
Substrate-induced phase transition within liquid condensates reverses the catalytic activity of nanoparticles†
S. M.
Rose
 a,
Silky
Bedi
a,
Sabyasachi
Rakshit
*b and
Sharmistha
Sinha
*a
a,
Silky
Bedi
a,
Sabyasachi
Rakshit
*b and
Sharmistha
Sinha
*a
aChemical Biology Unit, Institute of Nano Science and Technology, Sector 81, SAS Nagar, Punjab-140306, India. E-mail: sinhas@inst.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research, Sector 81, SAS Nagar, Punjab-140306, India. E-mail: srakshit@iisermohali.ac.in
First published on 12th July 2024
Abstract
Liquid–liquid phase separation is reported to enhance the catalytic reaction rates severalfold. Herein, we explored the interactions between a catalyst and a range of substrate concentrations to understand the impact on the droplet phase and catalytic reaction kinetics. We observed that the substrate above a critical concentration induces phase transitions within liquid condensates and restricts the free movement of both the substrate and products, resulting in an overall reduction of the reaction rate, an observation not reported earlier.
Introduction
Dynamic assemblies of biopolymers (protein/nucleic acid) orchestrated through liquid–liquid phase separation (LLPS) are referred to as biomolecular condensates. Investigations conducted in recent years reveal that these condensates serve as organisers for various biochemical processes, including cell signalling, transcription, stress response and enzyme catalysis. Condensates engaged in enzyme catalysis are further subclassed as catalytic condensates, which sequestrate catalysts inside a small volume, leading to a significantly elevated local concentration of the catalyst.1,2 Consequently, the turnover of the reaction is enhanced severalfold. For example, glycolytic enzymes in cells form catalytic condensates (glycolytic bodies/G bodies) under hypoxic conditions to increase the rate of glycolysis. Similarly, six enzymes involved in de novo purine biosynthesis assemble in purinosomes and increase the production of nucleotides.3,4In recent years, extensive research has been dedicated to mimicking catalytic condensates in vitro and have consistently demonstrated improved rate kinetics.5–8 This enhanced rate is attributed to the increased proximity or local concentration of reacting molecules in the condensate. Furthermore, the reported enhancement in catalytic reactions is based on the single-point concentration of the substrate and catalyst.1,5 Importantly, the concentration of small molecules (substrates) can significantly alter the driving forces of LLPS (transient and non-covalent interactions).9–11 Consequently, the phase of the condensate changes, potentially affecting its viscoelastic properties and the dynamics of substrate channelling during the reaction.2,12 However, the present body of literature lacks comprehensive studies elucidating the impact of substrate concentration on the kinetics of these catalytic condensates. Herein, we systematically vary the substrate concentration and derive the effect on catalytic reaction kinetics in liquid condensates. We obtained a non-linear change in the kinetics and correlated it with the phase behaviour of liquid condensates. We opted for a low-cost, readily synthesizable BSA-gold nanocluster (AuNC@BSA) as a catalyst and the oxidation of pyrogallol as a model reaction.13,14 Furthermore, the catalyst exhibits the ability to undergo LLPS and has demonstrated an enhanced reaction-rate in condensates for a particular substrate concentration.15 Overall, our findings here pave the way to understand the effect of small biological molecules (e.g., nucleotides, amino acids or other metabolites) on the reaction kinetics in the condensate phase.
Results and discussion
As reported, we synthesized AuNC@BSA through the reduction of Au3+ ions using BSA as a reducing and stabilising agent.16,17 The formation of AuNC@BSA was confirmed by an emission peak at 670 nm when excited at 450 nm (Fig. 1a) as well as by agarose gel electrophoresis (inset, Fig. 1a). We induced the LLPS of AuNC@BSA using 5% (w/v) PEG6000 in 500 mM of kosmotropic salt, Na2SO4. To follow the formation of liquid droplets, we labelled 1% of BSA with Texas Red (TR) and monitored fusion events under a fluorescence microscope (Fig. 1b).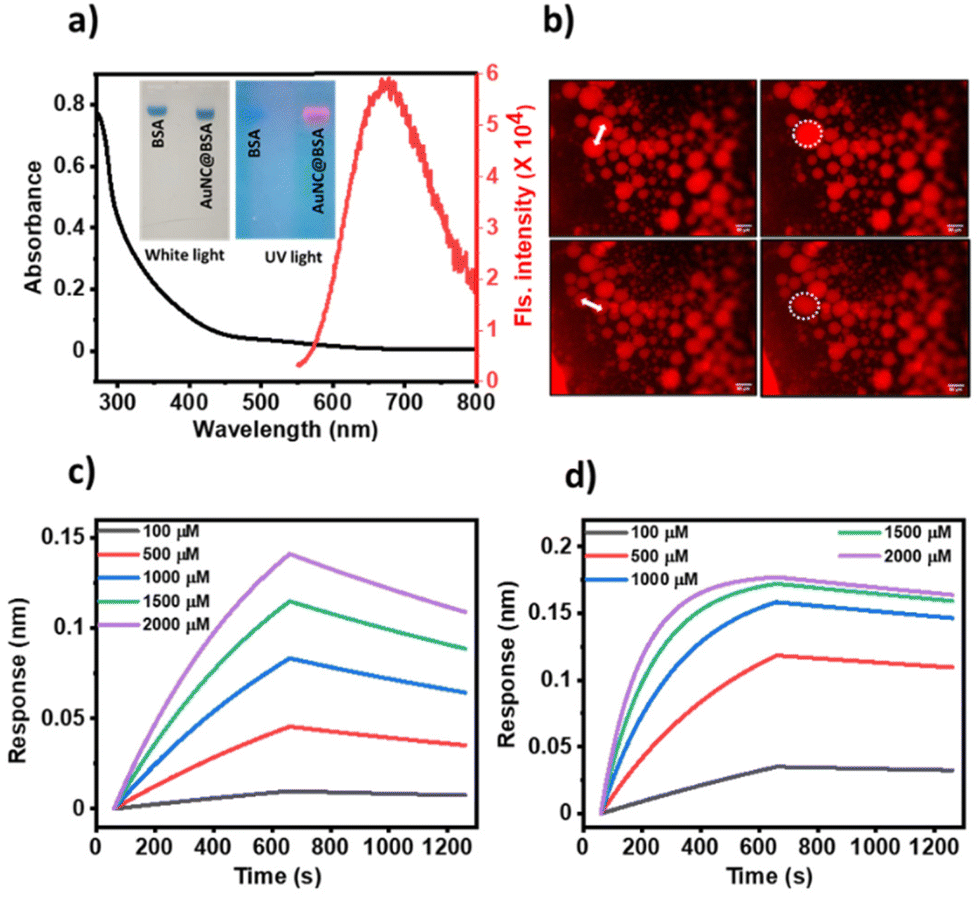 | ||
| Fig. 1 Characterisation and phase separation of AuNC@BSA: (a) UV-vis and fluorescence spectra of AuNC@BSA. Inset shows agarose gel electrophoresis of BSA and AuNC@BSA loaded with Coomassie blue under white and UV light. Colocalization of blue (from Coomassie blue) and red (from AuNCs) suggest the presence of AuNCs with BSA, (b) fluorescence images of TR-labelled AuNC@BSA condensates featuring fusion events (white double-headed arrow indicates fusing droplets). BLI studies of (c) BSA, (d) AuNC@BSA at different concentrations of pyrogallol showing high affinity of pyrogallol towards AuNC@BSA with a KD value of (35.1 ± 3.2) × 10−6 M in comparison to BSA with KD = (66.7 ± 2.0) × 10−5 M. | ||
Furthermore, using biolayer interferometry,18 we probed the interaction of the substrate with the catalyst. 1 mg ml−1 of AuNC@BSA or BSA was loaded on the activated amine reactive second generation (AR2G) sensor and the association and dissociation kinetics were recorded by immersing the receptor-loaded sensor in different concentrations of pyrogallol and water. We obtained dissociation constants (KD) of (35.1 ± 3.2) × 10−6 M for the interaction of pyrogallol-AuNC@BSA, and (66.7 ± 2.0) × 10−5 M for pyrogallol-BSA (Fig. 1c and d). The micromolar range of the dissociation constant indicates reversible binding between the substrate and the catalyst. These interaction studies also indicate that in addition to the catalytic sites on the AuNCs, the protein BSA can also harbour the substrate through at orders of magnitude weaker affinity.
AuNC@BSA catalysts exhibited Michaelis–Menten kinetics in the dispersed phase consistent with previous reports. However, under phase-separated conditions, the reaction rates followed a bell-shaped curve with increasing substrate concentration (Fig. 2a). We observed a notable 2 to 5-fold increase in the reaction rate within the 100 μM to 1000 μM substrate concentration range, followed by a gradual decline and saturation at 2000 μM to 3000 μM, respectively (Table S1† and Fig. 2a). While the initial increase in the catalytic rate can be attributed to the enhanced local concentrations of both, the substrate and catalyst within the condensates, the subsequent decrease in the reaction rate remains unclear. To decipher the underlying reason for the drop in the reaction rate, we first estimated the partitioning of the substrate between the dispersed phase and condensed phase. The partition coefficient remains unaltered with increasing substrate concentrations, indicating no bias in the substrate uptake by the condensates (Fig. S1†). Furthermore, we did not observe any change in the droplet integrity and size with increasing substrate concentration (Fig. 2b and Table S2†).
 | ||
| Fig. 2 (a) Kinetics of the oxidation of pyrogallol to purpurogallin catalysed by the BSA-Gold nanocluster (AuNC@BSA) in the presence of high excess H2O2 (0.1 M) in dispersed and phase separated (PS) conditions, (b) fluorescence microscopy images of Texas red labelled AuNC@BSA in the absence (control) and presence of different concentrations (100 μM, 1000 μM, 1500 μM and 2000 μM) of pyrogallol. Images are taken after starting the oxidation reaction by the addition of H2O2. Scale bar is 100 μm. (c) Change in fluorescence of AuNC@BSA with increasing concentration of pyrogallol. | ||
Next, we probed the effect of the substrate on catalysts and monitored the fluorescence intensity of the AuNC@BSA at 670 nm with increasing amounts of pyrogallol. We observed a typical sigmoidal feature, a gradual increase in the fluorescence intensity with increasing pyrogallol concentration (100–1500 μM), followed by a sudden jump above 1500 μM. The intensity reaches a saturation above 2500 μM (Fig. 2c). It is known that Au-nanoclusters exhibit higher emission in non-polar solvents compared to that in polar.19 We argue that pyrogallol binds with BSA and screens solvent accessibility for AuNC@BSA, thus gradually enhancing the hydrophobicity within the droplet and around AuNC@BSA. We already measured a weak interaction of pyrogallol with BSA using BLI. To understand the nature of interactions, we docked BSA (PDB ID 3V03) and pyrogallol using AutoDock Vina, and identified several possible sites of interactions that may support our argument (Fig. S2†). The observed docking complexes indicate that the hydroxyl groups of pyrogallol are engaged in hydrogen bonding interactions with several residues in BSA (bovine serum albumin). This interaction suggests that the hydroxyl groups are shielded by these interactions, potentially leading to an enhancement in hydrophobicity. This shielding effect could arise from the formation of hydrogen bonds between the hydroxyl groups of pyrogallol and specific amino acid residues in BSA, which might alter the overall hydrophobic character of the system.
Furthermore, it is reported that the fluorescence intensity of AuNC@BSA increases with the phase separation to a higher-order aggregation.22 We too observed a step-wise increase in fluorescence intensity beyond 1500 μM of the substrate concentration, followed by saturation, indicative of the phase transition. The catalytic rate also declines within this concentration range. Based on these findings, we propose that the hydrophobic environment within the droplets facilitates a phase transition from a liquid state to a crystalline phase. This crystalline phase could impede the efficient channelling of the substrate and product, resulting in a decrease in reaction kinetics (see Fig. 2a). To test our hypothesis, we first measured the rate of droplet formation of AuNC@BSA with and without pyrogallol from the turbidity assay (scattering at 700 nm). We found that the condensate forming rate is twice in the presence of 2000 μM pyrogallol (Fig. 3a). The faster rate indicates that the presence of the substrate molecule facilitates faster ageing of the condensates. Faster ageing may lead to the faster phase separation of the liquid condensates to a solid-like phase, a phenomenon commonly noticed in amyloids. Secondly, we measured the lower critical solution temperature (LCST) of the liquid condensates. For this, we gradually heated the phase-separated mixture and monitored the temperature above which the liquid condensates of AuNC@BSA separated into two immiscible phases as seen in Fig. 3b.
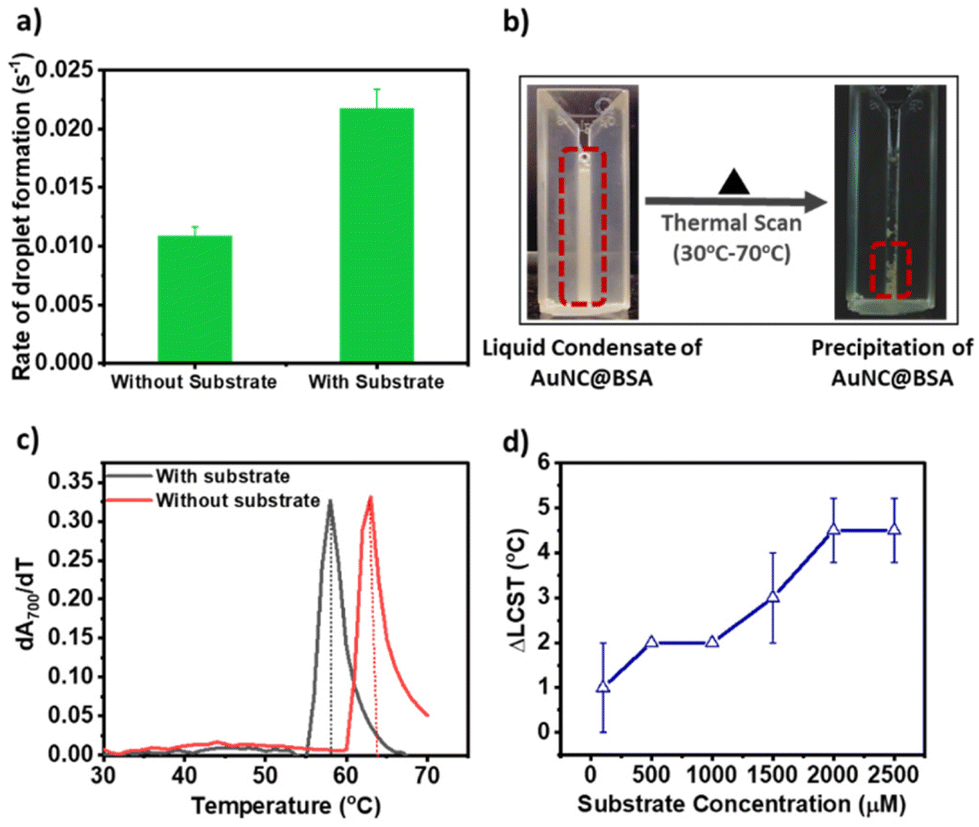 | ||
| Fig. 3 Effect of the substrate on the phase behaviour of AuNC@BSA: (a) rate of formation of the droplet monitored by measuring the change in turbidity with respect to time at 700 nm, (b) condensates of AuNC@BSA undergoes liquid to the solid phase transition on increasing temperature. The phase transition was monitored by measuring turbidity at 700 nm using a UV-vis spectrophotometer and the transition is LCST (lower critical solution temperature) type. (c) LCST of the liquid to the solid phase transition of AuNC@BSA in the absence and presence of the substrate (2000 μM). (d) The difference of LCST (ΔLCST) in the absence and presence of various concentrations of the substrate. | ||
At lower temperatures, robust solvent–solute interactions, primarily through hydrogen bonding, yield a negative enthalpy of mixing. Consequently, this results in an overall negative change in the free energy mixing, thus maintaining solubility at lower temperatures. However, upon heating beyond the LCST, these solvent–solute interactions are disrupted. Instead, entropically driven interactions, such as hydrophobic interactions between solute molecules, become dominant. This transition leads to phase separation, transforming the initially miscible liquid into an immiscible solid phase.20,21 Here, we estimated the LCST for AuNC@BSA condensates at each pyrogallol concentration by measuring turbidity at 700 nm while heating the mixture. LCST was calculated from the differential change in turbidity with temperature (as shown in (dA700/dT) vs. temperature plot in Fig. 3c). In the absence of any substrate, the LCST of the droplets was 62 ± 1 °C and decreased gradually upon increasing the substrate concentration (Fig. 3c). We measured the difference of LCST between the no-substrate to the substrate (ΔLCST = LCSTno substrate − LCSTsubstrate) and plotted the change in ΔLCST with increasing substrate concentration. We observed a gradual increase in the ΔLCST value up to 1000 μM, followed by a sudden jump thereafter until 2000 μM and saturation (Fig. 3d). The trend ΔLCST corresponds to the change in fluorescent intensity of AuNC@BSA with the increasing substrate, and supports our hypothesis of phase behaviour alteration within the condensates.
Our observations suggest that once the substrate concentration surpasses a critical threshold, the internal environment within the liquid condensates undergoes changes, influencing the dynamics of the solvent. This alteration impedes the free movement of both substrate and products, resulting in an overall reduction in the reaction rate.
Conclusions
This study investigated the intricate interaction dynamics between pyrogallol and AuNC@BSA catalysts across varying substrate concentrations. At lower concentrations, weaker interactions limit significant phase transitions, while reaction rates remain higher within the condensed phase. As the substrate concentration increases, stronger interactions initiate the phase transitions impacting both reaction kinetics and ΔLCST values. The sigmoidal increase in the fluorescence intensity observed from 1500 μM signifies a less sensitive response to initial phase transitions compared to ΔLCST and reaction rate measurements. Complete phase transition at higher substrate saturates fluorescence intensity and ΔLCST, correlating with decreased reaction rates due to altered dynamics in the condensed phase.Our studies have revealed that catalytic reactions occurring within liquid condensates do not adhere to the classical Michaelis–Menten kinetics model. This departure from the traditional enzymatic kinetics underscores the unique nature of biochemical processes within the condensed phases, where factors such as spatial confinement and altered microenvironments play significant roles in modulating reaction kinetics. These findings emphasize the critical importance of considering the substrate concentration when studying the reaction rates within catalytic condensates. Moreover, phase-separated catalytic condensates demonstrate significant potential for applications in energy,23 environment,24 biotechnology sectors,8 and enhancing reaction kinetics. However, for effective in vitro catalysis, modifying catalytic surfaces or supplementing systems with small molecules is crucial to restrict further phase transitions in condensates without compromising reaction rates. In sum, these findings not only advance a fundamental understanding of reaction dynamics within condensates but also offer practical insights into manipulating and emulating biological systems.
Author contributions
SM Rose: experimental design and execution, methodology, data analysis, investigation, original manuscript drafting and editing. Silky Bedi: experiment execution, manuscript editing. Sabyasachi Rakshit: conceptualization and manuscript writing. Sharmistha Sinha: supervision, conceptualization, writing – reviewing and editing.Data-availability
Part of the data supporting the findings of the manuscript is presented in the ESI.† Additional data if required will be available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SMR acknowledges INST for financial support and all the research facilities. SB acknowldeges DST-INSPIRE for fellowship.References
- B. Saini and T. K. Mukherjee, J. Phys. Chem. B, 2023, 127, 180–193 CrossRef CAS PubMed.
- B. G. O'Flynn and T. Mittag, Curr. Opin. Cell Biol., 2021, 69, 70–79 CrossRef PubMed.
- H. Zhao, C. R. Chiaro, L. Zhang, P. B. Smith, C. Y. Chan, A. M. Pedley, R. J. Pugh, J. B. French, A. D. Patterson and S. J. Benkovic, J. Biol. Chem., 2015, 290, 6705–6713 CrossRef CAS PubMed.
- A. M. Pedley, J. P. Boylan, C. Y. Chan, E. L. Kennedy, M. Kyoung and S. J. Benkovic, J. Biol. Chem., 2022, 298, 101845 CrossRef CAS PubMed.
- A. M. Küffner, M. Prodan, R. Zuccarini, U. Capasso Palmiero, L. Faltova and P. Arosio, ChemSystemsChem, 2020, 2, e2000001 CrossRef.
- M. Guan, M. V. Garabedian, M. Leutenegger, B. S. Schuster, M. C. Good and D. A. Hammer, Biochemistry, 2021, 60, 3137–3151 CrossRef CAS PubMed.
- M. Liu, S. He, L. Cheng, J. Qu and J. Xia, Biomacromolecules, 2020, 21, 2391–2399 CrossRef CAS PubMed.
- S. Lim and D. S. Clark, Trends Biotechnol., 2023, 42, 496–509 CrossRef PubMed.
- S. Li, Y. Wang and L. Lai, Acta Biochim. Biophys. Sin., 2023, 55, 1075–1083 CrossRef CAS PubMed.
- W. M. Babinchak, B. K. Dumm, S. Venus, S. Boyko, A. A. Putnam, E. Jankowsky and W. K. Surewicz, Nat. Commun., 2020, 11, 5574 CrossRef CAS PubMed.
- K. L. Saar, A. S. Morgunov, R. Qi, W. E. Arter, G. Krainer, A. A. Lee and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2019053118 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- Y. Song, J. Qiao, W. Liu and L. Qi, Microchem. J., 2020, 157, 104871 CrossRef CAS.
- R. Ahmad, J. Shanahan, S. Rizaldo, D. S. Kissel and K. L. Stone, Catalysts, 2020, 10, 499 CrossRef CAS.
- S. Bedi, G. Kumar, S. M. Rose, S. Rakshit and S. Sinha, Chem. Commun., 2022, 58, 8634–8637 RSC.
- S. Govindaraju, S. R. Ankireddy, B. Viswanath, J. Kim and K. Yun, Sci. Rep., 2017, 7, 40298 CrossRef CAS PubMed.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- R. L. Petersen, Biosensors, 2017, 7, 49 CrossRef PubMed.
- Z. Gan, J. Chen, J. Wang, C. Wang, M.-B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed.
- Q. Sun, Molecules, 2022, 27, 7009 CrossRef CAS PubMed.
- P. Paricaud, A. Galindo and G. Jackson, Mol. Phys., 2003, 101, 2575–2600 CrossRef CAS.
- A. Maity and A. Kumar, Nanoscale Adv., 2022, 4, 2988–2991 RSC.
- Q. Liang, Y. Huang, Y. Guo, X. Zhang, X. Hu, H. Zeng, K. Liang, D. Zhao, L. Jiang and B. Kong, Nat. Sustain., 2024, 7, 628–639 CrossRef.
- M. Yan, D. Ma, B. Qiu, T. Liu, L. Xie, J. Zeng, K. Liang, H. Xin, Z. Lian, L. Jiang and B. Kong, Chem. – Eur. J., 2022, 28, e202200307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01402b |
| This journal is © The Royal Society of Chemistry 2024 |