
Breaking hydrogen-bonds in aqueous electrolytes towards highly reversible zinc-ion batteries†
Yilong
Zhu
 ,
Qianru
Chen
,
Junnan
Hao
* and
Yan
Jiao
*
,
Qianru
Chen
,
Junnan
Hao
* and
Yan
Jiao
*
School of Chemical Engineering, The University of Adelaide, Adelaide, 5005, Australia. E-mail: yan.jiao@adelaide.edu.au; junnan.hao@adelaide.edu.au
First published on 30th June 2024
Abstract
Aqueous zinc-ion batteries (AZIBs) have attracted significant attention for their potential in large-scale energy storage. However, their practical application is limited by the poor zinc reversibility because of structural deterioration and side reactions induced by water molecules. Herein, we identified pentaerythritol (PTT) as an electrolyte additive to break the H-bond network of water in the conventional aqueous ZnSO4 electrolyte, after considering the cost, toxicity, conductivity, high H-bond donor number, and structure features among several options. The unique symmetry structure of PTT with four hydroxyl groups (–OH) significantly enhances its interaction with water molecules and changes the proportion of different hydrogen-bond (H-bond) types between water molecules. The introduction of PTT therefore could break the water H-bond network and change the Zn2+ solvation structure, as evidenced by both experimental findings and theoretical simulations. Consequently, water-induced side reactions and dendrite growth during cycling are significantly suppressed, leading to improved Zn reversibility and overall battery performance. Notable outcomes include the average coulombic efficiency reaching 99.7% and long-term stability exceeding 1000 h. This research contributes to the development of a cost-effective and efficient electrolyte strategy aimed at addressing water-induced issues in AZIBs.
 Junnan Hao | Junnan Hao received his PhD degree in 2020 from the University of Wollongong, Australia. Now he works as an ARC DECRA fellow at the University of Adelaide, Australia. His research interests are focused on energy storage and conversion, including aqueous Zn-ion batteries, high-voltage Li-ion batteries, and flexible energy storage devices. |
Introduction
The escalating demand for renewable energy sources has boosted a burgeoning market for high-performance energy storage devices in recent years.1–3 Within this context, aqueous electrolyte-based batteries have gained significant attention due to their low flame risk and high ionic conductivity compared to those with non-aqueous electrolytes.4,5 Particularly, aqueous Zn-ion batteries (AZIBs) have emerged as a promising alternative to traditional Li-ion batteries for large scale energy storage. The metallic Zn can be directly used as the anode because of its unique features such as a high theoretical anode capacity of 820 mA h g−1, a favourable redox potential (−0.76 V vs. standard hydrogen electrode) and relatively low cost (AU $4.88 per kg).6–10 In addition to the advantages offered by using zinc as the anode, the performance can be further improved by optimizing the cathode material; for example, AZIBs using vanadium-oxide-based cathodes have achieved a lifespan of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles while maintaining 91.4% of their original capacity, even at a high current density of 40.0 A g−1.11
000 charge–discharge cycles while maintaining 91.4% of their original capacity, even at a high current density of 40.0 A g−1.11
Despite their great potential for energy storage, one of the challenges that AZIBs face is related to the low coulombic efficiency (CE) of Zn anodes led by water-induced irreversible side reactions, including hydrogen evolution reaction (HER), corrosion reaction, by-product accumulation and Zn dendrite growth during charge and discharge cycles.12–17 The concurrent generation of hydrogen is unavoidable in aqueous batteries given the competing HER against the Zn electrochemical deposition.18–20 The HER also leads to an increased concentration of hydroxide ions (OH−) near the Zn electrode surface, corroding the Zn electrode. In addition, the OH− accumulation triggers the formation of an inactive Zn4SO4(OH)6·xH2O barrier, which hinders ion/electron diffusion at the Zn/electrolyte interphase, thereby reducing the reversibility of Zn anodes.21–25
Various strategies have been proposed to address the challenges related to AZIBs, which include the modification of electrolytes by adding high-concentration salts and organic additives.26–31 For instance, a highly concentrated electrolyte (HCE) of 1 M zinc di[bis(trifluoromethylsulfonyl)imide] (Zn(TFSI)2) + 20 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) has been demonstrated to reduce the H2O number in the Zn2+ solvation structure and suppress the side reactions.27 However, the high viscosity of HCE leads to sluggish Zn2+ diffusion and poor rate capability.32 Alternatively, a wide range of organic additives have been reported, such as methanol, dimethyl sulfoxide (DMSO), ethylene glycol (EG) and triethyl phosphate (TEP).33–35 As a representative, methanol has been widely studied in aqueous ZnSO4 electrolytes as an anti-solvent. However, since a single methanol molecule possesses only one H-bond donor, a large volume ratio (55%) is required to destroy the water H-bond network in the electrolyte. This high volume addition not only increases the cost and reduces the ionic conductivity, but also increases the risk of flammability and weakens the advantages of aqueous batteries.36 Therefore, it is crucial to identify cost-effective additives capable of forming strong H-bonds with water molecules at low concentrations. In addition, previous research has not thoroughly explored the screening process and mechanisms at the molecular level, highlighting the importance of further investigation.
Herein, we first selected pentaerythritol (PTT) as a promising additive candidate due to its cost-effectiveness, low toxicity, high conductivity, high H-bond donor number, and unique structure features. Density functional theory (DFT) calculations were performed to demonstrate the interaction forces between additive molecules and water, with comparison to several previously reported organic alcohol additives. Results indicated that PTT with unique four symmetry hydroxyl groups (–OH) showed greater interaction with water. To investigate the dynamic impact of PTT on water molecules in the electrolyte, theoretical simulations including both classical molecular dynamics (MD) and ab initio molecular dynamics (AIMD) were further performed. Results demonstrated that PTT could change the coordination chemistry of Zn2+ by entering its solvation structure. Additionally, PTT could also break the H-bond network of water molecules and the proportion of different H-bond types between water molecules. The influence of PTT on water activity was also confirmed by experiments. The results from in situ gas chromatography (GC) and ex situ wide angle X-ray scattering (WAXS) confirmed that the water-induced side reactions and dendrite growth during cycling are significantly suppressed after adding PTT in the electrolyte. Moreover, symmetric cells assembled using the PTT-containing electrolyte exhibited stable long-term cycling over 1000 h at a current density of 1 mA cm−2 with a capacity of 0.5 mA h cm−2. Moreover, they also exhibited a high average Zn plating/stripping CE of 99.7%. This work contributed to a cost-effective and efficient electrolyte strategy designed to tackle water-induced challenges in AZIBs.
Results and discussion
To screen a feasible additive candidate, several commonly used organic electrolyte additives were compared, such as methanol, DMSO, 1,3-dimethyl-2-imidazolidinone (DMI), EG, 1,4-dioxane (DIOXANE), N,N-dimethylformamide (DMF), and PPT. For comparison, six key parameters were selected: cost, conductivity, safety, dielectric constant, H-bond acceptor number, and H-bond donor number. To facilitate clear comparison, each parameter was normalized, as illustrated from the radar map in Fig. 1. Based on this, PTT was chosen after a thorough comparison with previously mentioned electrolyte additives. The two-dimensional structure of PTT highlights the four symmetrical hydroxyl groups, which facilitate interactions with water molecules (Fig. S1a†). Similar to the 2D diagram, the hydroxyl groups from a three-dimensional perspective are oriented in different directions, enabling PTT to interact with water from a wide range of angles (Fig. S1b†). Both figures illustrate the molecule's symmetry and the distribution of hydroxyl groups (–OH). Importantly, the unique structure of PTT (Table S1†) exhibits a highest H-bond donor number and acceptors in a single molecule among these additives, which is known to significantly impact the H-bond network of water molecules. Moreover, conductivity is a performance indicator for batteries as analysed by previous studies.37–46 The ZnSO4 electrolyte has been proven to have high biological safety and is one of the ideal zinc salts for biocompatible ZIBs.47 The conductivity of a 2 M ZnSO4 solution with 0.05 M PTT was found to be higher than that of other organic additives in the ZnSO4 electrolyte (excluding the 20% DMSO volume ratio in 2 M ZnSO4). In addition, considering the dielectric coefficient as an inherent physical property influencing the H-bonds in water, PTT features a high dielectric constant of 42.9, higher than those of most of the additives. | ||
| Fig. 1 Radar map screening seven different electrolyte additives for aqueous ZIBs by considering cost, conductivity, safety H-bond donor/acceptor number and dielectric coefficient. | ||
Safety assessment is based on the classification standards of the Globally Harmonized System of Classification and Labelling of Chemicals (GHS Rev.10, 2023) by the United Nations, with the acute poisoning (skin) indicator of five hazard categories used for classifying organic substances. The safety ranking reveals that PTT has a relatively less toxic category among organic additives. Moreover, cost comparison is based on quotations from the Fisher Scientific official website of Thermo Fisher Chem™ Company (AU$ per 500 ml or 500 g), revealing that PTT has a relatively low cost compared to other organic additives. In summary, a comprehensive evaluation of the six parameters suggests that PTT possesses overall advantages, making it a promising choice for the electrolyte additive in ZIBs.
DFT calculations were performed to verify the interaction between additives and water molecules at a microscopic level. The interaction forces were visualized by using the independent gradient model based on the Hirshfeld partition (IGMH) method.48 IGMH analysis in Fig. 2a–c shows regions of different interaction strengths, displayed through isosurfaces corresponding to different δg values (which indicates variations in electron density gradients at points of interest). Additionally, the function sign(λ2)ρ serves as a mapping tool to distinguish interaction types. Here, r represents the genuine electron density within the current system, and a higher value signifies a stronger interaction. Moreover, l2 is the second largest eigenvalue of the electron density Hessian matrix, with sign() retrieving the sign of a value. Therefore, positive sign(λ2) represents repulsion while negative sign(λ2) represents attraction. Overall, the sign(λ2)ρ value could be used to distinguish the interaction type and intensity.
 | ||
| Fig. 2 Visual analysis of intermolecular interactions, (a) 1H2O–1H2O, (b) 1PTT–1H2O, and (c) 1PTT–4H2O. Color code: red, O; white, H; cyan, C. (d) Average binding energy and average H-bond length between water molecules and alcohol-based additives from theoretical calculation, including methanol (MeTH), ethanol (ETH), PP, glycerol, EG, PG, and PTT. | ||
Since the height of the δg peak in the interaction region is positively correlated with the interaction strength, H-bonds represent the main interactions between two water molecules with an intensity of 0.05 a.u. (Fig. 2a). Similarly, the PTT molecule can also form H-bonds with a single water molecule with an intensity of 0.06 a.u., indicating higher interaction strength compared to those in pure water (Fig. 2b). Due to the high H-bond donor number of PTT (Fig. 2c), a single PTT molecule could form four stable H-bonds with water molecules, which shows that PTT molecules have strong bonding ability to attract water molecules in the electrolyte.
To evaluate the ability to bond with water, PTT and several previously reported additives were evaluated through DFT simulations, comparing their average binding energy and the average H-bond length. Here, methanol, ethanol, polypropylene (PP), glycerol, EG, and propylene glycol (PG) were selected because all of them share similar oxygen-containing groups and belong to organic alcohols (Fig. 2d). The H-bonds formed by many reported alcohol additives and water do have stronger binding energy and shorter bond length than those formed in the pure water molecules, indicating that they are able to reduce the water activity in the electrolytes (Fig. S2 and S3†). In comparison, the H-bond formed between PTT and water shows the shortest bond length and the strongest binding energy, which indicates that PTT is more promising as a potential additive candidate to reduce water activity.
Experiments were carried out to verify PTT's effectiveness for AZIBs. First, to explore the appropriate ratio of PTT, different concentrations of PTT additives (0.01–0.2 M) in 2 M ZnSO4 electrolyte were prepared. After a week of standing, it was found that when the concentration of PTT added was greater than 0.05 M, solutions become turbid with precipitation, indicating improper concentration (Fig. S4†). Therefore, the concentration of 0.05 M PTT was chosen for the following MD simulation and battery performance testing. To verify the effect of PTT addition on the Zn metal anode, the polished Zn foil was soaked into the ZnSO4 electrolytes containing different concentrations of PTT (Fig. 3a). After soaking for 7 days, the Zn foil was taken out and analyzed by the X-Ray diffraction (XRD) technique. As shown in Fig. 3b, the Zn foil in the pure ZnSO4 electrolyte showed a significant peak at approximately 9.8°, which is indexed to the (002) plane of the Zn4SO4(OH)6·xH2O by-product. In contrast, the peak intensity of this corrosion by-product in the ZnSO4 electrolyte with 0.05 M PTT was significantly reduced. This finding proved that the PTT additive could improve the anti-corrosion ability of Zn electrodes by reducing the activity of the electrolyte. In addition, the conductivity test showed that with the same concentration of additives, the conductivity of the ZnSO4 electrolyte with methanol addition dropped by 35.2%, while the conductivity of the ZnSO4 electrolyte with pentaerythritol only dropped by 4.1%, indicating that adding PTT to the ZnSO4 solution has little effect on the conductivity (Fig. S5†).
 | ||
| Fig. 3 Fundamental experiments on different electrolytes. (a) Snapshot of Zn foils soaking in 2 M ZnSO4 electrolyte with different PTT concentrations after 7 days. (b) XRD results of the pristine Zn foil and Zn foils soaked in the pure ZnSO4 electrolyte and ZnSO4 with 0.5 M PTT addition. The performance of pristine Zn foil is included for comparison purposes. (c) LSV curves of the pure ZnSO4 electrolyte and ZnSO4 with different PTT concentrations. Snapshots of contacting angles of the pure ZnSO4 electrolyte (d) and ZnSO4 with 0.05 M PTT(e). (f) FTIR spectra of the pure ZnSO4 electrolyte and ZnSO4 with different PTT concentrations. | ||
To evaluate the effect of PTT on the water activity, linear sweep voltammetry (LSV) curves were measured. As shown in Fig. 3c, at the same cut-off voltage, the 0.05 M PTT-added ZnSO4 electrolyte exhibited a lower current than the original ZnSO4 electrolyte. This phenomenon indicates that the addition of PTT can inhibit the HER to retard the water decomposition. Additionally, the sessile drop contact angle technique was used to study the wettability of the Zn electrode in various electrolytes (using pure Zn foil as the electrode), because this feature affects the energy barrier for Zn nucleation formation and evolution. Due to its hydrophobicity, the Zn metal exhibits a high contact angle of 85.6° in the pure ZnSO4 electrolyte (Fig. 3d). However, with the addition of PTT, the contact angle was significantly decreased to 57.2° (Fig. 3e). These findings emphasized the high wettability of Zn metal in the additive electrolyte because PTT has a higher adsorption than water molecules on the Zn metal surface in the additive electrolyte, thus affecting the water-induced HER and Zn nucleation formation during Zn deposition.49 Additionally, Fourier transform infrared (FTIR) spectra of the chemical environment of H2O in different electrolytes were collected to reveal the impact of PTT addition on H-bonds, as shown in Fig. 3f. The peak located at 3205 cm−1 corresponds to the strong H-bonds in H2O.50,51 It is worth noting that with the addition of PTT, the presence of the strong H-bonds between water molecules in the electrolyte decreased, which indicates that the addition of PTT can disturb the binding ability between water molecules.
In addition, Raman spectroscopy was utilized to investigate the changes in the H-bond network of water molecules and the solvation structure of Zn2+ (Fig. S6†). In the 2 M ZnSO4 with 0.05 M PTT electrolyte, the vibration peak of [Zn2+·OH2] shifted from 390 cm−1 to 382 cm−1 compared to the peak in the 2 M ZnSO4 electrolyte, indicating that the typical solvation structure of the [Zn(H2O)6]2+ complex was suppressed (Fig. S6a†). This implies that PTT addition influences Zn2+ solvation, lowering the energy barrier for Zn2+ detachment. Moreover, the intensity peak of [HOH·OH2] between 3264–3304 cm−1 in the 2 M ZnSO4 with 0.05 M PTT electrolyte was also lower than in the 2 M ZnSO4 electrolyte (Fig. S6b†), indicating weaker H-bond strength between water molecules. This suggests that PTT disrupts water H-bonds, facilitating Zn2+ transport in the battery. Similarly, the reduced intensity of the [HOH·OSO32−] vibration between 3387–3416 cm−1 indicates decreased H-bond strength between water and SO42−, reflecting stronger PTT–water interaction. These results above demonstrate the PTT effects on the hydrogen bonding network of water molecules and the solvation structure of Zn2+.
To further explore the impact of PTT on the ZnSO4 electrolyte at the atomic level, theoretical calculations were carried out to simulate the pure ZnSO4 electrolyte environment and ZnSO4 electrolyte with PTT addition (Table S2†). As shown in Fig. 4a and S7†, after relaxation for 100 ns, each component is evenly distributed in the ZnSO4 aqueous solution system. Moreover, with the presence of PTT in the electrolyte, as shown in the partially enlarged solvation sheath structure in Fig. 4b, PTT molecules can form H-bonds with water molecules. Moreover, the Zn2+ solvation structure changes after adding the additive, as confirmed by the radial distribution function (RDF, i.e. g(r)) and coordination number (Ncoor.) of Zn2+ with the oxygen in water or the additive (Fig. 4c). Results showed that the addition of PTT decreased the water molecules' coordination number from 5.3 to 4.1 in the first solvation sheath of Zn2+. This is also confirmed by the AIMD results. The root mean square deviation (RMSD) of the two systems shows that the addition of PTT could change the solution environment, which verifies that PTT contributes to the reshaping of the Zn2+ solvation structure as well. Additionally, the intermolecular interactions between PTT and surrounding ZnSO4 molecules in the model of 2 M ZnSO4 with 0.05 M PTT were investigated. As shown in Fig. 4d, the PTT molecule interacts with the Zn2+ and surrounding water molecules. The interaction is represented by the δg isosurface values (same color scale as in Fig. S8†), which indicates variations in electron density gradients at points of interest. Among them, the green isosurface represents the dispersion force, which is related to the van der Waals interaction, while the navy blue isosurface represents the prominent attractive interaction. This figure suggests that Zn2+ has a contribution to attracting PTT into the solvation sheath. Moreover, the PTT molecule in the system can also interact with surrounding water molecules without Zn2+ (Fig. S8†), which proves that PTT molecules not only can form H-bonds with water molecules, but also have a high strength attraction with Zn2+, thus affecting the solvation sheath of Zn2+. These findings indicate that the addition of PTT can affect the Zn2+ solvation equilibrium, leading to the de-solvation and rapid transport of Zn2+ with a low energy barrier.28,52,53
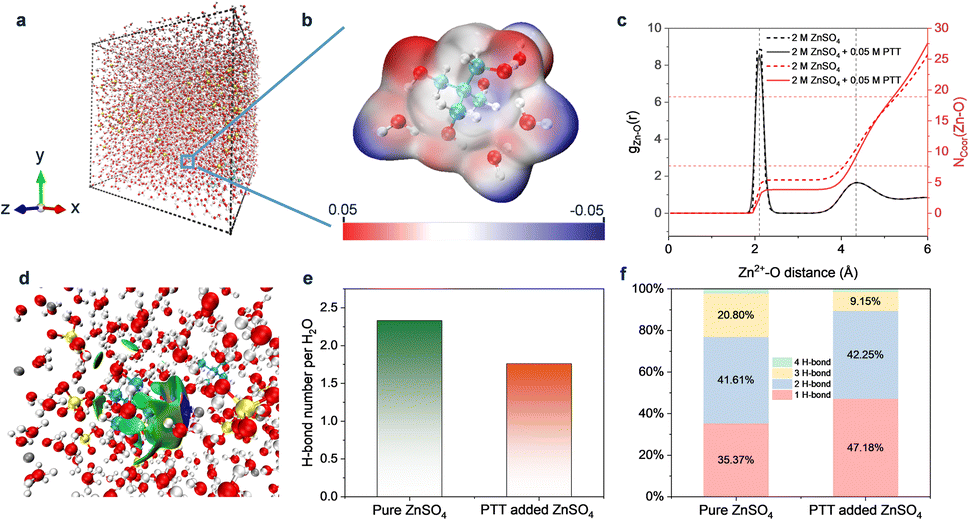 | ||
| Fig. 4 Theoretical MD simulation and analyses. Snapshot of (a) the model for pure ZnSO4 with 0.05 M PTT (b) the Electrostatic Potential (ESP) map of one PTT molecule with four water molecules. (c) Ncoor. and g(r) of 2 M ZnSO4 and 2 M ZnSO4 with 0.05 M PTT between Zn2+ and the first solvation sheath (last 5 ns within equilibrium simulations). (d) Snapshot of the intermolecular interactions between PTT and surrounding ZnSO4 molecules in the model of 2 M ZnSO4 with 0.05 M PTT. (e) The average H-bonds of two electrolyte systems. (f) Proportion of each H-bond type between water molecules of two electrolyte systems. Color code: red, O; white, H; yellow, S; cyan, C; grey, Zn. | ||
As shown in Fig. 4e, among the two models that contain the same number of water molecules, the average number of H-bonds per H2O in pure ZnSO4 solution is about 2.3. As for the ZnSO4 solution with PTT added, the average number of H-bonds was reduced to 1.7, which was a 28% reduction compared with the pure ZnSO4 solution. This confirmed that the original water–water H-bond network was disrupted, therefore indicating that the addition of PTT can effectively reduce the water molecules' activity, and thereby inhibit the HER. These simulation results were consistent with the experimental results (Fig. 3c). In addition, Fig. S9† shows the classification of H-bond numbers between water molecules; for example, the 1-H-bond means that the water molecule only contributes to one H-bond, and the 4-H-bond means that both H atoms and O atoms in the water molecules contribute to the formation of the H-bonds. In aqueous solutions, stronger H-bonds between water molecules result in a robust hydrogen bonding network that impedes the transport of Zn2+.
Additionally, water molecules tend to accumulate on the surface of the negative electrode during the charging progress, leading to the HER. By analyzing the classification of different numbers of H-bonds between water molecules, the types of 1-H-bond and 2-H-bond are related to the weak H-bond, while the types of 3-H-bond and 4-H-bond are related to the strong H-bond. As shown in Fig. 4f, the types of 1-H-bond and 2-H-bond in pure ZnSO4 accounted for 35.37% and 41.61%, respectively. The 3-H-bond and 4H-bond accounted for 20.8% and 2.22%, respectively. In contrast, in the ZnSO4 with 0.05 M PTT, the proportion of 1-H-bond type and 2-H-bond increased to 47.18% and 42.25%, respectively. The proportion of 3-H-bond and 4-H-bond dropped to 9.15% and 1.42%, respectively. Due to the interference of PTT on the H-bond between water molecules, some of the strong H-bonds were converted into weak H-bonds.54 Additionally, some PTT molecules will replace water molecules on the surface of the negative electrode, thereby inhibiting the occurrence of the HER. The changes proved that the addition of PTT can significantly break the H-bond network of the ZnSO4 electrolyte by affecting the number and type of H-bonds of water molecules in the solution, which is beneficial in reducing the HER in the solution and improving the Zn2+ transmission efficiency.
Electrochemical characterization was performed to evaluate the CE of Zn stripping/plating with different electrolytes. The reversibility of Zn chemistry was studied by conducting plating/stripping measurements on Zn//Cu coin cells at 1 mA cm−2 and 0.5 mA h cm−2. As shown in Fig. 5a, the Zn//Cu cell using the 2 M ZnSO4 electrolyte achieved an average CE of 97.8% in the first 170 cycles. The CE value fluctuated in subsequent cycles due to the cell failure, which is mainly related to interfacial passivation caused by dendrite deposition, HER and Zn4SO4(OH)6·xH2O by-products.55 In contrast, Zn//Cu cells with the addition of PTT showed significantly improved CE. Specifically, the Zn//Cu cell showed higher CE in the first 10 h and remained stable after 1000 h. Importantly, this Zn//Cu battery exhibited a higher average CE of approximately 99.68%, which is attributed to the reduced water molecule activity with PTT. When the current density increased to 5 mA cm−2, the cell with the 2 M ZnSO4 electrolyte had an average CE of 98.7%. In contrast, the ZnSO4 electrolyte with 0.05 M PTT addition still exhibited high Zn reversibility, with an average CE of 99.85% over 1000 h (Fig. 5b). These findings show that compared to the pure ZnSO4 electrolyte, the ZnSO4 electrolyte with PTT addition shows better electrochemical stability in stripping/plating.
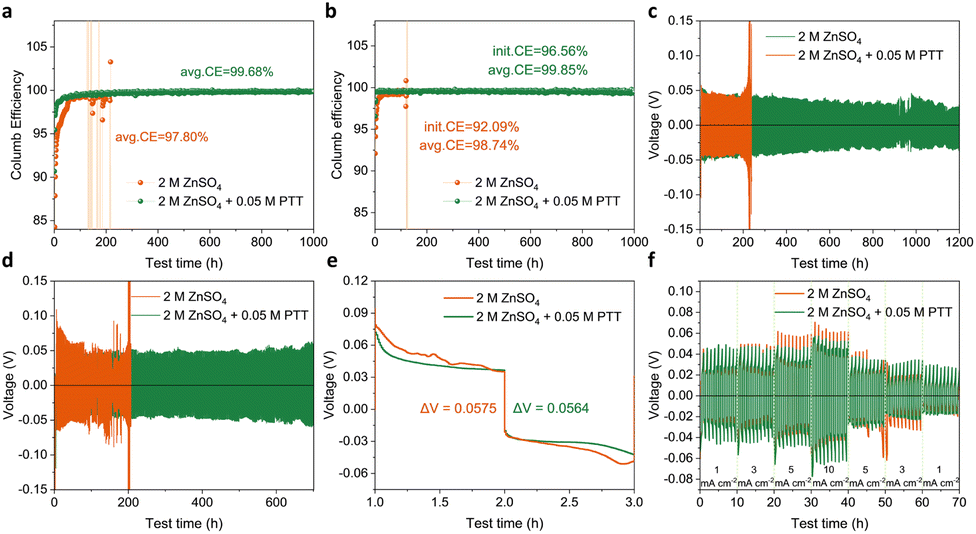 | ||
| Fig. 5 Coulombic efficiency of Zn//Cu cell plating/stripping in the pure ZnSO4 electrolyte and ZnSO4 with 0.05 M PTT: (a) at 1 mA cm−2 and a capacity of 0.5 mA h cm−2, (b) at 5 mA cm−2 and a capacity of 2.5 mA h cm−2. The cycling lifespan of Zn//Zn symmetric cells in the pure ZnSO4 electrolyte and designed electrolyte: (c) at 1 mA cm−2 and a capacity of 0.5 mA h cm−2, (d) at 5 mA cm−2 and a capacity of 2.5 mA h cm−2. (e) The first charge/discharge cycle of Zn//Zn symmetric cells. (f) Rate capability of Zn//Zn symmetric cells under different current densities from 1 mA cm−2 to 10 mA cm−2. | ||
Zn//Zn symmetric cells were assembled to test the effect of adding PTT on the cycling life and stability of Zn anode plating/stripping. At the current density of 1 mA cm−2, the voltage of the cell in the pure ZnSO4 electrolyte dropped significantly after about 230 h, indicating that the battery failed due to the occurrence of short circuit (Fig. 5c). Additionally, under the same conditions, the cell with the ZnSO4 electrolyte containing PTT showed superior cycle stability with a lifespan of over 1000 h. At 5 mA cm−2, the voltage of the cell in the pure ZnSO4 electrolyte became unstable after 150 h, indicating battery failure. However, the cell with the 0.05 M PTT added ZnSO4 electrolyte also had a lifespan over 700 h (Fig. 5d). Additionally, from the first charge/discharge cycle, the curves of the ZnSO4 with PTT addition are smoother. However, the polarization of the cells with different electrolytes is similar under low current density. Fig. 5f shows the rate performance of symmetric cells with the pure ZnSO4 electrolyte and the ZnSO4 with PTT electrolyte for 1 h per cycle at current densities of 1, 3, 5, and 10 mA cm−2, respectively. It can be seen that the symmetrical cell using ZnSO4 with PTT has lower polarization than that using the pure ZnSO4 electrolyte under high current densities, e.g. 5 mA cm−2. These findings indicated that PTT could promote stable and efficient operation of Zn-based batteries.
To further investigate the reason why PTT can improve the Zn reversibility, Zn electrodes stripped from cells with different electrolytes were evaluated after the 100th plating cycle. XRD measurements were performed to study the deposition behavior of Zn electrodes after cycling. In the ZnSO4 electrolyte, the Zn electrode showed a significant peak at approximately 9.8° (Fig. 6a), pointing to the (002) plane of the Zn4SO4(OH)6·xH2O by-product. This finding confirmed that significant corrosion occurs during battery cycling. In contrast, a significantly reduced peak appeared at about 9.8° in the XRD pattern of the Zn electrode after cycling in the ZnSO4 with 0.05 M PTT, indicating that the deposition orientation of the Zn electrode was significantly changed. These results help to improve the Zn reversibility in the electrolyte. Furthermore, for the Zn electrode in the pure ZnSO4 electrolyte, the peak intensity ratio of Zn(002)/Zn(101) was 30.58%. But for the Zn electrode in the ZnSO4 with 0.05 M PTT, the peak intensity ratio of Zn(002)/Zn(101) increased to 52.62%, indicating that PTT changes the preferred orientation for Zn (002) deposition. It has been widely accepted that the Zn (002) deposition helps to suppress the dendrite growth. In addition, WAXS measurements were performed to verify the deposition orientation of Zn electrodes after cycling. For the Zn electrode in the pure ZnSO4 electrolyte, the (101) plane showed a significant scattering peak intensity (Fig. 6b). As a comparison, the Zn electrode in the ZnSO4 electrolyte with PTT addition showed a decreased scattering peak intensity of the (101) plane while an increased peak for the (002) plane. This suggests the change of the Zn deposition direction in the ZnSO4 electrolyte with PTT addition as well (Fig. 6c). In contrast, in the ZnSO4 electrolyte added with PTT, the peak of the (002) plane of the Zn electrode appears significantly reduced, confirming that the deposition orientation of the Zn electrode was significantly changed, which is consistent with XRD results. In addition, the one-dimension of intensity counts showed the same results (Fig. S10†). It was observed from scanning electron microscopy (SEM) images that the surface of the stripped Zn electrode with the pure ZnSO4 electrolyte was corroded after 100th cycled plating/stripping at a current density of 1 mA cm−2 with a capacity of 0.5 mA h cm−2, with a large number of byproducts (such as Zn4SO4(OH)6·xH2O) and holes found on the Zn electrode surface, resulting in limited reversibility of Zn in the ZnSO4 electrolyte (Fig. 6d). In comparison, the Zn electrode after cycling in the ZnSO4 with PTT addition exhibited a clean and uniform surface; there are no obvious holes or by-products found (Fig. 6e).
 | ||
| Fig. 6 Zn plating behaviour studies. (a) XRD patterns of Zn electrodes after 100th plating in the pure ZnSO4 electrolyte and the ZnSO4 with PTT electrolyte. WAXS of the Zn electrode in the ZnSO4 electrolyte (b) and ZnSO4 with PTT addition (c) after the 100th plating. SEM images of Zn electrodes in different ZnSO4 electrolytes: after the 100th plating in the pure ZnSO4 electrolyte (d) and in the ZnSO4 electrolyte with PTT addition (e). In situ GC curves to dynamically evaluate the H2 amount during the Zn plating/stripping (f) in the pure ZnSO4 electrolyte and (g) in the ZnSO4 electrolyte with PTT addition. | ||
Chronoamperometry (CA) tests were performed on an electrochemical workstation; the change of current with time at a constant potential can sensitively reflect the nucleation process and surface changes (Fig. S11†). At a constant voltage of −150 mV, measured by CA, the current density for the coin cell using the pure 2 M ZnSO4 electrolyte continued to decrease over 150 s. This indicates ongoing 2D diffusion processes and rough deposition propagation. Zn2+ ions tend to aggregate and grow into dendrites to minimize the surface energy and exposed area. The current curve for 2 M ZnSO4 with 0.05 M PTT shows an almost constant value at −30 mA cm−2 after a brief 2D diffusion period of 20 s, reflecting a prolonged 3D compact diffusion process after nucleation. These results suggest that PTT can be beneficial for Zn surface deposition. This result shows the inhibition of the corrosion for the Zn electrolyte when adding PTT into the ZnSO4 electrolyte. It was observed from cyclic voltammetry (CV) tests that the nucleation overpotential (NOP) is the potential difference between the intersection point (A) and the point (B/B′), where Zn2+ ions begin to reduce on the substrate (Fig. S12†). It is considered a convenient parameter to explain the degree of polarization and shows the effect of electrode modification.56–59 Compared with the Zn/Cu battery using the pure ZnSO4 electrolyte, the Zn/Cu battery using the 2 M ZnSO4 with 0.05 M PTT electrolyte improved the NOP by 10 mV (−30 mV to −40 mV). This increased overpotential provides sufficient driving force for the nucleation and growth process of finer nuclei.60 In addition, the current density of the coin cell using 2 M ZnSO4 with 0.05 M PTT is higher compared to that of the coin cell using the pure ZnSO4 electrolyte, indicating that the latter has high electrochemical reactivity and higher capacity. Such observations clearly demonstrate the positive role of adding PPT in regulating the Zn deposition. The results from Energy Dispersive X-Ray Spectroscopy (EDX) analysis also confirmed that there are fewer by-products found in the stripped Zn electrode with the PTT addition (Fig. S13†). This finding also confirmed that the corrosion of the Zn electrode is significantly inhibited after the addition of PTT.
In addition, in situ GC analysis was conducted to dynamically assess hydrogen release during battery operation. As shown in the contour plots in Fig. 6f and g, the highest amount of H2 produced in the pure ZnSO4 aqueous electrolyte was 58.03 ppm during the repeated Zn plating/stripping process at a current density of 5 mA cm−2 (Fig. 6f). In the ZnSO4 electrolyte with PTT addition, the intensity of hydrogen release was significantly suppressed (Fig. 6g). The one-dimensional in situ GC curves are shown in Fig. S14†. By comparison, the dynamic hydrogen release of the ZnSO4 electrolyte with PTT addition decreased by 44.5%, indicating that the HER was significantly inhibited, which contributes to the enhancement of Zn reversibility.
Moreover, we assembled Zn–I2 full cells with/without the additive and tested their electrochemical performance, such as charge/discharge curves and cycling performance (Fig. S15†). As shown in Fig. S15a†, for the representative charge/discharge curves, the cell containing the 2 M ZnSO4 with 0.05 M PTT electrolyte demonstrates a higher CE of 92.7%, compared to 85.2% for the Zn–I2 cell with pure 2 M ZnSO4. This indicates that the addition of PTT enhances the charge/discharge reversibility of the Zn–I2 battery. Cycling performance was assessed to demonstrate the PTT effect. While having a higher CE, the Zn–I2 cell containing the 2 M ZnSO4 with 0.05 M PTT electrolyte also achieves a higher capacity retention of 98.9% compared to 86.2% for the cell with the pure ZnSO4 electrolyte after 200 cycles at 0.2 A g−1 (Fig. S15b†). It shows that the addition of PTT significantly inhibits the shuttling effect of polyiodide anions (I3−/I5−). These results indicate that the addition of PTT can achieve a highly reversible and shuttle-free Zn–I2 battery and prolong the battery lifespan.
Conclusions
In summary, we screen a promising electrolyte additive for sustainable AZIBs and provide atomic-level insights into exploring its impact on electrolyte performance by combining experiments and theoretical calculations. PTT has a unique symmetrical structure with four hydroxyl groups (–OH), which significantly enhances its interaction with water molecules and changes the ratio of different H-bond types (strong H-bonds decrease and weak H-bonds increase) between water molecules. These results prove that PTT as an additive can break the H-bond network of water molecules and change the solvation structure of Zn2+, thereby suppressing dendrite growth and side reactions on the Zn anode during cycling. As a result, water-induced side reactions and dendrite formation during cycling are significantly reduced, resulting in improved Zn reversibility and overall battery efficiency. Notable outcomes include the average CE reaching 99.7% and long-term stability exceeding 1000 h. This study contributes to the development of cost-effective and efficient electrolyte strategies aimed at solving the problems caused by water in AZIBs.Methods
Experimental
Electrochemical tests were carried out using CR 2032 coin-type cells with glass fiber filters serving as separators. Prior to usage, the Zn foil underwent polishing using softback sanding sponges (3M, USA) and was subsequently wiped with ethanol. Unless specified otherwise, Zn anodes were cut into disk-shaped electrodes with a 10 mm diameter for coin cell assembly. The volume of electrolyte addition was 70 μL for coin cells and 100 μL for the full cells. The volume was meticulously measured using a calibrated pipette to maintain high accuracy and repeatability across all experimental runs. Charge–discharge tests for coin cells were conducted using the LAND battery test system (CT2001A). For intermittent charge/discharge tests, Zn//Zn cells were sequentially charged/discharged at a current density of 1 mA cm−2, reaching a total capacity of 0.5 mA h cm−2. Zn//Cu asymmetric cells were subjected to charge–discharge cycles at 1 mA cm−2, with a capacity of 0.5 mA h cm−2 and an upper cut-off voltage of 0.8 V. These cycles were employed to assess the CE of different electrolytes.
LSV curves of ZnSO4 were collected using a three-electrode system, in which the glycerol electrode was used as the reference electrode, and stainless steel was used as the working electrode and counter electrode. The test was performed on an electrochemical working station CHI 760E within a voltage range from 0 V to −1.6 V with a scan rate of 1 mV s−1. Chronoamperometry was performed on Zn//Zn symmetric batteries on a CHI760E with a constant step potential of −150 mV. Raman spectra were collected with Labram HR Evolution (Horiba Scientific) using a 532 nm laser.
The forced field parameters for PTT, Zn2+ and SO42− were obtained from CHARMM36 force fields.68 The TIP3P water model was employed for H2O.69 The time step was set to be 2 fs. The cutoff radius for vdW was 12 Å and the electrostatic interactions were 10 Å. The standard periodic boundary condition was used in all simulations. After minimization of the initial structure for 50000 steps (100 ps), each system was heated from 100 K to 300 K by performing Langevin dynamics temperature control for 0.8 ns (400000 steps). The systems were further relaxed for another 9.2 ns under NPT by the Nosé–Hoover Langevin piston pressure control method70 at 1.01325 bar. After relaxation, each system was simulated for 100 ns under the canonical ensemble (NVT) for data collection and statistical analysis.71
AIMD simulations were performed with the CP2K computational suite (v. 2023.1),72 employing the DFT representation of the electronic structure, which is integrated within the Quickstep module of CP2K.73 The Perfect Bayesian Equilibrium (PBE) exchange–correlation functional74 was used.75 The molecularly optimized short-ranged double-zeta (DZVP-MOLOPT-SR-GTH) basis set for atomic orbitals was utilized,76 coupling with the auxiliary plane wave expansion of the electron density up to a 400 Ry cutoff. Core electrons were depicted using norm-conserving GTH pseudopotentials tailored for the PBE functional.77 Additionally, smoothing of the electron density and its derivative on the spatial integration grid was implemented (via keywords XC_SMOOTH_RHO NN50 and XC_DERIV NN50_SMOOTH in CP2K), as previous findings indicated a notable enhancement in the stability of local energetics for liquid water.78 Dispersion effects were accounted for using a two-body DFT-D3 empirical dispersion correction, with zero damping terms and a cutoff set to 10 Å.79
The 2 M ZnSO4 electrolyte system contained 200 water molecules, 20 Zn2+ ions and 20 SO42− anions. And the system of 2 M ZnSO4 with 0.05 M PTT contained 200 water molecules, 20 Zn2+, 20 SO42− and 1 PTT molecules. Each system was first equilibrated for 0.5 ps in an AIMD simulation in the NPT ensemble at T = 298.15 K, and the simulation time step was set to be 1 fs. After this equilibration period, the NVT ensemble was continued for 10 ps, with data collection every 1 fs. All MD analyses were performed using the VMD software package (v. 1.9.3).64
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge financial support by the Australian Research Council (FT190100636 and DE230100471). MD computations within this research were undertaken with the assistance of resources and services from the Phoenix High Performance Computing (HPC), which is supported by The University of Adelaide. DFT computations were undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI), which was supported by the Australian Government. We would like to thank Yanzhang Zhao for his contribution to developing the code for H-bond analysis from AIMD results.Notes and references
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang and B.-J. Hwang, Nature, 2015, 520, 324–328 CrossRef CAS PubMed
.
- H. Pan, Y. Shao, P. Yan, Y. Cheng, K. S. Han, Z. Nie, C. Wang, J. Yang, X. Li and P. Bhattacharya, Nat. Energy, 2016, 1, 1–7 Search PubMed
- Y. Liu, A. Gao, J. Hao, X. Li, J. Ling, F. Yi, Q. Li and D. Shu, Chem. Eng. J., 2023, 452, 139605 CrossRef CAS
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng and N. Zhao, Nat. Energy, 2020, 5, 440–449 CrossRef CAS
- L. Kang, J. Zheng, K. Yue, H. Yuan, J. Luo, Y. Wang, Y. Liu, J. Nai and X. Tao, Small, 2023, 19, 2304094 CrossRef CAS PubMed
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang and D. C. Bock, Science, 2019, 366, 645–648 CrossRef CAS
- X. Zeng, J. Mao, J. Hao, J. Liu, S. Liu, Z. Wang, Y. Wang, S. Zhang, T. Zheng and J. Liu, Adv. Mater., 2021, 33, 2007416 CrossRef CAS
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef
- R. Zhao, H. Wang, H. Du, Y. Yang, Z. Gao, L. Qie and Y. Huang, Nat. Commun., 2022, 13, 3252 CrossRef CAS
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan and E. Thimsen, Nat. Sustain., 2023, 6, 325–335 CrossRef
- S. Deng, Z. Yuan, Z. Tie, C. Wang, L. Song and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 22002–22006 CrossRef CAS
- J. Fu, D. U. Lee, F. M. Hassan, L. Yang, Z. Bai, M. G. Park and Z. Chen, Adv. Mater., 2015, 27, 5617–5622 CrossRef CAS
- M. Xu, D. Ivey, Z. Xie and W. Qu, J. Power Sources, 2015, 283, 358–371 CrossRef CAS
- Z. Liu, T. Cui, G. Pulletikurthi, A. Lahiri, T. Carstens, M. Olschewski and F. Endres, Angew. Chem., Int. Ed., 2016, 55, 2889–2893 CrossRef CAS PubMed
- X. Zeng, J. Hao, Z. Wang, J. Mao and Z. Guo, Energy Storage Mater., 2019, 20, 410–437 CrossRef
- Z. Li and A. W. Robertson, Battery Energy, 2023, 2, 20220029 CrossRef CAS
- J. Chen, Z. Yan, K. Li, A. Hu, B. Yang, T. Li, M. He, Y. Li, Z. Wei Seh and J. Long, Battery Energy, 2024, 3, 20230063 CrossRef
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS
- L. Ma, S. Chen, N. Li, Z. Liu, Z. Tang, J. A. Zapien, S. Chen, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 1908121 CrossRef CAS
- C. Liu, X. Xie, B. Lu, J. Zhou and S. Liang, ACS Energy Lett., 2021, 6, 1015–1033 CrossRef CAS
- F. Yang, J. A. Yuwono, J. Hao, J. Long, L. Yuan, Y. Wang, S. Liu, Y. Fan, S. Zhao and K. Davey, Adv. Mater., 2022, 34, 2206754 CrossRef CAS
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS
- L. Yuan, J. Hao, C.-C. Kao, C. Wu, H.-K. Liu, S.-X. Dou and S.-Z. Qiao, Energy Environ. Sci., 2021, 14, 5669–5689 RSC
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Joule, 2020, 4, 69–100 CrossRef CAS
- X. Li, Z. Chen, P. Ruan, X. Hu, B. Lu, X. Yuan, S. Tian and J. Zhou, Nanoscale, 2024, 16, 2923–2930 RSC
- X. Xie, J. Li, Z. Xing, B. Lu, S. Liang and J. Zhou, Natl. Sci., 2023, 10, nwac281 CAS
- X. Shi, J. Xie, J. Wang, S. Xie, Z. Yang and X. Lu, Nat. Commun., 2024, 15, 302 CrossRef CAS PubMed
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song and Y. Cui, Nano Energy, 2021, 80, 105478 CrossRef CAS
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS
- J. Hao, J. Long, B. Li, X. Li, S. Zhang, F. Yang, X. Zeng, Z. Yang, W. K. Pang and Z. Guo, Adv. Funct. Mater., 2019, 29, 1903605 CrossRef
- K. Lu, C. Chen, Y. Wu, C. Liu, J. Song, H. Jing, P. Zhao, B. Liu, M. Xia and Q. Hao, Chem. Eng. J., 2023, 457, 141287 CrossRef CAS
- S. Chen, Q. Nian, L. Zheng, B.-Q. Xiong, Z. Wang, Y. Shen and X. Ren, J. Mater. Chem. A, 2021, 9, 22347–22352 RSC
- Y. Ma, Q. Zhang, L. Liu, Y. Li, H. Li, Z. Yan and J. Chen, Natl. Sci., 2022, 9, nwac051 CAS
- D. Wang, Q. Li, Y. Zhao, H. Hong, H. Li, Z. Huang, G. Liang, Q. Yang and C. Zhi, Adv. Energy Mater., 2022, 12, 2102707 CrossRef CAS
- X. S. Lin, Z. R. Wang, L. H. Ge, J. W. Xu, W. Q. Ma, M. M. Ren, W. L. Liu, J. S. Yao and C. B. Zhang, Chemelectrochem, 2022, 9, e202101724 CrossRef CAS
- Y. Lv, Y. Xiao, L. Ma, C. Zhi and S. Chen, Adv. Mater., 2022, 34, 2106409 CrossRef CAS PubMed
- Y. Wu, Z. Zhu, D. Shen, L. Chen, T. Song, T. Kang, Z. Tong, Y. Tang, H. Wang and C. S. Lee, Energy Storage Mater., 2022, 45, 1084–1091 CrossRef
- Y. Yang, C. Huang, H. Li, Z. Teng, H. Zhang, X. Wei, H. Zhang, L. Wu, C. Zhang and W. Chen, J. Mater. Chem. C, 2023, 11, 9559 RSC
- W. Kao-ian, M. T. Nguyen, T. Yonezawa, R. Pornprasertsuk, J. Qin, S. Siwamogsatham and S. Kheawhom, Mater. Today Energy, 2021, 21, 100738 CrossRef CAS
- Y. Geng, L. Pan, Z. Peng, Z. Sun, H. Lin, C. Mao, L. Wang, L. Dai, H. Liu and K. Pan, Energy Storage Mater., 2022, 51, 733–755 CrossRef
- J. Li, Z. Liu, S. Han, P. Zhou, B. Lu, J. Zhou, Z. Zeng, Z. Chen and J. Zhou, Nano-Micro Lett., 2023, 15, 237 CrossRef CAS
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC
- Y. Dong, L. Miao, G. Ma, S. Di, Y. Wang, L. Wang, J. Xu and N. Zhang, Chem. Sci., 2021, 12, 5843–5852 RSC
- J.-B. Brubach, A. Mermet, A. Filabozzi, A. Gerschel, D. Lairez, M. Krafft and P. Roy, J. Phys. Chem. B, 2001, 105, 430–435 CrossRef CAS
- H. Du, R. Zhao, Y. Yang, Z. Liu, L. Qie and Y. Huang, Angew. Chem., 2022, 134, e202114789 CrossRef
- Y. Zhu, J. Hao, Y. Huang and Y. Jiao, Small Struct., 2023, 4, 2200270 CrossRef CAS
- Q. Sun, Vib. Spectrosc., 2009, 51, 213–217 CrossRef CAS
- C. Li, Z. Sun, T. Yang, L. Yu, N. Wei, Z. Tian, J. Cai, J. Lv, Y. Shao and M. H. Rümmeli, Adv. Mater., 2020, 32, 2003425 CrossRef CAS PubMed
- D. Mackinnon, J. Brannen and P. Fenn, J. Appl. Electrochem., 1987, 17, 1129–1143 CrossRef CAS
- B. Tripathy, S. Das, G. Hefter and P. Singh, J. Appl. Electrochem., 1997, 27, 673–678 CrossRef CAS
- Q. Zhang and Y. Hua, J. Appl. Electrochem., 2009, 39, 261–267 CrossRef CAS
- D. Mackinnon, R. Morrison, J. Mouland and P. Warren, J. Appl. Electrochem., 1990, 20, 728–736 CrossRef CAS
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS
-
M. Frish, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Paterson, Gaussian09, Gaussian, Inc., Wallingford CT, 2009, vol. 121, pp. 150–166 Search PubMed
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph. Model., 1996, 14, 33–38 CrossRef CAS
-
T. Williams, C. Kelley, C. Bersch, H.-B. Bröker, J. Campbell, R. Cunningham, D. Denholm, G. Elber, R. Fearick and C. Grammes, An Interactive Plotting Program, 2017, vol. 2, p. 1, available online: http://www.gnuplot.info/docs_5 Search PubMed
- S. Simon, M. Duran and J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef
- T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt and F. Schiffmann, J. Chem. Phys., 2020, 152, 194103 CrossRef
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- M. A. Marques, M. J. Oliveira and T. Burnus, Comput. Phys. Commun., 2012, 183, 2272–2281 CrossRef CAS
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703 CrossRef CAS PubMed
- R. Jonchiere, A. P. Seitsonen, G. Ferlat, A. M. Saitta and R. Vuilleumier, J. Chem. Phys., 2011, 135, 154503 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02316a |
| This journal is © The Royal Society of Chemistry 2024 |