
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From Li2NiO3 to high-performance LiNiO2 cathodes for application in Li-ion and all-solid-state batteries†
Leonhard
Karger‡
a,
Philip
Henkel‡
a,
Saravanakumar
Murugan
a,
Ruizhuo
Zhang
 a,
Aleksandr
Kondrakov
*ab and
Torsten
Brezesinski
*a
a,
Aleksandr
Kondrakov
*ab and
Torsten
Brezesinski
*a
aBattery and Electrochemistry Laboratory, Institute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131 Karlsruhe, Germany. E-mail: torsten.brezesinski@kit.edu
bBASF SE, Carl-Bosch-Str. 38, Ludwigshafen 67056, Germany. E-mail: aleksandr.kondrakov@basf.com
First published on 12th September 2024
Abstract
The synthesis of LiNiO2 (LNO) typically involves oxidizing Ni(II) to Ni(III), thus leading to NiLi point-defect formation. Here, materials containing Ni(IV) in the form of overlithiated Li1+xNi1−xO2 (0 ≤ x ≤ 1/3) are converted into LNO. This method produces low defect-density samples at high temperatures, offering an attractive route toward coarse-grained particles that are naturally coated by the byproduct Li2O. In thiophosphate-based solid-state batteries, this kind of self-coating effect shows a direct correlation between residual lithium content and cycling performance.
LiNiO2 (LNO) has the prototypical structure of layered Ni-rich LiNixCoyMnzO2 (NCM or NMC, with x ≥ 0.8) cathode active materials (CAMs),1 and seems particularly attractive for solid-state battery (SSB) applications. However, for successful implementation in thiophosphate-based SSBs, the CAM particle surface must be stabilized to prevent ion interdiffusion and lattice oxygen loss, as well as to suppress side reactions with the solid electrolyte.2,3 With regard to stabilization, coating and/or (surface) doping strategies have been reported for Ni-rich CAMs. Even though these strategies are successful to varying degrees, they often require additional processing steps.4–6
A promising strategy for mitigating mechanical degradation is the application of coarse-grained or even single-crystalline CAMs.7 Increasing the calcination temperature in the synthesis is one way to achieving desired particle morphologies.8 However, this is typically accompanied by a relatively high degree of substitutional defects (occupational disorder), due to reduced oxygen activity at elevated temperatures and potential lithium loss.9–11 Other methods for preparing single-crystalline LNO do exist, but either require long dwell times or the use of fluxes as reaction media, which need to be removed in subsequent processing steps.12,13
Herein, we describe a promising alternative route to LNO that combines the benefits of large grain size, low defect density, and the possibility for intrinsic self-coating. All of this is achieved through a simple two-step preparation (synthesis/decomposition) process. While conventional synthesis of LNO involves oxidation of Ni(II) to Ni(III), we make use of overlithiated Li1+xNi1−xO2 (0 ≤ x ≤ 1/3) containing Ni(III)/Ni(IV) species. Specifically, a series of Li-rich LNO (referred to as LRLNO) samples were prepared at ambient pressure and at 550 °C (see ESI† for details).14 In a second step, these LRLNOs were then thermally decomposed to yield LNO and Li2O. The latter may act as a protective coating or as a lithium source to target more complex coating chemistries.
Initially, the decomposition of Li1+xNi1−xO2 with 50 mol% lithium excess (x = 0.2) was probed at temperatures ranging from 725 to 900 °C (referred to as LRLNO 0.2–xxx, with xxx denoting the calcination temperature) using scanning electron microscopy (SEM) and X-ray diffraction (XRD), as shown in Fig. 1a–h and Fig. S1 (ESI†). Successful synthesis of monoclinic Li1.2Ni0.8O2 (see Fig. 1b), crystallizing in the C2/m space group, was confirmed by comparing the lattice parameters with data available in the literature.14 Only samples heated at temperatures ≥725 °C were found to convert into hexagonal LNO (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group). By contrast, those obtained at lower temperatures (of note, the commonly used calcination temperature for high-quality LNO is 700 °C) appear to still be monoclinically distorted, revealing Bragg reflections around 8.5 and 20.2° 2θ (see XRD patterns in Fig. S2, ESI†). These peaks are characteristic of Li2NiO3, reflecting the ordered distribution of lithium ions on the nickel positions, and vanish upon calcination at temperatures ≥725 °C. In particular, the split peaks at 20.2° evolve into a single one centered at 20.0°, indicating mitigation of monoclinic distortion.15 At 750 °C, single-phase LNO (ignoring Li2O) with both a coarse-grained particle structure and a low fraction of NiLi defects (1.3%) is obtained, as shown in Fig. 1f and g. The full structural refinement results are given in Table S1 (ESI†). Increasing the temperature beyond 750 °C results in grain growth while maintaining a relatively low degree of substitutional defects up to 800 °C. Micrometer-sized, single-crystalline particles are obtained at 850 and 900 °C (see Fig. S1, ESI†). Overall, the results indicate that the formation of LNO from LRLNOs necessitates higher calcination temperatures than that from Ni(II)-based precursor CAMs. However, the samples contain fewer point defects as compared to LNO prepared by conventional solid-state synthesis at 700 °C (see Table S1, ESI†). As mentioned previously, the particle morphology somewhat changes upon converting the Li1.2Ni0.8O2 (LRLNO 0.2) into LNO at 750 °C. In particular, the primary particles increase in size, while the secondary particles largely maintain their original morphology (see SEM images in Fig. 1a and f).
m space group). By contrast, those obtained at lower temperatures (of note, the commonly used calcination temperature for high-quality LNO is 700 °C) appear to still be monoclinically distorted, revealing Bragg reflections around 8.5 and 20.2° 2θ (see XRD patterns in Fig. S2, ESI†). These peaks are characteristic of Li2NiO3, reflecting the ordered distribution of lithium ions on the nickel positions, and vanish upon calcination at temperatures ≥725 °C. In particular, the split peaks at 20.2° evolve into a single one centered at 20.0°, indicating mitigation of monoclinic distortion.15 At 750 °C, single-phase LNO (ignoring Li2O) with both a coarse-grained particle structure and a low fraction of NiLi defects (1.3%) is obtained, as shown in Fig. 1f and g. The full structural refinement results are given in Table S1 (ESI†). Increasing the temperature beyond 750 °C results in grain growth while maintaining a relatively low degree of substitutional defects up to 800 °C. Micrometer-sized, single-crystalline particles are obtained at 850 and 900 °C (see Fig. S1, ESI†). Overall, the results indicate that the formation of LNO from LRLNOs necessitates higher calcination temperatures than that from Ni(II)-based precursor CAMs. However, the samples contain fewer point defects as compared to LNO prepared by conventional solid-state synthesis at 700 °C (see Table S1, ESI†). As mentioned previously, the particle morphology somewhat changes upon converting the Li1.2Ni0.8O2 (LRLNO 0.2) into LNO at 750 °C. In particular, the primary particles increase in size, while the secondary particles largely maintain their original morphology (see SEM images in Fig. 1a and f).
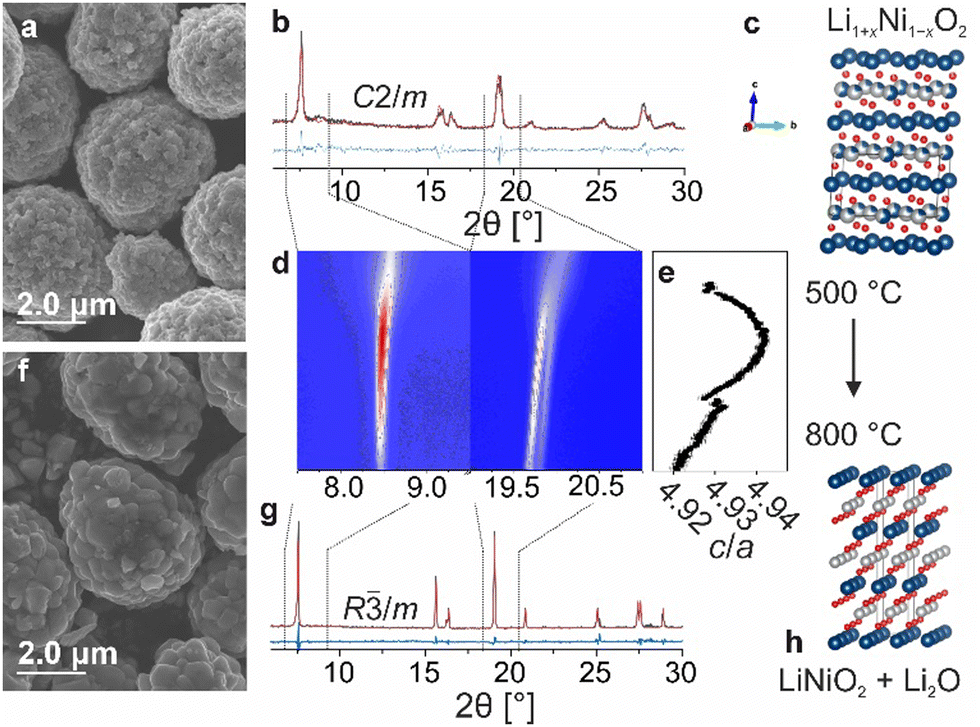 | ||
| Fig. 1 (a) SEM image, (b) XRD pattern including refinement plot, and (c) illustration of the crystal structure of Li1.2Ni0.8O2. (d) Zoomed-in view of the XRD raw data from an in situ heating experiment (from 500 to 800 °C) and (e) corresponding refined layering parameter c/a. (f) SEM image, (g) XRD pattern including refinement plot, and (h) illustration of the crystal structure of LiNiO2 obtained by thermal decomposition of Li1.2Ni0.8O2 at 750 °C. | ||
To gain more insight into the decomposition of Li1.2Ni0.8O2, an in situ XRD heating experiment was performed in the temperature range of 500 to 800 °C (see Fig. 1d). As evident from the Le Bail refinement results in Fig. 1e, the layering parameter c/a increases first up to 4.942 and then decreases. This suggests that the CAM quality strongly depends on the synthesis conditions, given that LNO itself is prone to degradation at high temperatures. It also means that LNO produced closer to the actual decomposition temperature of Li1.2Ni0.8O2 can be expected to exhibit the best electrochemical performance, due to low defect density resulting in high specific capacity because of more efficient CAM re-lithiation.16,17 Nevertheless, the lithium excess must be taken into account when evaluating the cyclability.18,19 Said lithium is present in the form of Li2O and can be clearly detected by XRD (see Fig. S3, ESI†). The amount of Li2O increases with increasing degree of overlithiation (i.e., x in Li1+xNi1−xO2), eventually leading to impedance growth and reducing the capacity and cycle life of Li-ion batteries (LIBs).1
In the present study, LRLNO 0.2–750 before and after washing and re-annealing was tested electrochemically in LIB half-cells (see Fig. S4, ESI†). As expected, the washed CAM delivered higher capacities and exhibited lower overpotentials during cycling. Therefore, for fair comparison, all LRLNO 0.2–xxx samples were washed and re-annealed, as described in the ESI.† The room-temperature cycling performance of the LRLNO 0.2–725, 0.2–750, and 0.2–800 cathodes with areal loadings of 7–10 mgCAM cm−2 was then examined in a potential range between 3.0 and 4.3 V versus Li+/Li and compared to that achieved with the reference LNO. The different samples delivered first-cycle specific discharge capacities exceeding 200 mA h gCAM−1 at 0.05C (see Fig. 2a), with the highest of 221 mA h gCAM−1 observed for LRLNO 0.2–750 being close to that of the reference LNO (222 mA h gCAM−1). As can be seen from the voltage profiles in Fig. 2a, the charge–discharge curves of the cells using LRLNO 0.2–725 are smoother with less pronounced plateaus, indicating solid-solution-like behavior and pointing toward incomplete conversion of the Li1.2Ni0.8O2 into LNO at 725 °C. By contrast, the voltage profiles of LRLNO 0.2–750 and 0.2–800 are similar, except for the end of discharge (shorter “plateau” for LRLNO 0.2–800). This is also apparent to some degree from the second-cycle differential capacity curves shown in Fig. 2b. The latter region, which is associated with poor lithium diffusion in layered oxide cathodes, is also called the kinetic-hindrance (KH) region.20,21 Overall, a lower differential capacity in the KH region is indicative of less mobile lithium and/or large grains.22,23 The initial specific charge/discharge capacities achieved with LRLNO 0.2–750 were slightly higher compared to those of LRLNO 0.2–800 (249/221 mA h gCAM−1vs. 246/213 mA h gCAM−1). This difference is primarily due to the larger primary particle size (increase in diffusion path length) at 800 °C. However, a higher concentration of NiLi defects also leads to slower lithium diffusion, and therefore to lower capacities.24 It should be noted that LRLNO 0.2–750 indeed exhibits a better first-cycle Coulomb efficiency than the reference LNO (89% vs. 87%), emphasizing the improved kinetics, which is also evident from the rate capability (see Fig. 2c). However, regarding long-term performance, LRLNO 0.2–800 was found to be the most stable, retaining 86% of its capacity – relative to the first cycle at 0.33C after rate performance testing – after 50 cycles, as shown in Fig. 2c. This is likely an attribute of the coarse-grained structure of LRLNO 0.2–800, as larger particles are expected to have a lower tendency for surface-related side reactions (lower reactivity) and are less prone to cracking.7,25 Interestingly, the cells using LRLNO 0.2–725 experienced lower capacity degradation than those with LRLNO 0.2–750. This result could suggest that a partially decomposed structure is beneficial from a stability perspective. Nevertheless, the state of charge (SOC) achieved in the initial cycles at low C-rates was different among the cells. Of note, the SOC is known to strongly affect the degradation behavior of layered Ni-rich cathodes. Overall, the data demonstrate that thermal decomposition of Li1.2Ni0.8O2 is effective in obtaining high-quality LNO that is capable of outperforming the reference CAM. Because LRLNO 0.2–750 delivered the highest specific discharge capacity in the initial cycle, a calcination temperature of 750 °C was chosen for further studies on the effect of x in Li1+xNi1−xO2 on cyclability.
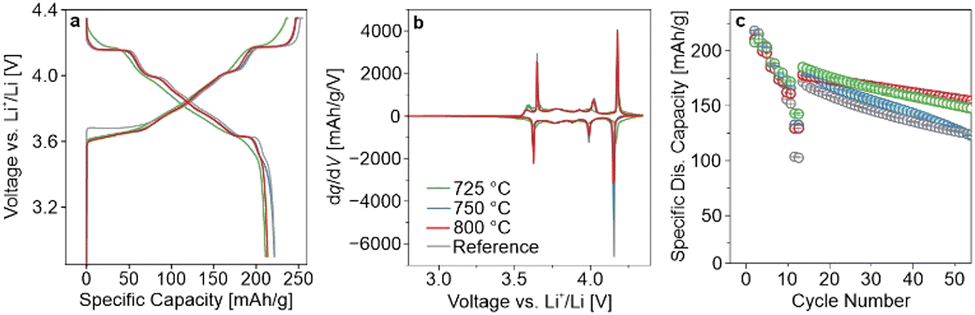 | ||
| Fig. 2 (a) First-cycle charge–discharge curves of LIB half-cells using LRLNO 0.2–725, 0.2–750, 0.2–800, or reference LNO at 25 °C and at 0.05C. (b) Respective second-cycle differential capacity curves. (c) C-rate testing at 0.05C, 0.1C, 0.33C, 1C, 2C, and 5C (two cycles each) followed by cycling at 0.33C. The error bars represent the standard deviation from three independent cells. | ||
Samples with x = 0, 0.1, 0.2, and 0.33, hereafter referred to as LRLNO 0–750, 0.1–750, 0.2–750, and 0.33–750, respectively, were synthesized following the procedure described above (see ESI† for details; structural refinement results are given in Table S2, ESI†). The different materials were also washed and re-annealed prior to electrochemical testing in LIB half-cells. The first-cycle voltage profiles, second-cycle differential capacity curves, and rate capabilities followed by long-term cycling at 0.33C are shown in Fig. 3a–c. LRLNO 0–750 delivered the highest initial specific discharge capacity of 225 mA h gCAM−1 at 0.05C and exhibited a capacity retention of 79% after 50 cycles (relative to the first cycle at 0.33C after rate performance testing). Increasing x in Li1+xNi1−xO2 to 0.33 leads to a lower first-cycle specific discharge capacity of 207 mA h gCAM−1. However, the capacity does not decrease linearly; it was found to be lower for LRLNO 0.1–750 than for LRLNO 0.2–750 (212 mA h gCAM−1vs. 221 mA h gCAM−1), as can be seen from Fig. 3a. SEM images collected from the different samples (see Fig. S5, ESI†) indicate primary particle growth with increasing x, especially when compared to LRLNO 0–750 (x = 0). This can be explained by excess lithium (Li2O) acting as a flux and accelerating grain growth.12 Surprisingly, somewhat larger primary particles were found for LRLNO 0.1–750 than for LRLNO 0.2–750. A possible reason could be the use of different tube furnaces in the synthesis, although all of them were calibrated to ±5 °C, suggesting that the CAM, in general, is very sensitive to the calcination conditions, with small deviations in temperature leading to different grain sizes. The differences in particle size are also reflected in the electrochemical data, with smaller-grained CAM delivering higher capacities, irrespective of C-rate (see Fig. 3c). Similarly, cells using LRLNO 0.1–750 achieved lower capacities than those with LRLNO 0.2–750 over the first 40 cycles or so. This is because larger particles (grains) inevitably lead to longer diffusion pathways for lithium and electron transport during charge and discharge, which in turn results in lower CAM utilization and compromised rate performance.26 However, the LRLNO 0.2–750 cells suffered much more severe capacity fading, as shown in Fig. 3c.
 | ||
| Fig. 3 (a) First-cycle charge–discharge curves of LIB half-cells using LRLNO 0–750, 0.1–750, 0.2–750, or 0.33–750 at 25 °C and at 0.05C. (b) Respective second-cycle differential capacity curves. (c) C-rate testing at 0.05C, 0.1C, 0.33C, 1C, 2C, and 5C (two cycles each) followed by cycling at 0.33C. The error bars represent the standard deviation from three independent cells. | ||
The higher lithium excess in LRLNO 0.33–750 should, in principle, result in more lithium being released (see Fig. S3, ESI†). This could be beneficial with regard to SSB applications, as Li2O may act as a protective coating on the secondary particles. From the SEM images in Fig. S5 (ESI†), it is evident that a surface layer covers the LRLNO 0.33–750 particles. To examine the protective nature of Li2O, the CAMs were also tested in pellet-stack SSB cells. However, unlike for LIBs, they were not washed prior to use, ensuring that the variation in x manifests in the form of a coating. Specifically, the unwashed samples were electrochemically tested in pellet-stack cells using a lithium thiophosphate (argyrodite Li6PS5Cl) solid electrolyte at 45 °C and at 81 MPa in a potential range between 2.3 and 3.7 V vs. In/InLi (see ESI† for details). The initial specific discharge capacity of LRLNO 0.33–750 at 0.1C was the lowest with 107 mA h gCAM−1 (see Fig. 4a). This was to be expected, since with 100 mol% lithium excess a rather thick layer is formed on the particle surface, which negatively affects the charge transfer and leads to impedance build-up. However, the cells still delivered reasonable capacities and displayed dq/dV peaks characteristic of LNO, despite losing capacity over the whole potential window, but especially so at the end of discharge (see Fig. 4b). Decreasing x leads to a significant increase in capacity (see also rate capability data in Fig. 4c), with the specific discharge capacity reaching about 160 mA h gCAM−1 for LRLNO 0–750 and 0.1–750 in the first cycle. Regarding long-term performance, LRLNO 0.33–750 was found to have the best stability, as shown in Fig. 4c. From this data, it can be concluded that the coating effectively mitigates adverse side reactions at the CAM|solid electrolyte interface, thus contributing to cycling stability, but also limits the capacity. By contrast, lack of lithium excess, as in LRLNO 0–750, results in increased capacities over the first few cycles while sacrificing stability.
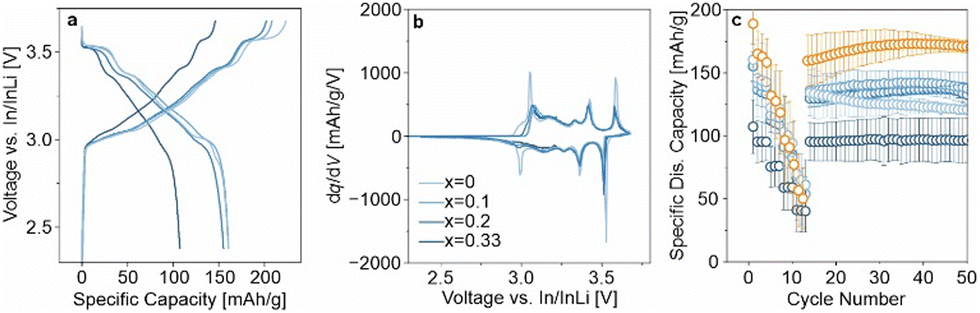 | ||
| Fig. 4 (a) First-cycle charge–discharge curves of SSB cells using LRLNO 0–750, 0.1–750, 0.2–750, or 0.33–750 at 45 °C and at 0.1C. (b) Respective second-cycle differential capacity curves. (c) C-rate testing at 0.1C (one cycle), 0.2C, 0.5C, 1C, und 2C (three cycles each) followed by cycling at 0.2C. The orange data points display the performance of LNO with a cobalt-based coating. The error bars represent the standard deviation from three independent cells. | ||
Ideally, the residual lithium should be controlled at a very low level or utilized in the formation of an ionically conductive coating for further improving the material performance.27 As a proof-of-concept, a facile approach has been developed by incorporating 5 mol% of Co(NO3)2 into the reaction sequence of Li1+xNi1−xO2 with x = 0.025. In fact, this kind of modification significantly enhanced the cyclability, with the SSB cells exhibiting a first-cycle specific charge capacity of about 235 mA h gCAM−1, even higher than that of LRLNO 0–750, and a specific discharge capacity of 189 mA h gCAM−1. The cells were also able to maintain 171 mA h gCAM−1 after 50 cycles, as indicated by the orange data points in Fig. 4c. The improvement can presumably be attributed to the formation of LiCoO2 (LCO) or spinel-type LiCo2O4 (from the lithium excess), both of which are well-established electrode materials for application in LIBs. Apparently, they also act as a protective coating and consume at least some, if not most, of the lithium (Li2O) released during thermal decomposition, offering a promising strategy to boost the cycling performance and stability of the formed CAM. However, XRD did not reveal detectable amounts of Li2O, LiCo2O4, or LCO, with only a minor additional reflection at 18.3° 2θ indicating the presence of an unknown impurity phase (see Fig. S6, ESI†). As can be seen from Table S2 (ESI†), the lattice parameter a of the hexagonal LNO phase is smaller compared to that of the LRLNO samples, hinting at cobalt substitution.28 SEM imaging (see Fig. S5, ESI†) further revealed that the cobalt is in fact segregating at the free surface, thus leading to the formation of a protective coating, which evidently plays a decisive role in stabilizing the CAM against degradation.
In summary, in this work, a facile method for synthesizing LNO from Li2NiO3-type compounds is reported. This approach yields single-phase LNO with larger primary particles and fewer substitutional defects than conventional solid-state synthesis routes. Notably, calcination temperatures above 700 °C still allow obtaining LNO of good quality. The samples were either washed and tested in LIBs or directly examined in SSBs, with the intrinsically formed Li2O serving as a kind of protective coating on the secondary CAM particles. In LIBs, 750 °C gave the best results with regard to first-cycle capacity, probably due to a trade-off between defect density and grain size. In SSBs, the degree of lithium excess was found to be crucial, as it determines the Li2O content (coating thickness). Larger amounts negatively affect the battery capacity while enhancing long-term cycling stability. Aside from that, we have demonstrated that the released lithium can be utilized as a source for more advanced coating chemistries.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declareNotes and references
- M. Bianchini, M. Roca-Ayats, P. Hartmann, T. Brezesinski and J. Janek, Angew. Chem., Int. Ed., 2019, 58, 10434–10458 CrossRef PubMed.
- R. Zhang, F. Strauss, L. Jiang, L. Casalena, L. Li, J. Janek, A. Kondrakov and T. Brezesinski, Chem. Commun., 2023, 59, 4600–4603 RSC.
- Y. Ma, J. H. Teo, F. Walther, Y. Ma, R. Zhang, A. Mazilkin, Y. Tang, D. Goonetilleke, J. Janek, M. Bianchini and T. Brezesinski, Adv. Funct. Mater., 2022, 32, 2111829 CrossRef.
- Q. Liu, Q. Chen, Y. Tang and H.-M. Cheng, Electrochem. Energy Rev., 2023, 6, 15 CrossRef.
- S. Kalluri, M. Yoon, M. Jo, H. K. Liu, S. X. Dou, J. Cho and Z. Guo, Adv. Mater., 2017, 29, 1605807 CrossRef PubMed.
- D. Weber, Đ. Tripković, K. Kretschmer, M. Bianchini and T. Brezesinski, Eur. J. Inorg. Chem., 2020, 2020, 3117–3130 CrossRef.
- W. van den Bergh, L. Karger, S. Murugan, J. Janek, A. Kondrakov and T. Brezesinski, ChemElectroChem, 2023, 10, e202300165 CrossRef.
- P. Kurzhals, F. Riewald, M. Bianchini, H. Sommer, H. A. Gasteiger and J. Janek, J. Electrochem. Soc., 2021, 168, 110518 CrossRef.
- L. Karger, D. Weber, D. Goonetilleke, A. Mazilkin, H. Li, R. Zhang, Y. Ma, S. Indris, A. Kondrakov, J. Janek and T. Brezesinski, Chem. Mater., 2023, 35, 648–657 CrossRef.
- E. McCalla, G. H. Carey and J. R. Dahn, Solid State Ionics, 2012, 219, 11–19 CrossRef.
- D.-h Lee, M. Avdeev, D.-i Kim, W. H. Shin, J. Hong and M. Kim, ACS Appl. Energy Mater., 2023, 6, 5309–5317 CrossRef.
- W. van den Bergh, R. Yao, R. Zhang, A. Kondrakov, J. Janek and T. Brezesinski, J. Mater. Chem. A, 2024, 12, 8683–8688 RSC.
- M. Kim, L. Zou, S.-B. Son, I. D. Bloom, C. Wang and G. Chen, J. Mater. Chem. A, 2022, 10, 12890–12899 RSC.
- M. Bianchini, A. Schiele, S. Schweidler, S. Sicolo, F. Fauth, E. Suard, S. Indris, A. Mazilkin, P. Nagel, S. Schuppler, M. Merz, P. Hartmann, T. Brezesinski and J. Janek, Chem. Mater., 2020, 32, 9211–9227 CrossRef.
- T. Ohzuku, A. Ueda, M. Nagayama, Y. Iwakoshi and H. Komori, Electrochim. Acta, 1993, 38, 1159–1167 CrossRef.
- Z. Lu, X. Huang, H. Huang, L. Chen and J. Schoonman, Solid State Ionics, 1999, 120, 103–107 CrossRef.
- J. Välikangas, P. Laine, M. Hietaniemi, T. Hu, P. Tynjälä and U. Lassi, Appl. Sci., 2020, 10, 8988 CrossRef.
- M. Tabuchi, N. Kuriyama, K. Takamori, Y. Imanari and K. Nakane, J. Electrochem. Soc., 2016, 163, A2312–A2317 CrossRef.
- A. Manthiram, J. C. Knight, S.-T. Myung, S.-M. Oh and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- L. Karger, S. Korneychuk, W. van den Bergh, S. L. Dreyer, R. Zhang, A. Kondrakov, J. Janek and T. Brezesinski, Chem. Mater., 2024, 36, 1497–1512 CrossRef.
- H. Nguyen, R. Silverstein, A. Zaveri, W. Cui, P. Kurzhals, S. Sicolo, M. Bianchini, K. Seidel and R. J. Clement, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202306168.
- R. Ruess, D. Gomboso, M. Ulherr, E. Trevisanello, Y. Ma, A. Kondrakov, T. Brezesinski and J. Janek, J. Electrochem. Soc., 2023, 170, 020533 Search PubMed.
- H. Li, N. Zhang, J. Li and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A2985–A2993 CrossRef.
- J. Li, G. Liang, W. Zheng, S. Zhang, K. Davey, W. K. Pang and Z. Guo, Nano Mater. Sci., 2023, 5, 404–420 CrossRef.
- S. Xia, L. Mu, Z. Xu, J. Wang, C. Wei, L. Liu, P. Pianetta, K. Zhao, X. Yu, F. Lin and Y. Liu, Nano Energy, 2018, 53, 753–762 CrossRef.
- Y. Han, S. H. Jung, H. Kwak, S. Jun, H. H. Kwak, J. H. Lee, S.-T. Hong and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2100126 CrossRef.
- S. Payandeh, F. Strauss, A. Mazilkin, A. Kondrakov and T. Brezesinski, Nano Res. Energy, 2022, 1, e9120016 CrossRef.
- D. Caurant, N. Baffier, B. Garcia and J. P. Pereira-Ramos, Solid State Ionics, 1996, 91, 45–54 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03873h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |