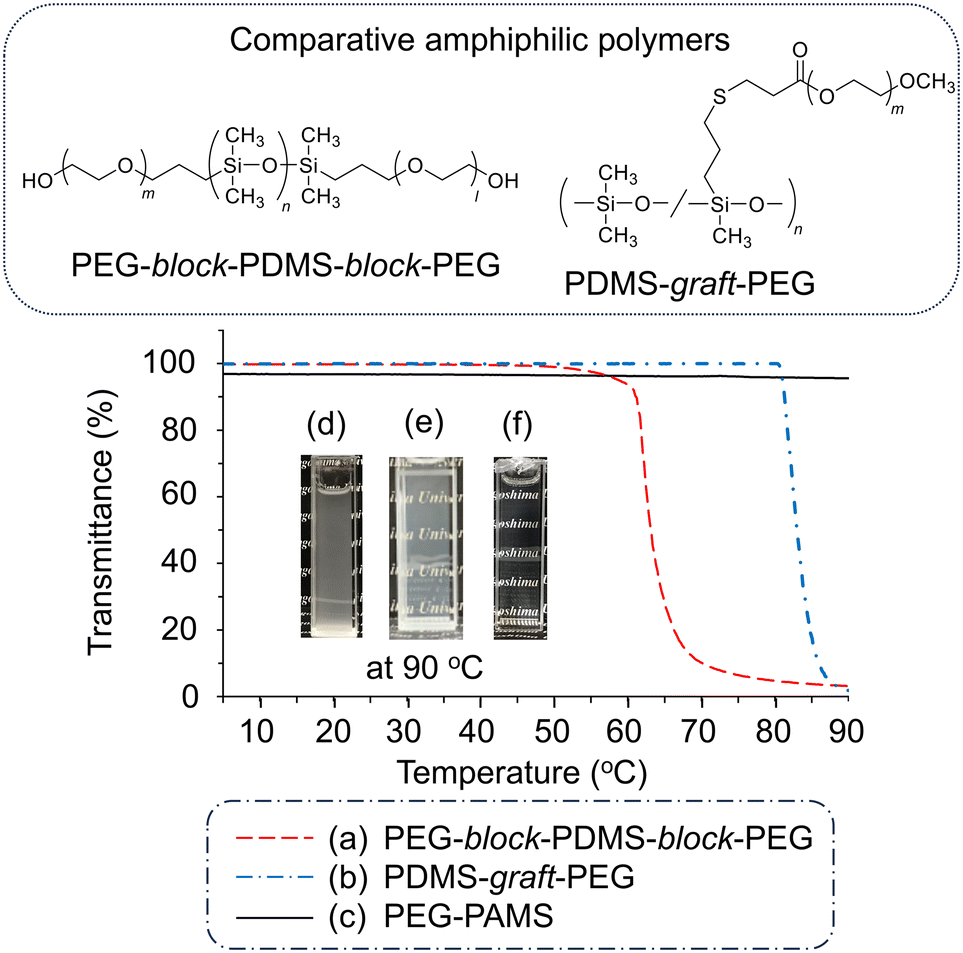 | ||
| Fig. 8 Transmittance curves (λ = 500 nm) of (a) PEG-block-PDMS-block-PEG, (b) PDMS-graft-PEG and (c) PGE–PAMS in water during heating. The polymer concentration was 0.1 w/v%. Inset: photographs of (d) PEG-block-PDMS-block-PEG, (e) PDMS-graft-PEG and (f) PGE–PAMS at 90 °C in water (0.1 w/v%). | ||
Furthermore, the volume-average particle size of PGE–PAMS was found to be 84.0 ± 29.8 nm by performing DLS measurements in water (0.1 w/v%) at high temperatures up to the limit of our instrument (80 °C), (Fig. 7b). This suggests that the rigid ladder-like structure of PGE–PAMS hinders conformational changes in the hydrophilic PGE chain, thereby preventing dehydration, which explains the lack of formation of large aggregates even at high temperatures of 80 °C–90 °C.
Solubilization of TPP by PGE–PAMS nanoaggregates
Amphiphilic molecules can encapsulate and solubilize hydrophobic molecules within the hydrophobic space of the nanoaggregates formed in water. Here, the encapsulation behaviour of the PGE–PAMS nanoaggregates in water containing a small amount of DMF was evaluated using the hydrophobic dye TPP in free base form as the guest molecule.Before investigating the solubilization of TPP by PGE–PAMS in a water/DMF (9/1, v/v) mixed solvent, the solubility of TPP in the mixed solvent was determined in the absence of PGE–PAMS. When 2 mL of a DMF solution containing TPP (0.5 mg) was slowly mixed with 18 mL of purified water under stirring, TPP gradually precipitated (Fig. 9a). In contrast, when a mixture of PEG–PAMS (2.0 mg) and TPP (0.5 mg) in DMF was slowly mixed with purified water (18 mL) under stirring, TPP remained soluble in the mixed solvent without precipitation (Fig. 9b). Additionally, DLS measurement of this mixture confirmed the formation of nanoaggregates with a particle size of ca. 65 nm (Fig. 9c). From these findings, it can be inferred that TPP was encapsulated within the hydrophobic space of the PGE–PAMS nanoaggregates.
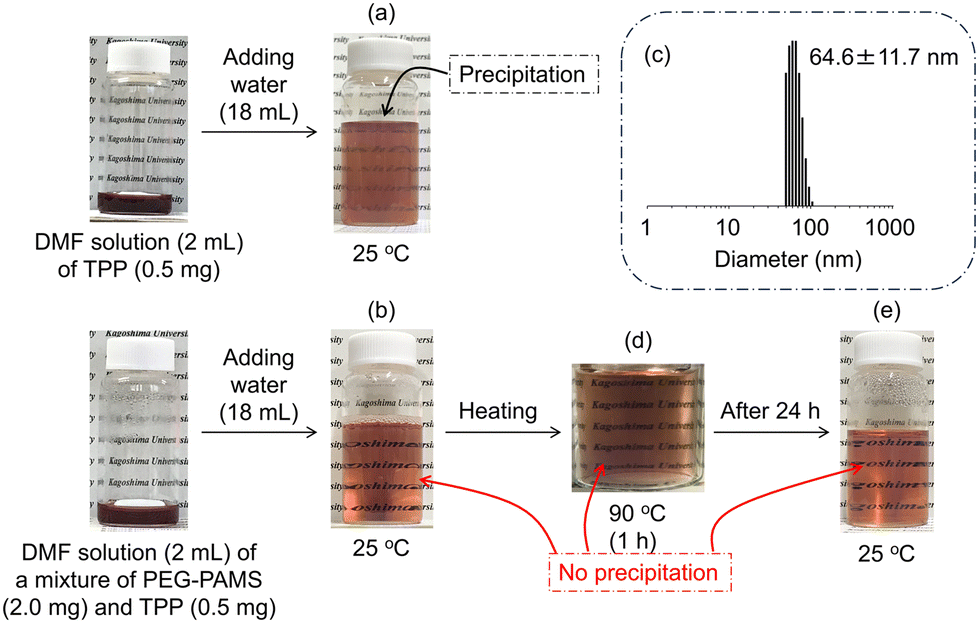 | ||
| Fig. 9 Photographs of water/DMF (9/1, v/v) solutions of (a) TPP at 25 °C and (b) a mixture of PGE–PAMS and TPP at 25 °C. (c) Volume-average particle size distribution of a mixture of PGE–PAMS and TPP measured by DLS in a water/DMF (9/1, v/v) mixed solvent at 25 °C (concentration: 1.0 × 10−2 w/v% for PGE–PAMS and 2.5 × 10−3 w/v% for TPP). Photographs of water/DMF (9/1, v/v) solutions of a mixture of PGE–PAMS and TPP (d) at 90 °C and (e) after being left at room temperature for 24 h. | ||
The UV–Vis spectrum of a mixed solution of PGE–PAMS (2.0 × 10−3 w/v%) and TPP (5.0 × 10−4 w/v%) revealed that TPP existed in a monomeric state (λmax = 420 nm) when solubilized by PGE–PAMS (Fig. S6†). This was confirmed by comparing this UV–Vis spectrum with that of a dilute solution of TPP alone in chloroform (a good solvent for TPP, 5.0 × 10−4 w/v%) and with a previous report50 (Fig. S7†).
In addition, to investigate whether the solubilized state of TPP could be maintained at high temperatures, the mixed solution of PGE–PAMS and TPP was heated to 90 °C, and it was found that no precipitate was formed (Fig. 9d). This suggests that TPP was stably encapsulated within the PGE–PAMS nanoaggregates even at 90 °C, since the collapse or aggregation of the nanoaggregates would cause the precipitation of either TPP or PGE–PAMS from the mixed solution. Additionally, after being left at room temperature for 24 h, no precipitate was observed, confirming that TPP remained solubilized (Fig. 9e).
In contrast, although PEG-block-PDMS-block-PEG and PDMS-graft-PEG could solubilize TPP in a water/DMF (9/1, v/v) mixed solvent at room temperature, precipitation occurred upon heating to 90 °C and standing for 1 h (Fig. S8†). These results indicate that the nanoaggregates formed by the block and graft copolymers can solubilize TPP but are unable to stably maintain the TPP encapsulation in the water/DMF (9/1, v/v) mixed solvent at high temperatures, despite containing similar components.
It has been reported that the intermolecular interactions of a rigid conjugated ladder-like polymer are considerably more stable at higher temperatures than those of a non-ladder-like polymer with similar structures owing to the low entropy nature of the rigid ladder-like polymer.23 Therefore, the stability of the PGE–PAMS nanoaggregates at high temperature can be attributed to the low entropy of its ladder-like structure as well as to the suppression of the conformational changes of the hydrophilic segment, which prevent dehydration.
Conclusions
In this study, PGE–PAMS, an amphiphilic ladder-like polymer, was successfully prepared via the thiol–ene reaction between MPDMMS and PAGE, followed by intramolecular polycondensation of the dimethoxysilyl groups in the side chains of the resulting polymer, PGE–DS. The results of solubility tests, GPC, 1H NMR, 29Si NMR and TEM suggest that PGE–PAMS possesses a ladder-like structure composed of PGE chains as hydrophilic segments and PAMS chains as hydrophobic segments, which are linked in parallel by sulfide bonds. This is the first report on the preparation of a ladder-like polymer with two chains exhibiting highly contrasting properties. In water, PGE–PAMS formed nanoaggregates with a diameter of ca. 60–70 nm, which remained stable even at high temperatures (80 °C–90 °C). This stability is likely due to the ladder-like structure of PGE–PAMS preventing conformational changes in the PGE chain, thereby inhibiting dehydration of the hydrophilic segments. Additionally, in water containing a small amount of DMF, the PGE–PAMS nanoaggregates could solubilize a hydrophobic dye (TPP), which remained encapsulated within the nanoaggregates even at a high temperature of 90 °C.Author contributions
Conceptualization, Y. K.; data curation, S. M., S. N. and A. M.; formal analysis, S. M., S. N. and A. M.; investigation, S. M.; methodology, S. M.; supervision, Y. K.; writing – original draft, S. M. and S. N.; writing – review & editing, Y. K.; TEM measurement, K. S. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844–6892 CrossRef CAS PubMed.
- T. Yoshida, M. Doi, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5704–5709 CrossRef CAS.
- T. Thurn-Albrecht, J. Schotter, G. A. Kästle, N. Emley, T. Shibauchi, L. Krusin-Elbaum, K. Guarini, C. T. Black, M. T. Tuominen and T. P. Russell, Science, 2000, 290, 2126–2129 CrossRef CAS PubMed.
- A. C. Edrington, A. M. Urbas, P. DeRege, C. X. Chen, T. M. Swager, N. Hadjichristidis, M. Xenidou, L. J. Fetters, J. D. Joannopoulos, Y. Fink and E. L. Thomas, Adv. Mater., 2001, 13, 421–425 CrossRef CAS.
- T. H. Vanghn, H. R. Suter, L. G. Lundsted and M. G. Kramer, J. Am. Oil Chem. Soc., 1951, 28, 294–299 CrossRef.
- C. Booth and D. Attwood, Macromol. Rapid Commun., 2000, 21, 501–527 CrossRef CAS.
- C. Y. Gong, S. Shi, P. W. Dong, B. Kan, M. L. Gou, X. H. Wang, X. Y. Li, F. Luo, X. Zhao, Y. Q. Wei and Z. Y. Qian, Int. J. Pharm., 2009, 365, 89–99 CrossRef CAS PubMed.
- M. Boffito, P. Sirianni, A. M. Di Rienzo and V. Chiono, J. Biomed. Mater. Res., Part A, 2015, 103A, 1276–1290 CrossRef CAS PubMed.
- H. Deng, A. Dong, J. Song and X. Chen, J. Controlled Release, 2019, 297, 60–70 CrossRef CAS PubMed.
- G. Bonacucina, M. Cespi, G. Mencarelli, G. Giorgioni and G. F. Palmieri, Polymers, 2011, 3, 779–811 CrossRef CAS.
- J. Shi, L. Yu and J. Ding, Acta Biomater., 2021, 128, 42–59 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae and S. W. Kim, Colloids Surf., B, 1999, 16, 185–193 CrossRef CAS.
- P. Wang, W. Chu, X. Zhuo, Y. Zhang, J. Gou, T. Ren, H. He, T. Yin and X. Tang, J. Mater. Chem. B, 2017, 5, 1551–1565 RSC.
- H. Kunieda, M. H. Uddin, M. Horii, H. Furukawa and A. Harashima, J. Phys. Chem. B, 2001, 105, 5419–5426 CrossRef CAS.
- M. J. Hey, S. M. Ilett and G. Davidson, J. Chem. Soc., Faraday Trans., 1995, 91, 3897–3900 RSC.
- K. Tasaki, J. Am. Chem. Soc., 1996, 118, 8459–8469 CrossRef CAS.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- P. Alexandridis and T. A. Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS.
- S. Honda, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 10251–10253 CrossRef CAS PubMed.
- C. Arnold Jr., J. Polym. Sci., Macromol. Rev., 1979, 14, 265–378 CrossRef.
- M. M. Tessler, J. Polym. Sci., Part A-1, 1966, 4, 2521–2532 CrossRef CAS.
- G. Ma, M. Leng, S. Li, Z. Cao, Y. Cao, D. P. Tabor, L. Fang and X. Gu, J. Mater. Chem. C, 2022, 10, 13896–13904 RSC.
- J. F. Brown Jr., L. H. Vogt Jr., A. Katchman, J. W. Eustance, K. M. Kiser and K. W. Krantz, J. Am. Chem. Soc., 1960, 82, 6194–6195 CrossRef.
- J. F. Brown Jr., L. H. Vogt Jr. and P. I. Prescott, J. Am. Chem. Soc., 1964, 86, 1120–1125 CrossRef.
- Y. Abe and T. Gunji, Prog. Polym. Sci., 2004, 29, 149–182 CrossRef CAS.
- Y. Kaneko, N. Iyi, K. Kurashima, T. Matsumoto, T. Fujita and K. Kitamura, Chem. Mater., 2004, 16, 3417–3423 CrossRef CAS.
- Y. Kaneko, N. Iyi, T. Matsumoto and K. Kitamura, Polymer, 2005, 46, 1828–1833 CrossRef CAS.
- X. Zhang, P. Xie, Z. Shen, J. Jiang, C. Zhu, H. Li, T. Zhang, C. C. Han, L. Wan, S. Yan and R. Zhang, Angew. Chem., Int. Ed., 2006, 45, 3112–3116 CrossRef CAS PubMed.
- S. S. Choi, H. S. Lee, S. S. Hwang, D. H. Choi and K. Y. Baek, J. Mater. Chem., 2010, 20, 9852–9854 RSC.
- Z. Ren, D. Sun, H. Li, Q. Fu, D. Ma, J. Zhang and S. Yan, Chem. – Eur. J., 2012, 18, 4115–4123 CrossRef CAS PubMed.
- H. Toyodome, Y. Kaneko, K. Shikinaka and N. Iyi, Polymer, 2012, 53, 6021–6026 CrossRef CAS.
- Y. Kaneko, H. Toyodome, T. Mizumo, K. Shikinaka and N. Iyi, Chem. – Eur. J., 2014, 20, 9394–9399 CrossRef CAS PubMed.
- A. Harada, K. Shikinaka, J. Ohshita and Y. Kaneko, Polymer, 2017, 121, 228–233 CrossRef CAS.
- Y. Kaneko, Polymer, 2018, 144, 205–224 CrossRef CAS.
- H. Kämmerer and S. Ozaki, Makromol. Chem., 1966, 91, 1–9 CrossRef.
- H. Kämmerer and A. Jung, Makromol. Chem., 1966, 101, 284–295 CrossRef.
- R. Jantas and S. Polowinski, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 1819–1827 CrossRef CAS.
- R. Jantas, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1973–1982 CrossRef CAS.
- R. Jantas, G. Janowska, H. Szocik and S. Polowinski, J. Therm. Anal. Calorim., 2000, 60, 371–376 CrossRef CAS.
- R. Jantas, J. Szumilewicz, G. Strobin and S. Polowinski, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 295–300 CrossRef CAS.
- R. Jantas, S. Polowinski and G. Strobin, Polym. Int., 1995, 37, 315–318 CrossRef CAS.
- R. Saito, Y. Iijima and K. Yokoi, Macromolecules, 2006, 39, 6838–6844 CrossRef CAS.
- R. Saito and Y. Iijima, Polym. Adv. Technol., 2009, 20, 280–284 CrossRef CAS.
- S. Kinoshita, S. Watase, K. Matsukawa and Y. Kaneko, J. Am. Chem. Soc., 2015, 137, 5061–5065 CrossRef CAS PubMed.
- M. Nobayashi, K. Shikinaka and Y. Kaneko, Polym. J., 2022, 54, 11–20 CrossRef CAS.
- S. Aoki, A. Koide, S. Imabayashi and M. Watanabe, Chem. Lett., 2002, 31, 1128–1129 CrossRef.
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS PubMed.
- Y. Sugahara, S. Okada, K. Kuroda and C. Kato, J. Non-Cryst. Solids, 1992, 139, 25–34 CrossRef CAS.
- M. Zhou, S. Ouyang, Z. Liu, G. Lu, S. Gao and Z. Li, Vib. Spectrosc., 2009, 49, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01316f |
| This journal is © The Royal Society of Chemistry 2025 |