
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic pathways to create asymmetric center at C1 position of 1-substituted-tetrahydro-β-carbolines – a review
Md. Moaz Ahmed Asif†
 a,
Susmita Roy Lisa†b and
Nazmul Qais*c
a,
Susmita Roy Lisa†b and
Nazmul Qais*c
aDepartment of Pharmacy, Faculty of Science & Engineering, University of Information Technology & Sciences, Holding 190, Road 5, Block J, Baridhara, Maddha Nayanagar, Vatara, Dhaka-1212, Bangladesh
bDepartment of Chemistry, Syracuse University, Syracuse, NY 13244, USA
cDepartment of Clinical Pharmacy and Pharmacology, Faculty of Pharmacy, University of Dhaka, Dhaka-1000, Bangladesh. E-mail: nqais@du.ac.bd
First published on 19th September 2024
Abstract
The 2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indoles or tetrahydro-β-carbolines (THβCs) are tricyclic compounds that are found in various natural sources that exhibit a wide range of important pharmacological activities. Chiral 1-substituted-THβCs, which have an asymmetric center at C1, have attained significant interest due to their possible Monoamine Oxidase (MAO) inhibitory activity, benzodiazepine receptor binding activity, and antimalarial effectiveness against chloroquine-resistant Plasmodium falciparum. This review highlights and summarizes various novel stereoselective approaches to introduce chirality at the C1 position of 1-substituted-THβCs in good yield and enantiomeric excess (ee) or diastereomeric excess (de). These methods include the Pictet–Spengler reaction, chiral auxiliary, Asymmetric Transfer Hydrogenation (ATH) with chiral catalysts, asymmetric addition reaction, and enzymatic catalysis. The syntheses of chiral THβCs are reviewed comprehensively, emphasizing their role in drug development from 1977 to 2024.
1. Introduction
The tetrahydro-β-carbolines (THβCs) are a group of compounds found in a variety of natural and synthetic compounds containing a unique tricyclic pyrido[3,4-b]indole ring and renowned for their promising biological actions. Originating from tryptamine or tryptophan, these compounds are widespread in nature and have been isolated from various sources including plants, fungi, animals, and marine organisms.1 THβCs exhibit a broad spectrum of pharmacological activities; including phosphodiesterase 5 (PDE5)-inhibitory,2 antitumor,3,4 antiviral,5,6 and antiprotozoal7 especially antimalarial effects.8,9 Chiral 1-substituted-THβCs 1 (Fig. 1), having an asymmetric center present at the C1 position, are still being sought even after being discovered more than a hundred years ago.10 They are mainly MAO inhibitors or work by binding to benzodiazepine receptors.11,12 They have gained particular interest due to their potential antimalarial efficacy against a Plasmodium falciparum strain (FcB1-Colombia) that is chloroquine-resistant.13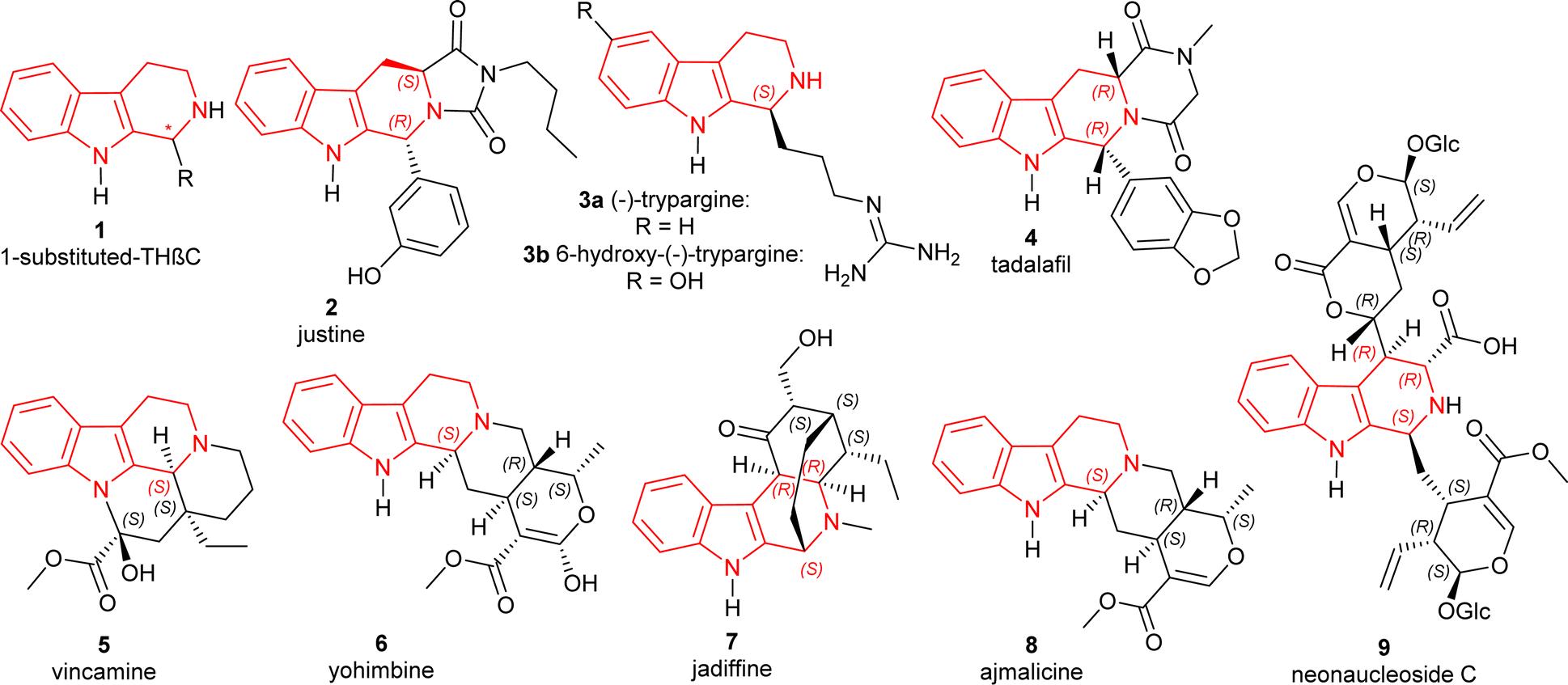 | ||
| Fig. 1 Structures of important chiral 1-substituted-THβCs. | ||
Some of the specific 1-substituted-THβCs (Fig. 1) that have biological importance are given below:
• Justine 2 (HR22C16) induces mitotic arrest and blocking cell division in taxol-resistant cancer cells.4,14
• The African rhacophorid frog Kassina senegalensis15 is the source of trypargine 3a, a highly poisonous THβC alkaloid. It was recently discovered in a hitherto unknown ground ascidian Eudistoma sp.16 A very similar chemical, 6-hydroxy-trypargine, was shown to be a strong neurotoxic in the venom of the Brazilian web spider Parawixia bistriata.17
• Tadalafil 4 is an orally active PDE5 inhibitor and also highly potent.2,18
• Vincamine 5 aided in mild to moderate dementia patients.19
• Yohimbine 6, an α2-adrenoceptor blocker that helps in erectile dysfunction.20,21
• Jadiffine 7 collected from Vinca difformis.22
• Ajmalicine 8 (ref. 23) and reserpine 10 (ref. 24) (Scheme 1) used as an antihypertensive.
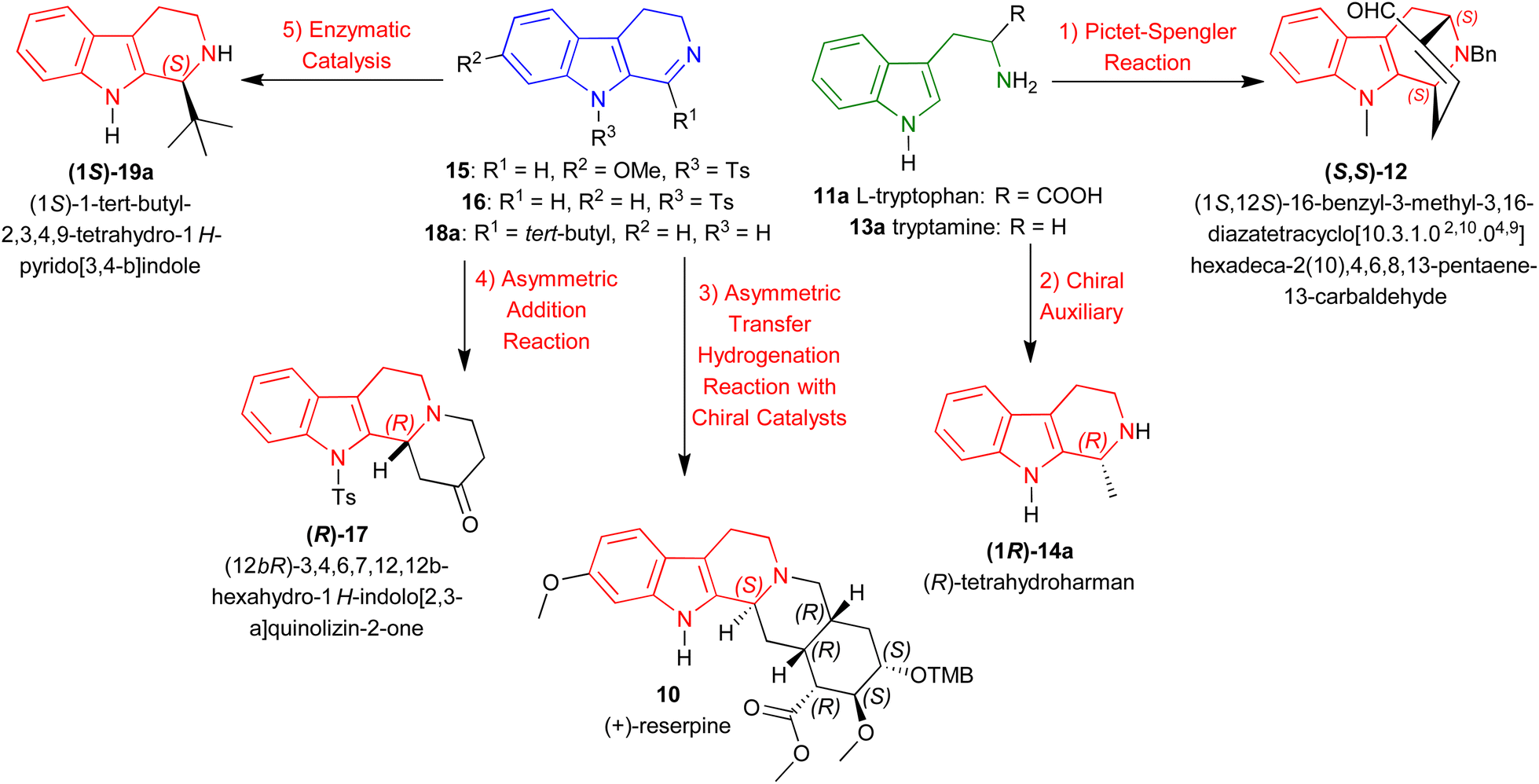 | ||
| Scheme 1 Representative examples of asymmetric methods for synthesizing chiral 1-substituted-tetrahydro-β-carbolines. | ||
• Neonaucleoside C 9 collected from Neonauclea sessilifolia.25
• Fumitremorgins are found in fungi that have antiviral26 and cell-cycle inhibitory activities.27 They also worked as protein kinase and topoisomerase II inhibitors.28
Synthetic methodologies to introduce chirality at the C1 position in THβCs have been extensively studied.29 These methods include the Pictet–Spengler reaction,30 asymmetric alkylation using N2-auxiliary as a directing group,31 and acid-induced epimerization in conjunction with the Pictet–Spengler reaction.32 Additionally, the Bischler–Napieralski reaction33 and classical Noyori ATH conditions34 have been highlighted as key synthetic routes to create chiral 1-substituted-THβCs. The C1 stereocenter in THβCs plays a crucial role in their pharmacological properties, influencing their activity in various therapeutic areas. With a ubiquitous presence in both natural sources and synthetic derivatives, these compounds have significant attention in medicinal chemistry for their potential therapeutic applications. The intricate interplay of their chemical structure and biological effects underscores their pivotal role in drug discovery endeavors, accentuating the paramount importance of advancing synthetic methodologies to access these compounds efficiently.
In the last 11 years, Laine et al., Maity et al., Szabó et al., Wang et al., and Du et al. published reviews that emphasized on the pharmacological importance, overall synthetic methods, biological activities, and applications of THβCs,35–39 but our review does not comprise any of the above-mentioned perspective wholeheartedly. This review neither talks about pharmacological importance, nor biological activities; neither gives all of the synthetic methodologies, nor the applications of THβCs also.
Instead, this review intends to offer a complete overview of the asymmetric synthesis of 1-substituted-THβCs, focusing on the synthetic methods to introduce chirality at the C1 position and their implications for drug development. Here, we discussed about five methods to create an asymmetric center at the C1 position of 1-substituted-THβCs reported from as early as 1977 to as latest as 2024. With representative examples (Scheme 1), they are:
(1) Pictet–Spengler reaction: From L-tryptophan 11a to synthesize (1S,12S)-16-benzyl-3-methyl-3,16-diazatetracyclo[10.3.1.02,10.04,9]hexadeca-2(10),4,6,8,13-pentaene-13-carbaldehyde (S,S)-12.40
(2) Chiral auxiliary: From tryptamine 13a to synthesize (R)-tetrahydroharman (1R)-14a.41
(3) ATH with chiral catalysts: From 7-methoxy-9-(4-methylphenyl)sulfonyl-3,4-dihydropyrido[3,4-b]indole (7-methoxy-9-tosyl-DHβC) 15 to synthesize (+)-reserpine 10.42
(4) Asymmetric addition reaction: From 9-tosyl-DHβC 16 to synthesize (12bR)-3,4,6,7,12,12b-hexahydro-1H-indolo[2,3-a]quinolizin-2-one (R)-17.43
(5) Enzymatic catalysis: From 1-tert-butyl-4,9-dihydro-3H-pyrido[3,4-b]indole 18a to synthesize (1S)-1-tert-butyl-THβC (1S)-19a.44
2. Enantioselective synthesis of 1-substituted-tetrahydro-β-carbolines
The enantioselective synthesis of 1-substituted-THβCs 1 can be conducted by the following five methods:Method 1. Pictet–Spengler reaction.
Method 2. Chiral auxiliary.
Method 3. Asymmetric transfer hydrogenation reaction with chiral catalysts.
Method 4. Asymmetric addition reaction.
Method 5. Enzymatic catalysis.
Method 1. Pictet–Spengler reaction
More than 113 years ago from now in 1911, Amé Pictet and Theodor Spengler devised a novel way to produce 1,2,3,4-tetrahydroisoquinoline by heating β-phenylethylamine and formaldehyde dimethylacetal in the presence of hydrochloric acid.10 This reaction is known as the Pictet–Spengler reaction. In 1928, Tatsui used the basis of this reaction to be the first to produce 1-methyl-THβC from tryptamine and ethanal in the presence of sulphuric acid.45First, L-tryptophan methyl ester 11b was condensed with methyl 4-oxobutanoate at 0 °C with excess 2,2,2-trifluoroacetic acid (TFA) in dichloromethane (DCM) to get (1S,3S)-20 (predominating than its (1R,3S)-diastereomer by 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio or dr) by Pictet–Spengler reaction under kinetic control49 with a total yield of 61%.
:1 diastereomeric ratio or dr) by Pictet–Spengler reaction under kinetic control49 with a total yield of 61%.
The N2 of (1S,3S)-20 was then protected by benzyl carbonochloridate producing 21, and N9 of 21 was protected by methyl iodide (CH3I)/sodium hydride (NaH) or benzyl iodide/NaH respectively at 0 °C giving 22a or 22b.
With NaH and protic methanol (MeOH), Dieckmann cyclization of 22a and 22b gave the β-keto ester 23a and 23b and their enolic form 24a and 24b. These esters were hydrolyzed and decarboxylated by heating at 130 °C with NaCl and H2O in N,N-dimethylformamide (DMF)50 producing the bridged ketone 25a (>95% ee) and 25b.
25a was then reacted with Tf2NPh/NaH/THF, and LiCN/benzene (PhH)/Pd-(PPh3)4 (ref. 51) to get benzyl (1S,12S)-13-hydroxy-3-methyl-3,16-diazatetracyclo[10.3.1.02,10.04,9]hexadeca-2(10),4,6,8,13-pentaene-16-carboxylate 26 which possesses an α,β-unsaturated nitrile for Michael addition of a C4 fragment, giving access to the full carbon skeleton of N9-methylated alkaloids of the ajmaline–sarpagine group. This overall route is more efficient than that of the N2-benzyl derivative of 26.52
On the other hand, catalytic hydrogenation of 25b with 10% Pd–C in MeOH produced (1S,12S)-3-benzyl-13-oxo-3,16-diazatetracyclo[10.3.1.02,10.04,9]hexadeca-2(10),4,6,8-tetraene 27 which is the antipode of the intermediate used in the syntheses of (+)-koumine, (+)-taberpsychine, and (+)-koumidine (Scheme 2).53
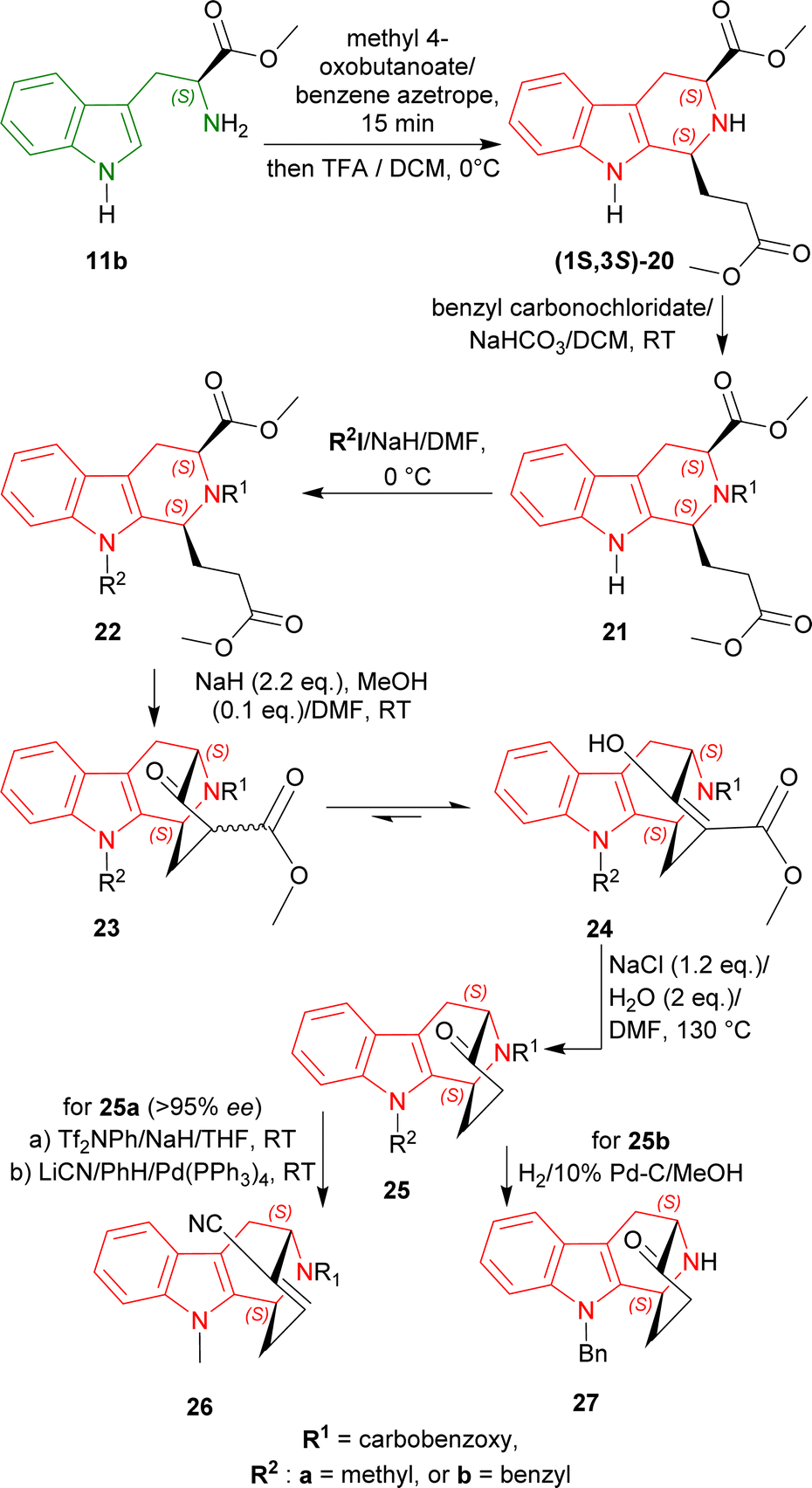 | ||
| Scheme 2 Asymmetric formal syntheses of (−)-koumine, (−)-taberpsychine, and (−)-koumidine intermediates from L-tryptophan methyl ester. | ||
So, methyl 4-oxobutanoate predominantly produced (1S,3S)-diastereomer with L-tryptophan methyl ester by Pictet–Spengler reaction under kinetic control.
L-Tryptophan 11a was converted to its homologated nitrile 28 in four steps in 50% overall yield.54 Modified Pictet–Spengler reaction of 28 with methyl prop-2-ynoate followed by treatment with TFA gave rise to a 60% yield of the acetate (1S,3S)-29 (54% de). In this reaction, Bailey et al. used a carbonyl-conjugated alkyne instead of the conventional aldehyde.52,55,56
N2 benzylation and N9 methylation of (1S,3S)-29 furnished the compound 30 in an overall 46% total yield. With lithium diethylamide at −78 °C, Dieckmann/Thorpe cyclisation57 of 30 gave 31 in 90% yield. The reduction of 31 with sodium borohydride in MeOH at room temperature (RT) afforded the corresponding hydroxynitrile58 and dehydration with POCl3 produced 32 in 87% yield. Finally, reduction with bis(2-methylpropyl)alumane (DIBAL) gave a 99% yield of (S,S)-12 (>97% ee) which was used in the synthesis of (−)-suaveoline, (−)-raumacline and (−)-Nb-methylraumacline (Scheme 3).59
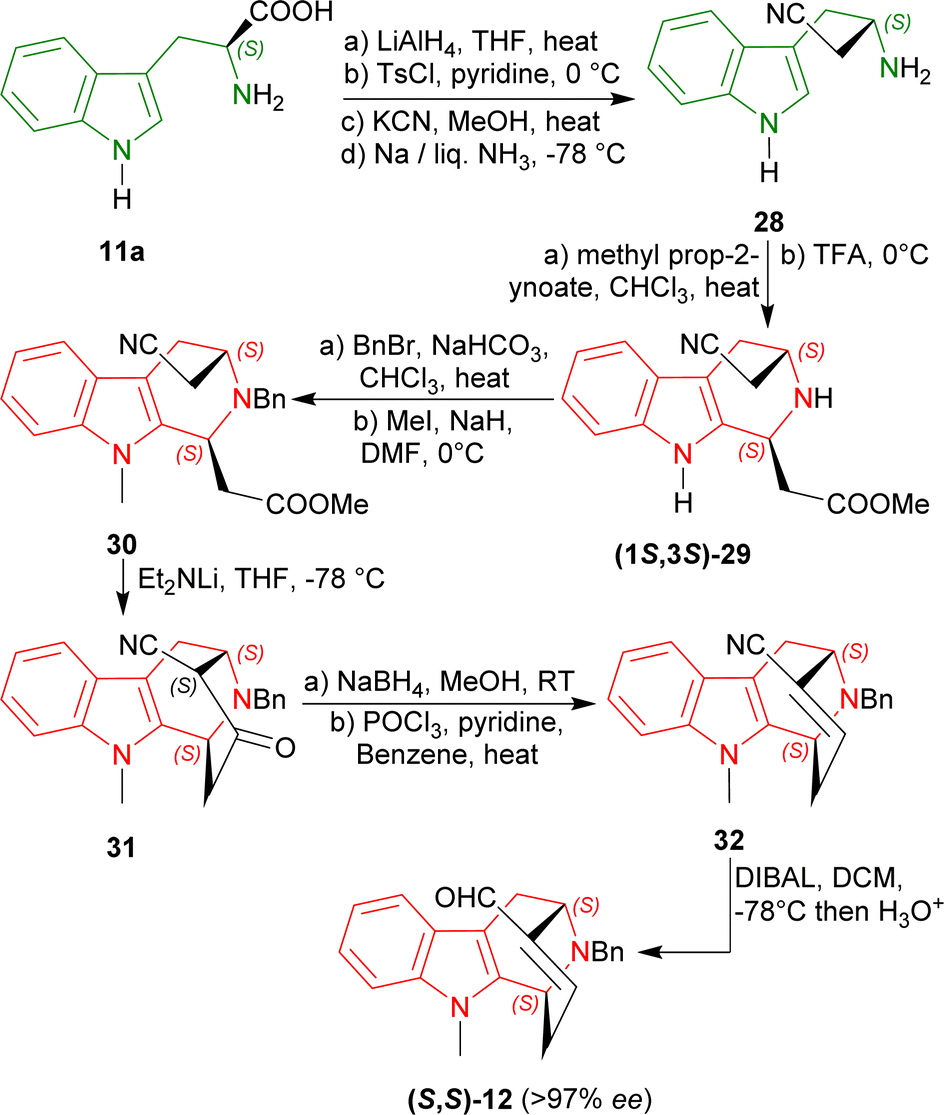 | ||
| Scheme 3 Asymmetric formal syntheses of (−)-suaveoline, (−)-raumacline, and (−)-Nb-methylraumacline intermediates from L-tryptophan. | ||
So, methyl prop-2-ynoate predominantly produced (1S,3S)-diastereomer with (3S)-3-amino-4-(1H-indol-3-yl)butanenitrile by modified Pictet–Spengler reaction.
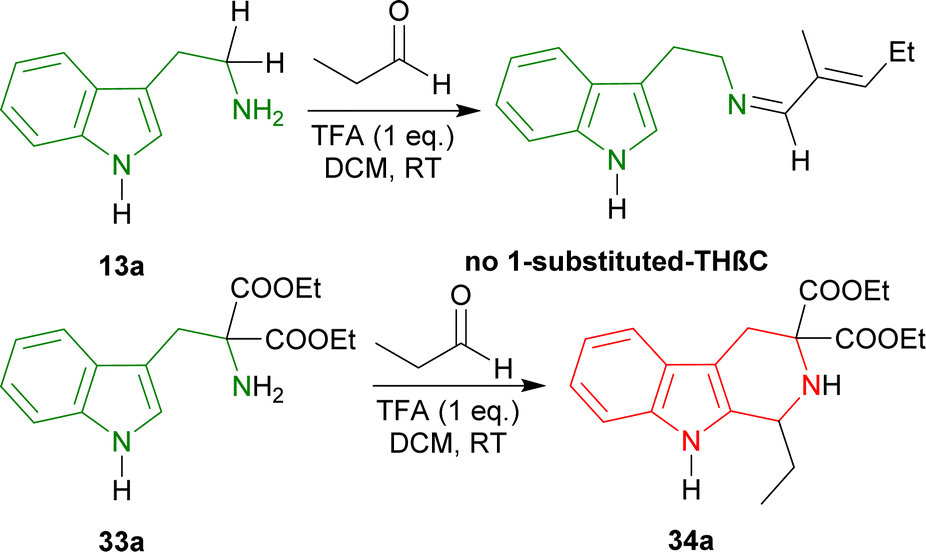 | ||
| Scheme 4 Tryptamine failed to produce any 1-substituted-THβC by Pictet–Spengler reaction but diethyl 2-amino-2-(1H-indol-3-ylmethyl)propanedioate was able to produce diethyl 1-ethyl-1,2,4,9-tetrahydropyrido[3,4-b]indole-3,3-dicarboxylate. | ||
To find an appropriate chiral organic Brønsted acid, 20 mol% 35a–f (Fig. 2) was then examined with Na2SO4 in toluene at RT for 1–3 hours. Among them, 35f gave the highest 66% ee with a good yield of 90% of (1R)-34a (Scheme 5).
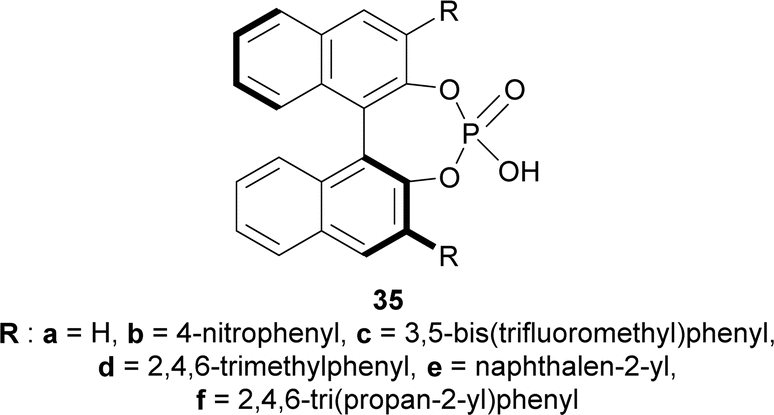 | ||
| Fig. 2 Chiral organic Brønsted acid for catalytic asymmetric Pictet–Spengler reaction. | ||
 | ||
| Scheme 5 Screening of chiral organic Brønsted acid for catalytic asymmetric Pictet–Spengler reaction. | ||
When the previous reaction was conducted with 35f at −30 °C for 3–5 days, the yield of (1R)-34a decreased to 76% while ee increased to 88%. 33b–d gave a similar ee of 86–90% with an excellent yield of 94–98% for (1R)-34b–d (Scheme 6).
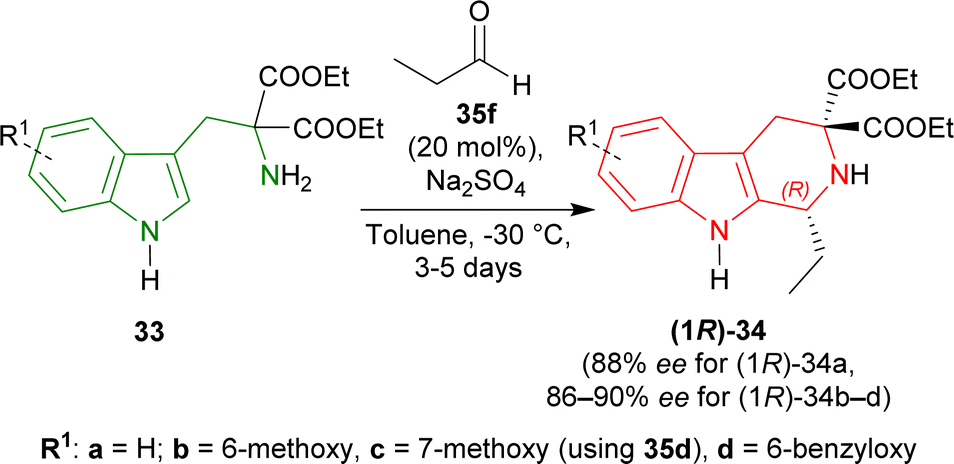 | ||
| Scheme 6 Screening of diethyl 2-amino-2-(1H-indol-3-ylmethyl)propanedioate derivatives. | ||
Various aldehydes were then reacted with 33a,b. Aliphatic unbranched and branched aldehydes produced (1R)-34e–j (58–98% yield, 72–88% ee) and (1R)-34k–m (50–93% yield, 81–91% ee) respectively at −30 °C in toluene for 3–6 days. When the temperature was decreased from −30 °C to −45 °C, ee of (1R)-34l increased slightly from 91% to 94% but yield decreased from 93% to 64%. Aromatic and electron-poor aromatic aldehydes also gave moderate to good yield (40–98%) and ee (62–96%) for (1R)-34n–r at −10 °C in DCM (Scheme 7).
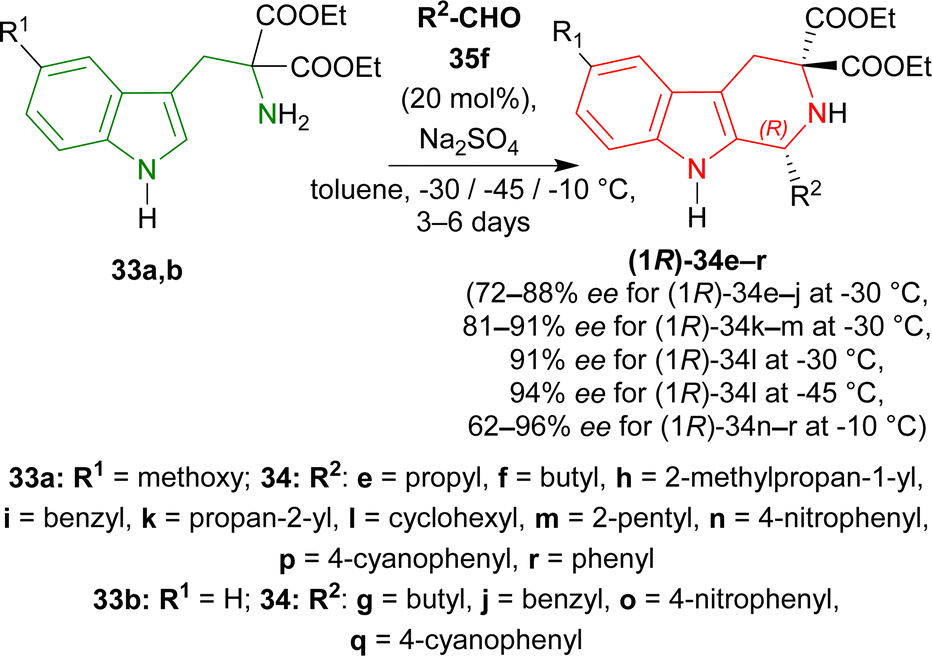 | ||
| Scheme 7 Screening of aldehydes. | ||
So, propionaldehyde and other aldehydes predominantly produced (1R)-enantiomer with diethyl 2-amino-2-(1H-indol-3-ylmethyl)propanedioate and its derivatives catalyzed by chiral organic Brønsted acid.
1H-indole 36 was reacted with different nitroalkenes and aldehydes in the presence of 2,4-dibromo-6-[[[(4S,5S)-1-(4-methylphenyl)sulfonyl-4,5-diphenyl-4,5-dihydroImidazol-2-yl]methyl-[(1S)-1-phenylethyl]amino]methyl]phenol (11 mol%) (Scheme 8), copper(I) trifluoromethanesulfonate (CuOTf or CF3SO3−Cu+, 10 mol%), fluoro(iodo)phosphane (HFIP, 2 equivalents or eq.)62 in toluene to produce (1S,2S,3R)-37a–d in 76–84% yield and 97–99% ee at 0 °C or RT which are (R,S,S)-Friedel–Crafts/Henry adducts. Next (1S,2S,3R)-37a, reduced with nickel boride,63,64 gave (1S,2S,3R)-3-(1H-indol-3-yl)-2-amino-1,3-diphenylpropan-1-ol 38a (20% yield) at 0 °C for 0.5 hour. But, Zn powder under acidic condition65 at 0 °C for 24 hours gave a 59% yield of 38a. At RT for 15–18 hours, this condition gave a 99% yield of 38a–b from (1S,2S,3R)-37a–b. Ultimately, Zn-nanopowder was used to reduce the reaction time to 3 hours at RT to give 98–99% yields of 38c–d from (1S,2S,3R)-37c–d (Scheme 8).
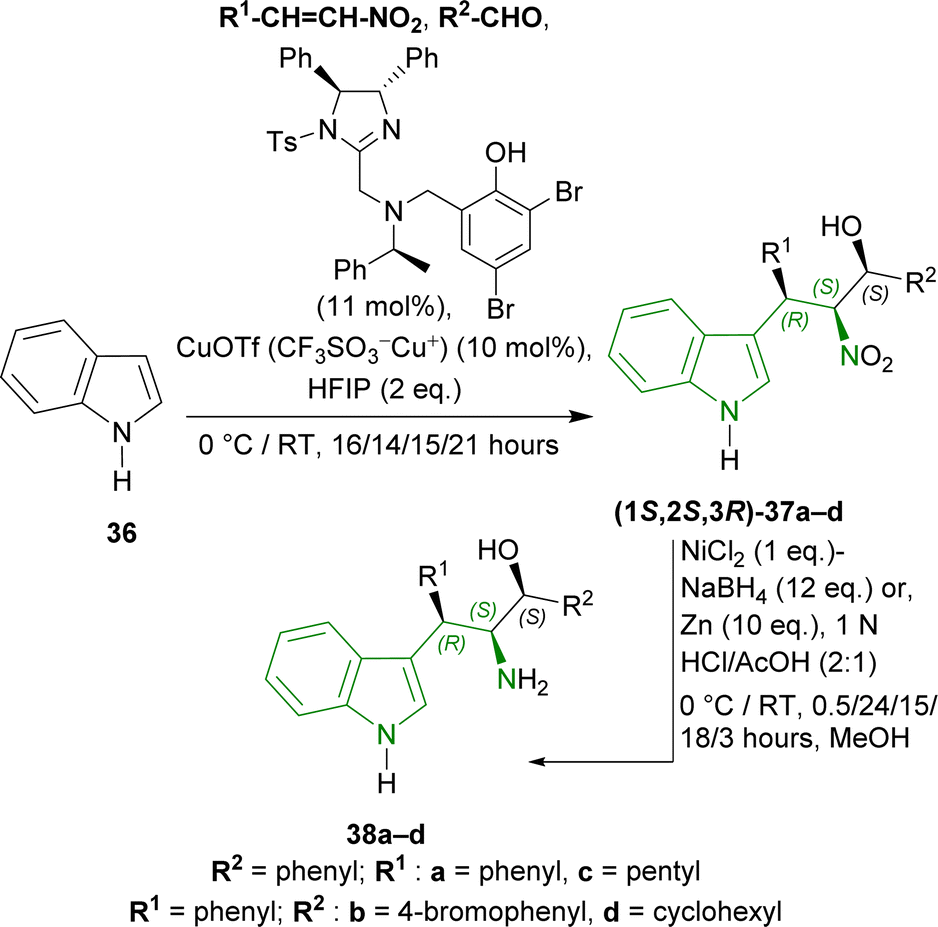 | ||
| Scheme 8 Synthesizing Friedel–Crafts/Henry adducts from 1H-indole and their reduction. | ||
Then, 38a was experimented with to optimize Pictet–Spengler reaction. 38a was converted into 39a with triethylsilyl chloride (TESCl) in DMF. 39a was cyclized with benzaldehyde and ethanoic acid, TFA, MgSO4, ytterbium(III) trifluoromethanesulfonate (Yb(OTf)3 or (CF3SO3−)3Yb3+); but TFA gave 30% yield of (S)-[(1S,3S,4R)-1,4-diphenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-3-yl]-phenylmethanol 40aa. After that, 39a with TFA (1.1 eq.) at RT for 19 hours following without and with MgSO4 at RT for 5 hours in CHCl3 gave 47% and 67% yield of 40aa respectively (Scheme 9).
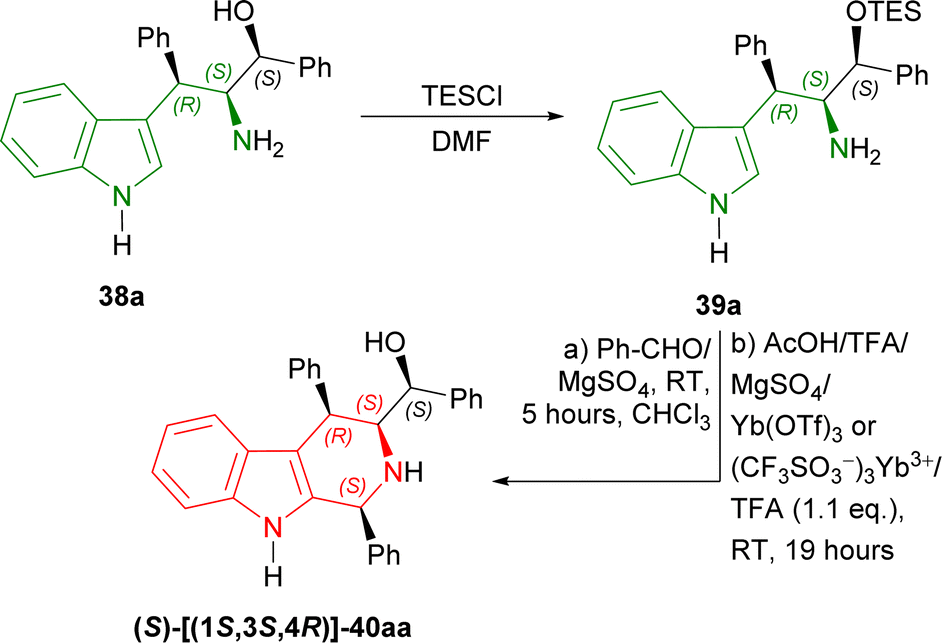 | ||
| Scheme 9 Protecting the OH group with TES group, Pictet–Spengler reaction, and removing the OH-protection. | ||
38a–d was tested with different aromatic aldehydes for 19–25 hours to give (S)-[(1S,3S,4R)]-40ab–ae,ba–bb,ca,da in 38–72% yields. Every product had 100% de except (S)-[(1S,3S,4R)]-40ca of which de was 91% (Scheme 10).
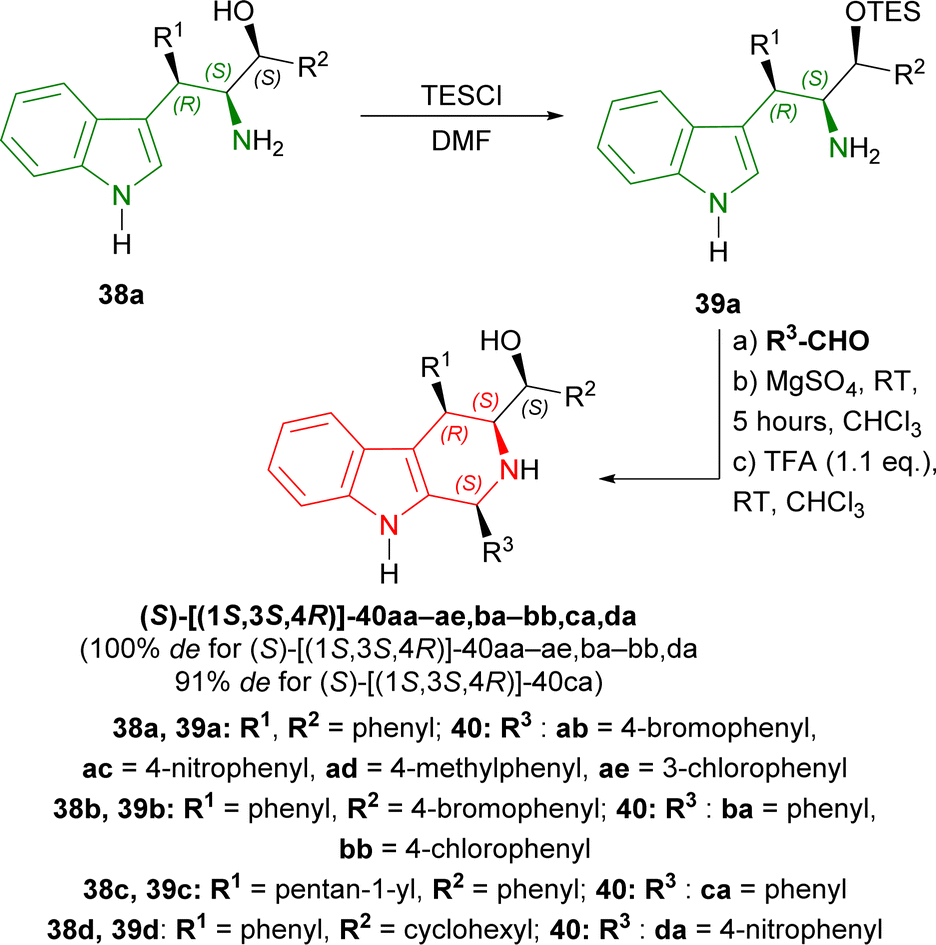 | ||
| Scheme 10 Screening of aldehydes. | ||
So, phenylaldehyde and other aldehydes produced (1S,3S,4R)-THβCs predominantly with (1S,2S,3R)-1-(1H-indol-3-yl)-1,3-diphenyl-3-triethylsilyloxypropan-2-amine and its derivatives (made from Friedel–Crafts/Henry adducts) by Pictet–Spengler reaction.
Firstly, (2S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-3-[1-[(2-methylpropan-2-yl)oxycarbonyl]indol-3-yl]propanoic acid 41 (10 mmol) was treated with prop-2-yn-1-ol (2 eq.), [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium;tetrafluoroborate (TBTU, 1.1 eq.), 1-hydroxybenzotriazole (HOBt·H2O, 1.1 eq.), N-ethyl-N-propan-2-ylpropan-2-amine (DIEA, 2.2 eq.) and in DMF to get 87% yield of 42. The Fmoc protection was then removed by diethylamine and acetonitrile and the Boc protection group was eliminated by cooled reagent K (TFA, water, phenol, ethanedithiol, triethylsilane, thioanisol) gaining a 66% yield of 43. The compound 43 was then reacted with various aromatic aldehydes and TFA in DCM at 0 °C. Benzaldehyde gave higher yield (73% for prop-2-ynyl (1S,3S)-1-phenyl-THβC-3-carboxylate (1S,3S)-44a) than 3- and 4-substituted-benzaldehydes (52–67% yields for prop-2-ynyl (1S,3S)-1-substituted-THβC-3-carboxylate (1S,3S)-44b–f) and thiophene-2-carbaldehyde (57% yield for prop-2-ynyl (1R,3S)-1-thiophen-2-yl-THβC-3-carboxylate (1R,3S)-44g) (Scheme 11).
 | ||
| Scheme 11 Synthesizing prop-2-ynyl ester of (2S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-3-[1-[(2-methylpropan-2-yl)oxycarbonyl]indol-3-yl]propanoic acid, removing Fmoc protection group, followed by Pictet–Spengler reaction. | ||
Secondly, L-tryptophan 11a is converted to 11b (95% yield) with thionyl chloride in MeOH at −10 °C for 24 hours. With 11b and hydrazine in MeOH at RT for 72 hours, 11c was obtained in 95% yield. 11c was then reacted with aromatic aldehydes in the presence of TFA as a catalyst in MeOH at RT for 24 hours. 4-Substituted-benzaldehydes gave similar yields (78–83% for (1S,3S)-1-(4-substituted-phenyl)-N-[(E)-(4-substituted-phenyl)methylideneamino]-THβC-3-carboxamide 45a–c); while 5-bromofuran-2-aldehyde had slightly better yield (85% for (1S,3S)-1-(5-bromofuran-2-yl)-N-[(E)-(5-bromofuran-2-yl)methylideneamino]-THβC-3-carboxamide 45d) (Scheme 12).
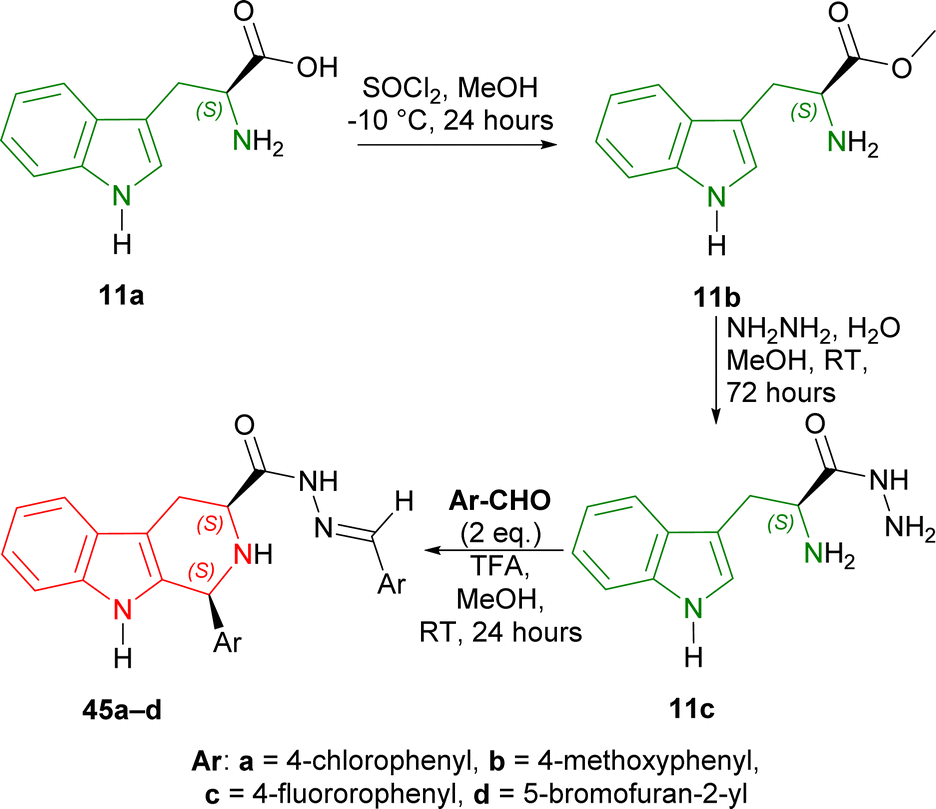 | ||
| Scheme 12 Synthesizing methyl ester of L-tryptophan, then L-tryptophan hydrazide, and Pictet–Spengler reaction. | ||
So, prop-2-ynyl (2S)-2-amino-3-(1H-indol-3-yl)propanoate and L-tryptophan hydrazide produced mainly (1S,3S)-THβCs with aromatic aldehydes by TFA-catalyzed Pictet–Spengler reaction; the only exception being prop-2-ynyl (1R,3S)-1-thiophen-2-yl-THβC-3-carboxylate.
Method 2. Chiral auxiliary
A chiral auxiliary is actually a pure enantiomeric organic chemical that is coupled with the starting material to generate a new product that may then undergo diastereoselective reactions using intramolecular asymmetric induction.67,68 At the end of the reaction, the auxiliary is removed under circumstances that ensures no racemization of the product. Then, it is often recovered and reused. Two of the widely used chiral auxiliaries are: Evans oxazolidinones,69 and Oppolzer sultams.70 There are many applications with the use of chiral auxiliaries.67,711-Benzyl-3-(2-bromoethyl)indole 46 underwent Vilsmeier–Haack reaction to get 47 (50% yield). With (2R)-2-amino-2-phenylethanol at RT for 1 hour, 47 formed the iminium salt 48 after azeotropic distillation with benzene. Treatment with triethylamine (Et3N) at −5 °C for 1 hour in chloroform/DCM then cyclized 48 into (3R,11bS)-49 (85% de); after recrystallization from ethanol, it was found in 100% de with 62% yield. Then, (3R,11bS)-49 was reacted with two Grignard reagents (MeMgI and PhMgI) at −78 °C for 1 hour to give 90% de of (S,R)-50a and (S,R)-50b. The purification process involved column chromatography on silica gel. Hydrogenolysis on Pd(OH)2–carbon at RT for 12 hours will remove the chiral auxiliary, and sodium in liquid ammonia removed the N-benzyl group with 100% ee of (1S)-1-methyl-THβC (1S)-14a and (1S)-1-phenyl-THβC (1S)-14b (Scheme 13).
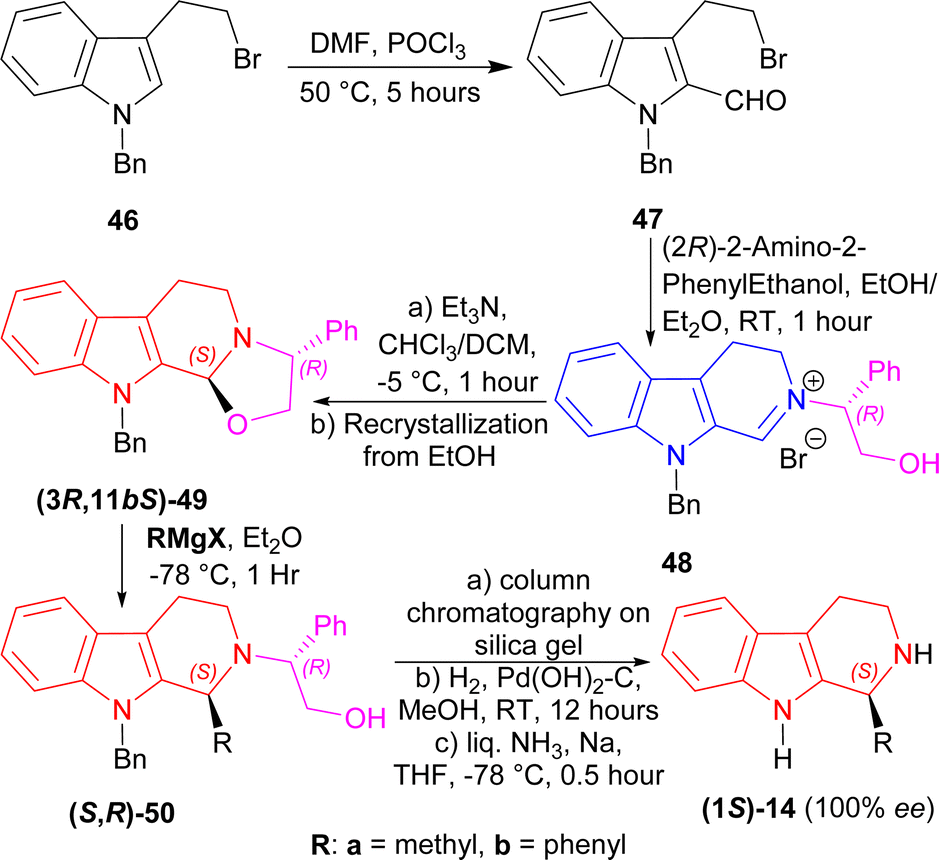 | ||
| Scheme 13 Synthesizing (1S)-1-methyl-THβC and (1S)-1-phenyl-THβC with (2R)-2-amino-2-phenylethanol as chiral auxiliary. | ||
So, the (R)-configured chiral auxiliary (2R)-2-amino-2-phenylethanol predominantly produced (1S)-1-substituted-THβCs.
At first, tryptamine 13a was added with (R)-51a,b to form 52a and 52b. Then 52a,b was cyclized with TFA or toluene-p-sulfonic acid (p-TsOH) to form 53a and 53b as a major compound. RANEY® nickel desulfurization of 53a,b then resulted in 80% yield of optically pure (100% ee) (R)-tetrahydroharman (1R)-14 (ref. 73 and 74) (overall 57% yield) (Scheme 14).
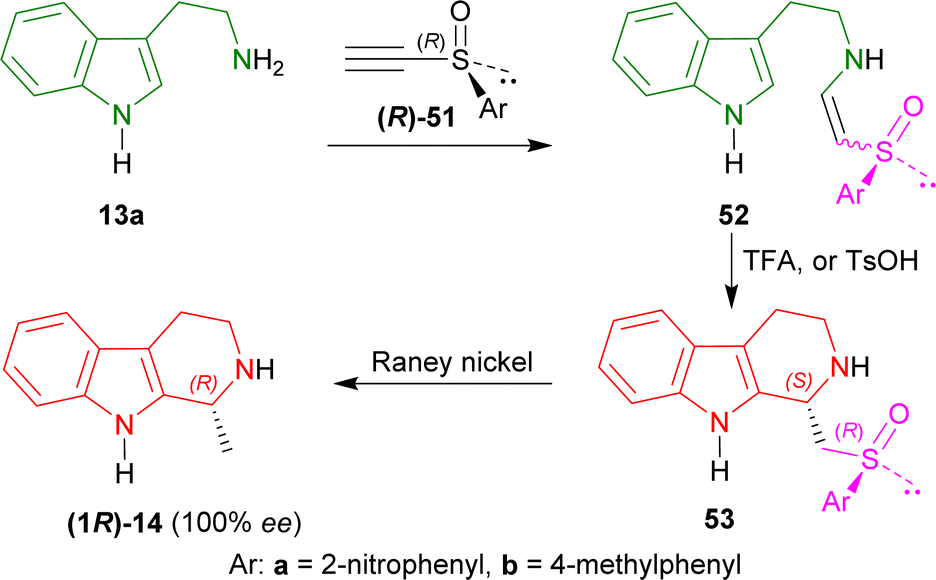 | ||
| Scheme 14 Synthesizing (R)-tetrahydroharman with chiral acetylenic sulfoxides as chiral auxiliary. | ||
So, (R)-configurated chiral acetylenic sulfoxides produced (R)-configurated THβC named (R)-tetrahydroharman.
tert-Butyl (S)-pyroglutamate (S)-54, NaH with various N-protecting reagents (R-X) producing (S)-55a–j (highest 94% yield for (S)-55a); which was converted to (2S)-1-substituted-5-oxopyrrolidine-2-carboxylic acid (S)-56a–j (highest 97% yield for (2S)-1-(2-naphthylmethyl)-5-oxopyrrolidine-2-carboxylic acid (S)-56d) using TFA at RT (Scheme 15).
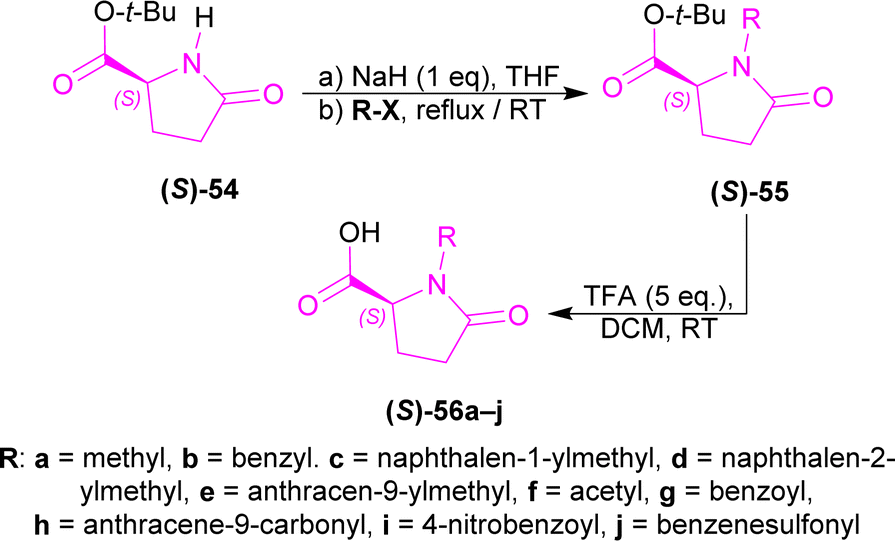 | ||
| Scheme 15 Synthesis of (S)-pyroglutamic acid derivatives. | ||
(S)-56a–j were reacted with β-carboline 57 and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) to get 58a–j. Among these, 58g reached 99% yield in 20 hours but, 58e reached 95% yield in 4 hours.
58a–j was reacted with 2,2,2-trichloroethylcarbonyl chloride (2 eq.), and tributyl(prop-2-enyl)stannane (allyltributyltin, 3 eq.) as a nucleophile in DCM at −40 °C for 24 hours to produce 59a–j. Among these, 59d,f,i were found in quantitative yields; and 59e,b,g,c in good yields of 98, 95, 92, and 87%. NaOH in THF-H2O at RT for 1.5–2.5 hours were needed to remove the chiral auxiliary to give 2,2,2-trichloroethyl 1-prop-2-enyl-1,9-dihydropyrido[3,4-b]indole-2-carboxylate 60.
For 59a–e, which had alkyl N-protecting groups, (S)-configuration product (1S)-2,2,2-trichloroethyl 1-prop-2-enyl-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-60 were found, and for 59f–j, which had acyl and sulfonyl N-protecting groups, (R)-configuration product (1R)-2,2,2-trichloroethyl 1-prop-2-enyl-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1R)-60 were noticed. Among 59a–e, the bulkier the N-protecting groups, the more ee was seen in (1S)-60 (e.g., highest 91% ee in (S)-60 for 59e having anthracene-9-ylmethyl substituent; lowest 7% ee in (1S)-60 for 59a having methyl substituent). Among 59f–j, the bulkiness of substituents did not affect ee of (1R)-60 that much, only lowered the % yields (Scheme 16).
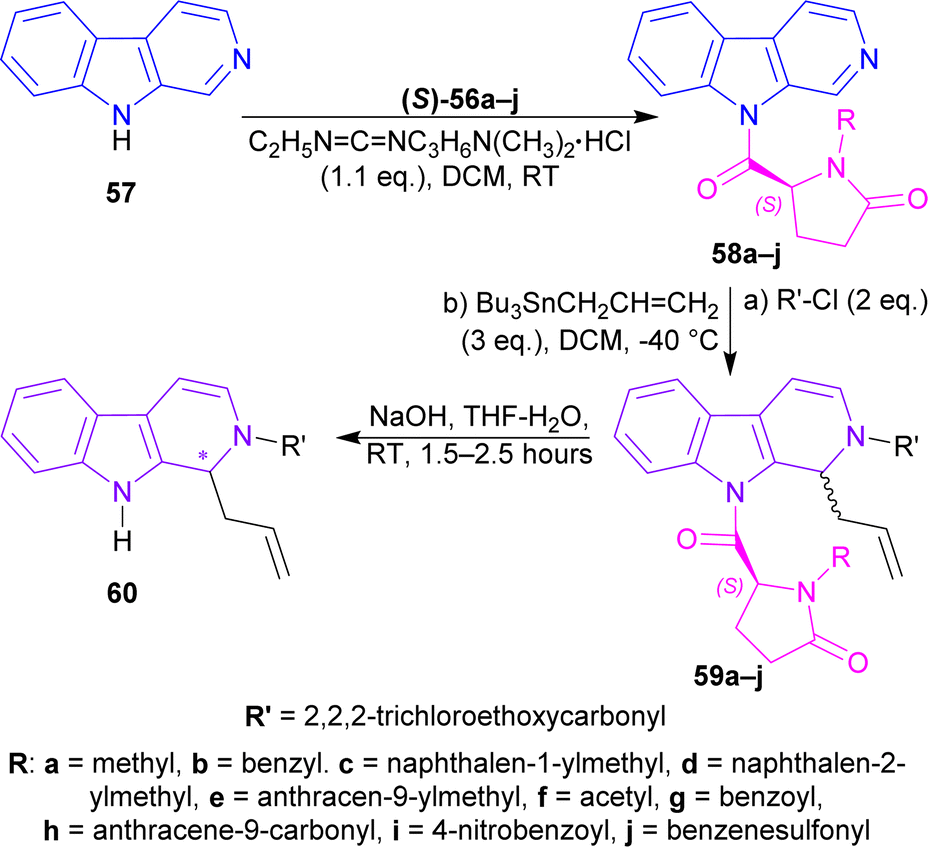 | ||
| Scheme 16 N9 addition of chiral auxiliary to the β-carboline, C1 addition of allyltributyltin and N2 protecting, then ultimately removal of the chiral auxiliary. | ||
After that, silyl enol ethers 61a–e76 were used as nucleophiles instead of allyltributyltin. At 0 °C, reaction with (5S)-1-(anthracene-9-ylmethyl)-5-(pyrido[3,4-b]indole-9-carbonyl)pyrrolidin-2-one 58e and 61a reached only 40% yield with 79% ee of 2,2,2-trichloroethyl (1S)-1-(2-oxopropyl)-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-62a in 24 hours; 61b needed 2.5 hours to reach 79% yield with 86% ee of 2,2,2-trichloroethyl (1S)-1-phenacyl-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-62b; 61c gained quantitative yield only at 30 minutes with 82% ee of 2,2,2-trichloroethyl (1S)-1-(1-methoxy-2-methyl-1-oxopropan-2-yl)-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-62c. Reducing the temperature to −40 °C reduced yields to 81 and 83% with 61d and 61e respectively even at reaction times of 12 and 19 hours but, increased ee slightly to 88 and 87% of 2,2,2-trichloroethyl (1S)-1-(2-oxo-2-phenylmethoxyethyl)-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-62d and 2,2,2-trichloroethyl (1S)-1-(2-benzylsulfanyl-2-oxoethyl)-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1S)-62e respectively.
61d was chosen to react with 58f,g,i at −78 °C for 40 hours to produce 2,2,2-trichloroethyl (1R)-1-(2-oxo-2-phenylmethoxyethyl)-1,9-dihydropyrido[3,4-b]indole-2-carboxylate (1R)-62d (75–76% ee). Less steric hindered acetyl-substitution 58f had a 93% yield of (1R)-62d but more steric hindered benzoyl-substitution 58g and 4-nitrobenzoyl-substitution 58i both had quantitative yields of (1R)-62d (Scheme 17).
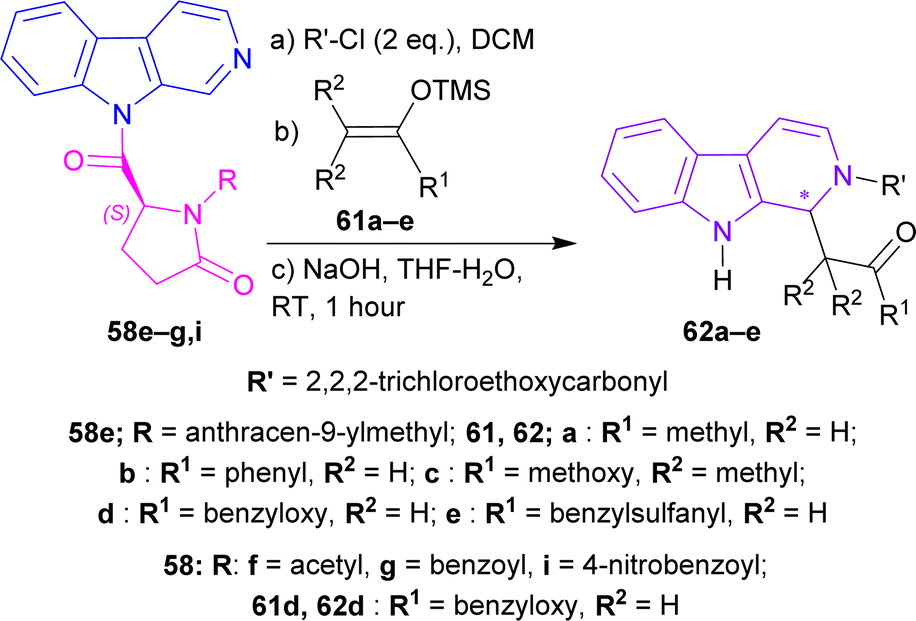 | ||
| Scheme 17 N2 protecting, C1 addition of silyl enol ether, and then ultimately removal of the chiral auxiliary. | ||
2,2,2-Trichloroethyl (1S)-1-(2-oxo-2-phenylmethoxyethyl)-THβC-2-carboxylate (1S)-62d was reduced with Et3SiH in DCM at RT for 15 minutes gave rise to (1S)-63 which was again reduced and N-2-deprotected with Zn-acetic acid (AcOH) to produce 92% yield of methyl 2-[(1S)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl]acetate (1S)-64 (88% ee calculated according to Tietze et al.77) (Scheme 18).
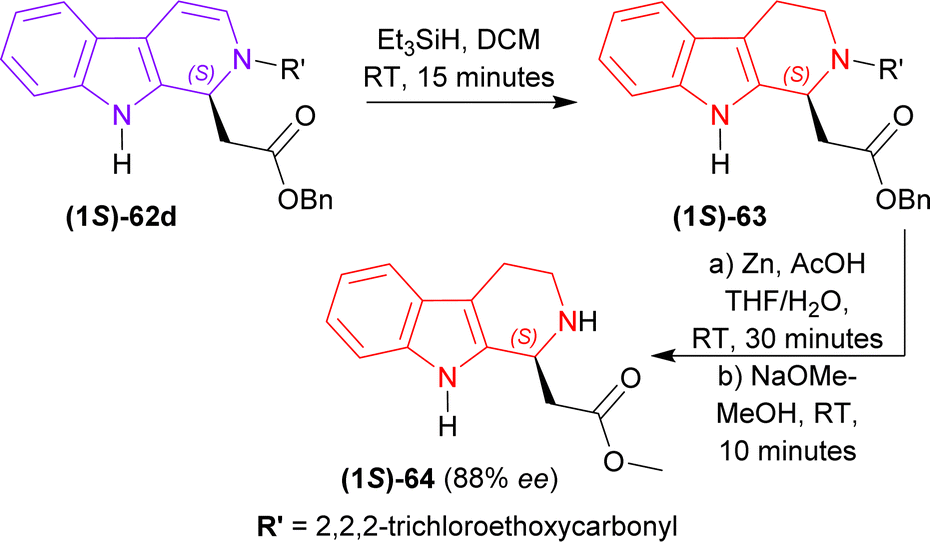 | ||
| Scheme 18 Reducing to THβC and removing N9 protection. | ||
So, (5S)-1-substituted-5-(β-carboline-9-carbonyl)pyrrolidin-2-one having alkyl N9-protecting groups as the chiral auxiliary ultimately produced (1S)-1-substituted-THβC.
Tryptamine 13a with diethyl oxalate produced 65. (1R)-1-Phenylethanamine (1R)-66a79 and (1R)-1-naphthalen-1-ylethanamine (1R)-66b80 with 65, produced (R)-67a and (R)-67b. Bischler–Napieralski cyclization of (R)-67a,b with POCl3 in refluxing DCM gave (R)-68a and (R)-68b.
After that, several reducing agents were experimented with e.g., sodium borohydride (NaBH4), sodium triacetoxy-borohydride (NaBH(AcO)3), sodium tris(2-methylpropanoyloxy)borohydride (NaBH(i-PrCOO)3), sodium tris(2,2-dimethylpropanoyloxy)borohydride (NaBH(t-BuCOO)3) in ethanol to produce dr of 62:38–78:22 for (1R)-N-[(1R)-1-phenylethyl]-THβC-1-carboxamide (R,R)-69a and (S,R)-69a from (R)-68a; and dr of 64:36–83:17 for (1R)-N-[(1R)-1-naphthalen-1-ylethyl]-THβC-1-carboxamide (R,R)-69b and (S,R)-69b from (R)-68b (Scheme 19).
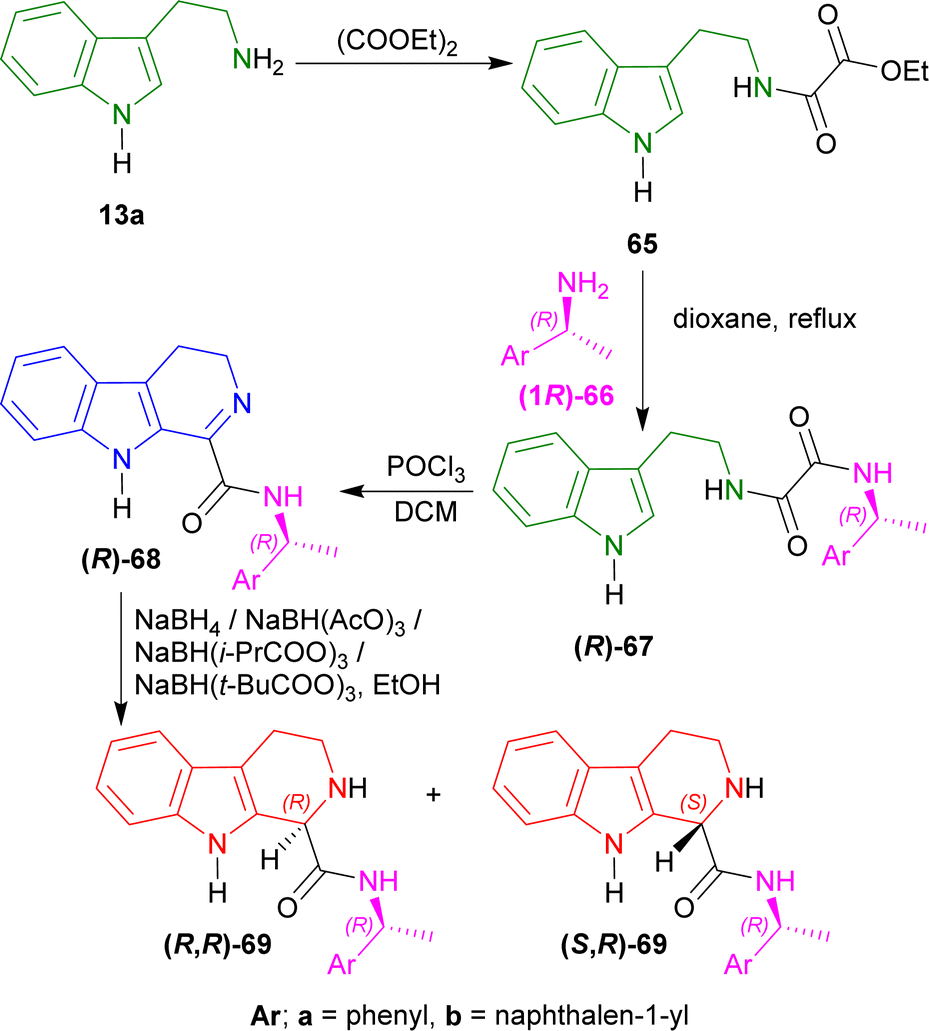 | ||
| Scheme 19 Addition of chiral auxiliary, Bischler–Napieralski cyclization, and reduction to THβC. | ||
So, the (R)-configured chiral auxiliary (1R)-1-phenylethanamine and (1R)-1-naphthalen-1-ylethanamine predominantly produced (1R)-1-substituted-THβCs.
2-(1-Benzylindol-3-yl)ethanol 70a and 2-(1-methylindol-3-yl)ethanol 70b was reacted with POCl3 in DMF at 60 °C for 12 hours to produce 71a and 71b. Then 71a,b was refluxed with Ellman's sulfinamide or (R)-2-methylpropane-2-sulfinamide as chiral auxiliary,82–85 and Ti(OEt)4 in DCM for 24 hours to have 78% yield of (R,E)-72a and 79% yield of (R,E)-72b.86
Then it was experimented with various Grignard reagents 73a–d e.g., MeMgI, PrMgBr, allyl magnesium bromide, and EtMgCl in DCM at −78 °C to have 77–84% yield and 84 to >98% de of (S,R)-74aa, (S,R)-74ab, (S,R)-74ac, (S,R)-74ba, (S,R)-74bd; among which (S,R)-74ac had the highest de of >98%.87 Base-catalyzed cyclization88 of (S,R)-74aa–ac,ba,bd with NaH in DMF at 0 °C to RT for 4 hours gave rise to 72–85% yield of (S,R)-75aa, (S,R)-75ab, (S,R)-75ac, (S,R)-75ba, (S,R)-75bd; among which (S,R)-75ac had the highest yield of 85%.
(S,R)-75aa,ab with Na in liquid NH3 (ref. 89) at −40 °C for 20 minutes removed the chiral auxiliary, N9-benzyl and produced 70% yield of (−)-tetrahydroharman (1S)-76a′a and 68% yield of (−)-komaroidine (1S)-76a′b. (S,R)-75ba,bd with 4 M HCl in dioxane in MeOH at 0 °C to RT for 30 minutes removed the chiral auxiliary and produced 87% yield of (+)-N-methyltetrahydroharman (1S)-76ba and 83% yield of (1S)-1-ethyl-9-methyl-1,2,3,4-tetrahydropyrido[3,4-b]indole (1S)-76bd.
(1S)-76a′b with acetyl chloride and Et3N in DCM at RT for 2 hours produced an 87% yield of (+)-N-acetylkomaroidine (1S)-77a′b (Scheme 20).
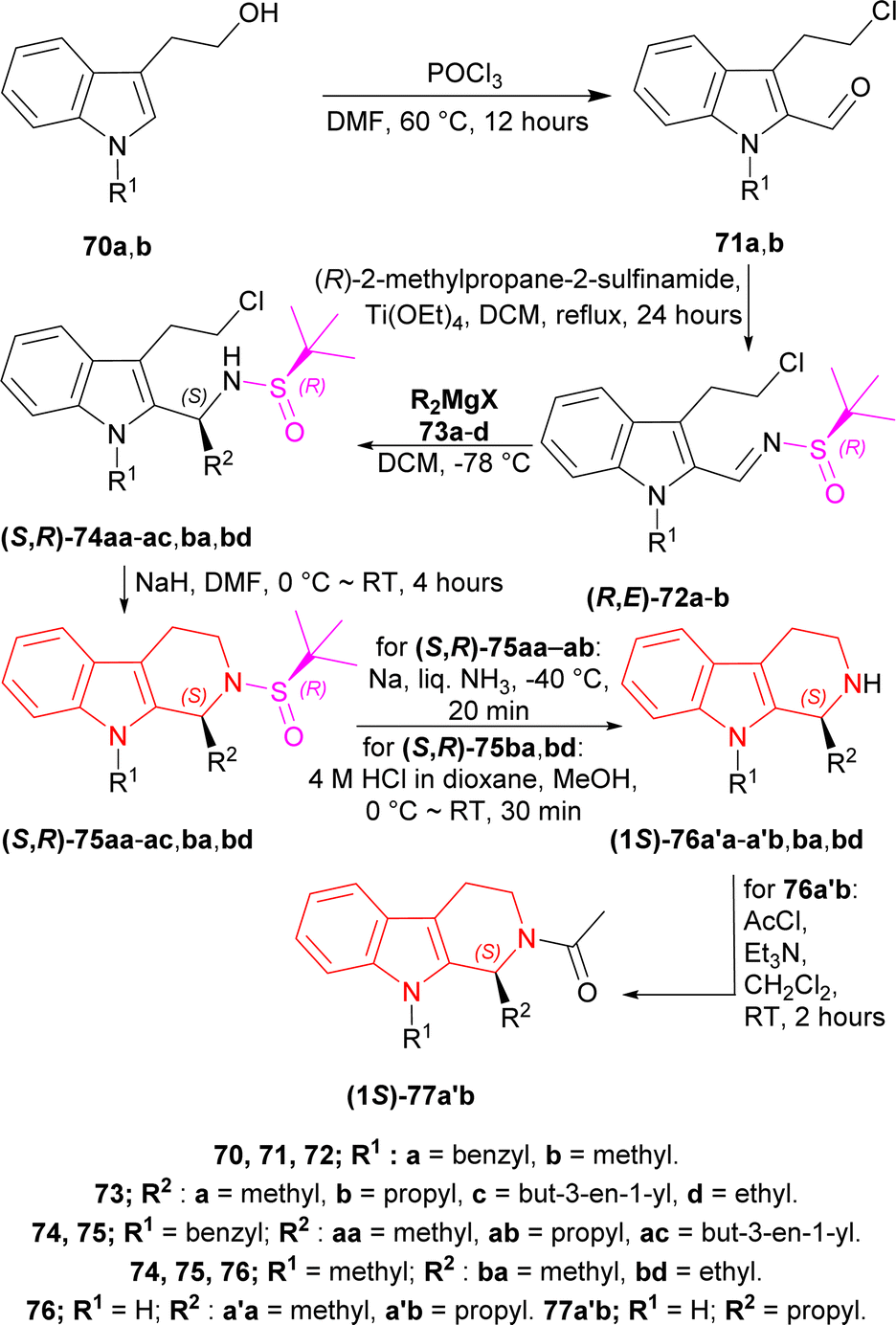 | ||
| Scheme 20 Addition of chiral auxiliary, base-catalyzed cyclization to THβC and removal of the chiral auxiliary. | ||
(1S)-9-Benzyl-2-[(R)-tert-butylsulfinyl]-1-prop-2-enyl-THβC (S,R)-75ac with 4 M HCl in dioxane in MeOH solvent at 0 °C to RT for 30 minutes; tert-butyl (2-methylpropan-2-yl)oxycarbonyl carbonate (Boc2O) and Et3N in DCM at RT for 1 hour produced 88% yield of (1S)-78. Then (1S)-78 with BH3·DMS in THF at −25 °C for 3 hours; H2O2 in NaOH at RT for 24 hours gave 83% yield of (1S)-79. After that, (1S)-79 with methanesulfonyl chloride (MsCl) and Et3N in DCM at RT for 2 hours; TMSOTf and NaHCO3 in DCM at RT for 3 hours had 70% yield of (11bS)-80. Lastly, (11bS)-80 with Na in liquid NH3 at −40 °C for 20 minutes gave the ultimate product (−)-harmicine (11bS)-81 of 72% yield (Scheme 21).
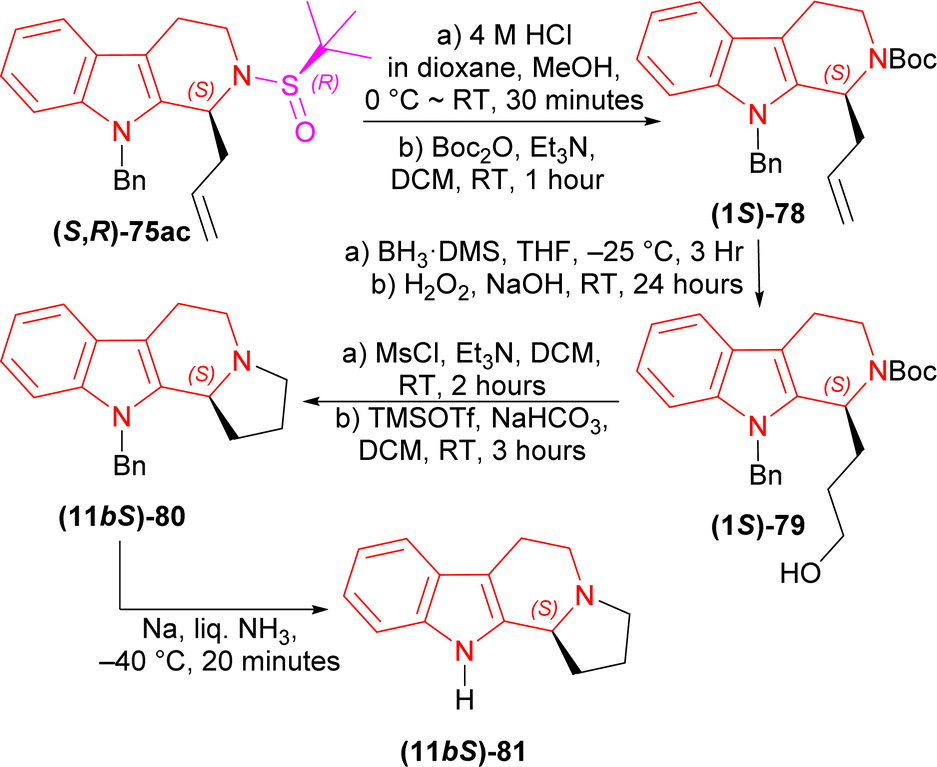 | ||
| Scheme 21 Removal of chiral auxiliary and protecting N-2, cyclization of the fourth ring, removal of N-9 protection. | ||
So, Ellman's sulfinamide as chiral auxiliary produced (1S)-1-substituted-THβCs e.g., (−)-tetrahydroharman, (−)-komaroidine, (+)-N-methyltetrahydroharman, (+)-N-acetylkomaroidine, and (−)-harmicine which all have various important pharmacological activities.12,90
Method 3. Asymmetric transfer hydrogenation reaction with chiral catalysts
The transfer hydrogenation reaction, which involves the addition of hydrogen to a molecule from a non-H2 source, is a versatile and effective approach for producing various hydrogenated compounds. This method is gaining popularity in hydrogenation research as an appealing alternative to direct hydrogenation. The key reasons for its growing interest include: (i) it eliminates the need for potentially hazardous pressurized H2 gas and complex experimental setups, (ii) the hydrogen donors used are typically affordable, easy to handle, and widely available, (iii) the main byproduct can often be recycled, and (iv) the catalysts involved are generally easy to obtain and not highly sensitive.91–101ATH emerged in the early 1980s. The first reports were of the Ru catalyzed ATH.93,102,103 ATH that used the late transition-metal catalysts has shown to be one of the most potent strategies for asymmetric reduction of diverse unsaturated substrates to create chiral chemicals.94,95,97,98,104–107
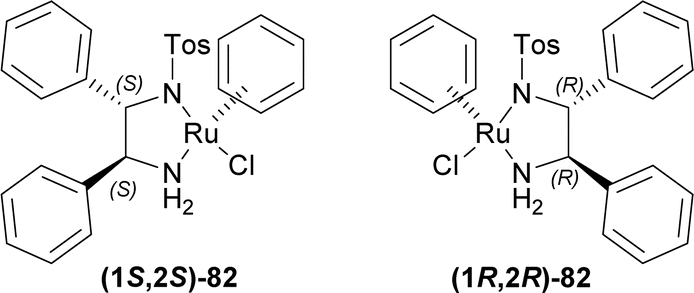 | ||
| Fig. 3 Chiral catalysts for ATH. | ||
Tryptamine 13a was reacted with acetic anhydride with Et3N, butyric acid in xylene, nonanoic acid in xylene, stearic acid in xylene, oleic acid, and arachidonic acid to produce the 83a–f.109 With P2O5 or POCl3, Bischler–Napieralski cyclization produces 84a–f which instantly underwent ATH34 with (1S,2S)-82 and (1R,2R)-82.77 All ATH products had >98% ee. For 84a–f, (1S,2S)-82 gave 70–85% yields of (1R)-1-substituted-THβCs (1R)-85a–f; and for 84a–e, (1R,2R)-82 gave 77–88% yields of (1S)-1-substituted-THβCs (1S)-85a–e. Highest yield of 88% was found for (1S)-1-propyl-THβC (1S)-85b and lowest yield of 70% for (1R)-1-[(4Z,7Z,10Z,13Z)-nonadeca-4,7,10,13-tetraenyl]-THβC (1R)-85f having highly sterically hindered substituents. Switching catalyst from (1S,2S)-82 to (1R,2R)-82 lowered the % yields of the products of (1S)-85a,c,d slightly by 2–4% from that of (1R)-85a,c,d. (1S)-85b had 9% more yield than (1R)-1-propyl-THβC (1R)-85b while (1S)-1-[(Z)-heptadec-8-enyl]-THβC (1S)-85e had 7% less yield than (1R)-1-[(Z)-heptadec-8-enyl]-THβC (1R)-85e (Scheme 22).
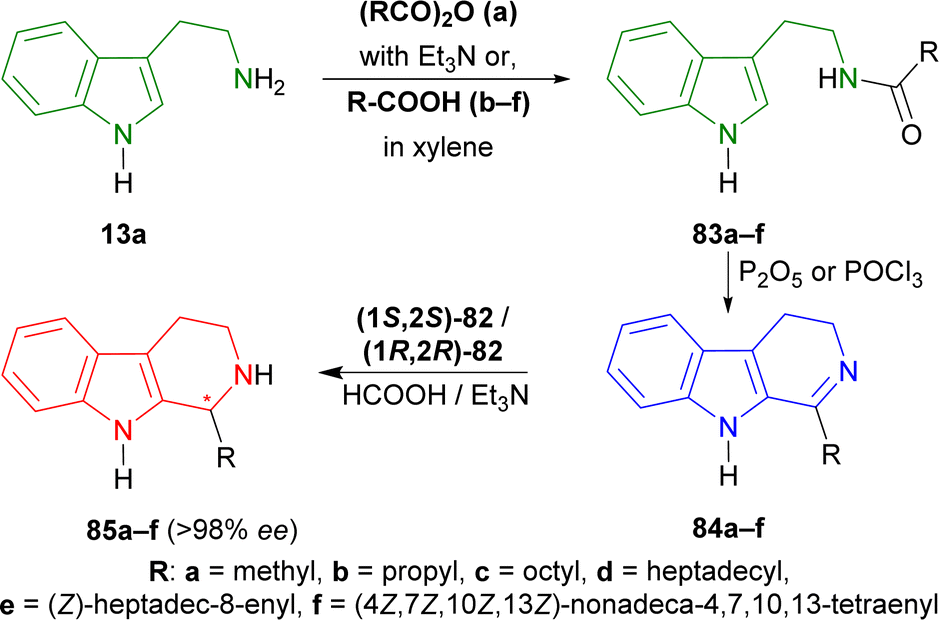 | ||
| Scheme 22 Synthesizing N-[2-(1H-indol-3-yl)ethyl]amides from tryptamine, then 1-substituted-DHβCs, and ultimately ATH to get 1-substituted-THβCs with chiral catalysts. | ||
So, (1S,2S)-82 chiral catalyst produced (1R)-1-substituted-THβCs and (1R,2R)-82 chiral catalyst produced (1S)-1-substituted-THβCs predominantly.
Oxolan-2-one (γ-butyrolactone) 86a1, and oxan-2-one (δ-valerolactone) 86a2 were treated with tryptamine 13a produced 87aa1 (ref. 111) (78% yield), and 87aa2 (ref. 112) (87% yield). Then Bischler–Napieralski cyclization in POCl3 gave iminium salts 88aa1 and 88aa2. Immediate ATH of 88aa1,2 with (1S,2S)-82 ultimately gave rise to (11bR)-2,3,5,6,11,11b-hexahydro-1H-indolizino[8,7-b]indole or (R)-harmicine (R)-89aa1 (81% yield, 79% ee) and (12bR)-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizine or (R)-desbromoarborescidine A (R)-89aa2 (84% yield, 90.5% ee) (Scheme 23).
 | ||
| Scheme 23 Synthesizing N-[2-(1H-indol-3-yl)ethyl]hydroxamides from tryptamine, then 1-substituted-DHβCs, and ultimately ATH to get 1-substituted-THβCs with chiral catalysts. | ||
So, (1S,2S)-82 chiral catalyst produced (1R)-1-substituted-THβCs predominantly.
4-Aminobutanoic acid 90 was treated with 2-benzofuran-1,3-dione (phthalic anhydride) 91 at 180 °C for 1 hour114 to give 92 which was turned into 93 with sulfonyl chloride at 80 °C for 30 minutes. This was reacted with 13a in DCM to get 82% yield of 94. It was reacted with POCl3 in refluxing acetone (MeCN) to give 85% yield of 95 via Bischler–Napieralski reaction. After that, ATH of 95 with (1S,2S)-82 (S:C ratio of 160:1) in 5:3 azeotropic solution of formic acid (HCOOH):Et3N34 afforded 92% yield of (1R)-96 (>98% ee).
(1R)-96 was reacted with hydrazine in ethanol at RT for 1 hour to remove the phthaloyl group which was readily subjected to tert-butyl N-[[(2-methylpropan-2-yl)oxycarbonylamino]-methylsulfanylmethylidene]carbamate in DMF at RT to get 53% yield of (1R)-97. At the last step, Boc group of (1R)-97 was removed by TFA in DCM at RT, and successive evaporation with methanolic HCl provided the final product HCl salt of (R)-trypargine (1R)-98 in quantitative yield. The isolated compound's analytical results were entirely consistent with what was previously published by Cesar et al.115 (Scheme 24).
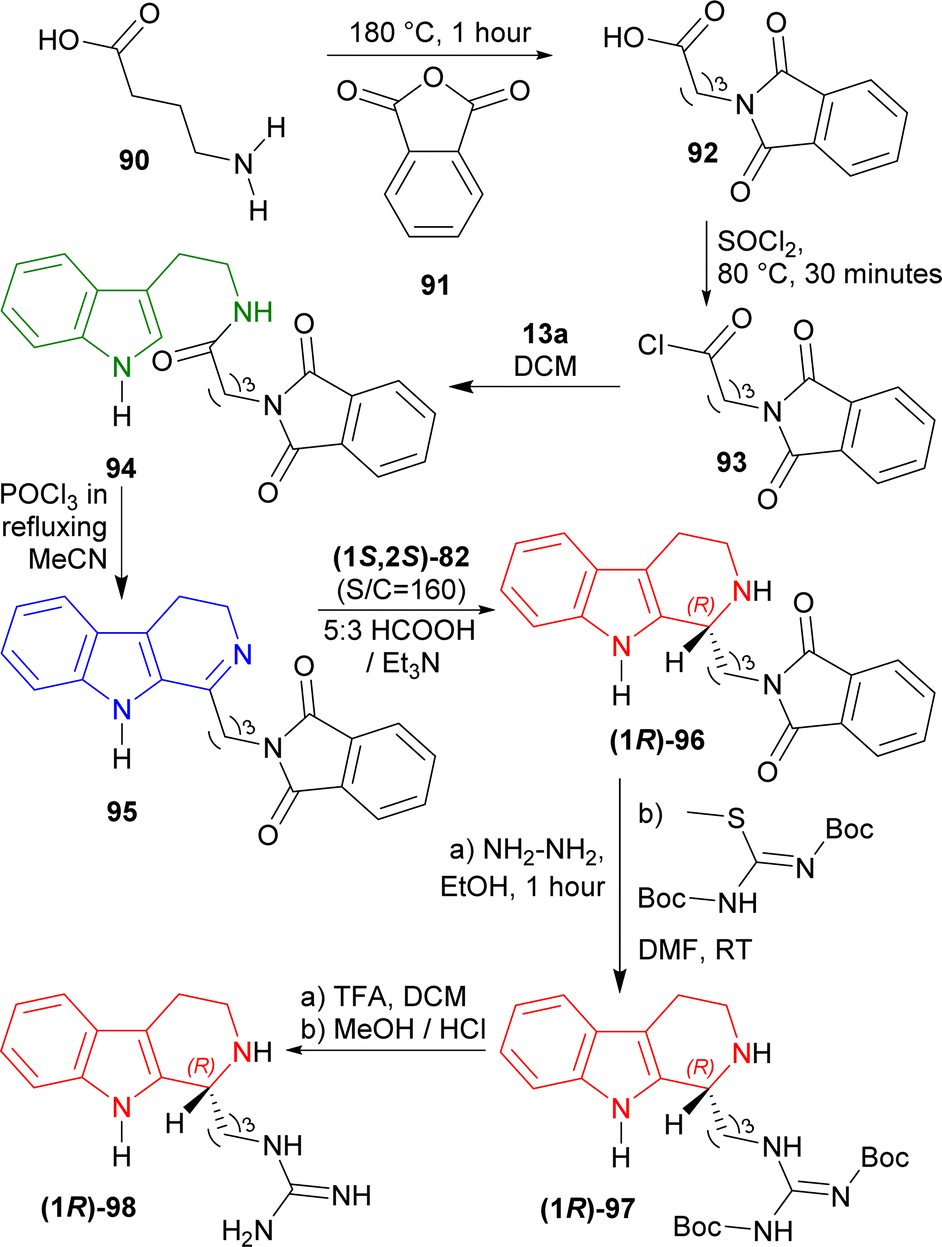 | ||
| Scheme 24 ATH with chiral catalysts to synthesize HCl salt of (R)-trypargine. | ||
So, (1S,2S)-82 chiral catalyst produced HCl salt of (R)-trypargine predominantly.
2-(5-Bromo-1H-indol-3-yl)ethanamine 99 (ref. 117) with (2S)-2-[9H-fluoren-9-ylmethoxycarbonyl(methyl)amino]-3-phenylpropanoic acid,1181,2-dichloroethane (EDC), and HOBt in DCM produced 100. Bischler–Napieralski cyclization119 of 100 in benzene gave rise to 101. The compound 101 was then treated with (1S,2S)-82 followed by 5:2 HCOOH/Et3N in DMF and N-2 was methylated with aq. HCHO, NaBH3CN, in CH3CN into (R,S)-102 with 89% yield (predominating (S,S)-102 by >10:1 dr). The Fmoc group was removed by 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine (DBU) in DCM to produce (1S)-1-[(1R)-6-bromo-2-methyl-1,3,4,9-tetrahydropyrido[3,4-b]indol-1-yl]-N-methyl-2-phenylethanamine (R,S)-103 (eudistomidin B) in 85% yield. But when (1R,2R)-82 was used, (1S)-1-[(1S)-6-bromo-2-methyl-1,3,4,9-tetrahydropyrido[3,4-b]indol-1-yl]-N-methyl-2-phenylethanamine (S,S)-103 (diastereomer of eudistomidin B) was found in 78% yield (Scheme 25).
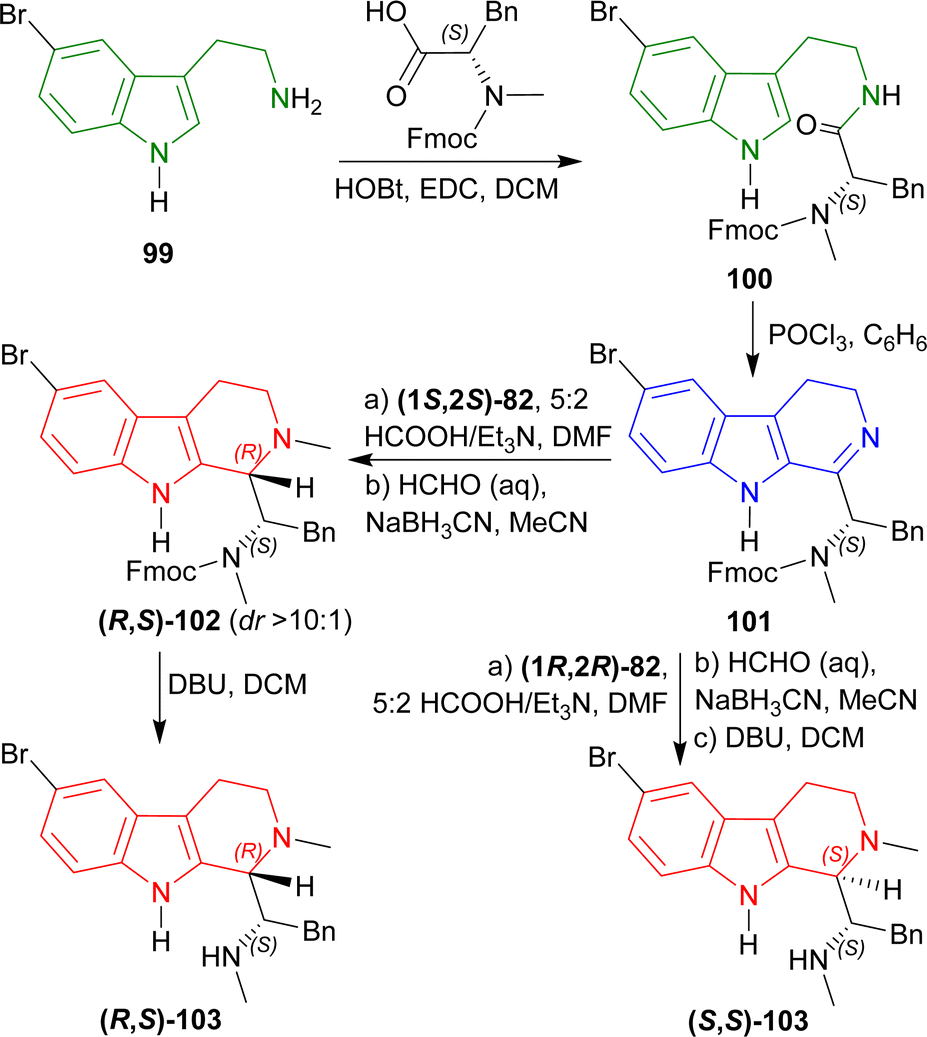 | ||
| Scheme 25 Asymmetric synthesis of eudistomidin B and it's diastereomer. | ||
So, (1S,2S)-82 chiral catalyst produced (1R)-1-substituted-THβC eudistomidin B; and (1R,2R)-82 chiral catalyst produced (1S)-1-substituted-THβC, the (S,S)-diastereomer of eudistomidin B.
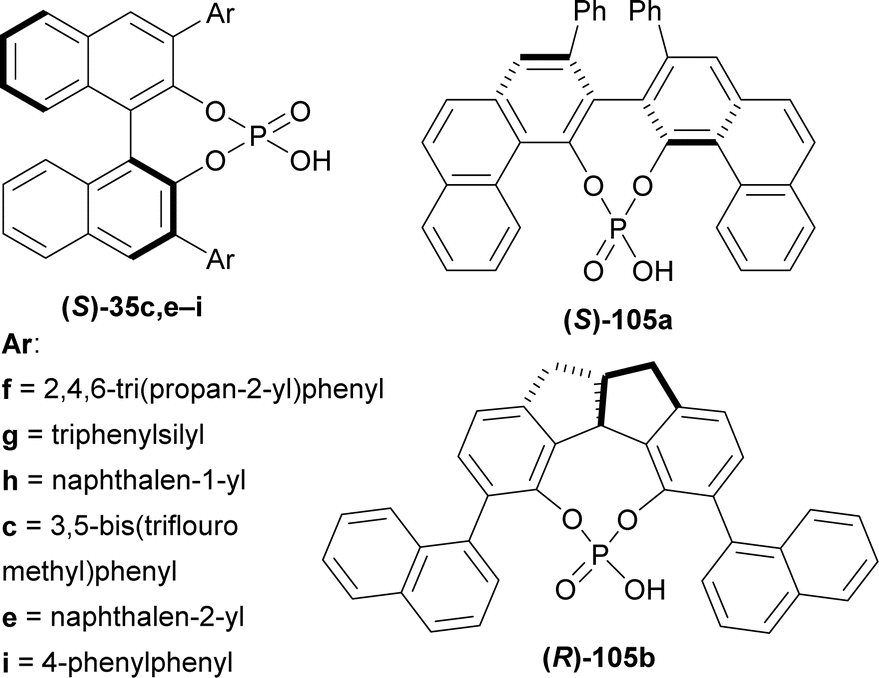 | ||
| Fig. 4 (S)-BINOL, VAPOL, and SPINOL derived chiral phosphoric acid. | ||
Tryptamine and its derivatives 13a–j were refluxed with phthalic anhydride 91 in toluene, and trifluoromethanesulfonic acid (TfOH) in DCM to give 2-hydroxy-10,20-diazapentacyclo[11.7.0.02,10.03,8.014,19]icosa-1(13),3,5,7,14,16,18-heptaen-9-one and its derivatives 104a–j in 39–63% yields (Scheme 26).121,122
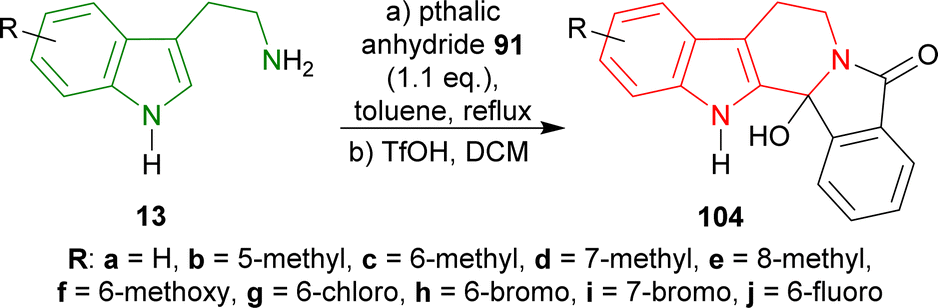 | ||
| Scheme 26 Synthesis of hydroxylactams from tryptamine and its derivatives. | ||
(S)-BINOL, VAPOL, and SPINOL-derived chiral phosphoric acid catalysts (S)-35c,e–i, (S)-105a, and (R)-105b123,124 (Fig. 4) respectively were tested for transfer hydrogenation reaction of 2-hydroxy-10,20-diazapentacyclo[11.7.0.02,10.03,8.014,19]icosa-1(13),3,5,7,14,16,18-heptaen-9-one 104a with a Hantzsch ester (diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate, 2 eq.) as the hydride source to give (2R)-10,20-diazapentacyclo[11.7.0.02,10.03,8.014,19]icosa-1(13),3,5,7,14,16,18-heptaen-9-one (R)-106a in DCM at RT. Among the catalysts, (S)-35g provided with highest ee of 52% with 88% yield of the product. After that, different solvents were tested among which dioxane had highest ee of 75% with 84% yield (Scheme 27).
 | ||
| Scheme 27 Screening of chiral phosphoric acid catalysts for the acid catalyzed ATH of hydroxylactam. | ||
Then, a different Hantzsch ester (ditert-butyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate, 2 eq.) was tested as hydride donor with the presence of (S)-35g that resulted in 91% yield and 80% ee of (R)-106a from 104a in 24 hours. But reactions using 3, 4, or 5 Å meshes and magnesium sulphate did not increase the yield or ee (Scheme 28).
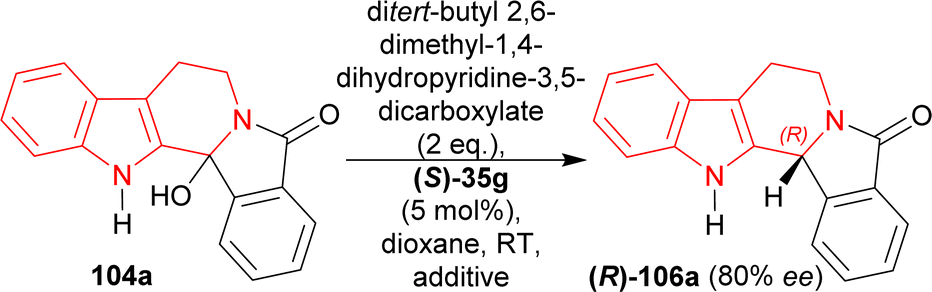 | ||
| Scheme 28 Screening of additive for the acid catalyzed ATH of hydroxylactam. | ||
Under the optimized conditions, 104b–j were converted to (R)-106b–j with (S)-35g; among which (R)-106b–f containing an electron-rich group had 68–93% yields with 77–85% ee, and (R)-106g–j containing an electron-poor group had 90–94% yields with 82–90% ee (Scheme 29).
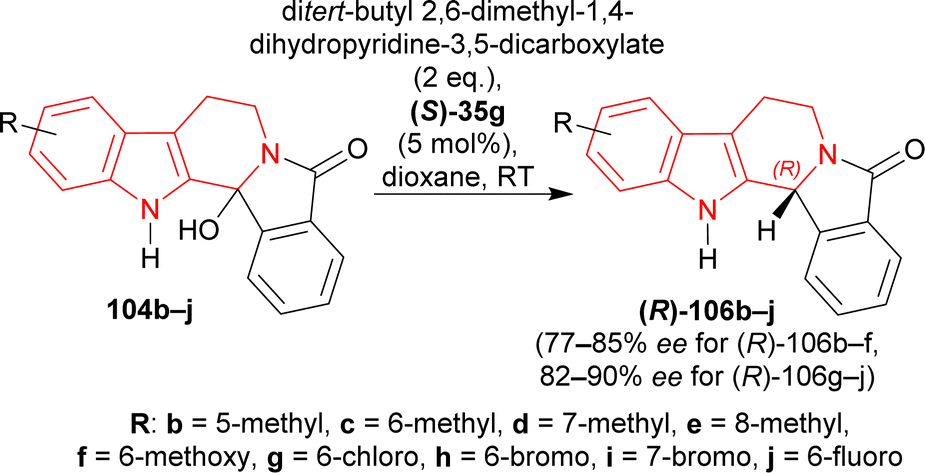 | ||
| Scheme 29 Acid catalyzed ATH reaction of hydroxylactams by (S)-35g under the optimized conditions. | ||
So, (S)-BINOL-derived chiral phosphoric acid produced (1R)-1-substituted-THβCs predominantly.
1° amine catalyst e.g., hexan-1-amine 107, (2S)-2-[[(1R,2R)-2-aminocyclohexyl]carbamothioylamino]-N-benzhydryl-N,3,3-trimethylbutanamide (S,R,R)-108 or (2R)-2-[[(1S,2S)-2-aminocyclohexyl]carbamothioylamino]-N-benzhydryl-N,3,3-trimethylbutanamide (R,S,S)-108 (Fig. 5) were used to couple 7-methoxy-9-(4-methylphenyl)sulfonyl-3,4-dihydropyrido[3,4-b]indole (7-methoxy-9-tosyl-DHβC) 15 with (S,S)-109 (ref. 125) (1.2 eq.)126,127 around acetic acid in toluene at 23 °C. 100 mol% of 107 had 90% conversion rate after 9 Days with 1:1 dr of the (S,bS,S,S)-110 and (R,bR,S,S)-110. 20 mol% of (S,R,R)-108 (ref. 125) produced >99% conversion after 6 days with 11.5:1:1.8 dr of the (S,bS,S,S)-110, (R,bR,S,S)-110, and (R,bS,S,S)-110; ultimately yielding 76% of the desired (S,bS,S,S)-110. Here, the use of 20 mol% (R,S,S)-108 produced greater dr for (R,bR,S,S)-110. The cleavage of the primary TBS ether of (S,bS,S,S)-110 was done in two steps by pyridine-buffered HF in pyridine at 0 °C to 23 °C; and then oxidation with the Dess–Martin periodinane in DCM producing (S,bS,S,S)-111. Piperidine and p-TsOH was treated with (S,bS,S,S)-111 to produce an intramolecular enamine aldol (S,S,S,S,S,R)-112. Pinnick oxidation followed by esterification with diazomethane of (S,S,S,S,S,R)-112 provided (S,S,S,S,S,R)-113. Trifluoroacetylation of (S,S,S,S,S,R)-113 and subsequent elimination by DBU gave (S,R,S,S)-114. Hydrogenation with H2 (1 atm) in DCM and [Ir(COD)(PCy3)(py)]BArF (ref. 128 and 129) gave 6:1 dr of (S,R,S,S,R,R)-115 (44% isolated yield) and (S,R,S,S,R,S)-115. Treating (S,R,S,S,R,R)-115 with TfOH, sodium–mercury amalgam, and 3,4,5-trimethoxy benzoyl chloride130 resulted in cleavage of PMB ether (86% yield), cleavage of tosyl protective group (69% yield), and esterification (90% yield) respectively; which ultimately gave (+)-reserpine 10 (Scheme 30).
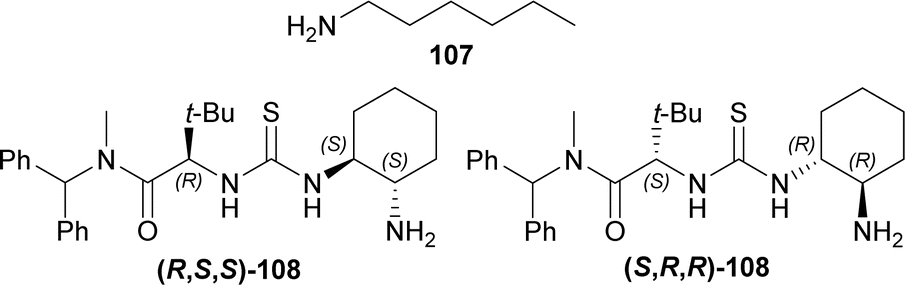 | ||
| Fig. 5 1° amine catalysts for coupling of 7-methoxy-9-tosyl-DHβC. | ||
 | ||
| Scheme 30 Total synthesis of (+)-reserpine by primary amine catalysts and [Ir(COD)(PCy3)(py)]BArF. | ||
So, 1° amine catalysts and [Ir(COD)(PCy3)(py)]BArF created (1S) and (19R,20R)-chiral centers predominantly to produce (+)-reserpine ultimately.
Method 4. Asymmetric addition reaction
(S)-Proline catalyzed asymmetric addition reactions131,132 are getting popularized day-by-day. In this method, unsaturated C1 and N2 get saturated and 1-substituted-THβCs are produced with the help of a ketone. Besides that, cycloaddition133 is also being used highly in synthetic organic chemistry,134,135 specially [3 + 3] cycloaddition to produce heterocyclic compounds.136–1409-Tosyl-DHβC 16 (ref. 141) was reacted with 20% (v/v) of MeCN, (S)-proline (30 mol%) in DCM, MeCN, or DMSO at RT for 1.5–2 hours to produce 1-[(1R)-9-(4-methylphenyl)sulfonyl-1,2,3,4-tetrahydropyrido[3,4-b]indol-1-yl]propan-2-one (1R)-116a in good to quantitative yield but ee was very low (5–34%). After adding 10 eq. of water in each solvent, ee increased significantly (67–80%) but reaction time also increased (2–3.5 hours). For DCM, increasing water to 50 eq. did not help at all (trace yield). For MeCN, increasing water to 50 eq. increased ee only 2% (with quantitative yield), though 100 eq. of water decreased both the yield and ee. Lastly for DMSO, increasing water gradually to 50, 100, and 150 eq. increased ee to 80, 86, and 87%. So, DMSO was chosen as the solvent.
Then at −2 °C, the lowest temperature at which the solvent remained liquid, 50 and 100 eq. of water produced similar ee (92–93%) with increasing yields (91 and 99% respectively). Decreasing (S)-Proline to 3 mol% did not decrease the yield, but increasing water from 2 to 10 eq. at RT increased ee from 4 to 60%; and increasing water to 50 eq. at −2 °C required 23 hours to get 99% yield with 94% ee (Scheme 31).
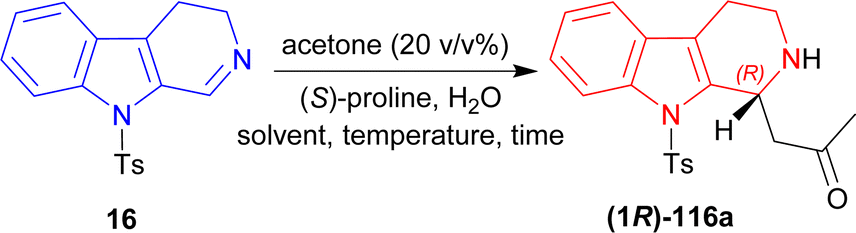 | ||
| Scheme 31 Screening of solvent, temperature, and time. | ||
Then butan-2-one, pentan-2-one, and 4-(2-methylpropoxy)butan-2-one was used instead of MeCN with 50 mol% of (S)-proline. Without water, the products had low ee of 7–28% with 57–78% yields at RT for reaction times of 4–26 hours. With 10 eq. of water, 1-[(1R)-9-(4-methylphenyl)sulfonyl-1,2,3,4-tetrahydropyrido[3,4-b]indol-1-yl]butan-2-one (1R)-116b and 1-[(1R)-9-(4-methylphenyl)sulfonyl-1,2,3,4-tetrahydropyrido[3,4-b]indol-1-yl]-4-(2-methylpropoxy)butan-2-one (1R)-116d had 51–80% ee with 65–81% yields at RT after 8–20 hours of reaction. For 50 eq. of water, products had 75–88% ee with 51–81% yields at RT after 7–20 hours reaction times; at −2 °C, (1R)-116b and 1-[(1R)-9-(4-methylphenyl)sulfonyl-1,2,3,4-tetrahydropyrido[3,4-b]indol-1-yl]pentan-2-one (1R)-116c had 89–92% ee with 76–85% yields after reacting 30–48 hours.
When 5 mol% of (S)-proline was used with 50 eq. of water at RT, (1R)-116b and (1R)-116c required 36–72 hours to reach 85% ee with 86–98% yield; at −2 °C, (1R)-116b and (1R)-116c required 120 hours to reach 66–81% ee with 91–92% yield (Scheme 32).
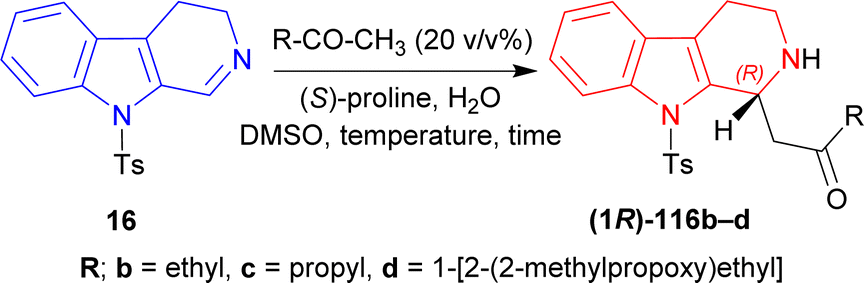 | ||
| Scheme 32 Screening of ketones. | ||
Then, 3 eq. of but-3-en-2-one was reacted with 16 in dry DMSO with (S)-proline at RT for 7 days to give (12bR)-116e (76% yield, 92% ee). Then (12bR)-116e was refluxed with 6 eq. of tetrabutylazanium;fluoride (TBAF) in dry THF for 1.5 hours to yield 74% (12bR)-3,4,6,7,12,12b-hexahydro-1H-indolo[2,3-a]quinolizin-2-one (R)-17 (ref. 142) (92% ee) which has been used as a precursor for the synthesis of yohimbine143 and deserpidine144 (Scheme 33).
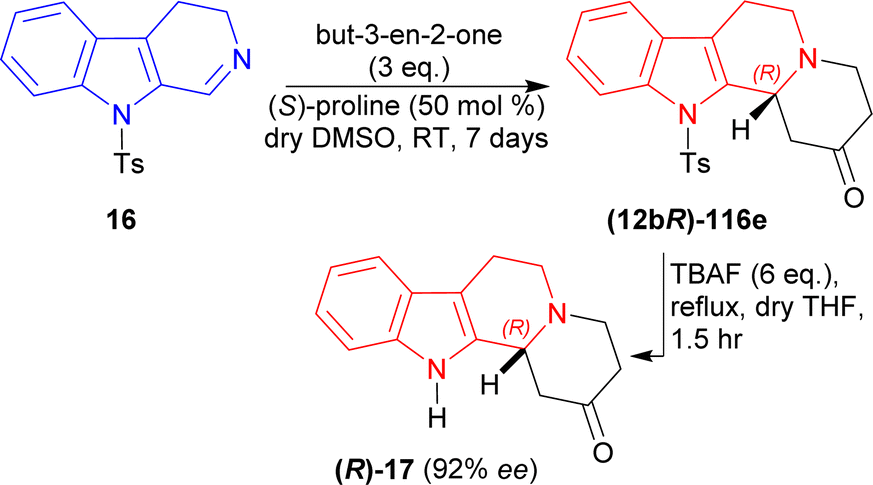 | ||
| Scheme 33 Synthesis of the precursor of yohimbine and deserpidine. | ||
So, but-3-en-2-one was asymmetrically added to 9-tosyl-DHβC by (S)-proline catalysis to synthesize a (1R)-1-substituted-THβC, the precursor of yohimbine and deserpidine.
16 was reacted with 30 eq. of 1-(cyclohexen-1-yl)ethenone, 50 mol% of (S)-proline in DMSO at RT for 12 day to produce 91% yield and 96% ee of (1R,14S,19R)-3-(4-methylphenyl)sulfonyl-3,13-diazapentacyclo[11.8.0.02,10.04,9.014,19]henicosa-2(10),4,6,8-tetraen-20-one (1R,14S,19R)-116f (Scheme 34).
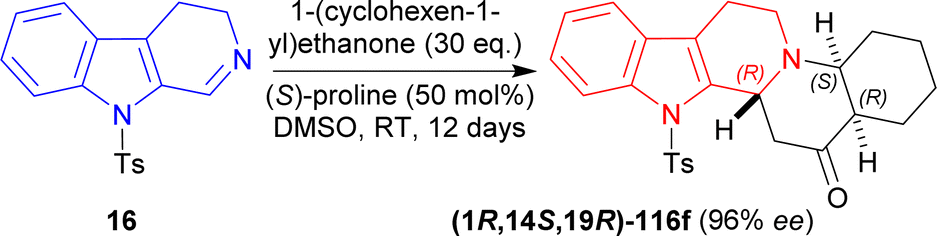 | ||
| Scheme 34 Generating three chiral centers in a single step (S)-proline catalyzed asymmetric addition reaction. | ||
16 was reacted with 30 eq. of 3-methylidenepentan-2-one,145 50 mol% of (S)-proline in DMSO at RT for 7 days to produce (3R,12bR)-116g (85% yield, 99% ee). After that, (3R,12bR)-116g was reacted with a Wittig reagent (sodium;methyl 2-dimethoxyphosphorylacetate) in benzene to have (2E,3S,12bR)-116h in very much higher ratio than its Z isomer (E:Z = 20:1). Then reflux with 5 eq. of Red-Al in DCM for 2 hours reduced (2E,3R,12bR)-116h to (2E,3S,12bR)-116i (63% yield). Lastly hydrogenation of (2E,3S,12bR)-116i with H2 in presence of 20 mol% Pd–C in methanol at RT for 13 hours yielded 74% (38% total yield) of 2-[(2S,3S,12bR)-3-ethyl-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl]ethanol (2S,3S,12bR)-116j (enantiomer of dihydrocorynantheol) (Scheme 35).
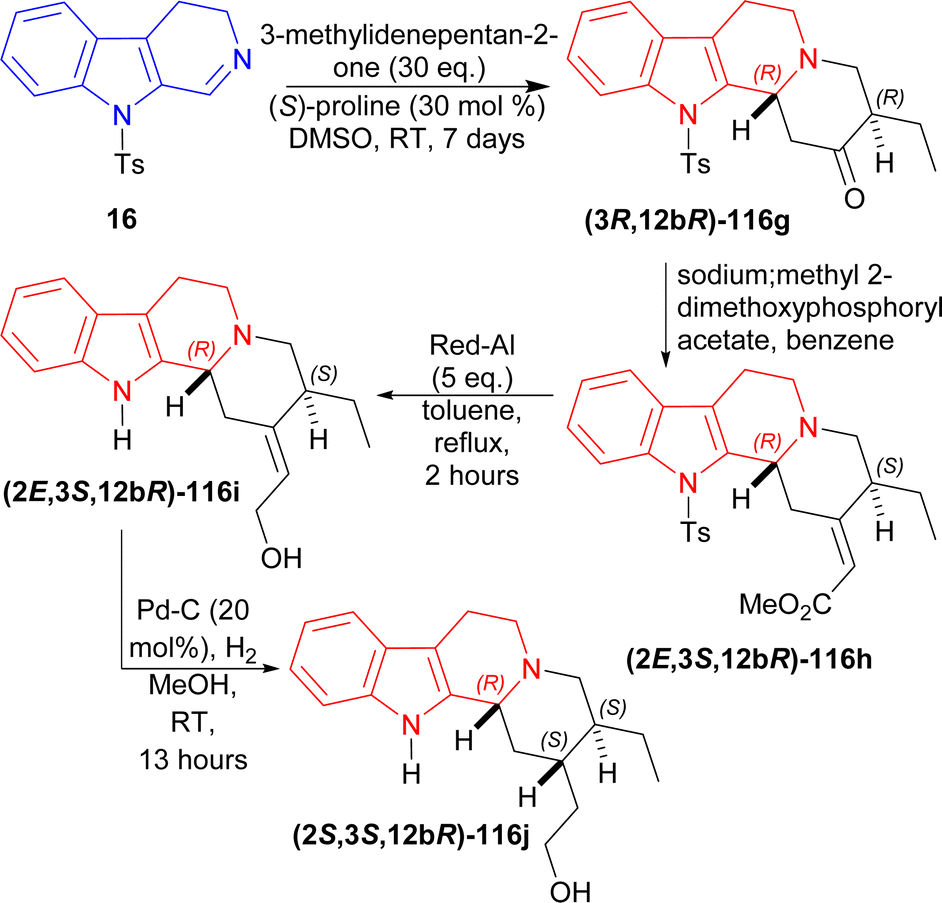 | ||
| Scheme 35 Synthesizing enantiomer of dihydrocorynantheol by (S)-proline catalyzed asymmetric addition reaction in four steps. | ||
So, 1-(cyclohexen-1-yl)ethenone and 3-methylidenepentan-2-one were asymmetrically added to 9-tosyl-DHβC by (S)-proline catalysis to synthesize (1R)-1-substituted-THβCs in one step and four steps respectively.
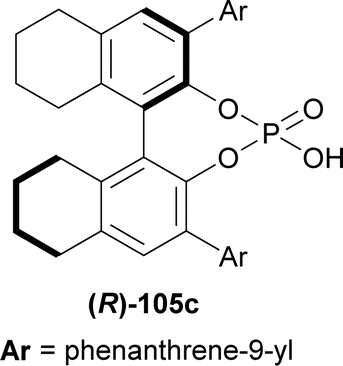 | ||
| Fig. 6 (R)-H8-BINOL derived chiral phosphoric acid. | ||
117 was stirred with 1.2 eq. 118 and 10 mol% (R)-105c for 5 hours in toluene at 0 °C to give catalytic asymmetric (3 + 3) cycloaddition product (6R,13R)-6-(2-methoxyphenyl)-12-methyl-6-phenyl-13-(2-methylphenyl)-6,13-dihydro-5H-pyrido[1,2-a:5,4-b′]diindole (R,R)-119 in 57% yield and 96% ee (Scheme 36).
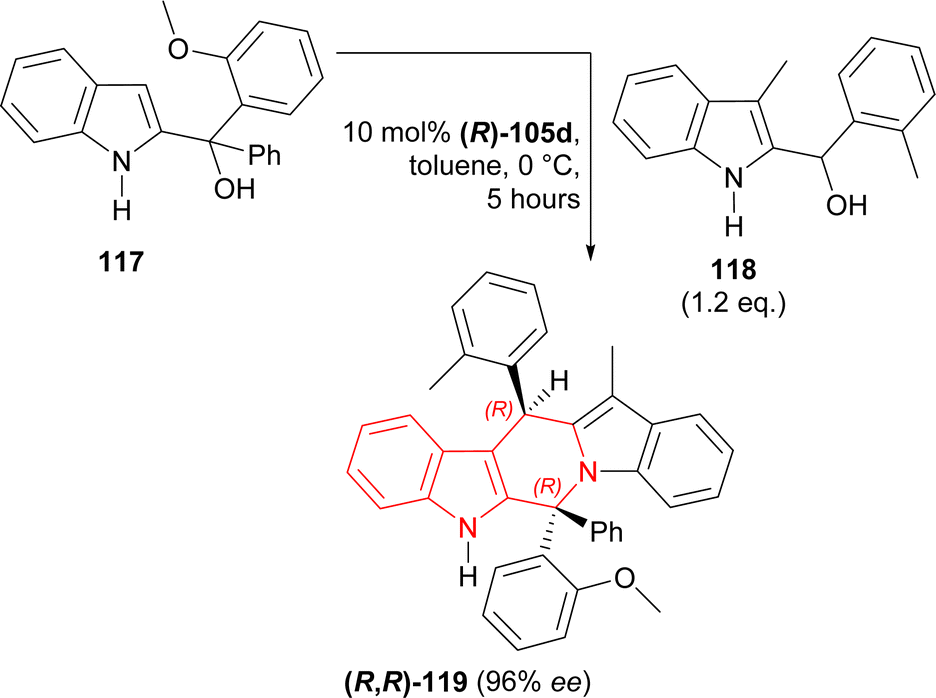 | ||
| Scheme 36 Catalytic asymmetric (3 + 3) cycloaddition of two different 2-indolylmethanols. | ||
So, (R)-configured H8-BINOL derived catalyst gave (R,R)-configured catalytic asymmetric (3 + 3) cycloaddition product.
Method 5. Enzymatic catalysis
Biocatalysis have already been employed in a variety of synthesis methods throughout the last few decades.147,148 Enzymes are proteins that activate any reaction process by binding to a particular location on the substrate. They are active at mild reaction conditions (pH, temperature, and reaction media e.g., water). These multifunctional catalysts enable several complex chemical processes to be performed in very mild conditions while maintaining excellent activity, selectivity, and specificity.149,150 For these reasons, enzymes are essential for the production of chiral building blocks, enantiopure medicines, and pharmaceuticals.150–154Then, six different variants of AoIRED were used to reduce 120c. Only AoIRED N241A produced (1R)-7-methoxy-1-methyl-THβC (1R)-121c with 96% conversion and 60% ee. AoIRED N171D, AoIRED Y179F, and AoIRED Y179A produced 99% ee of (1S)-121c with low conversion of 6, 9, and 10% respectively. AoIRED WT and AoIRED N171A slightly increased % conversion to 15 and 18 but ee decreased to 62 and 71 respectively (Scheme 37).
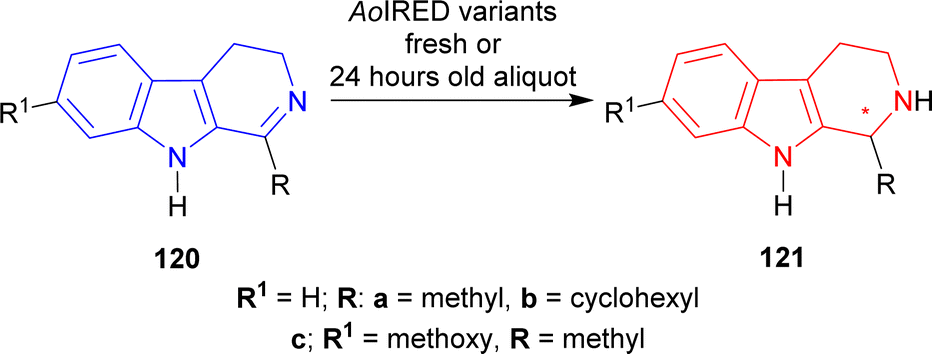 | ||
| Scheme 37 Reduction of 1-substituted-DHβC with fresh or 24 hours old aliquot of AoIRED variants. | ||
So, freshly purified AoIRED and AoIRED N241A produced (1R)-1-substituted-THβCs; 24 h old aliquot of AoIRED and AoIRED N171D, AoIRED Y179F, AoIRED Y179A, AoIRED WT, AoIRED N171A produced (1S)-1-substituted-THβCs predominantly.
Among the Y-type IREDs, IRED-J (UniProt: D2PR38, collected from Kribbella flavida DSM 17836),160 IRED-K (UniProt: D2AWI4, collected from Streptosporangium roseum DSM 43201), IRED-L (UniProt: K0F8R0, collected from Nocardia brasiliensis ATCC 700358), and IRED-M (UniProt: K0K4C6, collected from Saccharothrix espanaensis ATCC 51144) produced (1S)-1-methyl-THβC (1S)-121a of 91–92% conversion rate (96 to >99% ee) and (1S)-7-methoxy-1-methyl-THβC (1S)-121c of 95–88% conversion rate (92 to >99% ee). IRED-I (from Streptomyces sp. GF3546, UniProt: M4ZS15)157 produced >99% ee for both products but (1S)-121a had 90% conversion while (1S)-121c had only 9% conversion; and IRED-N (from Bacillus cereus, UniProt: J7YM26)161 showed very little conversion (5 and 2% respectively) for both.
The D-type IREDs did not show any good activity at all for 120c. For 120a, IRED-A (UniProt: M4ZRJ3, collected from Streptomyces sp. GF3587)156 and IRED-G (UniProt: L8EIW6, collected from Streptomyces rimosus ATCC 10970) gave (1S)-1-methyl-THβC (1S)-121a in 8, 21% conversion and 93, >99% ee respectively; IRED-D (UniProt: V7GV82, collected from Mesorhizobium sp. L2C089B000) and IRED-F (UniProt: V6KA13, collected from Streptomyces niveus NCIMB 11891) gave (1R)-1-methyl-THβC (1R)-121a in 27, 13% conversion and 78, >99% ee respectively. IRED-B (UniProt: Q1EQE0, collected from Streptomyces kanamyceticus)162 did not give any product at all for both reactions while IRED-C (UniProt: W7VJL8, collected from Micromonospora sp. M42) did the same as above for 120a and only had 1% conversion for its product. IRED-E (UniProt: J7LAY5, collected from Nocardiopsis alba) and IRED-H (UniProt: I8QLV7, collected from Frankia sp. QA3) gave only 1% conversion for both of their products (Scheme 38).
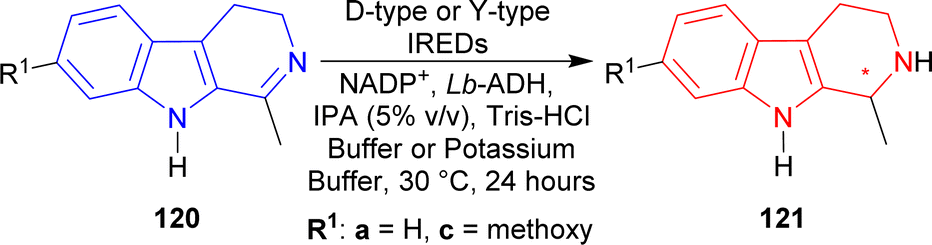 | ||
| Scheme 38 Reduction of 1-substituted-DHβC with D-type IREDs e.g., IRED-A–IRED-H, and Y-type IREDs e.g., IRED-I–IRED-N. | ||
So, IRED-A, IRED-G, IRED-I–IRED-N produced (1S)-1-substituted-THβCs; and IRED-D, IRED-F produced (1R)-1-substituted-THβCs predominantly.
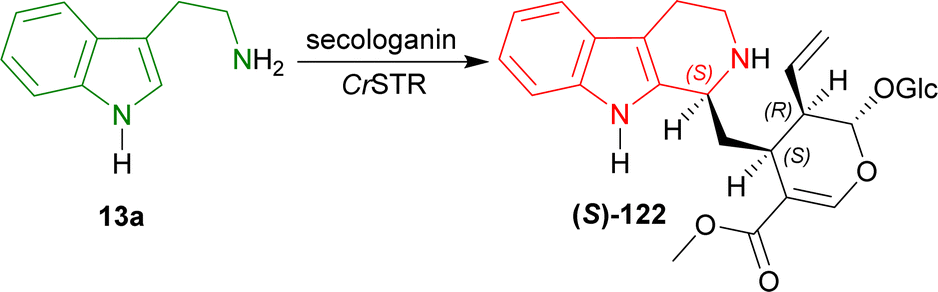 | ||
| Scheme 39 Stereoselective condensation of tryptamine and secologanin with CrSTR. | ||
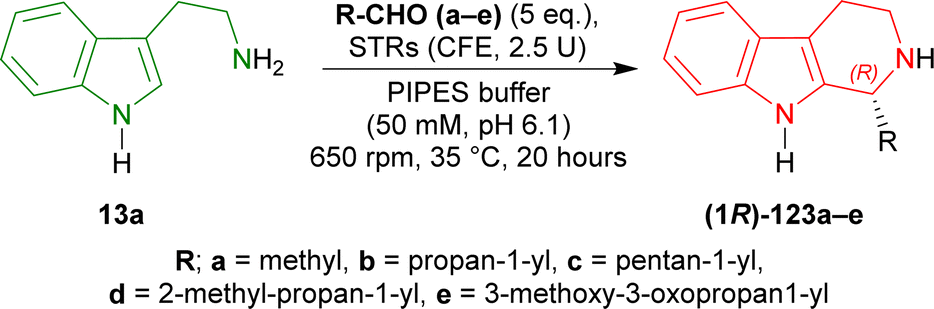
Pressnitz et al. tested CrSTR, OpSTR, RsSTR, and RvSTR for the stereoselective condensation of tryptamine 13a and five small aliphatic aldehydes that gave (1R)-1-substituted-THβCs (1R)-123a–e as products.164
14–38% conversion and 28–43% ee was seen for (1R)-123a in case of acetaldehyde as the aliphatic aldehyde. Even, racemic 123a was found for CrSTR and RsSTR. The ee improved to 61–91% for (1R)-123b when butanal was used. Overall conversion also increased to 7–45%. Decreased conversion of 4–8% and ee of 46–82% was seen for (1R)-123c when hexanal was used. So, carbon number in the aliphatic aldehyde was not increased further.
After that, steric hindrance in the aliphatic chain of the aldehyde was increased by the use of 3-methylbutanal instead of butanal. Improved 12–77% conversion and 88 to >98% ee was seen for (1R)-123d compared to (1R)-123b.
Lastly, methyl 4-oxobutanoate was used to asymmetrically condense with 13a. The product (1R)-123e showed >98% ee for all the STRs and overall increased conversion of 11–95% was seen compared to (1R)-123b.
CrSTR had generally much lower conversion and ee than any other STRs. While RvSTR showed highest ee in each product, RsSTR had highest conversion for only (1R)-123a,c,e. RsSTR and RvSTR had same or almost similar conversion and ee for (1R)-123b,c. RsSTR had higher conversion compared to OpSTR for each product except (1R)-123c. RvSTR had higher or same ee compared to OpSTR for each product (Scheme 40).
 | ||
| Scheme 40 Stereoselective condensation of tryptamine and small aliphatic aldehydes with CrSTR, OpSTR, RsSTR, and RvSTR. | ||
So, CrSTR produced (S)-strictosidine with secologanin, while CrSTR, OpSTR, RsSTR, and RvSTR produced (1R)-1-substituted-THβCs predominantly with small aliphatic aldehydes.
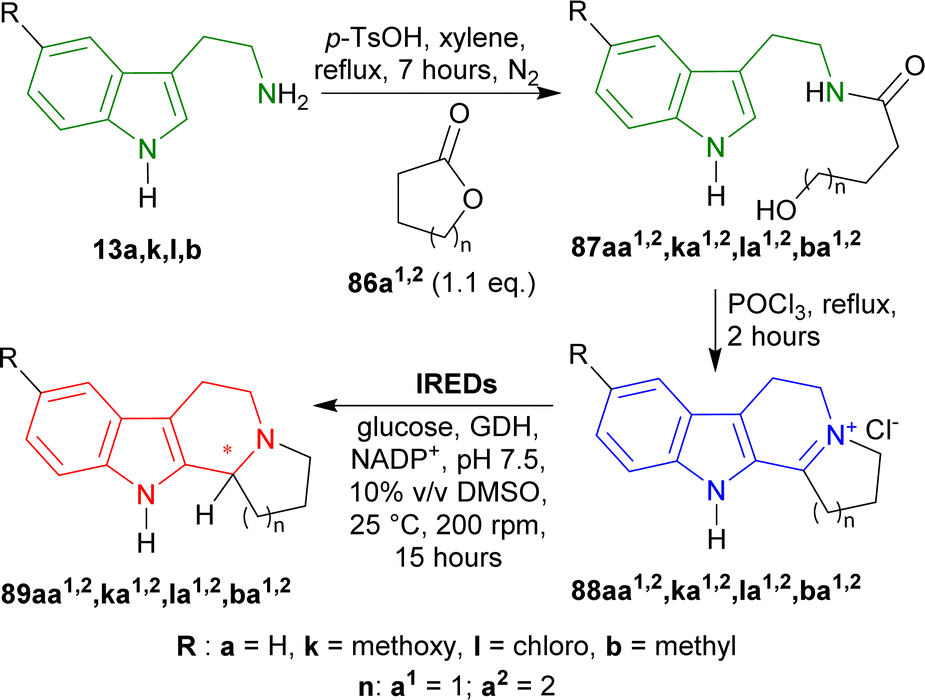
13a,b,k,l was stirred with 1.1 eq. of oxolan-2-one (γ-butyrolactone) 86a1 or oxan-2-one (δ-valerolactone) 86a2 and p-TsOH in toluene, refluxed for 7 hours to produce hydroxamides 87aa1,2,ba1,2,ka1,2,la1,2. Then they were stirred with POCl3 in toluene, refluxed for 2 hours to get iminium salts 88aa1,2,ba1,2,ka1,2,la1,2 in 58–72% yields by Bischler–Napieralski cyclization.110,166 After that, the iminium salts were asymmetrically reduced by IREDs named IR51 (from Myxococcus fulvus, Protein Identifier: WP_074958336.1), IR64 (from Actinocorallia populi, Protein Identifier: WP_106402132.1), IR86 (from Paenibacillus lactis, Protein Identifier: WP_007130043.1),167 and IR88 (Metagenome (pIR23)).168,169
IR51 and IR88 produced only (R)-configured products. IR51 had highest yield of >98% and 99% ee for (R)-89aa1; other products (R)-89aa2,ba1,2,ka1,2,la1,2 had 80–98% yields and 95–99% ee. IR88 showed the best result having 99% ee for all products (R)-89aa1,2,ba1,2,ka1,2,la1,2 with >98% yields except only (R)-89ka1 (80–98% yield).
On the other hand, IR64 and IR86 produced only (S)-configured products. IR64 did not even react with 88aa1,ka1. Among the other products, (S)-89la1 had the lowest ee of 29% with 50–80% yield and (S)-89aa2 had the highest ee of 99% with 50–80% yield. (S)-89ba2,ka2,la2 had the lowest yields of 10–50% with 82–95% ee. IR86 had 99% ee for (S)-89aa2 with 80–98% yield; 98–99% ee for (S)-89ba1,2,la1,2 with 50–98% yields; 76 and 70% ee for (S)-89ka1,2 with 10–50% and 80–98% yields respectively; 80–98% yield for (S)-89aa1 with no ee data reported.
Lastly, IR86 and IR88 were used for the preparative scale synthesis of chiral 89aa1,2,ba1,2,ka1,2,la1,2. IR86 achieved >98 to >99% ee, 77–95% conversion, and 57–82% yields for (S)-89aa2,ba1,2,la2 while, IR88 achieved >99% ee, 79–97% conversion, and 57–74% yields for (R)-89aa1,2,ba1,2,ka1,2,la1,2 (Scheme 41).
 | ||
| Scheme 41 Asymmetrically synthesizing fused-ring THβCs by imine reductases. | ||
So, IR86 produced (S)-configured products and IR88 produced (R)-configured products predominantly.
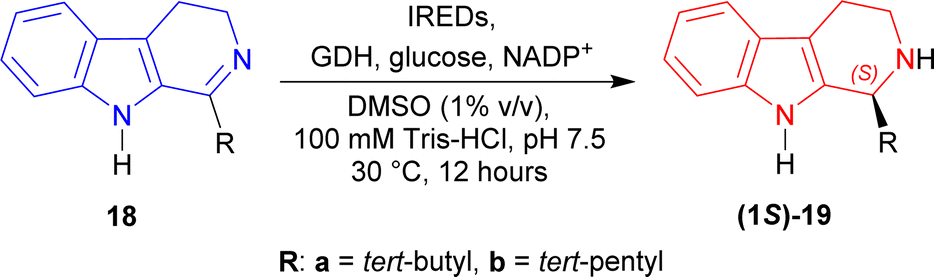
Except IRED-I, others had 97 to >99% conversion of (1S)-1-tert-butyl-THβC (1S)-19a; IRED-G (from Streptomyces, Accession No.: WP_003985113.1), IRED-I (from Streptomyces sp. GF3546, Accession No.: 4OQY) had 40–41% ee, IRED-J (from Kribbella flavida, Accession No.: WP_012921542.1) had 73% ee, IRED-M (from Saccharothrix espanaensis, Accession No.: WP_015105194.1) had 97% ee, and IRED-K (from Streptosporangium roseum, Accession No.: WP_012890722.1), IRED-L (from Nocardia brasiliensis, Accession No.: WP_014988976.1) had the highest 99% ee of (1S)-19a.
Then, IRED-K–IRED-M were tested on 1-tert-pentyl-4,9-dihydro-3H-pyrido[3,4-b]indole 18b. They produced 79–96% ee but conversion was only 44–70% of (1S)-1-tert-pentyl-THβC (1S)-19b (Scheme 42).
 | ||
| Scheme 42 Reduction of 1-substituted-DHβC with IRED-G, IRED-I–IRED-M. | ||
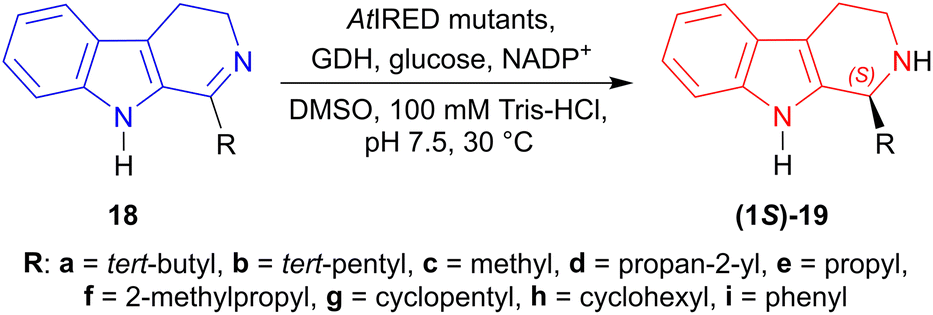
Site-saturation mutagenesis on AtIRED (from Amycolatopsis thermoflava, Accession No.: WP_027931120.1) produced two single mutants named as M118′L and P120′G; one double mutant named as M118′L/P120′G which reduced 18a–i to (1S)-19a–i in 98 to >99% ee.
M118′L reduced 1-propyl-4,9-dihydro-3H-pyrido[3,4-b]indole 18e and 1-cyclopentyl-4,9-dihydro-3H-pyrido[3,4-b]indole 18g to (1S)-1-propyl-THβC (1S)-19e and (1S)-1-cyclopentyl-THβC (1S)-19g respectively in 69% yields, while 1-(2-methylpropyl)-DHβC 18f to 1-(2-methylpropyl)-THβC (1S)-19f in 51% yield.
P120′G reduced 18b to (1S)-19b in 78% yield, 1-cyclohexyl-DHβC 18h to (1S)-1-cyclohexyl-THβC (1S)-19h in 64% yield, and 1-phenyl-DHβC 18i to (1S)-1-phenyl-THβC (1S)-19i in 30% yield.
M118′L/P120′G reduced 18a to (1S)-19a in 87% yield, 1-propan-2-yl-DHβC 18d to (1S)-propan-2-yl-THβC (1S)-19d in 62% yield, and 18c to (1S)-1-methyl-THβC (1S)-19c in 50% yield (Scheme 43).
 | ||
| Scheme 43 Reduction of 1-substituted-DHβC with AtIRED mutants. | ||
So, IRED-G, IRED-I–IRED-M and AtIRED mutants produced (1S)-1-substituted-THβCs predominantly.
3. Conclusion
Novel natural and synthetic THβC products continued to be discovered, with ongoing exploration of their biological activity directly related to the C1 chiral center. 1-Substituted-THβCs and their derivatives have diverse biological actions, indicating that they are a promising drug scaffold for treating various diseases. We discussed five synthetic methods with the purpose for creating C1 chiral center. For Pictet–Spengler reaction, the highest yield 99% and >97% ee was found from modified Pictet–Spengler reaction for formal syntheses of (−)-suaveoline, (−)-raumacline, and (−)-Nb-methylraumacline intermediates; for chiral auxiliary, the highest 97% yield and highest 91% ee was reported from asymmetric synthesis of 1-substituted-THβC using pyroglutamic acid derivatives; for ATH with chiral catalysts, the highest afforded 92% yield and highest >98% ee was observed from ATH to synthesize 1-alkyl-1,2,3,4-tetrahydropyrido[3,4-b]indole; for asymmetric addition reaction, the highest 91% yield and 96% ee was recorded from synthesis of enantiomer of dihydrocorynantheol; for enzymatic catalysis, the highest conversion of 95% with >98% ee was obtained from stereoselective condensation by STR from Rauwolfia serpentina.The methods that we have discussed here are the most used and widely found pathways for creating C1 chirality which is crucial for prominent biological activities. More efficient and economically feasible pathways should be revised so they could be applied for synthesizing new promising THβCs.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
M. M. A. Asif: writing – original draft, visualization, writing – review & editing. S. R. Lisa: writing – original draft, writing – review & editing. N. Qais: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.References
- M. Hesse, Alkaloids: Nature's Curse or Blessing?, WILEY-VCH, Weinheim, 2002 Search PubMed.
- A. Daugan, P. Grondin, C. Ruault, A.-C. Le Monnier de Gouville, H. Coste, J. M. Linget, J. Kirilovsky, F. Hyafil and R. Labaudinière, J. Med. Chem., 2003, 46, 4533–4542 CrossRef CAS PubMed.
- R. Skouta, M. Hayano, K. Shimada and B. R. Stockwell, Bioorg. Med. Chem. Lett., 2012, 22, 5707–5713 CrossRef CAS PubMed.
- S. Hotha, J. C. Yarrow, J. G. Yang, S. Garrett, K. V. Renduchintala, T. U. Mayer and T. M. Kapoor, Angew. Chem., Int. Ed., 2003, 42, 2379–2382 CrossRef CAS PubMed.
- J. H. Van Maarseveen, P. H. H. Hermkens, E. De Clercq, J. Balzarini, H. W. Scheeren and C. G. Kruse, J. Med. Chem., 1992, 35, 3223–3230 CrossRef CAS PubMed.
- J. F. Miller, E. M. Turner, R. G. Sherrill, K. Gudmundsson, A. Spaltenstein, P. Sethna, K. W. Brown, R. Harvey, K. R. Romines and P. Golden, Bioorg. Med. Chem. Lett., 2010, 20, 256–259 CrossRef CAS PubMed.
- A. Gellis, A. Dumètre, G. Lanzada, S. Hutter, E. Ollivier, P. Vanelle and N. Azas, Biomed. Pharmacother., 2012, 66, 339–347 CrossRef CAS PubMed.
- R. A. Davis, S. Duffy, V. M. Avery, D. Camp, J. N. A. Hooper and R. J. Quinn, Tetrahedron Lett., 2010, 51, 583–585 CrossRef CAS.
- M. Rottmann, C. McNamara, B. K. S. Yeung, M. C. S. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. González-Páez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H.-P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175–1180 CrossRef CAS PubMed.
- A. Pictet and T. Spengler, Ber. Dtsch. Chem. Ges., 1911, 44, 2030–2036 CrossRef CAS.
- B. T. Ho, W. M. McIsaac, K. E. Walker and V. Estevez, J. Pharm. Sci., 1968, 57, 269–274 CrossRef CAS PubMed.
- R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479–500 CrossRef CAS PubMed.
- J. Wang, A. N. Pearce, S. T. S. Chan, R. B. Taylor, M. J. Page, A. Valentin, M.-L. Bourguet-Kondracki, J. P. Dalton, S. Wiles and B. R. Copp, J. Nat. Prod., 2016, 79, 607–610 CrossRef CAS PubMed.
- A. I. Marcus, U. Peters, S. L. Thomas, S. Garrett, A. Zelnak, T. M. Kapoor and P. Giannakakou, J. Biol. Chem., 2005, 280, 11569–11577 CrossRef CAS PubMed.
- T. Akizawa, K. Yamazaki, T. Yasuhara, T. Nakajima, M. Roseghini, G. F. Erspamer and V. Erspamer, Biomed. Res., 1982, 3, 232–234 CrossRef CAS.
- R. M. Van Wagoner, J. Jompa, A. Tahir and C. M. Ireland, J. Nat. Prod., 1999, 62, 794–797 CrossRef CAS PubMed.
- L. M. M. Cesar, M. A. Mendes, C. F. Tormena, M. R. Marques, B. M. De Souza, D. M. Saidemberg, J. C. Bittencourt and M. S. Palma, Toxicon, 2005, 46, 786–796 CrossRef CAS PubMed.
- B. Elgoyhen, P. S. Lorenzo, M. T. Tellez-Iñón and E. Adler-Graschinsky, J. Pharmacol. Exp. Ther., 1992, 261, 534–539 CAS.
- P. K. Fischhof, R. Möslinger-Gehmayr, W. M. Herrmann, A. Friedmann and D. L. Ruβmann, Neuropsychobiology, 1996, 34, 29–35 CrossRef CAS PubMed.
- R. v. d. Heijden, D. I. Jacobs, W. Snoeijer, D. Hallard and R. Verpoorte, Curr. Med. Chem., 2004, 11, 607–628 CrossRef PubMed.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- J. Garnier, J. Mahuteau, M. Plat and C. Merienne, Phytochemistry, 1989, 28, 308–309 CrossRef CAS.
- N. Neuss, Indole and Biogenetically Related Alkaloids, Academic Press, London, 1980 Search PubMed.
- J. M. Müller, E. Schlittler and H. J. Bein, Experientia, 1952, 8, 338 CrossRef PubMed.
- A. Itoh, T. Tanahashi, N. Nagakura and T. Nishi, Phytochemistry, 2003, 62, 359–369 CrossRef CAS PubMed.
- J. H. van Maarseveen, H. W. Scheeren, E. De Clercq, J. Balzarini and C. G. Kruse, Bioorg. Med. Chem., 1997, 5, 955–970 CrossRef CAS PubMed.
- A. M. Deveau, M. A. Labroli, C. M. Dieckhaus, M. T. Barthen, K. S. Smith and T. L. Macdonald, Bioorg. Med. Chem. Lett., 2001, 11, 1251–1255 CrossRef CAS PubMed.
- H. Osada, C.-B. Cui, R. Onose and F. Hanaoka, Bioorg. Med. Chem., 1997, 5, 193–203 CrossRef CAS PubMed.
- A. M. P. Koskinen, Asymmetric Synthesis of Natural Products, Wiley, 1st edn, 2012 Search PubMed.
- P. D. Bailey, P. D. Clingan, T. J. Mills, R. A. Price and R. G. Pritchard, Chem. Commun., 2003, 2800–2801 RSC.
- H. Waldmann, G. Schmidt, H. Henke and M. Burkard, Angew. Chem., Int. Ed. Engl., 1995, 34, 2402–2403 CrossRef CAS.
- J. Li, T. Wang, P. Yu, A. Peterson, R. Weber, D. Soerens, D. Grubisha, D. Bennett and J. M. Cook, J. Am. Chem. Soc., 1999, 121, 6998–7010 CrossRef CAS.
- A. Bischler and B. Napieralski, Ber. Dtsch. Chem. Ges., 1893, 26, 1903–1908 CrossRef.
- N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS.
- A. Laine, C. Lood and A. Koskinen, Molecules, 2014, 19, 1544–1567 CrossRef PubMed.
- P. Maity, D. Adhikari and A. K. Jana, Tetrahedron, 2019, 75, 965–1028 CrossRef CAS.
- T. Szabó, B. Volk and M. Milen, Molecules, 2021, 26, 663 CrossRef PubMed.
- J. Wang, F. Gong, T. Liang, Z. Xie, Y. Yang, C. Cao, J. Gao, T. Lu and X. Chen, Eur. J. Med. Chem., 2021, 225, 113815 CrossRef CAS PubMed.
- Y. Du, A. Semghouli, H. Mei, L. Kiss and J. Han, Adv. Synth. Catal., 2024, 366, 3050–3084 CrossRef CAS.
- P. D. Bailey, I. D. Collier, S. P. Hollinshead, M. H. Moore, K. M. Morgan, D. I. Smith and J. M. Vernon, J. Chem. Soc., Chem. Commun., 1994, 1559–1560 RSC.
- A. W. M. Lee, W. H. Chan, Y. Tao and Y. K. Lee, J. Chem. Soc., Perkin Trans. 1, 1994, 477–481 RSC.
- N. S. Rajapaksa, M. A. McGowan, M. Rienzo and E. N. Jacobsen, Org. Lett., 2013, 15, 706–709 CrossRef CAS PubMed.
- T. Itoh, M. Yokoya, K. Miyauchi, K. Nagata and A. Ohsawa, Org. Lett., 2003, 5, 4301–4304 CrossRef CAS PubMed.
- Y. Li, X. Yue, Z. Li, Z. Huang and F. Chen, Org. Lett., 2023, 25, 1285–1289 CrossRef CAS PubMed.
- G. Tatsui, J. Pharm. Soc. Jpn, 1928, 48, 92–99 Search PubMed.
- Z.-J. Liu and R.-R. Lu, in The Alkaloids: Chemistry and Pharmacology, ed. A. Brossi, Academic Press, 1988, vol. 33, pp. 83–140 Search PubMed.
- P. R. Benoin, R. H. Burnell and J. D. Medina, Tetrahedron Lett., 1968, 9, 807–809 CrossRef.
- P. D. Bailey and N. R. McLay, Tetrahedron Lett., 1991, 32, 3895–3898 CrossRef CAS.
- P. D. Bailey, S. P. Hollinshead and N. R. McLay, Tetrahedron Lett., 1987, 28, 5177–5180 CrossRef CAS.
- A. P. Krapcho, Synthesis, 1982, 1982, 805–822 CrossRef.
- E. Piers and F. F. Fleming, J. Chem. Soc., Chem. Commun., 1989, 756–757 RSC.
- P. D. Bailey and S. P. Hollinshead, J. Chem. Soc., Perkin Trans. 1, 1988, 739–745 RSC.
- P. Magnus, B. Mugrage, M. R. DeLuca and G. A. Cain, J. Am. Chem. Soc., 1990, 112, 5220–5230 CrossRef CAS.
- J. P. Kutney, G. K. Eigendorf, H. Matsue, A. Murai, K. Tanaka, W. L. Sung, K. Wada and B. R. Worth, J. Am. Chem. Soc., 1978, 100, 938–943 CrossRef CAS.
- J. Vercauteren, C. Lavaud, J. Levy and G. Massiot, J. Org. Chem., 1984, 49, 2278–2279 CrossRef CAS.
- P. D. Bailey, S. P. Hollinshead and Z. Dauter, J. Chem. Soc., Chem. Commun., 1985, 1507–1509 RSC.
- P. D. Bailey and S. P. Hollinshead, Tetrahedron Lett., 1987, 28, 2879–2882 CrossRef CAS.
- P. D. Bailey, S. P. Hollinshead, M. H. Moore, K. M. Morgan, D. I. Smith and J. M. Vernon, Tetrahedron Lett., 1994, 35, 3585–3586 CrossRef CAS.
- X. Fu and J. M. Cook, J. Org. Chem., 1993, 58, 661–672 CrossRef CAS.
- J. Seayad, A. M. Seayad and B. List, J. Am. Chem. Soc., 2006, 128, 1086–1087 CrossRef CAS PubMed.
- T. Arai, M. Wasai and N. Yokoyama, J. Org. Chem., 2011, 76, 2909–2912 CrossRef CAS PubMed.
- T. Arai and N. Yokoyama, Angew. Chem., Int. Ed., 2008, 47, 4989–4992 CrossRef CAS PubMed.
- J. O. Osby and B. Ganem, Tetrahedron Lett., 1985, 26, 6413–6416 CrossRef CAS.
- S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 4900–4901 CrossRef CAS PubMed.
- W. Oppolzer, O. Tamura, G. Sundarababu and M. Signer, J. Am. Chem. Soc., 1992, 114, 5900–5902 CrossRef CAS.
- V. Vavsari, V. Dianati, S. Ramezanpour and S. Balalaie, Synlett, 2015, 26, 1955–1960 CrossRef CAS.
- Compendium of Chiral Auxiliary Applications, ed. G. Roos, Academic Press, New York, 2002 Search PubMed.
- Y. Gnas and F. Glorius, Synthesis, 2006, 2006, 1899–1930 CrossRef.
- D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- W. Oppolzer, C. Chapuis and G. Bernardinelli, Helv. Chim. Acta, 1984, 67, 1397–1401 CrossRef CAS.
- Handbook of Reagents for Organic Synthesis: Chiral Reagents for Asymmetric Synthesis, ed. L. A. Paquette, Wiley, Chichester, 2003 Search PubMed.
- N. Qais, N. Nakao, K. Hashigaki, Y. Takeuchi and M. Yamato, Chem. Pharm. Bull., 1991, 39, 3338–3340 CrossRef CAS.
- H. Akimoto, K. Okamura, M. Yui, T. Shioiri, M. Kuramoto, Y. Kikugawa and S. Yamada, Chem. Pharm. Bull., 1974, 22, 2614–2623 CrossRef CAS.
- K. Yamada, M. Takeda and T. Iwakuma, J. Chem. Soc., Perkin Trans. 1, 1983, 265–270 RSC.
- T. Itoh, M. Miyazaki, S. Ikeda, K. Nagata, M. Yokoya, Y. Matsuya, Y. Enomoto and A. Ohsawa, Tetrahedron, 2003, 59, 3527–3536 CrossRef CAS.
- E. W. Colvin, Silicon Reagents in Organic Synthesis, Academic Press, San Diego, 1988 Search PubMed.
- L. F. Tietze, Y. Zhou and E. Töpken, Eur. J. Org Chem., 2000, 2000, 2247–2252 CrossRef.
- A. Siwicka, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, A. Zawadzka and Z. Czarnocki, Can. J. Chem., 2007, 85, 1033–1036 CrossRef CAS.
- F. M. Cordero, F. Pisaneschi, K. Meschini Batista, S. Valenza, F. Machetti and A. Brandi, J. Org. Chem., 2005, 70, 856–867 CrossRef CAS PubMed.
- J. Biała, Z. Czarnocki and J. K. Maurin, Tetrahedron: Asymmetry, 2002, 13, 1021–1023 CrossRef.
- N. S. S. Reddy, R. A. Babu and B. V. S. Reddy, Synthesis, 2016, 48, 1079–1086 CrossRef.
- J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS PubMed.
- X.-W. Sun, M.-H. Xu and G.-Q. Lin, Org. Lett., 2006, 8, 4979–4982 CrossRef CAS PubMed.
- G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831–840 CrossRef CAS PubMed.
- F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Chem. Soc. Rev., 2009, 38, 1162–1186 RSC.
- F. A. Davis, J. Y. Melamed and S. S. Sharik, J. Org. Chem., 2006, 71, 8761–8766 CrossRef CAS PubMed.
- D. A. Cogan, G. Liu and J. Ellman, Tetrahedron, 1999, 55, 8883–8904 CrossRef CAS.
- J. L. García Ruano, J. Alemán and M. B. Cid, Synthesis, 2006, 2006, 687–691 CrossRef.
- J. Fujiwara, Y. Fukutani, H. Sano, K. Maruoka and H. Yamamoto, J. Am. Chem. Soc., 1983, 105, 7177–7179 CrossRef CAS.
- K. Pulka, Curr. Opin. Drug Discovery Dev., 2010, 13, 669–684 CAS.
- G. Brieger and T. J. Nestrick, Chem. Rev., 1974, 74, 567–580 CrossRef CAS.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051–1069 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045–2061 CrossRef CAS.
- K. Everaere, A. Mortreux and J. Carpentier, Adv. Synth. Catal., 2003, 345, 67–77 CrossRef CAS.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750–1770 CrossRef CAS PubMed.
- X. Wu and J. Xiao, Chem. Commun., 2007, 2449–2466 RSC.
- S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- M. Bianchi, U. Matteol, G. Menchi, P. Frediani, U. Pratesi, F. Piacenti and C. Botteghi, J. Organomet. Chem., 1980, 198, 73–80 CrossRef CAS.
- U. Matteoli, P. Frediani, M. Bianchi, C. Botteghi and S. Gladiali, J. Mol. Catal., 1981, 12, 265–319 CrossRef CAS.
- J. S. M. Samec, J.-E. Backvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- T. R. Ward, Acc. Chem. Res., 2011, 44, 47–57 CrossRef CAS PubMed.
- M. Bartok, Chem. Rev., 2010, 110, 1663–1705 CrossRef CAS PubMed.
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- P. Roszkowski, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, T. Lis and Z. Czarnocki, J. Mol. Catal. A: Chem., 2005, 232, 143–149 CrossRef CAS.
- Z. Czarnocki, M. P. Matuszewska and I. Matuszewska, Org. Prep. Proced. Int., 1998, 30, 699–702 CrossRef CAS.
- J. Szawkało, S. J. Czarnocki, A. Zawadzka, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, Z. Czarnocki and J. Drabowicz, Tetrahedron: Asymmetry, 2007, 18, 406–413 CrossRef.
- S. McLean, G. I. Dmitrienko and A. Szakolcai, Can. J. Chem., 1976, 54, 1262–1277 CrossRef CAS.
- M. Nakagawa, M. Kiuchi, M. Obi, M. Tonozuka, K. Kobayashi, T. Hino and Y. Ban, Chem. Pharm. Bull., 1975, 23, 304–312 CrossRef CAS.
- S. J. Czarnocki, K. Wojtasiewicz, A. P. Jóźwiak, J. K. Maurin, Z. Czarnocki and J. Drabowicz, Tetrahedron, 2008, 64, 3176–3182 CrossRef CAS.
- L. K. Vinograd and N. N. Suvorov, Chem. Heterocycl. Compd., 1984, 20, 984–988 CrossRef.
- L. M. M. Cesar, C. F. Tormena, M. R. Marques, G. V. J. Silva, M. A. Mendes, R. Rittner and M. S. Palma, Helv. Chim. Acta, 2005, 88, 796–801 CrossRef CAS.
- Y. Takahashi, H. Ishiyama, T. Kubota and J. Kobayashi, Bioorg. Med. Chem. Lett., 2010, 20, 4100–4103 CrossRef CAS PubMed.
- T. Ito, M. Kitajima and H. Takayama, Tetrahedron Lett., 2009, 50, 4506–4508 CrossRef CAS.
- E. Biron, J. Chatterjee, O. Ovadia, D. Langenegger, J. Brueggen, D. Hoyer, H. A. Schmid, R. Jelinek, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2008, 47, 2595–2599 CrossRef CAS PubMed.
- A. Bischler and F. J. Howell, Ber. Dtsch. Chem. Ges., 1893, 26, 1384–1399 CrossRef.
- Q. Yin, S.-G. Wang and S.-L. You, Org. Lett., 2013, 15, 2688–2691 CrossRef CAS PubMed.
- J. Selvakumar, A. Makriyannis and C. R. Ramanathan, Org. Biomol. Chem., 2010, 8, 4056–4058 RSC.
- J. Selvakumar and C. R. Ramanathan, Org. Biomol. Chem., 2011, 9, 7643–7646 RSC.
- F. Xu, D. Huang, C. Han, W. Shen, X. Lin and Y. Wang, J. Org. Chem., 2010, 75, 8677–8680 CrossRef CAS PubMed.
- I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370–17373 CrossRef PubMed.
- M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891–1894 CrossRef CAS PubMed.
- T. Itoh, M. Yokoya, K. Miyauchi, K. Nagata and A. Ohsawa, Org. Lett., 2006, 8, 1533–1535 CrossRef CAS PubMed.
- K. Nagata, H. Ishikawa, A. Tanaka, M. Miyazaki, T. Kanemitsu and T. Itoh, Heterocycles, 2010, 81, 1791–1798 CrossRef CAS.
- L. D. Vazquez-Serrano, B. T. Owens and J. M. Buriak, Inorg. Chim. Acta, 2006, 359, 2786–2797 CrossRef CAS.
- B. Wüstenberg and A. Pfaltz, Adv. Synth. Catal., 2008, 350, 174–178 CrossRef.
- G. Stork, Pure Appl. Chem., 1989, 61, 439–442 CrossRef CAS.
- B. List, Synlett, 2001, 2001, 1675–1686 CrossRef.
- B. List, Tetrahedron, 2002, 58, 5573–5590 CrossRef CAS.
- The IUPAC Compendium of Chemical Terminology: The Gold Book, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 4th edn, 2019 Search PubMed.
- W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon Press, Oxford, England, New York, 1st edn, 1990 Search PubMed.
- Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jørgensen, Wiley, 1st edn, 2001 Search PubMed.
- G. S. Buchanan, J. B. Feltenberger and R. P. Hsung, Curr. Org. Synth., 2010, 7, 363–401 CrossRef CAS PubMed.
- A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed.
- J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434–12446 RSC.
- J. Deng, X. Wang and R. P. Hsung, in Methods and Applications of Cycloaddition Reactions in Organic Syntheses, ed. N. Nishiwaki, Wiley, 1st edn, 2014, pp. 283–354 Search PubMed.
- R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed.
- A. W. Rey, W. A. Szarek and D. B. MacLean, Can. J. Chem., 1992, 70, 2922–2928 CrossRef CAS.
- H. Waldmann, M. Braun, M. Weymann and M. Gewehr, Tetrahedron, 1993, 49, 397–416 CrossRef CAS.
- T. Kametani, Y. Hirai, M. Kajiwara, T. Takahashi and K. Fukumoto, Chem. Pharm. Bull., 1975, 23, 2634–2642 CrossRef CAS PubMed.
- C. Szántay, G. Blaskó, K. Honty, L. Szabó and L. Töke, Heterocycles, 1977, 7, 155–160 CrossRef.
- D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1973, 95, 553–563 CrossRef CAS PubMed.
- T. Li, S. Liu, S. Wu, Q. Cheng, Q. Chen, Y. Jiao, Y. Zhang and F. Shi, Sci. China: Chem., 2024, 67, 2629–2636 CrossRef CAS.
- E.-L. Teo, G.-K. Chuah, A. R. J. Huguet, S. Jaenicke, G. Pande and Y. Zhu, Catal. Today, 2004, 97, 263–270 CrossRef CAS.
- K. Koch, R. Vandenberg, P. Nieuwland, R. Wijtmans, M. Wubbolts, H. Schoemaker, F. Rutjes and J. Vanhest, Chem. Eng. J., 2008, 135, S89–S92 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- D. Muñoz Solano, P. Hoyos, M. J. Hernáiz, A. R. Alcántara and J. M. Sánchez-Montero, Bioresour. Technol., 2012, 115, 196–207 CrossRef PubMed.
- J. Ogawa and S. Shimizu, Trends Biotechnol., 1999, 17, 13–21 CrossRef CAS PubMed.
- E. Quiroga, N. Priolo, D. Obregón, J. Marchese and S. Barberis, Biochem. Eng. J., 2008, 39, 115–120 CrossRef CAS.
- M. G. Moghaddam, F. B. H. Ahmad, M. Basri and M. B. Abdul Rahman, J. Appl. Sci., 2010, 10, 337–342 CrossRef CAS.
- M. G. Moghaddam, F. B. H. Ahmad, M. Basri and M. B. Abdul Rahman, Electron. J. Biotechnol., 2010, 13(3) DOI:10.2225/vol13-issue3-fulltext-9.
- K. Mitsukura, M. Suzuki, K. Tada, T. Yoshida and T. Nagasawa, Org. Biomol. Chem., 2010, 8, 4533–4535 RSC.
- K. Mitsukura, M. Suzuki, S. Shinoda, T. Kuramoto, T. Yoshida and T. Nagasawa, Biosci., Biotechnol., Biochem., 2011, 75, 1778–1782 CrossRef CAS PubMed.
- K. Mitsukura, T. Kuramoto, T. Yoshida, N. Kimoto, H. Yamamoto and T. Nagasawa, Appl. Microbiol. Biotechnol., 2013, 97, 8079–8086 CrossRef CAS PubMed.
- G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 CrossRef CAS.
- S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- D. Wetzl, M. Berrera, N. Sandon, D. Fishlock, M. Ebeling, M. Müller, S. Hanlon, B. Wirz and H. Iding, ChemBioChem, 2015, 16, 1749–1756 CrossRef CAS PubMed.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- M. Rodríguez-Mata, A. Frank, E. Wells, F. Leipold, N. J. Turner, S. Hart, J. P. Turkenburg and G. Grogan, ChemBioChem, 2013, 14, 1372–1379 CrossRef PubMed.
- J. Stöckigt and M. H. Zenk, J. Chem. Soc., Chem. Commun., 1977, 646–648 RSC.
- D. Pressnitz, E. Fischereder, J. Pletz, C. Kofler, L. Hammerer, K. Hiebler, H. Lechner, N. Richter, E. Eger and W. Kroutil, Angew. Chem., 2018, 130, 10843–10847 CrossRef.
- L. Yang, J. Li, Z. Xu, P. Yao, Q. Wu, D. Zhu and Y. Ma, Org. Lett., 2022, 24, 6531–6536 CrossRef CAS PubMed.
- G.-H. Hou, J.-H. Xie, P.-C. Yan and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 1366–1367 CrossRef CAS PubMed.
- M. Li, Y. Cui, Z. Xu, X. Chen, J. Feng, M. Wang, P. Yao, Q. Wu and D. Zhu, Adv. Synth. Catal., 2022, 364, 372–379 CrossRef CAS.
- J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, D. J. Cook, S. J. Charnock and N. J. Turner, Nat. Chem., 2021, 13, 140–148 CrossRef CAS PubMed.
- P. Yao, J. R. Marshall, Z. Xu, J. Lim, S. J. Charnock, D. Zhu and N. J. Turner, Angew. Chem., Int. Ed., 2021, 60, 8717–8721 CrossRef CAS PubMed.
Footnote |
| † Co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |