
Geometries and stabilities of chromium doped nitrogen clusters: mass spectrometry and density functional theory studies†
Zaifu
Jiang‡
a,
Peixin
Fu‡
ab,
Meicheng
Chen
c,
Chen
Chen
c,
Bole
Chen
d,
Wei
Dai
a,
Kewei
Ding
 *ef and
Cheng
Lu
*c
*ef and
Cheng
Lu
*c
aSchool of Mathematics and Physics, Jingchu University of Technology, Hubei 448000, China
bDepartment of Physics and Optoelectronic Engineering, Yangtze University, Jingzhou 434023, China
cSchool of Mathematics and Physics, China University of Geosciences (Wuhan), Wuhan 430074, China. E-mail: lucheng@calypso.cn
dSchool of Science, Chongqing University of Posts and Telecommunications, Chongqing 400065, China
eXi’an Modern Chemistry Research Institute, Xi’an 710065, China. E-mail: dkw204@163.com
fState Key Laboratory of Fluorine & Nitrogen Chemicals, Xi’an 710065, China
First published on 3rd May 2024
Abstract
Metal-doped nitrogen clusters serve as effective models for elucidating the geometries and electronic properties of nitrogen-rich compounds at the molecular scale. Herein, we have conducted a systematic study of VIB-group metal chromium (Cr) doped nitrogen clusters through a combination of mass spectrometry techniques and density functional theory (DFT) calculations. The laser ablation is employed to generate CrNn+ clusters. The results reveal that CrN8+ cluster exhibits the highest signal intensity in mass spectrometry. The photodissociation experiments with 266 nm photons confirm that the chromium heteroazide clusters are composed of chromium ions and N2 molecules. Further structural searches and electronic structure calculations indicate that the cationic CrN8+ cluster possesses an X shaped geometry with D2 symmetry and exhibits robust stability. Molecular orbital and chemical bonding analyses demonstrate the existence of strong interactions between Cr+ cation and N2 ligands. The present findings enrich the geometries of metal doped nitrogen clusters and provide valuable guidance for the rational design and synthesis of novel transition metal nitrides.
1 Introduction
Metal atoms, including transition metals (TMs), incorporated into nitrogen cluster nanoparticles are recognized as potential high-energy-density materials due to substantial energies released during the fragmentation reactions.1–6 Compared to pure nitrogen clusters, which suffer from poor stabilization and challenging preparation processes, metal-doped nitrogen clusters exhibit distinctive properties. Normally, the nitrogen skeletons are prone to enhance their stabilities through bonding with the metal atoms and their structures show diversity, making them environmentally friendly high-energy-density material candidates.7–10 Thus, the investigations on the varied geometries and structural stabilities of metal-doped nitrogen clusters have attracted much attention. Ongoing efforts focus on understanding the distinct structural patterns and evolutions guided by different metal-doped nitrogen clusters, with the goal of elucidating the interactions and bonding mechanisms between the metal atoms and the main nitrogen ligands.So far, researchers have conducted extensive studies on metal-doped nitrogen clusters through diverse experimental and theoretical methods. For example, N4 rings doped with alkaline earth metals (M = Ca2+, Sr2+, Ba2+) exhibit pyramidal structures.11 The syntheses of numerous binary azides, including group 4 elements (like Ti), group 5 elements (such as V, Nb and Ta),12–14 group 6 elements (Mo and W),15 group 15 element (Bi)16 and so on, were systematically explored through experimental methods. Gagliardi et al.17 identified a local minimum isomer with C7v symmetry for ScN7 in Sc-doped N clusters and demonstrated the local stability of sandwich geometric structures in TM-doped N5MN7 (M = Ti, Zr, Hf, Th) clusters. In addition, the geometries and stabilities of numerous metal-doped polyazides (e.g., tri-azides M(N3)3 (M = Sc, Y, La, Al, Ga, In, Tl)18–20 of group 3 and 13, tetra-azides M(N4)3 (M = Ti, Zr, Hf, Th, Ge, Sn, Pb)18,21 of group 4 and 14, as well as novel aromatic compounds with planar N6 rings of ScN6−, VN6+, Ca2N6, ScN6 Cu and M(η6-N6) (M = Ti, Zr, Hf, Th)22,23 and so on), have been extensively studied both experimentally and theoretically.
In particular, in 2017, Zhang et al.2 successfully synthesized the pentazolate anion cyclo-N5−, revealing its unexpected stability and potential application in energetic polynitrogen compounds. Subsequently, Sun et al.24 achieved a significant breakthrough in cyclo-pentazole chemistry by experimentally isolating the AgN5 of the cyclo-N5− metal complex. Wang et al.25 reported the synthesis of planar N62− hexazine dianions through potassium azide (KN3) under high pressure. Laniel et al.26 conducted a study on the synthesis of the K9N56 compound by laser heating under high pressure and a first principles calculations. The results revealed that the K9N56 compound comprises a complex arrangement of planar aromatic hexazine [N6]4− and [N5]− rings, along with neutral nitrogen dimers. Moreover, the diverse properties exhibited by metal-doped nitrogen clusters of varying sizes have stimulated scientists to systematically explore the size-dependent properties of these clusters. In recent years, there has been a surge of in-depth studies focusing on the alkali metal heteroazide clusters, specifically LiNn+ and Li2Nn+,27–29 as well as potassium heteroazide clusters (KNn+),30 and sodium heteroazide clusters (NaNn+).29 Substantial advancements and notable progress have been achieved.
Except for the main group elements, other attentions were directed towards the TM-doped nitrogen clusters, i.e. MNn+ (M = Sc, Zr, V, Cu, Fe, Co, Ni, Ti, Zn),31–35 which focus on the generations and geometries of these clusters, shedding light on their structure and electronic properties. These findings verify the imperative for further exploration of the structures and characteristics of metal-doped nitrogen clusters. A deeper comprehension of the bonding modes between metal and nitrogen atoms holds promise for the rational design of stabilizers and catalysts tailored for all-nitrogen materials. Overall, these studies reveal the profound impact of metal doping, including transition metals, in modulating the geometric and electronic properties of nitrogen clusters, unveiling unique properties distinct from their bulk materials.
Cr is the inaugural element in group 6 of the periodic table, which exhibits distinctive physical properties, presenting as a steely-grey, lustrous, hard, and brittle transition metal. The [Ar]3d54s1 electronic configuration of Cr fosters the formation of robust d–d bonds and notably short bond lengths (1.68 Å) in its dimers.36,37 Beyond its inherent electronic properties, Cr holds significance in industrial applications, engaging in interactions with non-metallic elements such as oxygen,38 carbon,39 and nitrogen.39–41 The versatility of Cr enhances its value, making it advantageous for applications as the catalyst or the coating for hard materials. Wang et al.42 uncovered the nitrogen-induced magnetic transition of Cr atom in their exploration of CrmN (m = 2–5) and Cr2N2. Specifically, the neighboring Cr atom exhibited antiferromagnetic coupling to nitrogen, causing the robust Cr–N bonds. The study of Cr doped nitrogen clusters also contributes to the understanding of the structural evolution and formation mechanisms of nitrogen rich compounds at the atomic and molecular levels. Some of them are expected to serve as precursors for energy-containing materials or the synthesis of nitrogen-riched materials. Consequently, there is an intriguing impetus to study Cr doped nitrogen clusters and explore their unconventional chemical bonding patterns. This work involves a systematic study of Cr doped nitrogen clusters using laser sputtering and photolysis techniques in combination with DFT calculations. The objective is to achieve a comprehensive understanding of the structural evolutions and stabilities of Cr doped nitrogen clusters.
2 Methods
The experiments were conducted employing a custom-designed laser sputtering-time-of-flight (TOF) mass spectrometer.43 The apparatus comprises a Nd:YAG laser, a laser sputtering ion source, a reflectance TOF mass spectrometer, a vacuum system, a carrier gas system, and a timing system, etc. It was successfully applied to prepare various metal doped nitrogen clusters.28,32–34 Firstly, solid samples with a diameter of 13 mm and a thickness of 2–5 mm were fabricated using a tablet press, employing a Cr/AlN mole ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Subsequently, the surface of the samples underwent pulsed laser sputtering on a laser sputtering-TOF mass spectrometer to generate chromium hetero nitrogen clusters. High-purity nitrogen served as the carrier gas during this process to facilitate the formation of chromium-hybrid nitrogen clusters, while liquid nitrogen was employed to cool the clusters generated by laser bombardment. The laser system was characterized by specific operational parameters, namely a wavelength of 532 nm, a pulse energy of approximately 10 mJ, and a repetition frequency of 10 Hz. At a pressure of approximately 4 atmospheres, nitrogen was introduced into the source region through a pulse valve (General Valve Series 9) to cool the clusters formed. The synthesized Cr doped nitrogen clusters were acceleration, deflection, and focusing with the carrier gas in the high-voltage electric field acceleration region, reaching the reflection region, and finally arriving at the microchannel plate (MCP) detector of the TOF mass spectrometer after reflection. Signals from the detectors were passed through a preamplifier and recorded by a 200 MHz digital acquisition card. The acquired digital data were subsequently organized and analyzed using self-programmed software. In addition, CrN8+ cluster, which exhibits the stronger peak in mass spectra of the generated chromium heteroazide clusters, was photolysed with a laser at a wavelength of 266 nm using a mass gate in a time-of-flight mass spectrometer, and mass spectra of the photolysed products were obtained.
:1. Subsequently, the surface of the samples underwent pulsed laser sputtering on a laser sputtering-TOF mass spectrometer to generate chromium hetero nitrogen clusters. High-purity nitrogen served as the carrier gas during this process to facilitate the formation of chromium-hybrid nitrogen clusters, while liquid nitrogen was employed to cool the clusters generated by laser bombardment. The laser system was characterized by specific operational parameters, namely a wavelength of 532 nm, a pulse energy of approximately 10 mJ, and a repetition frequency of 10 Hz. At a pressure of approximately 4 atmospheres, nitrogen was introduced into the source region through a pulse valve (General Valve Series 9) to cool the clusters formed. The synthesized Cr doped nitrogen clusters were acceleration, deflection, and focusing with the carrier gas in the high-voltage electric field acceleration region, reaching the reflection region, and finally arriving at the microchannel plate (MCP) detector of the TOF mass spectrometer after reflection. Signals from the detectors were passed through a preamplifier and recorded by a 200 MHz digital acquisition card. The acquired digital data were subsequently organized and analyzed using self-programmed software. In addition, CrN8+ cluster, which exhibits the stronger peak in mass spectra of the generated chromium heteroazide clusters, was photolysed with a laser at a wavelength of 266 nm using a mass gate in a time-of-flight mass spectrometer, and mass spectra of the photolysed products were obtained.
The systematic search of the potential energy surfaces (PESs) of neutral and cationic CrNn0/+ (n = 2–11) were performed using the CALYPSO package.44–46 The CALYPSO package is based on the particle swarm optimization (PSO) method, which is very efficient in prediction structure of various cluster systems,47–52 including boron clusters48 and nitrogen clusters.34,47 After cluster structural searches, numerous of neutral and cationic CrNn0/+ with were obtained. We then selected candidate isomers for re-optimization based on the following criteria: (a) the isomer with relative lower energy less than 3 eV compared to the lowest-energy structure. (b) The isomer with high symmetry. (c) The isomers with variegated geometries. Subsequently, the selected structures were re-optimized at the high-accuracy level of M06-2X/6-311+G(d,p), which was verified to be reliable in various DFT calculations of metal doped nitrogen clusters. The calculated bond length of N2 molecular is 1.090 Å which is in agreement with the experimental value of 1.097 Å.53 For each isomer, various spin multiplicities (doublet, quartet, sextet for neutrals and singlet, triplet, quintet for cations) were considered. During the geometric optimizations, the vibration frequencies were considered for all low-lying isomers to ensure that the structures are stable and without imaginary frequencies. Chemical bonding analyses were performed using the natural bond orbital (NBO) and adaptive natural density partitioning (AdNDP) methods54 to gain deeper insights into bonding characteristics. All computations were carried out using the Gaussian 09 package.55
3 Results and discussions
As depicted in Fig. 1(a), the chromium–nitrogen clusters produced by laser bombardment of Cr/AlN samples are mainly CrN6+, CrN8+, CrN9+, and CrN11+ clusters. Notably, the signal intensity of CrN8+ cluster is significantly higher than that of other chromium–nitrogen clusters. This observation suggests that CrN8+ cluster represents the most stable isomer among the synthesized chromium–nitrogen clusters. In addition, mass spectral peaks of aluminum–nitrogen clusters, specifically AlN2+, AlN4+ and AlN6+ are observed due to the presence of aluminum impurities. Then, laser photolysis of CrN8+ cluster is performed at 266 nm, as shown in Fig. 1(b). The cationic CrN8+ cluster exhibits two dissociation channels: one involving the loss of six N atoms, resulting in the production of CrN2+ fragment ions, and the other involving the loss of eight N atoms, leading to the generation of Cr+ fragment ions.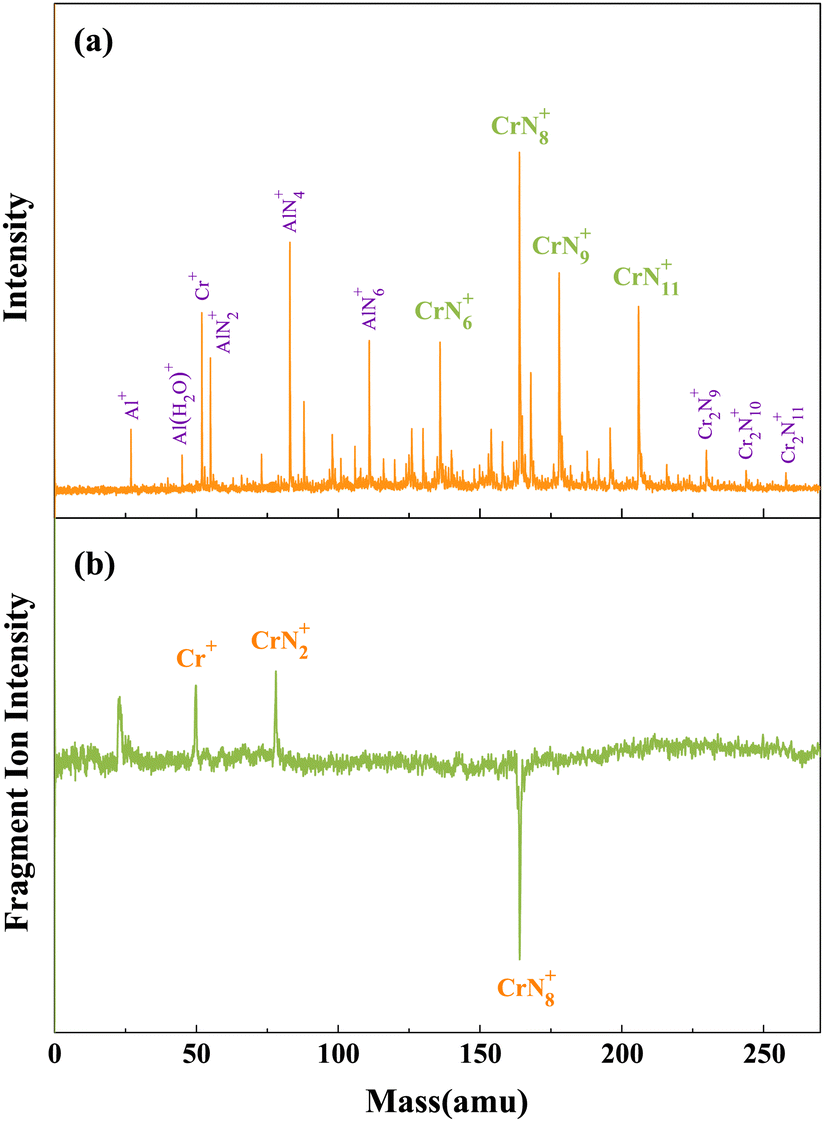 | ||
| Fig. 1 (a) Mass spectra of CrNn0/+ clusters produced by laser bombardment of Cr/AlN samples. (b) Photolysis mass spectra of CrN8+ cluster at 266 nm. | ||
The geometries, symmetries, spin multiplicities and relative energy differences of neutral and cationic chromium–nitrogen clusters are presented in Fig. 2 and 3, respectively. Each isomers of the neutral chromium–nitrogen cluster are labeled as na, nb, and nc (na+, nb+, and nc+ for cations), where n represents the number of N atoms in isomers. The letters a, b, and c (a+, b+, c+) denote the energy order of the isomers, respectively. By considering vibration frequencies, all the shown structures are identified to be stable. The calculated total energies and minimum frequencies of ground state and metastable isomers of chromium–nitrogen clusters are listed in Tables S1 and S2 of the ESI.† In addition, the electronic states of the ground state isomers are listed in Table 1.
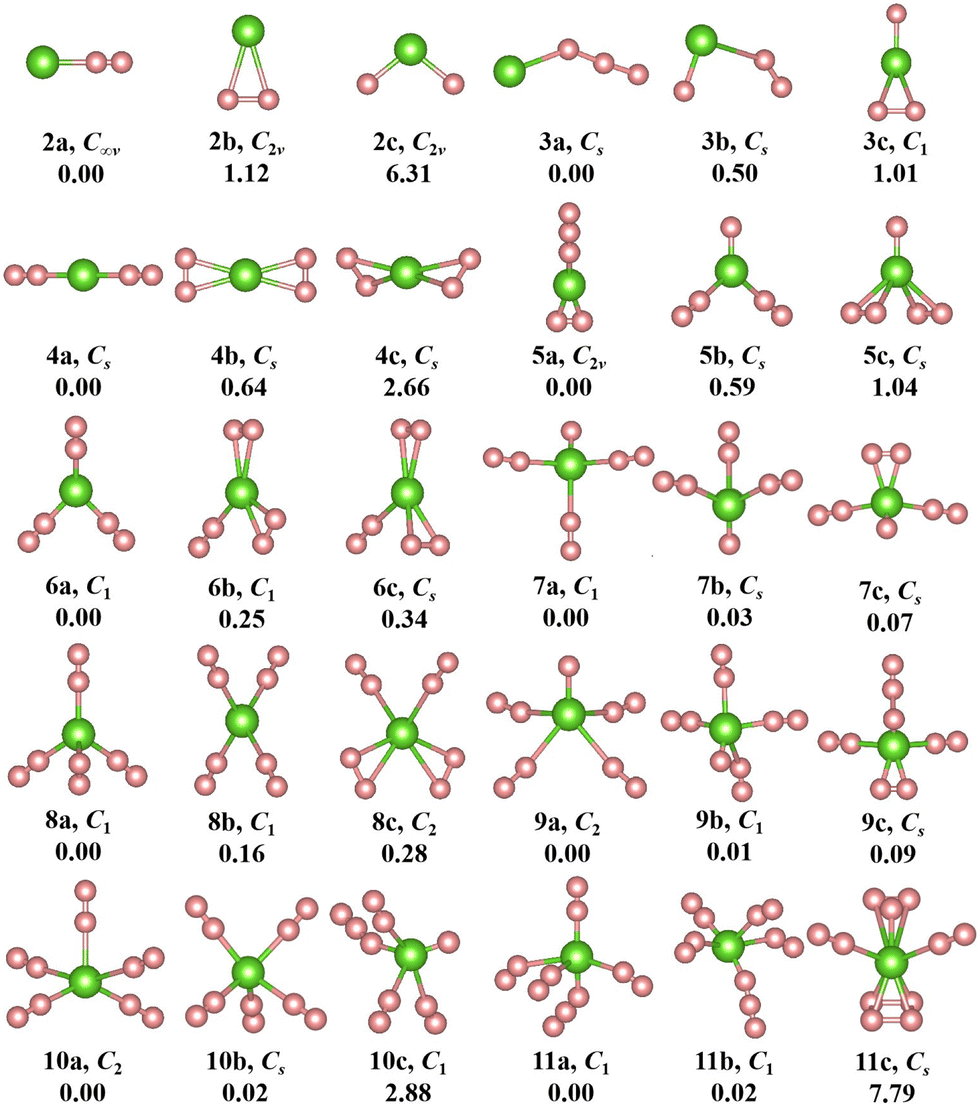 | ||
| Fig. 2 The geometries, symmetries and relative energies (in eV) of optimized neutral CrNn clusters (n = 2–11) at the M06-2X/6-311+G(d,p) level. The green and pink spheres represent Cr and N atoms, respectively. | ||
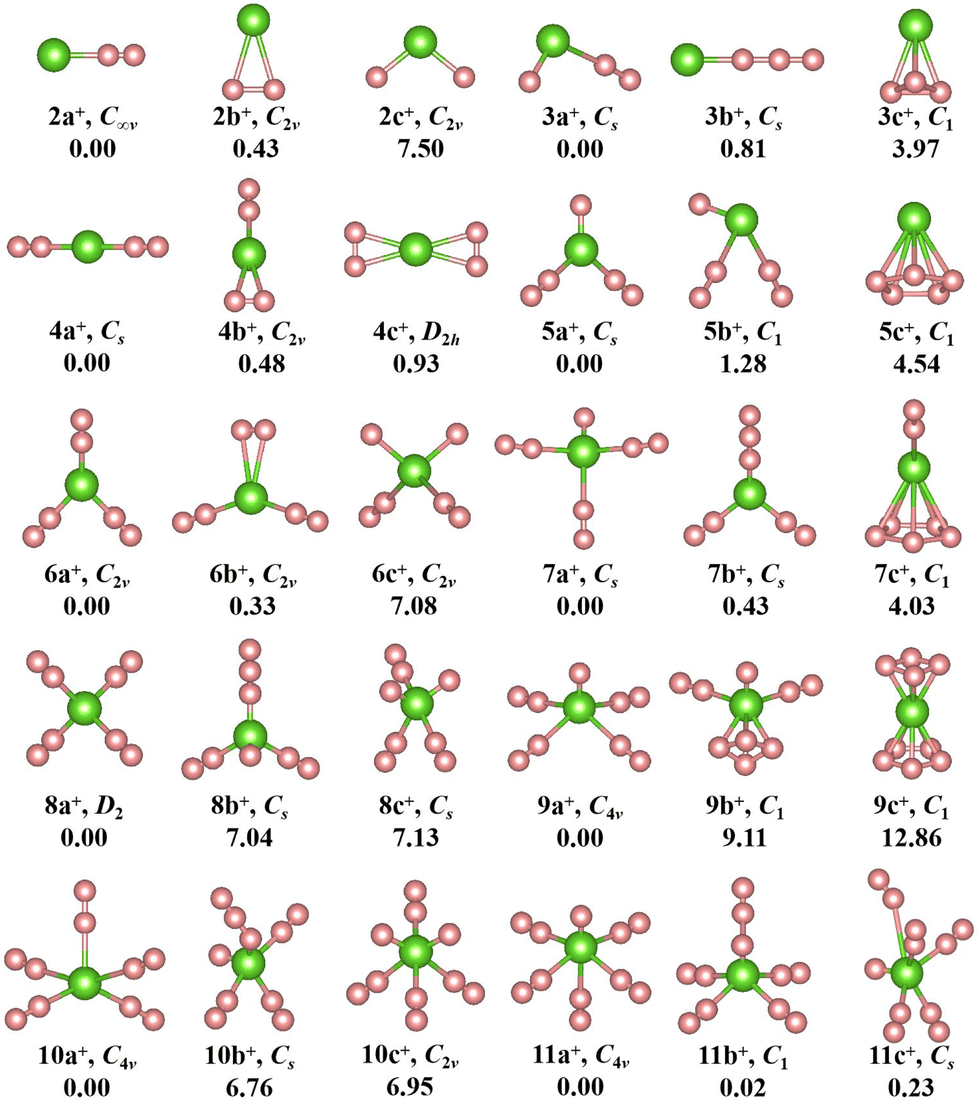 | ||
| Fig. 3 The geometries, symmetries and relative energies (in eV) of optimized cationic CrNn+ clusters (n = 2–11) at the M06-2X/6-311+G(d,p) level. The green and pink spheres represent Cr cation and N atoms, respectively. | ||
| n | CrNn | CrNn+ | ||||||
|---|---|---|---|---|---|---|---|---|
| State | E b | Δ2E | Q(Cr) | State | E b | Δ2E | Q(Cr+) | |
| 2 | 5Σ | 0.32 | 0.01 | 6Σ | 0.78 | 0.99 | ||
| 3 | 6A′ | −1.00 | −3.57 | 0.75 | 3A′′ | −1.37 | −6.57 | 1.06 |
| 4 | 5A′ | 0.13 | 3.01 | −0.08 | 6A′ | 0.84 | 6.55 | 0.89 |
| 5 | 6A1 | −0.40 | −2.65 | 0.80 | 3A′′ | −0.46 | −6.19 | 0.85 |
| 6 | 5A | 0.14 | 3.13 | 0.38 | 6A1 | 0.75 | 6.00 | 0.81 |
| 7 | 4A | −0.38 | −3.72 | 0.28 | 3A′′ | −0.11 | −5.80 | 0.61 |
| 8 | 5A′ | 0.17 | 3.86 | 0.35 | 6A | 0.70 | 6.00 | 0.67 |
| 9 | 4A | −0.27 | −3.74 | 0.26 | 3A2 | −0.01 | −5.91 | 0.52 |
| 10 | 5B | 0.14 | 2.59 | 0.27 | 6A2 | 0.61 | 5.82 | 0.57 |
| 11 | 6A | −0.01 | 0.58 | 3A2 | 0.06 | 0.41 | ||
The geometries of neutral CrNn (n = 2–11) clusters are illustrated in Fig. 2. As for CrN2 cluster, the 2a ground state is a stable linear Cr–N–N configuration with electronic state of 5Σ and C∞v symmetry. Two metastable 2b and 2c isomers with C2v symmetry are identified, representing a planar triangular geometry and an N–Cr–N nonlinear structure, respectively. The most stable structure of CrN3 cluster is 3a, wherein the Cr atom is nonlinearly attached to the N3 chain, exhibiting CS symmetry. The corresponding electronic state is 6A′. For n = 4, the ground state 4a (5A′ electronic state) also possesses CS symmetry and constitutes a linear N2–Cr–N2 geometry derived by adding the N2 unit to 2a. The replacement of the single N atom at the topmost part of 3c by the N3 chain leads to the formation of 5a. It is C2v symmetry with 6A1 electronic state. In the CrN6 cluster, the 5A-6a ground state exhibits low symmetry of C1, resembling a herringbone geometry similar to 5b, with the central Cr atom attached to three N2 units. The geometry of 7a (electronic state 3A) in the CrN7 cluster is characterized by C1 symmetry, where the central Cr atom is linked by three N2 units and an isolated N atom. For CrN8 cluster, the 5A′-8a ground state is CS symmetry and adopts an orthotetrahedral geometry containing four N2 ligands. In the case of n = 9, the lowest energy isomer 9a assumes the configuration of CrN(N2)4 with 4A electronic state and C1 symmetry. The ground state isomer of CrN10 cluster is 10a. It is C2 symmetry formed by the addition of the N2 unit to the 8b isomer, which exhibits a spread-eagled shape, representing the configuration of Cr(N2)5. As for CrN11 cluster, the most stable isomer is 11a (6A, C1) with the configuration of CrN3(N2)4, which contains a N3 chain similar to 3a and 5a.
The geometries of cationic CrNn+ (n = 2–11) clusters are depicted in Fig. 3. The ground state structure of cationic CrN2+ clusters, like neutral CrN2 cluster, is 2a+ isomer. It is stable linear Cr–N–N geometry with C∞v symmetry. Two additional low-lying isomers are also observed: the planar triangular structure 2b+ and the nonlinear N–Cr–N structure 2c+. Both exhibit C2v symmetry. In CrN3+ cluster, the ground-state 3a+ is similar to neutral 3b isomer, containing an isolated N atom and an N2 unit. It is CS symmetry and the corresponding electronic state is 3A′′. The 3a ground state of neutral CrN3 undergoes a structural transition from a nonlinear geometry to a linear structure 3b+ (Cr+–N3) after the loss of one electron. For CrN4+ clusters, like neutral isomer 4a, the ground state is 4a+ (6A′) with CS symmetry, which is also a linear N2–Cr–N2 structure. Similar to the neutral 5b isomer, the most stable structure of the CrN5+ cluster is 5a+. Its electronic state is 3A′′. It is characterized by a herringbone shape with CS symmetry. In the case of n = 6, the 6A1-6a+ ground state contains three N2 units, similar to the neutral 6a, but exhibits a herringbone geometry with C2v symmetry. For CrN7+ cluster, the geometry of the 3A′′-7a+ ground state assumes an X-like shape of Cr+ N(N2)3, characterized by higher CS symmetry compared to the neutral 7a. Similar to 7a, 8b, and 7a+, the ground state isomer 8a+ (electronic state 6A) with D2 high symmetry features the standard X-shape, where four N2 molecules interact equitably with the central Cr atom through their terminal nitrogen atoms. The ground state of CrNn+ (n = 9–11) clusters, 3A2-9a+, 6A2-10a+ and 3A2-11a+, exhibit a similar spread-eagled shape with the same C4v symmetry, which are characterized by Cr+ N(N2)4, Cr+ (N2)5, and Cr+ N(N2)5, respectively.
Interestingly, the most stable structures of CrNn0/+ (n = 2–11) clusters, where n is even, the distinctive conformations, Cr(N2)n/2 (or Cr+(N2)n/2), are observed, which are characterized by N atoms interacting with the central Cr ion through the N2 ligands. Meanwhile, it is noted that the most stable isomers of neutral CrNn (n = 2–11) clusters transition from one-dimensional linear (C∞v-2a, CS-4a) to two-dimensional branching (C1-6a) and further to three-dimensional tetrahedral (CS-8a) and square pyramid (C2-10a) configurations with the increase of number of N2 units, whereas the most stable isomers of cationic CrNn+ (n = 2–11) clusters transform from one-dimensional linear (C∞v-2a+, CS-4a+) to two-dimensional branching (C2v-6a+, D2-8a+) and finally to three-dimensional square pyramid (C4v-10a+) configurations. When the number of N atoms is odd (except for CrN3, CrN5 and CrN11), the N atoms are connected to the Cr atom to form the configurations of CrN(N2)(n−1)/2 (or Cr+N(N2)(n−1)/2). These findings indicate that the chromium–nitrogen clusters are composed of Cr atom (or Cr+ ion) and N2 molecules. The isomer with more number of N2 units usually exhibit relatively lower energies than the isomer with all-nitrogen units, such as the cationic CrN9+ cluster. Similar results were reported by Ding et al.28,33,34 for other metal doped nitrogen clusters. Notably, on the one hand, although the loss of an electron changes the spin multiplicity of chromium–nitrogen cluster, the geometrical symmetries and electronic states of 2a+ and 4a+ cations remain unchanged compared to their corresponding neutral ground states. On the other hand, in 3a, 5a and 11a, nitrogen units interact with Cr atoms in forms of N3 chains and N2 ligands. In addition, the spin multiplicities of ground states are mostly 4, 5 and 6 (6 for the CrNn clusters (CrN3, CrN5 and CrN11) possessing N3 ligand) for neutral CrNn clusters, and the symmetries of ground states in the large-sized CrNn clusters decreases to C1 symmetry, except for CrN10 cluster. However, unlike the neutral clusters, the geometries of ground state cationic isomers exhibit relatively high symmetries with spin multiplicities of 3 or 6, which suggest that the loss of one electron in large-sized chromium–nitrogen clusters can result in enhanced symmetry.
To estimate the strengths of the interactions between Cr atom and N ligands as well as the relative stabilities among the CrNn0/+ clusters, we have calculated the average binding energy and the second-order difference. The average binding energy Eb serves as an effective criterion for assessing the thermodynamic stability of cluster, while the second-order difference Δ2E is a crucial parameter reflecting the relative stability between adjacent clusters. The average binding energy and second-order difference values are calculated as below:
| Eb(CrNn0/+) = 2[E(Cr0/+) + n/2E(N2) − E(CrNn0/+)]/n | (1) |
| Δ2E(CrNn0/+) = E(CrNn+10/+) + E(CrNn−10/+) − 2E(CrNn0/+) | (2) |
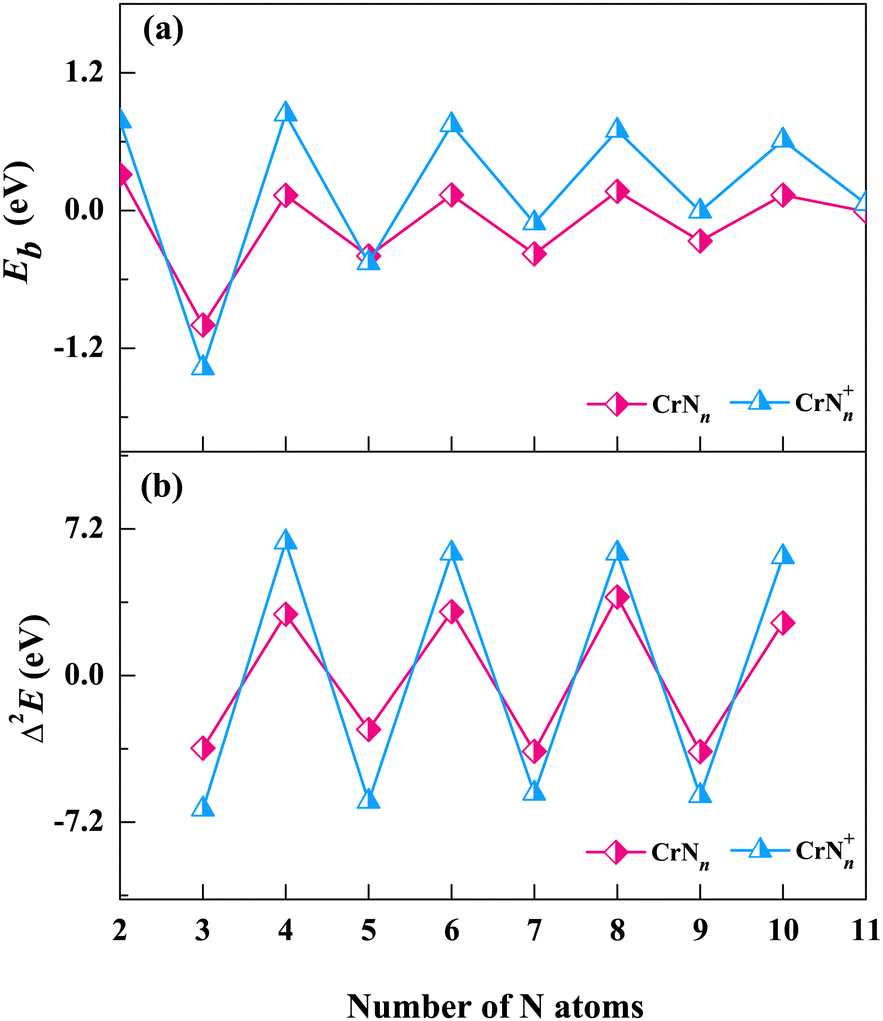 | ||
| Fig. 4 The average binding energy Eb and the second order difference Δ2E of the low-lying isomers of CrNn0/+ clusters. (a) average binding energy and (b) second order difference. | ||
The Eb curves show odd–even oscillations for both neutral and cationic chromium–nitrogen clusters with the increase of cluster sizes. Eb is positive when n is even, otherwise it is negative. The negative values indicate the absorption of the energies. Notably, the prominent peaks appear at n = even, and the peaks for cationic clusters are higher than those of neutral clusters. The odd–even oscillations observed in the Eb curves of CrNn0/+ clusters are attributed to the interplay of quantum mechanical effects, particularly the shell structures of clusters. This phenomenon arises due to the quantization of energy levels associated with the discrete arrangement of Cr atom/cation within the CrNn0/+ clusters. In CrNn0/+ clusters with an even number of N atoms, such as n = 4, 6, 8, 10, the presence of the unpaired Cr atom/cation leads to enhanced stability, as the unpaired Cr atom/cation occupies a lower energy level, resulting in a higher average binding energy. Meanwhile, geometric analyses reveal that the low-lying isomers of CrNn0/+ clusters predominantly adopt conformations of Cr(N2)n/2 (or Cr+(N2)n/2) when n is even, which suggests the propensities of Cr atom/cation in both neutral and cationic chromium–nitrogen clusters readily interact with N2 ligands, resulting in the formation of stable structures. It is worth noting that, except for n = 3 and 5, the Eb of cationic clusters are higher than those of the corresponding neutral clusters, especially pronounced when n = even. This observation implies that the removal of one electron enhances the stability of the most chromium nitrogen clusters.
The second-order difference values, calculated according to eqn (2), are presented in Table 1 and Fig. 4(b). The second-order difference curve also exhibits the obvious trend of odd–even oscillations, which is consistent with the average binding energy curves. Similarly, the significant peaks on the second-order difference curves are located at the points where n is even, with cationic clusters displaying greater amplitude of oscillations. In cationic chromium–nitrogen clusters, the maximum value of Δ2E is 6.55 eV (CrN4+), followed by 6.00 eV (CrN8+). In neutral clusters, the maximum value is 3.87 eV (CrN8). The calculated results of both cationic and neutral clusters show that the chromium–nitrogen clusters with even number of N atoms are more stable in comparison to their adjacent clusters, reconfirming the stabilities of clusters containing the N2 units are more stable than those containing the single N atom. However, mass spectral results reveal that CrN9+ and CrN11+ with an odd number of N atoms also exhibit relatively high intensities in their mass spectral peaks, which is possibly attributed to their “kinetically easier formation” under the present reaction conditions. In addition, the HOMO–LUMO gap (HLG) is another important parameter to describe the thermodynamic stability of cluster, which reflects the energy required for the electron to transition from the HOMO to the LUMO, i.e., the energy needed for an excited state transition. The HLGs of CrNn0/+ clusters are listed in Table S3 of the ESI.† It can be seen that the HLGs of CrN8+ clusters are 8.23 eV and 13.33 eV for α and β orbitals, respectively, indicating their remarkable stabilities.
We next explore the electronic structures of CrNn0/+ (n = 2–11) clusters by assessing the charge transfer of Cr atoms (or Cr+ cations) using natural population analysis (NPA). The natural charges on Cr atoms (or Cr+ cations) are determined for structures corresponding to the lowest energy isomers. As shown in Fig. 5, the charge curve of Cr atom is positive (between 0 and 1) for neutral CrNn clusters, except for CrN4. The most notable peaks occur at n = 3, 5 and 11, and the Cr atoms in these clusters are observed to interact with the N3 ligand. The Cr+ cation charge distributions of cationic CrNn+ clusters are also positive in the range of 0 to 1, except for n = 3, which show gradual decrease with n increasing. These results indicate that Cr atoms easily lose electrons in neutral CrNn clusters, on the contrary, the Cr+ cations in CrNn+ clusters are more likely to gain electrons. Notably, for n = 2, CrN2 and CrN2+ clusters share the same geometrical configuration of linear Cr–N–N. However, the charge of Cr atom in CrN2 cluster is 0, while the charge of Cr+ cation in CrN2+ cluster is approximately 1, which suggests the more charge exchanges between the N3 ligand and the Cr atom compared to the N2 ligand. The NPA results suggest that the factors such as the cluster size, the N ligand configurations, and the oxidation states of Cr (neutral or cationic) in CrNn0/+ (n = 2–11) clusters are likely to influence the interactions and charge transfers between Cr atoms (or Cr+ cations) and N ligands.
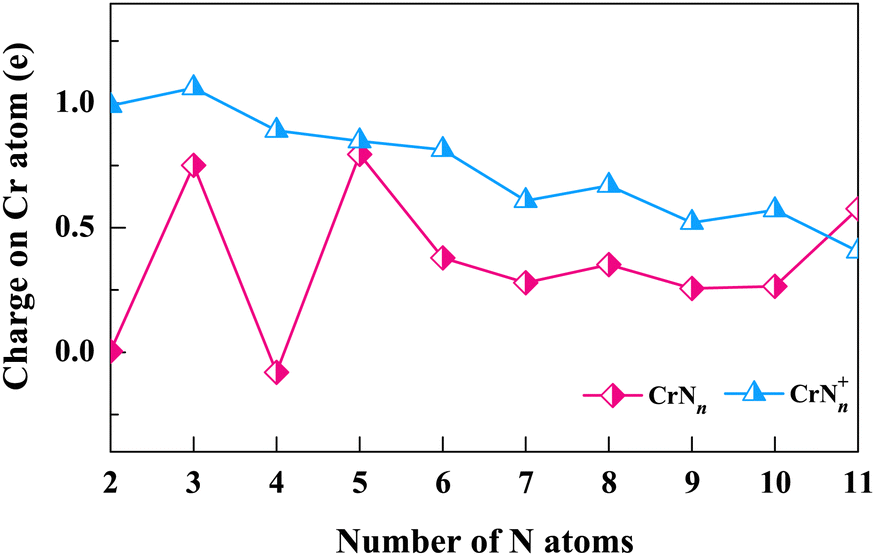 | ||
| Fig. 5 Charges on the Cr atoms (or Cr+ cations) in the ground state structures of CrNn0/+ clusters. | ||
Our experimental mass spectra of CrNn+ (n = 2–11) clusters reveal the predominant presence of the CrN8+ cluster. Additionally, the comprehensive theoretical analyses of the relative stabilities of CrNn0/+ (n = 2–11) clusters demonstrate that the CrN8+ cluster exhibits high D2 symmetry and excellent stability. Thus, we selected CrN8+ cluster as a example to explore the molecular orbitals and the chemical bonding patterns of chromium doped nitrogen clusters. Clearly, the CrN8+ cluster is open-shell structure. The diagrams of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) as well as their eigenvalues with nearest-neighbor molecular orbitals (MOs) of CrN8+ cluster are displayed in Fig. 6. The representations are segregated into α- (spin-up) and β-orbitals (spin-down). The HOMO–LUMO gap for CrN8+ cluster is 8.23 and 13.33 eV for α- and β-orbitals, respectively. In α-MOs, both LUMOs and HOMOs of CrN8+ cluster are triple degeneration. The LUMOs are mainly contributed by N-2p atomic orbitals (AOs), especially 2px, while Cr AOs contribute only 19.07–19.25%. The HOMOs are contributed by Cr-3d orbitals (88.93–88.98%), particularly 3dxy. Similarly, in β-MOs, triple degenerate LUMOs and HOMOs are also observed. The β-LUMOs are mainly contributed by Cr-AOs with 4p orbitals accounting for 35.71–36.06% and 3d orbitals for 14.59–14.75%. The largest contribution to β-HOMOs is the N-2p orbitals, with Cr-AOs accounting for only 2.67–2.68%. As mentioned above, the triple degeneracy of LUMOs and HOMOs at the α and β orbitals of cationic CrN8+ cluster is attributed to the fourfold collocation of Cr+ ion with the D2 symmetry, which leads to the splitting of the Cr+-4p orbitals into the triple degenerate px, py, and pz orbitals and the Cr+-3d orbitals into the triple degenerate dxy, dxz, and dyz orbitals, as well as the double degenerate dx2–y2 and dz2 orbitals. However, as shown in Fig. S4 (ESI†), no degeneracy orbitals appear in the MOs of neutral CrN8 cluster. It is found that the neutral CrN8 cluster possesses an additional electron in comparison to CrN8+ cluster, which induces the Jahn–Teller effect. The loss of an electron from the neutral CrN8 cluster leads to the degeneracy of AOs and the increase of the HLG values in CrN8+ cluster. Meanwhile, the symmetry changes from D2 to CS. The stability of CrN8+ cluster is attributed to the strong interactions between the Cr-3d AOs and the N-2p AOs.
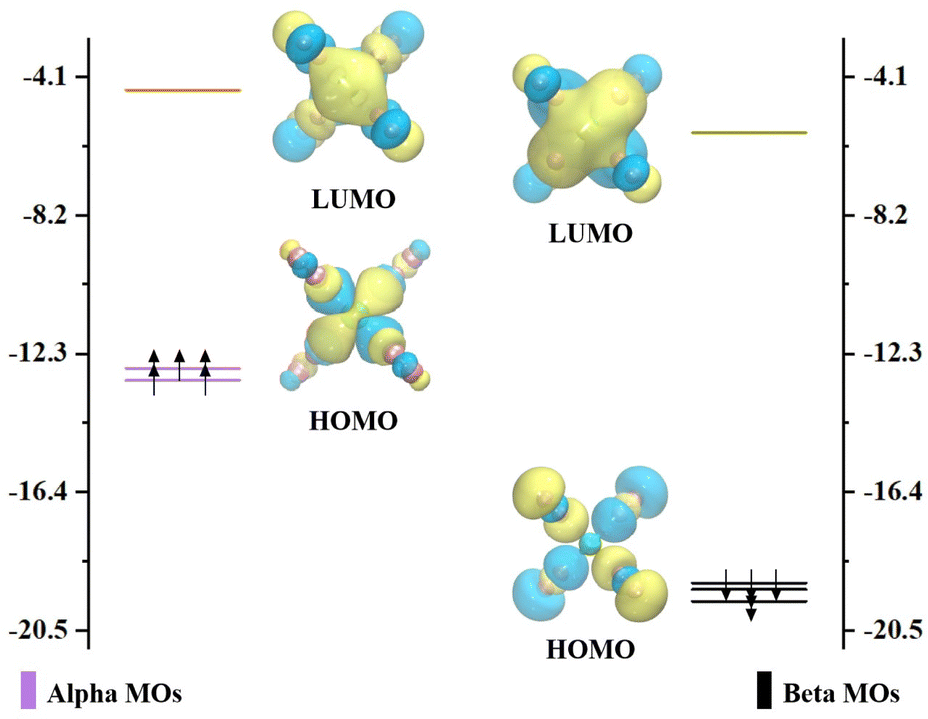 | ||
| Fig. 6 The molecular orbitals of the ground state structure of CrN8+ cluster. The purple and black lines indicate positions of the occupied α and β orbitals, while the orange and green lines indicate the unoccupied α and β orbitals, respectively. | ||
To explore the chemical bonding of CrN8+ cluster, the multicenter chemical bonds between Cr+ cation and N atoms are analyzed by AdNDP method. The multicenter bonding is denoted as the xc–2e, where x denotes the number of centers involved (x can range from one center (i.e., alone electrons on single atom) to the maximum number of atoms in the cluster (i.e., fully delocalization bonding)), and ON denotes the number of electrons occupying the cluster, with an ideal value of 2.00 |e|. The AdNDP results (Fig. 7) show that the chemical bonding patterns of CrN8+ clusters can be categorized into: five localized spin orbitals (LSOs) occupied by a single electron, four lone pairs (LPs), and 16 two center–two electron bonds (2c–2e). The five 3d subshell layers are occupied by Cr-3d electrons. These LSOs exhibit inert characteristics, devoid of participating in the chemical bonding. The four LPs with ON = 1.98 |e| mainly comprise 2s electrons of N atoms. The sixteen 2c–2e bonds, with ON values approximating the ideal value of 2.00 |e|, contain three types of bonds: the N–N σ bond, the N–N π bond, and the Cr–N σ bond. Among them, eight N–N π-bonds and four N–N σ-bonds contribute to the chemical bonding between the N2 units, while the remaining four σ-bonds between Cr and N describe the strong chemical bonds between the central Cr cation and the N2 units. These bonds are collectively responsible for the stability of cationic CrN8+ cluster. In addition, the chemical bonding patterns of neutral CrN8 cluster are shown in Fig. S5 (ESI†). Similarly, the fifteen 2c–2e bonds of CrN8 cluster are divided into three bonding modes, i.e. N–N σ bonds, N–N π bonds, and Cr–N σ bonds. Thus, it can be inferred that the presence of strong chemical bonds between Cr cation and N2 ligands enhances the stability of cationic CrN8+ cluster.
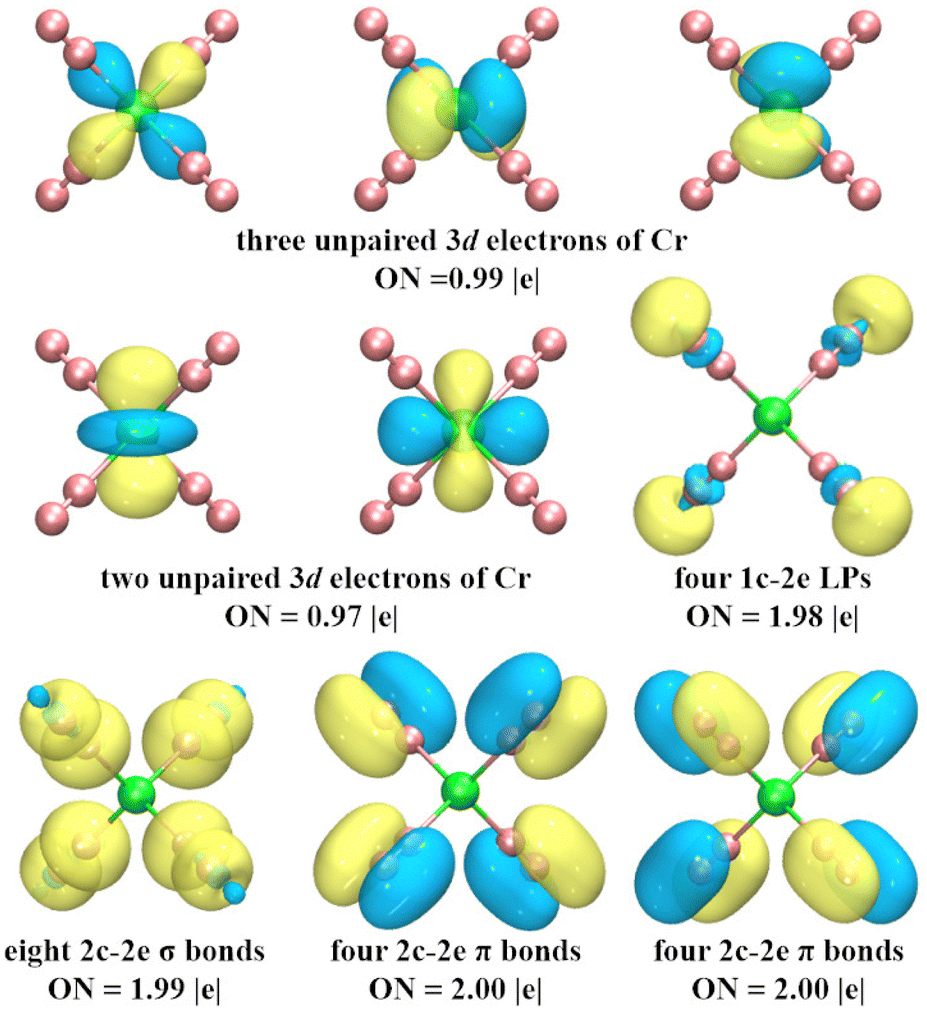 | ||
| Fig. 7 AdNDP analysis of the ground state structure of CrN8+ cluster. ON indicates the number of occupied electrons. | ||
4 Conclusions
In summary, we have produced chromium heteroazide clusters, namely CrN6+, CrN8+, CrN9+, and CrN11+, using laser sputtering techniques. The photodissociation results indicate the remarkable abundance of CrN8+ cluster compared to other chromium–nitrogen clusters. Subsequently, we have systematically studied the geometrics, stabilities and electronic properties of CrNn0/+ clusters (n = 2–11) by CALYPSO cluster structural search method and DFT calculations at the M06-2X/6-311+G(d,p) level. The obtained ground state structures of cationic chromium–nitrogen clusters are in good agreement with the mass spectra experiments. The calculated results reveal that the isomer with multiple N2 units tend to possess relatively lower energy than those with all-nitrogen units in chromium–nitrogen clusters. Cationic CrNn+ (n = 2–11) clusters, upon electron loss, exhibit enhanced symmetry and stability, especially for CrN8+ (6A, D2) cluster. Most ground state structures conform to configurations of Cr0/+ (N2)n/2 and Cr0/+ N(N2)(n−1)/2. Molecular orbital analyses of CrN80/+ clusters elucidate the emergence of the Jahn–Teller effect, primarily influenced by the interactions of the Cr-3d and N-2p orbitals. This effect is accompanied by a notable elevation in the HOMO–LUMO gap of cationic CrN8+ cluster. These results provide valuable insights for understanding the structural evolutions, chemical bonding and growth mechanisms of Cr doped nitrogen clusters, which provide an important avenue for the design and synthesis of novel transition metal nitrides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities, the China University of Geosciences (Wuhan) (Grant No. G1323523065), the National Natural Science Foundation of China (Grant No. 12304296), the Natural Science Foundation of Hubei (Grant No. 2022CFB527), Scientific Research Project of Jingchu University of Technology (YY202207, YB202212) and the Scientific and Technological Research of Chongqing Municipal Education Commission (KJQN202200620).References
- H. Hu, X. Wang, J. P. Attfield and M. Yang, Metal nitrides for seawater electrolysis, Chem. Soc. Rev., 2024, 53, 163–203 RSC.
- Y. Y. Wang, S. Liu, S. C. Lu, Y. Li and Z. Yao, Nitrogen-rich Ce-N compounds under high pressure, Phys. Chem. Chem. Phys., 2024, 26, 9601–9607 RSC.
- C. Zhang, C. Sun, B. Hu, C. Yu and M. Lu, Synthesis and characterization of the pentazolate anion cyclo-N5− in (N5)6(H3O)3(NH4)4Cl, Science, 2017, 355, 374–376 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. Bykov, S. Chariton, E. Bykova, S. Khandarkhaeva and T. Fedotenko, et al., High-pressure synthesis of metal-inorganic frameworks Hf4N20 center dot N2, WN8 center dot N2, and Os5N28 center dot 3N2 with polymeric nitrogen linkers, Angew. Chem., Int. Ed., 2020, 59(26), 10321–10326 CrossRef CAS PubMed.
- D. Laniel, B. Winkler, E. Koemets, T. Fedotenko, M. Bykov, E. Bykova, L. Dubrovinsky and N. Dubrovinskaia, Synthesis of magnesium-nitrogen salts of polynitrogen anions, Nat. Commun., 2019, 10, 4515 CrossRef PubMed.
- S. Lin, J. Chen, B. Zhang, J. Hao, M. Xu and Y. Li, Lanthanium nitride LaN9 featuring azide units: the first metal nine-nitride as a high-energy-density material, Phys. Chem. Chem. Phys., 2024, 26, 3605–3613 RSC.
- S. Niu, Y. Liu, Z. Yang, S. Liu and Z. Yao, Prediction of metastable phase of the Sc-N system in the N-rich region under high pressure, Phys. Chem. Chem. Phys., 2023, 25, 20009–20014 RSC.
- X. Xin, I. Douair, T. Rajeshkumar, Y. Zhao, S. Wang, L. Maron and C. Zhu, Photochemical synthesis of transition metal-stabilized uranium (VI) nitride complexes, Nat. Commun., 2022, 13, 3809 CrossRef CAS PubMed.
- T. Ye, S. Park, Y. Lu, J. Li, M. Sasase, M. Kitano and H. Hosono, Contribution of nitrogen vacancies to ammonia synthesis over metal nitride catalysts, J. Am. Chem. Soc., 2020, 142(33), 14374–14383 CrossRef CAS PubMed.
- L. P. Cheng and Q. S. Li, N4 ring as a square planar ligand in novel MN4 species, J. Phys. Chem. A, 2005, 109, 3182–3186 CrossRef CAS PubMed.
- R. Haiges, J. A. Boatz, T. Schroer, M. Yousufuddin and K. O. Christe, Experimental evidence for linear metal-azido coordination: the binary group 5 azides [Nb(N3)5], [Ta(N3)5], [Nb(N3)6]−, and [Ta(N3)6]−, and 1:1 acetonitrile adducts [Nb(N3)5(CH3CN)] and [Ta(N3)5(CH3CN)], Angew. Chem., Int. Ed., 2006, 45, 4830–4835 CrossRef CAS PubMed.
- R. Haiges, J. A. Boatz, M. Yousufuddin and K. O. Christe, Monocapped trigonal-prismatic transition-metal heptaazides: syntheses, properties, and structures of [Nb(N3)7]2− and [Ta(N3)7]2−, Angew. Chem., Int. Ed., 2007, 46, 2869–2874 CrossRef CAS PubMed.
- R. Haiges, J. A. Boatz and K. O. Christe, The syntheses and structure of the vanadium(IV) and vanadium(V) binary azides V(N3)4, [V(N3)6]2−, and [V(N3)6]−, Angew. Chem., Int. Ed., 2010, 49, 8008–8012 CrossRef CAS PubMed.
- R. Haiges, J. A. Boatz, R. Bau, S. Schneider, T. Schroer, M. Yousufuddin and K. O. Christe, Polyazide chemistry: the first binary group 6 azides, Mo(N3)6, W(N3)6, [Mo(N3)7]−, and [W(N3)7]−, and the [NW(N3)4]− and [NMo(N3)4]− ions, Angew. Chem., Int. Ed., 2005, 117(12), 1894–1899 CrossRef.
- C. Knapp and J. Passmore, On the way to “solid nitrogen” at normal temperature and pressure? binary azides of heavier group 15 and 16 elements, Angew. Chem., Int. Ed., 2004, 43, 4834–4836 CrossRef CAS PubMed.
- L. Gagliardi and P. Pyykkö, Scandium cycloheptanitride, ScN7: a predicted high-energy molecule containing an [η7-N7]3− ligand, J. Am. Chem. Soc., 2001, 123, 9700–9701 CrossRef CAS PubMed.
- Q. S. Li and H. X. Duan, Density functional theoretical study of a series of binary azides M(N3)n (n = 3, 4), J. Phys. Chem. A, 2005, 109, 9089–9094 CrossRef CAS PubMed.
- J. Müller, Azides of the heavier Group 13 elements, Coord. Chem. Rev., 2002, 235, 105–119 CrossRef.
- R. Haiges, J. A. Boatz, J. M. Williams and K. O. Christe, Preparation and characterization of the binary group 13 azides M(N3)3 and M(N3)3CH3 CN (M = Ga, In, Tl), [Ga(N3)5]2−, and [M(N3)6]3− (M = In, Tl), Angew. Chem., Int. Ed., 2011, 123, 8990–8995 CrossRef.
- L. Gagliardi and P. Pyykkö, Predicted group 4 tetra-azides M(N3)4 (M = Ti-Hf, Th): the first examples of linear M-NNN coordination, Inorg. Chem., 2003, 42, 3074–3078 CrossRef CAS PubMed.
- M. Straka, N6 ring as a planar hexagonal ligand in novel M(η6-N6) species, Chem. Phys. Lett., 2002, 358, 531–536 CrossRef CAS.
- H. X. Duan and Q. S. Li, A series of novel aromatic compounds with a planar N6 ring, Chem. Phys. Lett., 2006, 432, 331–335 CrossRef CAS.
- C. Sun, C. Zhang, C. Jiang, C. Yang, Y. Du, Y. Zhao, B. Hu, Z. Zheng and K. O. Christe, Synthesis of AgN5 and its extended 3D energetic framework, Nat. Commun., 2018, 9, 1269 CrossRef PubMed.
- Y. Wang, M. Bykov, I. Chepkasov, A. Samtsevich, E. Bykova, X. Zhang, S. Q. Jiang, E. Greenberg, S. Chariton and V. B. Prakapenka, Stabilization of hexazine rings in potassium polynitride at high pressure, Nat. Chem., 2022, 14, 794–800 CrossRef CAS PubMed.
- D. Laniel, F. Trybel, Y. Yin, T. Fedotenko, S. Khandarkhaeva, A. Aslandukov, G. Aprilis, A. I. Abrikosov, T. Bin Masood and C. Giacobbe, Aromatic hexazine [N6]4− anion featured in the complex structure of the high-pressure potassium nitrogen compound K9N56, Nat. Chem., 2023, 15, 641–646 CrossRef CAS PubMed.
- K. Ding, T. Li, H. Xu, Y. Li, Z. Ge, W. Zhu and W. Zheng, Mass spectrometry detection of LiN12+ cluster and theoretical investigation of its structures and stability, Chem. Phys. Lett., 2020, 747, 137310 CrossRef CAS.
- Z. Ge, K. Ding, Y. Li, H. Xu, Z. Chen, Y. Ma, T. Li, W. Zhu and W. Zheng, Structural evolution of LiNn+ (n = 2, 4, 6, 8, and 10) clusters: mass spectrometry and theoretical calculations, RSC Adv., 2019, 9(12), 6762–6769 RSC.
- K. Ding, T. Li, X. Hongguang, P. Jian, Y. Bin, W. Zheng and Z. Ge, Generation and detection of sodium heteronitrogen clusters with high nitrogen content, Chin. J. Explos. Propellants, 2020, 28, 597–602 CAS.
- T. Li, K. Ding, H. Xu, L. Qin, Q. Chen, W. Zheng and Z. Ge, Laser sputtering method for generating alkali metal heteronitrogen clusters NaNn+ and KNn+, Chem. Wld., 2021, 60(1), 28–32 Search PubMed.
- T. Li, K. Ding, H. Xu, L. Qian, J. Xiao, W. Zhu and Z. Ge, Formation and photodissociation of Cr doped nitrogen clusters CrNn+, Chin. J. Explos. Propellants, 2017, 40(06), 55–58 Search PubMed.
- K. Ding, H. Xu, Y. Yang, T. Li, C. Zhao, Z. Ge, W. Zheng and W. Zhu, Mass spectrometry and theoretical investigation of VNn+ (n = 8, 9, and 10) clusters, J. Phys. Chem. A, 2018, 122, 4687–4695 CrossRef CAS PubMed.
- K. Ding, L. Xiao, H. Xu, T. Li, Z. Ge, W. Qian and W. Zhu, Experimental observation of TiN12+ cluster and theoretical investigation of its stable and metastable isomers, Chem. Sci., 2015, 6, 4723–4729 RSC.
- K. Ding, H. Chen, H. Xu, B. Yang, Z. Ge, C. Lu and W. Zheng, Identification of octahedral coordinated ZrN12+ cationic clusters by mass spectrometry and structure searches, Dalton Trans., 2021, 50, 10187–10192 RSC.
- T. Li, K. Ding, L. Hong, P. Jian, Y. Bin, L. Wei and Z. Ge, Formation and laser photolysis of metal-doped nitrogen clusters FeNn+, CoNn+ and NiNn+, Chin. J. Explos. Propellants, 2022, 40, 200–204 Search PubMed.
- S. M. Casey and D. G. Leopold, Negative ion photoelectron spectroscopy of chromium dimer, J. Phys. Chem., 1993, 97, 816–830 CrossRef CAS.
- K. Andersson, The electronic spectrum of Cr2, Chem. Phys. Lett., 1995, 237, 212–221 CrossRef CAS.
- G. Gewinner, J. C. Peruchetti, A. Jaegle and A. Kalt, Photoemission study of the chromium (111) surface interacting with oxygen, Surf. Sci., 1978, 78, 439–458 CrossRef CAS.
- T. Jiang, I. Odnevall Wallinder and G. Herting, Chemical stability of chromium carbide and chromium nitride powders compared with chromium metal in synthetic biological solutions, Int. Scholarly Res. Not., 2012, 2012, 379697 Search PubMed.
- Z. Schauperl, V. Ivušić and B. Runje, Wear resistance of chromium-nitride and diamond-like carbon thin hard coatings, Mater. Test., 2008, 50, 326–331 CrossRef CAS.
- M. Khan, W. Cao, N. Chen, U. Asad and M. Z. Iqbal, Ab initio calculations of synergistic chromium-nitrogen codoping effects on the electronic and optical properties of anatase TiO2, Vacuum, 2013, 92, 32–38 CrossRef CAS.
- Q. Wang, Q. Sun, B. K. Rao, P. Jena and Y. Kawazoe, Nitrogen-induced magnetic transition in small chromium clusters, J. Chem. Phys., 2003, 119, 7124–7130 CrossRef CAS.
- Y. C. Zhao, Z. G. Zhang, J. Y. Yuan, H. G. Xu and W. J. Zheng, Modification of reflectron time-of-flight mass spectrometer for photodissociation of mass-selected cluster ions, Chin. J. Chem. Phys., 2009, 22, 655–662 CrossRef CAS.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Particle-swarm structure prediction on clusters, J. Chem. Phys., 2012, 137, 084104 CrossRef PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, CALYPSO: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal structure prediction via particle swarm optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 7174–7182 Search PubMed.
- B. L. Chen, K. H. He, W. Dai, G. L. Gutsev and C. Lu, Geometric and electronic diversity of metal doped boron clusters, J. Phys.: Condens. Matter, 2023, 35, 183002 CrossRef CAS PubMed.
- J. N. Zuo, L. L. Zhang, B. L. Chen, K. H. He, W. Dai, K. W. Ding and C. Lu, Geometric and electronic structures of medium-sized boron clusters doped with plutonium, J. Phys.: Condens. Matter, 2023, 36, 015302 CrossRef PubMed.
- B. L. Chen, L. J. Conway, W. Sun, X. Kuang, C. Lu and A. Hermann, Phase stability and superconductivity of lead hydrides at high pressure, Phys. Rev. B, 2021, 103, 035131 CrossRef CAS.
- C. Lu, C. Cui, J. Zuo, H. Zhong, S. He, W. Dai and X. Zhong, Monolayer ThSi2N4: an indirect-gap semiconductor with ultra-high carrier mobility, Phys. Rev. B, 2023, 108, 205427 CrossRef CAS.
- Q. Duan, L. Zhan, J. Shen, X. Zhong and C. Lu, Predicting superconductivity near 70 K in 1166-type boron-carbon clathrates at ambient pressure, Phys. Rev. B, 2024, 109, 054505 CrossRef CAS.
- B. L. Chen, K. H. He, W. Dai, G. L. Gutsev and C. Lu, Geometric and electronic diversity of metal doped boron clusters, J. Phys.: Condens. Matter, 2023, 35, 183002 CrossRef CAS PubMed.
- W. M. Haynes, CRC handbook of chemistry and physics, CRC press, 2014 Search PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Developing paradigms of chemical bonding: adaptive natural density partitioning, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. Kudin, J. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01203h |
| ‡ These authors contributed equally to this work and should be considered as co-first authors. |
| This journal is © the Owner Societies 2024 |