
Material design, development, and trend for surface-enhanced Raman scattering substrates
Yue
Ying
ab,
Zhiyong
Tang
 ab and
Yaling
Liu
*ab
ab and
Yaling
Liu
*ab
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: liuyl@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 7th June 2023
Abstract
Surface-enhanced Raman scattering (SERS) is a powerful and non-invasive spectroscopic technique that can provide rich and specific chemical fingerprint information for various target molecules through effective SERS substrates. In view of the strong dependence of the SERS signals on the properties of the SERS substrates, design, exploration, and construction of novel SERS-active nanomaterials with low cost and excellent performance as the SERS substrates have always been the foundation and the top priority for the development and application of the SERS technology. This review specifically focuses on the extensive progress made in the SERS-active nanomaterials and their enhancement mechanism since the first discovery of SERS on the nanostructured plasmonic metal substrates. The design principles, unique functions, and influencing factors on the SERS signals of different types of SERS-active nanomaterials are highlighted, and insight into their future challenge and development trends is also suggested. It is highly expected that this review could benefit a complete understanding of the research status of the SERS-active nanomaterials and arouse the research enthusiasm for them, leading to further development and wider application of the SERS technology.
 Yue Ying | Yue Ying received his bachelor's degree in chemistry from Jilin University in 2019. He is now a PhD candidate at National Center for Nanoscience and Technology in China under the supervision of Prof. Yaling Liu and Prof. Zhiyong Tang. His research covers active non-metal nanomaterials for surface-enhanced Raman scattering. |
 Zhiyong Tang | Zhiyong Tang obtained his Ph.D. degree in 1999 from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. After that he went to the Swiss Federal Institute of Technology Zurich, Oklahoma State University and University of Michigan for his postdoctoral research. In November 2006, he joined National Center for Nanoscience and Technology in China and took a full professor position. His current research interests are focused on the fabrication and application of nanomaterials. |
 Yaling Liu | Yaling Liu received her Ph.D. degree in 2008 from the Institute of Chemistry, Chinese Academy of Sciences. After graduation, she joined National Center for Nanoscience and Technology in China. Her current research is focused on the controllable synthesis and function regulation of nanostructured materials. |
1 Introduction
Raman spectroscopy, a powerful vibrational optical spectroscopic technique, is used to provide unique and detailed fingerprint information for target molecules on chemical structures, molecular interactions, etc.1–5 However, the inherent weakness of Raman signals due to the low probability that nearly one in a million scattered photons belongs to the Raman scattering restricts its development and further wider application, especially in the fields of trace analysis and surface science, which has been broken through in the 1970s with the discovery of surface-enhanced Raman scattering (SERS) by molecules adsorbed on nanostructured metal surfaces.6–10 Interestingly, the Raman enhancement factors by SERS can be up to 108 or even larger owing to the introduction of SERS-active nanomaterials as the substrates.11 For example, in 1997, the SERS detection of single rhodamine 6G molecules adsorbed on the selected silver colloidal nanoparticles (NPs) were achieved by Nie et al., revealing the ultra-sensitivity of SERS up to the single-molecule level and meanwhile prompting its huge application potential in more widespread fields, especially in surface science.12 At present, SERS has become a versatile analytical tool with ultra-sensitivity in material characterization, analytical science, biomedicine, and so on, driven by the continuous development of nanoscience and nanotechnology.13–15SERS is a surface-sensitive and intrinsically nanostructure-based phenomenon, and the SERS enhancement effect largely depends on the properties of the nanostructured SERS-active substrates, including the component, size, shape, structure, local environment, surface chemistry, and interaction with target molecules.16 Hence, the design, exploration, and construction of effective SERS-active nanomaterials as the SERS substrates are the foundation and the top priority for the development and applications of SERS technology all the time. Theoretically, any material that can support the activity of plasmons at the excitation wavelength can be used as the SERS substrates. In fact, for analysis purposes and further practical applications, high-quality SERS spectra are necessary, so the SERS substrates not only need to meet the basic requirements of high sensitivity, good reproductivity, and long-term stability but also need to overcome many challenges on how to improve the selectivity and multifunctionality for target molecules, to eliminate matrix interferences, and so on.13,17
In the past few decades, aiming to realize the highly-effective SERS enhancement, improve the SERS detection sensitivity, and widen the SERS application, SERS-active nanomaterials as the SERS substrates have undergone extensive development from original single-component materials (e.g., metals, semiconductors, graphene, and metal–organic frameworks) to abundant composite materials with functional multi-components.18–21 Meanwhile, the SERS enhancement mechanism has also mainly evolved from a single long-range electromagnetic or short-range chemical mechanism to a combination mechanism of both of them. In this review, the SERS mechanism is first briefly introduced owing to its guiding significance for material design. Subsequently, the design and development of a series of the reported SERS-active nanomaterials are highlighted according to their components, and at the same time the factors that influence the SERS signals are discussed. Finally, future challenges and development trends in the design, exploration, and construction of SERS-active nanomaterials are prospected.
2 SERS mechanism
Compared with the progress made in SERS experiments and applications, the research on the theory of SERS mechanisms has been relatively lagging behind and is still being debated owing to the complexity of the system with the SERS effect. So far, there are two theories largely accepted to explain the SERS effect, the physical electromagnetic enhancement mechanism, and the chemical enhancement mechanism. Moreover, the physical electromagnetic enhancement mechanism is believed to play a major role in the observed SERS signals.11,22When the incident light interacts with a molecule, the external electromagnetic field can change the charge distribution of the molecule and generate an induced dipole.23 Raman scattering is emitted by the induced dipole (p) oscillating at a frequency (ωR), which is different from the frequency of the incident light (ωL). The induced dipole depends on the electromagnetic field in its position (ELoc) and its Raman polarizability tensor (αR). In most cases, the induced dipole can be described within a linear approximation and expressed as:
| p(ωR) = αR(ωR, ωL)·ELoc (ωL) | (1) |
From classical electrodynamics, the intensity of Raman scattering (I) is proportional to |p|2:
| I ∝ |p|2 | (2) |
Thus, by changing the polarizability tensor and the local electromagnetic field, a more intensive Raman signal can be obtained. Generally, electromagnetic enhancement is achieved by enhancing the local electromagnetic field whereas chemical enhancement originates from changes in the Raman polarizability tensor.
The enhancement factor (EF) is one of the most important metrics used to quantitatively measure the enhancement. It can be expressed as EF = (ISERS/INRS) × (NNRS/NSERS), where I and N represent the signal intensity and the number of detected molecules of SERS or normal Raman scattering (NRS).24,25 Obviously, this definition focuses on the intrinsic characteristics of the SERS substrates and can measure the average SERS enhancements of each molecule on different substrates.26 However, the challenge of accurately estimating the number of detected molecules makes this EF parameter unable to directly reflect the changes of the SERS signal intensity in experiment results. Hence, for many applications, analytical enhancement factor (AEF) is defined for conveniently comparing SERS signals to normal Raman signals under given experimental conditions, which can be expressed as AEF = (ISERS/INRS) × (cNRS/cSERS), where c represent the concentration of the detected molecules of SERS or NRS.24,26
2.1 Electromagnetic mechanism
Electromagnetic mechanism (EM) refers to the enhancement of the local electric field caused by plasmon resonance excitation, which is the major origin of the enhancement of the SERS-active nanomaterials based on metal nanostructures.27–29 When the incident light interacts with plasmonic metal nanostructures, it can excite the conductive electrons of plasmonic metal nanostructures into collective oscillations, resulting in the amplification of the local electromagnetic field. Namely, the energy of the incident light is confined in a small region around plasmonic metal nanostructures with a strongly enhanced electromagnetic field.When a Raman process happens nearby plasmonic metal nanostructures, the enhanced local electromagnetic field leads to enhanced polarization.23 Based on the above eqn (1) and (2), the enhancement can be described with a factor MLoc(ωL) = |E(ωL)|2/|E0(ωL)|2, where the subscript “0” relates to the Raman process occurring without nearby plasmonic metal nanostructures. The enhanced local electromagnetic field also enhances the re-radiation process with another factor MLoc(ωR) = |E(ωR)|2/|E0(ωR)|2. Combining these two contributions, the overall EF of EM can be expressed as:
| EFEM ≈ MLoc(ωL)MLoc(ωR) | (3) |
Usually, EFEM can be further simplified by ignoring the Raman shift, so that ωL can be approximately equal to ωR and meanwhile the well-known |E|4-approximation can be obtained.
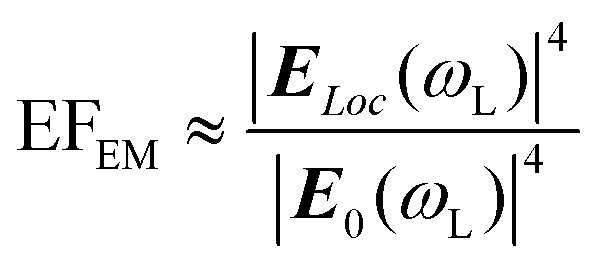 | (4) |
Thus, it can be seen that EM depends on the SERS substrates not on the type of target molecules, the intensity of which can be adjusted by modulating the property of the SERS substrates including the size, shape, constituent, and arrangement.30 Moreover, in addition to plasmon resonance, some other phenomena can also lead to electromagnetic enhancement, such as Mie resonance, which can enhance the local electromagnetic field and usually happens in particles with sizes comparable to the wavelength of the incident light, and “slow photons”, which can enhance the light adsorption owing to the increased effective optical path length and usually appear in the red edge of the photonic band gap in a photonic crystal.20,31–40
2.2 Chemical mechanism
Chemical mechanism (CM) refers to the enhancement in polarizability caused by chemical effects. Among them, the Raman enhancements induced by molecular resonance, ground-state or static chemical interaction, and charge transfer (CT) resonance are the three major contributions.41–43 Molecular resonance can enhance Raman signals by several orders when the incident wavelength is resonant or pre-resonant with the molecular transitions, which may be enhanced or quenched by the SERS substrates.44 Ground-state chemical enhancement comes from the changes in the structure and charge distribution of target molecules adsorbed or bonded on the SERS substrates owing to the interaction between target molecules and the nanostructured SERS substrates, which is independent of any excitation in the whole systems.45,46 Usually, the EF of this enhancement is usually under 102 and does not dominate in CM enhancements reported in many works.42,47,48 In comparison, the CT resonance between target molecules and the SERS substrates can more effectively magnify the polarizability of target molecules, which requires strong interactions between target molecules and the SERS substrates and energy level matching between the highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) of the target molecules and the conduction band/valence band of the semiconductor substrates or the Fermi level of the metal substrates.31,42,49 When the energy of the incident light matches with that of the CT transitions between target molecules and the SERS substrates, the molecular polarizability can be increased, and meanwhile, the enhanced Raman signals can be observed.More importantly, CT resonance can be further enhanced by molecular resonance and exciton resonance. According to the theory of SERS modelled by Lombardi and coworkers,31,50,51 the polarizability tensor can be expressed by the sum of three terms derived by Albrecht,52ασρ = A + B + C, where σ and ρ are the scattered and incident polarization direction. The A-term is normally related only to resonance Raman scattering, and the other two terms, B-term and C-term, involve molecule-to-substrate CT and substrate-to-molecule CT, respectively. Taking the semiconductor substrates, for example, a typical term in the sum for either B- or C-term looks like:51
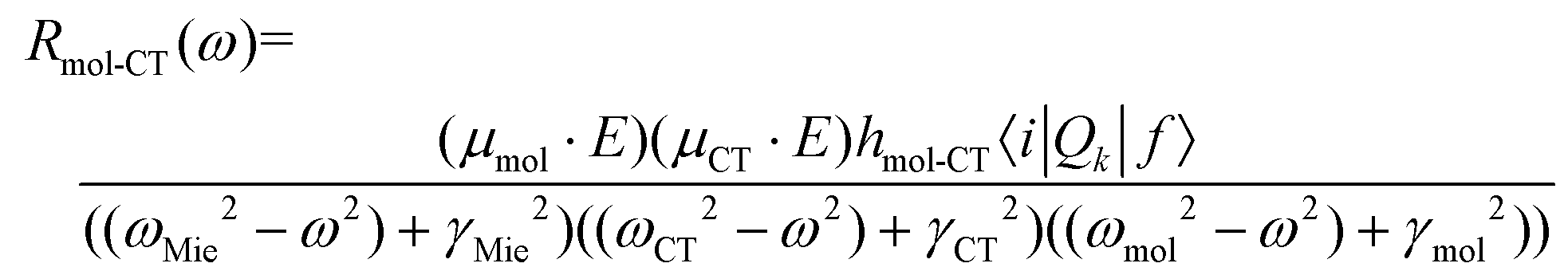 | (5) |
The denominator contains possible Mie resonance (ω = ωMie) belonging to EM, CT resonance (ω = ωCT) and molecular resonance (ω = ωmol). CT resonance (μCT) is coupled to molecular resonance (μmol) through the Herzberg–Teller coupling constant (hmol-CT), and “borrows” intensity from it, as shown in the numerator, where the last term (〈i|Qk|f〉) is the transition moment of a molecular normal mode. In a semiconductor-molecule system, exciton resonance in a semiconductor can also contribute to CT resonance in the same way. Therefore, if the frequency of molecular and exciton resonance matches with that of CT resonance, proper incident light can simultaneously excite these resonances, and higher CM enhancement can be obtained.
Compared to EM, CM relies on chemical interaction and energy level matching between target molecules and the SERS substrates, which inherently offers selective enhancement and can be improved by manipulating the properties of the SERS substrates, including the component, electronic structure, crystallinity, defect, and morphology.19,53 Moreover, through CM, desirable SERS signals can also be generated and observed by plasmon-free materials.
3 Single-component SERS-active nanomaterials
At the early stage, classical noble metal substrates including Au, Ag, and Cu were mainly selected as the SERS-active nanomaterials owing to the easy generation of surface plasmon resonance and excellent EM enhancement abilities in the visible range.54 Subsequently, considering the limitation of noble metal nanostructures for SERS detection, single-component SERS-active nanomaterials are gradually widened to transition metals, semiconductors, graphene, metal–organic frameworks (MOFs), etc. Their SERS activity is closely related to their properties.3.1 Metals
Among all the plasmonic metal substrates, both Au and Ag are the two most widely used substrates for SERS due to their relatively higher stability in ambient conditions. Relatively, Au exhibits excellent chemical stability and low biological toxicity, which make it a better choice for most complex conditions, especially in biological applications.55,56 Ag is less stable than Au, however, the lower imaginary part of the dielectric constant in the visible and near-infrared region of Ag makes it tend to get stronger local electromagnetic field and SERS enhancement.54 Theoretically, at a suitable laser frequency, the EM EF of 25 nm spherical Au single particles is about 103–104, while 105–106 for Ag.23,57There are two common strategies to tune the plasmon resonance of single-component metal substrates. One strategy is to precisely regulate the morphology of metal nanostructures. For single plasmonic metal NPs, their high curvature regions, such as tips and edges, present stronger local electromagnetic fields than other regions.58 Moreover, the frequency of the surface plasmon resonance can also be effectively modulated by morphology regulation, which enables the metal SERS substrates to meet the application requirements of different laser frequencies.59 The other strategy is to create the “hotspot” regions in the conjunctions and gaps among NPs by the assembly. Le Ru and Etchegoin have calculated that, compared with single Au nanospheres with a diameter of 25 nm, the local EF of the dimer with a 2 mm gap constructed by two identical 25 nm Au nanospheres in the “hotspot” region is around one hundred thousand times (∼3 × 109) that of single nanospheres, and its average EF is thousands of times that of single nanospheres.60 In fact, many experiments have confirmed that the local EFs in these created “hotspot” regions are at least several orders of magnitude higher than in other regions of plasmonic metal nanostructures.61–64 Hence, this strategy is more effective than morphology regulation and the resulting enhancement dominates the overall enhancement based on metal nanostructures.60,64
Spherical Au and Ag NPs are most commonly used as SERS substrates because of easy synthesis. Due to the isotropy, the plasmon resonances of Au and Ag nanospheres mainly depend on their size in a given dielectric environment. Small Au and Ag nanospheres (few tens of nanometers) exhibit an extinction band at around green and blue region (in hydrosol), respectively, which is related to their dipole plasmon resonance.65,66 With increase of the particle size, the dipole plasmon resonance red-shifts and broadens, accompanied by excitation of the higher-order multipole plasmon resonances (such as quadrupole and octupole) at shorter wavelengths (Fig. 1A and B).65,67,68 Correspondingly, the higher EM enhancement can be obtained by optimizing the size of Au and Ag nanospheres under a given laser frequency. Taking Ag quasi-spherical NPs for example, the highest SERS enhancement to rhodamine 6G was observed from NPs with the size of 150 nm under 532 nm laser excitation (Fig. 1C), while the maximum enhancements appeared in 175 nm and 225 nm Ag NPs under 633 and 785 nm laser excitation, respectively.65 Notably, compared with the extinction spectra, the plasma coupling effect between NPs under the SERS detection condition can lead to a redshift of the dipole, quadrupole, and higher-order modes in the extinction band.
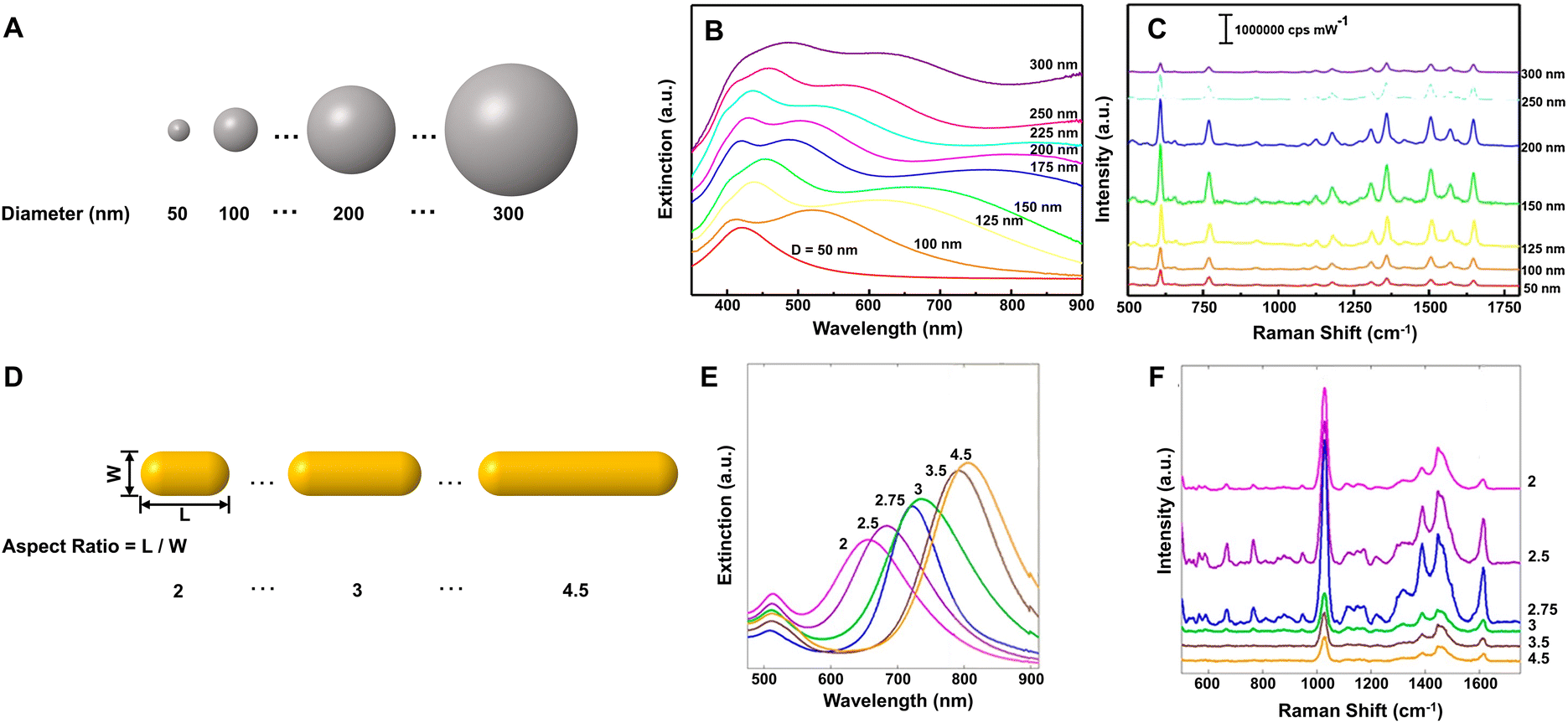 | ||
| Fig. 1 (A) Illustration of Ag NPs with increasing size, (B) their extinction spectra and (C) the corresponding SERS spectra of rhodamine 6G under 532 nm laser excitation. Reproduced from ref. 65 with permission from John Wiley & Sons, copyright 2016. (D) Illustration of Au nanorods with different aspect ratio, (E) their extinction spectra and (F) the corresponding SERS spectra of methylene blue under 785 nm laser excitation. Reproduced from ref. 73 with permission from American Chemical Society, copyright 2013. | ||
Compared to the isotropic spherical NPs, anisotropic Au and Ag NPs can offer more tunable plasmon resonance and higher EM enhancement owing to the existence of the high curvature regions. For example, Au or Ag nanorods have transverse plasmon resonance modes in the short-wavelength region and strong longitudinal plasmon resonance modes in the long-wavelength region. The longitudinal surface plasmon resonance is mainly dependent on the aspect ratio of nanorods and can be tuned from the visible to the near-infrared region (Fig. 1E).69–73 Sivapalan et al. carefully studied the dependence of plasmon resonance frequency on the aspect ratio of Au nanorods and found that the maximum SERS enhancement was observed for nanorods that have a plasmon band blue-shifted from the incident laser excitation wavelength (Fig. 1D–F).73 Similarly, other anisotropic Au and Ag NPs, such as nanotriangles, nanocubes, nanowires, nanostars, and nanoflowers also exhibit morphology-dependent plasmon resonances and can generate higher single-particle enhancement due to a large number of sharp tips, edges, and gaps on their surfaces.66–80 Xie et al. found that the SERS intensity of Au nanoflowers prepared by a simple one-pot method was around 10-fold that of Au nanospheres with similar size in an aqueous solution.81 Niu et al. revealed that Au nanostars with high symmetry exhibited nearly four times stronger single-particle enhancement and higher reproducibility than those of asymmetric nanostars.82 Subsequently, Harder et al. demonstrated that the SERS signals of Au nanostars to uranyl molecules were enhanced with an increase in their branches and aspect ratios, namely, an increase of the branches and aspect ratios could improve the plasmonic properties of Au nanostars for SERS enhancement, especially the electromagnetic enhancement associated with the bonding plasmonic modes and the plasmon resonance caused by branch–branch coupling.83 Of course, the enhancement of single NPs can also be improved by creating the roughened surface on the pre-prepared NPs, selectively depositing additional plasmonic NPs on the pre-prepared nanoparticle templates, etc.25,84,85
Furthermore, in order to obtain more effective enhancement, nanoparticle aggregates, and assemblies are gradually used for SERS detection because they can offer conjunctions and gaps as the “hotspot” regions.10,25,86,87 In the liquid phase, inorganic salts can be added as the aggregating agents to promote the appropriate aggregation of Au and Ag NPs and achieve high SERS enhancement with the formation of effective SERS hotspots.88–91 In 1997, Kneipp et al. discovered that the addition of NaCl solution could make the citrate-stabilized Ag colloids slightly aggregate to 100–150 nm-sized clusters, which exhibited single-molecule level sensitivity to crystal violet molecules.86 The inorganic salts can not only promote aggregation of NPs but also guide target molecules into the vicinity of the hotspots by electrostatic interactions between target molecules and specific adsorbed halide ions so that high sensitivity can be achieved.88,92 In 2019, Lu et al. found that despite NaCl and NaI with the same concentration could induce aggregation of Ag NPs to a similar degree, Ag NPs with NaI exhibited 4-order higher sensitivity than Ag NPs with NaCl in detecting positively charged tropane alkaloids in aqueous solution due to the coadsorption of I− and tropane alkaloids onto Ag NPs.88 Moreover, with the help of organic linkers, NPs, especially anisotropic NPs can be assembled into different configurations, which also plays an important role in affecting the SERS enhancement.93–95 Taking nanorods as an example, the end-to-end configuration tends to show higher EF than the side-by-side configuration because the gaps in the end-to-end configuration are located between high curvature regions with higher local electromagnetic fields.93
Although liquid-phase SERS detection with metal NPs has good reproducibility for quantitative analysis,96 its detection sensitivity is restricted seriously by the concentration of NPs. In contrast, the solid SERS substrates constructed by densely packed NPs can easily overcome this problem and has much better detection stability in complex environments, the prerequisite of which is controllable and reproducible construction of uniform highly ordered solid SERS substrates.96 Moreover, for practical applications, the solid SERS substrates also need to meet the requirements of reproducibility with relative standard deviation (RSD) under 20% either from spot-to-spot or substrate-to-substrate.97 In view of these, many methods have been developed for the preparation of the solid SERS substrates, including assembly, template, and lithography, and many uniform solid SERS substrates with excellent detection sensitivity, stability, and reproducibility have been reported.25,96,98–100 For example, the self-assembled large-scale monolayer of Au NPs with the hexagonal close-packed structure and interparticle gaps smaller than 2 nm (Fig. 2A) can be used as a suitable SERS substrate with high activity (EF ∼106), high stability (∼45 days) and high uniformity (RSD < 10%) due to the low spot-to-spot and substrate-to-substrate variations in intensity (Fig. 2B),87 while the large-area two-layer vertically close-packed arrays of Au nanorods exhibited a limit of detection (LOD) down to 10−15 M, an RSD under 20%, and exceptional detecting reproducibility for the detection of malachite green molecules (Fig. 2C and D).99
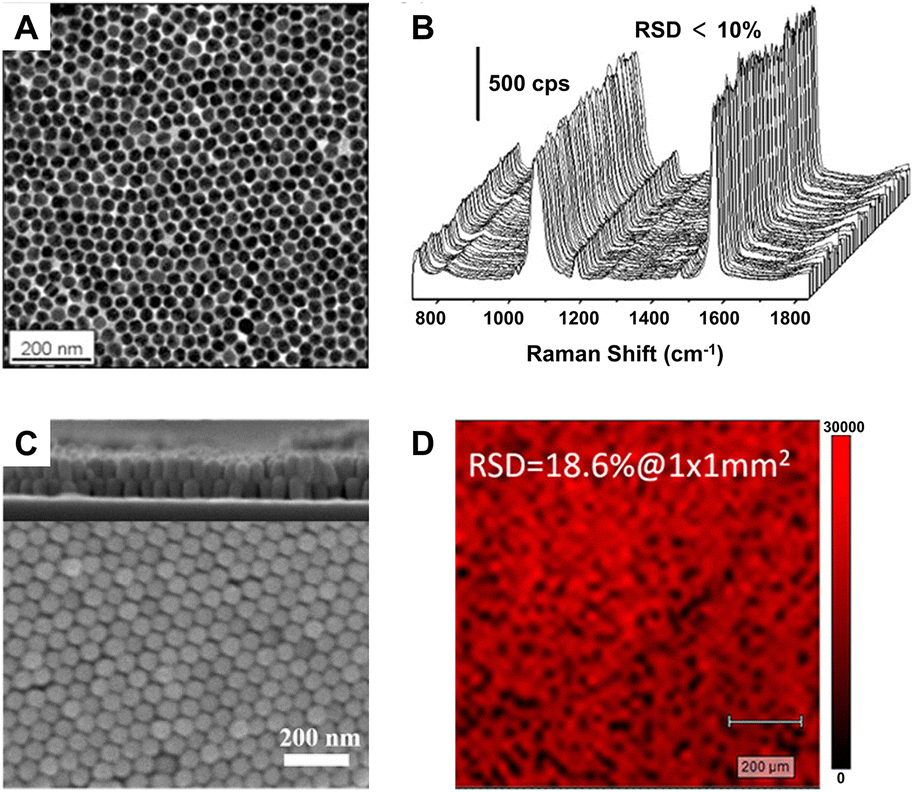 | ||
| Fig. 2 (A) Transmission electron microscope (TEM) image of the monolayer of Au NPs through interfacial assembly and (B) SERS mapping spectra of 1,4-benzenedithiol for 100 different spots on the same monolayer film. Reproduced from ref. 87 with permission from American Chemical Society, copyright 2016. (C) Top view scanning electron microscope (SEM) image of the two-layer vertically close-packed arrays of Au nanorods through controlled evaporation with a cross-sectional image (inset) and (D) Raman mapping image at peak 1616 cm−1 of malachite green on the arrays. Reproduced from ref. 99 with permission from American Chemical Society, copyright 2018. | ||
Uniform solid SERS substrates of metals can also be fabricated by using SERS-inactive materials as the template. Taking the metal-film-over-nanosphere (MFON) substrates as an example, they can be fabricated using polystyrene or silica nanospheres as a cost-effective template, and correspondingly, their surface roughness, stability, and reproducibility are determined by the close packing and arrangement of the templated spheres.101–103 Typically, gold or silver is evaporated or sputtered onto a closely packed monolayer of polystyrene or silica nanospheres to form the MFON substrates. Benefiting from the mature assembly processes for polystyrene or silica nanospheres and the deposition process, uniform and large-area MFON substrates up to square centimeters can be fabricated.104–106 The enhancement from MFON can be optimized by varying the size of nanospheres. As Lin et al. reported, for a range of nanosphere sizes from 430 nm to 1500 nm diameters, the optimum SERS signal with EF of 4.3 × 106 was obtained using the AgFON substrate fabricated by drop-coating polystyrene nanospheres with a diameter of approximately 1000 nm when the wavelength of the incident light was 532 nm.105 In addition to continuous MFON, the immobilized nanorod assembly (INRA) substrates containing nanoscale pillars over nanospheres with small gaps can also be obtained by manipulating the details of metal film deposition. Greeneltch and coworkers reported that the AgINRA substrates on polystyrene nanospheres have highly tunable LSPR from visible to near-infrared regions with an increase in the diameters of the support nanospheres.106 Thus, the enhancement ability of the AgINRA substrates can be optimized to fit different laser wavelengths by tuning the relative position of LSPR maxima with respect to the laser wavelength. A high EF of 1.0 × 108 under a 1064 nm laser was achieved when the LSPR maximum (∼1100 nm) was suitably red-shifted to the laser wavelength, which is even higher than the optimized EFs under shorter laser wavelength (1.3 × 107 at 633 nm and 4.9 × 107 at 785 nm).106
Apart from Au and Ag, some other metals (Cu, Al, Pt, Fe, Co, Ni, Ru, Rh, Pd, etc.) can also be used as the SERS-active substrates. Cu nanostructures can support strong surface plasmon resonance in the visible region and have high SERS enhancement comparable to Au and Ag.107 Al nanostructures have considerable EF up to 104–106 and a unique advantage that they can support surface plasmon resonance in the ultra-violet region, which is hard for the nanostructures of Au, Ag, and Cu.108–110 Notably, SERS in the deep ultra-violet region can excite many biological molecules including protein (e.g., α-helical peptide (LA)7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 111) and DNA (e.g., DNA base adenine112 and 12-mer single-standard DNA113) in this region so that more selective and sensitive biological detection can be achieved. However, both Cu and Al have low chemical stability because they are easy to be oxidized in air and form native oxide layers on their surfaces, which limits their further applications. For other transition metals (Pt, Fe, Co, Ni, Ru, Rh, Pd, etc.), the reported results show that their SERS enhancement is usually relatively weak (EFs are mostly 10–103), which cannot support the practical applications.30,114 Excitingly, with subtle construction, nanostructures of these transition metals may offer higher and sufficient enhancement for particular applications. A recent work by Wang et al. showed that the single dimer of Pd nanocubes with ∼1 nm gap exhibits ∼2.8 × 104 EF with the highest values computed for the top corners facing toward each other, which is enough for in situ detection of Suzuki–Miyaura coupling reaction catalyzed by the dimer of Pd nanocubes (Fig. 3).115
111) and DNA (e.g., DNA base adenine112 and 12-mer single-standard DNA113) in this region so that more selective and sensitive biological detection can be achieved. However, both Cu and Al have low chemical stability because they are easy to be oxidized in air and form native oxide layers on their surfaces, which limits their further applications. For other transition metals (Pt, Fe, Co, Ni, Ru, Rh, Pd, etc.), the reported results show that their SERS enhancement is usually relatively weak (EFs are mostly 10–103), which cannot support the practical applications.30,114 Excitingly, with subtle construction, nanostructures of these transition metals may offer higher and sufficient enhancement for particular applications. A recent work by Wang et al. showed that the single dimer of Pd nanocubes with ∼1 nm gap exhibits ∼2.8 × 104 EF with the highest values computed for the top corners facing toward each other, which is enough for in situ detection of Suzuki–Miyaura coupling reaction catalyzed by the dimer of Pd nanocubes (Fig. 3).115
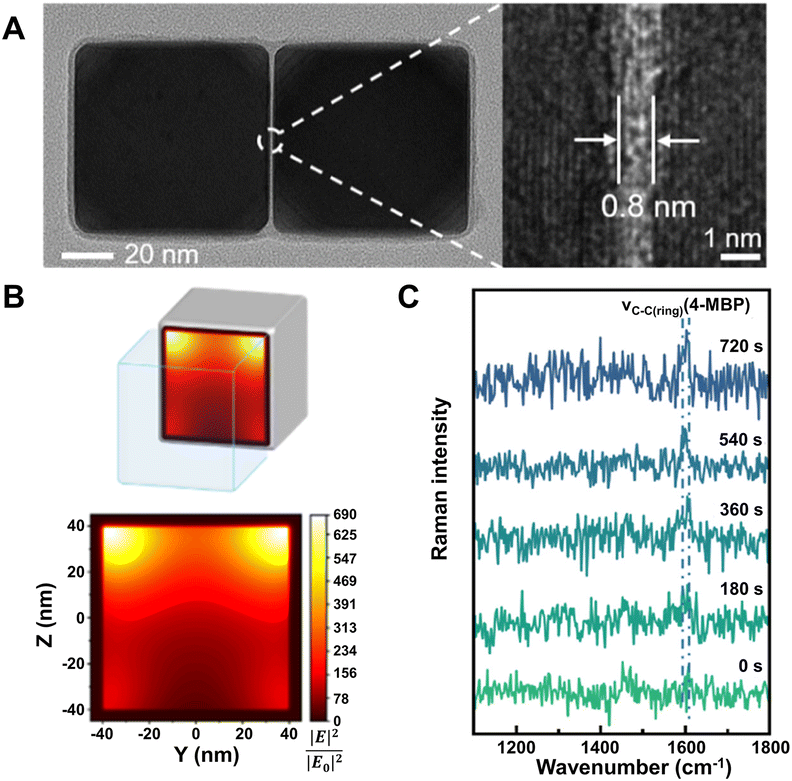 | ||
| Fig. 3 (A) High-resolution TEM image of a single Pd nanocube dimer with an enlarged view showing the gap (right). (B) Finite-difference time-domain calculation of the electric field intensity (YZ plane) inside the gap of the Pd nanocube dimer. (C) In situ SERS spectra of 4-mercaptobiphenyl (4-MBP) formation during Suzuki–Miyaura coupling reaction recorded at different reaction times. Reproduced from ref. 115 with permission from American Chemical Society, copyright 2022. | ||
3.2 Semiconductors
The surface research of SERS-active semiconductor nanomaterials can be traced back to the 1980s.53,116,117 After decades of development, more and more semiconductor materials have been reported to exhibit SERS enhancement, including a variety of inorganic semiconductors (e.g., metal oxides,47,118–122 metal sulfides,123–128 metal selenides,129–131 metal tellurides,132–134 metal halides,135–138 and Si,139,140) and a small number of organic semiconductors (e.g., α,ω-diperfluorohexylquaterthiophene141). In the meantime, the EFs of these semiconductors have improved from less than 103 to the EF value (105–107) equivalent to gold and silver nanostructures.48,119,134,142–145 In comparison with metal substrates, most semiconductor substrates not only can exhibit significant charge transfer enhancement owing to their additional optical and electrical properties but also have a lot more control over their properties, including constituents, defects, doping, crystallinity, size, and shape. Thus, more modulation methods can be applied to improve the enhancement ability of semiconductors.19Defect engineering is one of the effective strategies to modulate the electronic structures of semiconductors, which can even change the SERS activities of semiconductor materials.48,146,147 Taking non-SERS active α-MoO3 as an example, the introduction of oxygen vacancy defects can make it transform to the SERS-active substrates.48 More importantly, the SERS EF can be greatly enhanced by controlling the oxygen vacancy defect concentration and the SERS performance can also be optimized according to the detecting target molecules and the activating laser wavelength (Fig. 4A). For the detection of rhodamine 6G on α-MoO3−x nanobelts, the EF can be as high as 1.8 × 107 with a LOD of 10−8 M under the 532 nm laser excitation. Meanwhile, an effective electric current model based on the influence of oxygen vacancy defects was proposed by Li et al., which can quantitatively describe the photo-induced CT process between the target molecules and the semiconductor substrates and anticipate the SERS activity of metal oxide semiconductors such as CrO3, Cr2O3, and Ta2O5.48
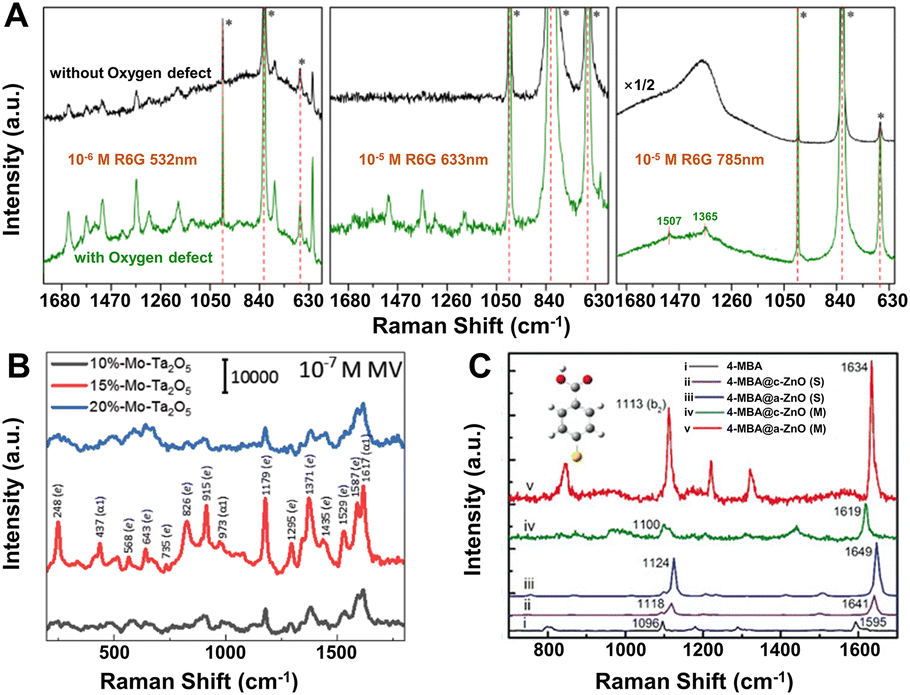 | ||
| Fig. 4 (A) SERS spectra of rhodamine 6G on α-MoO3 without (black) and with (green) oxygen vacancies excited with 532, 633 and 785 nm lasers. Reproduced from ref. 48 with permission from The Royal Society of Chemistry, copyright 2017. (B) SERS spectra of 10−7 M MV on Mo-doped Ta2O5 substrates (x = 10%, 15%, and 20% of Mo percentage) under the 532 nm laser excitation. Reproduced from ref. 148 with permission from John Wiley & Sons, copyright 2019. (C) Measured (M) and simulated (S) SERS spectra of adsorbed 4-MBA molecules on a single a-(amorphous) and c-(crystalline) ZnO nanocage under the 633 nm laser excitation. Reproduced from ref. 121 with permission from John Wiley & Sons, copyright 2017. | ||
Element doping is another promising method to optimize the SERS performance of semiconductor substrates by energy band engineering.148–152 Yang et al. found that Mo-doped Ta2O5 substrate could exhibit a remarkable SERS sensitivity with an EF of 2.2 × 107 and a low LOD 9 × 10−9 M for methyl violet (MV) molecules under the 532 nm laser excitation (Fig. 4B).148 The extraordinary SERS performance could be attributed to the synergistic resonance enhancement of three components under 532 nm laser excitation: (i) MV molecule resonance, (ii) photo-induced CT resonance between MV molecules and Ta2O5 nanorods adjusted by element doping, and (iii) EM enhancement around the “gap” and “tip” of the anisotropic Ta2O5 nanorods, which was realized by regulating the photo-induced CT resonance frequency of the Mo-doped Ta2O5 substrates to be quasi-equivalent to the electromagnetic resonance frequency nearby 532 nm and the given 532 nm laser by energy engineering through element doping.148 This “coupled resonance” strategy proposed by Yang et al. provides a new way to obtain the ultra-sensitive SERS-active semiconductor nanomaterials.
Besides those above, many other strategies can be applied to obtain higher EFs for SERS-active semiconductor substrates.153–156 For instance, (1) size control. Nanoparticle size plays an important role to affect the SERS intensity, especially when it is smaller than the exciton Bohr radius, quantum confinement effect can lead to a strong size dependence of the SERS spectra and higher enhancement by tuning the position of band edge in semiconductors.53,124,157 (2) Crystallinity regulation. In 2017, Guo et al. first observed the remarkable SERS activity in amorphous ZnO nanocages, the EF of which (up to 6.62 × 105) for 4-mercaptobenzoic acid (4-MBA) was higher than that of the crystalline counterparts (Fig. 4C).121 First-principles density functional theory (DFT) simulations further confirmed that the metastable electronic states of the amorphous surfaces of ZnO nanocages can improve the interfacial CT process by weaker constraint to surface electrons compared with their crystalline counterpart. (3) Shape control. Among various semiconductors, two-dimensional (2D) semiconductors, especially transition metal dichalcogenides (TMDs) and transition metal chalcogenides have received special attention owing to their unique thickness-dependent physico–chemical properties with enhanced chemical-based CT processes.126,158–162 Excitingly, the 2D 1T′-WTe2 atomic layers exhibit a very high EF of 1.8 × 109 for the detection of rhodamine 6G with femtomolar level concentration.132 (4) Environmental change. Recently, the low temperature has been proved to boost the SERS activity of porous ZnO nanosheets, which can be attributed to the efficient photo-induced CT process enhanced by suppressing phonon-assisted non-radiative recombination on surface defect states in the semiconductor-molecule system.163
Compared with inorganic semiconductors, the research on the SERS enhancement of pure organic semiconductors is still very limited. Until 2017, the SERS signals on nanostructured organic semiconductor films for molecular detection were observed for the first time by Yilmaz and co-workers.141,164 When methylene blue is used as a probe molecule, the nanostructured α,ω-diperfluorohexylquaterthiophene (DFH-4T) films without any additional plasmonic layer exhibit the unprecedented EF of 3.4 × 103, indicating the successful extension of the SERS-active substrates to pure organic semiconductors.141 Moreover, by adjusting the number of the thiophene rings in the central chain of DFH-4T (for instance, to DFH-5T), or by replacing the fluorines with hydrogen (such as with DH-4T), the exact location of the band edges can be finely tuned, so that more precise control over the location of CT transitions to and from a specific molecule to be detected can be realized and correspondingly the selectivity of organic semiconductors to the specific molecules in the mixture can be further improved.165 Subsequently, Demirel et al. reported another nanostructured film of the small molecule 5,5′′′-diperfluorophenyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (DFP-4T) that consisted of a fully π-conjugated diperfluorophenyl-substituted quaterthiophene structure as an efficient SERS platform for detection of methylene blue.166 DFP-4T exhibits a higher EF of 2.7 × 105 and a LOD of as low as 10−9 M for methylene blue, which is comparable to those reported for the best inorganic semiconductors and even intrinsic plasmonic metal-based SERS platforms.166 Recently, Deneme et al. revealed the influence of π-backbone structure design of organic semiconductors on SERS enhancement.167 After carbonyl functionalization of the fused thienoacene π-system in 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT) the resulting nanostructured 1,10-(benzo[b]benzo[4,5]thieno[2,3-d]thiophene-2,7-diyl)bis(octan-1-one) (D(C7CO)-BTBT) film exhibited nearly twenty-fold stronger SERS signal for methylene blue molecules than C8-BTBT and additional enhancement for other three dye molecules (crystal violet, rhodamine 6G, and malachite green) (Fig. 5A and B). The low-lying LUMO and the face-on π-backbone of D(C7CO)-BTBT film enabled CT resonance through strong π-orbital interactions between analyte and semiconductor molecules, which originates from dipolar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO interactions, hydrogen bonds, and strengthened π-interactions in D(C7CO)-BTBT compared to C8-BTBT (Fig. 5C).167 Apart from small molecular organic semiconductors, a few semiconductive organic substrates, including π-conjugated polymers168,169 and peptide nanotubes,170 are also reported.
O⋯CO interactions, hydrogen bonds, and strengthened π-interactions in D(C7CO)-BTBT compared to C8-BTBT (Fig. 5C).167 Apart from small molecular organic semiconductors, a few semiconductive organic substrates, including π-conjugated polymers168,169 and peptide nanotubes,170 are also reported.
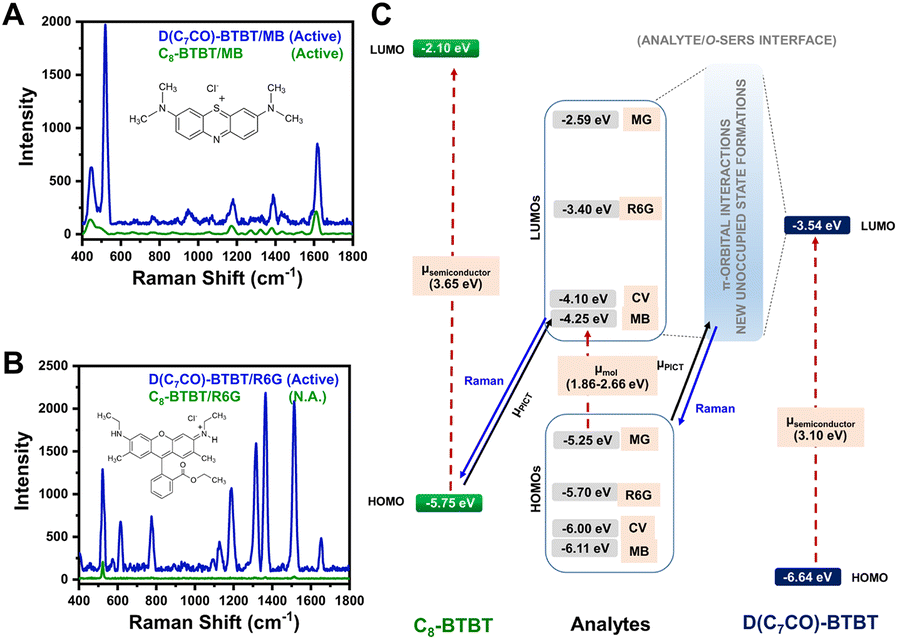 | ||
| Fig. 5 SERS spectra of (A) methylene blue and (B) rhodamine 6G analytes on D(C7CO)-BTBT and C8-BTBT films. (C) Energy level diagram for the current analyte/o-SERS systems for D(C7CO)-BTBT and C8-BTBT showing the frontier molecular orbital (HOMO/LUMO) energies and plausible (shown with solid black arrow)/non-plausible (shown with dashed red arrows) transitions. μPICT stands for photoinduced charge-transfer between analyte and semiconductor under the excitation of 785 nm. Raman signals were produced during the transitions shown with blue solid arrows. Reproduced from ref. 167 with permission from Springer Nature, copyright 2021. | ||
3.3 Graphene
Since the first report about the Raman enhancement on the surface of monolayer graphene in 2010 by Zhang, Liu, and coworkers,171 graphene has developed into one of the most important and widely studied 2D SERS materials.53,158,164,172 Many factors such as the number of layers, the surface properties and the interaction between graphene and target molecules have a remarkable impact on the CM enhancement effect of graphene.158 Moreover, owing to the unique electronic structure and morphology, graphene can quench fluorescence and suppress self-absorption of molecules adsorbed on its surface, leading to the low LOD in the magnitude of 10−8–10−10 M realized for the detection of some specific analytes on pristine graphene.45,173 In 2015, Huang et al. investigated the molecular selectivity of graphene-enhanced Raman scattering toward a variety of different molecules with different molecular properties and discussed the selection rules with reference to two main features of the molecules, namely the molecular energy levels and molecular structures.174 They found that when the HOMO or LUMO levels of the molecules are on a suitable energy range with respect to the fermi level of graphene, strong interaction, and CT between graphene and molecules can lead to high Raman enhancement.174 If the molecule can further meet the symmetric requirement, including Dnh symmetry (the dihedral groups of molecular symmetry, adapted from group theory), the interaction and CT between graphene and molecules will be stronger and correspondingly a much higher Raman enhancement will be achieved. Furthermore, the Fermi level of graphene can shift by controlling the nitrogen doping levels, and if the shift can align with the LUMO level of target molecules, CT can be enhanced, leading to the lower LOD of 10−11 M for dye molecules.1753.4 Metal–organic frameworks
In recent years, MOFs, hybrid organic–inorganic materials with unique periodic framework structures, have attracted a lot of attention as independent SERS substrates owing to their excellent properties such as molecular enrichment ability, selectivity, gas sensing capability, and additional chemical enhancement ability.13,17 The first example was discovered and reported by Yu et al. in 2013. They successfully observed the adsorption orientation-dependent SERS effect of methyl orange on two types of MOF substrates, MIL-100 and MIL-101, although the EF value was only 120.176 In 2019, the high Raman enhancement on MOFs (EF = 1.9 × 106 for rhodamine 6G on ZIF-67) was realized.177 More importantly, because of the high tailorability, MOFs can be used as the SERS substrates with molecular selectivity, which is very difficult to realize for the traditional SERS plasmonic-nanoparticle substrates relying on EM.13,177 For instance, Sun et al. confirmed the strong impact of three different types of structural engineering on the SERS performance of MOFs, including (i) metal ion replacement (Fig. 6A and B), (ii) pore-structure optimization (Fig. 6C and D), and (iii) surface modification.177 Furthermore, Xu et al. revealed that the enhancement effect could be improved by adjusting the electronic structure of MOFs, and specifically, after doping with Cu2+, the doped ZIF-67 substrates showed a high EF of 6.07 × 106, an exceptional detection sensitivity with 10−8 M of methylene blue as the probe molecule, and good signal reproducibility with an RSD of 14.1%, as well as outstanding signal stability with an RSD of 2.59%.178 In 2020, MIL-100(Fe) was identified for the first time as an excellent SERS-active substrate for the capture and recognition of diverse volatile organic compounds with high sensitivity (e.g., LOD of 2.5 ppm for toluene) owing to its unique organic–inorganic structures, in which the aromatic ligand could bind with the aromatic volatile organic compounds through π–π interaction and the metallic nodes could coordinate with polarized small molecules.179 In 2021, Chen et al. reported a mixed valence state Mo-MOF with high SERS activity triggered by UV irradiation (EF = 1.33 × 105 for the detection of crystal violet), demonstrating that the creation of oxygen vacancies could induce the formation of the SERS-active sites in MOFs.180 Recently, more strategies for higher enhancement in MOFs substrates including reducing the thickness of 2D-MOFs and stabilizing photo-induced vacancy defects in MOFs are gradually reported.181,182 All these results have proven that the SERS-active MOF platform presents great modifiability and expandability, which is of great significance for further wide applications of MOF materials for SERS detection.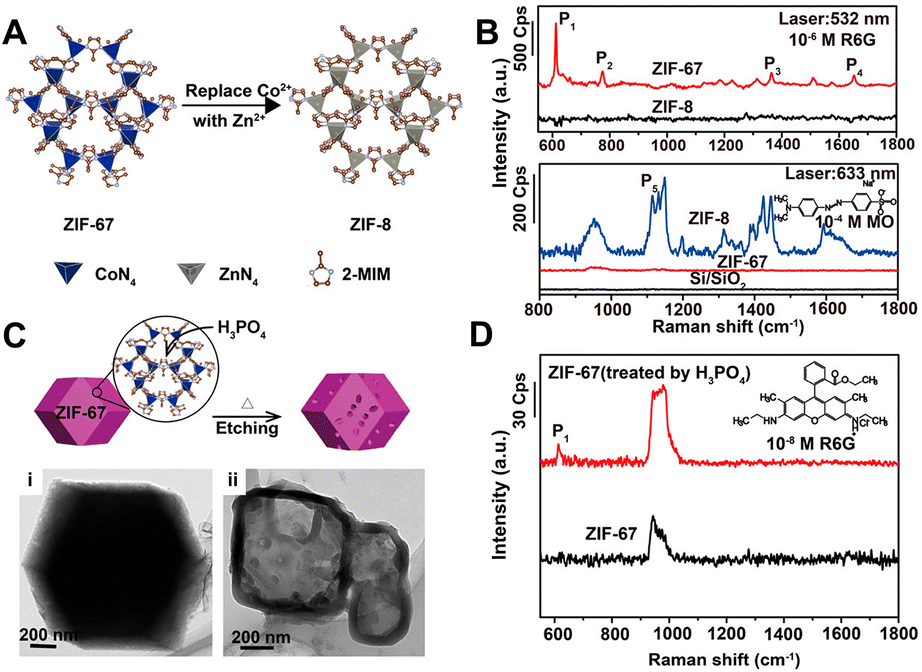 | ||
| Fig. 6 (A) Illustration of metal ion replacement from ZIF-67 to ZIF-8. (B) SERS spectra of rhodamine 6G (R6G) (10−6 M) on the ZIF-67 and ZIF-8 substrates excited by 532 nm laser, and SERS spectra of methyl orange (MO) (10−4 M) on the ZIF-67 and ZIF-8 substrates excited by 633 nm laser. (C) Illustration of pore-structure optimization of ZIF-67 by phosphoric acid etching with TEM image of (i) raw ZIF-67 and (ii) acid-treated ZIF-67. (D) SERS spectra of R6G (10−8 M) on the raw ZIF-67 and acid-treated ZIF-67 substrates excited by 532 nm laser. Reproduced from ref. 177 with permission from American Chemical Society, copyright 2019. | ||
4 Composite SERS-active nanomaterials
The extended construction of the SERS-active nanomaterials from a single component to two-or-more components is an inevitable trend for material development. On one hand, the limitation of single-component SERS-active nanomaterials, such as the reproducibility, stability, selectivity, and durability of plasmonic metal nanostructures, can be effectively overcome by controllable integration with other functional components. On the other hand, collective properties and improved performance in the composite SERS platforms can be obtained through the property synergy or complementarity among different functional components, which is more conducive to the realization of advanced applications in wider fields. Furthermore, the difficulty and complexity of mechanism revelation for composite SERS substrates owing to the complicated electronic structures and charge transfer are also increased exponentially. Up to now, many types of composite SERS-active nanomaterials with improved SERS performance have been developed by optimizing each component of the whole system. For instance, the combination of plasmonic metal nanostructures and inert oxides can result in higher stability and durability;183–186 Integration of plasmonic metal nanostructures with porous materials can provide additional advantages on selectivity, sensitivity, and specific recognition;17,187–189 Composites of two-or-more plasmonic metals can provide wider control over their plasmon properties than single metals.190–192 Hybridization of plasmonic metal nanostructures with semiconductors can effectively boost the light harvesting and conversion and enable high sensitivity.20,193–197 Besides those metal-based composite SERS-active materials, the SERS-active composites without plasmonic metals have also been exploited, which provides more possibilities for further enrichment and construction of multi-functional SERS substrates.119,198–202 Mechanism of excess enhancement ability on composite SERS-active materials is complicated and under debate. Most existing studies suggest that the charge separation and transfer at the interface of heterostructures composed of the SERS-active nanomaterials play a key role in improving the CT process between substrates and target molecules to achieve stronger CM enhancement and increasing the electron density of metal nanostructures to achieve stronger EM enhancement.4.1 Nanocomposites constructed by multicomponent metals
SERS-active nanocomposites constructed by multicomponent metals can exhibit more advantageous chemical and physical properties than single-component nanomaterials due to the synergetic effects associated with different metals and the corresponding change of the electronic structure and spatial arrangement mode induced by precise manipulation of the composition, structure, distribution, shape, etc.191,203,204 Typically, Au–Ag bimetallic nanocomposites exhibit broader tunable surface plasmon resonance (SPR) properties compared to pure Au NPs and higher chemical stability than pure Ag NPs, and, moreover, their spectral characteristics are closely dependent on their structure and composition.190,192,205,206 As demonstrated by Rituraj Borah and Sammy W. Verbruggen, the spectral shift is considerably different for both Au–Ag alloy and core–shell configurations, namely, the red shift is proportionate to the amount of Au for alloy NPs when moving from pure Ag to Au whereas the red shift is not gradual for core–shell NPs (Fig. 7A and B), and the position of localized SPR (LSPR) peaks for Au@Ag core–shell NPs are located at larger wavelengths than for alloy NPs of the same overall composition.190 Kitaev and coworkers further found that a wide range of LSPR peak tuning from 470–800 nm could be achieved for the obtained Au–Ag NPs through the formation of Au shells and rebuilding of Ag.205 Definitely, the influence of the adjustable LSPR properties of Au–Ag bimetallic nanocomposites on their SERS performance is notable and worthy of attention. Cui et al. revealed that the SERS activity of the core–shell Ag100−x@Aux bimetallic NPs toward thiophenol and p-aminothiophenol is critically dependent on the molar ratio of Ag to Au.207 With the increase of the Au molar fraction (x), the SERS activity enhances first and then weakens, and the maximum signal intensity appears at x = 18 and is 10 times stronger than that of pure Ag NPs owing to the presence of some pinholes that act as hotspots for the electromagnetic field enhancement on the surfaces of the core–shell Ag100−x@Aux bimetallic NPs. Similarly, Fan et al. explored the SERS properties of the Aux-Ag10−x alloy NPs, the enhancement trends of which are closely related to both the composition of the alloy NPs and the chemical nature of the probe molecules.208 As shown in Fig. 7C, for the probes 4-hydroxythiophenol (HTP) and thiophenol (TP), the best SERS performance was obtained for the highest Ag ratio, whereas for the two positively charged SERS probes, oxazine 720 and Nile Blue A, the alloy NPs with higher Au content provided the largest SERS signal. The relationship between the enhancement trends and the chemical nature of the probe molecules was assigned to the selective binding of probe molecules to different metallic domains on the alloy surface promoted by charge donation from Ag to Au atoms in the alloys. Besides the structure and composition, the SERS activity is also closely related to the morphology and the crystal facets of the core–shell Au–Ag composites. For instance, the Ag@Au concave cuboctahedra synthesized by titrating aqueous HAuCl4 into a suspension of Ag cuboctahedra enclosed by both {100} and {111} facets exhibit excellent SERS activity that was more than 15-fold stronger than that of Ag@Au cuboctahedra and 70-fold stronger than that of the original Ag cuboctahedra at an excitation wavelength of 785 nm.209 That is because the deposition of Au on Ag cuboctahedra that are covered by a mix of {111} and {100} facets is at a ratio of 1:1.7 in terms of surface area, and correspondingly more sharpened corners and edges on concave cuboctahedra that can concentrate the electromagnetic field and lead to enormous SERS signals are formed compared to conventional cuboctahedra.
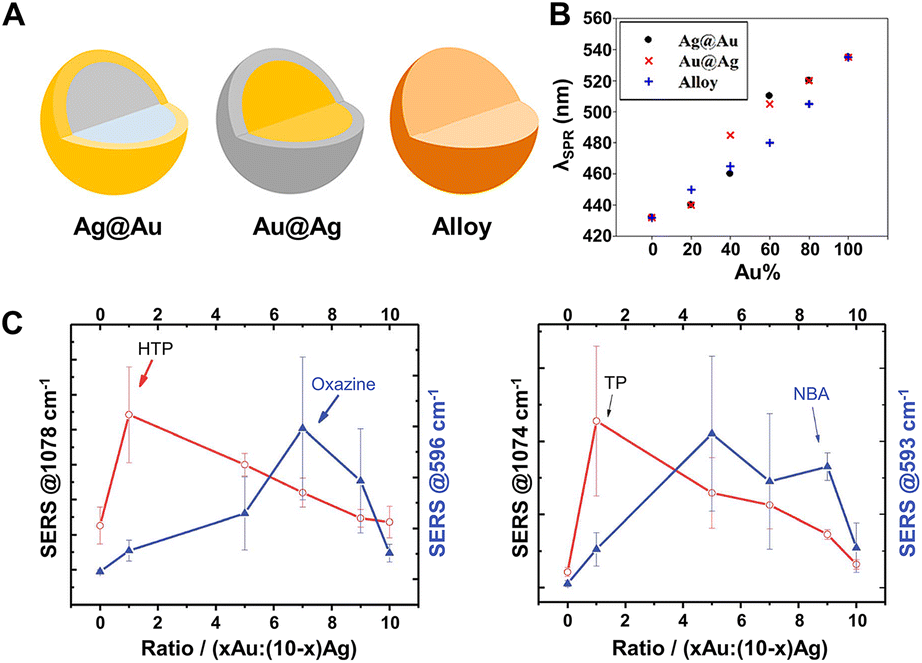 | ||
| Fig. 7 (A) Structure illustrations of gold and silver bimetallic NPs. (B) Absorption peak shift with the incorporation of Au for Ag@Au, Au@Ag, and alloy NPs of 60 nm in diameter. Reproduced from ref. 190 with permission from American Chemical Society, copyright 2020. (C) SERS intensity dependence of 4-hydroxythiophenol (HTP), oxazine 720, thiophenol (TP) and nile blue A (NBA) on the Au:Ag ratio of Au–Ag alloy NPs. Reproduced from ref. 208 with permission from The Royal Society of Chemistry, copyright 2013. | ||
In order to improve the SERS enhancement for target molecules adsorbed on transition metals, fabrication of NPs with high SERS-active metal (such as Au and Ag) core and ultrathin transition metal shell is an effective way, so that transition metals can “borrow” the SERS activity from the Au or Ag core and ultimately achieve the average SERS enhancement of 104–105.9,210–212 Taking the 55 nm Au@1.4 nm Pt NPs as an example, the peak intensity of the SERS signals from this type of Au@Ptcore–shell NPs film electrode toward CO is about 40-fold stronger than 12 nm Pt nanocubes and about 200-fold stronger than the roughened Pt electrode.212 Moreover, effective integration of the catalytic property of transition metals and the enhanced SERS activity of noble metals enables in situ detection or monitoring of various catalytic reactions by SERS. Recently, Ze et al. synthesized Au@PtNi NPs by catalytically depositing active PtNi alloy shells on Au NP cores, on which the processes of the oxygen reduction reaction could be explored by using in situ electrochemical SERS.213 By monitoring the direct signal of the intermediary species, *OOH, and its red-shift with the increase of the Ni content in the Au@PtNi NPs via SERS, the critical role of the Ni doping for efficient electron transfer as well as the enhanced oxygen reduction reaction activity was revealed, beneficial for clarification of the catalytic mechanism and further design of catalyst with high efficiency.213 Ulteriorly, the design, and adoption of the mixed shells of transition metals with plasmonic metals can further enhance the SERS signals of adsorbents on transition metals. The Ag@Pd–Ag nanocubes synthesized by Qin's group were evidenced not only for in situ quantitative SERS monitoring the Pt-catalyzed reactions of the reduction of 4-nitrothiophenol (4-NTP) to 4-aminothiophenol (4-ATP) by NaBH4 but also exhibited two-fold higher enhancement than that by Ag@Pd nanocubes.214
In order to achieve more quantitative SERS analysis, the internal standards can also be embedded into the interface inside the core–shell metal nanocomposites for correcting the signal fluctuation between samples and measuring conditions.14,215–220 In 2015, Shen et al. successfully prepared one type of core-molecule-shell (CMS) NPs for quantitative SERS analysis, composed of Au nanospheres as the uniform core, Ag as the shell for higher enhancement, and both cysteamine and 4-mercaptopyridine in the molecular layer as the framework and the SERS internal standards, respectively.215 By normalizing the raw signals of target molecules, 1,4-phenylene diisocyanide, to the intensity of the feature peak of 4-mercaptopyridine, an RSD of less than 8% and better linearity of the working curve was achieved. More importantly, in addition to 1,4-phenylene diisocyanide, the CMS NPs can also be applied to the quantitative SERS analysis of basic red 9 molecules with weak affinity to Ag over a large concentration range from 0.5 nM to 100 nM.215 That is because, compared to directly adding internal standards, the embedding method can avoid the influence from outer environments to internal standards and the competitive absorption between target molecules and internal standards, thus extending its versatility to target molecules with a larger range of concentrations and even different affinity to the SERS substrates. Moreover, the CMS NPs can be further assembled into large-scale close-packed nanoparticle arrays to create more intensive and abundant SERS “hotspots” for lower RSD and more sensitive detection.216,217 Taking the monolayer array of Au@4-MBA@Ag CMS NPs fabricated through a commonly used interfacial self-assembly method by Wang and Li as an example (Fig. 8A), compared to the Au@Ag array without internal standard, the point-to-point RSD was decreased from 10.08% to 8.84% (Fig. 8B) and the batch-to-batch RSD was impressively decreased from 35.57% to 14.97% (Fig. 8C) for the detection of thiram.216 Meanwhile, the LOD using the Au@4-MBA@Ag array for thiram detection could reach 0.38 ppb,216 better than that of 1.1 ppb using the Au@Ag array without the internal standards and 72 ppb using SiO2@Au@4-MBA@Ag NPs without assembly.221,222
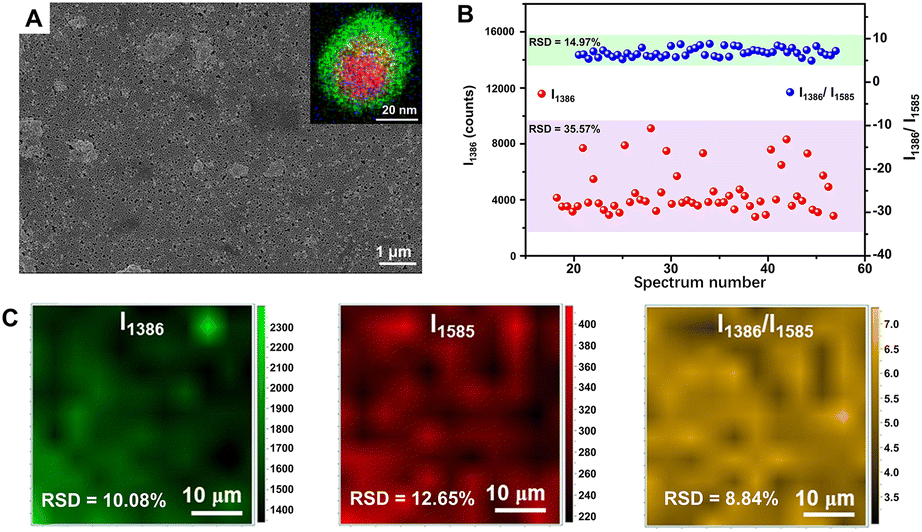 | ||
| Fig. 8 (A) SEM image of interfacial self-assembled Au@4-MBA@Ag array on silicon wafer. Inset is the superimposed image of high angle annular dark field-scanning transmission electron microscopy-electron dispersive X-ray spectroscopy element mapping (red to Au, green to Ag, blue to S) of an Au@4-MBA@Ag CMS NP. (B) The random 54 SERS signals of thiram at 1386 cm−1 and its ratio to 4-MBA at 1585 cm−1 distributions from 5 batches of Au@4-MBA@Ag array. (C) Raman mapping of thiram with its signal at 1386 cm−1, 4-MBA with its signal at 1585 cm−1, and the ratio of them. Reproduced from ref. 216 with permission from Elsevier, copyright 2021. | ||
4.2 Nanocomposites composed of metal and SERS-inactive non-metal nanomaterials
Development of the nanocomposite SERS substrates composed of plasmonic metals and SERS-inactive non-metallic nanomaterials such as various oxides and organic polymers has attracted much attention for the realization of superior SERS performance and more advanced applications through function integration. Although these non-metal nanomaterials do not have SERS activity, they can endow more functions to the nanocomposite SERS substrates, such as offering different interactions and selectivity from metal NPs toward target molecules, controlling the assembly of metal NPs and protecting metal NPs from aggregation and sintering, which completely depend on the intrinsic characteristics of the introduced non-metallic nanomaterials.9,17,223,224The shell-isolated NPs with an ultrathin inert dielectric shell surrounding the plasmonic metal nanoparticle core such as Au@SiO2 are one of the most representative examples.15,184–186,225 In this system, the inert shells can not only prevent the direct contact of metal particles with the chemical environment, the surface of interest, and the target molecules but also can protect them from aggregation.9,224 Hence, compared to bare metal NPs, the shell-isolated NPs can exhibit better chemical stability and long-term performance. Notably, the intensity of the SERS signal is a function of the shell thickness, namely, with the increase of the shell thickness, the electromagnetic field enhancement as well as the SERS signal decreases exponentially with the increase of the distance between target molecules and the plasmonic metal nanoparticle core.49,212,226 Systematic study on the shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS) of Au@SiO2 and Au@Al2O3 with shell thickness ranging from 2 to 20 nm by Tian et al. confirmed that the experimental data were in good agreement with the three-dimensional finite-difference time-domain (3D-FDTD) calculated results and strong optical signals could be detected only when the shell thickness was 2–4 nm.184 More importantly, SHINERS extends the application of SERS to the atomically flat single-crystal surfaces, using which, the behavior of the adsorbed hydrogen on Pt (111) single crystals was examined for the first time.184 Of course, in addition to adsorption on the shell surface, the molecules such as dyes or aromatic molecules can also be embedded between the plasmonic metal core and the inert shell of the shell-isolated NPs as SERS tags for immunoassay, multi-channel detection, and so on.227,228
How to effectively and selectively capture target molecules onto the surface of the SERS substrates is one of the keys to achieving high-performance SERS detection.223 Aiming to solve this problem, MOFs have been introduced and gradually developed into an ideal material for combination with the plasmonic metal NPs owing to their high specific surface area, tunable pore size and volume, multiple coordination sites, strong adsorption capacities, beneficial to selectively capture the specific target molecules.13,17 For example, single core–shell Au@MOF-5 nanoparticle with a shell thickness of 3.2 ± 0.5 nm exhibited highly selective sensing properties toward CO2 in gas mixtures, whereas no SERS signals were observed for bare Au NPs, pure MOF-5 spheres, and Au@MOF-5 NPs with a thicker MOF-5 shell (Fig. 9A–C).188 Obviously, these sensing characteristics were attributed to both the near-field electromagnetic enhancement of the Au nanoparticle core (mostly within a distance of no more than 3 nm) and the specific adsorption capability of the uniform MOF-5 shell toward CO2 molecules.229 Moreover, such SERS detection can be easily applied to other analytes such as dimethylformamide, ethanol, aromatic dyes, and volatile organic compounds. Thanks to the selective pre-concentration of the target molecules by the MOF shell to make them in close proximity to the plasmonic Au NPs, the substrate of Au nanoparticle-embedded MIL-101 (AuNPs/MIL-101) exhibited much stronger enhancement toward a series of aromatic amine compounds, especially benzidine (Fig. 9E), than Au colloids for liquid phase detection.189 Selective SERS enhancement to molecules with different sizes was also realized due to the sieving effect from the nanoscopic pores of MIL-101. Selecting 4,4′-bipyridine and poly(4-vinylpyridine) with different molecular sizes as model analytes and using AuNPs/MIL-101 as the substrate, it was found that much stronger SERS signals were readily detected for 4,4′-bipyridine while almost no Raman response was observed for poly(4-vinylpyridine) (Fig. 9F), which was not able to diffuse into nanopores of the MIL-101 shell and to interact with the embedded Au NPs.189 Besides MOFs, other materials such as porous polymer shells can also be used to highly selectively recognize target molecules, resulting in improvement of the SERS activity of the substrates.230,231
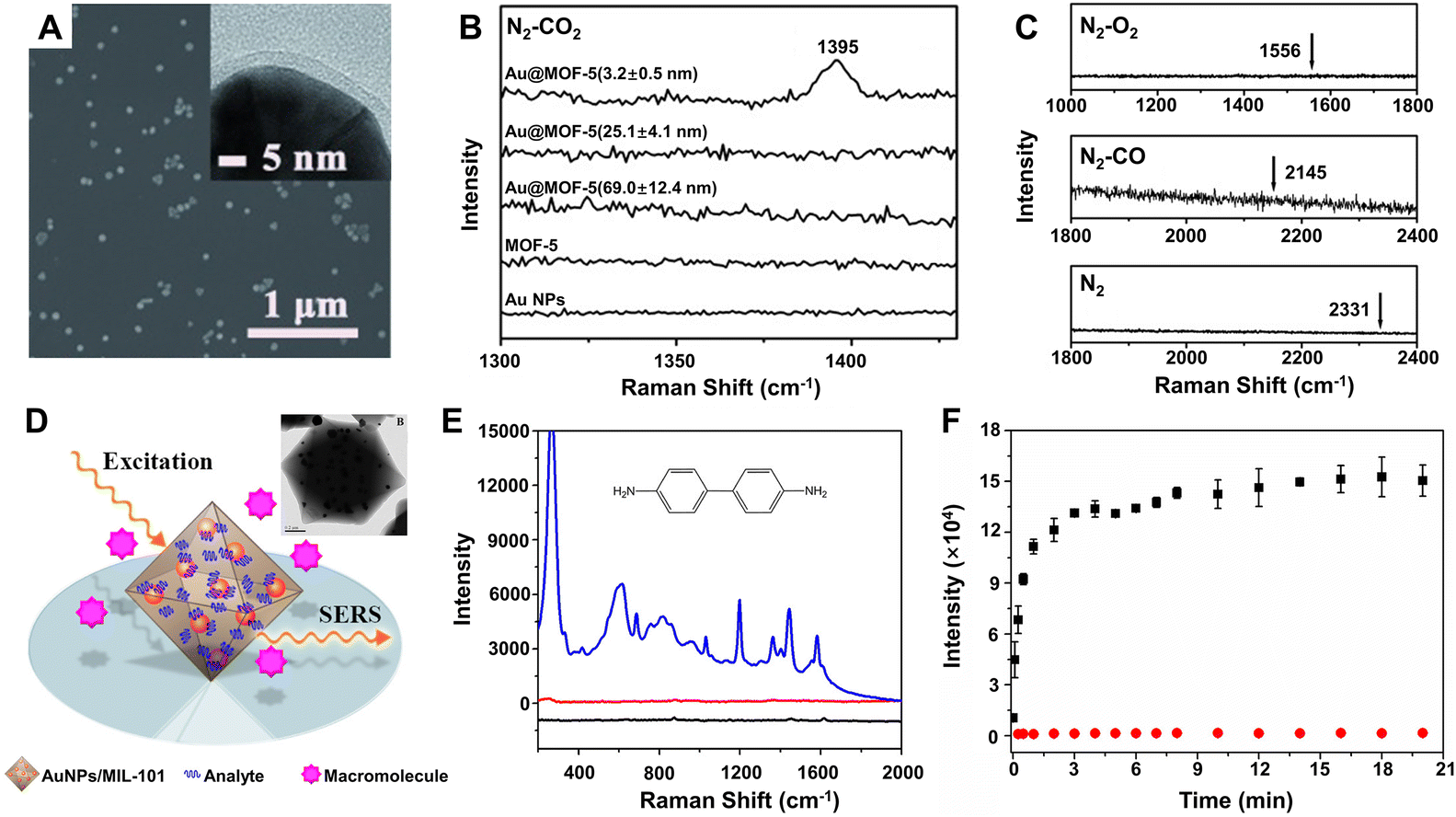 | ||
| Fig. 9 (A) SEM and TEM (inset) images of the core–shell Au@MOF-5 NPs with shell thicknesses of 3.2 ± 0.5 nm. (B) SERS spectra of single Au NPs, single MOF-5 spheres, and single core–shell Au@MOF-5 NPs with different shell thickness toward CO2 in the CO2/N2 gas mixture (the ratio is 1:5 for CO2vs. N2) at room temperature. (C) SERS spectra of single Au@MOF-5 NPs with shell thicknesses of 3.2 ± 0.5 nm toward N2, CO, and O2 with arrows point to their characteristic SERS peak positions. Reproduced from ref. 188 with permission from John Wiley & Sons, copyright 2013. (D) Illustration of AuNPs/MIL-101 for selective SERS detection with TEM image of AuNPs/MIL-101 (inset). (E) SERS spectra of benzidine on the Au NPs/MIL-101 substrate (blue lines), Au colloids substrate (red line), and virgin MIL-101 (black line). (F) Raman intensity plotted against time at 1030 and 1292 cm−1 for 4,4′-bipyridine (black square) and poly(4-vinylpyridine) (red circle). Reproduced from ref. 189 with permission from American Chemical Society, copyright 2014. | ||
How to effectively collect the SERS-active colloids based on plasmonic metals to improve the SERS enhancement is also a major challenge for in-liquid molecular detection via SERS. The combination of plasmonic metals with magnetic materials can enable the position control and collection of the magnetic-plasmonic NPs by applying an external magnetic field in an analyte solution, and thus the enhanced SERS enhancement can be achieved by molecules located at the junction between the aggregated magnetic-plasmonic NPs, induced by collection.232–235 Usually, the magnetic-plasmonic NPs are constructed using iron oxides (Fe3O4 or Fe2O3) as the cores and Au with different shapes as the shells. After dispersing in fresh very diluted (10−8 M) aqueous solution of thiram and subsequent depositing on a silicon sheet via collection by magnets, the star-shaped Fe3−xO4-Au core–shell NPs have been successfully used as SERS substrates for thiram trace detection owing to their high saturation magnetization at room temperature and rough enough plasmonic surface, highlighting their capability to be used as chemical trace sensors.232
To further improve the SERS performance of the composite substrates in practical applications, different functional components being able to enrich target molecules and SERS-active colloids can be integrated into more complex composites for sample preparation and detection all-in-one.236–239 For instance, Zhang et al. reported CoFe2O4@halloysite nanotubes (HNTs)/Au NPs composites with magnetic CoFe2O4 beads inside HNTs and SERS-active Au NPs on the surface of HNTs.238 The magnetism of CoFe2O4 beads, molecular enrichment of HNTs, and SERS-activity of Au NPs are all integrated into one composite nanomaterials. The resulting CoFe2O4@HNTs/Au NPs substrates have good pH stability from 3.0 to 11.0 and can realize rapid (within 5 min), sensitive (LOD down to 10−2 mg L−1 and EF up to 107) and reproducible (RSD < 10%) detection of 4,4′-thioaniline and nitrofurantoin in fish feed and aquatic samples. Besides, other functional components, such as internal standards for more quantitative detection can also be integrated into all-in-one composite substrates.239
4.3 Nanocomposites consisted of metal and SERS-active non-metal nanomaterials
The efficient combination of both the EM and CM enhancement into one SERS substrate is of great significance in boosting overall SERS sensitivities and strengthening the SERS theories. The hybridization of plasmonic metals with SERS-active non-metal nanomaterials creates an ideal tunable platform for the exploration of the synergistic effect between EM and CM. Importantly, in order to realize the effective combination of EM and CM, the design of this type of nanocomposite needs to fully consider the distance dependence of EM enhancement and the interaction between target molecules and substrates related to CM enhancement so that different resonances belonging to EM and CM can be simultaneously excited under the same incident light as well as the ideal SERS enhancement can be observed. So far, many SERS-active non-metal nanomaterials have been introduced to combine with plasmonic metals, including MOFs, organic and inorganic semiconductors, and graphene, and correspondingly, the overall SERS performance has been significantly improved by optimization of each component in the hybrid nanocomposites. For example, after simply depositing concentrated gold nanoparticle colloids onto the MIL-100(Fe) substrate, the Au/MIL-100(Fe) composite substrate provided a remarkable EF up to 1010, enabling a LOD down to 0.48 ppb for toluene detection, which was nearly 4 orders of magnitude lower than that of the MIL-100(Fe) substrate (2.5 ppm) without a covering of Au colloids.179 Obviously, the improved sensing performance is closely related to the employed hotspots between colloidal Au NPs onto the MIL-100(Fe) substrate and attributed to both the CM enhancement from the charge transfer between toluene and MIL-100(Fe) and the EM enhancement from the surface plasmon resonance of Au. Similarly, after coating a thin Au layer on the superhydrophobic nanostructured organic DFH-4T film, remarkable Raman EFs (∼1010) and extremely low analyte detection (<10−21 M) for methylene blue were also achieved on the Au/DFH-4T composite substrate.141 More recently, Zhang et al. reported a general approach to a hybrid platform of Au nanoparticles on monolayer semiconductors by gold-mediated mechanical exfoliation followed by appropriate etching in KI/I2 solution, which leaves randomly distributed and high-density Au NPs on the surface of monolayer semiconductor. The resulting Au NPs/ReSe2, Au NPs/MoS2, and Au NPs/PdSe2 have LOD approximately three to four orders of magnitude lower than their bare 2D materials counterparts.240Both the distribution and density of carriers and the frequency of the plasmonic resonance can significantly affect the SERS performance of the nanocomposites, which can be modulated by adjusting many factors, including the characteristics of each component in the nanocomposites and the excitation energy.195 Zhang et al. fabricated an ordered Ag/Cu2S substrate with the hemispherical dual-shell structure (Ag monolayer as the inside shell and Cu2S layer as the outside shell) onto the self-assembled monodispersed polystyrene nanostructures by magnetron sputtering and found that the carrier distribution and density could be easily monitored by controlling the thickness of the Cu2S shell under different laser excitations and ultimately led to different SERS performance to be measured.195 As shown in Fig. 10A, it is obvious that the Ag/Cu2S substrate with 30 min sputtering time showed higher enhancement toward 4-MBA than pure silver and other Ag/Cu2S substrates with longer sputtering time under the laser wavelength of 514 nm or 633 nm, whereas under the laser wavelength of 785 nm, the signal intensity monotonously decreased with an increase of the sputtering time. That is because the excitation energy from the laser can also influence the intensity of the probe molecule 4-MBA adsorbed on the Cu2S surface. The laser energies for both 514 and 633 nm excitations are higher than the band-gap energy between the HOMO of 4-MBA and the valence band of Cu2S (Fig. 10B), hence the intensity is mainly attributed to the synergistic effect between the EM and CT enhancement. In comparison, the laser energy of 785 nm excitation is lower than the band-gap energy, so the intensity is mainly contributed to the EM enhancement that comes from Ag. The decrease of the intensity after the sputtering time of Cu2S is longer than 30 min is due to the great suppression of the EM enhancement with the thickness increase of the outer Cu2S layer.
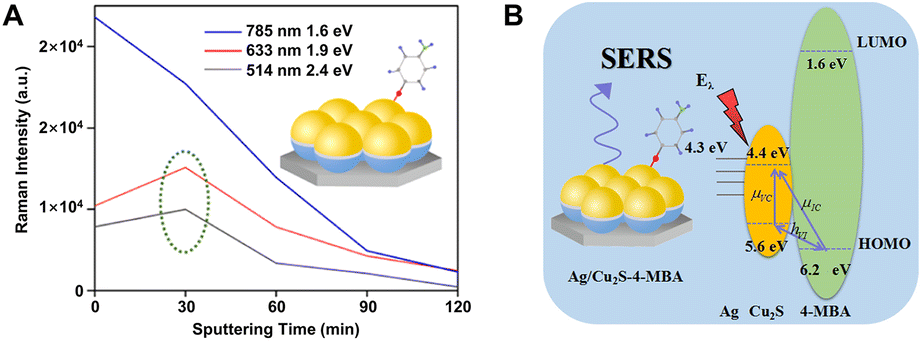 | ||
| Fig. 10 (A) Intensity of Raman peak at 1586 cm−1 of 4-MBA on Ag/Cu2S hemispherical dual-shell with various excitation wavelengths and sputtering times and (B) the corresponding coupling scheme in this Ag/Cu2S/4-MBA system. Reproduced from ref. 195 with permission from American Chemical Society, copyright 2018. | ||
The plasmon-induced CT from metals to SERS-active non-metal nanomaterials can also enhance the chemical contributions to the overall SERS signals. Taking the Au–TiO2 system prepared by photocatalytic reduction of HAuCl4 on TiO2 NPs as an example, it showed a higher enhancement than the pure TiO2 substrate while using 4-MBA as the probe molecule, however, Raman shifts and Raman peaks exhibited no change compared with the pure TiO2 substrate but were significantly different from the Au nanoparticle substrate.20,241 Clearly, the additional considerable enhancement of the Au–TiO2 composites is closely associated with the deposition of Au, which dramatically improves the CT-induced SERS enhancement of TiO2 NPs. Namely, in the Au/TiO2/4-MBA composites, the “donor–bridge–acceptor” CT occurs more easily than the direct CT between Au and 4-MBA due to energy level matching, and the additional CTs were assisted by LSPR of Au and the intrinsic TiO2-to-molecule CT are together responsible for the considerable SERS enhancement of adsorbed molecules.241
Especially, the enhancement ability of the composite substrates of plasmonic metals and photo-activated semiconductors can be further improved by an emerging technique called photo-induced enhanced Raman scattering (PIERS).196,242–245 In PIERS, the substrates need to be irradiated with a second light before or during the Raman testing in order to foster the CT processes between semiconductors and metals, enabling strong Raman enhancement and improved sensitivity beyond the normal SERS effect.196,242 In 2016, Parkin's group reported the PIERS effect on Au/AgNP-TiO2 substrates, which showed stronger enhancement with an order of magnitude to target molecules including dyes, explosives, and biomolecules over SERS (e.g., rhodamine 6G shown in Fig. 11A).196 Noteworthily, this PIERS effect requires pre-irradiation on the substrates with a second light for a period of time before detection of target molecules and can be slowly decayed due to surface healing upon exposure to air (Fig. 11B), and the substrate can be recycled via re-irradiation without loss of analyte signal intensity (Fig. 11C). The authors suggested that the additional enhancement originates from oxygen vacancy defects on TiO2 surfaces generated by pre-irradiation, namely, the vacancy species below the conduction bond edge enabled electrons to be excited into the conduction bond and then transferred into metal energy levels upon Raman laser illumination, leading to a stronger local electromagnetic field of metal NPs.196,246 Besides inorganic semiconductors, organic semiconductors can also be used to combine with plasmonic metals for PIERS applications. Almohammed et al. demonstrated that the UV-induced charge transfer in the substrate of Ag nanoparticle-decorated aligned diphenylalanine peptide nanotubes (FF-PNTs) facilitated CM that provided up to a 10-fold increase in SERS intensity and allowed the detection of a wide range of small molecules and low Raman cross-section molecules at concentrations as low as 10−13 M.243
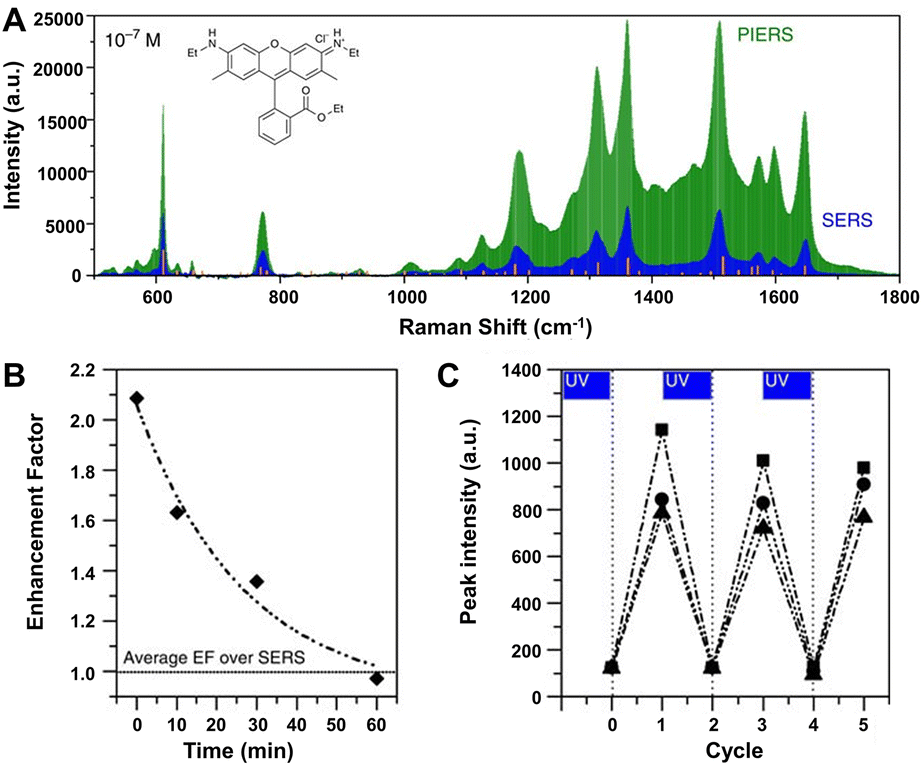 | ||
| Fig. 11 (A) PIERS and SERS spectra of rhodamine 6G (10−7 M), with Raman modes of the solid shown by the orange lines. (B) Average EF of PIERS over SERS for dinitrotoluene (10−9 M) over time. Horizontal line indicates the normal SERS intensity. (C) The recyclability of the substrate on irradiation to clean and re-charge for measuring dinitrotoluene (10−9 M) at three wavelengths, 1315 cm−1 (squares), 1367 cm−1 (circles) and 1385 cm−1 (triangles). Reproduced from ref. 196 with permission from Springer Nature, copyright 2016. | ||
4.4 Nanocomposites without plasmonic metals
Compared with plasmonic metal-based SERS platforms, the metal-free SERS-active nanomaterials present many attractive properties, including lower cost, more flexible controllability, higher stability and uniformity, better biocompatibility, and better inherent selectivity from CM enhancement. Furthermore, in contrast to single-component non-metal nanomaterials, the metal-free nanocomposites not only can present stronger CM enhancement owing to their complex electronic structures, which can more efficiently interact with excitation light by more possible exciton resonance inside the substrates and charge separation at the interface,119,198–202,247 but also can introduce EM enhancement through elaborate control of the morphology and interaction of the nanocomposites.248Strong coupling in the heterojunction interface can improve the separation of electron–hole pairs and further facilitate effective charge transfer, which is beneficial to CM enhancement in SERS. For example, the TiO2/ZnO heterojunction composed of two classic inorganic semiconductors showed strong SERS enhancement (EF up to 6.8 × 105) for a non-resonant molecule of 4-MBA, higher than the performance measured with pure TiO2 NPs, pure ZnO NPs, and their simple mixture as the substrates, respectively (Fig. 12A).202 Through calculation, Jiang et al. found that the order of the SERS intensity ratio was as follows: TiO2/ZnO heterojunction composite > mixture of TiO2 and ZnO > TiO2 > ZnO, which is consistent with the order of the intensity ratio between a non-totally symmetric mode (b2, assigned to the band at 1144 cm−1) and a totally symmetric mode (a1, attributed to the band at 1075 cm−1) in the SERS spectra of 4-MBA on these substrates, suggesting that the charge-transfer contribution significantly increased in the heterojunction (Fig. 12B).202 Additionally, the charge-transfer process in the heterojunction can be effectively modulated using the engineering methods such as element doping and defect introduction. Zhou et al. designed and created a sponge-like Cu-doping NiO-SnO2 p–n semiconductor heterostructure (SnO2-NiOx/Cu) as a CM-based SERS substrate with extremely high EF (1.46 × 1010 for copper phthalocyanine), mainly attributed to the enhanced charge separation efficiency of the SnO2-NiOx/Cu heterojunction, the charge transfer resonance caused by Cu doping, and enrichment of the probe molecules by the porous nanosponge structure of the SnO2-NiOx/Cu heterojunction.119 The doping of Cu introduced vacant oxygen into NiO and narrowed the band gap by shifting the Fermi level towards the conduction band, leading to the conversion of type-I SnO2-NiO heterojunction into type-II SnO2-NiOx/Cu heterojunction with more efficient charge separation and the improved charge transfer between adsorbed molecules and SnO2-NiOx/Cu heterojunction and thus resulting in the incredible SERS performance improvement on SnO2-NiOx/Cu with an EF 105 times higher than that on SnO2-NiO (Fig. 12C and D).
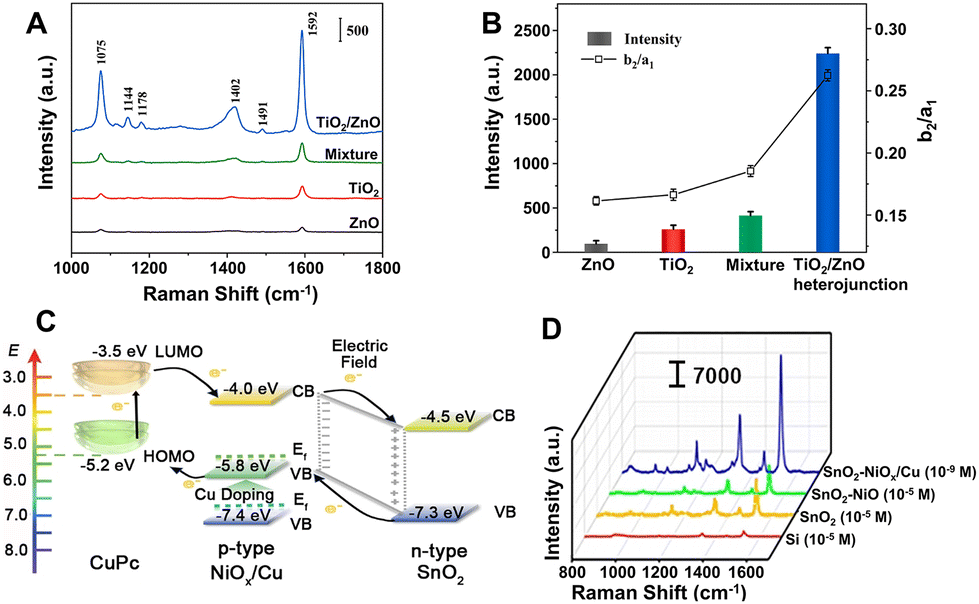 | ||
| Fig. 12 (A) SERS spectra of 4-MBA (10−3 M) adsorbed on the surface of pure TiO2 NPs, pure ZnO NPs, mixture of TiO2 and ZnO NPs, and TiO2/ZnO heterojunction composites. (B) The SERS intensity of the band at 1592 cm−1 and the ratio of I1144/I1075 (Ib2/Ia1) for different substrates. Reproduced from ref. 202 with permission from Elsevier, copyright 2022. (C) Energy level diagram of copper phthalocyanine (CuPc) molecule adsorbed on SnO2-NiOx/Cu SERS substrate. (D) Raman spectra of CuPc on Si (10−5 M), SnO2 (10−5 M), SnO2-NiO (10−5 M), and SnO2-NiOx/Cu (10−9 M) substrates under 785 nm laser excitation. Reproduced from ref. 119 with permission from John Wiley & Sons, copyright 2021. | ||
The van der Waals (vdW) heterostructures, a type of vertical 2D heterostructures, in which the interlayer interaction is mainly the vdW force and no or little requirement for lattice matching between the different layers needs to be met, are able to combine various 2D materials with more complicated electronic structures and possibly enhanced interfacial charge transfer, providing a unique platform for SERS.198,201,248–250 In 2017, Tan et al. constructed a series of vdW heterostructures composed of the monolayer graphene (G) and WSe2 (W) with artificially designed sequences using the wet-chemical transfer technique and found that the Raman enhancement effect on those heterostructures was dependent on the stacked methods.198 Among them, the G/W (graphene on top) vdW heterostructure exhibited stronger SERS enhancement toward copper phthalocyanine than the isolated WSe2 monolayer, the graphene monolayer, the W/G heterostructure, and the W/G/G/W heterostructure, but is equivalent to the G/W/G/W heterostructure. These differences were due to the different interlayer couplings in heterostructures related to electron transition probability rates, whereas the magnitude of the enhancement fact mainly depended on the top two layers. For the G/W heterostructure, the interlayer distance could be decreased from ∼4 nm to ∼0.4 nm to reach the optimized combination of graphene and the WSe2 monolayer via carbon ion irradiation, enriching the electronic state density of graphene through the interlayer coupling and meanwhile promoting the charge transfer between copper phthalocyanine molecules and the substrate. In addition to CM enhancement, EM enhancement has also been reported in vdW heterostructures. In 2019, Ghopry and coworkers reported a novel TMD (MoS2 and WS2) nanodomes/graphene van der Waals heterostructure SERS substrate with the extraordinary SERS sensitivity of 5 × 10−11 to 5 × 10−12 M to rhodamine 6G, more than three orders of magnitude higher than that on single-layer TMD and graphene substrates.248 The SERS sensitivity was closely related to the nanodome morphology of the TMD layer and reached the maximum value when the nanodomes of MoS2 have a lateral dimension on the order of 200–500 nm and a height in the range of 3–5 nm, and the peak intensity of the graphene's Raman signature with the TMD nanodomes was 8–10 times higher than that of graphene only. So, the authors suggested that the increased free carriers due to charge transfer across the vdW interface and optical excitation, and the confined morphology of MoS2 (WS2) allowed LSPR in nanodomes, which offered extra EM enhancement. Correspondingly, they attributed the high SERS sensitivity on TMD/graphene vdW heterostructure substrates to the combination of CM enhancement due to the enhanced dipole–dipole interaction at the TMD/graphene interface and the EM enhancement through the LSPR onto the photodoped TMD nanodomes/graphene.248
5 Conclusion and outlook
A series of SERS substrates have been developed and reported in the past half century since the discovery of SERS and meanwhile, SERS technology has also attracted extensive attention from different fields such as chemistry, materials, physics, energy, and life science, owing to its application potentials in these areas. Unfortunately, all the existing SERS substrates have their own inherent disadvantages that are difficult to overcome, mainly focusing on generality, uniformity, application compatibility, sensitivity, selectivity, long-term stability, reproducibility, and other aspects, which seriously limits the further widespread practical application and even commercialization of SERS technology. Hence, designing, exploration, and construction of high-performance SERS substrates, especially those that can meet the requirements for practical applications still remains a great challenge and the core for the development of SERS technology for a long period of time. Based on the progress in the SERS substrates described above, further efforts can be devoted to the following promising directions, aiming to create more desirable and effective SERS substrates to address the increasingly stringent application requirements.First of all, for the existing SERS substrates, the key to their further development is to explore and develop effective methods to solve the bottlenecks encountered by them so that improved SERS performance suitable for real-world applications can be achieved. Although the development of the methods with generality applicable to all types of SERS substrates is very challenging, some methods, according to the characteristics of the nanostructured materials and the SERS enhancement mechanism, including morphology and size control, structure design and optimization, surface modification and functionalization, and hotspot engineering, can be adopted to realize the effective improvement of the SERS performance. Among them, one of the critical difficulties is how to construct the nanostructures with highly ordered, abundant uniform, and reproductive hotspots for SERS enhancement by adjusting their size, spacing, shape, arrangement, and distribution. Hence, further development of simple, reliable, and novel nano processing techniques and hierarchical construction methods is highly expected, leading to the introduction of smaller nano gaps (less than 5 nm) as well as the construction of denser hotspot meshes and thereby addressing the limitations of the existing techniques or methods.
Secondly, the system of SERS-active nanomaterials needs to be further expanded and enriched. It is a long-standing key challenge to develop ideal SERS-active nanomaterials with low cost and excellent performance for SERS applications. (i) Finding new low-cost nanomaterials with SERS enhancement comparable to plasmonic noble metals especially Au and Ag is highly desirable. In contrast to plasmonic noble metals, the study of non-metal SERS-active nanomaterials is still at its early stage, and more non-metal nanomaterials with high enhancement ability, including semiconductors and MOFs need to be explored extensively and deeply. In particular, only a few examples of SERS-active organic semiconductors are reported although their π-conjugated frameworks can provide infinite possibilities for optimizing charge transfer to specific target molecules through special customization. (ii) Combination or integration of different components is always one of the most effective ways to obtain ideal SERS substrates. The difficulty lies in how to effectively integrate the advantages of each component and make full use of their multiple contributions to induce and enhance the SERS signals through careful selection of functional components and reasonable design of the composite structures. For composite substrates, the combination of strong EM enhancement and relatively weak CM enhancement with intrinsic selectivity is an attractive way to achieve both highly sensitive and selective SERS enhancement. However, all the currently-developed methods to obtain the improved SERS substrates through effective integration of EM and CM (such as simple depositing and “borrowing” strategy) have their own limitations and face many challenges including controllable construction and complex mechanism revealment. It can be expected that if CM and EM enhancement can be combined in a multiplicative way, ultra-trace detection down to fM scale is very promising to be achieved for SERS. In addition, some new types of nanomaterials with specific functions, such as dielectrics and covalent organic frameworks, can be introduced to broaden the range of material selection and further make the foundation for the expansion of the SERS application field.
Last but not least, the design and development of SERS-active nanomaterials also need to fully consider their adaptability and applicability, needing to meet the standards for specific applications in most cases. For quantitative analysis, precise screening of internal standard molecules with no characteristic Raman peaks overlapped with those of target molecules in advance is very critical to achieve accurate, effective, and reliable quantitative SERS measurements. For biomedical applications, the biocompatibility, toxicity, and stability of SERS substrates should be in line with the requirements of biological systems. For the detection of special target molecules, such as chemical warfare agents, toxic vapors, explosives, and volatile organic compounds, the key challenge is the exploration of SERS-active nanomaterials for rapid and highly selective identification of these dangerous materials in a complex environment. For the detection of weakly adsorbed molecules, low sensitivity is an important issue, and it is urgent to focus on how to achieve stronger and more effective affinity between target molecules and SERS substrates by strengthening the interaction between them via the design of SERS-active nanomaterials. Of course, in practical applications, it is highly expected to overcome molecular university, the most challenging bottleneck of SERS, and develop SERS-active nanomaterials that can detect a series of target molecules, especially those with similar structures, in complex mixtures and environments.
In summary, although great progress has been made, more efforts are still needed to be made to overcome the challenges and problems encountered in the design, development, and application of SERS-active nanomaterials. This is also of great significance to further promote the enrichment and improvement of SERS theory, update the SERS spectrum library, and expand the SERS applications. It is highly hoped that the research enthusiasm on SERS-active nanomaterials can make the SERS technology develop into a powerful and universal tool in nanoscience, surface science, and analytical science and lead to many exciting applications in wider fields including energy, environment, and biomedicine.
Author contributions
Yue Ying: investigation, writing – original draft; Zhiyong Tang: writing – review and editing; Yaling Liu: conceptualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (2022YFB3805203, Y. L. L.; 2021YFA1200302, Z. Y. T.), Strategic Priority Research Program of Chinese Academy of Sciences (XDB36000000, Z. Y. T., and Y. L. L.), and National Natural Science Foundation of China (22073021, Y. L. L.; 92056204, 21890381 and 21721002, Z. Y. T.).References
- E. Smith and G. Dent, Modern Raman Spectroscopy – A Practical Approach, Wiley, Chichester, UK, 2004, vol. 78 Search PubMed.
- J. B. Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- C. E. Harvey and B. M. Weckhuysen, Catal. Lett., 2015, 145, 40–57 CrossRef CAS PubMed.
- P. Mosier-Boss, Nanomaterials, 2017, 7, 142 CrossRef PubMed.
- R. Pilot, R. Signorini, C. Durante, L. Orian, M. Bhamidipati and L. Fabris, Biosensors, 2019, 9, 57 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- J. F. Li, Y. J. Zhang, S. Y. Ding, R. Panneerselvam and Z.-Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS.
- V. Shvalya, G. Filipič, J. Zavašnik, I. Abdulhalim and U. Cvelbar, Appl. Phys. Rev., 2020, 7, 031307 CAS.
- J. Langer, D. Jimenez de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. J. García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. Le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L. L. Tay, K. G. Thomas, Z. Q. Tian, R. P. Van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- S. Nie, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- H. Lai, G. Li, F. Xu and Z. Zhang, J. Mater. Chem. C, 2020, 8, 2952–2963 RSC.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- J. F. Li, X. D. Tian, S. B. Li, J. R. Anema, Z. L. Yang, Y. Ding, Y. F. Wu, Y. M. Zeng, Q. Z. Chen, B. Ren, Z. L. Wang and Z. Q. Tian, Nat. Protoc., 2013, 8, 52–65 CrossRef CAS PubMed.
- Z. Q. Tian, B. Ren and D. Y. Wu, J. Phys. Chem. B, 2002, 106, 9463–9483 CrossRef CAS.
- P. Wang, Y. Sun, X. Li, L. Wang, Y. Xu and G. Li, Molecules, 2021, 26, 209 CrossRef CAS PubMed.
- S. Fateixa, H. I. S. Nogueira and T. Trindade, Phys. Chem. Chem. Phys., 2015, 17, 21046–21071 RSC.
- L. Lan, Y. Gao, X. Fan, M. Li, Q. Hao and T. Qiu, Front. Phys., 2021, 16, 43300 CrossRef.
- Y. Liu, H. Ma, X. X. Han and B. Zhao, Mater. Horiz., 2021, 8, 370–382 RSC.
- X. Meng, L. Qiu, G. Xi, X. Wang and L. Guo, SmartMat, 2021, 2, 466–487 CrossRef CAS.
- B. Sharma, R. R. Frontiera, A. I. Henry, E. Ringe and R. P. Van Duyne, Mater. Today, 2012, 15, 16–25 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, in Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, 2009, pp. 265–297 Search PubMed.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794–13803 CrossRef CAS.
- H. K. Lee, Y. H. Lee, C. S. L. Koh, G. C. Phan-Quang, X. Han, C. L. Lay, H. Y. F. Sim, Y. C. Kao, Q. An and X. Y. Ling, Chem. Soc. Rev., 2019, 48, 731–756 RSC.
- E. C. Le Ru and P. G. Etchegoin, MRS Bull., 2013, 38, 631–640 CrossRef.
- J. Gersten and A. Nitzan, J. Chem. Phys., 1980, 73, 3023–3037 CrossRef CAS.
- S. Y. Ding, E. M. You, Z. Q. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042–4076 RSC.
- P. Alonso-González, P. Albella, M. Schnell, J. Chen, F. Huth, A. García-Etxarri, F. Casanova, F. Golmar, L. Arzubiaga, L. E. Hueso, J. Aizpurua and R. Hillenbrand, Nat. Commun., 2012, 3, 684 CrossRef PubMed.
- S. Y. Ding, J. Yi, J. F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z. Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2014, 118, 11120–11130 CrossRef CAS.
- I. Alessandri and J. R. Lombardi, Chem. Rev., 2016, 116, 14921–14981 CrossRef CAS PubMed.
- T. Ochiai, K. Sakoda and T. Sawada, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 245101 CrossRef.
- W. Ji, L. Li, W. Song, X. Wang, B. Zhao and Y. Ozaki, Angew. Chem., Int. Ed., 2019, 58, 14452–14456 CrossRef CAS PubMed.
- K. Frizyuk, M. Hasan, A. Krasnok, A. Alú and M. Petrov, Phys. Rev. B, 2018, 97, 085414 CrossRef CAS.
- I. Rodriguez, L. Shi, X. Lu, B. A. Korgel, R. A. Alvarez-Puebla and F. Meseguer, Nanoscale, 2014, 6, 5666–5670 RSC.
- L. Shi, T. U. Tuzer, R. Fenollosa and F. Meseguer, Adv. Mater., 2012, 24, 5934–5938 CrossRef CAS.
- S. Hayashi, R. Koh, Y. Ichiyama and K. Yamamoto, Phys. Rev. Lett., 1988, 60, 1085–1088 CrossRef CAS PubMed.
- M. Inoue and K. Ohtaka, J. Phys. Soc. Jpn., 1983, 52, 1457–1468 CrossRef CAS.
- B. J. Messinger, K. U. von Raben, R. K. Chang and P. W. Barber, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 649–657 CrossRef CAS.
- M. E. Lippitsch, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 29, 3101–3110 CrossRef CAS.
- L. Jensen, C. M. Aikens and G. C. Schatz, Chem. Soc. Rev., 2008, 37, 1061 RSC.
- J. R. Lombardi, R. L. Birke, T. Lu and J. Xu, J. Chem. Phys., 1986, 84, 4174–4180 CrossRef CAS.
- F. S. Ameer, C. U. Pittman and D. Zhang, J. Phys. Chem. C, 2013, 117, 27096–27104 CrossRef CAS.
- X. Ling, L. G. Moura, M. A. Pimenta and J. Zhang, J. Phys. Chem. C, 2012, 116, 25112–25118 CrossRef CAS.
- N. John and S. George, Raman Spectroscopy, Elsevier Inc., 2017, vol. 2 Search PubMed.
- Y. Wang, W. Ruan, J. Zhang, B. Yang, W. Xu, B. Zhao and J. R. Lombardi, J. Raman Spectrosc., 2009, 40, 1072–1077 CrossRef CAS.
- H. Wu, H. Wang and G. Li, Analyst, 2017, 142, 326–335 RSC.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2008, 112, 5605–5617 CrossRef CAS.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
- J. R. Lombardi, Faraday Discuss., 2017, 205, 105–120 RSC.
- A. C. Albrecht, J. Chem. Phys., 1961, 34, 1476–1484 CrossRef CAS.
- X. X. Han, W. Ji, B. Zhao and Y. Ozaki, Nanoscale, 2017, 9, 4847–4861 RSC.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- A. Kunzmann, B. Andersson, T. Thurnherr, H. Krug, A. Scheynius and B. Fadeel, Biochim. Biophys. Acta, Gen. Subj., 2011, 1810, 361–373 CrossRef CAS PubMed.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- D.-S. Wang, H. Chew and M. Kerker, Appl. Opt., 1980, 19, 2256 CrossRef CAS PubMed.
- E. C. Le Ru, J. Grand, I. Sow, W. R. C. Somerville, P. G. Etchegoin, M. Treguer-Delapierre, G. Charron, N. Félidj, G. Lévi and J. Aubard, Nano Lett., 2011, 11, 5013–5019 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- E. C. Le Ru and P. G. Etchegoin, in Principles of Surface-Enhanced Raman Spectroscopy, Elsevier, 2009, pp. 299–365 Search PubMed.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616–12617 CrossRef CAS PubMed.
- J. Jiang, K. Bosnick, M. Maillard and L. Brus, J. Phys. Chem. B, 2003, 107, 9964–9972 CrossRef CAS.
- D. K. Lim, K. S. Jeon, H. M. Kim, J. M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS.
- Y. Fang, N. H. Seong and D. D. Dlott, Science, 2008, 321, 388–392 CrossRef CAS PubMed.
- Y. Zhao, Y. J. Zhang, J. H. Meng, S. Chen, R. Panneerselvam, C.-Y. Li, S. B. Jamali, X. Li, Z. L. Yang, J. F. Li and Z. Q. Tian, J. Raman Spectrosc., 2016, 47, 662–667 CrossRef CAS.
- H. Xia, Y. Xiahou, P. Zhang, W. Ding and D. Wang, Langmuir, 2016, 32, 5870–5880 CrossRef CAS PubMed.
- F. Zhang, P. Chen, X. Li, J. T. Liu, L. Lin and Z. W. Fan, Laser Phys. Lett., 2013, 10, 045901 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- C. J. Orendorff, L. Gearheart, N. R. Jana and C. J. Murphy, Phys. Chem. Chem. Phys., 2006, 8, 165–170 RSC.
- K. Q. Lin, J. Yi, S. Hu, B. J. Liu, J. Y. Liu, X. Wang and B. Ren, J. Phys. Chem. C, 2016, 120, 20806–20813 CrossRef CAS.
- J. V. Jokerst, A. J. Cole, D. Van de Sompel and S. S. Gambhir, ACS Nano, 2012, 6, 10366–10377 CrossRef CAS PubMed.
- H. He, C. Wu, C. Bi, Y. Song, D. Wang and H. Xia, Chem. – Eur. J., 2021, 27, 7549–7560 CrossRef CAS PubMed.
- S. T. Sivapalan, B. M. DeVetter, T. K. Yang, T. van Dijk, M. V. Schulmerich, P. S. Carney, R. Bhargava and C. J. Murphy, ACS Nano, 2013, 7, 2099–2105 CrossRef CAS PubMed.
- M. Rycenga, X. Xia, C. H. Moran, F. Zhou, D. Qin, Z. Y. Li and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 5473–5477 CrossRef CAS PubMed.
- L. J. Sherry, S. H. Chang, G. C. Schatz, R. P. Van Duyne, B. J. Wiley and Y. Xia, Nano Lett., 2005, 5, 2034–2038 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS PubMed.
- J. C. Hulteen, D. A. Treichel, M. T. Smith, M. L. Duval, T. R. Jensen and R. P. Van Duyne, J. Phys. Chem. B, 1999, 103, 3854–3863 CrossRef CAS.
- X. Geng, W. Leng, N. A. Carter, P. J. Vikesland and T. Z. Grove, J. Mater. Chem. B, 2016, 4, 4182–4190 RSC.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 4597–4601 CrossRef CAS PubMed.
- J. Xie, Q. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473–2480 CrossRef CAS PubMed.
- W. Niu, Y. A. A. Chua, W. Zhang, H. Huang and X. Lu, J. Am. Chem. Soc., 2015, 137, 10460–10463 CrossRef CAS PubMed.
- R. A. Harder, L. A. Wijenayaka, H. T. Phan and A. J. Haes, J. Raman Spectrosc., 2021, 52, 497–505 CrossRef CAS PubMed.
- M. S. Goh, Y. H. Lee, S. Pedireddy, I. Y. Phang, W. W. Tjiu, J. M. R. Tan and X. Y. Ling, Langmuir, 2012, 28, 14441–14449 CrossRef CAS PubMed.
- Y. Liu, S. Pedireddy, Y. H. Lee, R. S. Hegde, W. W. Tjiu, Y. Cui and X. Y. Ling, Small, 2014, 10, 4940–4950 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- Q. Guo, M. Xu, Y. Yuan, R. Gu and J. Yao, Langmuir, 2016, 32, 4530–4537 CrossRef CAS PubMed.
- J. Lu, Z. Cai, Y. Zou, D. Wu, A. Wang, J. Chang, F. Wang, Z. Tian and G. Liu, ACS Appl. Nano Mater., 2019, 2, 6592–6601 CrossRef CAS.
- M. Y. Chan, W. Leng and P. J. Vikesland, ChemPhysChem, 2018, 19, 24–28 CrossRef CAS PubMed.
- T. Mostowtt, J. Munoz and B. McCord, Analyst, 2019, 144, 6404–6414 RSC.
- K. Kneipp, E. Roth, C. Engert and W. Kiefer, Chem. Phys. Lett., 1993, 207, 450–454 CrossRef CAS.
- L. Xie, J. Lu, T. Liu, G. Chen, G. Liu, B. Ren and Z. Tian, J. Phys. Chem. Lett., 2020, 11, 1022–1029 CrossRef CAS PubMed.
- E. Y. Hwang, J. H. Lee and D. W. Lim, J. Mater. Chem. B, 2021, 9, 5293–5308 RSC.
- M. U. Amin and J. Fang, ACS Appl. Nano Mater., 2022, 5, 10421–10430 CrossRef CAS.
- P. A. Mercadal, J. C. Fraire and E. A. Coronado, J. Phys. Chem. C, 2022, 126, 10524–10533 CrossRef CAS.
- X. M. Lin, Y. Cui, Y. H. Xu, B. Ren and Z. Q. Tian, Anal. Bioanal. Chem., 2009, 394, 1729–1745 CrossRef CAS PubMed.
- M. J. Natan, Faraday Discuss., 2006, 132, 321 RSC.
- R. Bhardwaj, X. Fang, P. Somasundaran and D. Attinger, Langmuir, 2010, 26, 7833–7842 CrossRef CAS PubMed.
- W. Wei, Y. Wang, J. Ji, S. Zuo, W. Li, F. Bai and H. Fan, Nano Lett., 2018, 18, 4467–4472 CrossRef CAS PubMed.
- T. P. Bigioni, X.-M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed.
- L. A. Dick, A. D. McFarland, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2002, 106, 853–860 CrossRef CAS.
- U. S. Dinish, F. C. Yaw, A. Agarwal and M. Olivo, Biosens. Bioelectron., 2011, 26, 1987–1992 CrossRef CAS PubMed.
- W. M. Ingram, C. Han, Q. Zhang and Y. Zhao, J. Phys. Chem. C, 2015, 119, 27639–27648 CrossRef CAS.
- P. I. Stavroulakis, N. Christou and D. Bagnall, Mater. Sci. Eng., B, 2009, 165, 186–189 CrossRef CAS.
- W. C. Lin, L. S. Liao, Y. H. Chen, H. C. Chang, D. P. Tsai and H. P. Chiang, Plasmonics, 2011, 6, 201–206 CrossRef CAS.
- N. G. Greeneltch, M. G. Blaber, G. C. Schatz and R. P. Van Duyne, J. Phys. Chem. C, 2013, 117, 2554–2558 CrossRef CAS.
- A. V. Markin, N. E. Markina, J. Popp and D. Cialla-May, TrAC, Trends Anal. Chem., 2018, 108, 247–259 CrossRef CAS.
- C. L. Lay, C. S. L. Koh, J. Wang, Y. H. Lee, R. Jiang, Y. Yang, Z. Yang, I. Y. Phang and X. Y. Ling, Nanoscale, 2018, 10, 575–581 RSC.
- S. Tian, O. Neumann, M. J. McClain, X. Yang, L. Zhou, C. Zhang, P. Nordlander and N. J. Halas, Nano Lett., 2017, 17, 5071–5077 CrossRef CAS PubMed.
- T. Dörfer, M. Schmitt and J. Popp, J. Raman Spectrosc., 2007, 38, 1379–1382 CrossRef.
- X. Wei, R. D. Jiji, A. Zare, B. Lada, X. Li and C. M. Greenlief, J. Raman Spectrosc., 2022, 53, 58–68 CrossRef CAS.
- B. Sharma, M. F. Cardinal, M. B. Ross, A. B. Zrimsek, S. V. Bykov, D. Punihaole, S. A. Asher, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2016, 16, 7968–7973 CrossRef CAS PubMed.
- A. Dubey, R. Mishra, C. W. Cheng, Y. P. Kuang, S. Gwo and T. J. Yen, J. Am. Chem. Soc., 2021, 143, 19282–19286 CrossRef CAS PubMed.
- G. V. Naik, V. M. Shalaev and A. Boltasseva, Adv. Mater., 2013, 25, 3264–3294 CrossRef CAS PubMed.
- D. Wang, F. Shi, J. Jose, Y. Hu, C. Zhang, A. Zhu, R. Grzeschik, S. Schlücker and W. Xie, J. Am. Chem. Soc., 2022, 144, 5003–5009 CrossRef CAS PubMed.
- H. Yamada, Y. Yamamoto and N. Tani, Chem. Phys. Lett., 1982, 86, 397–400 CrossRef CAS.
- H. Yamada and Y. Yamamoto, Surf. Sci., 1983, 134, 71–90 CrossRef CAS.
- Y. Ye, C. Chen, W. Li, X. Guo, H. Yang, H. Guan, H. Bai, W. Liu and G. Xi, Anal. Chem., 2021, 93, 3138–3145 CrossRef CAS PubMed.
- Y. Zhou, Q. Gu, T. Qiu, X. He, J. Chen, R. Qi, R. Huang, T. Zheng and Y. Tian, Angew. Chem., Int. Ed., 2021, 60, 26260–26267 CrossRef CAS PubMed.
- J. Lin, Y. Shang, X. Li, J. Yu, X. Wang and L. Guo, Adv. Mater., 2017, 29, 1604797 CrossRef PubMed.
- X. Wang, W. Shi, Z. Jin, W. Huang, J. Lin, G. Ma, S. Li and L. Guo, Angew. Chem., Int. Ed., 2017, 56, 9851–9855 CrossRef CAS PubMed.
- W. Yan, W. Ai, W. Liu, Z. Zhao, X. Hu, S. Cui and X. Shen, J. Alloys Compd., 2023, 943, 169042 CrossRef CAS.
- Y. Peng, C. Lin, Y. Li, Y. Gao, J. Wang, J. He, Z. Huang, J. Liu, X. Luo and Y. Yang, Matter, 2022, 5, 694–709 CrossRef CAS PubMed.
- X. Fu, Y. Pan, X. Wang and J. R. Lombardi, J. Chem. Phys., 2011, 134, 024707 CrossRef PubMed.
- X. Fu, T. Jiang, Q. Zhao and H. Yin, J. Raman Spectrosc., 2012, 43, 1191–1195 CrossRef CAS.
- C. Muehlethaler, C. R. Considine, V. Menon, W.-C. Lin, Y.-H. Lee and J. R. Lombardi, ACS Photonics, 2016, 3, 1164–1169 CrossRef CAS.
- A. D. Shutov, Z. Yi, J. Wang, A. M. Sinyukov, Z. He, C. Tang, J. Chen, E. J. Ocola, J. Laane, A. V. Sokolov, D. V. Voronine and M. O. Scully, ACS Photonics, 2018, 5, 4960–4968 CrossRef CAS.
- X. Ling, W. Fang, Y. Lee, P. T. Araujo, X. Zhang, J. F. Rodriguez-Nieva, Y. Lin, J. Zhang, J. Kong and M. S. Dresselhaus, Nano Lett., 2014, 14, 3033–3040 CrossRef CAS PubMed.
- Y. Tian, H. Wei, Y. Xu, Q. Sun, B. Man and M. Liu, Nanomaterials, 2020, 10, 1910 CrossRef CAS PubMed.
- Y. Lee, H. Kim, J. Lee, S. H. Yu, E. Hwang, C. Lee, J. Ahn and J. H. Cho, Chem. Mater., 2016, 28, 180–187 CrossRef CAS.
- S. K. Islam, M. Tamargo, R. Moug and J. R. Lombardi, J. Phys. Chem. C, 2013, 117, 23372–23377 CrossRef CAS.
- L. Tao, K. Chen, Z. Chen, C. Cong, C. Qiu, J. Chen, X. Wang, H. Chen, T. Yu, W. Xie, S. Deng and J.-B. Xu, J. Am. Chem. Soc., 2018, 140, 8696–8704 CrossRef CAS PubMed.
- B. Adhikari, T. B. Limbu, K. Vinodgopal and F. Yan, Nanotechnology, 2021, 32, 335701 CrossRef CAS PubMed.
- W. Li, R. Zamani, P. R. Gil, B. Pelaz, M. Ibáñez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098–7101 CrossRef CAS PubMed.
- L. Dawei, W. Jian, X. Houwen, S. Xu and L. Fan-chen, Spectrochim. Acta, Part A, 1987, 43, 379–382 CrossRef.
- H. Zhang, H. Xin, T. He and F. Liu, Spectrochim. Acta, Part A, 1991, 47, 927–932 CrossRef.
- Z. Yu, W. Yu, J. Xing, R. A. Ganeev, W. Xin, J. Cheng and C. Guo, ACS Photonics, 2018, 5, 1619–1627 CrossRef CAS.
- M. Xin, Y. Fu, Y. Zhou, J. Han, Y. Mao, M. Li, J. Liu and M. Huang, New J. Chem., 2020, 44, 17570–17576 RSC.
- X. Wang, W. Shi, G. She and L. Mu, J. Am. Chem. Soc., 2011, 133, 16518–16523 CrossRef CAS PubMed.
- K. Mantey, L. Quagliano, A. Rezk, S. Palleschi, L. Abuhassan, A. Nayfeh, E. Bahceci and M. H. Nayfeh, AIP Adv., 2021, 11, 105206 CrossRef CAS.
- M. Yilmaz, E. Babur, M. Ozdemir, R. L. Gieseking, Y. Dede, U. Tamer, G. C. Schatz, A. Facchetti, H. Usta and G. Demirel, Nat. Mater., 2017, 16, 918–924 CrossRef CAS PubMed.
- D. Maznichenko, K. Venkatakrishnan and B. Tan, J. Phys. Chem. C, 2013, 117, 578–583 CrossRef CAS.
- Y. Wang, Z. Sun, H. Hu, S. Jing, B. Zhao, W. Xu, C. Zhao and J. R. Lombardi, J. Raman Spectrosc., 2007, 38, 34–38 CrossRef CAS.
- L. Yang, X. Jiang, W. Ruan, B. Zhao, W. Xu and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 20095–20098 CrossRef CAS.
- S. Cong, Y. Yuan, Z. Chen, J. Hou, M. Yang, Y. Su, Y. Zhang, L. Li, Q. Li, F. Geng and Z. Zhao, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- L. Lan, X. Fan, C. Zhao, J. Gao, Z. Qu, W. Song, H. Yao, M. Li and T. Qiu, Nanoscale, 2023, 15, 2779–2787 RSC.
- L. Jiang, Y. Hu, H. Zhang, X. Luo, R. Yuan and X. Yang, Anal. Chem., 2022, 94, 6967–6975 CrossRef CAS PubMed.
- L. Yang, Y. Peng, Y. Yang, J. Liu, H. Huang, B. Yu, J. Zhao, Y. Lu, Z. Huang, Z. Li and J. R. Lombardi, Adv. Sci., 2019, 6, 1900310 CrossRef PubMed.
- Y. Zhang, T. Yang, J. Li, Q. Zhang, B. Li and M. Gao, Adv. Funct. Mater., 2023, 33, 2210939 CrossRef CAS.
- Q. Lv, J. Tan, Z. Wang, L. Yu, B. Liu, J. Lin, J. Li, Z. Huang, F. Kang and R. Lv, Adv. Funct. Mater., 2022, 32, 2200273 CrossRef CAS.
- J. Seo, Y. Kim, J. Lee, E. Son, M. H. Jung, Y. M. Kim, H. Y. Jeong, G. Lee and H. Park, J. Mater. Chem. A, 2022, 10, 13298–13304 RSC.
- Y. Luo, L. Niu, Y. Wang, P. Wen, Y. Gong, C. Li and S. Xu, Nanoscale, 2022, 14, 16220–16232 RSC.
- C. He, L. Jiang, R. Yuan and X. Yang, Sens. Actuators, B, 2023, 374, 132777 CrossRef CAS.
- C. Liang, Z. A. Lu, M. Zheng, M. Chen, Y. Zhang, B. Zhang, J. Zhang and P. Xu, Nano Lett., 2022, 22, 6590–6598 CrossRef CAS PubMed.
- Y. Feng, J. Wang, J. Hou, X. Zhang, Y. Gao and K. Wang, Int. J. Mol. Sci., 2022, 23, 15930 CrossRef CAS PubMed.
- F. Li, X. Mu, X. Tang, G. Song, H. Sun, X. Zha, P. Sun, J. Fang, D. Hu, S. Cong and Z. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202218055 CrossRef CAS PubMed.
- X. Xue, W. Ji, Z. Mao, H. Mao, Y. Wang, X. Wang, W. Ruan, B. Zhao and J. R. Lombardi, J. Phys. Chem. C, 2012, 116, 8792–8797 CrossRef CAS.
- P. Karthick Kannan, P. Shankar, C. Blackman and C. Chung, Adv. Mater., 2019, 31, 1803432 CrossRef PubMed.
- X. Zhou, D. Wu, Z. Jin, X. Song, X. Wang and S. L. Suib, J. Mater. Sci., 2020, 55, 16374–16384 CrossRef CAS.
- Y. Yin, P. Miao, Y. Zhang, J. Han, X. Zhang, Y. Gong, L. Gu, C. Xu, T. Yao, P. Xu, Y. Wang, B. Song and S. Jin, Adv. Funct. Mater., 2017, 27, 1606694 CrossRef.
- P. Miao, J. K. Qin, Y. Shen, H. Su, J. Dai, B. Song, Y. Du, M. Sun, W. Zhang, H. L. Wang, C. Y. Xu and P. Xu, Small, 2018, 14, 1–8 Search PubMed.
- Z. Lei, X. Zhang, Y. Zhao, A. Wei, L. Tao, Y. Yang, Z. Zheng, L. Tao, P. Yu and J. Li, Nanoscale, 2022, 14, 4181–4187 RSC.
- J. Lin, J. Yu, O. U. Akakuru, X. Wang, B. Yuan, T. Chen, L. Guo and A. Wu, Chem. Sci., 2020, 11, 9414–9420 RSC.
- G. Demirel, H. Usta, M. Yilmaz, M. Celik, H. A. Alidagi and F. Buyukserin, J. Mater. Chem. C, 2018, 6, 5314–5335 RSC.
- J. R. Lombardi, Nat. Mater., 2017, 16, 878–880 CrossRef CAS PubMed.
- G. Demirel, R. L. M. Gieseking, R. Ozdemir, S. Kahmann, M. A. Loi, G. C. Schatz, A. Facchetti and H. Usta, Nat. Commun., 2019, 10, 5502 CrossRef CAS PubMed.
- I. Deneme, G. Liman, A. Can, G. Demirel and H. Usta, Nat. Commun., 2021, 12, 6119 CrossRef CAS PubMed.
- X. Y. Zhang, S. Yang, L. Yang, D. Zhang, Y. Sun, Z. Pang, J. Yang and L. Chen, Chem. Commun., 2020, 56, 2779–2782 RSC.
- T. Wang, Y. Lu, L. Xu and Z. J. Chen, J. Mater. Sci., 2022, 57, 16965–16973 CrossRef CAS.
- S. Almohammed, A. Fularz, M. B. Kanoun, S. Goumri-Said, A. Aljaafari, B. J. Rodriguez and J. H. Rice, ACS Appl. Mater. Interfaces, 2022, 14, 12504–12514 CrossRef CAS PubMed.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2010, 10, 553–561 CrossRef CAS PubMed.
- M. Li, Y. Qiu, C. Fan, K. Cui, Y. Zhang and Z. Xiao, Acta Pharm. Sin. B, 2018, 8, 381–389 CrossRef PubMed.
- W. Xu, N. Mao and J. Zhang, Small, 2013, 9, 1206–1224 CrossRef CAS PubMed.
- S. Huang, X. Ling, L. Liang, Y. Song, W. Fang, J. Zhang, J. Kong, V. Meunier and M. S. Dresselhaus, Nano Lett., 2015, 15, 2892–2901 CrossRef CAS PubMed.
- S. Feng, M. C. dos Santos, B. R. Carvalho, R. Lv, Q. Li, K. Fujisawa, A. L. Elías, Y. Lei, N. Perea-López, M. Endo, M. Pan, M. A. Pimenta and M. Terrones, Sci. Adv., 2016, 2, e1600322 CrossRef PubMed.
- T. H. Yu, C. H. Ho, C. Y. Wu, C. H. Chien, C.-H. Lin and S. Lee, J. Raman Spectrosc., 2013, 44, 1506–1511 CrossRef CAS.
- H. Sun, S. Cong, Z. Zheng, Z. Wang, Z. Chen and Z. Zhao, J. Am. Chem. Soc., 2019, 141, 870–878 CrossRef CAS PubMed.
- J. Xu, C. Cheng, S. Shang, W. Gao, P. Zeng and S. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 49452–49463 CrossRef CAS PubMed.
- J. Fu, Z. Zhong, D. Xie, Y. Guo, D. Kong, Z. Zhao, Z. Zhao and M. Li, Angew. Chem., Int. Ed., 2020, 59, 20489–20498 CrossRef CAS PubMed.
- Z. Chen, L. Su, X. Ma, Z. Duan and Y. Xiong, New J. Chem., 2021, 45, 5121–5126 RSC.
- H. Sun, W. Gong, S. Cong, C. Liu, G. Song, W. Lu and Z. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 2326–2334 CrossRef CAS PubMed.
- H. Sun, G. Song, W. Gong, W. Lu, S. Cong and Z. Zhao, Nano Res., 2022, 15, 5347–5354 CrossRef CAS.
- C. Li, C. Yang, W. Li, M. Cheng and Y. Liu, Nano, 2020, 15, 2050122 CrossRef CAS.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- X. D. Lin, V. Uzayisenga, J. F. Li, P. P. Fang, D. Y. Wu, B. Ren and Z. Q. Tian, J. Raman Spectrosc., 2012, 43, 40–45 CrossRef CAS.
- C. Y. Li, M. Meng, S. C. Huang, L. Li, S. R. Huang, S. Chen, L. Y. Meng, R. Panneerselvam, S. J. Zhang, B. Ren, Z. L. Yang, J. F. Li and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 13784–13787 CrossRef CAS PubMed.
- Q. Q. Chen, R. N. Hou, Y. Z. Zhu, X. T. Wang, H. Zhang, Y. J. Zhang, L. Zhang, Z. Q. Tian and J. F. Li, Anal. Chem., 2021, 93, 7188–7195 CrossRef CAS PubMed.
- L. He, Y. Liu, J. Liu, Y. Xiong, J. Zheng, Y. Liu and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 3741–3745 CrossRef CAS PubMed.
- Y. Hu, J. Liao, D. Wang and G. Li, Anal. Chem., 2014, 86, 3955–3963 CrossRef CAS PubMed.
- R. Borah and S. W. Verbruggen, J. Phys. Chem. C, 2020, 124, 12081–12094 CrossRef CAS.
- Y. Liu and A. R. Hight Walker, Angew. Chem., Int. Ed., 2010, 49, 6781–6785 CrossRef CAS PubMed.
- Y. Yang, J. Shi, G. Kawamura and M. Nogami, Scr. Mater., 2008, 58, 862–865 CrossRef CAS.
- S. Wang, J. Yao, Z. Ou, X. Wang, Y. Long, J. Zhang, Z. Fang, T. Wang, T. Ding and H. Xu, Nanoscale, 2022, 14, 4705–4711 RSC.
- X. Jiang, X. Sun, D. Yin, X. Li, M. Yang, X. Han, L. Yang and B. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 11212–11219 RSC.
- X.-Y. Zhang, D. Han, Z. Pang, Y. Sun, Y. Wang, Y. Zhang, J. Yang and L. Chen, J. Phys. Chem. C, 2018, 122, 5599–5605 CrossRef CAS.
- S. Ben-Jaber, W. J. Peveler, R. Quesada-Cabrera, E. Cortés, C. Sotelo-Vazquez, N. Abdul-Karim, S. A. Maier and I. P. Parkin, Nat. Commun., 2016, 7, 12189 CrossRef CAS PubMed.
- G. Barbillon, T. Noblet and C. Humbert, Phys. Chem. Chem. Phys., 2020, 22, 21000–21004 RSC.
- Y. Tan, L. Ma, Z. Gao, M. Chen and F. Chen, Nano Lett., 2017, 17, 2621–2626 CrossRef CAS PubMed.
- M. Li, X. Fan, Y. Gao and T. Qiu, J. Phys. Chem. Lett., 2019, 10, 4038–4044 CrossRef CAS PubMed.
- X. Jiang, Q. Sang, M. Yang, J. Du, W. Wang, L. Yang, X. Han and B. Zhao, Phys. Chem. Chem. Phys., 2019, 21, 12850–12858 RSC.
- J. Seo, J. Lee, Y. Kim, D. Koo, G. Lee and H. Park, Nano Lett., 2020, 20, 1620–1630 CrossRef CAS PubMed.
- X. Jiang, L. Xu, W. Ji, W. Wang, J. Du, L. Yang, W. Song, X. Han and B. Zhao, Appl. Surf. Sci., 2022, 584, 152609 CrossRef CAS.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed.
- H. J. Ryu, H. Shin, S. Oh, J. H. Joo, Y. Choi and J. S. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 2842–2853 CrossRef CAS PubMed.
- N. Cathcart, J. I. L. Chen and V. Kitaev, Langmuir, 2018, 34, 612–621 CrossRef CAS PubMed.
- X. Dong, J. Zhou, X. Liu, D. Lin and L. Zha, J. Raman Spectrosc., 2014, 45, 431–437 CrossRef CAS.
- Y. Cui, B. Ren, J. Yao, R. Gu and Z. Tian, J. Phys. Chem. B, 2006, 110, 4002–4006 CrossRef CAS PubMed.
- M. Fan, F. Lai, H. Chou, W. Lu, B. Hwang and A. G. Brolo, Chem. Sci., 2013, 4, 509–515 RSC.
- J. Zhang, S. A. Winget, Y. Wu, D. Su, X. Sun, Z.-X. Xie and D. Qin, ACS Nano, 2016, 10, 2607–2616 CrossRef CAS PubMed.
- Y. J. Zhang, S. B. Li, S. Duan, B. A. Lu, J. Yang, R. Panneerselvam, C. Y. Li, P. P. Fang, Z. Y. Zhou, D. L. Phillips, J. F. Li and Z. Q. Tian, J. Phys. Chem. C, 2016, 120, 20684–20691 CrossRef CAS.
- J. F. Li, Z. L. Yang, B. Ren, G. K. Liu, P. P. Fang, Y. X. Jiang, D. Y. Wu and Z. Q. Tian, Langmuir, 2006, 22, 10372–10379 CrossRef CAS PubMed.
- Z. Q. Tian, B. Ren, J. F. Li and Z. L. Yang, Chem. Commun., 2007, 34, 3514–3534 RSC.
- H. Ze, X. Chen, X. T. Wang, Y. H. Wang, Q. Q. Chen, J. S. Lin, Y.-J. Zhang, X. G. Zhang, Z. Q. Tian and J. F. Li, J. Am. Chem. Soc., 2021, 143, 1318–1322 CrossRef CAS PubMed.
- J. Li, J. Liu, Y. Yang and D. Qin, J. Am. Chem. Soc., 2015, 137, 7039–7042 CrossRef CAS PubMed.
- W. Shen, X. Lin, C. Jiang, C. Li, H. Lin, J. Huang, S. Wang, G. Liu, X. Yan, Q. Zhong and B. Ren, Angew. Chem., Int. Ed., 2015, 54, 7308–7312 CrossRef CAS PubMed.
- K. Wang and J. Li, Spectrochim. Acta, Part A, 2021, 263, 120218 CrossRef CAS PubMed.
- Y. Xu, S. He, W. Zhang, M. Xu, B. Zhou and H. Liu, J. Mater. Chem. C, 2020, 8, 13213–13219 RSC.
- X. Dai, Z. L. Song, W. Song, J. Zhang, G. C. Fan, W. Wang and X. Luo, Anal. Chem., 2020, 92, 11469–11475 CrossRef CAS PubMed.
- Y. Feng, Y. Wang, H. Wang, T. Chen, Y. Y. Tay, L. Yao, Q. Yan, S. Li and H. Chen, Small, 2012, 8, 246–251 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, J. H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J. M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- L. Wu, H. Pu, L. Huang and D. W. Sun, Food Chem., 2020, 328, 127105 CrossRef CAS PubMed.
- X. H. Pham, E. Hahm, K. H. Huynh, B. S. Son, H. M. Kim, D. H. Jeong and B. H. Jun, Int. J. Mol. Sci., 2019, 20, 4841 CrossRef CAS PubMed.
- S. Wang, B. Sun, J. Feng, F. An, N. Li, H. Wang and M. Tian, Anal. Methods, 2020, 12, 5657–5670 RSC.
- J. F. Li, J. R. Anema, T. Wandlowski and Z. Q. Tian, Chem. Soc. Rev., 2015, 44, 8399–8409 RSC.
- J. E. S. van der Hoeven, H. Gurunarayanan, M. Bransen, D. A. M. de Winter, P. E. de Jongh and A. van Blaaderen, Adv. Funct. Mater., 2022, 32, 2200148 CrossRef CAS.
- Q. Ye, J. Fang and L. Sun, J. Phys. Chem. B, 1997, 101, 8221–8224 CrossRef CAS.
- W. E. Doering and S. Nie, Anal. Chem., 2003, 75, 6171–6176 CrossRef CAS PubMed.
- S. P. Mulvaney, M. D. Musick, C. D. Keating and M. J. Natan, Langmuir, 2003, 19, 4784–4790 CrossRef CAS.
- Y. Liu and Z. Tang, Adv. Mater., 2013, 25, 5819–5825 CrossRef CAS PubMed.
- A. Castro-Grijalba, V. Montes-García, M. J. Cordero-Ferradás, E. Coronado, J. Pérez-Juste and I. Pastoriza-Santos, ACS Sens., 2020, 5, 693–702 CrossRef CAS PubMed.
- T. Shahar, T. Sicron and D. Mandler, Nano Res., 2017, 10, 1056–1063 CrossRef CAS.
- T. T. Nguyen, S. Lau-Truong, F. Mammeri and S. Ammar, Nanomaterials, 2020, 10, 294 CrossRef CAS PubMed.
- F. Bao, J. L. Yao and R. A. Gu, Langmuir, 2009, 25, 10782–10787 CrossRef CAS PubMed.
- Q. Zhang, J. Ge, J. Goebl, Y. Hu, Y. Sun and Y. Yin, Adv. Mater., 2010, 22, 1905–1909 CrossRef CAS PubMed.
- E. A. Kwizera, E. Chaffin, X. Shen, J. Chen, Q. Zou, Z. Wu, Z. Gai, S. Bhana, R. Oconnor, L. Wang, H. Adhikari, S. R. Mishra, Y. Wang and X. Huang, J. Phys. Chem. C, 2016, 120, 10530–10546 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, R. Deng, L. Yang, S. Yu, S. Xu and W. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 14160–14168 CrossRef CAS PubMed.
- V. Maurer, A. Zarinwall, Z. Wang, S. Wundrack, N. Wundrack, D. Ag Seleci, V. Helm, D. Otenko, C. Frank, F. Schaper, R. Stosch and G. Garnweitner, Sens. Diagn., 2022, 1, 469–484 RSC.
- H. Zhang, H. Lai, X. Wu, G. Li and Y. Hu, Anal. Chem., 2020, 92, 4607–4613 CrossRef CAS PubMed.
- Y. Weng, X. Hu, L. Jiang, Q. Shi and X. Wei, Anal. Bioanal. Chem., 2021, 413, 5419–5426 CrossRef CAS PubMed.
- X. Zhang, Z. Lei, Z. Pan, J. Hu, L. Tao, Z. Zheng, X. Feng, J. Xue, L. Tao and Y. Zhao, J. Alloys Compd., 2023, 938, 168468 CrossRef CAS.
- X. Jiang, X. Sun, D. Yin, X. Li, M. Yang, X. Han, L. Yang and B. Zhao, Phys. Chem. Chem. Phys., 2017, 19, 11212–11219 RSC.
- K. Abid, N. H. Belkhir, S. B. Jaber, R. Zribi, M. G. Donato, G. Di Marco, P. G. Gucciardi, G. Neri and R. Maâlej, J. Phys. Chem. C, 2020, 124, 20350–20358 CrossRef CAS.
- S. Almohammed, F. Zhang, B. J. Rodriguez and J. H. Rice, Sci. Rep., 2018, 8, 41–44 CrossRef PubMed.
- J. Zhao, Z. Wang, J. Lan, I. Khan, X. Ye, J. Wan, Y. Fei, S. Huang, S. Li and J. Kang, Nanoscale, 2021, 13, 8707–8721 RSC.
- A. Brognara, B. R. Bricchi, L. William, O. Brinza, M. Konstantakopoulou, A. L. Bassi, M. Ghidelli and N. Lidgi-Guigui, Small, 2022, 18, 2201088 CrossRef CAS PubMed.
- X. Tang, X. Fan, L. Yao, G. Li, M. Li, X. Zhao, Q. Hao and T. Qiu, J. Phys. Chem. Lett., 2022, 13, 7816–7823 CrossRef CAS PubMed.
- S. Jia, A. Bandyopadhyay, H. Kumar, J. Zhang, W. Wang, T. Zhai, V. B. Shenoy and J. Lou, Nanoscale, 2020, 12, 10723–10729 RSC.
- S. A. Ghopry, M. A. Alamri, R. Goul, R. Sakidja and J. Z. Wu, Adv. Opt. Mater., 2019, 7, 1–11 Search PubMed.
- Z. Cai, B. Liu, X. Zou and H. M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS PubMed.
- L. Xu, W. Q. Huang, W. Hu, K. Yang, B. X. Zhou, A. Pan and G. F. Huang, Chem. Mater., 2017, 29, 5504–5512 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |