
Direct coupling of two inert CO2 molecules to form a C–C bond on the Cu0 atomic interfaces of the nitrogen-doped graphene-supported Cu4 cluster†
Xin-Jia
Cui
a,
Yong-Qing
Qiu
 b,
Hong-Qiang
Wang
a and
Chun-Guang
Liu
*a
b,
Hong-Qiang
Wang
a and
Chun-Guang
Liu
*a
aDepartment of Chemistry, Faculty of Science, Beihua University, Jilin City, 132013, P. R. China. E-mail: liucg407@163.com; Tel: +86 043264606919
bInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China
First published on 1st November 2023
Abstract
The electrochemical reduction reaction of CO2 (CO2RR) to C2+ products is strongly related to the C–C coupling reaction. In general, deoxidation productions of CO2 have been assigned as the active species for the C–C coupling reaction. For example, the direct coupling of two *CO, *CO and *CHO, and two *CHO species are always proposed to be the three main pathways for the formation of the C–C bond in CO2RR. In this case, a direct coupling of two inert CO2 molecules to form a CO2 dimer with strong C–C bond over the Cu0 atomic interfaces of the Cu4 cluster, which was anchored on a nitrogen-doped graphene support (Cu4/N3GN), has been proposed based on our periodic density functional theory (DFT) calculations. The mechanistic investigation shows that the atomic interface formed by the three Cu0 species of Cu4/N3GN can simultaneously adsorb two CO2 molecules, and two adsorbed CO2 molecules could be reduced to a CO2−˙ anionic radical with [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C˙O]− configuration via an electron-transfer process from the three Cu0 species to the two adsorbed CO2 molecules. Furthermore, the direct coupling of two CO2−˙ anionic radicals results in the formation of a C–C bond. The calculated free energy profiles reveal that the direct coupling of two CO2 molecules should be the main reaction relevant to the coupling of *OCHO and CO, *COOH and *CO, and two CO molecules over the catalyst surface. Electronic structural analysis indicates that the bent arrangement of two adsorbed CO2 molecules is a key factor in the determination of the formation of the C–C bond in the CO2 dimer via effective orbital interactions. In addition, the CO2 dimer could be reduced to ethane in the series of electrochemical elementary steps. The high ethane selectivity of the Cu4/N3GN catalyst studied here mainly arises from the strong Cu–O bonding interaction. Regarding the direct coupling of two inert CO2 molecules over the atomic interface of the Cu4 cluster, these findings would be very useful to guide the search for potential catalysts for the formation of the C–C bond in CO2RR into the subnanometer cluster with various active sites.
C˙O]− configuration via an electron-transfer process from the three Cu0 species to the two adsorbed CO2 molecules. Furthermore, the direct coupling of two CO2−˙ anionic radicals results in the formation of a C–C bond. The calculated free energy profiles reveal that the direct coupling of two CO2 molecules should be the main reaction relevant to the coupling of *OCHO and CO, *COOH and *CO, and two CO molecules over the catalyst surface. Electronic structural analysis indicates that the bent arrangement of two adsorbed CO2 molecules is a key factor in the determination of the formation of the C–C bond in the CO2 dimer via effective orbital interactions. In addition, the CO2 dimer could be reduced to ethane in the series of electrochemical elementary steps. The high ethane selectivity of the Cu4/N3GN catalyst studied here mainly arises from the strong Cu–O bonding interaction. Regarding the direct coupling of two inert CO2 molecules over the atomic interface of the Cu4 cluster, these findings would be very useful to guide the search for potential catalysts for the formation of the C–C bond in CO2RR into the subnanometer cluster with various active sites.
1. Introduction
Subnanometer copper (Cu)-based clusters have received considerable attention in the conversion of CO2 to valuable chemicals because of their unique electronic structure and excellent catalytic performance.1–6 Compared with bulk Cu surfaces and large Cu-based nanoparticles, subnanometer Cu–based clusters possess a larger surface-to-volume ratio and more active sites.7 Owing to the high surface energy, they have several advantages in the adsorption of the inert CO2 molecule. The adsorption and activation of the CO2 molecule by catalysts is the first step for the conversion of CO2 to any valuable chemicals.8,9 An activated CO2 molecule, which has the lengthening of the C–O bond and the bending of the O–C–O bond angle, is always generated from an efficient electron-transfer process between catalysts and the CO2 molecule.10–12 For this purpose, the oxidation state of the active Cu cluster has been well investigated by using experimental and theoretical methods. It has been reported that the ionic form of Cu+ was the active center for the activation of CO2 molecule by the Cu-based clusters;13–15 however, other investigations suggested that the measured Cu0 species is strongly correlated to the activation of CO2 molecule.16–18 Meanwhile, the cluster size and support composition are also key factors in the determination of the catalytic activity for the reported subnanometer Cu-based clusters.19–22 To date, a clear assignment of the active Cu site has not been achieved yet, and is still under debate for the subnanometer Cu-based clusters.Recently, several theoretical and experimental studies have been reported for the reduction of CO2 to methanol/ethanol catalyzed by a four-atom Cu cluster (Cu4) on the surface of various supports.23–25 Liu and co-workers reported that a carbon-supported Cun (n = 3, 4) cluster achieved a single-product faradaic efficiency (FE) of 91% at −0.71 V (versus the reversible hydrogen electrode).26 The FE of ethanol was found to be closely associated with the initial dispersion of Cu atoms. In their density functional theory (DFT) calculations, a supported Cu3 cluster was assigned as a model to probe the possible reaction mechanism, assuming a Cu4 cluster would share the similar mechanism. They found that the HCOOH* intermediate was reduced to CH3* + H2CO*, which underwent a C–C bond formation to form ethanol. Alternatively, the conventional CO* pathway was found to have a higher reaction free energy in several steps than the HCOO* pathway. In another case, the same group reported that a size-selected Cu4 cluster is the most active low-pressure catalyst for the reduction of CO2 to methanol.27 DFT calculations showed that the unsaturated Cu sites in the Cu4 clusters resulted in a strong adsorption of the adsorbates, which leads to an energetically low-lying reaction pathway. A systematic study of the size and support effects was carried out by using X-ray adsorption spectroscopy, catalytic activity measurement, and DFT calculations. They found that the catalytic activity for methanol synthesis varied strongly as a function of the Cu cluster size. The Cu4/Al2O3 catalyst shows the highest turnover rate for methanol among all reported catalysts.27,28 By contrast, the Cu3/Al2O3 catalyst showed less than 50% activity because the stronger charge transfer interaction with Al2O3 support for Cu3 than Cu4 leads to a less favorable energetic pathway for the reduction of CO2 to methanol.27 In addition to methanol and ethanol, the Cu4 cluster was found to be an efficient catalyst for the conversion of CO2 to CO and acetylene trimerization.29 Rodriguez et al. reported that a small Cu4 cluster in contact with TiC(001) displays a very high activity for the conversion of CO2 to CO.30 DFT calculations showed that the Cu4/TiC(001) catalyst can bind CO2 well, with the adsorption energy of −25.6 kcal mol−1, which is about 2 times as large as that of the Cu9/TiC(001) catalyst (−13.6 kcal mol−1). Schlexer and co-workers found that the interaction between the Cu4 cluster and a silica-surfaces can be enhanced by introduction of the hydroxyl groups,31 whereas the dopants have only a small effect on the interaction. Meanwhile, the possible reaction mechanism of acetylene trimerization catalyzed by the silica-supported Cu4 cluster has been probed based on their DFT calculations. All of these instructive studies showed that (i) the catalytic activity of the Cu4 clusters is unique and relevant to its analogues, such as Cu3, Cu9, Cu13 and C29 clusters, etc.; (ii) the reactivity of the Cu4 cluster may be tuned in a desired manner by the support composition; (iii) the active Cu site in the Cu4 cluster is strongly sensitive to the interaction between the Cu4 clusters and support surfaces. These factors encouraged us to probe the potential catalytic properties of the Cu4 cluster for C2 product synthesis.
Nitrogen-doped carbon materials have attracted considerable attention in the field of CO2RR because of their adjustable pore structure, high specific surface area, and good electrochemical stability.32–37 Especially in the study of M–N–C catalysts based on metal elements such as Cu, Ni, Fe, and Co, etc., it has been demonstrated that these catalysts have high activity and selectivity for the reduction of CO2 to CO.38–42 Among the various reported metals, the carbon-loaded Cu-based catalyst shows high activity in CO2RR.43–46 These reported catalysts are of particular interest because of their ability to generate large amounts of hydrocarbons, including HCOOH, CH4, C2H4, etc.47–51 Shen and co-workers found that the anchoring of Cu atoms on defective diamond graphene results in excellent catalytic performance for the selective conversion of acetylene to ethylene.52 Baturina et al. reported that introduction of the Cu atomic chains into nitrogen-doped carbon materials can break the scaling relationship by providing secondary adsorption sites, thus effectively reducing the overpotential of the electrocatalytic reduction of CO2 to methanol.53 Shi and co-workers proposed that the morphology of the carbon support has a significant effect on the catalytic performance of these Cu-based materials in CO2RR.54 They found that the selectivity for CO2RR is very sensitive to the carbon support morphologies. An onion-like carbon support can effectively improve the stability, activity, and selectivity for the reduction of CO2 to C2H4.55 DFT calculations show that the defect site of the nitrogen-doped carbon support not only promotes the electron transport to the key intermediate corresponding to the potential limiting step, but also prevents the sintering of Cu atoms because the defect sites can bind the Cu atom with the strong Cu–N bonding interaction.56,57
In previous investigations, the formation of the C–C bond in CO2RR generally comes from the coupling reactions between two deoxidation productions of CO2, such as the direct coupling of two *CO species, direct coupling of the *CO and *CHO species, and direct coupling of two *CHO species.28,58–61 In this case, a direct coupling of two inert CO2 molecules to form a CO2 dimer with a C–C bond over the atomic interface of the Cu4 cluster, which was anchored on a nitrogen-doped graphene substrate (Cu4/N3GN), has been proposed based on our DFT calculations. Our mechanistic investigation shows that the atomic interface formed by the three Cu0 species of Cu4/N3GN can simultaneously adsorb two CO2 molecules. The two adsorbed CO2 molecules were firstly reduced to a CO2−˙ anionic radical with [OC˙O]− configuration via an electron-transfer process from the three Cu0 species of Cu4/N3GN catalyst to the two adsorbed CO2 molecules. The direct coupling of the two CO2˙− anionic radicals then resulted in the formation of a CO2 dimer with a C–C bond. Furthermore, the produced CO2 dimer could be reduced to ethane in the series of electrochemical elementary steps. This study provided a new mechanism for the formation of the C–C bond, and shed light on further improving the CO2RR catalysts through the rational design of a subnanometer Cu-based catalyst.
2. Computation details
All of the first-principles calculations are based on DFT62 to calculate the Perdew–Burke–Ernzerhof (PBE) functional and projector augmented wave (PAW)63,64 pseudopotential using the generalized gradient approximation (GGA)65 realized in the Vienna ab initio simulation program (VASP).66,67 For all graphene supporting structures, a 6 × 6 × 1 original graphene supercell was used for the doping of nitrogen atoms and loading of metal clusters, and the geometrically optimized convergence threshold was set to an effective 0.03 eV Å−1. The cutoff energy of the plane wave basis group is 500 eV. In order to prevent the interaction between periodic images along the z-direction, the vacuum of 26.7 Å is used for the flat plate calculation, and a dipole correction is applied. The Brillouin zone of the surface is sampled with the Monkhorst-K point grid of 2 × 2 × 1 γ center, while only γ points are sampled for molecular calculations and higher 7 × 7 × 1 for the projected density of states (pDOS) calculations.68 The climbing image miniature elastic band method is used to calculate the transition state (TS).69,70 Some atomic charges are calculated by the Bader charge analysis method.71The computational hydrogen electrode (CHE) method was applied in free energy diagrams, which assumes that half of the free energy of gaseous hydrogen is equivalent to the free energy of a proton–electron pair at 0 V, all pH values, and 1 atm. Thus, the energy of H2(g) is used as an electrochemical ref. 72 The adsorption energy (Ead) is obtained from the following formula:
| Ead = Etotal − Eslab − Eadsorbate. |
Among them, Etotal represents the total energy of the catalyst surface adsorbed by the adsorption species, Eslab represents the surface energy of the catalysts without the adsorption species, and Eadsorbate represents the energy of the adsorption species. Therefore, the more negative the adsorption energy, the stronger the adsorption capacity. The binding energy Eb was calculated as:
| Eb = EAB − EA − EB |
| ΔG = EDFT + ΔZPE − TS |
where h is the Planck constant, vi is the calculated actual frequency of the system, and EDFT is the calculated total energy. The total entropy (S) is calculated from ref. 75
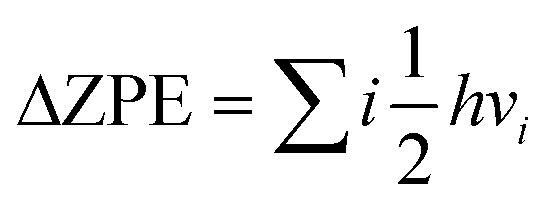
The frequency of the optimized adsorption species at γ point is determined by the VASP method, and the convergence accuracy is improved. When calculating the frequency of the adsorbent, the whole catalyst system is fixed and only the adsorbent is released. For the free energy correction of adsorbed matter, VASPKIT attributes the contribution of vibration to the translational or rotational part; in other words, the 3N vibration of the surface-adsorbed molecules (except the imaginary frequency) is used to calculate the correction of the thermal energy. In addition, the solvation program VASPsol was used to implicitly describe the effects of electrolytes and solvents.76 The solvent molecule was regarded as a continuous medium model, and the dielectric constant (ε = 78.4) was used to express the solvent. In this work, the free energy of all isomers, intermediates, transition state (TS), and products along the reaction pathways was calculated in the solvent.
3. Results and discussion
3.1. Geometric and electronic structure of Cu4/N3GN catalyst
In order to ascertain the adsorption and activation of CO2 by the Cu4/N3GN catalyst, we first need to determine the stable structure of the Cu4/N3GN catalyst. In the present paper, all of the possible structures of the initial geometries for the Cu4 cluster, which already were reported recently in the literature for a study of the adsorption of CO2 on Cu-based clusters, have been considered.77,78As shown in Fig. 1, our periodic DFT calculations obtained five possible geometric arrangements as anchoring of these Cu4 clusters onto the nitrogen-doped graphene supports (1, 2, 3, 4, and 5). Based on these optimized geometries, we calculated the free energy of these isomers (see Fig. 1). The calculated relative free energy of these isomers increased in the following order: 4 (0.00 eV) < 1 (0.19 eV) < 2 (0.21 eV) < 5 (5.80 eV) < 3 (10.92 eV), indicating that the most stable species comes from anchoring the Cu4 cluster with a rhombus-like structure onto the nitrogen-doped graphene supports, in which one Cu atom residing at the vertices of the rhombus-like structure coordinates to the three nitrogen atoms of the nitrogen-doped graphene support. The second higher energy isomer for the Cu4 cluster is an inverted-tetrahedron-like structure, and is only 0.19 eV higher in energy than the lowest energy isomer optimized with the PBE functional. The third higher energy isomer is found to be another rhombus-like structure for the anchored Cu4 cluster, which is only 0.21 eV higher in energy than the most stable isomer.
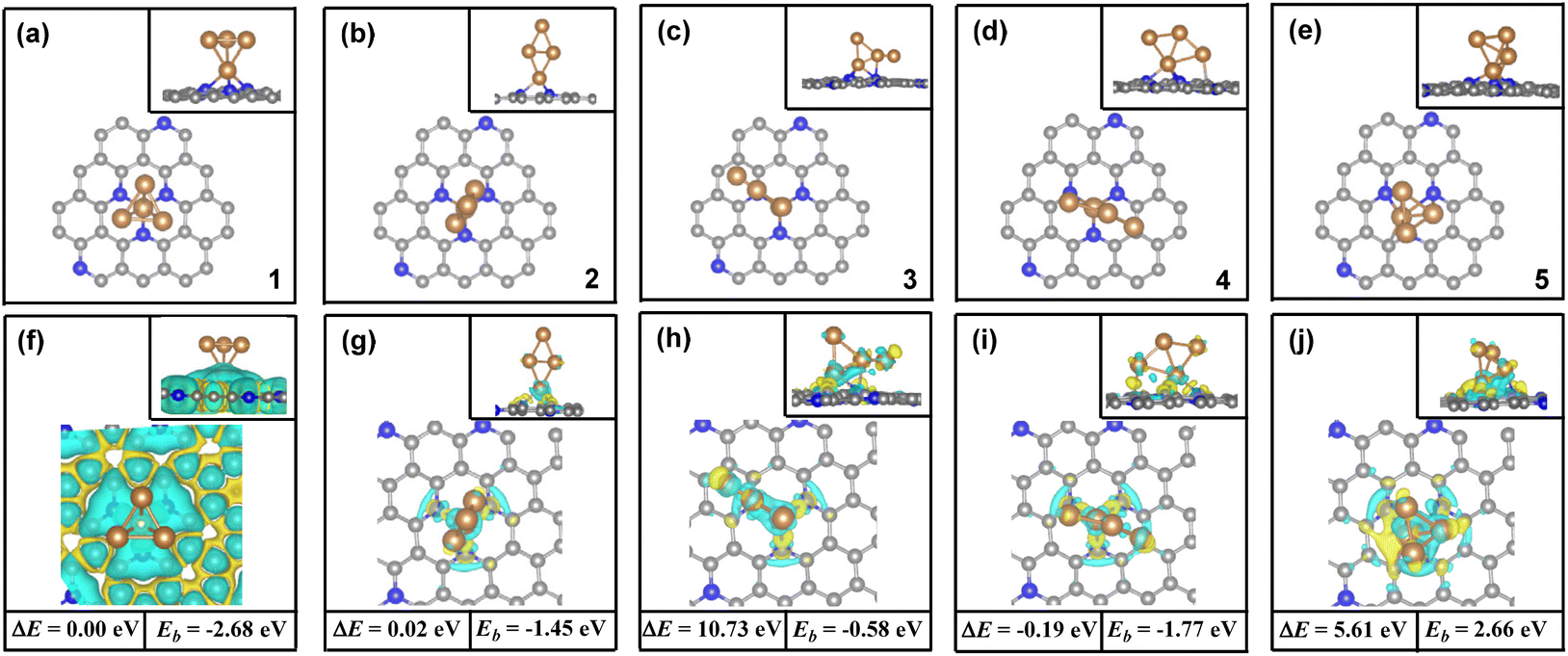 | ||
| Fig. 1 The geometric arrangement and charge density difference of all possible isomers for the Cu4/N3GN catalysts studied here, where the calculated relative free energy of these isomers and binding energy of the Cu4 cluster and nitrogen-doped graphene support also have been compared.((a) and (f) for 1, (b) and (g) for 2, (c) and (h) for 3, (d) and (i) for 4, (e) and (j) for 5.). | ||
To further evaluate the stability of the Cu4/N3GN catalyst, we carried out calculations of the binding energy (Eb) between the Cu4 cluster and nitrogen-doped graphene supports. The calculated Eb value of all possible isomers also has been compared in Fig. 1. It can be found that the calculated Eb value increases in the following order: 1 (−2.68 eV) < 4 (−1.77 eV) < 2 (−1.45 eV) < 3 (−0.58 eV) < 5 (2.66 eV). The negative Eb value indicates a strong interaction of the Cu4 cluster and nitrogen-doped graphene supports, and thus a good stability. Thus, the isomer 1 with inverted-tetrahedron-like structure possesses the strongest interaction of the Cu4 cluster and nitrogen-doped graphene supports among all isomers for the Cu4/N3GN catalysts studied here. The calculated Eb value of the inverted-tetrahedron-like isomer is about two times as large as that for the two rhombus-like isomers 2 and 4 (−2.68 vs. −1.45 and −1.77 eV). Our optimization calculations also provide the same results, in which the average Cu–N bond length of the inverted-tetrahedron-like isomer is optimized to be shorter than that of the two rhombus-like isomers (2.00 vs. 2.04 Å and 2.01 Å). Meanwhile, the Bader charge analysis shows that the charges of the coordinated Cu atom binding to the three N atoms in the inverted-tetrahedron-like isomer is about −0.57e. Furthermore, the charge density difference shows that electrons accumulate on the three N atoms and adjacent C atoms on the support surface of the inverted-tetrahedron-like isomer 1, which indicates an electron-transfer process from the coordinated Cu atom to the nitrogen-doped graphene support. Moreover, the extra electrons were entirely delocalized over the surface of nitrogen-doped graphene support. Such delocalized effects effectively improve the stability of the Cu4/N3GN catalyst. Meanwhile, these results support a donor–acceptor bonding interaction of the coordinated Cu atom and the nitrogen-doped graphene supports in the inverted-tetrahedron-like isomer. By the same logic, the donor–acceptor bonding interaction also can be found in two rhombus-like isomers. The charge density difference also shows that electrons accumulate between the three N atoms and coordinated Cu atom in the two rhombus-like isomers. However, no delocalization effects of the extra electrons have been observed on the nitrogen-doped graphene supports in the rhombus-like isomers. Thus, the stability of both isomers is not better than that of the inverted-tetrahedron-like isomer. All of these results indicate that the strong interaction of the Cu4 cluster with the inverted-tetrahedron-like structure and the nitrogen-doped graphene substrate experimentally provide good feasibility in terms of synthesis of the Cu4/N3GN catalyst. We will focus on the inverted-tetrahedron-like isomer in the following discussions.
3.2. Adsorption and activation of CO2 over the Cu4/N3GN catalyst
In the inverted-tetrahedron-like isomer, the four Cu atoms are tightly coupled through metal bonds to form a cluster (see ESI Table S1†). One Cu atom in the Cu4 cluster is anchored to the defects of the nitrogen-doped graphene substrate, and interacts with the three N atoms on the substrate. The calculated Bader charge of the Cu atoms, which does not directly attach to the N atom, is about zero (see ESI Table S2†). Thus, the Cu4 cluster in the inverted-tetrahedron-like isomer may be described as a Cu+–Cu03 configuration, in which the three Cu0 species form an atomic interface over the Cu4 cluster. The transition metal center in a lower oxidation state always displays stronger ability to bind the adsorbates, according to previous investigations. Furthermore, the atomic interface formed by the unsaturated three Cu0 species in the Cu4/N3GN catalyst may have the ability to interact with molecules. Thus, they should be viewed as potential sites for the adsorption and activation of the inert CO2 molecule. The effective combination of three possible Cu0 sites may adsorb one or two CO2 molecules over the surface of the Cu4/N3GN catalyst.For the adsorption of the first CO2 molecule, our DFT calculations provide three possible adsorption configurations (top, bridge_1, and bridge_2 models). For the top model, the CO2 molecule was adsorbed on the top of three Cu0 sites, in which the C and two O atoms of the CO2 molecule directly interact with three Cu0 sites. Meanwhile, in the bridge_1 model, the CO2 molecule was localized at a position between two Cu atoms, in which two O atoms of the CO2 molecule directly interact with the two Cu0 sites. On the other hand, in the bridge_2 model, the C atom and one O atom of the CO2 molecule directly interact with two Cu0 sites.
In order to identify the stability of these adsorption configurations, the adsorption energy (Ead) has been calculated by using our DFT calculations. The calculated Ead values also have been compared in Fig. 2. It was found that the calculated Ead value increases in the following order: Ead (bridge_2) (−0.87 eV) < Ead (bridge_1) (−0.19 eV) < Ead (top) (0.01 eV), indicating a strong adsorption of CO2 on the Cu4/N3GN catalyst in the bridge _2 model. By contrast, the calculated Ead values of the top and bridge_1 model are both higher than bridge_2, indicating that both adsorption configurations are not stable species. Such results indicate that the two Cu0 sites in the most stable adsorption configuration (bridge_2) are not occupied as the adsorption of the first CO2 molecule, which is available for adsorption of the second CO2 molecule.
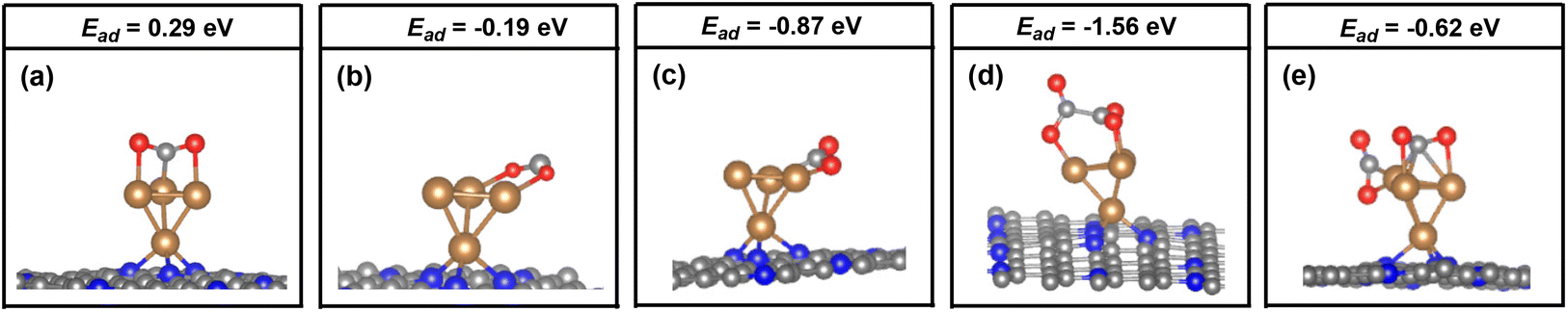 | ||
| Fig. 2 The key geometric parameters and adsorption energy analysis of the possible adsorption configuration of the CO2 molecule on the Cu4/N3GN catalyst with the (a) top, (b) bridge_1, (c) bridge_2, (d) coupling and (e) isolating models. | ||
For the adsorption of the second CO2 molecule, two possible adsorption configurations have been obtained (coupling and isolating models). The most stable configuration is found to be the coupling model (see Fig. 2d), in which the three O atoms of the two CO2 molecules direct interact with the three Cu0 sites. Meanwhile, a direct coupling of two adsorbed CO2 molecules to form a CO2 dimer with a C–C bond length of 1.55 Å has been found, according to our optimization calculations. The calculated Ead value of the coupling model is about 0.94 eV lower in energy than that of the isolating model (−1.56 vs. −0.62 eV). It is well known that the selectivity of the catalyst to the C2+ products in CO2RR is closely related to the formation of C–C bonds. According to previous reports, the formation of the C–C bond arises from three major pathways, including the *CO dimerization, *CO and *CHO dimerization, and *CHO dimerization over the catalyst surface. However, the direct coupling of two inert CO2 molecules to form a CO2 dimer with the strong C–C bond has not been found over the Cu-based catalysts. Interestingly, as shown in Fig. 2d, our optimization calculations provide a unique geometric arrangement, in which the two adsorbed CO2 molecules are bound to each other via a strong C–C single bond over the atomic interface of the Cu4/N3GN catalyst. Such results strongly suggest a direct coupling of two inert CO2 molecules to form a CO2 dimer over the atomic interface of the Cu4/N3GN catalyst.
3.3. CO2 dimerization
Electrochemical reduction of CO2 is a very complicated system. The CO2 molecule could be reduced to *OCHO, *COOH, *CO, *CHO, *CH, and *CH2, and others in the series of electrochemical elementary steps, which would become the potential coupling species for the formation of the C–C bond in CO2RR.49,79,80 These species may occupy the active sites of the catalyst and suppress the direct coupling of two CO2 molecules over the surface of the Cu4/N3GN catalyst. Owing to the low surface concentrations of *CHO, *CH, *CH2, and other multiple reduced species of CO2 molecule over the catalytic surface, the reaction pathway for the coupling of these species to form the C–C bond is not important when compared with the high concentration species *CO2, *OCHO, *COOH, and *CO. In the present paper, staring from the *CO2, *OCHO, *COOH, and *CO species, all possible coupling reactions for the combination of these species have been considered. According to our optimization calculations, four possible coupling pathways have been obtained. The calculated free energy curves for these reaction pathways are shown in Fig. 3.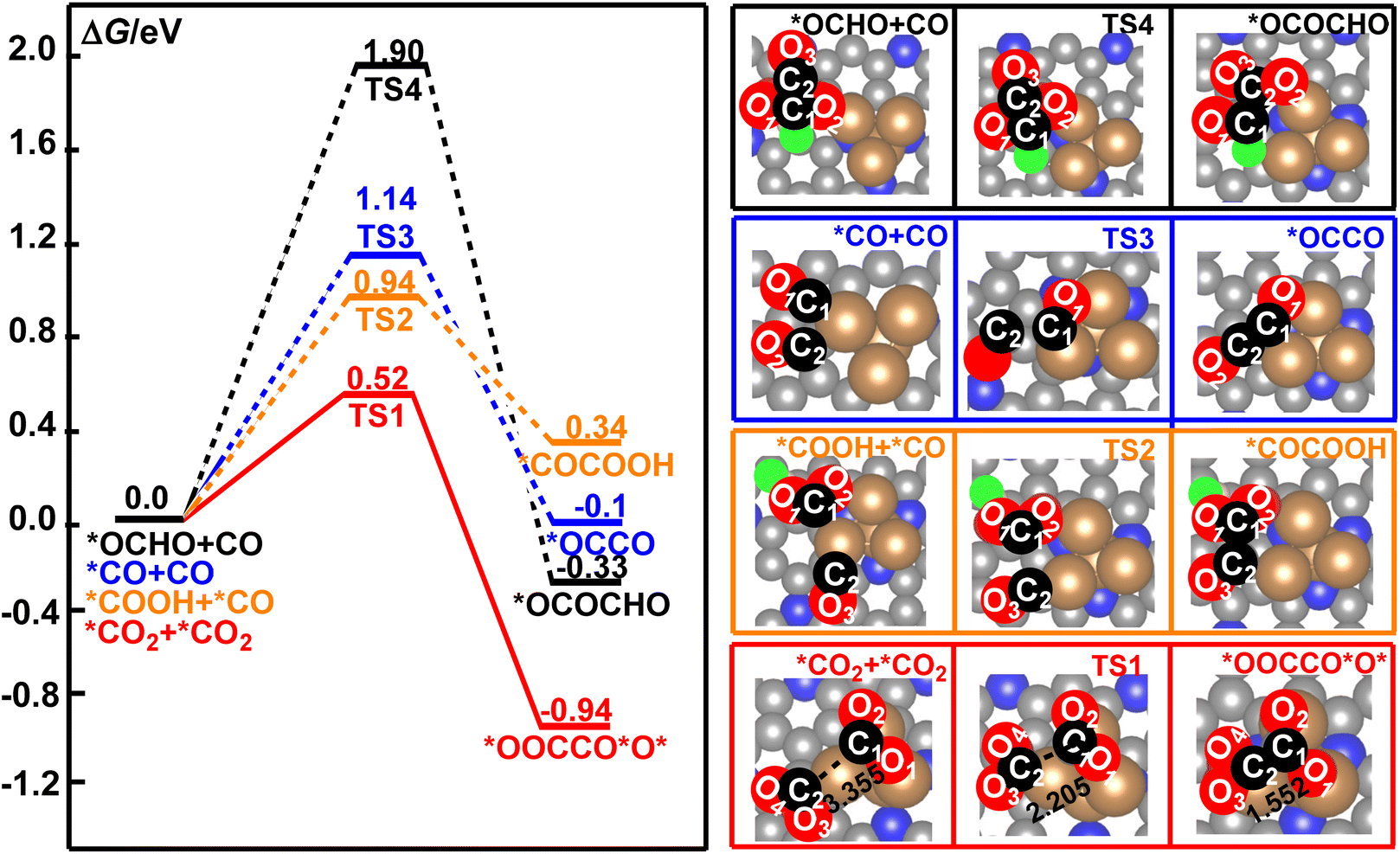 | ||
| Fig. 3 The calculated free energy profiles for the direct coupling of two *CO2, *OCHO and CO, *COOH and *CO, and two CO molecules to form a C–C bond on the surface of the Cu4/N3GN catalyst. | ||
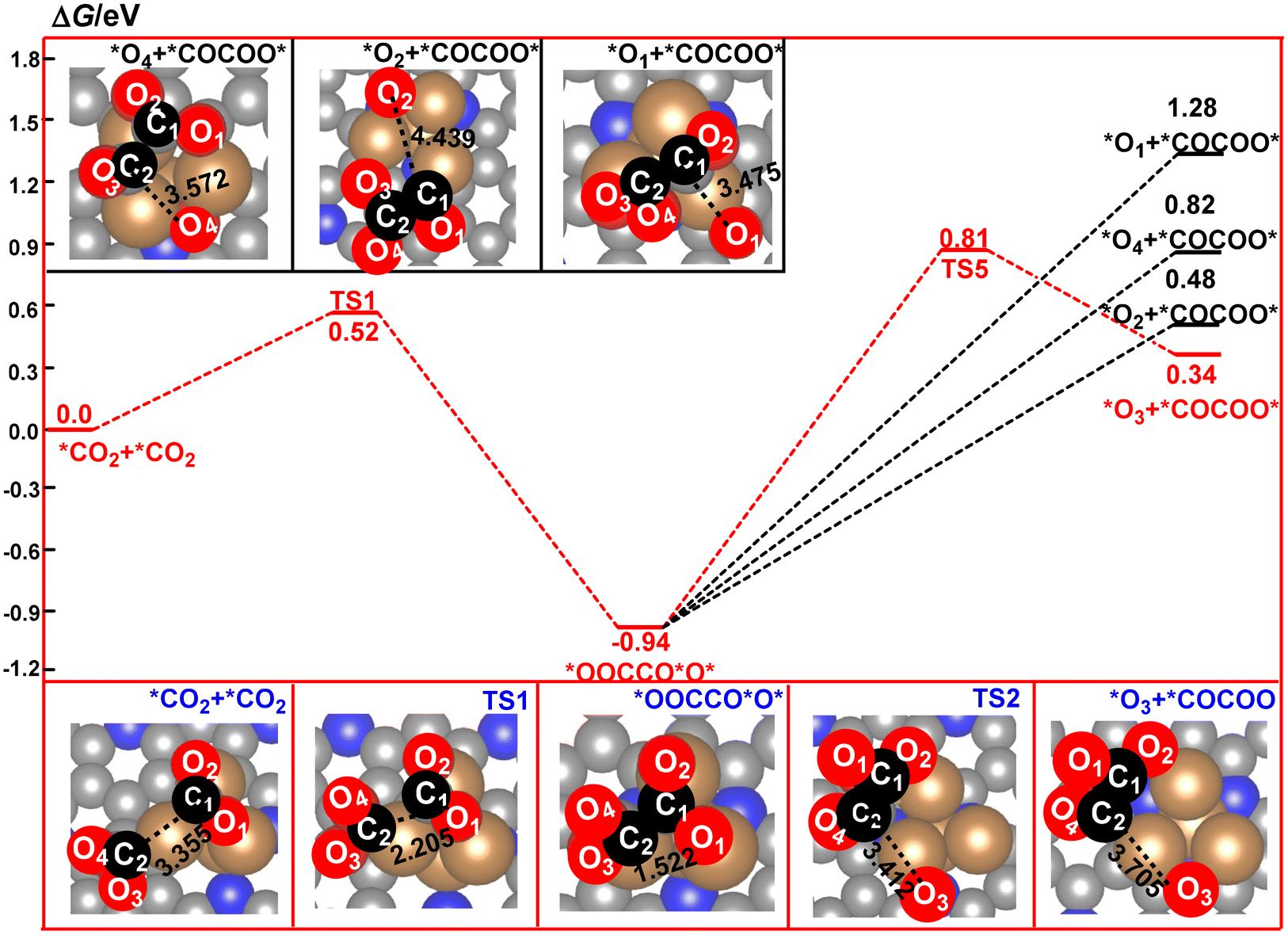
It was found that the calculated free energy curve for the direct coupling of two *CO2 molecules is located at the bottom, and has the lowest free energy barrier. By contrast, the calculated free energy curves of the direct coupling of two CO molecules are located in a third high-energy place, in which the calculated free energy barrier is about 0.62 eV higher in energy than that of the direct coupling of two CO2 molecules. The calculated relative energy of the CO dimer is about 0.84 eV higher in energy than that of the CO2 dimer. Thus, the direct coupling of two CO molecules over the surface of the Cu4/N3GN catalyst is both thermodynamically and kinetically unfavorable when compared with that of the two CO2 molecules. As shown in Fig. 3, the calculated free energy curve of the coupling of *CO and *OCHO is located at the top, and has the highest energy barrier among them. Thus, such coupling reaction is not significant both thermodynamically and kinetically. Although the calculated free energy curve for the coupling of *CO and *COOH is located in a second high-energy place, the calculated free energy barrier is about 2 times as high as that of the direct coupling of two CO2 molecules (0.94 vs. 0.52 eV). Thus, the competition effects of this coupling reaction may not be significantly relevant to the direct coupling of two CO2 molecules. All these results indicate that the direct coupling of two CO2 molecules over the surface of the Cu4/N3GN catalyst could be the main reaction for the formation of the C–C bond in the complicated electrochemical system.
The optimized geometric arrangements of the key transition states and intermediates for the direct coupling of two CO2 molecules are shown in Fig. 3. It was found that the two CO2 molecules firstly approach the three Cu0 species to form an adsorption complex *CO2+ *CO2 (isolating models). In *CO2+ *CO2, the optimized O2C⋯CO2 distance is 3.355 Å, indicating a very weak interaction between the two CO2 molecules over the surface of the Cu4/N3GN catalyst. The calculated Bader charge of the two adsorbed CO2 molecules is +0.78 e and +0.49 e in *CO2+ *CO2, respectively. The calculated Bader charge of the three Cu atoms in *CO2+ *CO2, which are not attached to the N atom of the nitrogen-doped graphene substrate, is −0.25, −0.44, and −0.25 e, respectively. The calculated Bader charge of the Cu atom coordinated to the three N atoms of the nitrogen-doped graphene substrate is −0.46 e in *CO2+ *CO2. Compared with the neutral CO2 molecule and Cu4 cluster with Cu+–Cu30 configuration on the clear surface of the Cu4/N3GN catalyst, such results indicate that the two adsorbed CO2 molecules have been activated via an electron-transfer process from three Cu0 species in the Cu4 cluster of the Cu4/N3GN catalyst to the two adsorbed CO2 molecules. Thus, the two CO2 moieties in *CO2+ *CO2 have been reduced and possess the bent structure with elongated C–O bond length relative to the free CO2 molecule, and can be viewed as two CO2˙− anionic radicals. A detailed analysis of the calculated Bader charge shows that the C atom is −2.10, −1.38, and −1.61 e for the free CO2 molecule and two adsorbed CO2 molecules, respectively. The calculated Bader charge of the two O atoms is (1.05 and 1.05 e), (1.08 and 1.08 e), and (1.04 and 1.06 e) for the free and two adsorbed CO2 molecules, respectively. Such results indicate that the reduced electrons are mainly localized on the C atom adjacent to the O atoms for the two adsorbed CO2 molecules in this electron-transfer process (Δq (C) = 0.72 and 0.49 e for the two adsorbed CO2 molecules in this electron-transfer process, respectively). Thus, the electron configuration of the two adsorbed CO2 molecules should be described as [OC˙O]−. The C atom of the two adsorbed CO2 molecules would significantly contribute to the formation of a C–C single bond in the process of CO2 dimerization. Then, our optimization calculation shows the direct coupling of two anionic CO2 radicals with [OC˙O]− configuration over the Cu4/N3GN catalyst viaTS1, which needs an activation energy of 0.52 eV with an imaginary frequency of 244.83 cm−1. At TS1, the O2C⋯CO2 distance was shortened to 2.205 Å, indicating that a new bond may form between the two adsorbed CO2 molecules. In TS1, the calculated Bader charge of the two C atoms in the adsorbed two CO2 molecules is −1.47 and −1.72 e, respectively, compared with the Bader charge of the C atoms in free CO2 (−2.10 e), indicating the distinct radical feature for the two adsorbed CO2 molecules in TS1. After TS1, the two CO2 anionic radicals come together to form a CO2 dimer, *OOCCO*O*, over the Cu4/N3GN catalyst. This coupling process is calculated to be exoergic by 0.94 eV, indicating that *OOCCO*O* has a high thermodynamic stability. Furthermore, the direct coupling of two CO2 molecule over the surface of Cu4/N3GN catalyst has a large thermodynamic driving force. All of these results indicate that the CO2 dimerization reaction over the Cu4/N3GN catalyst should be viewed as a free-radical-coupling reaction.
As mentioned above, the two adsorbed CO2 molecules over the atomic interface of Cu4/N3GN catalyst were reduced and possess bent structure. The reduced electron is mainly localized over the C atom of the adsorbed CO2 molecule, according to the Bader charge analysis. The frontier molecular orbitals (FMOs) of the free CO2 molecule are listed in Fig. 4. It can be seen that the two degenerated LUMOs of the free CO2 molecule consist of 2p orbitals of the C atom and two O atoms with π* antibonding feature, and mainly localized over the C atom adjacent to the O atoms. Thus, injection of extra electrons into the LUMOs would localize on the C atom of the CO2 molecule, which are in good agreement with our Bader charge analysis.
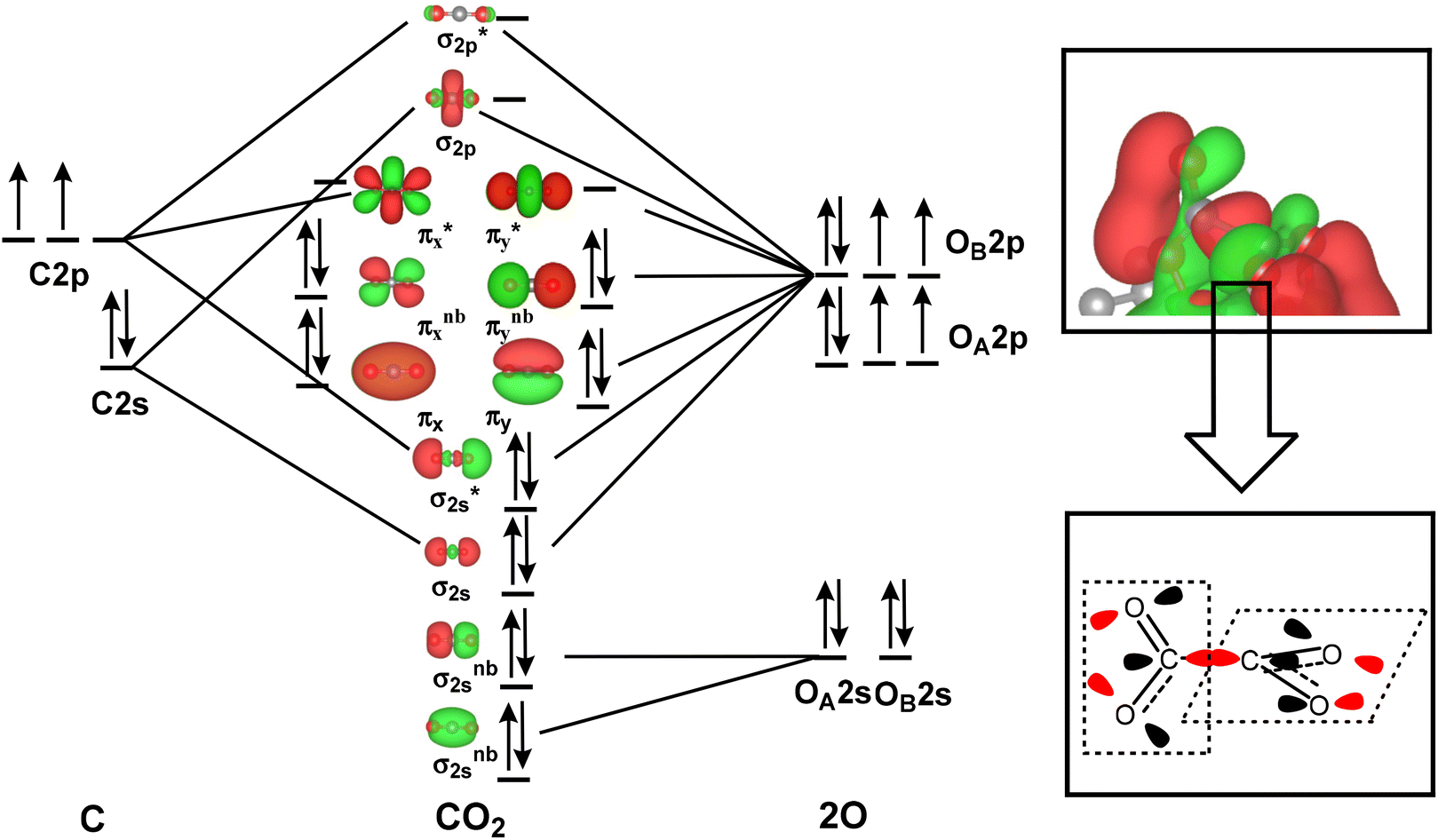 | ||
| Fig. 4 The frontier molecular orbitals of free carbon dioxide molecules and the HOMO of the *OOCCO*O* dimer, highlighting that the effective overlapping of two distorted π* antibonding orbitals of two bent CO2 molecules results in the formation of the C–C bond in the CO2 dimer. | ||
Meanwhile, our electronic structural analysis shows that *OOCCO*O* provided a unique molecular orbital topology for the formation of the C–C bond in the CO2 dimer. As shown in Fig. 4, our optimization calculations show that the two adsorbed CO2 molecules with bent structure definite two planes, in which the dihedral angle of the two planes is calculated to be 121.9°. Such geometric arrangement could effectively avoid the repulsion effects from the O atoms of the CO2 molecule, and make the two C atoms of the adsorbed CO2 molecules get closer in the process of CO2 dimerization. The molecular orbital topology (HOMO) of *OOCCO*O* represents the C–C bonding interaction. It can be seen that HOMO could be viewed as an effective overlapping of two distorted π* antibonding orbitals of the CO2 molecule. The bent arrangement of the adsorbed CO2 molecule leads to a mixing of 2p orbitals of the two O atoms in the CO2 molecule, which effectively polarizes the 2p orbital of the C atom in the CO2 molecule. The polarized 2p orbital of the C atom promotes the overlapping of two distorted π* antibonding orbitals, and significantly contributes to the formation of the C–C bond in the CO2 dimer. All of these results indicate that the bent arrangement of the CO2 molecule is the key factor in the determination of the formation of the C–C bond via effective orbital interactions.
The nature of the CO2 dimer, *OOCCO*O*, is a key factor in the determination of the formation of the C2 product in CO2RR. As shown in Fig. 5, the four oxygen atoms in *OOCCO*O* are the potential sites in the following elementary steps. In the present paper, we firstly considered the possible dissociated products of *OOCCO*O*via cleavage of the four C–O bonds, respectively. The calculated relative energy of the four dissociated products has been compared in Fig. 5. It can be found that the calculated relative energy of the four dissociated products increases in the following order: O3 (0.34 eV) < O2 (0.48 eV) < O4 (0.82 eV) < O1 (1.28 eV), indicating that these four dissociated processes are all endergonic. Furthermore, the most stable species among them comes from dissociation of the O3 atom from the *OOCCO*O* intermediate to form a *OCCOO* species and a remaining *O species. Owing to the high thermodynamic stability of *OOCCO*O*, this dissociated process is calculated to have a high free energy barrier of 1.75 eV. However, this high barrier can be compensated by the following reduction reaction of the remaining *O species to H2O over the Cu4/N3GN catalyst.
 | ||
| Fig. 5 Calculated reaction pathways for the formation and decomposition of the CO2 dimer on the surface of the Cu4/N3GN catalyst. | ||
After TS5, the O3 atom moves from the top site of the Cu atom into the bridge site between the two Cu atoms. Meanwhile, the dissociated fragment of *OCCOO* was bound over the surface of the Cu4/N3GN catalyst via a new Cu–C2 bond and a Cu–O2 bond. The remaining O3 atom would be reduced to H2O and leave the surface of the Cu4/N3GN catalyst. Although a lot of efforts have been made, we did not find any rational geometric arrangement for the hydrogenation of *OOCCO*O*. As shown in Fig. 3, three out of the four oxygen atoms in *OOCCO*O* were bound to the three Cu atoms of the Cu4/N3GN catalyst via three strong Cu–O bonds. Thus, the three O atoms are located in the Cu–O–C units and in the chemical saturated states. Such structural arrangement of the three O atoms significantly weakens the reactivity with the proton–electron pairs. The O4 atom, which was bound to the *OOCCO*O*via a C2–O4 bond, also cannot react with the proton–electron pair to form a hydrogenation product because of the weak reactivity. All of these results indicate that the conversation of the stable *OOCCO*O* to *OCCOO* over the surface of the Cu4/N3GN catalyst could be triggered by a heating process because dissociation of the stable CO2 dimer is an endergonic process with a high free energy barrier. By contrast, implementing of the electroreduction process at room temperature may not produce the C2 product for our studied system in this work.
3.4. Formation of ethane
In previous literature, the number of oxygen atoms in the C–C coupling product is up to two, such as *OCCO.81,82 In general, the proton–electron pair preferentially attacks at the O atom of the C–C coupling product because of the high basicity of the O atom relative to the C atom.82,83 Thus, the nature of the O atom in the C–C coupling product would significantly affect the selectivity of the catalyst in CO2RR. For our studied system, the decomposition product of the CO2 dimer, *O3+ *OCCOO*, contains four O atoms. The possible reaction pathways corresponding to the generation of the C2 products entirely differ from the reported mechanism. The calculated free energy profiles of the whole reaction pathway for the hydrogenation of *O3+ *OCCOO* to ethane is shown in Fig. 6 and 7. All of the O sites of the *O3+ *OCCOO* intermediate have been considered for the first hydrogenation step. As expected, the reduction of the *O3 atom to a *OH group is calculated to be energetically much more favorable among them. According to our free energy calculations, hydrogenation of *O3+ *OCCOO* to the *COCOO* + *OH intermediate is calculated to be exoergic by 1.19 eV, indicating that the *COCOO* + *OH intermediate is a thermodynamically stable species, and will remain on the catalyst surface long enough for further reduction to H2O.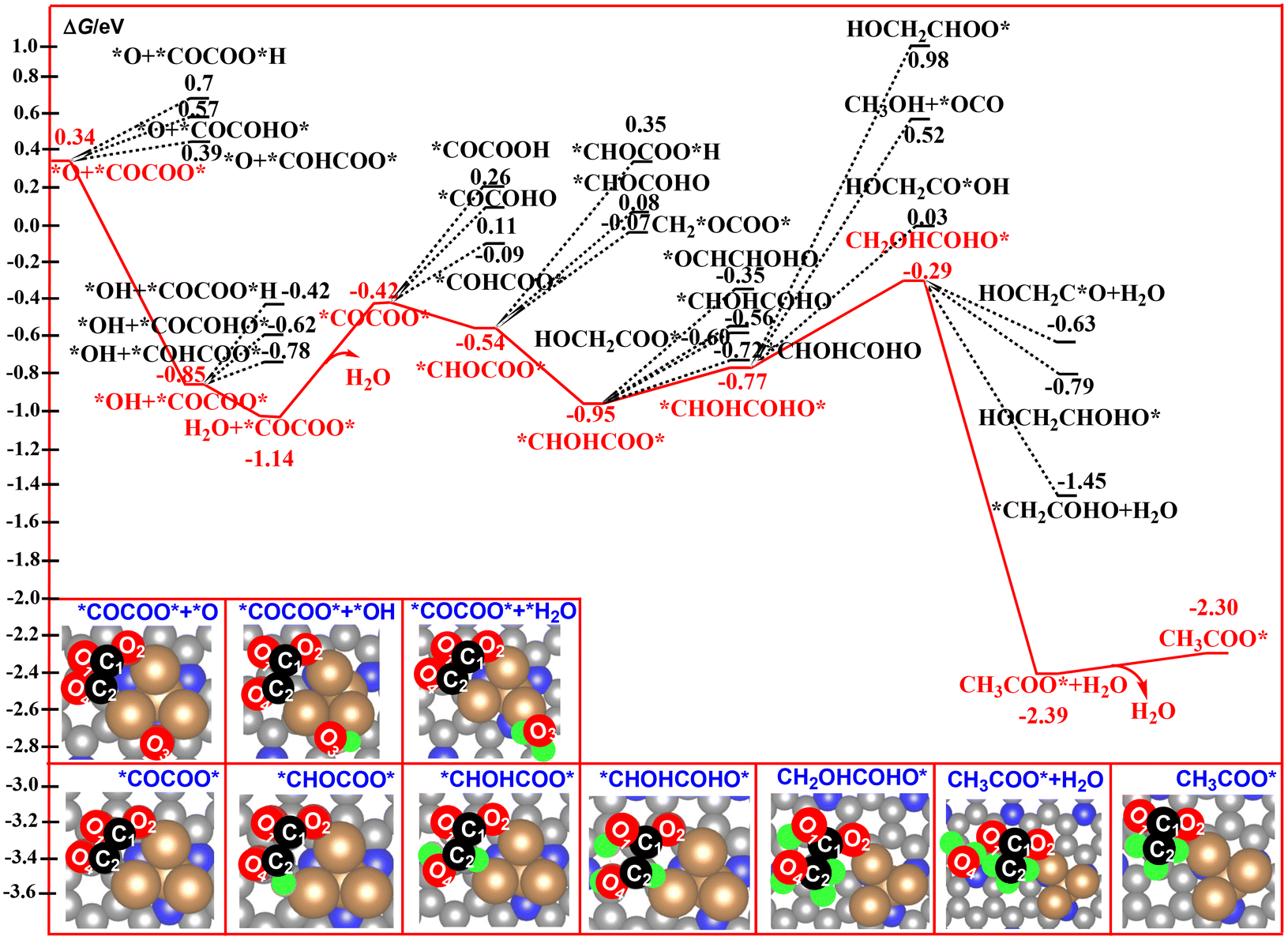 | ||
| Fig. 6 Calculated free energy profiles for the electrocatalytic reduction of *O3+ *OCCOO* to CH3COO*. | ||
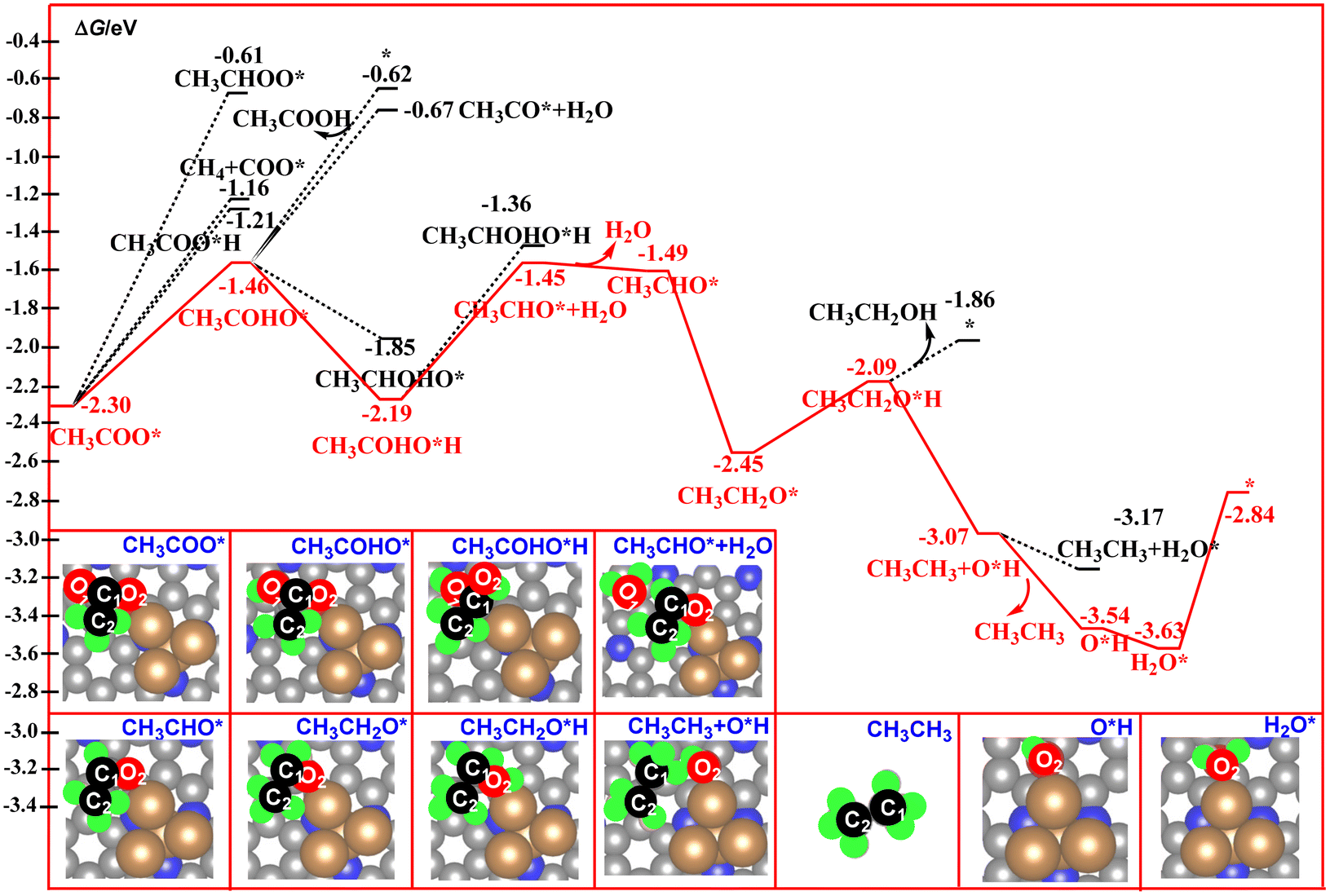 | ||
| Fig. 7 Calculated free energy profiles for the electrocatalytic reduction of CH3COO* to acetic acid, ethanol, and ethane. | ||
For the second reduction step, the hydrogenation of the *COCOO* + *OH intermediate to *COCOO* + *H2O is calculated to be exoergic by 0.29 eV. Next, desorption of the produced H2O molecules from the catalyst surface to form a *COCOO* intermediate is calculated to be endergonic by 0.72 eV.
Subsequently, the proton–electron pair preferentially attacks at the C2 atom of *COCOO*, which binds to the Cu atom, to generate *CHOCOO* intermediates with a free energy downhill of 0.12 eV. This step leads to a complete cleavage of the Cu–C2 bond, and the CHOCOO* moiety was bound to the catalyst surface via a Cu–O2 bond. Owing to the strong Cu–O2 bonding interaction, the hydrogenation of the O2 site in the conversion from *COCOO* to *COCOOH is calculated to be energetically unfavorable. Then, the proton–electron pair preferentially attacks at the O4 atom of *CHOCOO* to produce *CHOHCOO* with a free energy downhill of 0.41 eV. The optimized Cu–O2 bond length increases from 1.892 Å in *CHOCOO* to 1.896 Å in *CHOHCOO*, indicating that this hydrogenation step does not largely affect the Cu–O2 bonding interaction. In the following two hydrogenated steps, the *CHOHCOO* was hydrogenated to form CH2OHCOHO* with two exothermic steps by 0.18 and 0.48 eV, respectively. For the thermodynamically unstable CH2OHCOHO* species, it was immediately hydrogenated to form the second H2O molecule in CH3COO* + H2O over the Cu4/N3GN catalyst. This hydrogenation step is calculated to be most exoergic by 2.10 eV among all of the reaction steps studied here. Desorption of the second H2O molecule from the catalyst surface leads to the formation of CH3COO*. The calculated desorption energy for this process is 0.09 eV, indicating a very weak interaction of the produced H2O molecule and the catalyst surface.
For the CH3COO* intermediate, it could be reduced to CH3COOH via hydrogenation of the O2 atom. As shown in Fig. 7, the C2 atom in CH3COO* is in a saturated state via formation of three C–H bonds and one C–C bond. Thus, the O1, O2, and C1 atoms have been assigned as the potential sites for the reaction with the proton–electron pairs. According to our free energy calculations, hydrogenation of the O1 atom to form CH3COHO* is much more favorable compared to the O2 and C1 atoms.
This step is calculated to be exoergic by 0.84 eV. By contrast, hydrogenation of the O2 atom to form a CH3COOH molecule over the catalyst surface is calculated to be exoergic by 1.09 eV, and the release of the produced CH3COOH molecule from the catalyst surface via cleavage of the Cu–O2 bond needs a desorption energy of 0.59 eV. Such results indicate that the strong Cu–O2 bonding interaction suppresses the formation of the CH3COOH molecule. By contrast, the continuous hydrogenation of the CH3COHO* to CH3COHO*H is a favorable path, in which this step is calculated to be exoergic by 0.73 eV. Next, hydrogenation of CH3COHO*H to CH3CHO* + H2O results in the formation of the third H2O molecule over the catalyst surface with a free energy downhill of 0.74 eV. After desorption of the third H2O molecule from the catalyst surface, a CH3CHO* intermediate is formed with a free energy downhill of 0.04 eV. Further the hydrogeneration of CH3CHO* results in the formation of the CH3CH2O* intermediate with a free energy downhill of 0.96 eV. For the CH3CH2O* intermediate, the hydrogeneration of the O2 atom of the CH3CH2O* intermediate would produce the CH3CH2OH molecule. This step is calculated to be exoergic by 0.36 eV. For the CH3CH2O*H intermediate, the pathway bifurcates into ethanol– and ethane–forming routes. As shown in Fig. 7, the releasing of the CH3CH2OH molecule via cleavage of the Cu–O2 bond is found to be more unfavorable than the formation of the ethane molecule in CH3CH3+ *OH, as the calculated free energy of the former is 1.21 eV higher than that of the latter. Furthermore, desorption of the CH3CH3 molecule from the catalyst surface is calculated to be exoergic by 0.47 eV. These results indicate that ethane is the main product of direct CO2 reduction catalyzed by the Cu4/N3GN catalyst. Furthermore, the hydrogeneration of the produced *OH group results in the formation of the fourth H2O molecule with a free energy downhill of 0.09 eV. Finally, the catalyst was resumed via release of the fourth H2O with a free energy uphill of 0.79 eV. In the whole reaction pathway, the potential-limiting step is still the decomposition of the CO2 dimer to *O + *OCCOO* with an energy of 1.75 eV, which is lower than the direct coupling of two CO on the Cu(111) surface (1.81 eV). Thus, the Cu4/N3GN catalyst possesses excellent catalytic activity and high ethane selectivity for direct CO2 reduction.
4. Conclusions
We have systematically investigated the nitrogen-doped graphene-supported Cu4 cluster with various structures. Our optimization calculations show that anchoring the Cu4 cluster with an inverted-tetrahedron-like structure onto the nitrogen-doped graphene supports results in a catalyst with strong interaction between them, which provides high feasibility in terms of the experimental synthesis of the catalyst. The electronic structure analysis shows that the atomic interface of the inverted-tetrahedron-like Cu4 cluster possesses a Cu+–Cu03 configuration. Furthermore, the three unsaturated Cu0 species in the Cu4 cluster are the potential sites for the adsorption and activation of the CO2 molecules. Our optimization and free energy calculations reveal that the adsorption of two CO2 molecules over the catalyst surface results in a direct coupling of two inert CO2 molecules to form a CO2 dimer with a strong C–C bond. The calculated free energy profiles show that the CO2 dimerization should be the main reaction relevant to the coupling reaction of *OCHO and CO, *COOH and *CO, and two CO molecules. The mechanistic investigation shows that the two adsorbed CO2 molecules were firstly reduced to CO2˙− anionic radicals with [OC˙O]− configuration via an electron transfer process from the surface Cu0 species to the two CO2 molecules. Furthermore, the direct coupling of two anionic radicals, CO2˙−, results in the formation of the C–C bond in the CO2 dimer. The orbital analysis indicates that the bent arrangement of the CO2 molecule effectively promotes the overlap of two distorted π* antibonding orbitals, which significantly contribute to the formation of the C–C bond in the CO2 dimer. The free energy calculation shows that the CO2 dimer could be reduced to ethane in the series of electrochemical elementary steps. The high ethane selectivity of the Cu4/N3GN catalyst mainly arises from the strong Cu–O bonding interaction. This study calls for more fundamental investigations on the mechanism for the formation of the C–C bond in CO2RR.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (32130073, 21373043) and National Natural Science Foundation of Jilin Province (YDZJ202201ZYTS540).References
- H. Zhang, Y. Yang, Y. Liang, J. Li, A. Zhang, H. Zheng, Z. Geng, F. Li and J. Zeng, Molecular Stabilization of Sub–Nanometer Cu Clusters for Selective CO2 Electromethanation, ChemSusChem, 2022, 15, e202102010 CrossRef CAS PubMed.
- H. Dai, H. Zou, T. Song, L. Gao, S. Wei, H. Liu, H. Xiong, C. Huang and L. Duan, Pyridyl-Containing Graphdiyne Stabilizes Sub-2 nm Ultrasmall Copper Nanoclusters for the Electrochemical Reduction of CO2, Inorg. Chem. Front., 2023, 10, 2189–2196 RSC.
- H. Xiong, A. K. Datye and Y. Wang, Thermally Stable Single–Atom Heterogeneous Catalysts, Adv. Mater., 2021, 33, 2004319 CrossRef CAS.
- B. Zhang, Y. Chen, J. Wang, H. Pan and W. Sun, Supported Sub–Nanometer Clusters for Electrocatalysis Applications, Adv. Funct. Mater., 2022, 32, 2202227 CrossRef CAS.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, Carbon Dioxide Conversion to Methanol over Size-Selected Cu4 Clusters at Low Pressures, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern and F. Mehmood, Subnanometre Platinum Clusters as Highly Active and Selective Catalysts for the Oxidative Dehydrogenation of Propane, Nat. Mater., 2009, 8, 213–216 CrossRef CAS.
- G. Sun, A. N. Alexandrova and P. Sautet, Structural Rearrangements of Subnanometer Cu Oxide Clusters Govern Catalytic Oxidation, ACS Catal., 2020, 10, 5309–5317 CrossRef CAS.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard III, J. Yano and E. J. Crumlin, Subsurface Oxide Plays a Critical Role in CO2 Activation by Cu (111) Surfaces to Form Chemisorbed CO2, the First Step in Reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CrossRef CAS.
- X. Su, X.-F. Yang, Y. Huang, B. Liu and T. Zhang, Single-Atom Catalysis toward Efficient CO2 Conversion to CO and Formate Products, Acc. Chem. Res., 2018, 52, 656–664 CrossRef.
- A. Mazheika, Y.-G. Wang, R. Valero, F. Viñes, F. Illas, L. M. Ghiringhelli, S. V. Levchenko and M. Scheffler, Artificial-Intelligence-Driven Discovery of Catalyst Genes with Application to CO2 Activation on Semiconductor Oxides, Nat. Commun., 2022, 13, 419 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State–of–the–Art Accomplishment, Challenges, and Prospects, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- N. Aguilar, M. Atilhan and S. Aparicio, Single Atom Transition Metals on MoS2 Monolayer and Their Use as Catalysts for CO2 Activation, Appl. Surf. Sci., 2020, 534, 147611 CrossRef CAS.
- L. Yuan, S.-F. Hung, Z.-R. Tang, H. M. Chen, Y. Xiong and Y.-J. Xu, Dynamic Evolution of Atomically Dispersed Cu Species for CO2 Photoreduction to Solar Fuels, ACS Catal., 2019, 9, 4824–4833 CrossRef CAS.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Electrochemical CO2 Reduction (CO2RR) to Multi-Carbon Products over Copper-Based Catalysts, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Catalyst Electro-Redeposition Controls Morphology and Oxidation State for Selective Carbon Dioxide Reduction, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Cu Metal Embedded in Oxidized Matrix Catalyst to Promote CO2 Activation and CO Dimerization for Electrochemical Reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, Dopant-Induced Electron Localization Drives CO2 Reduction to C2 Hydrocarbons, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, Hydrogenation of CO2 to Methanol: Importance of Metal–Oxide and Metal–Carbide Interfaces in the Activation of CO2, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- A. G. Saputro, A. L. Maulana, F. Fathurrahman, G. Shukri, M. H. Mahyuddin, M. K. Agusta, T. D. K. Wungu and H. K. Dipojono, Density Functional and Microkinetic Study of CO2 Hydrogenation to Methanol on Subnanometer Pd Cluster Doped by Transition Metal (M = Cu, Ni, Pt, Rh), Int. J. Hydrogen Energy, 2021, 46, 14418–14428 CrossRef CAS.
- J. Timoshenko, A. Halder, B. Yang, S. Seifert, M. J. Pellin, S. Vajda and A. I. Frenkel, Subnanometer Substructures in Nanoassemblies Formed from Clusters Under A Reactive Atmosphere Revealed Using Machine Learning, J. Phys. Chem. C, 2018, 122, 21686–21693 CrossRef CAS.
- M. Gallego, A. Corma and M. Boronat, Influence of the Zeolite Support on the Catalytic Properties of Confined Metal Clusters: A Periodic DFT Study of O2 Dissociation on Cun Clusters in CHA, Phys. Chem. Chem. Phys., 2022, 24, 30044–30050 RSC.
- A. Halder, C. Lenardi, J. Timoshenko, A. Mravak, B. Yang, L. K. Kolipaka, C. Piazzoni, S. Seifert, V. Bonacic-Koutecky and A. I. Frenkel, CO2 Methanation on Cu-cluster Decorated Zirconia Supports with Different Morphology: A Combined Experimental in situ GIXANES/GISAXS, ex situ XPS and Theoretical DFT Study, ACS Catal., 2021, 11, 6210–6224 CrossRef CAS.
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang and W. Chen, Complementary Operando Spectroscopy Identification of in-situ Generated Metastable Charge-Asymmetry Cu2-CuN3 Clusters for CO2 Reduction to Ethanol, Nat. Commun., 2022, 13, 1322 CrossRef CAS PubMed.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, Dynamic Stability of Copper Single-Atom Catalysts Under Working Conditions, J. Am. Chem. Soc., 2022, 144, 17140–17148 CrossRef CAS.
- B. Yang, X. Yu, A. Halder, X. Zhang, X. Zhou, G. R. J. Mannie, E. Tyo, M. J. Pellin, S. Seifert and D. Su, Dynamic Interplay between Copper Tetramers and Iron Oxide Boosting CO2 Conversion to Methanol and Hydrocarbons under Mild Conditions, ACS Sustainable Chem. Eng., 2019, 7, 14435–14442 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean and R. E. Winans, Highly Selective Electrocatalytic CO2 Reduction to Ethanol by Metallic Clusters Dynamically Formed from Atomically Dispersed Copper, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- B. Yang, C. Liu, A. Halder, E. C. Tyo, A. B. Martinson, S. N. Seifert, P. Zapol, L. A. Curtiss and S. Vajda, Copper Cluster Size Effect in Methanol Synthesis from CO2, J. Phys. Chem. C, 2017, 121, 10406–10412 CrossRef CAS.
- J. Chen, Z. Wang, J. Zhao, L. Ling, R. Zhang and B. Wang, The Active Site of Ethanol Formation from Syngas over Cu4 Cluster Modified MoS2 Catalyst: A Theoretical Investigation, Appl. Surf. Sci., 2021, 540, 148301 CrossRef CAS.
- Y. Sun, S. Wang, J. Jia, Y. Liu, Q. Cai and J. Zhao, Supported Cu3 Clusters on Graphitic Carbon Nitride as An Efficient Catalyst for CO Electroreduction to Propene, J. Mater. Chem. A, 2022, 10, 14460–14469 RSC.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, CO2 Hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC Catalysts: Production of CO, Methanol, and Methane, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- F. Maleki, P. Schlexer and G. Pacchioni, Support Effects and Reaction Mechanism of Acetylene Trimerization over Silica-Supported Cu4 Clusters: A DFT Study, Surf. Sci., 2018, 668, 125–133 CrossRef CAS.
- I. C. Gerber and P. Serp, A Theory/Experience Description of Support Effects in Carbon-Supported Catalysts, Chem. Rev., 2019, 120, 1250–1349 CrossRef.
- T. Tang, Z. Wang and J. Guan, Electronic Structure Regulation of Single-Site MNC Electrocatalysts for Carbon Dioxide Reduction, Acta Phys.–Chim. Sin., 2022, 39, 10–3866 Search PubMed.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, Enhanced Catalytic Performance of Pt-Free Iron Phthalocyanine by Graphene Support for Efficient Oxygen Reduction Reaction, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- T. Wang, Q. Zhao, Y. Fu, C. Lei, B. Yang, Z. Li, L. Lei, G. Wu and Y. Hou, Carbon–Rich Nonprecious Metal Single Atom Electrocatalysts for CO2 Reduction and Hydrogen Evolution, Small Methods, 2019, 3, 1900210 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. R. Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Understanding Activity and Selectivity of Metal-Nitrogen-Doped Carbon Catalysts for Electrochemical Reduction of CO2, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- A. Azoulay, A. G. Baldovi, J. Albero, N. Azaria, J. Tzadikov, A. Tashakory, N. Karjule, S. Hayun, H. García and M. Shalom, Carbon-Phosphorus-Nitrogen Materials as Highly Thermally Stable Catalyst Supports for CO2 Hydrogenation to Methanol, ACS Appl. Energy Mater., 2022, 6, 439–446 CrossRef.
- Y. Yang, Y. Yang, Z. Pei, K.-H. Wu, C. Tan, H. Wang, L. Wei, A. Mahmood, C. Yan and J. Dong, Recent Progress of Carbon-Supported Single-Atom Catalysts for Energy Conversion and Storage, Matter, 2020, 3, 1442–1476 CrossRef.
- T. Tang, Z. Wang and J. Guan, Optimizing the Electrocatalytic Selectivity of Carbon Dioxide Reduction Reaction by Regulating the Electronic Structure of Single–Atom M–N–C Materials, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- M. Jia, C. Choi, T.-S. Wu, C. Ma, P. Kang, H. Tao, Q. Fan, S. Hong, S. Liu and Y.-L. Soo, Carbon-Supported Ni Nanoparticles for Efficient CO2 Electroreduction, Chem. Sci., 2018, 9, 8775–8780 RSC.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu and C. Wu, Exclusive Ni–N4 Sites Realize Near-Unity CO Selectivity for Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- L.-Y. Xiao, Z. Wang and J. Guan, Optimization Strategies of High-Entropy Alloys for Electrocatalytic Applications, Chem. Sci., 2023 10.1039/D3SC04962K.
- N. M. Julkapli and S. Bagheri, Graphene Supported Heterogeneous Catalysts: An Overview, Int. J. Hydrogen Energy, 2015, 40, 948–979 CrossRef CAS.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li and R. Zhang, Synergistic Effect of Atomically Dispersed Ni–Zn Pair Sites for Enhanced CO2 Electroreduction, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- F. Li, H. Wen and Q. Tang, Reaction Mechanism and Kinetics for Carbon Dioxide Reduction on Iron–Nickel Bi-Atom Catalysts, J. Mater. Chem. A, 2022, 10, 13266–13277 RSC.
- S. Wang, L. Li, J. Li, C. Yuan, Y. Kang, K. S. Hui, J. Zhang, F. Bin, X. Fan and F. Chen, High-Throughput Screening of Nitrogen-Coordinated Bimetal Catalysts for Multielectron Reduction of CO2 to CH4 with High Selectivity and Low Limiting Potential, J. Phys. Chem. C, 2021, 125, 7155–7165 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at A Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- T. Tang, Z. Wang and J. Guan, Structural Optimization of Carbon-Based Diatomic Catalysts towards Advanced Electrocatalysis, Coord. Chem. Rev., 2023, 492, 215288 CrossRef CAS.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang and B. Pang, Atomically Precise Dinuclear Site Active toward Electrocatalytic CO2 Reduction, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- H. Wang, G. Ren, Y. Zhao, L. Sun, J. Sun, L. Yang, T. Yu, W. Deng and L. Sun, In Silico Design of Dual-Doped Nitrogenated Graphene (C2N) Employed in Electrocatalytic Reduction of Carbon Monoxide to Ethylene, J. Mater. Chem. A, 2022, 10, 4703–4710 RSC.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, Precisely Constructing Orbital Coupling-Modulated Dual-Atom Fe Pair Sites for Synergistic CO2 Electroreduction, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- H. Shen, Y. Li and Q. Sun, Cu Atomic Chains Supported on β-Borophene Sheets for Effective CO2 Electroreduction, Nanoscale, 2018, 10, 11064–11071 RSC.
- O. Baturina, Q. Lu, F. Xu, A. Purdy, B. Dyatkin, X. Sang, R. Unocic, T. Brintlinger and Y. Gogotsi, Effect of Nanostructured Carbon Support on Copper Electrocatalytic Activity toward CO2 Electroreduction to Hydrocarbon Fuels, Catal. Today, 2017, 288, 2–10 CrossRef CAS.
- Z. Shi, W. Yang, Y. Gu, T. Liao and Z. Sun, Metal–Nitrogen–Doped Carbon Materials as Highly Efficient Catalysts: Progress and Rational Design, Adv. Sci., 2020, 7, 2001069 CrossRef CAS.
- Y. Yan, L. Ke, Y. Ding, Y. Zhang, K. Rui, H. Lin and J. Zhu, Recent Advances in Cu-based Catalysts for Electroreduction of Carbon Dioxide, Mater. Chem. Front., 2021, 5, 2668–2683 RSC.
- D.-H. Lim, J. H. Jo, D. Y. Shin, J. Wilcox, H. C. Ham and S. W. Nam, Carbon Dioxide Conversion into Hydrocarbon Fuels on Defective Graphene-Supported Cu Nanoparticles from First Principles, Nanoscale, 2014, 6, 5087–5092 RSC.
- Y. X. Duan, F. L. Meng, K. H. Liu, S. S. Yi, S. J. Li, J. M. Yan and Q. Jiang, Amorphizing of Cu Nanoparticles toward Highly Efficient and Robust Electrocatalyst for CO2 Reduction to Liquid Fuels with High Faradaic Efficiencies, Adv. Mater., 2018, 30, 1706194 CrossRef PubMed.
- K. Yao, J. Li, H. Wang, R. Lu, X. Yang, M. Luo, N. Wang, Z. Wang, C. Liu and T. Jing, Mechanistic Insights into OC–COH Coupling in CO2 Electroreduction on Fragmented Copper, J. Am. Chem. Soc., 2022, 144, 14005–14011 CrossRef CAS.
- X. Bai and J. Guan, Applications of MXene–Based Single–Atom Catalysts, Small Struct., 2023, 4, 2200354 CrossRef CAS.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Potential Link between Cu Surface and Selective CO2 Electroreduction: Perspective on Future Electrocatalyst Designs, Adv. Mater., 2020, 32, 1908398 CrossRef CAS.
- J. Li, S. U. Abbas, H. Wang, Z. Zhang and W. Hu, Recent Advances in Interface Engineering for Electrocatalytic CO2 Reduction Reaction, Nano-Micro Lett., 2021, 13, 1–35 CrossRef CAS.
- R. Haunschild, A. Barth and W. Marx, Evolution of DFT Studies in View of A Scientometric Perspective, J. Cheminf., 2016, 8, 1–12 Search PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for ab initio Total-Energy Calculations Using A Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved Grid–Based Algorithm for Bader Charge Allocation, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the Overpotential for Oxygen Reduction at A Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- T. Cheng, H. Xiao and W. A. Goddard III, Full Atomistic Reaction Mechanism with Kinetics for CO Reduction on Cu (100) from ab initio Molecular Dynamics Free-Energy Calculations at 298 K, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A pre- and Post-Processing Program for VASP Code. 2019, arXiv.org e-Print archive. https://arxiv.org/abs/1908.08269.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC press, 2004 Search PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. Arias and R. G. Hennig, Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- Megha, K. Mondal, T. K. Ghanty and A. Banerjee, Adsorption and Activation of CO2 on Small-Sized Cu–Zr Bimetallic Clusters, J. Phys. Chem. A, 2021, 125, 2558–2572 CrossRef CAS.
- X. Liang, X. Ren, M. Guo, Y. Li, W. Xiong, W. Guan, L. Gao and A. Liu, CO2 Electroreduction by AuCu Bimetallic Clusters: A First Principles Study, Int. J. Energy Res., 2021, 45, 18684–18694 CrossRef CAS.
- J. Zhao, P. Zhang, T. Yuan, D. Cheng, S. Zhen, H. Gao, T. Wang, Z.-J. Zhao and J. Gong, Modulation of *CHxO Adsorption to Facilitate Electrocatalytic Reduction of CO2 to CH4 over Cu-Based Catalysts, J. Am. Chem. Soc., 2023, 145, 6622–6627 CrossRef CAS PubMed.
- X. Ma, Y. Zhang, T. Fan, D. Wei, Z. Huang, Z. Zhang, Z. Zhang, Y. Dong, Q. Hong and Z. Chen, Facet Dopant Regulation of Cu2O Boosts Electrocatalytic CO2 Reduction to Formate, Adv. Funct. Mater., 2023, 33, 2213145 CrossRef CAS.
- J. Yin, Z. Yin, J. Jin, M. Sun, B. Huang, H. Lin, Z. Ma, M. Muzzio, M. Shen and C. Yu, A New Hexagonal Cobalt Nanosheet Catalyst for Selective CO2 Conversion to Ethanal, J. Am. Chem. Soc., 2021, 143, 15335–15343 CrossRef CAS PubMed.
- H. Bao, Y. Qiu, X. Peng, J.-A. Wang, Y. Mi, S. Zhao, X. Liu, Y. Liu, R. Cao and L. Zhuo, Isolated Copper Single Sites for High-Performance Electroreduction of Carbon Monoxide to Multicarbon Products, Nat. Commun., 2021, 12, 238 CrossRef CAS PubMed.
- F. Cai, X. Hu, F. Gou, Y. Chen, Y. Xu, C. Qi and D.-K. Ma, Ultrathin ZnIn2S4 Nanosheet Arrays Activated by Nitrogen-Doped Carbon for Electrocatalytic CO2 Reduction Reaction Toward Ethanol, Appl. Surf. Sci., 2023, 611, 155696 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi02000b |
| This journal is © the Partner Organisations 2024 |