
Uranium extraction from seawater: material design, emerging technologies and marine engineering
Yi
Xie
a,
Zeyu
Liu
b,
Yiyun
Geng
a,
Hao
Li
ac,
Ning
Wang
d,
Yanpei
Song
 e,
Xiaolin
Wang
*c,
Jing
Chen
a,
Jianchen
Wang
a,
Shengqian
Ma
*e and
Gang
Ye
*a
e,
Xiaolin
Wang
*c,
Jing
Chen
a,
Jianchen
Wang
a,
Shengqian
Ma
*e and
Gang
Ye
*a
aCollaborative Innovation Center of Advanced Nuclear Energy Technology, Institute of Nuclear and New Energy Technology, Tsinghua University, Beijing 100084, China. E-mail: yegang@mail.tsinghua.edu.cn
bAVIC Manufacturing Technology Institute, Beijing 100024, China
cChina Academy of Engineering Physics, Mianyang 621900, China
dState Key Laboratory of Marine Resource Utilization in South China Sea, Hainan University, Haikou 570228, China
eDepartment of Chemistry, University of North Texas, Denton, TX 76201, USA
First published on 30th November 2022
Abstract
Uranium extraction from seawater (UES), a potential approach to securing the long-term uranium supply and sustainability of nuclear energy, has experienced significant progress in the past decade. Promising adsorbents with record-high capacities have been developed by diverse innovative synthetic strategies, and scale-up marine field tests have been put forward by several countries. However, significant challenges remain in terms of the adsorbents’ properties in complex marine environments, deployment methods, and the economic viability of current UES systems. This review presents an up-to-date overview of the latest advancements in the UES field, highlighting new insights into the mechanistic basis of UES and the methodologies towards the function-oriented development of uranium adsorbents with high adsorption capacity, selectivity, biofouling resistance, and durability. A distinctive emphasis is placed on emerging electrochemical and photochemical strategies that have been employed to develop efficient UES systems. The most recent achievements in marine tests by the major countries are summarized. Challenges and perspectives related to the fundamental, technical, and engineering aspects of UES are discussed. This review is envisaged to inspire innovative ideas and bring technical solutions towards the development of technically and economically viable UES systems.
 Yi Xie | Yi Xie is currently studying for a PhD degree from the Institute of Nuclear and New Energy Technology (INET) at Tsinghua University, China. She received her BS and MS degrees from the Southwest University of Science and Technology in 2017 and the University of Science and Technology of China in 2020, respectively. Her research interests focus on the preparation and properties of porous organic materials and their application for uranium extraction from seawater. |
 Xiaolin Wang | Xiaolin Wang obtained his BS degree in Radiochemistry from Sichuan University, China, in 1985. Then, he joined the China Academy of Engineering Physics (CAEP), where he obtained his MS degree in Nuclear Chemistry and PhD degree in Nuclear Material in 1988 and 1997, respectively. Now he is a Full Professor of CAEP. His current research interests focus on nuclear chemistry, radioanalytical chemistry and nuclear materials. |
 Jianchen Wang | Jianchen Wang is a professor at the Institute of Nuclear and New Energy Technology (INET) at Tsinghua University, China. His research interests are in the areas of nuclear chemistry, separation chemistry in actinides and other nuclear fission elements. As one of the contributors, he set up the Chinese partitioning process for high-level liquid waste (HLLW), including the trialkyl phosphine oxide (TRPO) process to separate actinides, the crown ether process to separate strontium, and the calixcrown process to separate cesium from HLLW. As the author and co-author, he has published more than 70 articles and holds over 40 patents. |
 Shengqian Ma | Shengqian Ma obtained his BS degree from Jilin University in 2003, and graduated from Miami University with a PhD degree in 2008. After finishing a two-year Director's Postdoctoral Fellowship at Argonne National Laboratory, he joined the University of South Florida as an Assistant Professor in 2010, and was promoted to Associate Professor in 2015 and Full Professor in 2018. In August 2020, he joined the University of North Texas as the Robert A. Welch Chair in Chemistry. His current research interests focus on the development of functional porous materials including metal–organic frameworks, covalent organic frameworks, and porous organic polymers for energy/biological/environmental-related applications. |
 Gang Ye | Gang Ye received his BS and PhD from the Department of Chemical Engineering (DCE), Tsinghua University, in 2005 and 2010, respectively. He was then qualified to join the Institute of Nuclear and New Energy Technology (INET), Tsinghua University, as an Assistant Professor. He visited the Department of Chemical and Environmental Engineering, Yale University (M. Elimelech's group), in 2014. He is currently a tenured Associate Professor at INET, Tsinghua University. He has broad research interests in materials chemistry, and is particularly passionate about the function-oriented development of advanced adsorbents and membranes for challenging chemical separations and environment-related applications. |
1. Introduction
The transition towards a deeply decarbonized and sustainable energy system has become a defining challenge for human society in the 21st century.1 An increasingly tight global supply of traditional non-renewable energy supplies (e.g., oil, coal and natural gas) has provided an incentive for countries worldwide to develop new technologies to secure the energy supply.2 To exploit alternative sources to fossil fuels, renewable energy sources such as wind, solar, tidal and geothermal power have set off a boom in recent years.3 However, these kinds of green energy are still subject to constraints in terms of reliability, economy and environmental impact. By contrast, nuclear fission has been universally acknowledged as a mature and safe technology that can produce energy with consistent output and minimal carbon footprints. The vigorous development of nuclear energy has made a vital contribution towards global low-carbon energy needs.4Uranium (U), as an essential resource for nuclear energy production, is of strategic importance to the sustainability of the global nuclear industry.5 In recent years, the increasing scale of nuclear power has driven significant growth in reactor-related uranium demand. The dwindling availability of terrestrial uranium reserves, which are estimated to afford only about a century of consumption,6 thus has motivated considerable interest in identifying and utilizing unconventional uranium reserves. Uranium extraction from seawater (UES) has been highlighted as a potential approach to this end.7,8 There is about 4.5 billion tU in the ocean, nearly 1000 times more plentiful than the estimated amount of terrestrial uranium reserves (7.6 million tU) at present.9 This reserve presents a huge alternative reserve that can meet at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years of global demand for uranium resources, provided that the development of chemical separation techniques allows an economically viable recovery. Meanwhile, an additional advantage of UES lies in the environmental benefit, which has a less negative impact on ecological systems as compared with terrestrial uranium mining.
000 years of global demand for uranium resources, provided that the development of chemical separation techniques allows an economically viable recovery. Meanwhile, an additional advantage of UES lies in the environmental benefit, which has a less negative impact on ecological systems as compared with terrestrial uranium mining.
Natural seawater is a complex biogeochemical system with an ultra-low concentration of U (∼3.3 ppb), accompanied by a great amount of competing metal ions, high salinity, specific pH conditions, and substantial marine biofouling,10 making UES a rather challenging task. To enrich the extremely dilute U from the vast oceans, an energy-saving and cost-efficient method is required. Among the well-established chemical separation techniques,11 adsorption has been demonstrated to be an adaptable and reliable approach with environmental friendliness for marine uranium enrichment.12 As with every new technology that begins to gain widespread adoption, innovations in the design and optimization of new materials play a critical role in the advancement and development of this technology.13 Accordingly, the core job of developing adsorption techniques for UES has been focused on exploring high-performance adsorbent materials for uranium capture. The challenge as it stands is that adsorbent materials should have robust structures and exhibit remarkable properties, including adsorption capability, selectivity, biofouling resistance, and durability in seawater.14,15 Undoubtedly, the deliberate design and development of adsorbent materials are of central importance to guarantee the stable harvesting of marine uranium while accelerating the commercialization of UES.12
The origin of UES dates back to the “Project Oyster” initiated by the United Kingdom in the early 1950s. Since the first public report by Davies et al. in 1964,16 UES has spurred considerable interest among research entities worldwide. Initially, numerous studies were performed to screen potential adsorbent materials. Inorganic adsorbents, particularly hydrous titanium dioxide, were one of the earliest materials that drew attention.9,17–21 These materials were ruled out in the 1980s due to their minimal adsorption capacity and poor mechanical resistance. The focus has then been turned to synthetic polymers, which represent the most promising adsorbents applicable for the large-scale marine test of UES. Specifically, the development of amidoxime (AO) derived polymers, ‘state-of-the-art materials’ for UES, has aroused a research upsurge that has continued over the past decades.22–29 Up to now, ongoing efforts have been made to construct AO-based polymeric adsorbents with improved properties. Meanwhile, emerging nanostructured materials in recent years, such as porous carbon materials, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), porous organic polymers (POPs), porous aromatic frameworks (PAFs) and genetically-engineered proteins, have opened up a new avenue for the development of potential uranium adsorbents.30–39 Overall, with the evolution of adsorbent materials, significant achievements of UES, in terms of fundamental studies and marine engineering, have been made by scientists worldwide (Fig. 1). In particular, the past decade has been regarded as a ‘Golden Age’ of UES, as evidenced by the abundance of adsorbents studied in the literature and a series of milestone events in marine field tests.
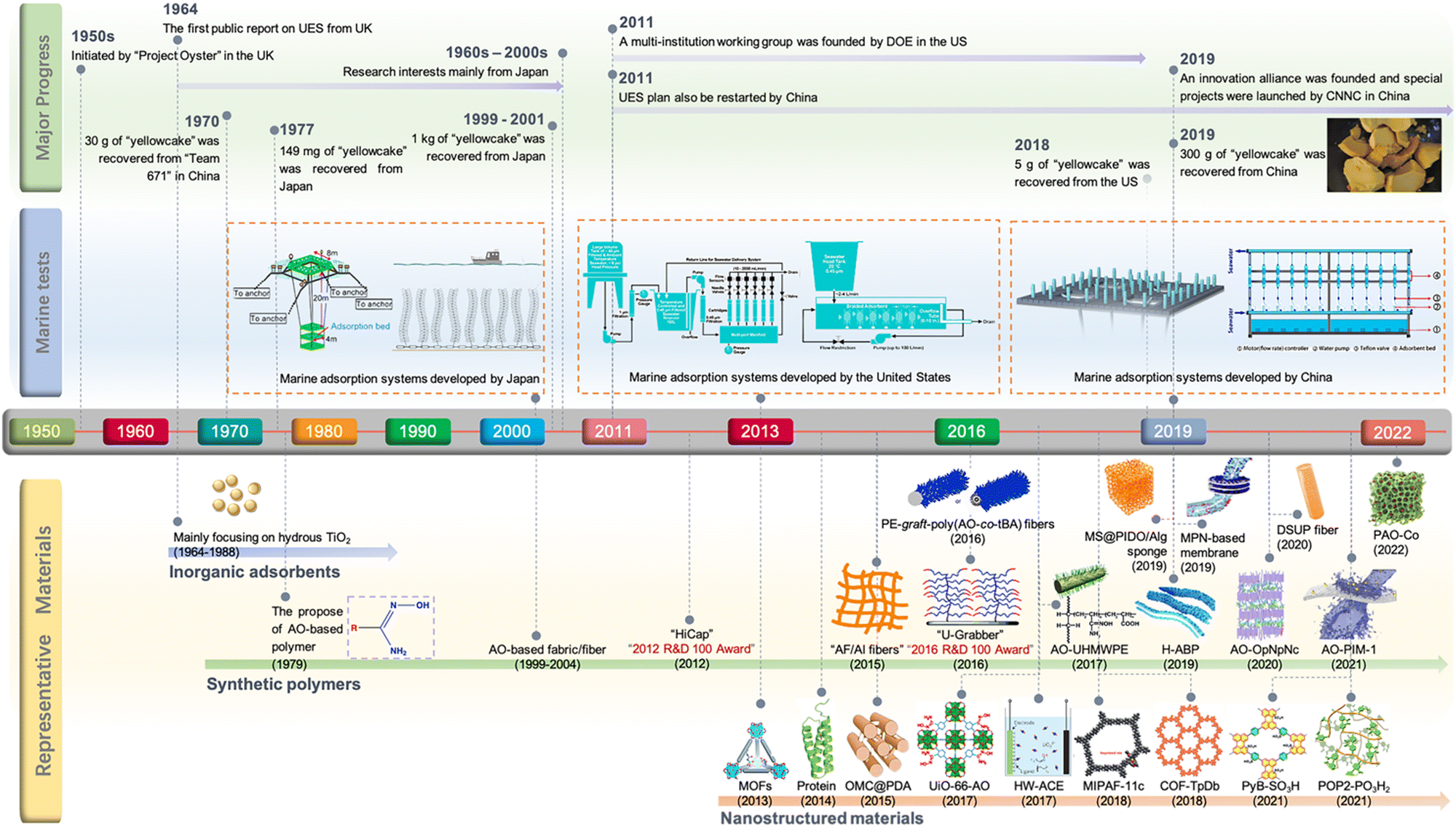 | ||
| Fig. 1 The development of UES presented in the form of a timeline of the major advances in milestone events. The schematic diagrams of adsorption platforms and materials were reproduced from the literature with copyright permissions from the respective publishers. They include: MOFs,32 U-Grabber,75 H-ABP,132 AO-OpNpNc,133 MPN-based membranes,144 PyB-SO3H,252 Royal Society of Chemistry; AF/AI fibers,110,111 PE-graft-poly(AO-co-tBA) fibers,76 OMC@PDA,39 UiO-66-AO,82 American Chemical Society; AO-UHMWPE,69 AO-PIM-1,145 Protein,178 HW-ACE,276 Springer Nature; MS@PIDO/Alg sponge,134 DSUP fibers,141 PAO-Co,245 MIPAF-11c,171 COF-TpDb,83 POP2-PO3H2,193 Wiley-VCH; marine adsorption systems developed by Japan,7 Elsevier; marine adsorption systems developed by the United States,155 American Chemical Society; and marine adsorption systems developed by China,137 Wiley-VCH. | ||
Several reviews summarizing the research on UES were published in the past decade. However, most of the reviews merely discussed one specific class of adsorbents (e.g., carbon materials,31 AO-based materials,25 and magnetic nanoparticles40) or just focused on their solution chemistry aspects,41 rather than presenting a comprehensive landscape of UES. A high-level overview was provided by Abney and co-workers,10 but primarily focused on the miscellaneous adsorbents developed from 2000 to 2016 without discussing the engineering aspects. In the past five years, the drive for new materials has generated a staggering number of research studies dealing with high-performance adsorbents for UES. According to the retrieval from the Web of Science database, more than 300 articles referring to UES have been published from 2017 to 2022. A notable trend is that novel electrochemical and photochemical approaches have been exploited to boost UES. Meanwhile, considerable achievements in UES have been made by locating adaptable materials and building infrastructures for scale-up marine tests. This urges a timely review to present the state-of-the-art of UES, with an emphasis on the updated insights into the design philosophy of materials and the emerging strategies to develop advanced UES systems. In addition, the latest achievements made in marine field tests are encouraging while suggesting technical and economic obstacles before we can be optimistic about the commercialization of UES. Overall, an up-to-date and comprehensive overview will expand our knowledge and understanding of the critical issues in this field, in terms of scientific, engineering, and commercial feasibility, which is envisaged to inspire innovative ideas and bring technical solutions towards a promising future for UES.
Thus, we will provide in the following a comprehensive summary of the advancements in the field of UES, highlighting the latest design philosophy of adsorbent materials and the emerging strategies towards more efficient UES systems in the past five years (Fig. 2). To rationally design effective adsorbents for uranium harvesting from the oceans, identifying the dominant species of uranium and their solvation structures under realistic seawater conditions is a prerequisite.42–44 Therefore, we will first start with a concise yet insightful introduction of the solution coordination chemistry of uranium in seawater. We will then focus on summarising the state-of-the-art strategies for the function-oriented design and synthesis of uranium adsorbents with elevated performance, aiming to provide insights into the development of next-generation uranium adsorbents. This will be followed by the introduction of emerging electrochemical and photochemical strategies that have been exploited for marine uranium harvesting. These non-passive strategies are envisioned to inspire the development of more efficient UES systems. In addition, we will present an overview of the implementation of marine tests in major countries including Japan, the United States, and China, which contributes valuable insights from an engineering point of view. Finally, we will take a deep dive into the challenges facing the further development of UES, and conclude this review with the opportunities and perspectives of UES in hopes of inspiring more advancements in this prospective field.
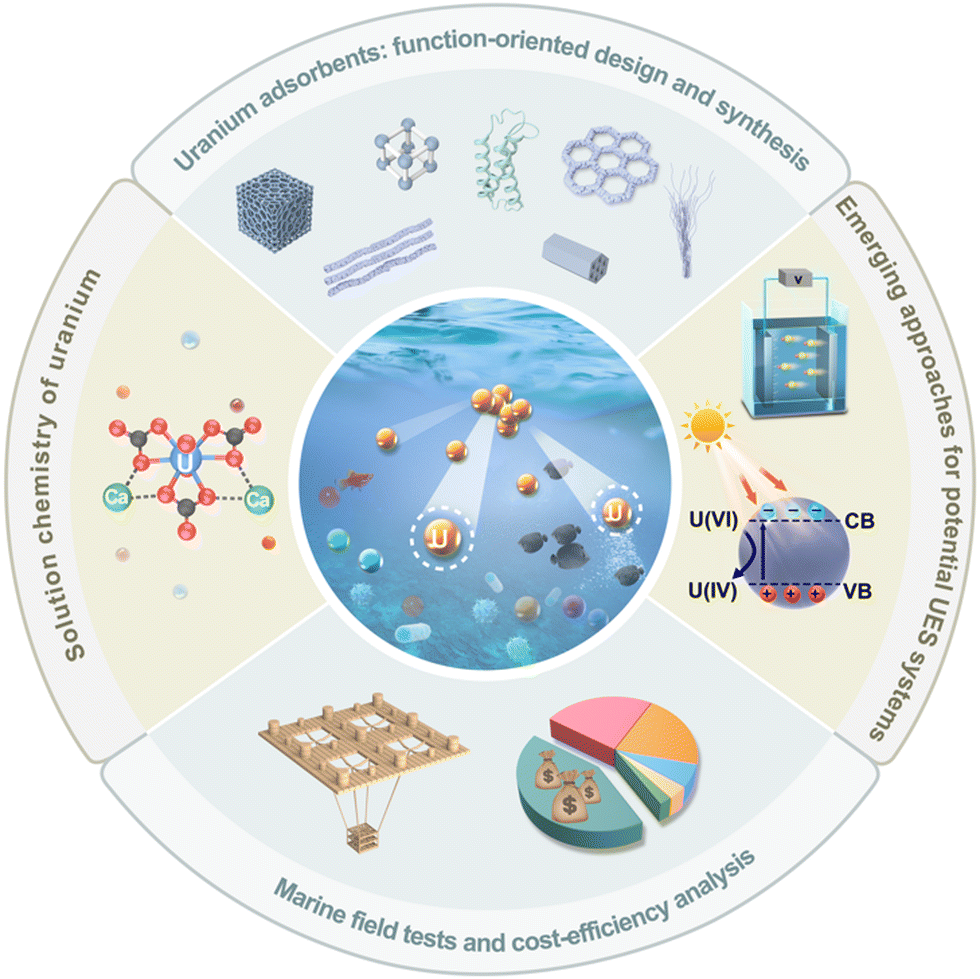 | ||
| Fig. 2 Overview of topics discussed in this review. | ||
2. Solution coordination chemistry of uranium in seawater
A molecular-level understanding of the solution coordination chemistry of uranium in seawater is an essential requirement for the development of achievable adsorbent materials.45 In this regard, thermodynamic studies, theoretical calculations and spectroscopic techniques43,44,46,47 are the primary approaches to identifying the speciation of uranium in seawater while investigating the coordination behaviours between uranium and various ligands (Fig. 3).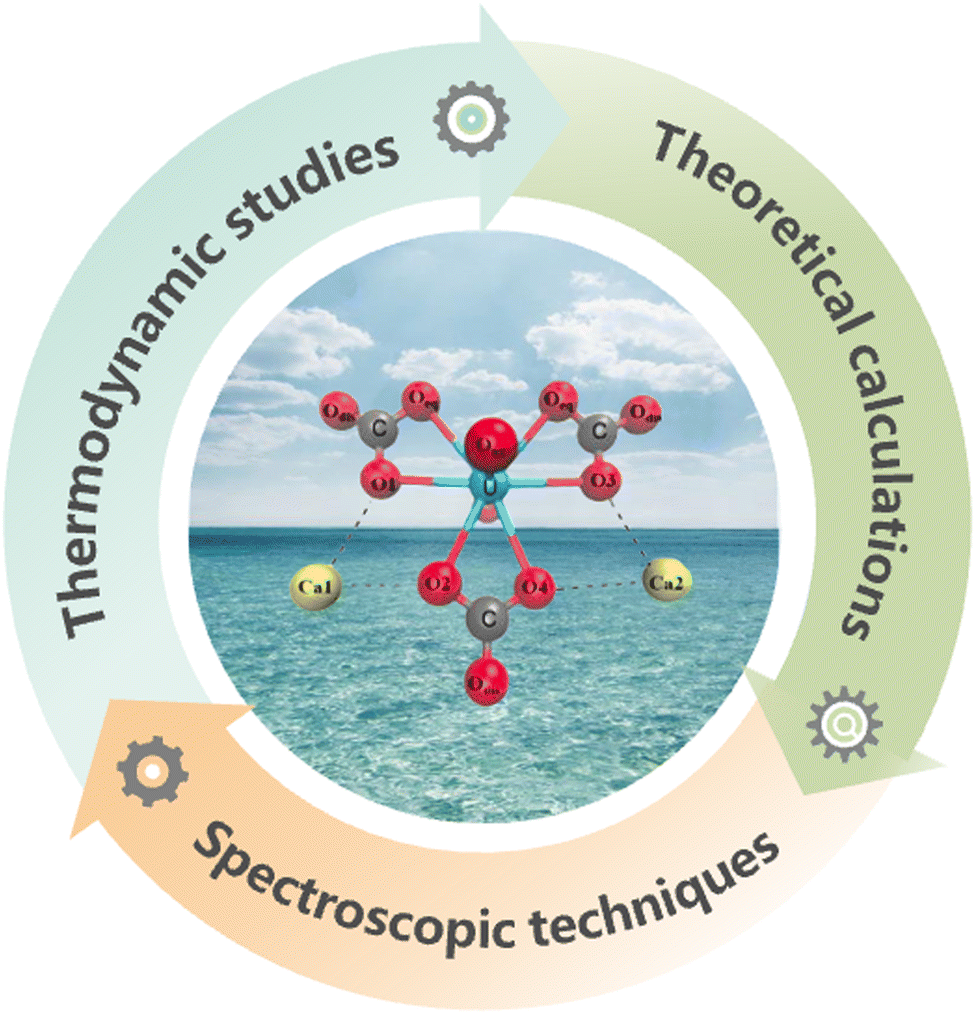 | ||
| Fig. 3 Summary of the approaches used for investigating the speciation of uranium in seawater. | ||
It has been found that more than 90% of the uranium in seawater exists in the stable ionic form of [UO2(CO3)3]4− and the neutrally charged derivative form of Ca2UO2(CO3)3.42 The uranyl ion ([UO2]2+), as the basic form of uranium, has a linear, center-symmetric structure with both U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths of about 180 pm. The double bonds between the uranium center and oxygen atoms are chemically inert, making uranyl an exceptionally stable molecular species. The perturbations of the O–U–O angle in [UO2]2+ are usually small (about 5°) and multiple ligands may coordinate with the uranyl ion in an equatorial plane around the uranium center.48
O bond lengths of about 180 pm. The double bonds between the uranium center and oxygen atoms are chemically inert, making uranyl an exceptionally stable molecular species. The perturbations of the O–U–O angle in [UO2]2+ are usually small (about 5°) and multiple ligands may coordinate with the uranyl ion in an equatorial plane around the uranium center.48
As a member of actinides, uranium is a strong electron acceptor and can be considered as a kind of hard acid according to the Hard–Soft-Acid–Base theory.49 Because of the electronic effect, the introduction of electron-donating groups can enhance the complexation ability between ligands and the uranyl. In addition, steric hindrance and weak interactions (e.g., hydrogen bond interaction, hydrophilic interaction, and hydrophobic interaction) in the coordination environment affect the stability of the uranyl complexes. Specifically, the extra hydrogen bonds can increase the bond length of UO, while shortening the length of the bonds between the coordination atom and the uranium in the near-equatorial plane.50
Integrating the solution chemistry of uranium with density functional theory (DFT) calculations provides a viable approach for the design and screening of potential uranyl ligands. This significantly promotes the development of various promising adsorbents, especially for prevailing organic framework materials (e.g., MOFs, COFs, POPs, and PAFs) with built-in functionalities and structural diversity. Meanwhile, the solution chemistry and the ligand-binding behaviours of competing ions in seawater, such as vanadium (V), are also worthy of attention. Substantial investigations in this regard have provided a comprehensive understanding of the coordination behaviours in marine environments, which has implications for the development of novel uranium adsorbents with better selectivity.
3. Uranium adsorbents: function-oriented design and synthesis
As aforementioned, adsorption is recognized as the most promising technique for UES because of its adaptability to marine engineering with minimum energy consumption. Researchers worldwide have achieved significant progress in this area by developing various adsorbents to harvest uranium in oceans. Considerable efforts have been made to screen potential candidates while improving their (1) adsorption ability, (2) selectivity, (3) biofouling resistance and (4) durability in seawater. In this section, the state-of-the-art strategies towards the function-oriented design and synthesis of uranium adsorbents with elevated performance will be comprehensively summarized, aiming to provide insights into the development of next-generation uranium adsorbents. Emphasis will be put on the design philosophies and inspiring strategies. Meanwhile, the underlying interaction mechanism responsible for the effective binding of uranium under marine conditions will also be discussed.The section is organized into four subsections, highlighting each of the functions outlined above (Fig. 4). Prior to this, it should be noted that due to the large quantity of literature, only milestone studies and impressive achievements in the past five years will be highlighted, rather than a detailed description of each reported material. Moreover, this review only concerns the materials developed for uranium extraction from environmental seawater; that is, it does not cover the investigations that deal with uranium only from aqueous solutions without considering seawater conditions.
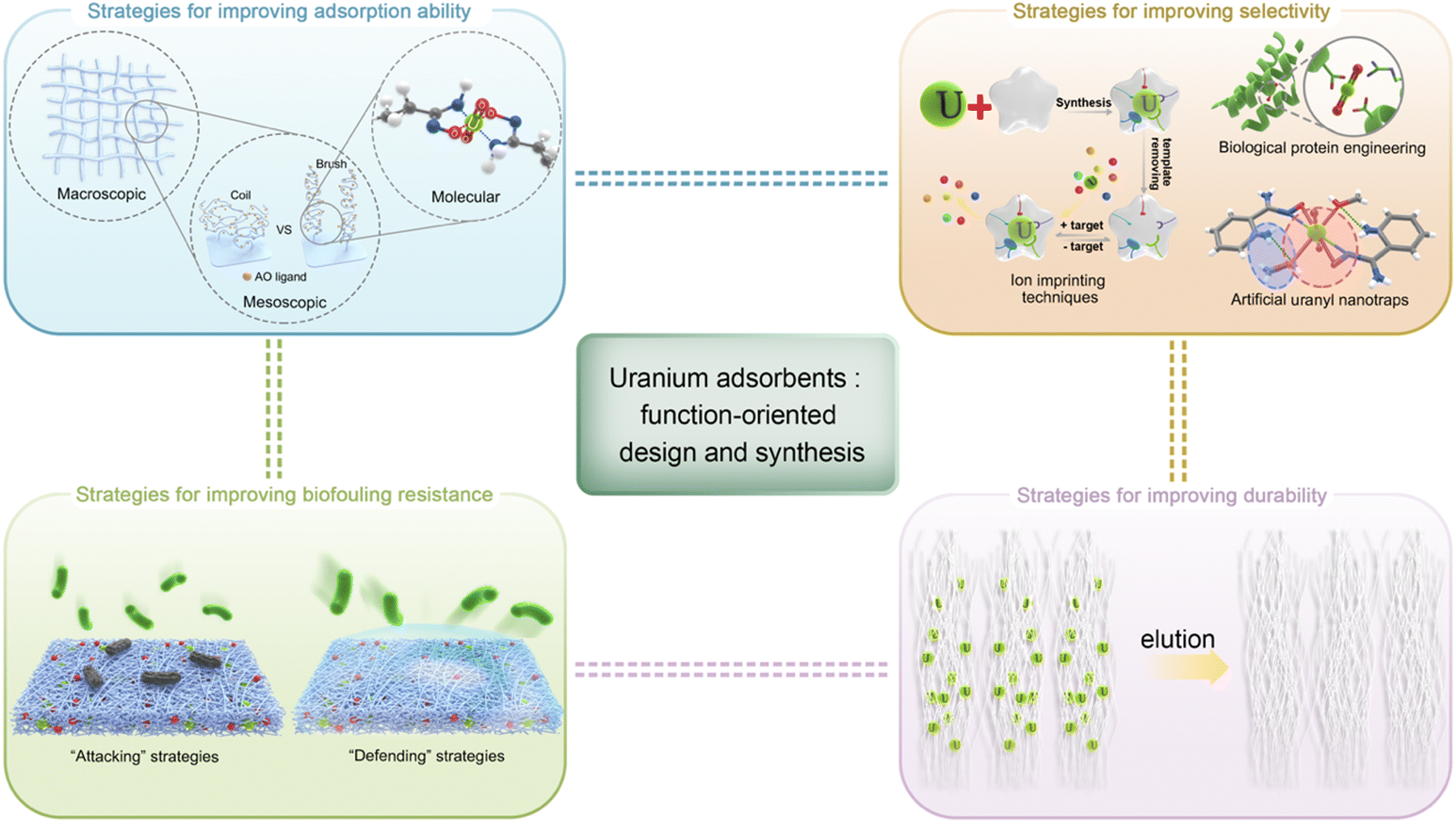 | ||
| Fig. 4 Overview of strategies for the function-oriented development of uranium adsorbents. | ||
3.1. Strategies for improving the adsorption ability
Strong adsorption towards U with a high capacity is the foremost important property of a potential adsorbent for UES. As the adsorption capacity towards uranium has been considered to be the main cost driver of UES, it has always been a top priority to improve the uranium uptake capability of adsorbent materials. In this subsection, based on an updated mechanistic understanding of the adsorption behaviours of well-developed adsorbents at the molecular, mesoscopic, and macroscopic levels, the strategies established for improving the adsorption ability of uranium adsorbents will be elaborated. Specifically, the methods to promote the ligand–uranyl interactions, regulate the spatial conformation of polymeric adsorbents and improve the structural features of uranium adsorbents will be discussed.Firstly, due to the high affinity towards uranyl ions in a chelating coordination mode, amidoxime (AO) has been identified as one of the most effective ligands of uranium.51–54 A considerable number of AO-containing adsorbent materials, especially those with polymer matrixes, have been developed for UES.25 The most studied AO-based polymers are generally prepared by grafting acrylonitrile (AN) monomers onto polyolefin supports with robust backbones, such as polyethylene (PE) and polypropylene (PP) (Fig. 5A). The classical radiation-induced graft polymerization (RIGP) method is widely employed to this end,26,55,56 followed by post-amidoximation to convert AN groups to AO ligands (Fig. 5B).57 The degree of grafting (DOG) is thus a direct reflection of the number of ligands, which is influenced by irradiation parameters (e.g., irradiation dose, dose rate) and grafting reaction conditions (e.g., atmosphere, retention time, temperature) during the RIGP process.58–60
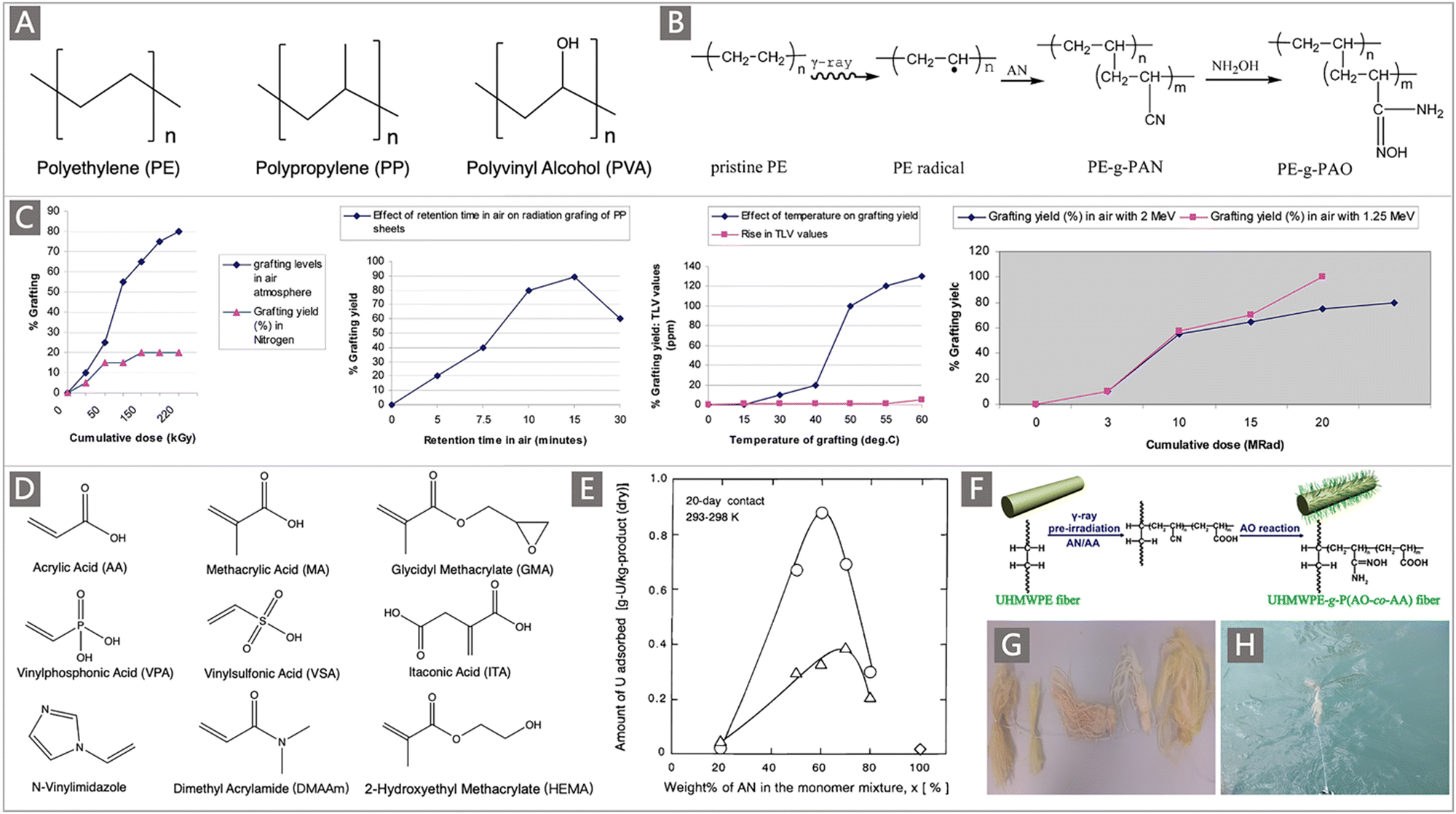 | ||
| Fig. 5 (A) The commonly used trunk materials for the preparation of uranium adsorbents by RIGP. (B) A typical scheme depicting the preparation of AO adsorbents by RIGP of AN and subsequent amidoximation. Reproduced with permission.57 Copyright 2012, American Chemical Society. (C) The DOG of PAN as a function of reradiation dose, atmosphere, temperature and retention time. Reproduced with permission.58 Copyright 2010, American Chemical Society. (D) A subset of comonomers studied for the preparation of uranium adsorbents using RIGP. (E) The uranium adsorption amount as a function of wt.% of AN in the monomer mixture. Circles correspond to MA-co-grafted PAO PE fibers, triangles correspond to HEMA-co-grafted-PAO PE fibers, and diamonds correspond to the bare PAO-graft-PE adsorbent. Reproduced with permission.65 Copyright 2000, American Chemical Society. (F) Schematic diagram of the preparation process of UHMWPE-g-P(AO-co-AA) fibers. Reproduced with permission.69 Copyright 2017, Springer Nature. (G) Photographs of the resultant AO-UHMWPE fibers and their floating in the seawater (H). Reproduced with permission.71 Copyright 2016, American Chemical Society. | ||
The Prasad group provided a detailed investigation of the effect of parameters including the cumulative dose, surrounding atmosphere, reaction temperature and duration on the DOG of AO polymers on the surfaces of PP fibers.58 The results showed that an increased DOG was obtained with the increase of cumulative dose (Fig. 5C). When irradiated in air, the DOG can reach 80 wt% around the cumulative dose of 220 kGy, and was still on the rise. However, when irradiated in an N2 atmosphere, the DOG was only up to 25 wt%. The DOG increased to 90 wt% when the irradiated PP sheet was exposed to the air for 15 min and then contacted with the monomer to induce polymerization. At the same time, a DOG of 130 wt% was obtained when the temperature of the grafting polymerization was raised to 60 °C. Besides, the effect of cumulative dose on the DOG was also varied with the energy beam. The as-prepared AO-functionalized PP sheet with a DOG of 110 wt% was subsequently tested in a mooring facility located in the Trombay estuary, with a uranium adsorption capacity (qU) of 0.61 mg g−1 obtained over an adsorption period of 13 d at the pilot scale.
In addition to the irradiation parameters, the DOG is also influenced by the feed monomer and comonomer ratio.60–67 Earlier studies suggested that comonomers such as acrylic acid (AA), methacrylic acid (MA), and glycidyl methacrylate (GMA) (Fig. 5D) had a certain influence on the adsorption performance of uranium adsorbents. So, continuous research has been performed to select suitable comonomers and elucidate the underlying relationship governing the adsorption capacity of the adsorbents.61,62,64,65
Choi and Nho64 conducted comprehensive studies on the graft copolymerization process of monomers including AA, MA, GMA and AN onto PE films. It was found that the increase of the AN component led to an increased content of AN in the AN:AA and AN:GMA copolymers, while the AN component of the AN:MA copolymer was almost independent of the AN component of the feed monomer. The maximum DOG of 125 wt% was obtained under the conditions of a feed ratio of AN:AA (40:60, mol%). Nevertheless, a subsequent adsorption study revealed that the polymer with a monomer ratio of AN:AA (50:50, mol%) was determined to acquire a maximum uptake of uranium.61 Saito and co-workers compared the adsorption performances of adsorbents prepared by RIGP of AN with two different hydrophilic comonomers (MA and 2-hydroxyethyl methacrylate (HEMA)) from a PE matrix.65 Varying the contents of the comonomers endowed adsorbents with different hydrophilicities and adsorption capability. The MA copolymer was found to exhibit a higher adsorption rate than the HEMA copolymer (Fig. 5E). Lab-based flow-through experiments and marine test in a submerged mode at an ocean site were both carried out to investigate the uranium adsorption performance, where a 60:40 AO:MA copolymer on a fibrous PE trunk was observed to achieve the optimal recovery, with a qU of 0.9 mg g−1 following 20 d deployment in the open ocean.
The nature of substrate materials subject to radiation for radical formation also plays a role in achieving higher DOG while exerting an influence on the ligand–uranyl interaction. To overcome the poor mechanical properties demonstrated by conventional polymeric substrates after irradiation,61,68 an ultra-high molecular weight polyethylene (UHMWPE) was utilized as a support and was grafted with AN and AA to prepare UHMWPE-g-(PAO-co-PAA) fibers through the RIGP approach (Fig. 5F).69–72 The DOG of γ-ray irradiated UHMWPE fibers (UHMWPE-g-(PAN-co-PAA)) can reach higher than 300 wt% after 3 h of graft polymerization with an absorbed dose of 50 kGy. After amidoximation treatment, the resultant UHMWPE-g-(PAO-co-PAA) fibers achieved a qU of 2.3 mg g−1 in the filtered seawater from Sequim Bay, Washington, US, over 42 d.70 However, the qU decreased to 0.25 mg g−1 when the fibers were deployed in the East China Sea for 60 d (Fig. 5G and H), which was ascribed to the competitive ions and biofouling in a real marine environment.71 In addition, when the grafting monomers on UHMWPE fibers were changed to GMA and MA, the maximum grafting rate could be improved to 553 wt% by adjusting the monomer concentration and radiation dose.73 The high DOG obtained by using UHMWPE fibers as the host was attributed to the high stability and a long half-life (26 d in a vacuum, 13 d in air) of free radicals generated in the fibers under irradiation.74
Atom-transfer radical polymerization (ATRP) represents a significant milestone in the development of polymer chemistry. The application of this controlled/living radical polymerization (CRP) technique broadens horizons for uranium adsorbent synthesis with record-setting performances.75–78 In particular, ATRP allows for the incorporation of monomers which are not suitable for RIGP due to radical quenching and is especially well-suited for the control polymerization of AN.79 A pioneering study utilizing ATRP to prepare a uranium adsorbent was reported by Saito and co-workers. Poly(vinylbenzyl chloride, VBC) (Fig. 6A) was first grafted onto a PE trunk through RIGP to install ATRP initiation sites. A fiber adsorbent PE-g-PVBC-g-(PAN-co-PtBA) with a “brush-on-brush” structure was then created by copolymerizing AN and tert-butyl acrylate (t-BA) (Fig. 6B) through ATRP, followed by amidoximation.75 Considerably high DOG ranging from 585 to 2818 wt% could be achieved by tuning the AN:tBA feed ratio while optimizing the synthetic conditions (e.g., reaction time, solvent, catalyst and temperature) in the ATRP process. The obtained adsorbents exhibited a qU of 141–179 mg g−1 when immersed in a 6 ppm uranium brine solution, and a qU of 0.99–1.56 mg g−1 when packed in a column adsorption system for uranium enrichment in real seawater. More significantly, by chain-extension of the grating polymer chains to further introduce the hydrophilic block of PtBA in the ATRP system, followed by hydrolysis to obtain the PE-g-PVBC-g-(PAO-co-PAA)-b-PAA adsorbent, the uranium uptake capacity in real seawater was doubled (qU ∼3.02 mg g−1) after 56 d of exposure. These results motivated the focus on the exploration of the polymer chain conformation and optimization of hydrophilic comonomers in the development of uranium adsorbents, which will be elaborated in Section 3.1.2.
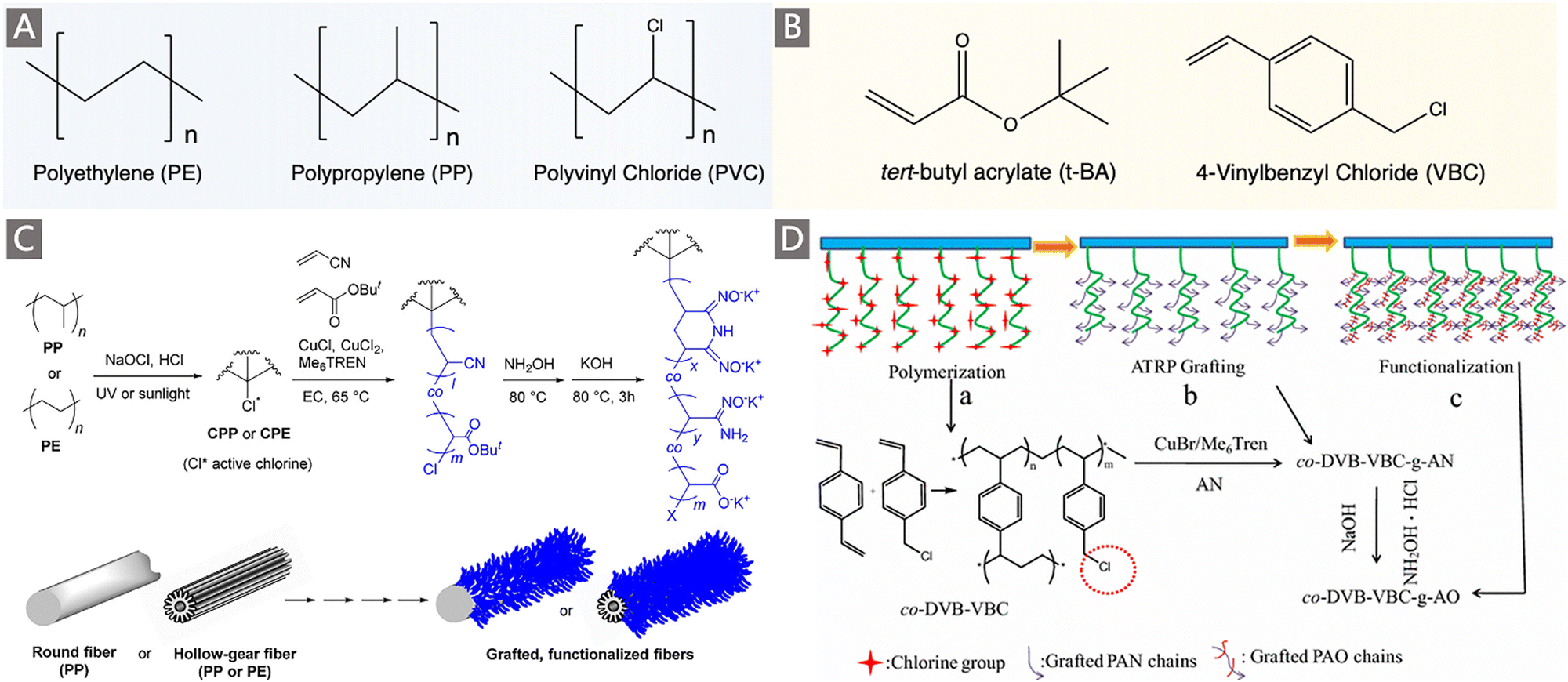 | ||
| Fig. 6 (A) The commonly used trunk materials for the preparation of uranium adsorbents by ATRP. (B) Two commonly used comonomers for the preparation of uranium adsorbents by ATRP. (C) A scheme depicting the preparation of CPE/CPP-g-poly(AO-co-tBA) fibers by ATRP. Reproduced with permission.76 Copyright 2016, American Chemical Society. (D) A scheme depicting the preparation of poly-DVB-VBC-g-AO resin adsorbents. Reproduced with permission.81 Copyright 2016, Springer Nature. | ||
Building on this work, Brown et al. reported the synthesis of adsorbents through ATRP without RIGP assistance.76 Especially, new trunk fibers namely chlorinated PE (CPE) and PP (CPP) fibers with various shapes were employed to afford ATRP initiation sites (Fig. 6C). Then, AN and tBA were successfully grafted onto the halogenated PE via ATRP copolymerization, and amidoximation was completed through the common approach. By changing the structure of active chlorine sites on trunk fibers and ATRP conditions such as the AN:tBA:RCl ratio and chlorination time, varied DOG values were yielded, and a high value of 2570 wt% was achieved. When immersed in 750 mL of simulated seawater containing 5–7 ppm added U, the maximum adsorption capacity of 146.6 mg g−1 was obtained. In a concurrent report from the same group, a poly-(vinyl chloride)-co-chlorinated poly(vinyl chloride) (PVC-co-CPVC) fiber was used as a backbone to graft AN and tBA via ATRP copolymerization.80 The optimal functional fiber with the tBA/AN ratio of 0.356 and a high DOG of 1390 wt% exhibited a recovery capacity of 5.22 mg g−1 after 49 d of exposure to seawater.
Additionally, ATRP demonstrated its versatility in the preparation of PAO functionalized mesoporous polymer adsorbents.79,81 Typically, mesoporous polymer monoliths containing chlorinated initiation sites were first synthesized through a free radical polymerization of VBC in the presence of DVB as the crosslinking agent. Then, ATRP of AN was initiated from the chloride in the poly(VBC-co-DVB), and an amidoximation reaction was performed to generate AO ligands (Fig. 6D).81 Likewise, the DOG of PAN, as well as the surface area and pore architectures of the final poly(VBC-co-DVB), could be tuned as a function of initiator density, catalyst concentration and the DVB:VBC ratio during the ATRP process. While the adsorbent with a 1:1 DVB:VBC achieved the highest DOG of PAN, the material synthesized with 2:1 DVB:VBC exhibited the highest adsorption capacity for uranium, indicating the important role of the structural properties of adsorbents, which will be elaborately discussed in Section 3.1.3. Finally, a qU of 75 mg g−1 was observed when tested in a 100 ppm uranium-spiked solution, while a dramatic decrease of uranium uptake (qU ∼2 mg g−1) was observed when the uranium solution was reduced to 10 ppb.
In addition to developing AO-functionalized polyolefin polymers, scientists have also made considerable efforts to exploit the expanding family of porous organic frameworks, such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), porous organic polymers (POPs) and porous aromatic frameworks (PAFs), to be potential uranium adsorbents in recent years.37,82–92 The flexible molecular design, programmable pore structures, broad chemical and reticular/topological variety, and especially ultrahigh surface area of these porous organic materials provide excellent opportunities to introduce a high density of chelating groups to promote the ligand–uranyl interaction.30 Typically, Wang and colleagues performed the first use of AO as a functional ligand of classical UiO-66 MOF material for UES.82 The adsorbent was prepared by a microwave-assisted cyanation reaction, followed by amidoximation treatment with hydroxylamine hydrochloride under basic conditions (Fig. 7A). The resultant UiO-66-CN and UiO-66-AO exhibited a high surface area of 878 m2 g−1 and 711 m2 g−1, respectively. Rapid adsorption of UiO-66-AO was observed in a kinetic investigation, with almost 99.0% of the total uranium adsorption achieved within 10 min from real seawater containing an extra 500 ppb U, and 94.8% of U could be recovered within 120 min from real seawater. Although the DOG of AO in the as-prepared UiO-66-AO was only 19.1 wt%, it afforded a qU of 2.68 mg g−1 in the seawater from the Bohai Sea within 3 d.
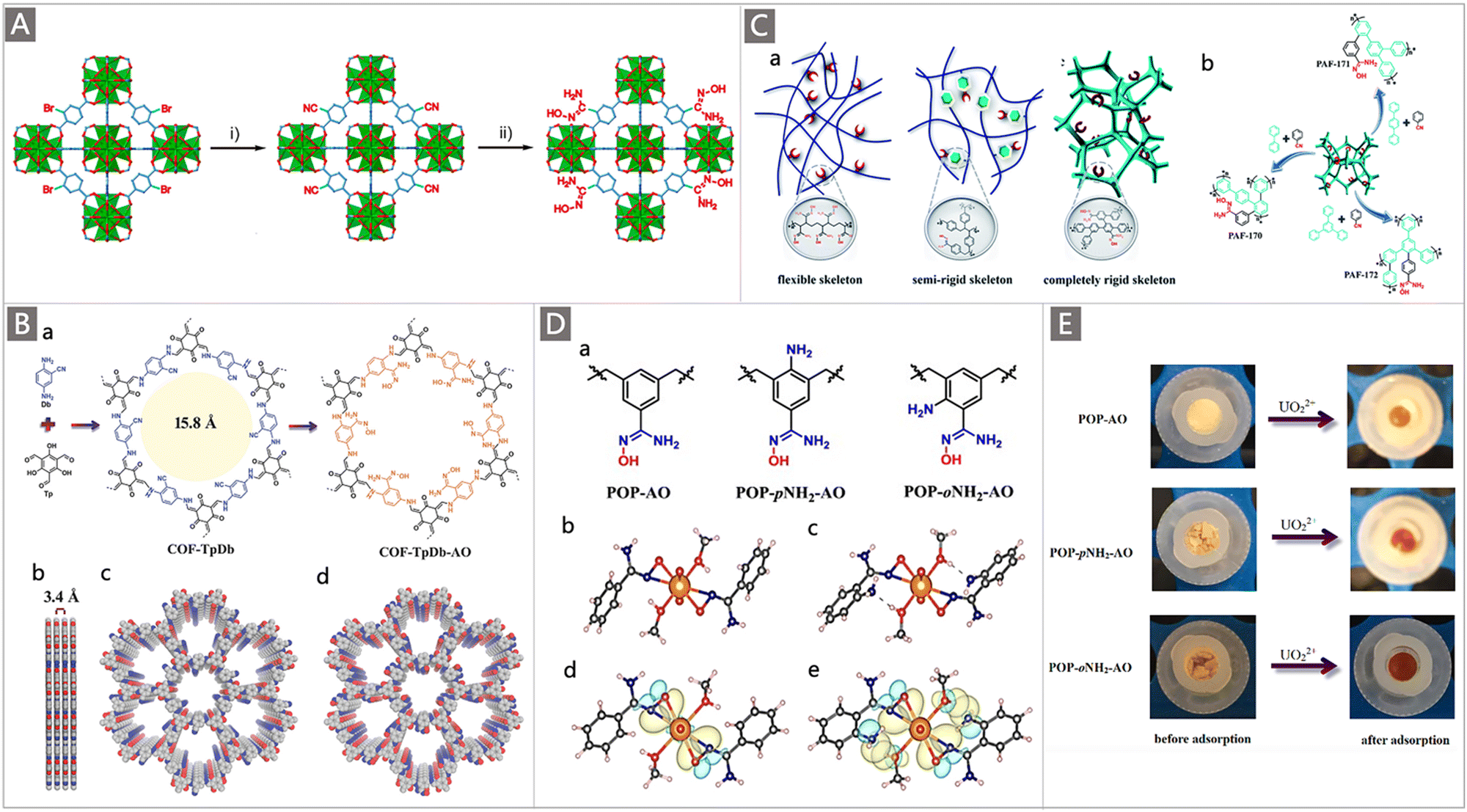 | ||
| Fig. 7 (A) Synthetic route of UiO-66-AO: (i) the synthesis of UiO-66-AN by microwave-assisted cyanation in the presence of CuCN; and (ii) the conversion of UiO-66-AN to UiO-66-AO through hydroxylamine hydrochloride treatment. Reproduced with permission.82 Copyright 2017, American Chemical Society. (B) Synthetic route of COF-TpDb-AO: (a) the condensation of Db with Tp and the corresponding transformation process of the cyano to AO group; and graphic view of the eclipsed AA stacking structure of COF-TpDb (b and c) and graphic view of COF-TpDb-AO (d). Reproduced with permission.83 Copyright 2018, Wiley-VCH. (C) (a) Schematic representation of AO-modified adsorbents including a flexible skeleton, semi-rigid skeleton, and completely rigid skeleton; and (b) scheme of synthesis for PAF-170-AO, PAF-171-AO and PAF-172-AO adsorbents. Reproduced with permission.88 Copyright 2020, Royal Society of Chemistry. (D) (a) Structure of building units of POP-AO, POP-oNH2-AO and POP-pNH2-AO polymers; single crystal structures and the corresponding DFT optimized structures of UO2(AO)2(MeOH)2 (b and d) and UO2(oNH2-AO)2(MeOH)2 (c and e). Reproduced with permission.87 Copyright 2018, Springer Nature. (E) Photos of various adsorbent materials before and after uranium inclusion. Reproduced with permission.87 Copyright 2018, Springer Nature. | ||
In comparison to MOFs, COFs have more robust structures supported by covalent bonds, making them a favourable platform to accommodate AO ligands for uranium capture from seawater. A two-dimensional (2D) COF-TpDb was first prepared by condensing 2,5-diaminobenzonitrile (Db) with triformylphloroglucinol (Tp), which was then processed to convert the cyano groups to AO ligands to generate the AO-functionalized COF adsorbent (COF-TpDb-AO) (Fig. 7B).83 A stable 2D crystalline architecture with a high surface area of 826 m2 g−1 and open 1D pore channel structures was determined, affording a saturation qU of 408 mg g−1 at pH 6. When tested in a spiked seawater sample containing 20 ppm U, a high capacity (127 mg g−1) and fast kinetics (90 min) were observed. It is noteworthy that, compared with materials with disordered pore networks, the well-defined ordered pore channels in these crystalline porous materials enable the decorated AO ligands to be more conveniently accessible. High exposure of the binding sites allows a higher interaction efficiency, thereby resulting in a high affinity and rapid adsorption kinetics towards uranium.
PAFs represent a new category of porous materials that consist of extended organic structures in which the light elements (H, B, C, N, and O) are interlinked by strong covalent bonds. Due to their high chemical stability and fascinating structural topologies, they have attracted tremendous attention in the field of UES.37,93–95 Dai and co-workers pioneered the synthesis of PAFs functionalized with AO ligands for UES.95 A highly porous PAF material (PPN-6) was grafted with AN monomer through ATRP reaction with a DOG of 150 wt%. After conditioning with KOH, the resultant PAO-functionalized PPN-6 (PPN-6-PAO) demonstrated a maximum uranium uptake capacity of 4.81 mg g−1 after 42 d of contact with the uranium-spiked filtered environmental seawater. It is expected that the introduction of rigid fragments into the skeleton could improve the availability of AO groups, whereas soft polymers with a flexible or semi-rigid skeleton may lead to a dense structure that masks the binding sites (Fig. 7Ca).
To supplement this effort, AO-modified building units were recently cross-linked into the rigid framework of PAFs via an AlCl3 catalysed Scholl reaction.88 Subsequent amidoximation treatment was performed to convert the cyano groups to AO ligands in the frameworks, generating PAF-170 series adsorbents (Fig. 7Cb). It was found that the adsorption capacity of these adsorbents varied with the molar ratio of the cyanphenyl and biphenyl feed monomers. The adsorption capacity of PAF-170-AO-4 (cyanphenyl:biphenyl = 2:1) was superior in the same series of adsorbents, with a high qU of 702 mg g−1 in a 7 ppm uranium solution at pH 6, which was attributed to its highest utilization ratio of the AO ligands. To further improve the porosity of adsorbents for promoting the availability of the adsorption sites, rigid monomers (p-terphenyl and 1,3,5-triphenylbenzene) with expanded structures were employed to prepare PAF-171-AO and PAF-172-AO adsorbents (Fig. 7Cb). While PAF-171-AO and PAF-172-AO showed an increased utilization ratio of the AO ligands, they afforded a decreased qU of 608 mg g−1 and 569 mg g−1, respectively. This was explained by the increased mass per structural unit by the incorporation of building blocks. Due to its open architecture, the PAF-170-AO-4 adsorbent also exhibited good selectivity for uranium with a large coefficient Kd of 9.37 × 106. Furthermore, considering that the powder form of PAF-170-AO-4 was not suitable for the practical marine experiment, it was further coated on the porous ceramic sheet (denoted PAF-CS) with the mass ratio ranging from 1 wt% to 10 wt%. When exposed to an electrically-driven flow seawater system containing 3.3 ppb uranium, a remarkable uptake of 8.92 mg g−1 was achieved after 60 d. These works put forth a promising design strategy to promote the performances of uranium adsorbents.
Another intriguing strategy to promote the binding ability of AO-based adsorbents towards uranyl ions is the introduction of collaborative functionalities.46,61,69,87,96–100 For instance, bifunctional AO-based fibers were prepared by attaching various auxiliary diethylenetriamine (EDTA) ligands on commercially-available PAN fibers based on the amination chemistry, followed by the conversion of the residual nitrile species to AO through hydroxylamine treatment.96 Adsorption studies were carried out in filtered environmental seawater in a flow-through column at PNNL. After a contact time of 21 d, the bifunctional AO-EDTA and AO-Phon-DETA showed a higher uranium extraction capacity of 0.54 mg g−1 and 0.79 mg g−1, respectively. In comparison, the pristine PAO and non-amidoximated Phon-DETA only exhibited a capacity of 0.12 and 0.14 mg g−1, respectively. This work provided a good illustration that bifunctional polymers display a potential synergistic effect between the incorporated functionalities and AO ligands in enhancing uranium adsorption.
A particularly interesting study by the Ma group highlighted the elevated extraction ability of judiciously designed bio-inspired nano-traps. The concept was illustrated by constructing chelating systems into POPs with the simultaneous introduction of amino and AO groups.87 Specifically, to evaluate the effect of the spatial distribution of amine and AO groups on their collaborative binding ability, a series of vinyl-functionalized monomers with an amino substituent in different positions relative to AO were designed for self-polymerization into highly porous materials, followed by post-transformation of the cyano into AO group, generating the POP-AO adsorbent without the amino group, POP-oNH2-AO adsorbent bearing an amino group in the ortho position and POP-pNH2-AO adsorbent bearing an amino group in the para position (Fig. 7Da). Upon exposure to a 7.56 ppm uranium solution, a dramatic colour change was observed for all three adsorbents, indicating the effective uptake of uranium (Fig. 7E). An uptake capacity of 440 mg g−1, 580 mg g−1 and 530 mg g−1 was obtained for POP-AO, POP-pNH2-AO and POP-oNH2-AO at pH 6, respectively. Further adsorption experiments in seawater indicated that POP-oNH2-AO exhibited a superior affinity for U, with a uranium uptake of 4.36 mg g−1 after 56 d, whereas POP-pNH2-AO and POP-AO showed lower extraction capacities of 2.27 and 1.32 mg g−1, respectively. The structure–property relationship of these adsorbents was then explored through X-ray crystallographic studies and DFT calculations (Fig. 7Db–d). It was demonstrated that the introduction of auxiliary amino substituents facilitated lowering the charge on uranyl in the complex and could serve as a hydrogen bond acceptor, thus resulting in an increased affinity of AO towards uranyl. This work offers inspiration for further exploiting the collaborative coordination interactions to promote the ligand–uranyl binding of uranium adsorbents.
Basically, well-established approaches to elevate the adsorption capacity of polymeric adsorbents towards uranium in seawater involve the tuning of the number, accessibility and synergistic binding ability of the functional ligands. The advancement of polymerization technology has broken through the bottleneck in developing well-defined uranium adsorbents with high DOG.79,81,101,102 However, substantial research has demonstrated that a maximum number of the ligands in the uranium adsorbent does not promise a high adsorption capacity. Without effective contact with uranyl ions, most of the ligands are in the ‘dormant’ state. Only a small portion (<1%) of the ligands participate in the coordination to uranyl ions.103 Thus, scientists have urged recently to address the huge gap between the theoretical DOG and realistic utilization ratio of the ligands by examining their accessibility and the kinetic issues. More discussion about the influence of the accessibility of binding sites on the uranium adsorption ability of the adsorbents will be presented in Section 3.1.2.84 Besides, special emphasis should be placed on the solution chemistry of uranyl ions by investigating their coordination to more ligands with collaborative functionalities. This helps bring forward new design philosophies for developing more advanced adsorbents with synergistic binding to uranyl ions.
On the other hand, an important question that should be raised here is whether AO is the most optimal ligand for binding uranyl ions in seawater. As we know, AO ligands have been investigated for over 40 years, and have proved their high affinity towards uranyl ions in the marine environment. Meanwhile, polymeric adsorbents bearing AO ligands have been readily produced on a pilot scale for marine tests. Thus, AO ligands are still considered to be one category of the most effective ligands for UES. However, benefiting from the latest advancement in uranium coordination chemistry, new effective ligands and promising design principles, such as the preorganization of collaborative ligands, have been developed recently.104–107 More discussion in this regard will be presented in Section 3.2.3. With further studies on the mechanistic insights into the uranyl–ligand interactions, next-generation ligands with improved affinity and selectivity to uranyl ions in ocean environments could be envisioned.
Since uranium adsorption is carried out in aqueous solutions, a basic approach to tune the chain conformation of polymeric adsorbents is to introduce a second monomer containing hydrophilic groups in the RIGP system. The hydrophilic groups provide a significantly-hydrated local environment around the functional ligands, promoting their accessibility to uranyl ions by improving the mass transfer resistance. The introduction of hydrophilic comonomers to synthesize polymeric adsorbents for UES was first conducted by Omichi and co-workers.55,56 AO-functionalized PE fibers prepared by co-grafting of AN and hydrophilic monomers (AA or N,N-dimethyl-acrylamide (DMAAm)) were found to show a significantly improved ability for uranium uptake, compared to that of pristine AO-grafted polymers without hydrophilic chain segments.
In another representative work, Oyola and Dai systematically studied uranium recovery as a function of different hydrophilic components including AA, MA, vinyl sulfonic acid (VSA), vinyl phosphonic acid (VPA) and itaconic acid (ITA), which were co-grafted with AN through RIGP onto PE fibers with a “hollow-gear” shape (Fig. 8A).109 When exposed to simulated seawater, the uranium adsorption in terms of comonomer follows the order: AA < VSA < MAA < ITA < VPA (Fig. 8B), while when contacted with real seawater after ∼11 weeks, a change in the performance was observed: MAA < AA (Mohr's salt) < VSA < ITA (Mohr's salt) < ITA < VPA (Fig. 8C). Obviously, the uranium adsorption capacity was related to the hydrophilic comonomers, and the PE fibers grafted with AN and ITA or VPA produced the highest performances for uranium. Building on this work, two classical series of adsorbents called “AF”110 and “AI”111 series containing AN along with ITA/VPA were developed (Fig. 8D). The study on the parameters influencing the DOG was carried out by varying the comonomer ratio. The DOG of the AF series was found to be 154–354 wt% when the AN:ITA ratio ranged from 3.76:1 to 23.26:1. The uranium uptake was unrelated to the DOG, with a maximum qU of 3.90 mg g−1 obtained for the adsorbent at a 7.57:1 AN:IA ratio after 1 h KOH conditioning and 56 d of the marine test (Fig. 8E). For the AI series, the AN:VPA ratio was adjusted from 1.91:1 to 7.39:1, leading to DOG of 110–300 wt%. Similarly, the amount of adsorption was independent of DOG, and an adsorbent with a 3.52:1 ratio of AN:VPA showed the best adsorption performance (3.35 mg g−1, after 3 h KOH conditioning) after 56 d of exposure to natural seawater (Fig. 8F). The improved adsorption performance by the inclusion of hydrophilic comonomers was confirmed by these reports, revealing the effectiveness of the optimization of hydrophilic comonomers.75 It is worthy of note that treating the nitrile-based polymeric adsorbent with KOH to generate deprotonated carboxylic acids has been considered as another approach to increase the hydrophilicity of the adsorbent. As indicated in Fig. 8E and F, in addition to the chemical composition, KOH conditioning time plays an important role in controlling the uranium adsorption capacity, where for all the AI series adsorbents, 3 h of KOH conditioning (0.44 mol L−1, at 80 °C) was better than 1 h with respect to uranium adsorption capacity; in contrast, for the AF series adsorbents, the adsorption capacity obtained after 1 h of KOH conditioning was better than that of the 3 h treatment.
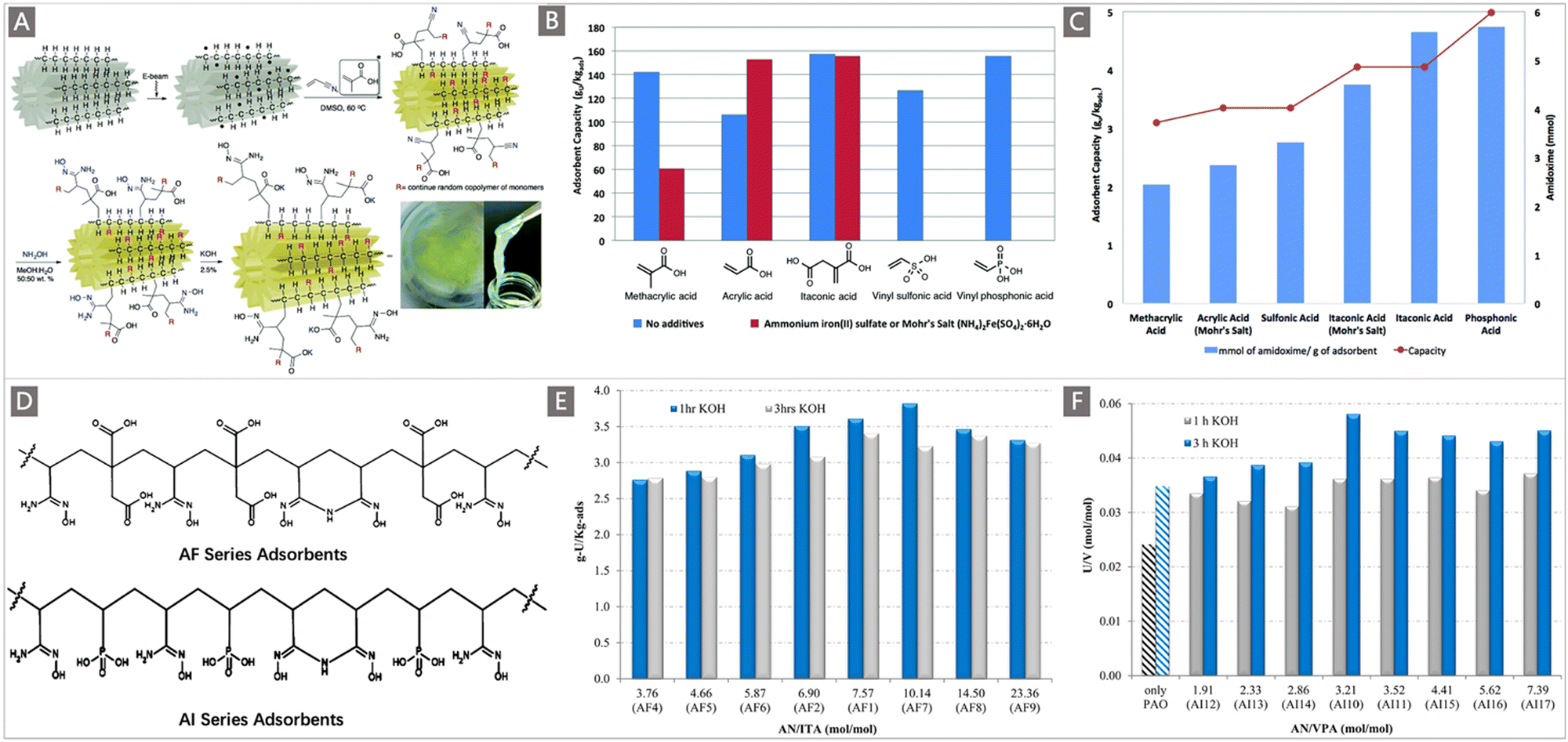 | ||
| Fig. 8 (A) The grafting, amidoximation and KOH conditioning of different hydrophilic comonomers to a PE fiber with a “hollow-gear” shape via RIGP, and the digital photo of the final adsorbent. (B) Comparison of adsorption performance between the adsorbents functionalized with different hydrophilic comonomers in simulated seawater using the laboratory screening protocol (red bars indicate the cases with Mohr's salt additives). (C) Comparison of the AO contents and adsorption performances of the adsorbents functionalized with different hydrophilic monomers in natural seawater after ∼11 weeks of exposure. A–C reproduced with permission.109 Copyright 2016, Royal Society of Chemistry. (D) Structure diagrams of AF series and AI series adsorbent materials. (E) Adsorption performances of AF series and (F) AI series adsorbents after 56 days of contact with Sequim Bay seawater in a flow-through column. The AF and AI series adsorbents were both treated with 0.44 mol L−1 KOH at 80 °C for 1 and 3 h. D–F reproduced with permission.110,111 Copyright 2016, American Chemical Society. | ||
To regulate the chain conformation of polymeric adsorbents to promote the accessibility and binding ability of tethered functional ligands, a brilliant alternative approach is to develop block copolymers (BCPs) containing hydrophilic comonomers by using diverse CRP techniques. Because of their well-defined structures with extraordinary compositional versatility, BCP-type adsorbents hold tremendous potential to afford maximum ligand availability while enabling a quantitative investigation of their structure–property relationship. A seminal work about BCP-type adsorbents was reported by Saito and co-workers combining the use of RIGP and ATRP. As mentioned in Section 3.1.1, for the preparation of the adsorbent PE-g-PVBC-g-(PAN-co-PtBA), PVBC was first grafted onto a PE trunk through RIGP to install initiation sites, and then a “brush-on-brush” structure was created by copolymerizing AN and tBA through ATRP, and followed by amidoximation reaction. To explore the effect of the chain conformation on the adsorption towards U, a chain extension of the grafting brushes through ATRP was performed where short hydrophilic PtBA chains were added to the tail of the PAN-co-PtBA brushes, followed by further hydrolysis to obtain PE-g-PVBC-g-(PAO-co-PAA)-b-PAA (Fig. 9A). When immersed in natural seawater, strikingly, the adsorbents after chain-extension introduction of the hydrophilic PAA block afforded nearly doubled seawater adsorption capacity from 1.56 mg g−1 to 3.02 mg g−1 under the same conditions.75
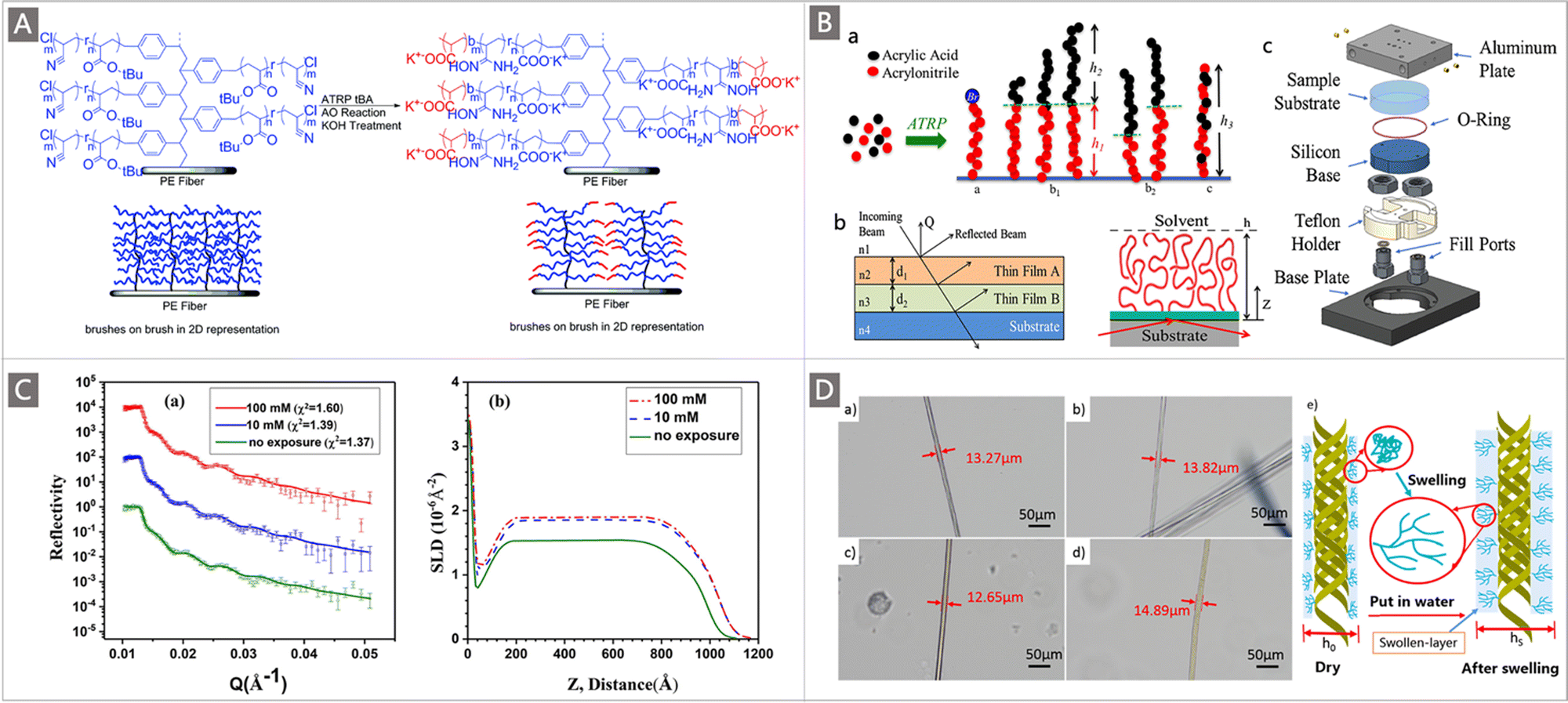 | ||
| Fig. 9 (A) Illustration of the addition of the hydrophilic group to the tail of the PE-g-PVBC-g-(PAN-co-PtBA) adsorbent. Reproduced with permission.75 Copyright 2014, Royal Society of Chemistry. (B) Illustration of a pure PAO monoblock polymer, deuterated-PAO polyacrylic acid diblock copolymers (d-PAO-b-PAA) and the random copolymer configuration (PAO-co-PAA). Reproduced with permission.112 Copyright 2018, Elsevier. (C) (a) The NR profiles of PAAO brushes measured before and after exposure to the UO2(NO3)2 solutions with different concentrations; and (b) SLD profiles obtained from the best fittings of the NR data. Reproduced with permission.113 Copyright 2018, Elsevier. (D) (a–d) Optical microscope photos of PAN and PAN-(OH)n-P fibers before and after swelling; and (e) swelling diagram of the swollen-layer on the PAN-(OH)n-P surface. Reproduced with permission.115 Copyright 2020, Elsevier. | ||
Since bare hydrophilic comonomers have been verified to have no adsorption ability for uranium, their influence on the polymer chain conformation was further investigated. Preliminary results revealed that the enhancement in uranium absorption capacity was attributed to the more stretched conformation of the polymer brushes with the incorporation of hydrophilic PAA blocks. In their follow-up study, by using the neutron reflectometry (NR) technique, the effects of hydrophilic groups, alkali conditioning and metal-ion loading on the conformation of polymer adsorbents were further verified.112 Specifically, three series of adsorbents namely the pure PAO monoblock polymer, deuterated-PAO polyacrylic acid diblock copolymers (d-PAO-b-PAA) and the random copolymer configuration (PAO-co-PAA) were prepared through ATRP of AN or AN with tBA at different molar concentrations (Fig. 9Ba). These adsorbents were deposited onto quartz substrates and were characterized by the NR technique in air and heavy water (D2O) (Fig. 9Bb), in which the NR data of solvated samples were collected using a liquid cell (Fig. 9Bc). The obtained NR revealed that the initial film thickness of pure PAO and PAO-co-PAA samples with similar molecular weights and grafting densities extended from 95 Å thickness in the air to 180 and 280 Å in D2O, respectively, suggesting that the incorporation of hydrophilic segments on the polymer would cause swelling of the polymer, which reduced the barrier to uranium diffusion, and in turn increased the uranium adsorption capacity. Moreover, the d-PAO thickness in both diblock samples increased by ∼100% and ∼150% in D2O, while the PAA swelled by ∼180% and ∼170%, respectively, relative to their thickness in the air. Additionally, the shrinking of the diblock copolymer was observed after alkali treatment and during metal ion adsorption, although it was not yet clear which metal ion was responsible for it.
In a concurrent report, Xie and colleagues conducted a study employing the in situ NR technique to probe the conformational behaviour of poly(acrylamidoxime) (PAAO) brushes in water, focusing on the change in polymer chain conformation before and after uranyl ion adsorption.113 The NR profiles were obtained by the measurements of the PAAO film both in air and in an aqueous solution. While no apparent interference fringes were found when the PAAO film was immersed in pure D2O solution, fringes were observed when the film was immersed in a solution containing UO2(NO3)2. After exposure to 10 mmol L−1 UO2(NO3)2 solution, the height of the PAAO brushes increased significantly by 5 nm, while continuing to increase the UO2(NO3)2 concentration to 100 ppm, only a slight increase was observed (Fig. 9C). A speculation was provided by the authors based on these results that due to the strong complexation between uranyl ions and AO groups, a shrinkage of the PAAO brushes occurred when uranyl ions were adsorbed, resulting in a collapse or more compact conformation of the polymer brushes. Concomitantly, the expulsion of the uranium solution from PAAO brushes was also observed, which further hindered the diffusion and adsorption of uranium. However, the uranium adsorption capacity increased with the increase in UO2(NO3)2 concentration, indicating that a new balance between the adsorption and expulsion of uranium solution induced by shrinkage of PAAO brushes was established before reaching the theoretical adsorption capacity. These results increased the mechanistic understanding of the adsorption of uranium to PAAO polymers. Besides, since there are more coexisting ions in the actual seawater environment, the influence of competitive ions on the conformation of polymer brushes is worth noting.
Motivated by these findings, the Wang research group investigated the effect of the molecular chain conformation and swelling behaviour on the adsorption of uranium by the polymer grafted with hydrophilic hyperbranched ligands.114,115 In their recent study, amino trimethylene phosphoric acid (ATMP) was chosen as a terminal group grafted onto the surface of a PAN fiber, generating adsorbents labeled PAN-(OH)n-P.115 When immersed in an aqueous solution, the swelling behaviour of the gel-like swollen-layer was captured with an optical microscope where both the dried PAN and PAN-(OH)n-P exhibited an increase in diameter (Fig. 9D). The swelling behaviour was expected to facilitate the uranium adsorption by decreasing the resistance of uranium into the swollen-layer. More importantly, the conformation of the molecular chain underwent a significant change, from the collapsed conformation to an extended conformation, as indicated in Fig. 9De, leading to an increased collision probability between uranium and the functional group. Accordingly, the PAN-(OH)n-P polymer demonstrated an improved adsorption for uranium compared to PAN-(OH)n adsorbent without grafting of ATMP under all tested adsorption conditions. The uranium adsorption performance of PAN-(OH)n-P was further determined in both simulated seawater and real seawater. When contacted with a simulated seawater system, the adsorbent acquired an uptake of 7.4 mg g−1 after 45 d, and 209.8 mg g−1 was recovered following 5 consecutive days of adsorption from 8 ppm uranium spiked seawater. These studies highlighted the importance of the polymer chain conformation in controlling the adsorption ability of polymeric adsorbents towards uranium.
Fundamental insights into the relationship between the chain conformation of polymers and their adsorption behaviours would accelerate the development of new-generation uranium adsorbents. To this end, based on the quantitative analysis of the mesoscopic scenario of metal ion adsorption by polymeric adsorbents, our group recently established a new paradigm for smart design while customizing polymeric adsorbents for UES, by integrating multi-scale computational simulation with precise synthesis.116 The concept and strategy used in this work are illustrated in Fig. 10. Specifically, by using the computational modeling involving both the dissipative particle dynamics (DPD) and molecular dynamics (MD) approaches, the theoretical assessment of the conformational dynamics of a series of model AO-functionalized block copolymers, PAOm-b-PPEGMAn, and their adsorption behaviours toward uranium was first performed. The simulation results revealed the effect of the block ratio (n/m) on the adsorption properties of materials, where the maximum adsorption capacity with atomic economy was predicted to achieve with a preferred block ratio of 0.18. In accordance with the computer-aided design, a series of model block copolymers with different block ratios were synthesized using a photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization117,118 in a continuous droplet-flow platform. Subsequently, the adsorption performances of the obtained polymeric adsorbents were investigated in spiked solutions, which exhibited predicted uranium adsorption behaviour, in conformity with that revealed by the computational simulation. Significantly, a high qU of 11.4 mg g−1 could be achieved when the optimal polymer adsorbent was exposed to real seawater within 28 d, demonstrating the great potential of AO-functionalized BCPs with structural and conformational benefits for UES. This study offers a new research perspective to study the adsorption of metal ions by polymers, bridging the specific metal–ligand interactions at the molecular level and the spatial conformational properties at the mesoscopic level.
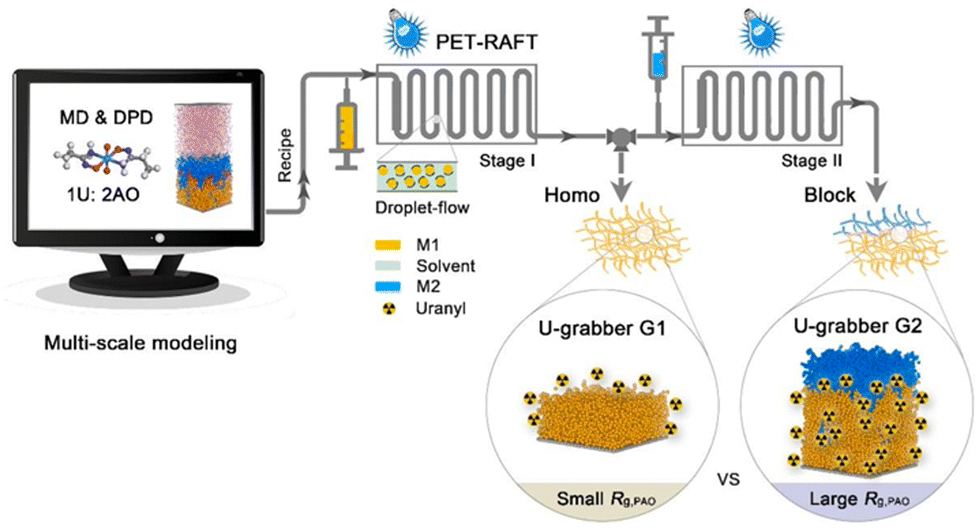 | ||
| Fig. 10 The illustration of multi-scale computational modeling combined with droplet-flow PET-RAFT polymerization for the target-oriented development of new-generation uranium grabbers. Reproduced with permission.116 Copyright 2022, Springer Nature. | ||
In brief, the polymer chain conformation exerts a significant influence on the adsorption ability. The hydrophilic component in the polymer affects the structure of grafting chains, putatively leading to interesting morphological phenomena, which have received increasing attention in the past five years. As clearly demonstrated, tuning the chain conformation of polymers can help to afford maximum ligand availability, and thus improve the interaction with uranium. However, the studies reported so far have only superficially grazed the influence of the chain conformation on the adsorption of uranium. In future work, it is necessary to combine the fundamental analysis based on polymer solution physics and classical adsorption theories to establish the relationship between the structure, conformation and adsorption behaviours of polymers. In addition, pivotal information on the polymer chain conformation in aqueous environments can be obtained with the aid of computational simulation and NR techniques, which would inspire the design of high-performance polymeric adsorbents. Meanwhile, more efforts are needed to develop controllable polymerization techniques to achieve the precise synthesis of polymers with structural and conformational benefits.76,80
First, it is generally accepted that the substrate materials of adsorbents with high SSA can afford maximum ligand availability. Accordingly, changing the cross-sectional shape of fibers is considered to be an effective way to increase the SSA of the adsorbents. This was demonstrated by Omichi and co-worders119 who synthesized adsorbents by grafting PAN onto PP fiber substrates with either a round or cross-shaped sectional view. Results showed that the adsorbent with the cross-shaped support with a larger SSA exhibited a superior uranium uptake to the adsorbent with the round-shaped support. Moreover, higher recovery of uranium was achieved by decreasing the diameter of adsorbents with round-shaped fiber supports, verifying the effect of SSA on uranium adsorption. Subsequently, the role of the geometry of trunk fibers in uranium recovery was also evaluated in Oyola's work,120 in which AN:MA at a ratio of 70:30 was first grafted onto a series of PE fibers with a round morphology and varying diameters, followed by amidoximation and adsorption tests in 8 ppm uranium solution. It was generally found that smaller-diameter fibers exhibited a higher adsorption capacity than larger-diameter fibers. The adsorption of uranium increased from 30 mg g−1 to 140 mg g−1 when the diameter decreased from 20 mm to 0.24 mm. Additionally, the uptake capacity of non-round-shaped trunk fibers was also explored where a fiber with a hollow gear shape as a PE trunk outperformed other non-round-shaped fibers, affording a uranium uptake of 188.1 mg g−1, while quasi-trilobal shaped adsorbents only showed an adsorption capacity of 20 mg g−1. In addition to the high SSA of the hollow gear adsorbent, the hollow interior also allowed increasing access to the AO groups, which increased the adsorption sites, thereby improving the uranium adsorption capacity.
Commercially available trunk fibers are mainly produced by the melt-spinning method. These microfiber adsorbents still suffer from relatively large diameters and small SAA.120 The development of spinning technologies, including but not limited to electrospinning,121,122 forcespinning123 and solution blow spinning approaches,124–129 has gained increasing attraction in recent years, providing reliable ways to produce fibers with diameters at nanometer levels and adsorbents with high SSA. Typically, electrospinning is a versatile method used to fabricate nanofiber mats in large quantities.130 In this process, a viscoelastic polymer fluid overcomes its surface tension through electrostatic traction, creating a continuous jet that is drawn out into ultrafine fibers with diameters down to tens of nanometers, which are further assembled directionally or randomly into fiber mats with sizes ranging from centimeters to meters.131 Taking advantage of this powerful technique, Xie and colleagues fabricated nanofibrous PAO/polyvinylidene fluoride (PVDF) composite mats through a two-nozzle electrospinning process,121 in which PAO and PVDF solutions were separately loaded into two syringes and connected to a high-voltage power supply, then PAO and PVDF nanofibers were simultaneously drawn out from their respective nozzles and interwoven into the final composite mats (Fig. 11Aa). By tuning the injection rate of the PVDF solution, fibrous composite mats with different PAO contents and diameters ranging from 150 nm to 400 nm were obtained (Fig. 11Ab–d). After blending with PVDF, the mechanical properties, porosity and utilization ratio of AO groups of the electrospun mats were highly enhanced. The adsorption performance was further evaluated in artificial seawater at pH 8.0 with 3.5 wt% sea salt and ten commonly-found interfering ions. The resultant PAO/PVDF composite mats demonstrated a uranium uptake capacity of 1.85 mg g−1 after 24 h of batch adsorption. To enhance the U adsorption capacity, the subsequent work from the same group introduced a carboxylic (AC−) group into the electrospun system where PAO and PVDF-g-PAAc were selected as the starting materials and were electrospun into the nanofibrous composite mats by the same technique.122 The addition of AC− groups enormously increased the U adsorption capacity (2.86 mg g−1 in simulated seawater) of the obtained composite mat adsorbents because of the synergistic effect between the two functional groups.
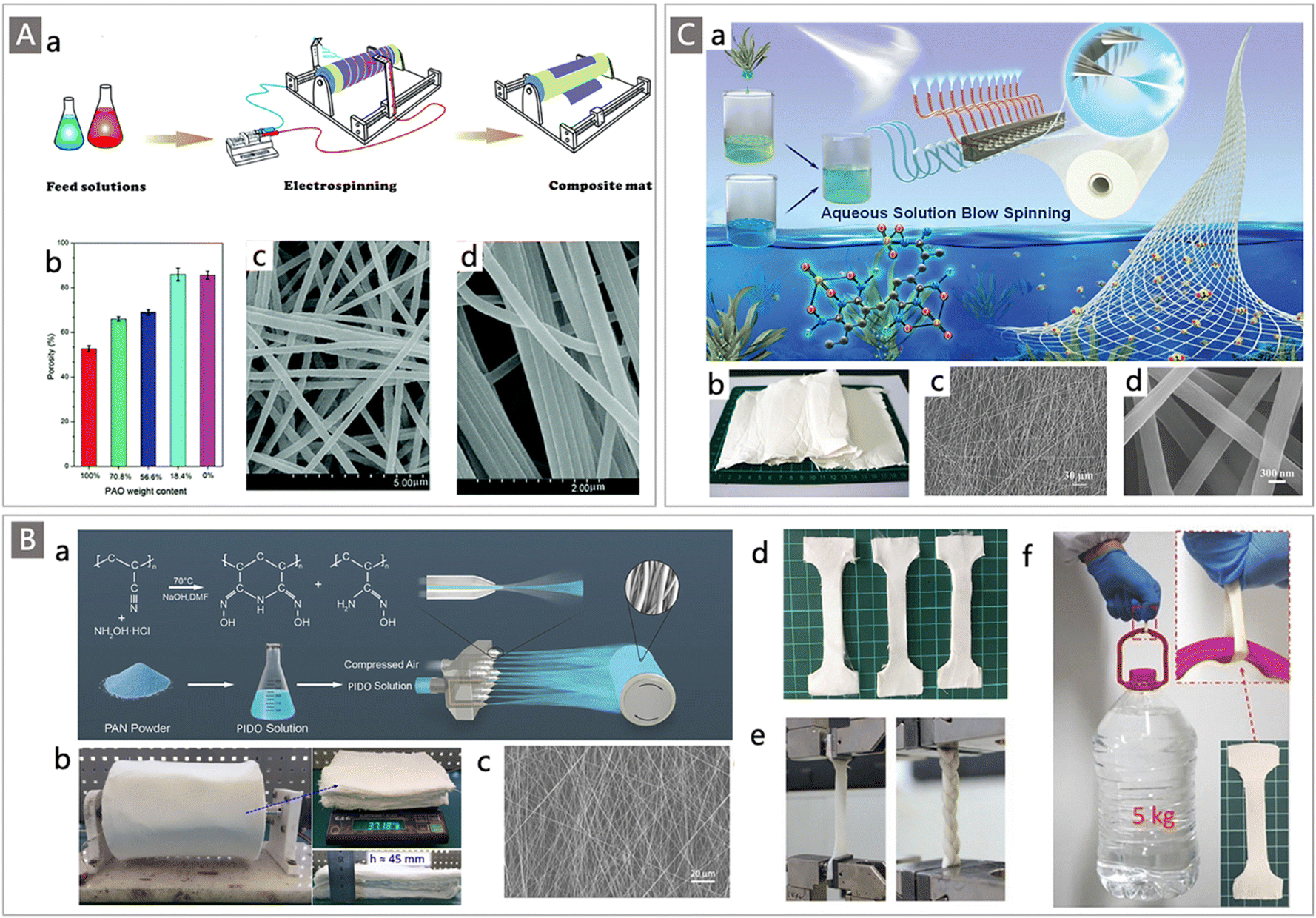 | ||
| Fig. 11 (A) (a) Schematic diagram of the two-channel parallel-blend electrospinning method for PAO/PVDF composite mat preparation. Reproduced with permission.122 Copyright 2016, Royal Society of Chemistry. (b) The change in porosity of the PAO/PVDF mats as a function of PAO content; SEM images of electrospun PAO/PVDF mats containing 56.6% (c) and 100% PAO (d). Reproduced with permission.121 Copyright 2015, Royal Society of Chemistry. (B) (a) Schematic diagram of the pre-amidoximation with a SBS strategy for fabricating PIDO nanofibers; (b) photograph of a PIDO NF mat collected with an air-permeable roller; SEM image (c) and photograph (d) of the PIDO NF mat; (e) photos of the PIDO mat and braid mounted in a Zwick universal testing machine; and (f) photograph of a representative mechanical test for the wet PIDO NF mat. Reproduced with permission.125 Copyright 2018, Wiley-VCH. (C) (a) Illustration of the ASBS process for PAO/Alg NF mat preparation; (b) photograph of the obtained typical PAO/Alg NF mats; and (c and d) SEM images of PAO/Alg NFs with different magnifications. Reproduced with permission.126 Copyright 2020, Wiley-VCH. | ||
Likewise, solution blow spinning (SBS) is also a versatile method for preparing nanofiber adsorbents, owing to its unique advantages such as no requirement of a high direct current voltage source and electrically conductive collector, and no limitation for the thickness of nanofibers deposited on the collector.125–127 In a landmark example, Wang and co-workers developed a novel pre-amidoximation with a SBS strategy to massively fabricate poly(imide dioxime) nanofiber (labelled PIDO NF) adsorbents for UES (Fig. 11Ba).125 Unlike the conventional AO-based adsorbents obtained by post-amidoximation treatment of nitrile functionalized fibers, in this work, the amidoximation of PAN raw material was first carried out through hydroxylamine treatment in organic solvent (e.g., DMF, DMSO). The obtained PIDO solution was then injected into a multi-nozzle system and stretched by the compressed air flow, and aligned fibers were continuously deposited on a rolling drum to form a uniform fiber mat (Fig. 11Bb). The obtained nanofibers have an average diameter of 406.13 ± 28.35 nm and a BET surface area of 28.42 m2 g−1 (Fig. 11Bc). Good extendibility and mechanical properties of the wet PIDO NF mat samples were also observed from an obvious necking phenomenon (Fig. 11Bd–f), which were beneficial for practical adsorption application in seawater. Subsequently, the adsorption performance of the obtained PIDO NF was tested in both uranium spiked and non-spiked seawater, which was collected from coastal water near the Boundary Island of the South China Sea. Combined with the full coverage of imide dioxime sites, the favourable characteristics of 3D porous architecture, excellent hydrophilicity, and high SSA, the PIDO NF showed an adsorption capacity up to 951 mg g−1 in 8 ppm uranium-spiked natural seawater. More importantly, the adsorption capacity of the PIDO NF for uranium reached 8.74 mg g−1 after 56 d contact with real seawater in the field column test, which represented a high adsorption value among the fabric adsorbents.
Building on this work, a modified aqueous solution blow spinning (ASBS) strategy was developed by the same team for the synthesis of PAO-based nanofibers (Fig. 11Ca).126 Similarly, a pre-amidoximation process of PAN was carried out to prepare the PAO precursor. While the amidoximation product PAO is not soluble in deionized water, the good solubility of PAO in alkaline solutions enables the possibility for the preparation of PAO-based nanofibers from aqueous solutions, rather than an organic solvent, which could avoid environmental impacts and reduce the cost. Alginate (Alg) was then chosen as a supporting component for hybrid nanofibers and a small amount of polyethylene oxide (PEO) was employed as an accessory ingredient to increase the spinnability of the polymer mixture. Accordingly, PAO was mixed with Alg and PEO in an alkaline aqueous solution to prepare the feed polymer solution, which was fed to the nozzles and then stretched rapidly; the PAO/Alg/PEO nanofibers were continuously deposited on a rolling collector through the ASBS process (Fig. 11Cb). Furthermore, an in situ crosslink treatment was performed by introducing calcium chloride (5 wt%) solution into the PAO/Alg/PEO NFs to enhance their water-stability and mechanical stability. At the same time, PEO was washed out during the crosslinking process due to its water-soluble property. Significantly, obvious changes in morphology and structure were observed after crosslinking, where the diameter of the dry fibers decreased from 402.15 ± 10.75 nm to 300.25 ± 16.35 nm (Fig. 11Cc and d), resulting in an increase of SSA from 5.8 m2 g−1 and 33.7 m2 g−1 for PAO/Alg/PEO NFs and PEO removed PAO/Alg NFs, respectively. When investigated for uranium recovery, a saturation capacity of 892.77 mg g−1 and 8.42 mg g−1 was obtained for PAO/Alg NFs from 8 ppm U-spiked seawater and 8 tons of non-spiked natural seawater, respectively. These novel SBS strategies show great potential in the preparation of new generation uranium adsorbents for UES.
Aside from reducing the diameter of trunk fibers, a novel strategy to increase the SSA of the grafting chain was proposed by Xu and co-workers to prepare high SSA AO-based polymeric (H-ABP) fibers to increase the uranium adsorption capacity while maintaining their mechanical properties.132 Specifically, a 3D hierarchically porous H-ABP fiber was fabricated via self-assembly of axial grafting chains. A two-step graft polymerization was involved, including the RIGP of 2-hydroxyethyl acrylate (HEA) onto a PE-coated PP fiber at a very low adsorbed dose (10 kGy) followed by the Ce4+ initiated graft polymerization (CIGP) of AN and AA onto the poly-HEA (PHEA) chains (Fig. 12A). The latter process brought about axial grafting chains along the trunk fibers rather than radial direction grafting chains generated by RIGP, and the self-assembly of axial grafting chains and subsequent amidoximation resulted in high SSA H-ABP fibers (12.7 m2 g−1) with uniform functional polymer particles (∼500 nm in size) and a 3D hierarchically porous structure (Fig. 12B). Remarkably, the adsorbent reached a record high qU of 11.5 mg g−1 within 90 d of contact in natural seawater, and a long service life of at least ten adsorption–desorption cycles. The authors believed that this would contribute to reducing the final uranium production cost.
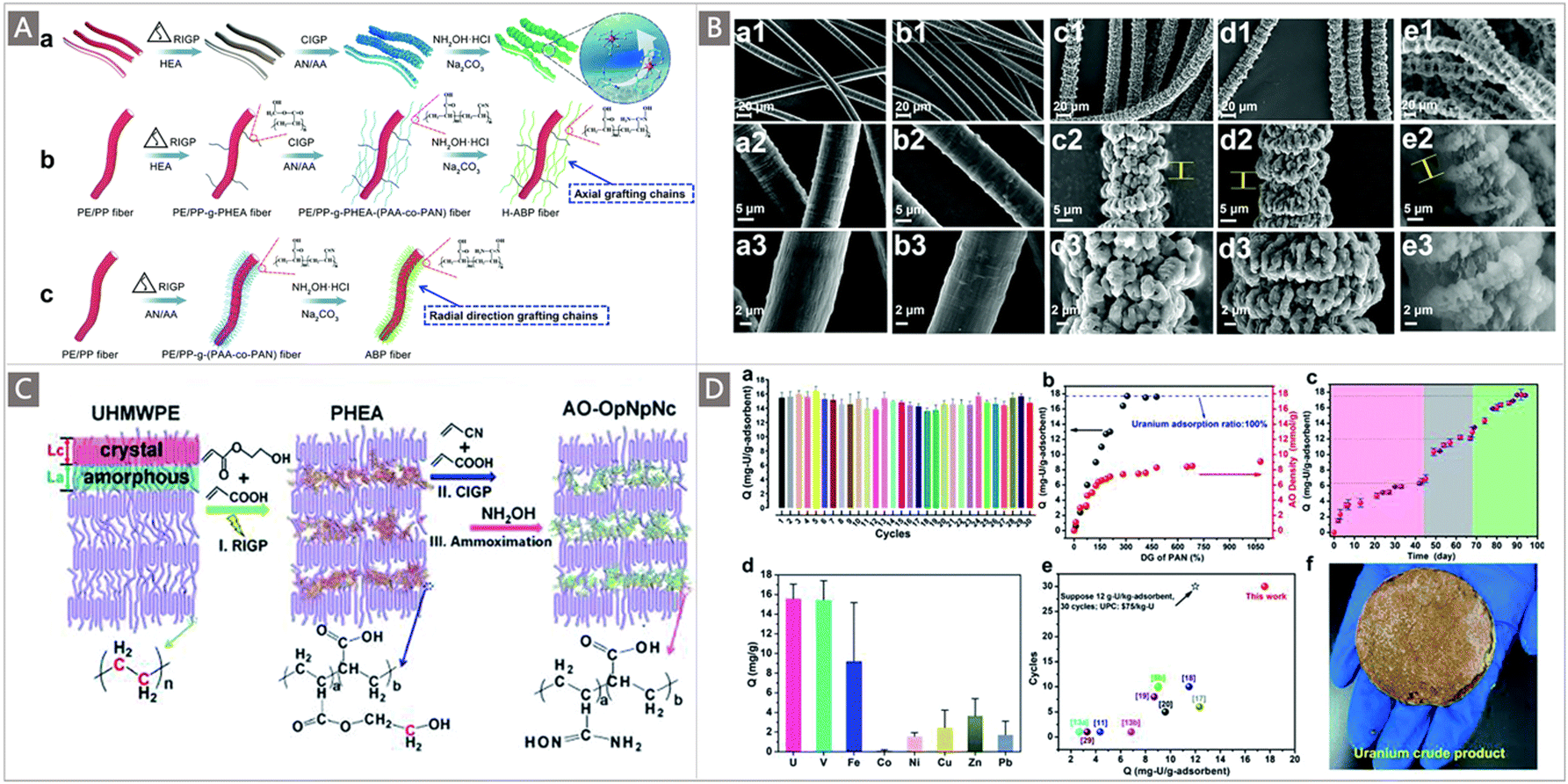 | ||
| Fig. 12 (A) Schematic illustration of the synthesis process for H-ABP fibers: (a) preparation of H-ABP fibers with a controllable order morphology; (b) structural evolution of grafted chains in H-ABP fibers induced by RIGP and CIGP; and (c) structural evolution of grafted chains in the ABP fiber induced by RIGP. (B) SEM images of PE/PP (a1–3), PE/PP-g-PHEA (b1–3), PE/PP-g-PHEA-(PAA-co-PAN) (c1–3) and H-ABP (d1–3) fibers at different magnifications, and ESEM image of the wet H-ABP fiber after being soaking in deionized water for three days (e1–3). (A and B) reproduced with permission.132 Copyright 2019, Royal Society of Chemistry. (C) Schematic illustration of the synthesis process for AO-OpNpNc fibers. (D) Adsorption performances of AO-OpNpNc fibers: (a) uranium adsorption capacity during 30 adsorption–desorption cycles with an initial uranium concentration of 330 ppb and coexisting ions; (b) the changes in uranium adsorption capacity and AO density as functions of different DOG of PAN; (c) uranium adsorption capacities after different contact periods in natural seawater; (d) adsorption capacities of uranium and other co-existing ions (including V, Fe, Co, Ni, Cu, Zn and Pb) after 90 d of adsorption in natural seawater; (e) comparison of uranium adsorption capacity and service life between AO-OpNpNc fibers and other representative reported adsorbents for UES; and (f) photo of 5.6 g of uranium crude product recovered from the ocean. (C and D) reproduced with permission.133 Copyright 2020, Royal Society of Chemistry. | ||
In a follow-up study, in order to maximize the functionality of trunk materials, the same team selected UHMWPE fibers as the backbone materials, and again employed the two-step co-grafting polymerization involving the RIGP of HEA and AA on the UHMWPE fibers followed by the CIGP of AN and AA onto poly-HEA side chains (Fig. 12C),133 resulting in poly-AN-co-AA (PAN) fibers with an interconnected open-pore architecture, PAN nanoparticles and nano-channels. After amidoximation, the fibrous AO-OpNpNc adsorbent was constructed. The fibers exhibited properties of high SSA (11.1 m2 g−1) and interconnected open-pore architecture, providing numerous nano-channels for the continuous mass transport of uranium. The uranium adsorption capacity and stability of the AO-OpNpNc fibers were investigated. When immersed in a lab-scale simulated seawater system,71 a high qU of 15.98 mg g−1 was achieved by the AO-OpNpNc fibers, which was higher than those of the competing ions including V, Mg and Ca in the system (Fig. 12Da). The adsorption capacity was found to increase with the increase of DOG of poly-AN and AO density of the AO-OpNpNc fibers, where an adsorbent with a DOG of 307 wt% and an AO density of 7.7 mmol g−1 afforded a 100% uranium uptake with a qU of 17.70 mg g−1 (Fig. 12Db). It was worthwhile to mention that the adsorbent exhibited an extremely long service life of at least 30 cycles in the simulated seawater system (Fig. 12Da), in which the adsorption capacity remained in the range of 16.33–13.28 mg g−1. This value represented the longest service life of the currently reported uranium adsorbents (Fig. 12De). The batch adsorption tests were also carried out in natural seawater; a uranium capacity of 17.57 mg g−1 was observed after three rounds of adsorption (Fig. 12Dc). Adsorption was also performed using a flow-through flume system developed in the coastal marine areas of Raoping, Guangdong Province, China. While the contamination of marine microorganisms was noted on the adsorbent's surface after 90 d of adsorption, an exceptional qU of 15.42 mg g−1 was still obtained, which was higher than those of the competing ions Fe, Co, Ni, Cu, Zn and Pb, except for V (Fig. 12Dd). Encouragingly, a total of 5.6 g crude product of uranium was recovered by the AO-OpNpNc fibers in a three-month test performed at the Tropical Marine Biological Research Station in Hainan, Chinese Academy of Sciences, Sanya, China (Fig. 12Df).
Hydrogels functionalized with appropriate ligands have recently emerged as a new class of uranium adsorbents. In contrast to most fibrous adsorbents with relatively hydrophobic and dense structures, the loose hydrophilic 3D network of hydrogel-based adsorbents could facilitate the diffusion of uranyl ions into their inner structures.134,135 Moreover, the relatively simple preparation process makes them attractive in the field of UES.136 Wang and colleagues developed a series of hydrogel-based adsorbents to explore their utility for UES.137–140 Typically, a PAO-embedded hydrogel membrane was prepared through eco-friendly sunlight (UV) polymerization.137 The obtained hydrogel membrane exhibited a semi-interpenetrating structure and a hydrophilic poly(acrylamide) 3D network to accommodate PAO (Fig. 13A). Adsorption tests of the resultant semi-IPN-PAO hydrogels were carried out in 8 ppm and 32 ppm uranium-spiked seawater, yielding an outstanding qU of 718 ± 16.6 mg g−1 and 1279 ± 14.5 mg g−1, respectively. When immersed in natural seawater in a flow-through system for 4 weeks, the hydrogel achieved an uptake capacity of 4.87 ± 0.38 mg g−1, indicating its potential for UES.
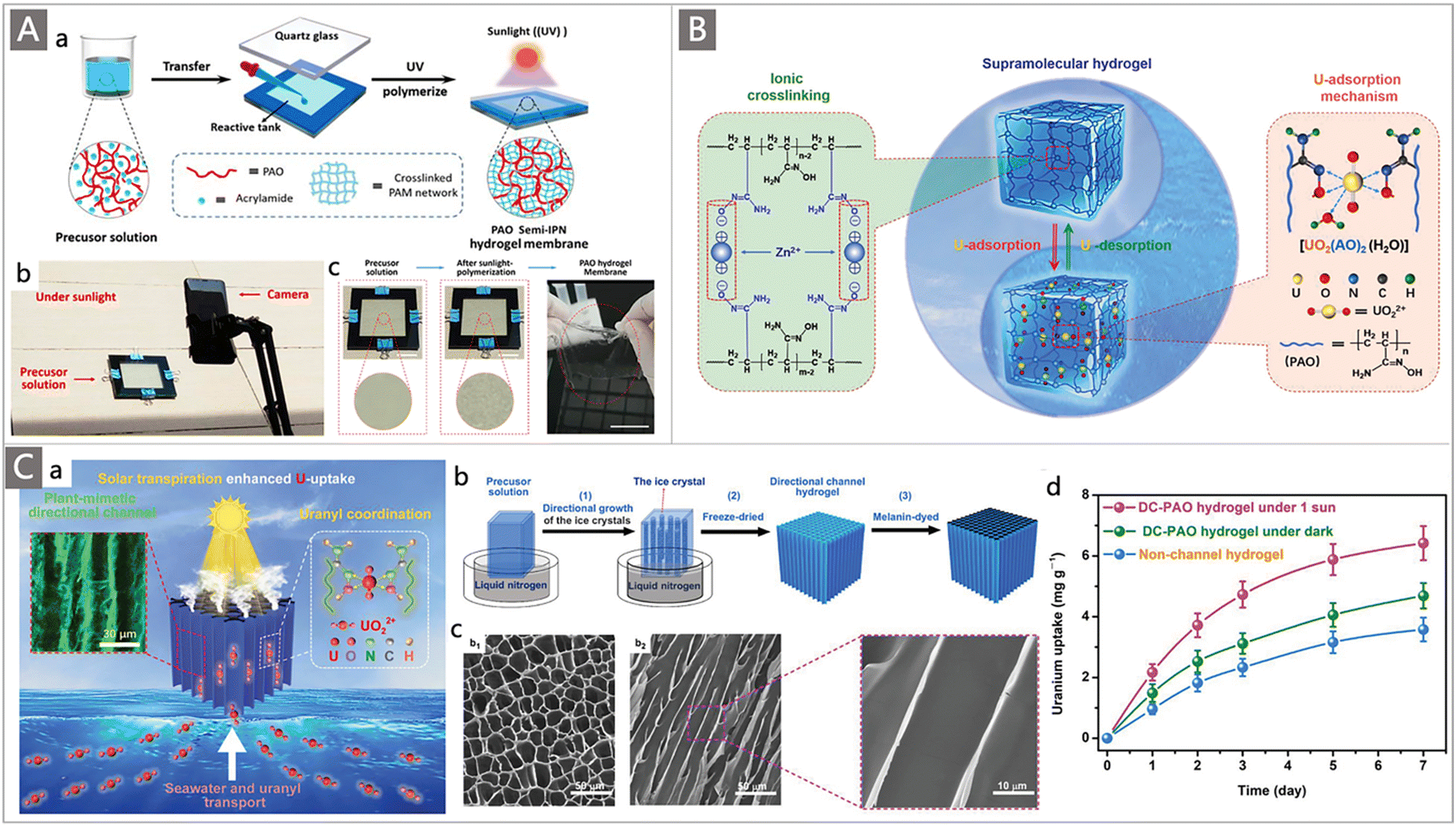 | ||
| Fig. 13 (A) Schematic illustration of the fabrication process of PAO hydrogel membranes: (a) illustration of fabricating PAO semi-IPN hydrogel membranes; (b) massively and directly fabricating PAO hydrogel membranes under sunlight; and (c) photos of self-supporting PAO hydrogel membranes. Reproduced with permission.137 Copyright 2019, Wiley-VCH. (B) Schematic illustration of the ionic cross-linking of Zn2+-PAO supramolecular hydrogel and the uranium-binding mechanism. Reproduced with permission.138 Copyright 2020, Wiley-VCH. (C) (a) Schematic of transpiration-enhanced uranium extraction from seawater using a plant-mimetic DC-PAO hydrogel; (b) the preparation process of the DC-PAO hydrogel; (c) SEM images of the (b1) cross-sectional view and (b2) longitudinal view of the dry hydrogel; and (d) uranium extraction performance of the DC-PAO hydrogel in natural seawater. Reproduced with permission.139 Copyright 2021, Wiley-VCH. | ||
In a follow-up study of the team,138 in order to increase the content of PAO in the hydrogel's structure to enhance the U adsorption capacity, an ion-crosslinked supramolecular Zn2+-PAO hydrogel membrane was designed and synthesized by simply mixing the solutions of zinc chloride and PAO. On account of the strong interaction between Zn2+ and two AO anions (AO−), the PAOs were directly cross-linked by the super-hydrophilic zinc ions that served as the cross-linker, forming a 3D hydrogel network with a high content of well-dispersed PAO (≈96 wt% of the dry gel) and good hydrophilicity (Fig. 13B). Consequently, the Zn2+-PAO hydrogel with different ratios of mZn2+:mPAO displayed high uranium adsorption capacities, and the sample with mZn2+:mPAO = 4:100 exhibited a superior uptake of 1188 ± 18.9 mg g−1 in 32 ppm U-spiked water and 830 ± 16.5 mg g−1 in 32 ppm U-spiked seawater. The lower adsorption of the hydrogel in 32 ppm U-spiked seawater was attributed to the existence of various competing metal ions (e.g., Na, Ca, Mg, K, V, Fe, Co, Ni and Cu) and high ionic strength. While the Zn2+-PAO hydrogel showed a weaker affinity for other trace metal elements, it showed high adsorption for V even more than uranium. This may be ascribed to the transformation of a portion of AO groups to cyclic imide-dioxime groups that have a high affinity for V. Finally, the ability of this hydrogel membrane to recover uranium from real seawater was examined with a continuous-flowing system. After 4 weeks of adsorption, an enhanced uranium adsorption performance (9.23 ± 0.52 mg g−1) was acquired, as compared with the semi-IPN-PAO hydrogel (4.87 ± 0.38 mg g−1).137
Very recently, inspired by the biological channels in plants, a directional-channel PAO (DC-PAO) hydrogel was created to improve the uranium extraction efficiency via the active pumping of uranyl ions into the adsorbent (Fig. 13Ca).139 Oriented channels were introduced into PAO through an ice-crystal growth technique (Fig. 13Cb). The diameter of the channels can be controlled by changing the solid contents of the precursor solution (Fig. 13Cc). Additionally, the directional channels of the DC-PAO hydrogel could be well-maintained in seawater. This carefully-designed structure provided favourable transport channels for the rapid migration of uranyl ions into the hydrogel networks, affording a high transpiration-enhanced uranium adsorption ability together with an ultra-efficient solar desalination performance. A significantly higher adsorption capacity of 6.42 ± 0.56 mg g−1 was achieved in the natural seawater for 7 d, as compared to that of the channel-less hydrogel (3.58 ± 0.39 mg g−1) and the DC-PAO hydrogel not exposed to irradiation (4.69 ± 0.42 mg g−1) (Fig. 13Cd). Of particular interest, owing to the introduction of the directional channels, this plant-mimetic transpiration-enhanced hydrogel exhibited a uranium adsorption rate of 0.917 mg g−1 d−1, which was the fastest uranium adsorption rate among the AO-based adsorbents reported at present. It is worthy of note that hydrogel-based adsorbents containing some biological entities such as specific proteins with ultrahigh selectivity for uranyl ion adsorption were also developed by the Wang team,141,142 which will be elaborately discussed in Section 3.2.
Furthermore, the rapid growth of membrane technology in recent years offers an alternative choice of adsorbents for UES. Novel porous membrane-based adsorbents have been constructed for UES due to their diverse operating modes, collection and recycle potential.143–145 A representative study by Guo and co-workers reported the design and preparation of a microporous polyphenol membrane which exhibited excellent performances for uranium extraction in both laboratory and field examinations.144 A biomass-derived microporous membrane was synthesized by incorporating polyphenol functionalities onto a commercially-available microporous polyamide (PA) membrane. When applied for uranium recovery, the selective adsorption of uranium mainly occurs through the formation of supramolecular uranium–phenolic networks on the surface of the microporous PA membrane (Fig. 14A and B). The potential application of the as-prepared polyphenol-functionalized membrane for uranium extraction from simulated seawater was evaluated. The uranium extraction efficiency of the membrane was not significantly affected by the multiple competing ions even at excess concentrations in simulated seawater (Fig. 14C), demonstrating its satisfactory selectivity. Based on the results of the laboratory test, marine field experiments were further carried out in different sea areas in China. Similarly, the polyphenol-functionalized membrane displayed high selectivity, high extraction efficiency and high permeate flux in the actual seawater test. After processing 10 L of seawater from the East China Sea, a total uranium mass of 27.81 μg with an extraction rate of 84% was acquired, and the extraction rate could also reach more than 80% even after 10 extraction cycles (Fig. 14D–F). The techno-economic analysis suggested that the utilization of natural polyphenols significantly reduced the cost of membrane synthesis (Fig. 14G). The deployment and cost of uranium extraction from real seawater using a polyphenol-functionalized membrane were estimated under the assumption that different potential energy drives were used. Compared with the passive adsorption of uranium in seawater, the integration of additional energy-driven systems with adsorbents could improve the uranium extraction efficiency, offering a promising solution to UES. Meanwhile, the results showed that the final cost of uranium production could be significantly reduced by integrating the membrane-based separation system with a natural tide-driven process or a desalination plant-integrated process (Fig. 14H).
 | ||
| Fig. 14 The preparation and adsorption performances of polyphenol-functionalized membranes. (A) SEM image of PIM membranes before and after polyphenol-functionalization and scheme highlighting uranium adsorption and recycle of the membranes. The uranium–phenolic network constructed from uranium and polyphenol building blocks is also presented. (B) Schematic illustration of the complex seawater environment containing various co-existing metal ions. (C) Extraction efficiency for uranium in the presence of various competing ions. (D) Marine field study site location on the local map of the East China Sea. (E) Extraction rate and uranium extraction mass by three layers of polyphenol-functionalized membranes under different seawater flow volumes. The half-wave rectified alternating current electrochemical (HW-ACE) method and conventional physicochemical adsorption approaches were used to compare polyphenol-functionalized membranes. (F) Extraction mass/moles of various metal ions after processing 10 L seawater. The inset displays the change in uranium extraction rate during 10 extraction cycles. (G) Scheme and material cost analysis of a polyphenol-functionalized membrane system for uranium extraction. (H) Schematic representations and combined cost analysis of three different routes that could potentially be applied in practical applications of polyphenol-functionalized membranes for uranium extraction. Reproduced with permission.144 Copyright 2019, Royal Society of Chemistry. | ||
Inspired by the high mass transfer efficiency of ubiquitous natural fractal networks, a biomimetic porous membrane based on AO-functionalized polymers of intrinsic microporosity (PIMs)146 with hierarchically porous structures and rich binding sites was developed by Wen and colleagues.145,147 The membrane, labeled AO-PIM-1, was fabricated by a non-solvent-induced phase separation method, resulting in a large number of interconnected multiscale channels (Fig. 15A). When employed for uranium extraction, a two-step adsorption behaviour was observed where uranium first entered the membrane through the artificial macropores on the surface, and then diffused into smaller biomimetic branching channels. Ultimately, uranium was conveyed to the intrinsic micropores of the polymer where adsorption occurred. This design allowed the high mobility of uranium in the AO-PIM-1 membrane while maintaining the high SSA for improving the uranium adsorption potential. Adsorption experiments were subsequently carried out to evaluate the practical performances of the AO-PIM-1 membrane. Upon being immersed in uranium-spiked water, an obvious colour change from white to yellow by the naked eye was captured due to the successful uranium uptake. The emergence of characteristic double peaks of U 4f5/2 and U 4f7/2 in the XPS spectrum of the membrane after adsorption further verified the presence of uranium on the membrane surface (Fig. 15B). The adsorption kinetics results confirmed the two-step adsorption process of uranium on the hierarchically porous AO-PIM-1 membrane, and the saturated adsorption capacities of the membrane with respect to the 8 ppm, 16 ppm and 32 ppm uranium-spiked solutions were 124.17 mg g−1, 197.92 mg g−1 and 345.94 mg g−1, respectively, after 50 h adsorption (Fig. 15C). Compared with the solution-cast membrane with only intrinsic microporosity, a 20-fold higher uranium adsorption capacity was observed owing to the rapid diffusion of uranium species in the biomimetic membrane with hierarchical structures and high SSA (Fig. 15D). Additionally, the AO-PIM-1 membrane exhibited an adsorption capacity of 9.03 mg g−1 from natural seawater after four weeks, and no perceptible decrease of the capacity was observed after five adsorption/desorption cycles. This work provides a new concept for membrane design inspired by biological structures for uranium enrichment.
 | ||
| Fig. 15 Uranium adsorption on the biomimetic porous membrane based on AO-functionalized PIM-1 with a hierarchically porous structure inspired by natural fractal networks. (A) Schematic illustration of the bioinspired hierarchically porous membrane, and mechanism of the two-step adsorption behaviour of uranium by the membrane where aqueous uranium molecules first entered the membrane through the artificial macropores on the surface, and then diffused into the smaller biomimetic branching channels, and ultimately conveyed to the intrinsic micropores of the polymer where adsorption occurred. (B) Digital photographs (top) and XPS profiles (bottom) of the AO-PIM-1 membrane before and after uranium loading. (C) Adsorption kinetics data of uranium on the AO-PIM-1 membrane in uranium-spiked water samples with different initial uranium concentrations. (D) Comparison of adsorption kinetics data between the AO-PIM-1 membrane and two types of solution-cast membrane in 32 ppm uranium-spiked solution. Reproduced with permission.145 Copyright 2022, Springer Nature. | ||
As discussed above, AO-functionalized polymeric adsorbents for UES can be shaped into diverse forms by various polymerization methods and processing techniques.148–153 Constant efforts are being made to improve their structures and morphologies for promoting their adsorption ability towards uranium in seawater. Currently, polymer fiber-based adsorbents are considered to be the most ideal form for UES because of their robust structure and easy deployment for marine engineering. The employment of advanced processing techniques allows the preparation of ultrafine AO-functionalized fibers with high SSA. However, the trade-off between these structural advantages and mechanical strength needs to be considered. The development of hydrogel-based adsorbents for UES is an active attempt, considering the simple synthesis of hydrogels with hydrophilic 3D networks. Nevertheless, owing to their low mechanical strength and salt resistance, whether hydrogel materials can meet the requirements of field tests of UES remains to be further explored. In addition, exploiting the membrane technology for UES is still in its infancy. On the one hand, functionalized membranes with high affinity and selectivity to uranium are scarcely reported, not to mention the biofouling problem. On the other hand, energy-consuming systems are required to be integrated with the membrane modules. Thus, a rigorous techno-economic analysis of the membrane-assisted UES system needs to be made.
3.2. Strategies for improving selectivity
The ocean is a high-salinity ecosystem due to the existence of various metal ions (e.g., Na+, K+, Mg2+, Ca2+) with much higher concentrations than uranium. The binding ability of uranium adsorbents is found to be significantly inhibited in such a competitive environment.154 Particular attention has been given to vanadium (V), which represents one of the strongest competitors of uranium in seawater. Even with a relatively lower concentration (∼1.9 ppm) than uranium in seawater,155 V is more strongly bound to AO-functionalized adsorbents, leading to a decrease in their adsorption capacity towards uranium.110 Thus, shaping adsorption materials with selectivity to uranium remains one of the fundamental challenges for UES. In recent years, several intriguing strategies have been developed to endow adsorbents with high selectivity to uranyl ions, including utilization of ion imprinting technology, bio-inspired ligands based on protein engineering, and synergistic chelation approaches, which will be comprehensively described and analyzed in this section.To create ion-imprinted polymers (IIPs), two steps are typically involved in which the imprinted ions are first introduced during the cross-linking polymerization of the chelating monomers, followed by the elution of the imprinted ions, yielding polymeric network materials with pores that are complementary in shape, size and ligand orientation to those of the template ions (Fig. 16A).158 The memory effect produced by the preparation process of IIPs endows the adsorbent materials with specific recognition sites, high binding capacity and selectivity towards the target uranyl ions. Numerous efforts have been made for the preparation of uranyl ion-imprinted polymers while evaluating influencing factors, such as the type of chelating ligand, cross-linker, and initiator. Chelating ligands play a pivotal role in the synthesis of IIPs by providing functional groups to bind with uranyl ions to form pre-polymerization complexes.159 The affinity of the ligand for uranyl ions directly affects the affinity of IIPs, which determines the selectivity of the recognition site.160
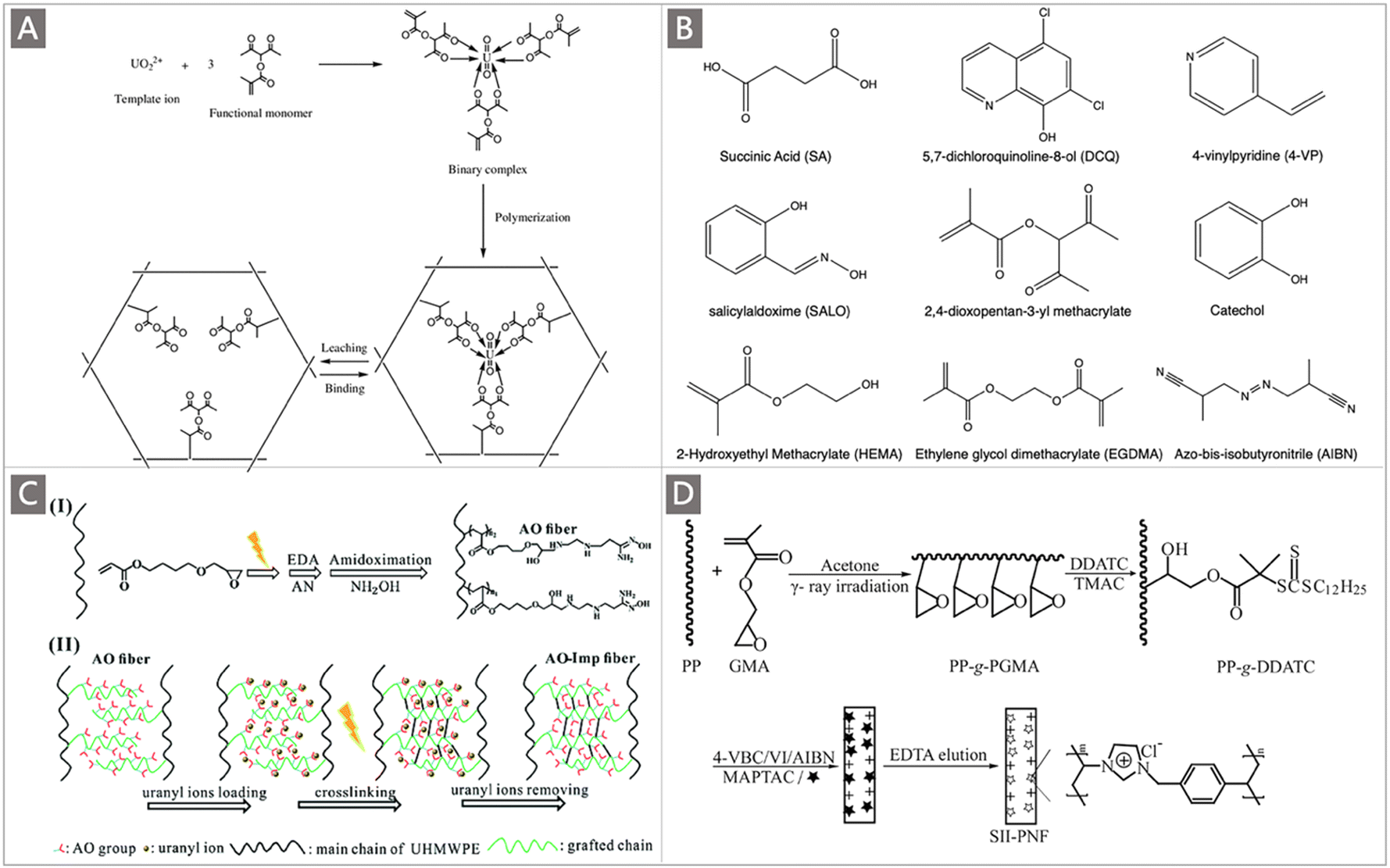 | ||
| Fig. 16 (A) Schematic illustration of the typical preparation process for UO22+-templated IIP. Reproduced with permission.162 Copyright 2013, Springer Nature. (B) Common chelating ligands, cross-linkers, and initiators used in IIP synthesis for UES. (C) Schematic illustration of the preparation of AO-Imp fibers based on radiation-induced crosslinking. Reproduced with permission.165 Copyright 2019, Royal Society of Chemistry. (D) Schematic illustration of the fabrication of the SII-PNF adsorbent. Reproduced with permission.166 Copyright 2017, American Chemical Society. | ||
In earlier studies, several typical chelating ligands such as succinic acid (SA), 5,7-dichloroquinoline-8-ol (DCQ), 4-vinylpyridine (4-VP), salicylaldoxime (SALO), and 2,4-dioxopentan-3-yl methacrylate (Fig. 16B) were screened for UES.161–163 In all cases, these IIPs showed improved uranium extraction capacity compared with the corresponding non-imprinted polymers. Good selectivity to uranium was observed, achieving >99% extraction of uranium over a wide range of cations with different valence states, including Ni2+, Co2+, Mn2+, Zn2+, Cu2+, Fe3+ and Th4+. Unfortunately, the bonding force of these functional monomers towards uranium was not strong enough, and the resultant IIPs only showed moderate uptake for uranium. In the polymerization process, a cross-linker is utilized to fix the monomers around the templates, which also play an important role in delivering mechanical stability and controlling the particle size, porosity and uniformity of IIPs. Divinylbenzene (DVB), 2-hydroxyethyl methacrylate (HEMA) and ethylene glycol dimethacrylate (EGDMA) were three commonly used cross-linkers for the preparation of uranyl ion-imprinted polymers.162,164
Interestingly, a novel method for preparing IIP adsorbents without introducing a crosslinking reagent was proposed.165 Firstly, AO-functionalized UHMWPE fibers were loaded with template uranyl ions, and then radiation-induced crosslinking was performed instead of using a crosslinking reagent. The AO-Imp fibers were obtained after the elution of uranyl ions (Fig. 16C). Because of the high binding capacity of AO ligands to uranium and the special complementary structure brought by IIT, the AO-Imp fibers exhibited lower adsorption for other metal ions, such as V, Fe, and Co, compared with uranium. Nevertheless, a limited adsorption capacity (1.0 mg g−1) of the AO-Imp fibers for uranium was observed during the test in the simulated seawater.
Additionally, the selection of an appropriate polymerization procedure is vital to produce IIPs with desirable properties. Bulk polymerization has been widely applied in the imprinting process. However, the resultant IIPs always suffer from the nonuniform distribution of particle size, swelling and shrinkage effects, and skeleton distortion.157 Much attention has been shifted to improving polymerization techniques to overcome these problems. For instance, Zhang and colleagues prepared a surface ion-imprinted PP nonwoven fabric (SII-PNF) by copolymerizing 4-vinylbenzyl chloride (4-VBC) and 1-vinylimidazole (VI) in the presence of template ions ([UO2(CO3)3]4−), and then eluting the template ions with EDTA solution (Fig. 16D).166 After contact with a uranium aqueous solution, the recognition cavities complementary to the template ions formed in the polymer matrix provided a maximum adsorption capacity of 133.3 mg g−1 within 15 h at pH 8.0. Besides, the SII-PNF indicated higher selectivity towards uranium with a high Kd value of 201 over vanadium (Kd = 98.8) and other competing ions such as Zn2+, Cu2+, Co2+, Ni2+, and Fe3+ (Kd < 10).
As mentioned above, owing to their large surface areas, well-defined porosities, modular structure and reticular variety, porous organic frameworks are favourable platforms to incorporate functionalities for developing adsorbents.167 The adsorbents employing porous frameworks as the scaffolds and chemically grafting uranyl complexing moieties inside the pores have revealed good performance in terms of adsorption capacity and adsorption rate.95,168 However, they lack selectivity in the presence of interfering ions in seawater. In this regard, the introduction of molecular imprinting technology (MIT) into porous skeletons to afford molecularly imprinted polymers with tailor-made binding sites provides an intriguing route to overcome the limitation (Fig. 17A).169,170
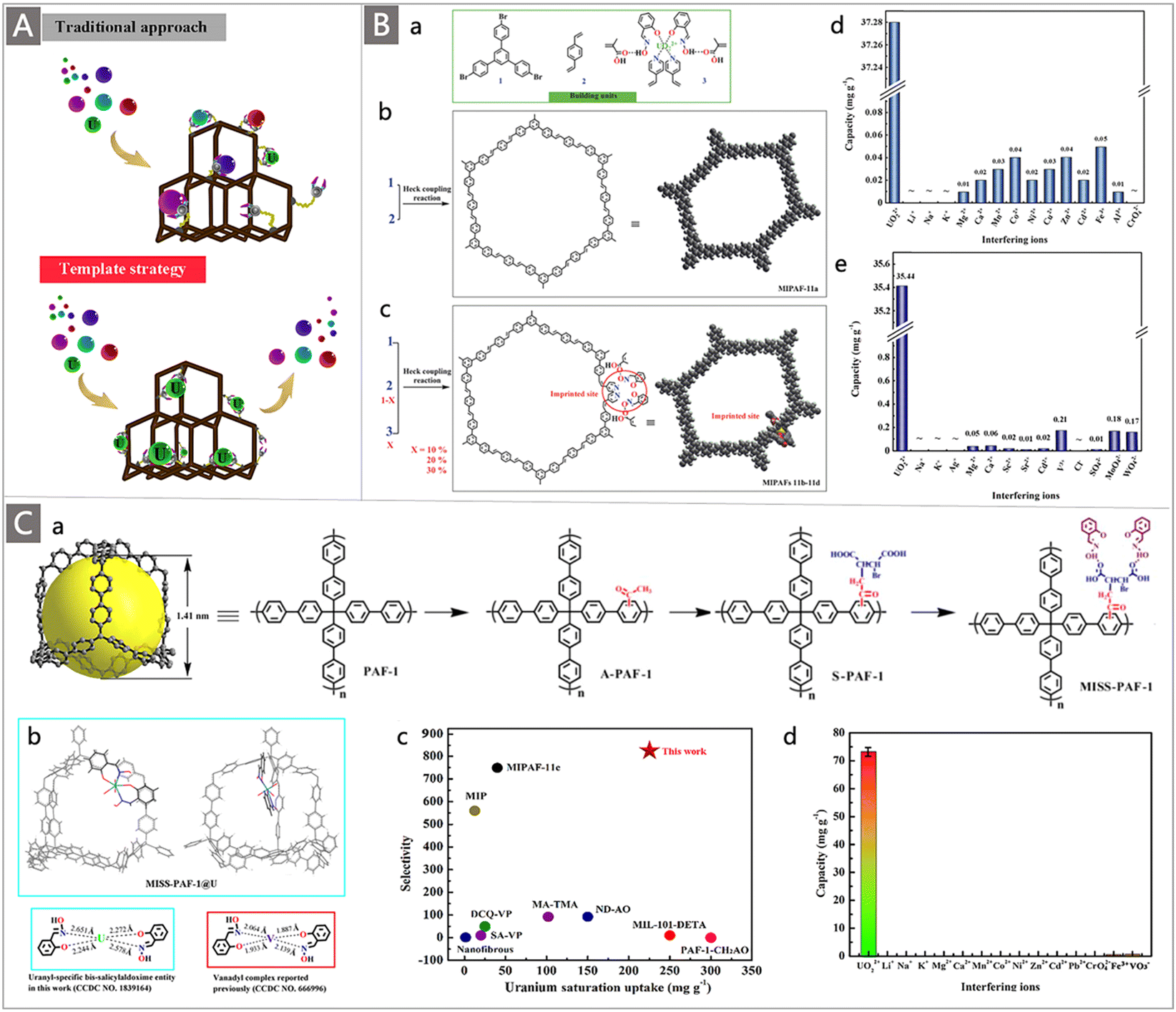 | ||
| Fig. 17 (A) Illustration of a traditional approach using simple coordination ligands and a template strategy using a coordination complex for capturing uranium. Reproduced with permission.172 Copyright 2019, American Chemical Society. (B) (a) Building units used for the construction of MIPAFs; schematic illustration of the preparation route for MIPAF-11a (b) and MIPAF 11b–11d (c) via the Heck-coupling reaction; adsorption capacities of UO22+ against different interfering ions in a mixed aqueous solution (d) and in the simulated seawater (salinity of 30) (e). Reproduced with permission.171 Copyright 2018, Wiley-VCH. (C) (a) Schematic illustration of the preparation route for MISS-PAF-1; and (b) possible patterns of a coordination complex fixed onto the MISS-PAF-1 skeleton (top) and comparison of coordination bond lengths for uranium and vanadium complexes (bottom); (c) comparison of uranium saturation uptake and ion selectivity between MISS-PAF-1 and other reported materials; and (d) adsorption selectivity of UO22+ against various interfering ions for MISS-PAF-1 in simulated seawater. Reproduced with permission.172 Copyright 2019, American Chemical Society. | ||
The first study on the integration of MIT into PAF structures (MIPAF) via state-of-the-art coupling chemistry was reported by Zhu and co-workers,171 where a uranyl-coordinating complex was doped into a PAF framework to give a MIPAF. Specifically, a UO22+-imprinted complex consisting of SALO, MA, and 4-VP through free assembly was first prepared, and a series of PAF networks were subsequently constructed using the prepared complex together with 1,3,5-tris(4-bromophenyl)benzene and p-DVB via the Heck coupling reaction. After removing template UO22+ ions, the resulting structures (MIPAF-11c) provided suitable anchors able to achieve a desired local bonding geometry that was specific to UO22+ (Fig. 17Ba–c). Combined with the large accessible surface area (182 m2 g−1), MIPAF-11c afforded a uranium uptake of 37.28 mg g−1 in a standard UO22+ solution containing 20 ppm UO22+, which was nearly 4-fold higher than the capacity of traditional MIPs prepared in the control experiment. A uranium capacity of 35.44 mg g−1 was also captured from the 7.05 ppm UO22+ spiked simulated seawater. Remarkably, the MIPAF-11c material achieved an unprecedented selectivity coefficient (Kd) over 746 (UO22+/Fe3+) in a mixed aqueous solution with a variety of interfering ions (Li+, Na+, K+, Ca2+, Mg2+, Co2+, Mn2+, Zn2+, Cu2+, Cd2+, Ni2+, Al3+, Fe3+, and CrO42+) (Fig. 17Bd), and a high Kd of 171 (UO22+/V3+) in a simulated seawater (salinity of 30) with various inorganic interferents (Na+, K+, Ag+, Ca2+, Mg2+, Sr2+, Sc2+, Cd2+, V3+, Cl−, SO42−, MoO42− and WO42−) (Fig. 17Be).
Building on this work, for improving the adsorption capacity while maintaining high selectivity, an imprinted complex (uranyl-specific bis-SALO entity) with a predetermined configuration of oxime fragments was designed by the same group,172,173 which was then tethered onto the PAF skeleton through hydrogen bonding (Fig. 17Ca). In terms of spatial structure, the configuration of the as-prepared imprinted complex is consistent with the fragment of PAF-1, and after releasing template uranyl species, the imprinted sites preserved their structural integrity via π–π interactions and direction for uranium rebinding (Fig. 17Cb). The tailor-made binding configuration where two SALO molecules bound with one uranyl species and a high SSA of 412 m2 g−1 endowed the resultant MISS-PAF-1 adsorbent with a maximum uranium adsorption uptake of 79.80 mg g−1 and an unprecedented selectivity coefficient of over 821 for (UO22+/Co2+) in an aqueous solution containing 7 ppm uranium and various interfering metal ions. The selectivity performance was much higher than those of the aforementioned IIPs (Fig. 17Cc). When contacted with 7.05 ppm UO22+ spiked simulated seawater, MISS-PAF-1 exhibited an uptake of 73.26 mg g−1 with a selectivity coefficient of 113 (UO22+/V3+) (Fig. 17Cd). Finally, an adsorption capacity of 5.79 mg g−1 for uranium was acquired after incubation for 56 days under shaking in the non-spiked real seawater from the Bohai Sea, China.
More recently, a particularly interesting study from the Wang team reported the integration of MIT into MOF structures to investigate their performances for UES.174 In this work, uranyl-imprinted MOF materials (MUUim) were designed through the in situ synthesis strategy where uranyl ions were introduced into a structure containing carboxyl and hydroxyl functionalities derived from the classic UiO-66 type MOF during the synthesis process. The uranyl ions in MUUim and MUUad were found to coordinate with the carboxyl group by forming four coordination bonds, and the formation of hydrogen bonds between the axial oxygen of the uranyl and the phenolic hydroxyl group was demonstrated, endowing the adsorbents with potential binding capacity. After the imprinted ions were eluted, the MOF material (MUUre) showed a high uranium extraction capacity of 7.35 mg g−1 after contacting with natural seawater for 16 days, which was 18.38 times higher than that of vanadium, revealing its good selectivity. Attributed to the pre-addition of uranyl, the most suitable local nanocage structure for targeting uranyl ion binding was acquired, which endowed the adsorbents with significant uranyl binding affinity. Meanwhile, the tunability of porous framework materials offers opportunities for future optimization. It is envisioned that the attractive IIT/MIT synthetic strategies could be utilized in diverse porous materials that have tunable structures and varying pore shapes, generating specific binding sites to complement template uranyl ions and thus allowing for improvement in adsorption capacity and selectivity.
For this concept, Zhou et al. developed a computational program named URANTEIN to screen proteins with potential binding affinity for UO22+ in the Protein Data Bank (PDB).178 After the initial screening, more than 5000 hits were identified. Then the hits were further selected according to their potential stability, steric clashes in the predicted coordination site, and accessibility of the binding site. Ten promising candidate proteins were screened, of which nine proteins were well expressed and four proteins exhibited binding to uranyl. Finally, a super uranyl binding protein (labeled SUP) was designed by inducing several mutations in Methanobacterium thermoautotrophicum, offering high selectivity to uranyl with a Kd of 7.4 fM and an excess of 10000-fold selectivity over other metal ions (e.g., Na+, Mg2+, K+, Ca2+, Sr2+, Rb+, Ba2+, VO2+, Pb2+, Ni2+, Zn2+).
A subsequent study was performed to study the uranyl-SUP system via a combination of DFT, MD, and free-energy simulations.180 It was reported that the binding affinity and selectivity of SUP to uranium were determined by three factors, including the interaction of the amino acid residues in the first shell, the integrity of the second-sphere hydrogen bond network, and the number of water molecules in the first coordination sphere. The advantage of this novel mechanism for UO22+ binding led to the design of a further optimized SUP, specifically the GLU64ASP mutant, which was discovered to show a stronger binding capacity and selectivity towards UO22+ than SUP.180 However, fundamental challenges remain regarding the viable deployment of these engineered proteins.
In this sense, a strategy to integrate SUP proteins with high selectivity into high-order molecular architectures offers a plausible solution to this challenge.184 Benefiting from the concept of covalent protein assembly tools such as genetically encoded click chemistry (GECC) and genetically encoded SpyTag–SpyCatcher chemistry,182,185,186 Sun and co-workers demonstrated the feasibility of covalently stitching SUPs into hydrogel materials181,187 and protein nanofilms188 capable of selective metal ion sequestration. Specifically, SUP proteins were integrated into macroscopic hydrogel materials through the “click chemistry” of thiol-maleimide, and droplets of SUP hydrogels were subsequently processed and cured into monodisperse SUP microbeads with a uniform radius of 165 μm via microfluidic technologies.181 The final monodisperse SUP hydrogel microbeads displayed active binding to uranium, affording a higher enrichment index for uranium over other metal ions (Na+, Mg2+, K+, Ca2+, and Sr2+). However, these SUP hydrogel microbeads showed moderate adsorption for uranium (0.0092 mg g−1) upon contact with the spiked seawater containing 13.7 nmol L−1 uranium.
In a later work, Yang et al. designed a genetically encoded protein polymer through the embedding of the SUP and the monomeric streptavidin into the polymer based on the SpyTag–SpyCatcher chemistry.182 By optimizing the number of Spy motifs in each monomer and the molar ratio between SpyTag (a 13-residue peptide) and SpyCatcher (a 116-residue domain), a Spy-SUP network with a 3D structure and a relatively low cross-linking degree was obtained, which was more permissive to the passage of uranyl ions. The polymer network demonstrated enhanced uranium adsorption by 10% through the immobilization of the SUP, which was ascribed to the electrostatic interaction and hydrodynamic resistance of the polymer in solution. Finally, another functional module (mSA) was introduced and the Spy-SUP-mSA polymers achieved 50–60% uranium removal rates after incubation and magnetic bead treatment in the uranium spiked water samples. Additionally, the embedding of mSA allowed uranyl retrieval without the need for centrifugation.
Wang and colleagues recently reported a series of modified SUP-based adsorbents for selective binding of uranium.141,183 Inspired by the high tensile strength and high elasticity of spider silk fiber, a chimeric spidroin-based SUP (SSUP) was constructed by fusing the SUP with spidroin, and further spun into protein fibers via a biomimetic spinning technology (Fig. 18Aa–c).183 The freshly prepared wet SSUP fiber exhibited a water-rich hydrogel-like structure and a diameter of 220 ± 8 μm with a white colour, which shrank to 21 ± 8 μm after drying in air due to the change in water content and protein conformation (Fig. 18Ad and e). Moreover, the spider silk protein component gave the SSIP fiber high mechanical properties, with a tensile breaking strength of up to 128.26 MPa. The water-rich hydrogel-like structure of the wet SSUP fiber could provide abundant hydrophilic intermolecular space for the entry of uranium (Fig. 18Af), affording fast uranium adsorption within 30 min with a saturation capacity of 15.91 mg g−1 in 16 ppm uranium-containing simulated seawater. When tested in a flow-column system with natural seawater, a fast saturation time of 3.5 days with a uranium uptake of 12.33 mg g−1 was observed, which was more than 6 times that of vanadium, indicating its high selectivity to uranium.
 | ||
| Fig. 18 Strategies for developing a spidroin-based protein fiber for selective binding of uranium: (A) (a) the domain composition of the SSUP fiber; (b and c) the synthesis of the SSUP fiber via biomimetic spinning technology; (d) morphology of the wet SSUP fiber; (e) SEM images of the dried SSUP fiber and the fracture surface of the dry fiber; and (f) the water-rich hydrogel-like structure of the wet SSUP fiber and its shrinkage after drying in air. Reproduced with permission.183 Copyright 2019, Wiley-VCH. (B) (a) Comparison of the 3D structures of spidroin and SUP; (b) the domain composition and uranium binding mechanism of the DSUP fiber; (c) the spinning process of the DSUP fiber; (d) photo of the loofah-shape of the DSUP fiber; (e) adsorption kinetics data for uranium and other competing ions in natural seawater; and (f) comparison of the uranium uptake capacities of different adsorbents in natural seawater. Reproduced with permission.141 Copyright 2020, Wiley-VCH. | ||
Inspired by the similarity of the 3D structure of SUP to that of spidroin (Fig. 18Ba), the same group replaced the spidroin in SSUP with SUP and constructed a novel dual-SUP (DSUP) chimeric protein containing two copies of SUP, which was subsequently spun into a protein fiber (Fig. 18Bb and c).141 The freshly prepared wet DSUP fiber exhibited a white colour with a diameter of 300 ± 11 μm, which shrank to 22 ± 2 μm after drying in air. Through the cross-interaction of protein nanofibers, DSUP hydrogel fibers presented a loofah-like structure (Fig. 18Bd), which exposed numerous functional uranium chelating sites, and the abundant connecting points between the nanofiber shared the tension, endowing the DSUP with outstanding mechanical properties. Remarkably, a significant improvement in the maximum adsorption capacity of the DSUP (25.73 mg g−1, obtained at pH 5.0) for uranium was observed, compared with that of SSUP (11.92 mg g−1, obtained at pH 6.0), which was ascribed to the increasing uranium binding sites by the replacement of spidroin. Fast uranium adsorption within 30 min but with a higher saturation capacity of 26.40 mg g−1 in 16 ppm uranium-containing simulated seawater was captured for the DSUP. Moreover, when the DSUP was placed into columns filled with natural seawater, it achieved an ultra-short saturation time of 3 days and an unprecedented uranium uptake capacity of 17.45 mg g−1 (Fig. 18Be and f). To date, this value has remained the highest uranium adsorption capacity reported in natural seawater. Meanwhile, the DSUP also shows excellent selectivity to uranium in the presence of other interfering metal ions (e.g., V, Cu, Fe and Zn) in natural seawater (Fig. 18Be).
In their follow-up work, owing to the specific recognition of uranyl, the DNAzyme was utilized as an aptamer polymerized into a DNA hydrogel (DNA-UEH) through rolling circle amplification (RCA) technology, for selective recognition of uranyl ions in seawater.142 Measurements were made in uranium spiked simulated seawater with a uranium concentration of 8 ppm at pH 5.0, with an equilibrium adsorption capacity of 13.71 mg g−1 obtained after interacting for 24 min. Besides, impressive selectivity was observed for uranyl over other competing ions present in seawater, even when added in more than 100 times excess. Deployment of the hydrogel DNA-UEH in natural seawater resulted in a high adsorption capacity of 6.06 mg g−1 in 6 days, which was 17.95 times higher than that of vanadium. Finally, mechanistic studies demonstrated that the oxygen atoms from the phosphate and carbonyl groups of the DNA molecule coordinate with uranium atoms to form a five-coordination DNA nano-pocket structure, which was responsible for its high affinity and high selectivity to uranyl.
To sum up, considerable efforts have been put into investigating the potential of biological substances for developing effective adsorbents with specific recognition of uranyl ions. The diversified functional groups, such as carboxyl, amino, phosphate and hydroxyl, in proteins, DNA and some other biological macromolecules participate in uranyl binding in a synergistic mode, which affords a unique ability for uranium recognition. Nevertheless, for a realistic application in UES, there remain significant challenges for the feasible deployment of these biomacromolecule-containing adsorbents. Besides, obstacles also lie in their high cost caused by the complicated preparation procedures and the depletion in the biological systems of oceans. Thus, more research is needed to demonstrate the feasibility of this class of biology-based adsorbents.
Meanwhile, an intriguing bio-inspired route that emerged recently for developing selective uranium adsorbents is based on biological protein engineering as detailed in Section 3.2.2. Since the identified uranyl binding biomacromolecules rely on the collaborative chelating effect of diverse functional groups, researchers have realized that, instead of relying on single strong binding from a randomly oriented site, the rational design and appropriate spatial arrangement of binding sites that allow collaborative chelation could be a promising strategy for improving the affinity to uranium.193–195 Specifically, the preorganization of the binding sites enables the formation of artificial uranyl nanotraps to achieve the specific recognition of uranium (Fig. 19A).196
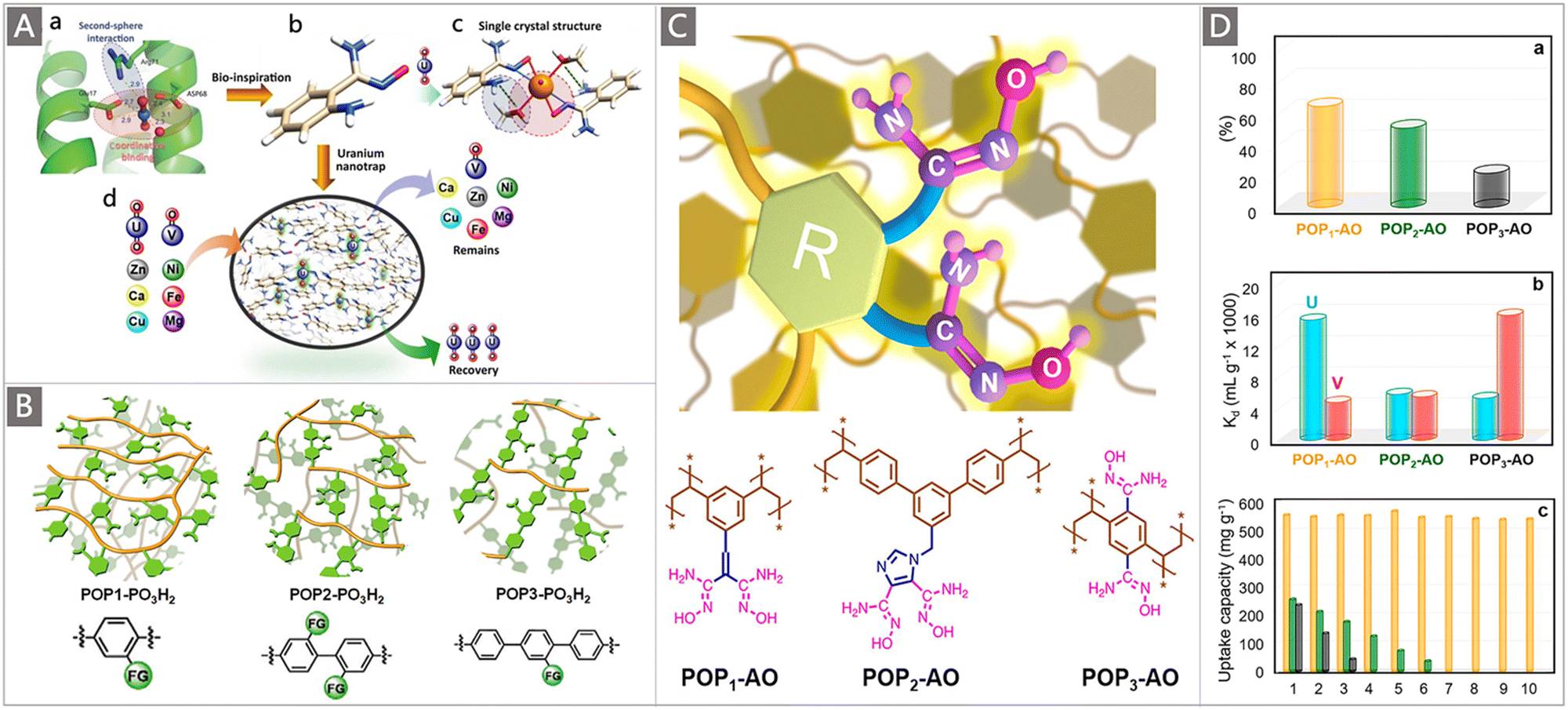 | ||
| Fig. 19 (A) Schematic illustration of the design of a high-affinity and selective uranium nanotrap inspired by the protein biological system. (a) Uranyl-binding pocket details of the SUP protein. (b) The designed structure of a uranyl-binding moiety inspired from (a). (c) Overview of the crystal structure of the uranyl complex in (b). (d) The construction of a selective uranium nanotrap that allows the enrichment of uranium over other metals. Reproduced with permission.196 Copyright 2019, Elsevier. (B) Schematic illustration of the structures of various phosphorylurea functionalized polymers including POP1-PO3H2, POP2-PO3H2 and POP3-PO3H2. Reproduced with permission.193 Copyright 2021, Wiley-VCH. (C) Schematic view of diamidoxime ligands with various R groups as specific “hooks” decorated on porous frameworks for uranyl recognition, and the corresponding structures of diamidoxime-functionalized POPs. (D) The competitive adsorption and recyclability performances of these diamidoxime-functionalized POPs: (a) the remaining uranium adsorption capacity of various POPs in the presence of equal concentrations of vanadium; (b) the Kd values for uranium and vanadium for various POPs; and (c) the recyclability of various POP adsorbents for ten consecutive cycles (orange indicates POP1-AO; olive corresponds to POP2-AO; and black represents POP3-AO). (C and D) reproduced with permission.194 Copyright 2021, American Chemical Society. | ||
Intrigued by this concept, Ma and colleagues experimentally demonstrated the management of the spatial distribution of binding sites in adsorbents and realized their synergistic binding towards uranium.193 Several adsorbents were designed, aiming at establishing a correlation between the adsorption performances and the spatial distribution of chelating groups in adsorbents. In this contribution, phosphorylurea ligands were chosen as chelating groups and POPs as the support, which allowed manipulating the compositions and local environments with high fidelity. Accordingly, various amine moieties including aniline, 2,2′-biphenyldiamine, and [1,1′:4′,1′′-terphenyl]-2′-amine were first installed on the polymerizable vinyl group, followed by the polymerization of these monomers to obtain amino polymers, which were subsequently converted into phosphorylurea-derived polymers (denoted POP1-PO3H2, POP2-PO3H2 and POP3-PO3H2, respectively) (Fig. 19B) through treatment with diethoxyphosphinyl isocyanate and hydrolysis reactions. The coordination between the phosphorylurea functionalities and uranium species was first confirmed through spectroscopic techniques. Then, the adsorption performances for uranium of the as-prepared adsorbents were evaluated. It was observed that the adsorbent with chelating groups populated adjacently in a spatially locked manner (POP2-PO3H2, Kd ∼2.5 × 108 mL g−1) displayed up to two orders of magnitude improvement in selectivity towards uranium compared to a random and isolated manner (Kd ∼1.8 × 107 and ∼1.5 × 106 mL g−1 for POP1-PO3H2 and POP3-PO3H2, respectively). This discrepancy in binding affinity to uranium confirmed the role of ligand distribution in the adsorbents. Furthermore, the rational arrangement of ligands can exert a synergistic binding effect, and thus enhance binding affinity. Upon contact with synthetic seawater, the POP2-PO3H2 adsorbent displayed superior extraction efficiency, affording an uptake capacity of 304 mg g−1, while POP1-PO3H2 and POP3-PO3H2 showed lower uptake capacities of 216 mg g−1 and 105 mg g−1. After 56 d in natural seawater, a uranium recovery capacity of 5.01 mg g−1 was achieved for POP2-PO3H2.
Building on this work, the same group further improved the strategy of collaborative binding through experimental and theoretical analyses. A series of smart adsorbents with uranyl-specific “hooks” were designed and fabricated by precisely manipulating the functional moieties in the adsorbents at the molecular level to achieve high affinity and selectivity for uranium.194 POPs were selected again as adsorbent materials, while AO ligands were chosen as chelating sites. A series of diamidoxime ligands with various R groups as specific “hooks” for uranyl recognition were decorated on the nanospace of POPs to afford uranium nanotraps, namely POP1-AO, POP2-AO, and POP3-AO, respectively (Fig. 19C). Upon exposure to uranium solutions, the resulting POPs displayed dissimilar coordination abilities towards uranium. Steeper adsorption for POP1-AO was observed at low uranium concentration and a higher Kd value was acquired for POP1-AO (Kd = 1.1 × 106 mL g−1), compared to those of POP2-AO (Kd = 3.2 × 105 mL g−1) and POP3-AO (Kd = 1.4 × 105 mL g−1), indicating the stronger binding affinity of POP1-AO towards uranium. Competitive adsorption experiments were subsequently carried out by exposing the POPs to mixed solutions spiked with uranium and vanadium. The decreasing uranium concentration was detected and 65%, 51%, and 22% uptake capacities were demonstrated by POP1-AO, POP2-AO, and POP3-AO, respectively (Fig. 19Da). The Kd value of POP1-AO for U was determined to be more than three times greater than that for V, while POP2-AO exhibited comparable Kd values for U and V, and POP3-AO showed a higher binding affinity towards V than that for U (Fig. 19Db), confirming the effectiveness of controlling the R group to enhance the collaborative binding to uranium. The recycle performances were also tested where the performance of POP1-AO could be maintained for ten consecutive cycles, while obviously decreased uptake performance was observed for POP2-AO and POP3-AO (Fig. 19Dc). Additionally, the adsorption capacity in natural seawater was evaluated and the results indicated that the optimized uranium nanotrap could extract more than one-third of the uranium in seawater. The qU of POP1-AO could reach 8.4 mg g−1 after 56 d, affording an enrichment index of 3836. These results suggested that the collaborative chelation of ligands might be a promising design principle for creating highly-selective uranium adsorbents.197
An interesting follow-up study by the Wang group involved MOF adsorbents decorated with carboxyl and amino groups, which was inspired by the high binding affinity of carboxyl and amino groups in the SUP protein for uranium sequestration.195 To demonstrate the role of the spatial arrangement of functional ligands for selective binding to uranyl ions, the classical UiO-66 MOF material served as a supporter and carboxyl and amino groups were introduced into the UiO-66 skeleton at different positions to construct nano-pockets of different spatial structures in MOFs (Fig. 20A). These six different combinations all formed cubic octahedron shaped particles with a similar morphology and crystal structure to UiO-66, with the particle size ranging from 368 nm to 429 nm (Fig. 20B). Especially, UiO-66-3C4N acquired the highest SSA of 1222 m2 g−1 among the MOFs, which achieved the highest adsorption capacity of 393.97 mg g−1 at pH 8.0 in 8 ppm uranyl-containing aqueous solution (Fig. 20Ca). When immersed in 8 ppm U-spiked simulated seawater with a pH of 8.0, UiO-66-3C4N still displayed the highest qU of 190.27 mg g−1 (Fig. 20Cb). Also, a high qU of 380.30 mg g−1 with an uptake rate of 23.77% within a saturation time of 8 h in 16 ppm U-spiked simulated seawater was detected for UiO-66-3C4N (Fig. 20Cc). The adsorption capacity was further promoted by increasing the initial uranium concentration, where a maximum adsorption of 642.92 mg g−1 was obtained in a 128 ppm uranyl nitrate aqueous solution (Fig. 20Cd). High selectivity to uranyl ions even against 104 excess interfering ions with a uranyl-binding Kd of 1.67 pM at pH 8.9 was achieved (Fig. 20Ce and f). Moreover, the created UiO-66-3C4N nano-pocket demonstrated high selectivity towards uranium in natural seawater in the presence of competing ions including V, Co, Cr, Cd, As, Ag, Ba, Hg, Ti, and Pb. A saturation capacity of 6.85 mg g−1 was acquired after 28 d, which was around 17 times higher than that of vanadium (Fig. 20Cg). In this work, the MOF adsorbents decorated with carboxyl and amino groups with appropriate arrangements provided specific nano-pockets, enhancing the effectiveness and selectivity of the coordination to uranyl ions.
 | ||
| Fig. 20 (A) Schematic illustration of the nano-pocket in the MOF for uranyl capture. (B) The ligand compositions and morphologies of UiO-66 and the derived MOFs. (C) The adsorption performances of UiO-66-3C4N MOF: (a) optimal pH for uranyl adsorption in water; (b) comparison of the uranium adsorption capacities of UiO-66-3C4N and the other MOFs in 8 ppm U-spiked simulated seawater at pH 8.0; (c) adsorption kinetics and the corresponding fitting data in U-spiked simulated seawater at pH 8.0; (d) adsorption isotherms and the corresponding fitting data in water at pH 6.0; (e) adsorption selectivity to uranium against the other competing metal ions; (f) competition assay versus total carbonate for uranyl at pH 8.9; and (g) adsorption capacities of uranium and other competing metal ions in natural seawater. Reproduced with permission.195 Copyright 2020, Wiley-VCH. | ||
Fundamentally, improving the selectivity of uranium adsorbents relies on the engineering of uranyl–ligand interactions to reach a more thermodynamically stable (energetically favoured) state. Based on the latest insights into the coordination chemistry of uranium, promising approaches have been proposed to address the traditional concern for the competition of vanadium ions. More importantly, a nascent concept to tune the preorganization of the binding sites has uncovered a new paradigm in the design of uranium adsorbents with high selectivity. By regulating the spatial arrangement of ligands and their coordinative binding to uranium, a series of customized uranyl nanotraps mimicking the binding motifs found in biomacromolecules have been designed, exhibiting precise recognition of uranyl ions. This principle for material design is important but still in its infancy. More in-depth studies are needed to reveal the subtle structures of the uranyl–ligand complexes in biological substances to optimize the preorganization principle for developing more advanced uranium adsorbents.
3.3. Strategies for improving biofouling resistance
Marine biofouling is a serious issue associated with the deployment of adsorbents in the ocean, arising from the undesirable accumulation of marine microorganisms, plants and animals on submerged surfaces.198 The growth of biofouling in marine environments is usually divided into four stages, as illustrated in Fig. 21.199,200 The process begins with the rapid formation of a conditioning film within a few seconds of immersion in seawater due to the initial adsorption of organic molecules (e.g., proteins, glycoproteins and polysaccharides). The second stage involves the creation of a biofilm matrix by the settlement and colonization of pioneer bacteria. Then, microfouling occurs since the biofilm provides nutrients for diatoms and macroalgal spores. The final stage includes the increased capture of larvae of marine microorganisms (e.g., macroalgae, barnacles and invertebrates) on the fouled surface.201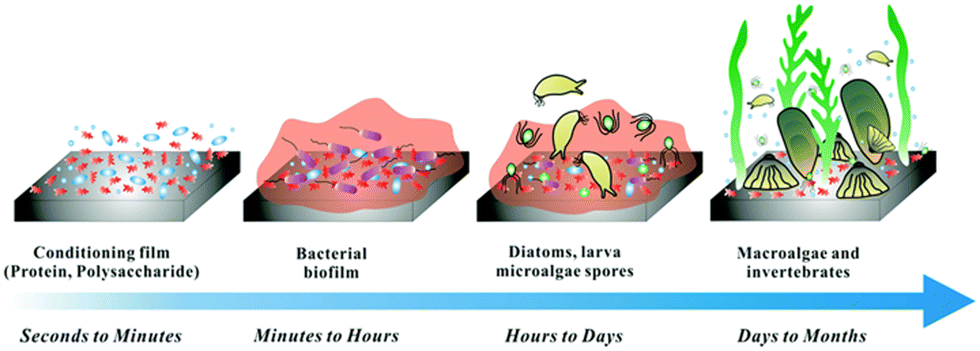 | ||
| Fig. 21 The typical growing process of marine biofouling. Reproduced with permission.199 Copyright 2019, Royal Society of Chemistry. | ||
Due to prolonged immersion in the ocean, adsorbents are inevitably subjected to biofouling. This unwanted colonization produces detrimental effects on the adsorbents for UES, with deterioration of adsorbent surfaces, increased roughness, and limited accessibility to the functional ligands, ultimately resulting in a significant reduction in uranium uptake.198 Early experiments have confirmed that marine biofouling could cause a loss of uranium adsorption of up to 30%.202 Another adverse impact is that fouling may lead to a decrease in the reusability of the adsorbents. Owing to the biocorrosion, harsh treatment is required to remove fouling, or damage may be caused by added weight and drag.202 These facts highlight the need for developing adsorbents with strong antibiofouling properties to stabilize uranium extraction in real ocean environments. In the last couple of years, the biofouling associated with adsorbents is of increasing concern, and a variety of adsorbents with antifouling performance have been constructed to combat biofouling. Two strategies called the “attacking” approach and the “defending” approach are popularly used to impart biofouling resistance to uranium adsorbents.203 In this section, we elaborate these strategies utilized in the development of biofouling-proof adsorbents for UES, which have never been systematically summarized elsewhere.
Inorganic nano-particles are well-known antibacterial materials with broad-spectrum antibacterial and persistent characteristics. Among them, silver (Ag) nanoparticles, as a common silver-based antibacterial agent, are widely used in the preparation of marine adsorbents for UES.204–209 Researchers from India first reported the biofouling resistance of a poly(ethylene glycol methacrylate phosphate) (PEGMP) based uranium adsorbent anchored with Ag nanoparticles.204 The Ag nanoparticles exerted bacterial repulsion and bactericidal properties to biofouling control. When the as-prepared Ag@PEGMP was incubated with pre-grown Escherichia coli (E. coli) cells, no growth of the bacteria was observed. Several subsequent reports by the Wang research team combined the Ag nanoparticles with the abundant oxygen-containing functional groups of graphene oxide (GO) to prepare composite adsorbents for minimizing the biofouling effect.206,207,209 Typically, a Ag ion doped ZIF-67 adsorbent (labeled GZA) with GO as a substrate was designed and synthesized.206 The combined effects of GO and Ag ions on algal inhibition and uranium adsorption were assessed, using P. tricornutum ACCC 01625 as model diatoms. The bifunctional adsorbent exhibited an obvious inhibitory behaviour for P. tricornutum with a more than 80% alga death rate after 7 days. The authors attributed the effect to the co-assistance of the functional groups on the GO and the positive charges with Ag ions. The adsorption behaviour of GZA was also investigated where a maximum qU of 602.41 mg g−1 was obtained in a uranium-spiked solution at pH 7.0. However, the GZA adsorbents exhibited a minimal uptake of 4.974 μg g−1 for uranium when contacted with natural seawater collected from the Huanghai Sea, China.
A majority of the inorganic nanoparticles with bactericidal activity are physically loaded onto the matrix for preparing antibiofouling adsorbents. The architectures are facing serious challenges in the ocean because the strong tidal flows could exfoliate the inorganic nanoparticles. Given this limitation, a strategy for constructing antibiofouling adsorbents via covalent cross-linking reactions between antibacterial components and substrate materials was purposefully developed.210–212,222 As a demonstration, Wang and co-workers prepared an antifouling adsorbent by introducing the aminoglycoside antibiotic neomycin into a UiO-66 MOF matrix (labeled Anti-UiO-66).210 The tight covalent cross-linking between the amino groups in neomycin and the carboxyl groups from the MOF matrix endowed the adsorbent with antimicrobial persistence and high reusability (Fig. 22A). Benefitting from the broad-spectrum antibacterial activity of neomycin, Anti-UiO-66 displayed more than 80.0% inhibition rate when co-cultivated with ten bacterial strains from different species at a concentration of 5 mg mL−1 in Luria broth (LB) (Fig. 22B), whereas the bare UiO-66 showed no significant influence on the growth of the bacteria (Fig. 22C). The inhibition rate of 87.0% to the growth of marine bacteria by Anti-UiO-66 was also noted, while no significant inhibition activity was observed for UiO-66, indicating the high antibacterial activity of Anti-UiO-66 (Fig. 22D). When a test was done for recovering uranium from real seawater after 30 d, a qU of 4.62 ± 0.09 mg g−1 was achieved for Anti-UiO-66, which was 24.4% higher than that of bare UiO-66 (3.71 ± 0.07 mg g−1). However, the introduction of antibacterial compounds also led to increased uptake of other competing metal ions, making it lose the selectivity to uranium. Moreover, neomycin was also introduced into two other carboxyl group-containing adsorbents including PIDO fibers and AO-functionalized UHMWPE fibers (labeled AO fibers) to investigate the adaptability of the strategy. After a simple one-step cross-linking reaction, both the PIDO nanofibers and AO fibers displayed inhibitory activity to the marine bacteria, with an inhibition rate of 80.9% and 86.6%, respectively (Fig. 22E). This strategy provides ideas for constructing antibiofouling uranium adsorbents by using antimicrobial compounds containing amino groups.
 | ||
| Fig. 22 (A) Schematic illustration for the construction and function of the Anti-UiO-66 adsorbent for UES. (B) Antibacterial spectrum of Anti-UiO-66. Antibacterial activity towards indicator bacteria (C) and marine bacteria (D) of UiO-66 and Anti-UiO-66. (E) Uranium adsorption capacities of UiO-66 and Anti-UiO-66 in uranium spiked seawater containing different concentrations of V. alginolyticus. Reproduced with permission.210 Copyright 2019, Wiley-VCH. | ||
Antimicrobial polymers are a class of candidates for developing antibiofouling adsorbents.236 They have either an inherent capacity to exhibit antimicrobial activity such as chitosan (CS), polyguanidines, and compounds with quaternary nitrogen groups, or they can act as backbones to fix other bactericides to display their activity.236 Due to the abundant functional groups in each polymer chain, antimicrobial polymers exhibit enhanced and broader-spectrum antimicrobial activity in contrast to single-molecule antibacterial agents or antibiotics.237 Additionally, their robust structures endow antimicrobial polymer-containing adsorbents with stability for long-term use.238
Chitosan (CS), as one of the most representative natural antimicrobial polymers, can effectively kill a variety of fungi and proteins through electrostatic action.239 Besides its intrinsic disinfection ability, the wide availability, nontoxicity, low cost, and strong biocompatibility of CS have attracted extensive attention. More importantly, the abundant NH2 and OH groups of CS endow them with more cross-linking sites and promote the chelation to uranium. Therefore, much effort has been made towards developing CS-containing antibacterial adsorbents for UES.128,211–213,221 For example, a series of robust and antibiofouling PAO aerogels (labeled Anti-PAO) were synthesized by a simple cross-linking reaction of the polymers (Fig. 23A). The resultant Anti-PAO aerogels exhibited high elasticity and flexibility, with marked recoverability even after repeated compressions for 100 cycles under large strains (Fig. 23B). The SEM images recorded the porous structure with massive macropores of the Anti-PAO aerogels (Fig. 23C), which was attributed to the randomly covalent cross-linking and partial overlapping by hydrogen bonding between CS and PAO nanosheets. When contacted with an 8 ppm uranium-spiked solution, the maximum adsorption capacity of the Anti-PAO aerogels reached 1013 mg g−1. The antibacterial activity was determined by incubating PAO and Anti-PAO with bacteria-containing nutrient agar plates for 12 h. Significant inhibition zones were produced around the Anti-PAO aerogels, whereas no inhibition zones were observed for the pure PAO aerogels (Fig. 23D). In the presence of 102 CFU mL−1 marine bacteria-spiked seawater with 8 ppm uranium, Anti-PAO displayed a qU of 694 mg g−1 (Fig. 23E), two times higher than that of the Anti-UiO-66 adsorbent (285 mg g−1) reported by the same team.210 Finally, the Anti-PAO aerogels achieved an uptake of 9.26 ± 0.59 mg g−1 when exposed in a continuous flow-through system with 1 ton of natural seawater after 30 d (Fig. 23F). In addition to CS, other natural antimicrobial agents such as cellulose paper140 and bamboo charcoal214 have also been exploited to prepare antibacterial adsorbents for UES. Benefiting from their broad-spectrum antibacterial activity and good hydrophilicity, these natural composite systems were observed to exhibit satisfactory performances.
 | ||
| Fig. 23 (A) Schematic illustration of the preparation process of Anti-PAO aerogels. (B) Repeated compression tests demonstrating the high elasticity and flexibility of the Anti-PAO aerogels. (C) SEM images of the Anti-PAO aerogels. (D) Antibacterial activities of the Anti-PAO aerogels towards different marine bacteria. (E) Uranium adsorption capacities of the Anti-PAO aerogels in the simulated seawater and in the non-filtered seawater containing different concentrations of marine bacteria. (F) Uranium adsorption capacity in natural seawater from the South China Sea. Reproduced with permission.211 Copyright 2020, Elsevier. | ||
As an important type of antimicrobial agent, guanidine has also been employed in adsorbent materials for UES because of its high water-solubility and excellent bactericidal efficiency.127,159,215–219 Guanidine is generally considered to be one of the strongest organic bases (pKa = 13.6),240 and can inhibit bacterial growth via adhesion to the negatively charged bacterial cell walls and subsequent destruction of Ca2+ salt bridges required for bacterial plaque adhesion or bacterial cell death.203,236 The Hua group was the first to report the antibiofouling properties of a guanidine and AO co-functionalized PP nonwoven fabric, in which GMA was first grafted onto the PP nonwoven fabric via RIGP, followed by the reaction with dicyandiamide (DC) to introduce guanidine groups. After amidoximation, the functionalized adsorbent (PP-g-AO) was obtained.215 The bactericidal function of guanidine for Gram-negative E. coli was observed since there were much fewer bacteria on the fabric surface after modification. Unfortunately, the functionalized fabric showed a moderate uptake of 0.1 mg g−1 after 10 d of exposure to real seawater.
In a later report, a typical antimicrobial polymer, polyhexamethylene guanidine hydrochloride (PHGC), with plenty of amine groups, was first grafted into commercial PAN fibers, and followed by amidoximation and conditioning with KOH to obtain guanidine-containing adsorbents (labeled PAO-G-A) (Fig. 24A).216 A saturation qU of 514.0 mg g−1 was acquired in 20 ppm uranium solution at pH 8, and a qU of 261.1 mg g−1 was obtained in 8 ppm uranium-spiked natural seawater. The tests in bacterial solution cultivated from seawater showed remarkable antimicrobial activity of PAO-G-A against marine bacteria with an inhibition rate of 99.46% at a high concentration of 108 CFU mL−1, whereas the bare PAN fibers showed no antimicrobial activity on the bacteria (Fig. 24B). More strikingly, the antimicrobial properties of the adsorbents were evaluated in open seawater for the first time. Kilogram-grade PAO-G-A was deployed in the South China Sea as shown in Fig. 24D. After 40 d of exposure, a qU of 1.144 mg g−1 was obtained and no marine microorganism attachment was observed (Fig. 24C), demonstrating excellent antibiofouling properties. The selectivity of PAO-G-A was also investigated. Although PAO-G-A showed superior adsorption for U over most coexisting ions including Li, Na, L, Cr, Mn, Co, Ni, Cu, Zn, and Cd, higher uptakes by PAO-G-A were observed for Ca (20.970 mg g−1), Mg (9.617 mg g−1), Fe (2.267 mg g−1) and V (1.216 mg g−1).
 | ||
| Fig. 24 (A) Schematic diagram of the preparation process and uranium adsorption of the PAO-G-A adsorbent. (B) Antimicrobial activity of PAN and PAO-G-A against marine bacteria. (C) Photographs and SEM images of the PAO-G-A adsorbent before and after uranium uptake in natural seawater. (D) Photos of adsorbent deployment (inset) and buoy in marine tests. Reproduced with permission.216 Copyright 2020, Elsevier. | ||
To supplement this effort, two other guanidine polymers, i.e., polyhexamethylene biguanidine (PHMB) and polyaminopropyl biguanide (PAPB), were grafted into commercial acrylic fibers to produce antibiofouling adsorbents by the same team.218 Through a similar synthetic method to PAO-G-A, PAO-PHMB-A and PAO-PAPB-A were obtained. When exposed to bacterial solution cultivated from seawater, PAO-PHMB-A exhibited better antibacterial activity than PAO-G-A and PAO-PAPB-A, with a 100% inhibition rate at a high concentration of 108 CFU mL−1. When immersed in 8 ppm uranium-containing simulated seawater, PAO-PHMB-A exhibited the highest adsorption capacity (227.33 mg g−1) among the three adsorbents. Therefore, PAO-PHMB-A was chosen for further marine tests in the South China Sea for uranium recovery. After 30 d of exposure, no evidence of organisms on the surface of PAO-PHMB-A was found, confirming its excellent antibacterial activity. A qU of 3.19 mg g−1 was achieved, which was higher than that of PAO-G-A.216 More importantly, while the uptake of Ca and Mg was still higher than that of U owing to their high concentrations in seawater, lower adsorption was observed for Na (2.84 mg g−1), V (2.23 mg g−1) and Fe (1.91 mg g−1), indicating the improved selectivity of PAO-PHMB-A.
Biological entities (e.g., plants, fungi, algae, and bacteria) that have both uranium uptake capacity and antimicrobial activity appear to be another category of potential candidates for UES.223,224,226,241 Yuan et al. discovered that a Bacillus velezensis strain, namely UUS-1, showed highly-efficient and ultrafast uranium immobilization capability, based on the screening of seventeen kinds of strains.241 The adsorption of uranium was determined in 10 ppm uranium-spiked LB broth. An uptake capacity of 48.25 ± 5.61 mg U g−1 dry bacterial cells was acquired after 24 h, which increased up to 352 ± 15.31 mg U g−1 dry bacterial cells when the uranium concentration increased to 100 ppm. Notably, UUS-1 reached the high immobilization capacity of 9.46 ± 0.39 mg g−1 in real seawater within 2 d. Moreover, with the broad-spectrum antimicrobial activity by producing fermentation liquid, UUS-1 exerted innate resistance to the biofouling of several marine microbiomes, including the Gram positive bacterium B. amyloliquefaciens, the Gram negative bacterium Vibrio alginolyticus, and the fungus Fusarium sp., with a growth inhibition rate of 99.01%.
Inspired by the excellent performance of bacterial strain UUS-1 in uranium capture and in biofouling control, a recent report by the same group demonstrated the potential of polypeptides with antimicrobial properties for UES.223 A biosafe omiganan peptide (OP, ILRWPWWPWRRK-NH2) was selected from the database of antimicrobial peptides, and subsequently covalently introduced into a polymeric peptide hydrogel (labeled PPH-OP) using glutaraldehyde as a cross-linking agent (Fig. 25A). The PPH-OP hydrogel exhibited a brown colour (Fig. 25B), and the resultant dry hydrogel showed a porous structure with a SSA of 17.97 m2 g−1 (Fig. 25C). For uranium adsorption in simulated seawater at pH 6, an equilibrium was reached in 70 min (Fig. 25D), and adsorption capacities of 76.35 mg g−1, 129.16 mg g−1 and 139.47 mg g−1 were obtained at uranium concentrations of 4 ppm, 8 ppm and 16 ppm, respectively. The equilibrium adsorption isotherms of PPH-OP were found to fit well with the Freundlich adsorption model (Fig. 25E). In the presence of various common competitive ions (e.g., Na, K, Mg, Ca, V, Co, Fe, Ni and Cu), impressive selectivity to uranium over other competing ions was presented with a high enrichment index of 89.17, while other metal ions were rarely adsorbed (Fig. 25F), even with a 1000-fold excess. The binding affinity of PPH-OP to uranium against the carbonate group was also assessed, and a low dissociation constant (Kd) of 3.5 nM for uranium was obtained in a carbonate-existing environment (Fig. 25G), suggesting the high affinity of PPH-OP to uranium. Because of the inherent antibacterial properties of the omiganan peptide, PPH-OP exhibited an inhibition rate higher than 89.9% against the tested microbial strains (Fig. 25H and I). When exposed to non-filtered natural seawater containing marine microorganisms, a qU of 7.12 mg g−1 was obtained by PPH-OP over 21 d. In comparison, a slightly-increased qU of 7.63 mg g−1 was achieved in filtered natural seawater, demonstrating that the influence of marine microorganisms was nearly negligible.
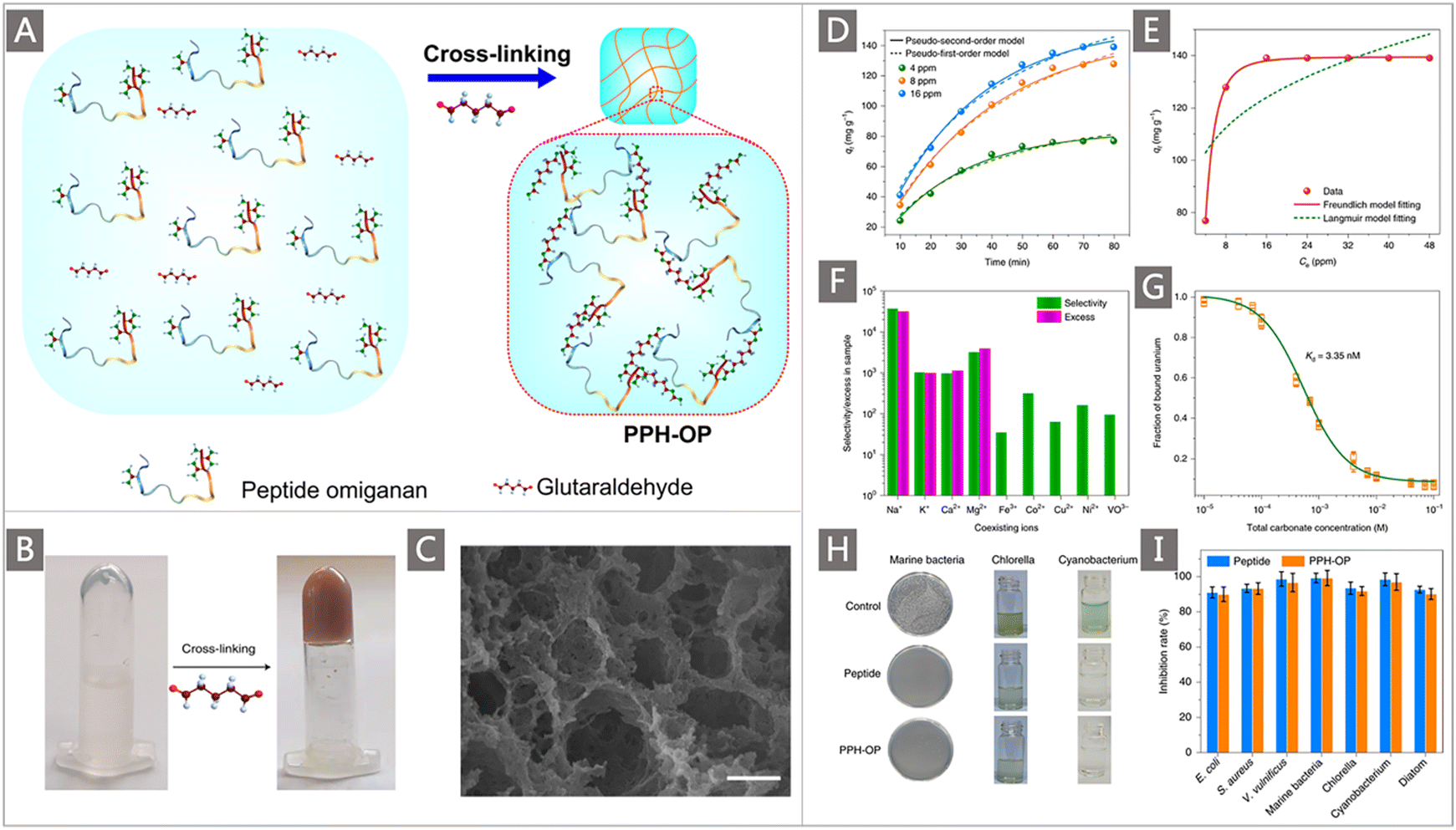 | ||
| Fig. 25 (A) Schematic diagram for the fabrication of PPH-OP. (B) Morphology of the wet PPH-OP hydrogel after cross-linking. (C) SEM image of a dry PPH-OP hydrogel; the scale bar represents 1μm. Extraction performances of the PPH-OP hydrogel for uranium. (D) Adsorption kinetics and the corresponding fitting data. (E) Adsorption isotherms and the corresponding fitting data. (F) Adsorption selectivity to uranium in 100 times concentrated seawater. (G) Binding affinity for uranium against total carbonate. (H) Antibiofouling activity against the marine strains. (I) Inhibitory activity of the peptide and PPH-OP against microbial strains. Reproduced with permission.223 Copyright 2021, Springer Nature. | ||
Recently, photoactivated sterilization, as an effective antibacterial strategy, has garnered colossal interest in the field of UES. It utilizes light irradiation at appropriate wavelengths, ranging from ultraviolet (UV) to near-infrared (NIR), to activate photo-responsive materials that absorb light energy to kill bacteria within a short time by producing radical oxygen species (ROS) and/or hyperthermic conditions.242 ROS mainly involves hydrogen peroxide (H2O2), superoxide anions (˙O2−), singlet oxygen (1O2), and hydroxyl radicals (˙OH), which can destroy the bacterial defence system by adhering to the membranes and cell walls of the bacteria, or directly or indirectly disrupting cellular respiration and other physiological activities by penetrating the bacterial membranes and entering cells, thereby killing the bacteria efficiently.243
Various available photo-responsive antibacterial materials including photocatalysts, photothermal materials and photosensitizers have been exploited to recover uranium from seawater while minimizing the biofouling effect. Firstly, traditional semiconductor materials, such as TiO2 and ZnO, were used as antibacterial components to construct antibiofouling adsorbents for UES owing to their optical response characteristics and antibacterial activity.228–231 For instance, a TiO2 functionalized composite was developed by in situ coprecipitation of nano-TiO2 particles onto AO-functionalized wool fibers prepared through RIGP.228 The resultant Wool-AO@TiO2 adsorbent exhibited good antibacterial activity against the model bacteria E. coli and Staphylococcus aureus (S. aureus), with maximum inhibition rates of 90.0% and 95.2%, respectively, at TiO2 dosages of 1.5–2.0 wt%. Meanwhile, the introduction of nano-TiO2 increased the adsorption of uranium, and the maximum adsorption capacity of Wool-AO@TiO2 was calculated to be 112 mg g−1, which was higher than that of Wool-AO (49.6 mg g−1) in artificial seawater containing 50 ppm uranium. Likewise, the introduction of ZnO nanoparticles can impart the wool fibers with good antimicrobial properties, effectively inhibiting the growth of E. coli and S. aureus with both 99.9%, sulfate reducing bacteria (SRB) with 95.0%, and fungus Candida albicans (C. albicans) with 92.6%.230 In the presence of water vapour, nano-TiO2/ZnO can be activated when irradiated with UV light, resulting in the production of highly reactive ROS that damage the organic compounds/cells, further causing the death of microorganisms.244
In a recent study, Co single atoms with a ROS-producing ability were anchored in the PAO matrix to fabricate antibiofouling adsorbents for UES.245 The adsorbent PAO-Co was synthesized by pre-loading cobalt ions (Co2+) into PAO by interacting with N and O atoms in PAO, and then Co2+ was transformed into Co single atoms via ball-milling (Fig. 26A). The antimicrobial activity of PAO-Co was first assessed under natural light irradiation. A significant inhibition to the growth of the tested marine bacteria and algae with the inhibition rate ranging from 77.0% to 93.4% was observed for PAO-Co, whereas PAO showed limited inhibition to the growth of these microbial species (Fig. 26B and C). The electron spin resonance (ESR) spectrum recorded the production of ROS with light irradiation, and more ROS was produced by PAO-Co than by PAO (Fig. 26D), which was ascribed to the presence of Co atoms in PAO-Co. With the light irradiation, the Co atom in PAO-Co served as an electron acceptor and in turn promoted the transfer of electrons in PAO. The electron received by the Co single atom was further transferred to O2 to produce ROS (including ˙O2− and 1O2). Simultaneously, H2O and OH− were oxidized by the residual electron holes to form ˙OH (Fig. 26E). The ROS generated by PAO-Co destroyed the cells of the test bacteria (e.g., E. coli), as illustrated by Fig. 26F, and eventually resulted in the death of microorganisms. In uranium adsorption experiments, a qU of 366 mg g−1, 443 mg g−1 and 687 mg g−1 was achieved in 8 ppm, 16 ppm and 32 ppm uranium-spiked simulated seawater at pH 5.0, respectively. After soaking in the unfiltered natural seawater for 49 d under light irradiation, the PAO-Co exhibited a high uranium uptake of 9.7 mg g−1, which was lower than that obtained in seawater without marine biological entities by only 11%, indicating the high resistance of the adsorbent to marine biofouling. However, as discussed above, due to the strong tidal flows in the ocean and relatively higher binding affinity of PAO to U relative to Co, the long-term stability and antimicrobial activity of PAO-Co remain to be further verified.
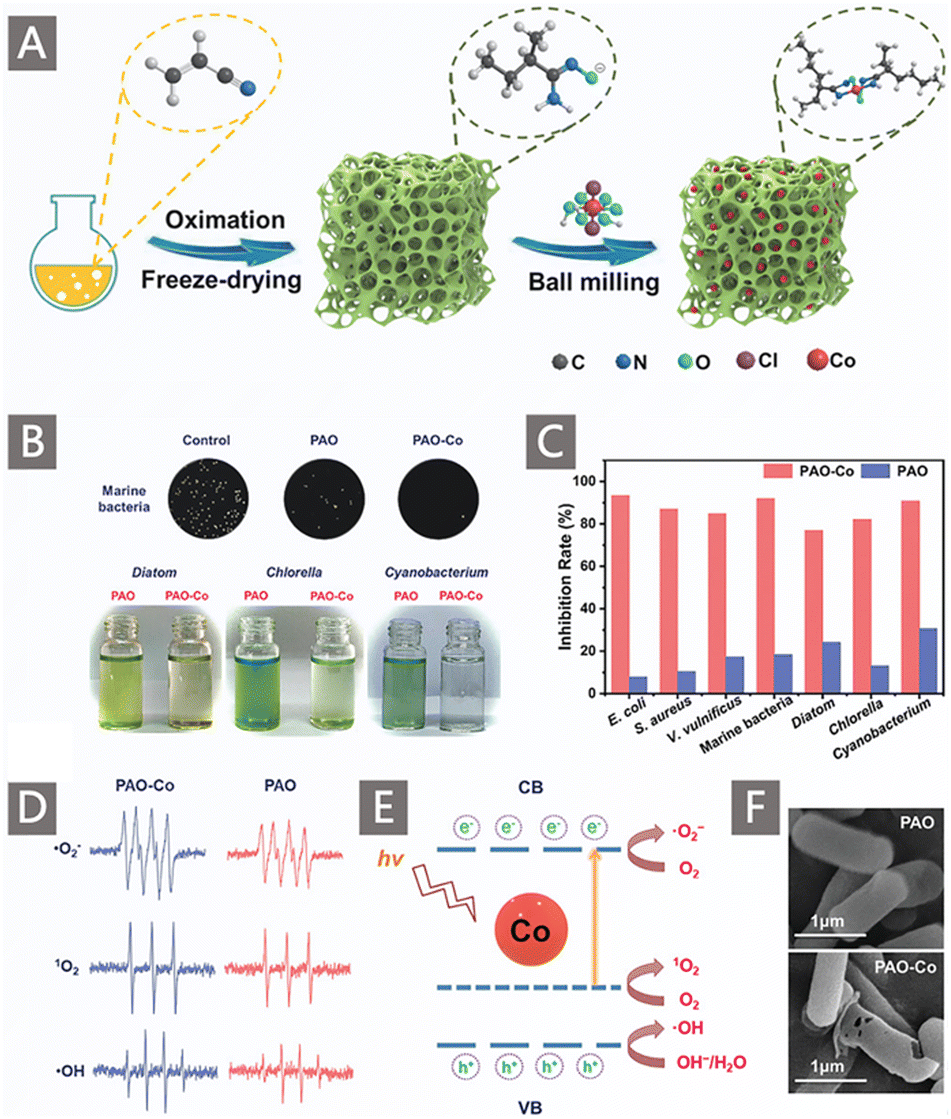 | ||
| Fig. 26 (A) Schematic diagram for the synthesis of the PAO-Co adsorbent. (B) Antimicrobial activity of PAO and PAO-Co against the tested marine bacteria and algae. (C) Inhibitory activity of PAO and PAO-Co against the growth of the microorganisms. (D) Detection of the ROS (O2−, 1O2 and ˙OH) produced by PAO-Co or PAO under light irradiation. (E) Illustration for the production mechanism of ROS. (F) SEM images of E. coli cells after treatment with PAO and PAO-Co adsorbents. Reproduced with permission.245 Copyright 2022, Wiley-VCH. | ||
In addition to their photocatalytic effects, the photothermal and photoelectric effects of semiconductor materials under light stimulation have also been exploited for constructing adsorbents with high uranium extraction capacity and antibiofouling properties.232–234 A black phosphorus (BP) nanosheet-loaded PAO adsorbent (labeled BP-PAO) was recently reported.232 With the ability to produce biotoxic ROS induced by the photocatalytic effect, the as-prepared BP-PAO displayed high antibiofouling activity by destroying the organic components of the microbial species. On the other hand, the photothermal effect of the BP nanosheet could lead to a temperature increase in local areas of the BP-PAO, which enhanced the interaction between uranium and BP-PAO. Moreover, the unique photoelectric effect of BP enabled the adsorbent to release electrons and form a positive electric field around the fiber, which could electrostatically attract negative uranium species [UO2(CO3)3]4− in the seawater environment, thereby enhancing the adsorption performances towards uranium. Combined with the above photoinduced multiple effects of the BP nanosheet (Fig. 27), the BP-PAO exhibited high antibacterial activity to the growth of the Gram-positive bacterium S. aureus, and the Gram-negative bacteria E. coli and Vibrio vulnificus. Especially, a growth inhibition ratio of up to 96.67% was observed for the marine bacterial community, indicating the broad antibiofouling activity of BP-PAO. Strikingly, when uranium recovery performance was determined in bacteria-containing natural seawater under simulated sunlight illumination, the photo-responsive BP-PAO fibers demonstrated a high uranium uptake of 11.76 ± 0.43 mg g−1 over 56 d, which was 1.5 times that of the PAO fibers. Overall, harnessing the photocatalytic and photoelectronic activities of materials is promising to mitigate biofouling while promoting adsorption performances in UES.233,234
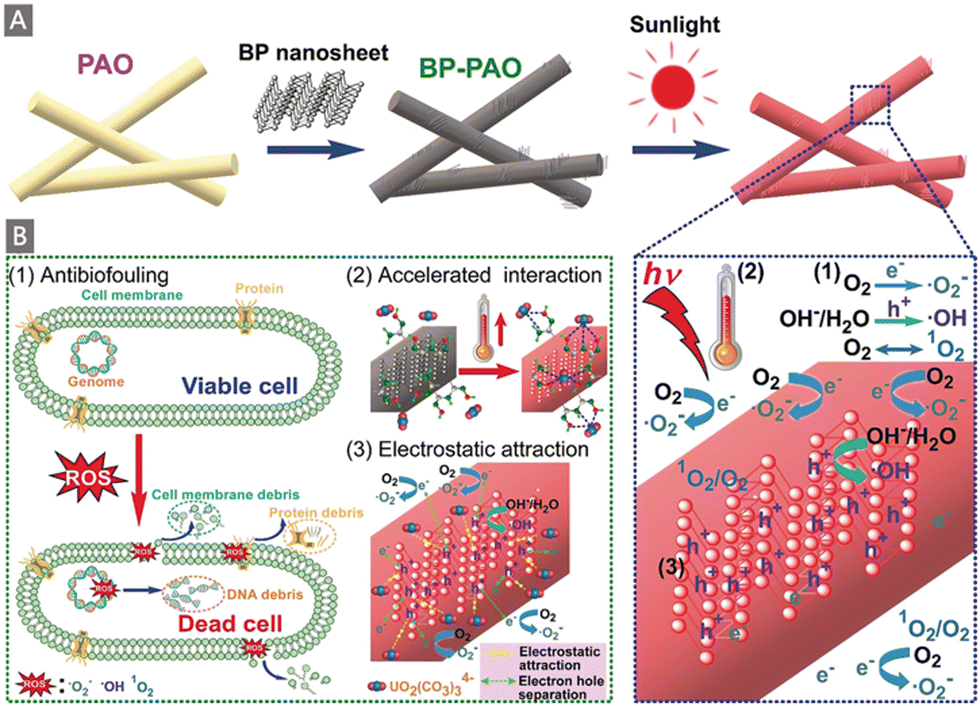 | ||
| Fig. 27 (A) Schematic diagram of the preparation process of the BP-PAO fiber. (B) Uranium adsorption/antifouling mechanism of the BP-PAO fiber. Reproduced with permission.232 Copyright 2020, Wiley-VCH. | ||
The last category of photo-responsive materials that has received particular interest in UES recently is semiconducting COFs. In this regard, the Qiu group has devoted to exploring new strategies for photo-enhanced uranium extraction and antibiofouling from seawater.85,246–251 To improve the adsorption capacity of COFs for uranium recovery, a highly planar conjugated sp2 carbon COF (labeled NDA-TN) was constructed by condensing the building blocks of naphthalene-2,6-dicarbaldehyde (NDA) and 2,2′,2′′-(benzene-1,3,5-triyl)triacetonitrile (TN) (Fig. 28Aa). Simultaneously, to highlight the significance of fully planar conjugated naphthalene-based COFs, another sp2-carbon COF (labeled BDA-TN) with an interrupted π-conjugation structure was also synthesized by reacting 4,4′-biphenyldicarboxaldehyde (BDA) with TN monomers (Fig. 28Ab). Subsequently, NDA-TN and BDA-TN were reacted with N(CH2CH3)3 and NH2OH·HCl to gain NDA-TN-AO and BDA-TN-AO, respectively. Benefiting from the stable CC bonds, both NDA-TN-AO and BDA-TN-AO exhibited superior stability under harsh conditions, compared to imine-based COFs. Moreover, the fully extended π-conjugation structure of NDA-TN-AO endowed it with unique physicochemical and semiconductor characteristics that contribute to uranium extraction.
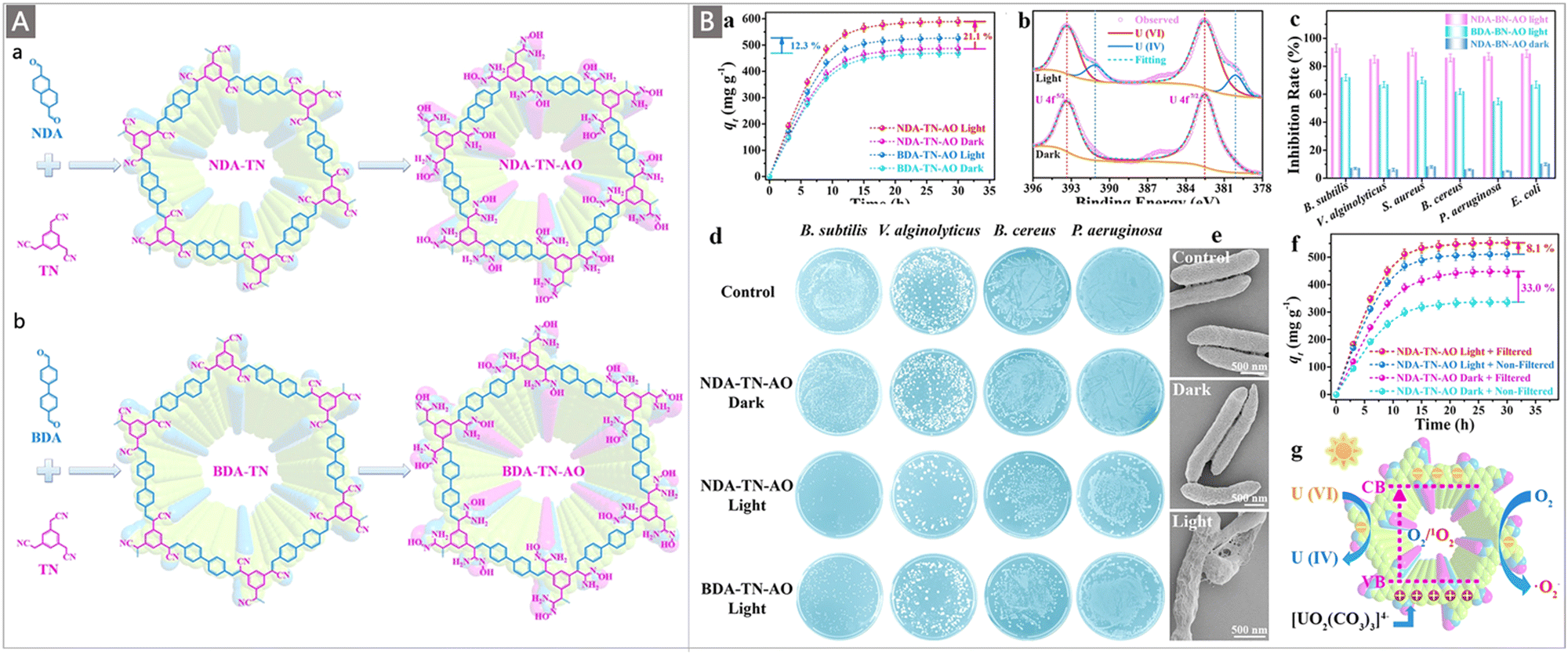 | ||
| Fig. 28 (A) Synthetic routes of semiconducting COFs: (a) fully planar π-conjugated NDA-TN-AO and (b) interrupted π-conjugated BDA-TN-AO. (B) Uranium extraction/antifouling performances of the as-prepared COFs under under light and dark conditions: (a) adsorption kinetics of NDA-TN-AO and BDA-TN-AO; (b) U 4f XPS spectra of NDA-TN-AO after uranium uptake; (c and d) antibacterial activity of NDA-TN-AO and BDA-TN-AO towards the tested bacteria; (e) SEM images of V. alginolyticus bacterial cells treated with NDA-TN-AO; (f) uranium extraction capacities of NDA-TN-AO in uranium spiked filtered and non-filtered natural seawater; and (g) proposed mechanism of NDA-TN-AO for the photo-enhanced uranium extraction. Reproduced with permission.85 Copyright 2020, Wiley-VCH. | ||
Specifically, the photoelectric effect could efficiently cause the release of electrons from the NDA-TN-AO skeletons and form a positive surface electric field, which displayed strong electrostatic attraction to the negatively charged species of [UO2(CO3)3]4−. Of particular interest, the photocatalytic activity of NDA-TN-AO not only enables the NDA-TN-AO to exhibit a high antibiofouling performance by producing ROS including ˙O2− and 1O2 under light irradiation, but also endows it with the capacity to induce the reduction of adsorbed UVI to insoluble UIV species, thereby enhancing the uranium extraction capacity. Consequently, with the help of these appealing photoinduced effects, the qU of NDA-TN-AO increased by 21.2% (from 486.4 mg g−1 to 589.1 mg g−1) after irradiation with simulated sunlight, while the BDA-TN-AO only showed a 12.3% increase in uranium adsorption in a 10 ppm U-spiked simulated seawater at pH 5.0 (Fig. 28Ba). The photo-induced reduction of uranium from UVI to UIV was verified by the existence of U 4f7/2 and U 4f5/2 peaks in the XPS spectrum (Fig. 28Bb), which allowed efficient regeneration of binding sites for further binding of additional UVI. Obvious antibacterial activities of NDA-TN-AO against Gram-positive B. subtilis, B. cereus, and S. aureus and Gram-negative E. coli, V. alginolyticus and P. aeruginosa were also observed (Fig. 28Bc and d), indicating its wide-spectrum anti-biofouling activity. SEM characterization of the V. alginolyticus suggested that the biological entities were completely damaged and their internal contents were released after light irradiation, whereas there was no significant change in the dark (Fig. 28Be). When contacted with uranium-spiked filtered and non-filtered natural seawater, it was found that the marine bacteria exerted a serious effect on uranium adsorption by NDA-TN-AO in the dark; by contrast, little effect of the bacteria on uranium adsorption was noted under light irradiation, demonstrating the role of the photocatalytic and photoelectric activities of NDA-TN-AO (Fig. 28Bf). Finally, the adsorption tests of NDA-TN-AO and BDA-TN-AO were carried out in natural seawater, yielding uranium adsorption capacities of 4.56 mg g−1 and 4.33 mg g−1 in the dark after 27 d. Upon exposure to the light irradiation, NDA-TN-AO reached an adsorption capacity of 6.07 mg g−1, which was 1.33 times that in the dark, further disclosing the effectiveness of the photoinduced effects in enhancing uranium adsorption and antibacterial properties (Fig. 28Bg). Along the same line, more novel AO-functionalized COFs (BD-TN-AO),247 AF Anti-COF248 and Tp-DBD249 were constructed by the same team for photo-enhanced uranium recovery. Additionally, COF-based sponges246 and hydrogels250,251 were also designed as a platform for improving the uranium adsorption efficiency and antibiofouling activity. The strategy of utilizing the photo-responsible performances of these appealing photocatalysts and photothermal materials provides an intriguing route to develop uranium extraction adsorbents with outstanding adsorption and antibiofouling properties.252
Taking that into account, Bai and colleagues reported an adsorbent with anti-adhesion properties for UES.253 A robust montmorillonite-polydopamine/polyacrylamide (MMT-PDA/PAM) nanocomposite hydrogel was prepared via a two-step process in which PDA was first intercalated into MMT nanosheets, and then a solution composed of monomers AM, initiator APS and crosslinking agent BIS was subjected to free radical polymerization to obtain the final hydrogel adsorbent (Fig. 29A). The best performing adsorbent had a 10 wt% content of MMT and 0.6 wt% of DA content, and was denoted as MMT10-PDA0.6/PAM. By comparison, the MMT10-PDA0.6 and MMT10/PAM were also prepared without the addition of the AM and DA, respectively. The adsorption capacity of these adsorbents was examined as a function of pH over the range of 3.0–8.5 in deionized water. The uranium uptake increased from pH 3.0 to 6.5, with the maximum capacity achieved at pH 6.5, then dropped slightly with a further increase in pH. A saturation capacity of 229.08 mg g−1 of the MMT10-PDA0.6/PAM hydrogel was obtained, which was higher than those of MMT10-PDA0.6 and MMT10/PAM. At pH 8.0 and 8.5, the adsorption of MMT10-PDA0.6/PAM decreased to 200.45 and 195.53 mg g−1 (Fig. 29Ba), respectively, due to the formation of [UO2(CO3)2]2− and [UO2(CO3)3]4−. Adsorption isotherms were obtained at pH 6.5 at 25 °C, 35 °C and 45 °C, which yielded saturation capacities of 540.54 mg g−1, 502.21 mg g−1 and 617.28 mg g−1 for MMT10-PDA0.6/PAM when fitted with the Langmuir model (Fig. 29Bb). Good selectivity to uranium was observed in the presence of a variety of competing ions, affording a high Kd of nearly 1.7 × 104 for uranium (Fig. 29Bc). When deployed in a lab-scale flow-through the column in contact with simulated seawater, the adsorption capacity of the MMT10-PDA0.6/PAM hydrogel was determined to be 44 mg g−1 after 63 d (Fig. 29Bd). A slight decrease in adsorption efficiency was observed for the adsorbent over six recycles, with uranium eluted with (NH4)2CO3 solution, suggesting its good reusability (Fig. 29Be). The antibiofouling activity of MMT10-PDA0.6/PAM was subsequently investigated. When cultured with Nitzschia in alga-containing simulated seawater, the algae adhered to the surfaces of MMT10-PDA0.6/PAM and MMT10-PDA0.6 were much fewer than those of MMT10/PAM, which proved its improved anti-algal adhesion properties. The introduction of PDA and MMT into the hydrogel endowed the adsorbent with high hydrophilicity, and the hydrated film formed by water molecules on the surface weakened the interaction between Nitzschia and the hydrogel, thereby enhancing the anti-adhesion performance. Moreover, the MMT10-PDA0.6/PAM hydrogel exhibited a higher adsorption amount (1.986 mg g−1) for uranium with an equilibrium time of 4 d in alga-containing simulated seawater, while it took 5 d and 6 d for MMT10-PDA0.6 and MMT10/PAM to reach equilibrium, and the adsorption capacities were 1.524 and 1.077 mg g−1, respectively (Fig. 29Bf).
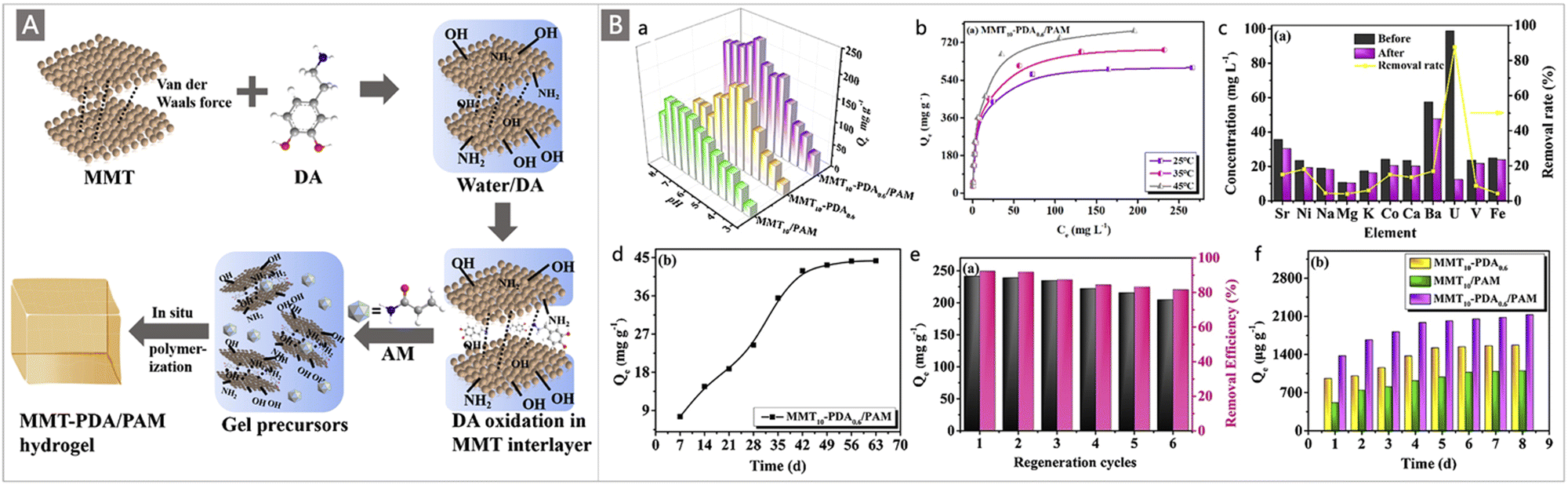 | ||
| Fig. 29 (A) Schematic diagram of the preparation process of the MMT-PDA/PAM hydrogel. (B) Uranium extraction performances of the as-prepared MMT10-PDA0.6/PAM hydrogel: (a) effects of pH values on the uranium adsorption capacities; (b) adsorption isotherms at 298.15 K, 308.15 K and 318.15 K; (c) adsorption selectivity towards uranium against other competing ions; (d) adsorption kinetics in 400 L of circulated simulated seawater; (e) uranium adsorption capacities and removal efficiency during six adsorption–desorption cycles; and (f) uranium adsorption capacities in alga-containing simulated seawater. Reproduced with permission.253 Copyright 2020, Elsevier. | ||
It is well known that polyethylene glycol (PEG) and zwitterionic polymers are commonly used as antibiofouling coatings to resist non-specific proteins as well as to minimize bacterial or mammalian cell adhesion.260,261 A physical barrier formed by hydrogen bonding or electrostatically induced hydration makes them exhibit remarkable protein impedance and bacterial anti-adhesion.262 Accordingly, the potential application of these candidates for antibiofouling in the field of UES has been explored. Phosphorylcholine (PC), one of the representative zwitterionic polymers, was employed as a building block for constructing a polymeric adsorbent.258 A PVC–PC adsorbent with superhydrophilicity was prepared by grafting 2-methacryloyloxyethyl phosphorylcholine (MPC) onto the polyvinyl chloride–chlorinated polyvinyl chloride (PVC–CPVC) fiber via ATRP. The bacterial test was carried out in a 1 × 104 CFU mL−1 vibrio solution, where the quantity of vibrio observed on the surface of PVC–PC was 9 times lower than that observed on the PVC–CPVC surface, indicating the excellent anti-adhesion properties of PC. A uranium adsorption study of the PVC–PC fiber demonstrated that the adsorption increased with increasing pH, with the maximum adsorption obtained at pH 7.0, then decreased with a further increase of pH, affording adsorption capacities of 338.7 mg g−1 and 240.0 mg g−1 at pH 7.0 and 8.0 in 20 ppm uranium-spiked solution, respectively. The high affinity between PVC–PC and uranium also led to excellent selectivity for uranium over multiple competing metal ions.
Recently, regulating the surface charge of materials has emerged as a new strategy for the preparation of anti-adhesion adsorbents, since the surface charge exerts a significant effect on attracting or repelling the charged uranyl carbonate species and marine microbes.255–257 In this respect, the Bai group introduced polyethyleneimine (PEI) as a positively charged poly(cationic) electrolyte and sodium alginate (SA) as a negatively charged electrolyte into the MOF ZIF-67 support.255 A series of MOF/SAP composite hydrogels with different charge systems were prepared by regulating the ratio of PEI and SA. According to the zeta potential test, the balanced charged hydrogel can be obtained when the doping amount of PEI was 0.45 wt% (labeled ZIF-67/SAP0.45). After being soaked in diatom-containing uranium solution, large areas of Nitzschia diatoms were observed on the most positively charged ZIF-67/SAP0.85 and the most negatively charged ZIF-67/SA composite hydrogels, while no live Nitzschia was observed on the balanced charged ZIF-67/SAP0.45 surface, suggesting that the electrically neutral composite hydrogels can effectively resist the settlement and adhesion of diatoms. Owing to the synergistic effect of the anti-adhesion properties and the hierarchically porous architectures that could accelerate the diffusion of uranium to the binding sites, ZIF-67/SAP0.45 reached a high uranium extraction capacity of 6.99 ± 0.26 mg g−1 with a 35 d test in natural seawater collected from the Bohai Sea, China. Besides, competitive adsorption analysis of U(VI) from spiked seawater with other 10 major metal ions (including Pb, Mn, V, Fe, Co, Cu, Ni, Zn, Ba and Sr) revealed desirable selectivity to U(VI), with a Kd of 77.81 ± 1.67 mg L−1. Notably, the balanced charged strategy can also be applied for modifying other types of MOF materials (e.g., ZIF-8, HKUST-1 and MIL-100) to prepare adsorbents with a good adsorption ability and antibiofouling properties.
Along the same line, Yuan and colleagues constructed a charged balanced hydrogel membrane (labeled PAO-CB) by cross-linking charge-negative PAO with charge-positive tetraethylenepentamine (TEPA) (Fig. 30A).256 The surface potential of the material was adjusted by varying the addition amount of TEPA. When the addition amount of TEPA was 20%, the material exhibited a nearly neutrally charged state (Fig. 30B). The best adsorption performance was achieved by the charged balanced adsorbent PAO-CB both in a fouling environment and in a non-fouling environment (Fig. 30C). When soaked in natural seawater within 56 d, a high uranium extraction capacity of 8.59 mg g−1 was recorded by the charged balanced adsorbent PAO-CB, which was enhanced by 45% compared with the bare PAO adsorbent. The SEM observation of the adsorbents immersed in natural seawater for one week suggested that serious fouling occurred on both the surfaces of the charge-negative PAO membrane and the charge-positive PAO membrane containing 40% TEPA, while only a few contaminants were observed on the surface of the charged balanced PAO-CB membrane, which was attributed to the decrease of the charge attraction between the charged contaminants and the adsorbent (Fig. 30D). The stable covalent linking between PAO and TEPA also endowed the PAO-CB with good reusability. These studies disclosed that the construction of charge balanced adsorbents provided an effective strategy to control marine biofouling in UES.
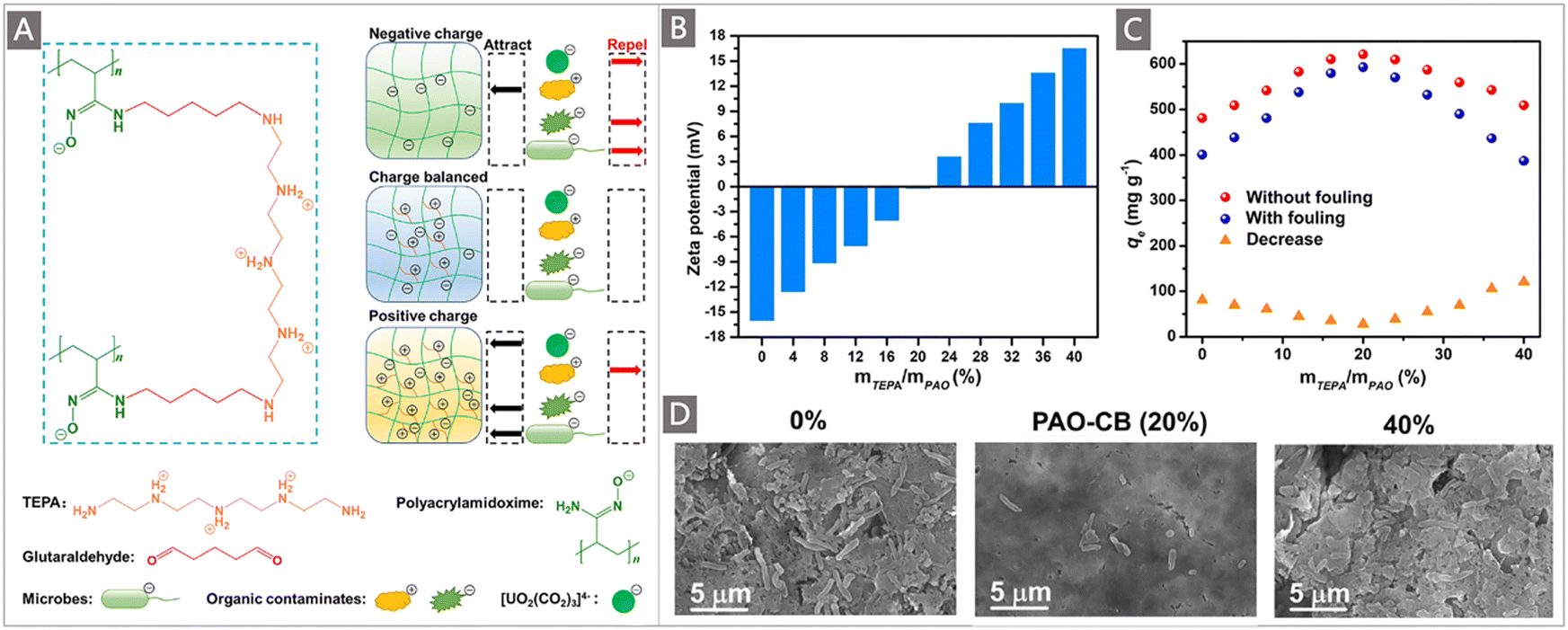 | ||
| Fig. 30 (A) Schematic diagram of the preparation process of the charge balanced PAO-CB adsorbent and the proposed anti-adhesive mechanism for marine biofouling. (B) Zeta potentials of PAO membranes containing different contents of TEPA. (C) The effects of fouling including protein and DNA on uranium adsorption amounts using PAO membranes containing different contents of TEPA. (D) SEM images showing the morphology of bacteria adhered onto PAO membranes containing different contents (0, 20 and 40%) of TEPA after uranium extraction from natural seawater. Reproduced with permission.256 Copyright 2021, Elsevier. | ||
Altogether, the ubiquitous microorganisms in the oceans bring serious biofouling to materials applied in marine engineering, which has been a tremendous challenge for UES. Various “attacking” and “defending” strategies mentioned above have been proposed to cope with the challenge. As for the former active bactericidal strategy, one concern is that killed bacteria can act as nutrients for bacterial growth, while the latter passive antifouling strategy may lack the ability to resist the dead bacterial cells that are already deposited on the surface.203 Therefore, adsorbents relying on a single antifouling mechanism cannot always meet the needs of different occasions. To alleviate the problem, the combined use of passive antifouling and active bactericidal strategies is expected to achieve greater effectiveness. Meanwhile, versatile antibacterial components can be incorporated into the adsorbents to generate multifunctional surfaces.
On the other hand, since the dominant biofouling species affecting uranium recovery from the oceans are unclear, antibacterial agents with broad-spectrum antibacterial characteristics are usually selected. The Gram-negative E. coli and Gram-positive S. aureus are often used as model bacteria for antibacterial research. However, the dominant target biofouling species are variable corresponding to the surface chemistry and structural features of the different uranium adsorbents. Identifying dominant bacterial species affecting uranium recovery is conducive to designing antifouling adsorbents for targeting specific biofouling bacteria. Last but not least, current antimicrobial evaluation experiments are basically carried out on a laboratory scale. However, in fact, biofouling is derived from the oceans, and the complex marine biological system brings more uncertainties and challenges. Therefore, future research on antimicrobial assessment should be performed in the open marine environment to verify the practical application performance of the adsorbents.
3.4. Strategies for improving durability
The stability of an adsorbent under realistic marine conditions is an essential issue that needed to be taken into consideration in the practical deployment of adsorbents.14 The adsorbents without structural stability might undergo degradation or the loss of functional components over prolonged seawater exposure, which will compromise their extraction ability towards uranium and raise the cost of UES. On the other hand, it may also bring about environmental concerns owing to the leaching of the components of adsorbents. To date, the potential of numerous inorganic adsorbents, polymers and nanostructured materials for UES has been extensively reported, while few results concerning their structural stability have been obtained in realistic marine tests.58,65,69,71,114,216,218,231 It is well documented that, for inorganic materials, their poor durability and their instability under basic conditions limit their large-scale application in ocean environments. Besides, although the various appealing characteristics have made many nanostructured materials (typically, porous framework materials, e.g., MOFs) interesting and potential in UES application, their chemical stability remains a tough challenge since a few of dative bonds in these materials could survive in the seawater environment with high-ionic-strength. To address this problem, strategies have been proposed to engineer the organic struts of the frameworks. However, the synthesis of these materials on an industrial scale is a huge challenge. From the perspective of durability, it is more practical to use polymer adsorbents for uranium recovery from seawater because of their stable covalently-bonded structures. Specifically, polymeric hosts with high mechanical strength are preferred.Another issue related to material durability that must be considered is the structural stability of adsorbents in the elution process.263 Since the ultimate goal of UES is to obtain high-concentration uranium products, uranium must be effectively eluted from the adsorbents.264 Meanwhile, the adsorbent should be regenerated for cycle use in marine tests, so as to minimize the cost of UES. This requires the development of relatively mild but efficient elution procedures. Conventionally, uranium adsorbed onto AO-based fibers was eluted through strong acid leaching (e.g., HCl, H2SO4 and HNO3).265,266 The advantage of the acid elution approach is that the uranium recovery rate is high, but it causes significant damage to the fibers, leading to the decrease of adsorption performances in cycle use.70 On the other hand, though AO ligands exhibit high stability in seawater environments, they are susceptible to decomposition through hydrolysis during the acid elution process, which is responsible for the degraded extraction performances of recycled AO-based adsorbents.267,268
After acid elution, an alkaline (typically KOH) reconditioning process at elevated temperature (usually 80 °C) is always required for the regeneration of the adsorbent. It has been proved that alkali treatment in the adsorption–elution cycles can reduce chemical damage and deterioration of the adsorption performance caused by acid elution.68,269–271 Nevertheless, alkali treatment also causes physical damage to the adsorbents, so that the tensile and mechanical strengths of the fibers decrease with conditioning time, which compromises the durability of the adsorbents for cycle use in seawater.120,272
To overcome the drawbacks of acid elution, an alternative elution system using a mixture of sodium carbonate and hydrogen peroxide (Na2CO3–H2O2) as the eluent was proposed by Pan and co-workers for recovering uranium from AO-based adsorbents.273 The elution system of Na2CO3–H2O2 can effectively remove the adsorbed-uranium from the adsorbents by the formation of a stable uranyl-peroxo-carbonato [(UO2)(O2)(CO3)2]4− complex. This approach was presumably milder than acid elution, but the presence of minor amounts of H2O2 may still lead to a certain reduction in uranium adsorption capacity. Moreover, the feasibility of leaching uranium from AO-based adsorbents by employing potassium bicarbonate solution (KHCO3) was also explored by the same group.274,275 The elution experiment revealed that uranium adsorbed onto amidoxime-based fibers in simulated seawater can be efficiently eluted using 3 mol L−1 KHCO3 as the eluent via the formation of the stable uranyl tris-carbonato complex [(UO2)(CO3)3]. The use of 3 mol L−1 KHCO3 solution is quite mild to the adsorbent since its pH (8.0–8.3) is close to the real seawater. More importantly, no more KOH reconditioning step was needed for the reuse of the adsorbent, and no loss of uranium uptake capacity was observed after 6 cycles of repeated use. The high efficiency of this elution strategy was attributed to the formation of the stable uranyl tris-carbonato complex in the pH range of 7–9. This non-acidic elution process that creates less damage to adsorbents could increase the reusability of the adsorbents, thus providing a cost-saving path for UES.
It is generally accepted that economically viable recovery of uranium from seawater requires the development of a technology that allows the cyclic deployment of adsorbents followed by elution/regeneration without significant deterioration and loss of adsorption capacity. Compared with the research on the strategies to promote the adsorption process, there are few reports on the improvement of the desorption process. More efforts are needed to improve the stability of ligands and the durability of the adsorbents, as well as to avoid the destruction of the physicochemical properties of the adsorbents during the uranium elution process. The development of non-acidic elution strategies offers a feasible method to alleviate the chemical damage to the trunk materials. There is still more room for innovation in new elution/regeneration protocols used to maximize the reusability of the adsorbents while maintaining their adsorption capacity.
4. Emerging approaches for potential UES systems
In addition to the well-established adsorption approaches relying on the surface-based physicochemical adsorption nature of various adsorbents, recently, novel strategies such as electrochemical and photochemical approaches have been exploited in emerging UES systems (Fig. 31). With the aid of an extra electric field or light energy, the adsorption–desorption equilibria controlled by the thermodynamics will be broken, and the mass transfer diffusion of uranium will be promoted. Consequently, these electrically-driven or light-driven systems have demonstrated higher capacity and faster kinetics in uranium extraction, compared with the current passive adsorption approach. These appealing methods open up new perspectives for uranium extraction and may lead to a more efficient yet economically viable process. In this section, the current representative case studies on electrochemical and photochemical methods for uranium extraction are summarized, and the involved mechanisms are also covered.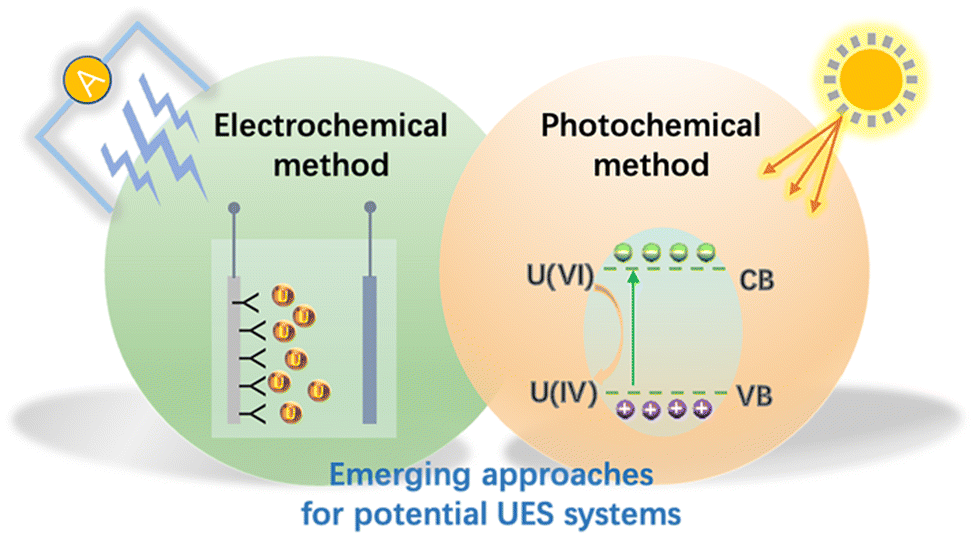 | ||
| Fig. 31 Overview of topics covered in this section. | ||
4.1. Electrochemical approaches
An impressive study by Cui and co-workers to manipulate an external electric field for promoting UES was reported in 2017.276,277 A marvellous uranium extraction capacity of 1932 mg g−1 was achieved by a half-wave rectified alternating current electrochemical (HW-ACE) method. The HW-ACE method is interpreted in five steps (Fig. 32) with a voltage alternating between −5 V and 0 V. In step I, all the ions are randomly dispersed in the aqueous solution. In step II, with the negative voltage, the ions start migrating because of the external electric field and generate an electric double layer on the surface of the AO electrode. In step III, the uranyl ions which are bound by the AO groups can be reduced and deposited on the surface of the electrode. In step IV, when the external electric field is removed, the ions which are neither chelated by the AO group nor electrodeposited are redispersed in the solvent. Step V is the cycle repeat; further uranyl ions are attached to the electrode and deposited as UO2 particles. Furthermore, the authors tested the HW-ACE method in 4 L filtered, unspiked seawater, and the uptake of the uranium was 1.62 μg, while the physicochemical adsorption methods extracted 0.56 μg uranium. Cui's work shows that deploying classical uranium extraction adsorbents in an electrochemical system can break the complexation balance by converting U(VI) to U(IV) and achieve high-efficiency UES.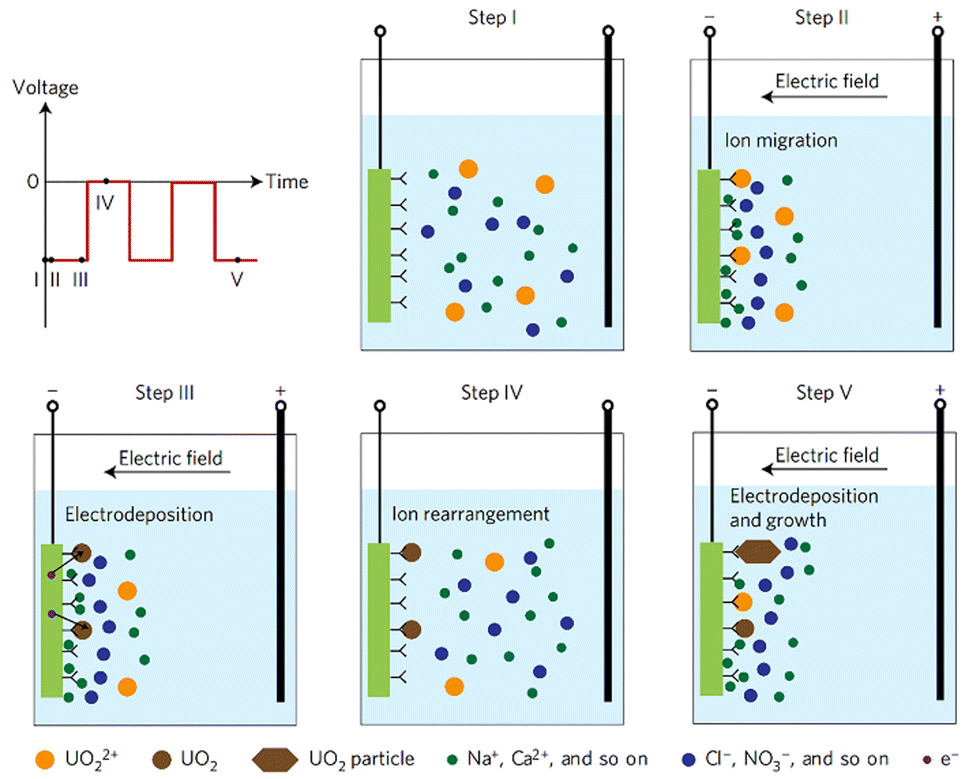 | ||
| Fig. 32 The illustration of the HW-ACE, where an external electric field is applied to the amidoxime-functionalized electrode with a voltage alternating between −5 V and 0 V, resulting in the electrodeposition and growth of uranium. Reproduced with permission.276 Copyright 2017, Springer Nature. | ||
The subsequent year, Chi and colleagues278 performed a similar work by using chitosan-coated graphite felt as electrodes. The authors replaced the alternating current with a direct current with a voltage of −3 V and uranyl ions could also be reduced as charge-neutral species (e.g., UO2). Meanwhile, Chi's work demonstrated the adaptability of the electrochemical method for UES by introducing an external electric field applied to different uranyl chelating groups. Tang et al.279 constructed S-terminated MoS2 nanosheets with abundant electrochemically active S-edge sites for uranium binding and reduction. Under an applied voltage of −3 V, the MoS2 nanosheets exhibited a uranium extraction capacity of 1823 mg g−1 in 100 ppm of uranium-spiked seawater. However, there are still two intrinsic limitations restricting the electrochemical extraction of uranium. First, the concentrations of various coexisting cations are several orders of magnitude higher than that of uranyl ions in oceans, which would considerably affect the uranium separation efficiency by electrochemical strategies. Second, the electric double layer (EDL) thickness or Debye length (ID) of the confined external electric field of the charged medium is less than 1 nm, and that is negligible to work for the enrichment of uranyl ions in the micrometer-sized adsorbent particles. The Zhu team designed and synthesized a novel core–shell structure PAF (PPA@MISS-PAF-1), in which the conductive chains of polyphenylacetylene (PPA) were incorporated into the porous channels of the PAF to serve as the pathways for ion transportation.93 Under the asymmetrical alternating current electrochemical (AACE) method, the charged PPA chains provided an expanded electric field to guide the migration of metal ions. Because of the enhanced electrically driven ion transport, PPA@MISS-PAF-1 achieved a uranium uptake capacity of 16.5 mg g−1 in 90 d from natural seawater.
Capacitive deionization (CDI) is a kind of novel technique for water treatment, which utilizes the mechanism of electrosorption of charged ions in EDLs, and plays an important role in valuable tasks such as wastewater purification and seawater desalination.280 Applying electrosorption for UES was first reported by Smail and Yim in 2015.281 The authors used a carbon slurry, which was prepared by using PVDF as a matrix to immobilize the activated carbon powder, as the electrode. With an applied potential of 0.4 V, the uranyl could be adsorbed by the electrode from seawater with a capacity of 3.4 mg g−1. The authors contributed the selectivity of electrosorption to the differences in the charge density of ions. This conclusion was limited since only high-valence metals (Na, K, Ca, and Sr) were discussed in the study. Further experiments are needed for a more in-depth understanding of the electrosorption mechanism for UES. In subsequent years, several studies were carried out for removing uranium from aqueous solutions by the CDI technique.282–286 Liao and co-workers first synthesized a highly porous phosphate-functionalized graphene hydrogel (HGP) and used it as an electrode material for uranium(VI) electrosorption by CDI (Fig. 33).282 The maximum adsorption capacity of their work reached 545.7 mg g−1 in 60 min. Zhou et al. developed a floriform WO3/C composite pseudocapacitive electrode material for uranium(VI) extraction from water by electrosorption.284 The WO3/C electrode exhibited a qU of 449.9 mg g−1 in 4 h running time. Tang and coworkers used a composite membrane consisting of chitosan and biomass-resourced porous activated carbons as the electrodes for the electrosorption of uranium from water.285 This kind of electrode can remove uranium(VI) from water at a lower applied potential of 0.9 V compared to previous work, and the best adsorption capacity was 207.6 mg g−1 in 350 min. Chen and Tong synthesized a kind of boron phosphide nanosheet with a large specific surface area (221.7 m2 g−1) and an average thickness of 8.422 nm.283 This material could serve as an electrode material for uranium extraction from the aqueous solution, with a maximum adsorption capacity of 2584 mg g−1. The authors also demonstrated the feasibility of the CDI method by conducting uranium adsorption tests in natural seawater, and a qU of 60.83 mg g−1 was achieved in 8 h.
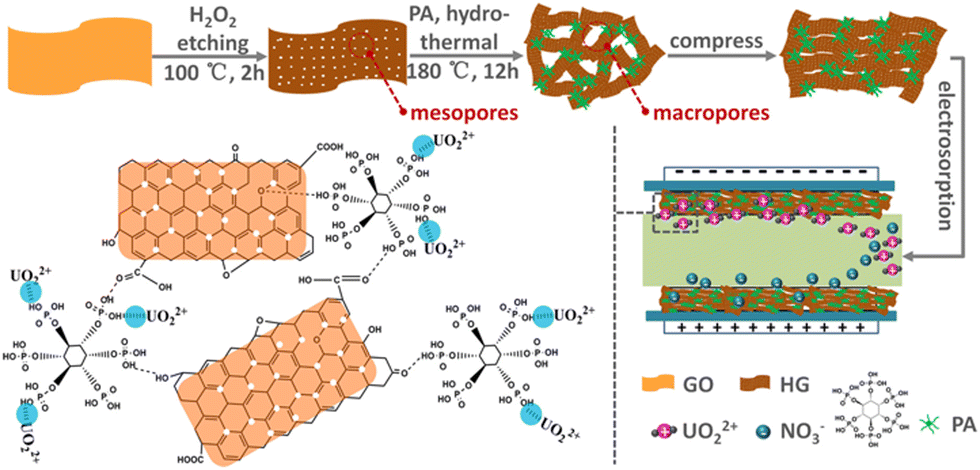 | ||
| Fig. 33 The illustration of the fabrication of HGP electrodes and their application in electrosorption of uranyl ions. Reproduced with permission.282 Copyright 2019, Elsevier. | ||
Electrocatalysis, combined with rational materials design, has been demonstrated recently to be a feasible approach for efficient uranium extraction from seawater.287,288 For example, a functionalized iron–nitrogen–carbon catalyst that consisted of N-doped carbon capsules supporting FeNx single-atom sites and surface chelating amidoxime groups was developed by Ma and co-workers.287 Specifically, ZIF-8 nanocrystals were used as precursors to prepare ZIF-8@Fe-TA (core–shell composite) by the coordination of potassium-tannic acid and the ion exchange of Fe3+ ions. Then, ZIF-8@Fe-TA was heated at 950 °C in an argon atmosphere to yield carbon capsules, Fe–Nx–C. Subsequently, the surface of the Fe–Nx–C was functionalized with the AO groups with a chelating ability to uranyl ions, and the resultant material was labeled Fe–Nx–C–R (Fig. 34A). The uranium extraction experiments were conducted using an alternating voltage between −5 V and 0 V at a frequency of 400 Hz, with a graphite rod serving as the positive electrode and Fe–Nx–C–R serving as the negative electrode. The experiment showed that Fe–Nx–C–R removed 99% of uranium within 24 h from a 10 ppm U-spiked seawater (Fig. 34B). To understand the mechanism, the authors conducted the extraction experiment in a freshly prepared 1000 ppm of U-spiked seawater. After 24 h, the qU was determined to be 14302 mg g−1 (Fig. 34C), and the yellow electrodeposited precipitate could be easily collected from the electrode (Fig. 34D). The composition of the yellow precipitate was determined to be Na2O(UO3·H2O)x by PXRD and XPS analyses. Further EAXFS spectra showed that the coordination number of the uranium in Na2O(UO3·H2O)x was 6. This novel electrocatalysis strategy for UES prevents the laborious and costly uranium elution procedures faced by traditional adsorbents, as well as their regeneration processes.
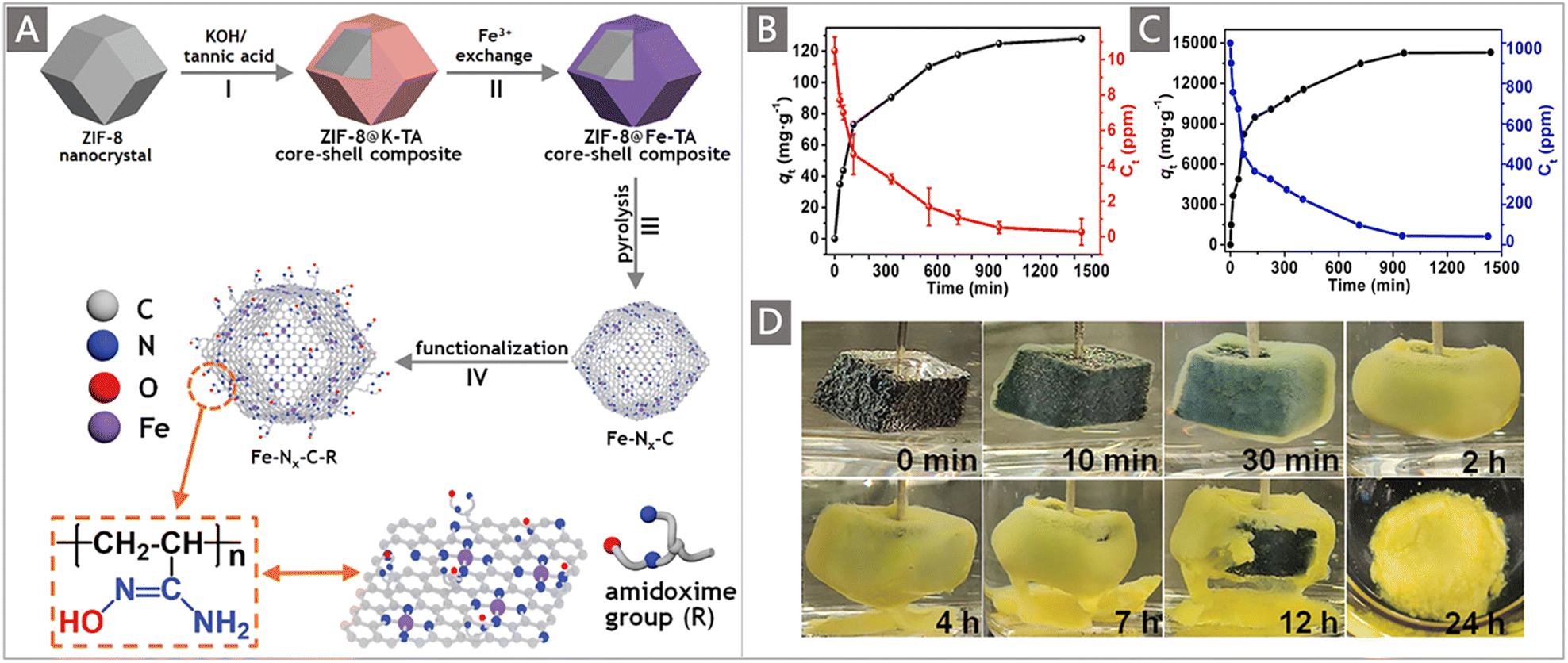 | ||
| Fig. 34 (A) Schematic illustration of the synthesis of Fe–Nx–C–R. (B) Uranium extraction using Fe–Nx–C–R from spiked seawater with an initial uranium concentration (C0) of ∼10 ppm. (C) Uranium extraction using Fe–Nx–C–R from spiked seawater with a C0 of ∼1000 ppm. (D) Photographs of the Fe–Nx–C–R electrode in spiked water (C0 = ∼1000 ppm) during electrocatalytic extraction at different times. Reproduced with permission.287 Copyright 2021, Wiley-VCH. | ||
In addition to electrochemical reduction deposition, a recent study by the Li group attempted to deposit uranium in aqueous solutions by an electrolytic strategy.289 The authors synthesized a functionalized reduced graphene oxide foam (3D-FrGOF) electrode by a hydrothermal process. During the hydrothermal reaction, the functional groups on graphene oxide reacted with ethylenediamine (EDA), polysulfides, polythiobisamines, and free polysulfide radical species, leading to a sulfur and amine-modified graphene foam. When a negative potential of −0.9 V vs. saturated calomel electrode (SCE) was applied to the 3D-FrGOF electrode, the uranyl ions moved to the working electrode, thereby forming complexes with the functional groups on the graphene foam. Then, because of the hydrogen evolution reaction (HER) happening on the electrode, the local OH− concentration increased, and the uranium was deposited because of the increase in pH. Through the electrolytic deposition strategy with the 3D-FrGOF electrode, the uranium concentration of uranium-spiked seawater could be decreased from 3 ppm to 19.9 ppb within 14 h.
The potential of electrochemical strategies in the practical application of UES is being continuously explored. In general, the marriage between electrochemical approaches and UES offers notable advantages related to both the kinetics and thermodynamics of the chemical separation process. First, assisted by an external electric field, the extraction efficiency of UES is increased because of the accelerated diffusional mass transfer of uranyl ions towards the functional ligands. Second, through the electrochemical reduction process, the uranyl ions bound to the functional ligands are converted to neutral precipitation (e.g., UO2, Na2O(UO3·H2O)x). This breaks the conventional adsorption–desorption equilibrium and makes it shift towards the generation of more uranium products. Third, unlike the traditional adsorption method, the uranium products obtained by the abovementioned electrochemical approaches are in the solid form which is convenient for recovery. This eliminates the tedious elution process and reduces the cost.
To further demonstrate the feasibility of the electrochemical approaches for UES, major problems that need to be solved include the following aspects. (1) Most of the above studies were carried out in an acidic system (pH = 4.0–5.0). The dominant species of uranium in aqueous systems in the pH range is UO22+ with a positively charged state. But the natural seawater is weakly alkaline (pH ∼8.3) and the uranium exists in the form of (UO2)(CO3)32− with a negatively charged state. Whether excellent adsorption performances can be maintained is a question that needs to be investigated. (2) Since the electrochemical methods need an external electric field, the deployment of such separation systems in the ocean requires more consideration at the engineering design level (such as the method safety, and the construction cost and operating cost). (3) Due to the extremely low concentration of uranium in seawater, the electrochemical devices need to be immersed in seawater for quite a long time to obtain recoverable uranium products. The corrosion of the electrochemical devices by the high concentration of salts and a wide variety of microorganisms in the ocean thus needs to be evaluated and addressed.
4.2. Photochemical approaches
Sunlight is a primary energy source that can be directly obtained from nature. Exploiting solar energy to improve the efficiency of UES has attracted extensive attention recently, since additional energy consumption could be prevented. In this respect, a series of photocatalytic approaches have been integrated with uranium extraction systems to promote their performances. Primarily, the reduction potentials of UO22+/UO2 and UO22+/U4+ are 0.411 V and 0.327 V compared with standard hydrogen electrodes, respectively, while the typical semiconductor photocatalyst SiO2 has a reduction potential of 2.7 V in aqueous solutions at pH 7. This means that the photocatalytic reduction of uranyl ions by the semiconductor is theoretically feasible.290,291 For the first time, Li et al. tested the potential of TiO2 as a photocatalyst for UES.292 The maximum uranium extraction capacity of this photocatalytic strategy was over 3960 mg g−1, and the spectrum analysis confirmed that UO2 was the species of the extracted uranium. Considering the inherently high salt concentration in saline lake brine or seawater, Wang et al. assessed the influence of salt ionic strength in aqueous solutions on the performance of TiO2 for photocatalytic reduction of uranium.293 The results showed that the extraction efficiency of uranium was only 50.2% in the uranium spiked pure saline lake brine. When the brine was diluted with the same volume of saline lake water, the extraction efficiency could reach 91.1%. Basically, due to the extremely low concentration of uranium in seawater and the complex ocean environment, it remains critical to further improve the photocatalytic reactivity of TiO2.For instance, to enhance the photocatalytic activity of TiO2 under visible light, Wang et al. hydrogenated TiO2 with NaBH4 to generate Ti3+ self-doped TiO2 (so-called black TiO2, BTn).294 By increasing the addition amount of NaBH4, three hydrogenation degrees of TiO2 were obtained (BT1, BT2, and BT3). The uranium adsorption experiments showed that increasing the amount of reduced TiO2(Ti3+) could improve the uranium extraction efficiency, and BT2 exhibited a uranium extraction capacity 4 times higher than that of pristine TiO2. The authors attributed the high response of black TiO2 under visible light to the efficient formation of oxygen vacancies and surface disordered layers during the reduction of Ti4+. Xu et al. synthesized a composite material (TiO2/CPAN-AO) by a hydrothermal and cyclization hydrogenation method (Fig. 35A).295 The surface of the TiO2/CPAN-AO composite is hybridized with a π-conjugated structure and Z-type heterojunction, prolonging the lifetime of photo-generated charges. The qU of TiO2/CPAN-AO reached 2380 mg g−1 under light irradiation (Fig. 35B), while in the dark, the qU was only 10% of that under illumination. The charge carrier transfer process made the TiO2/CPAN-AO composite a heterojunction with high redox capability (Fig. 35C–E), which greatly improved the photocatalytic reduction efficiency of U(VI).
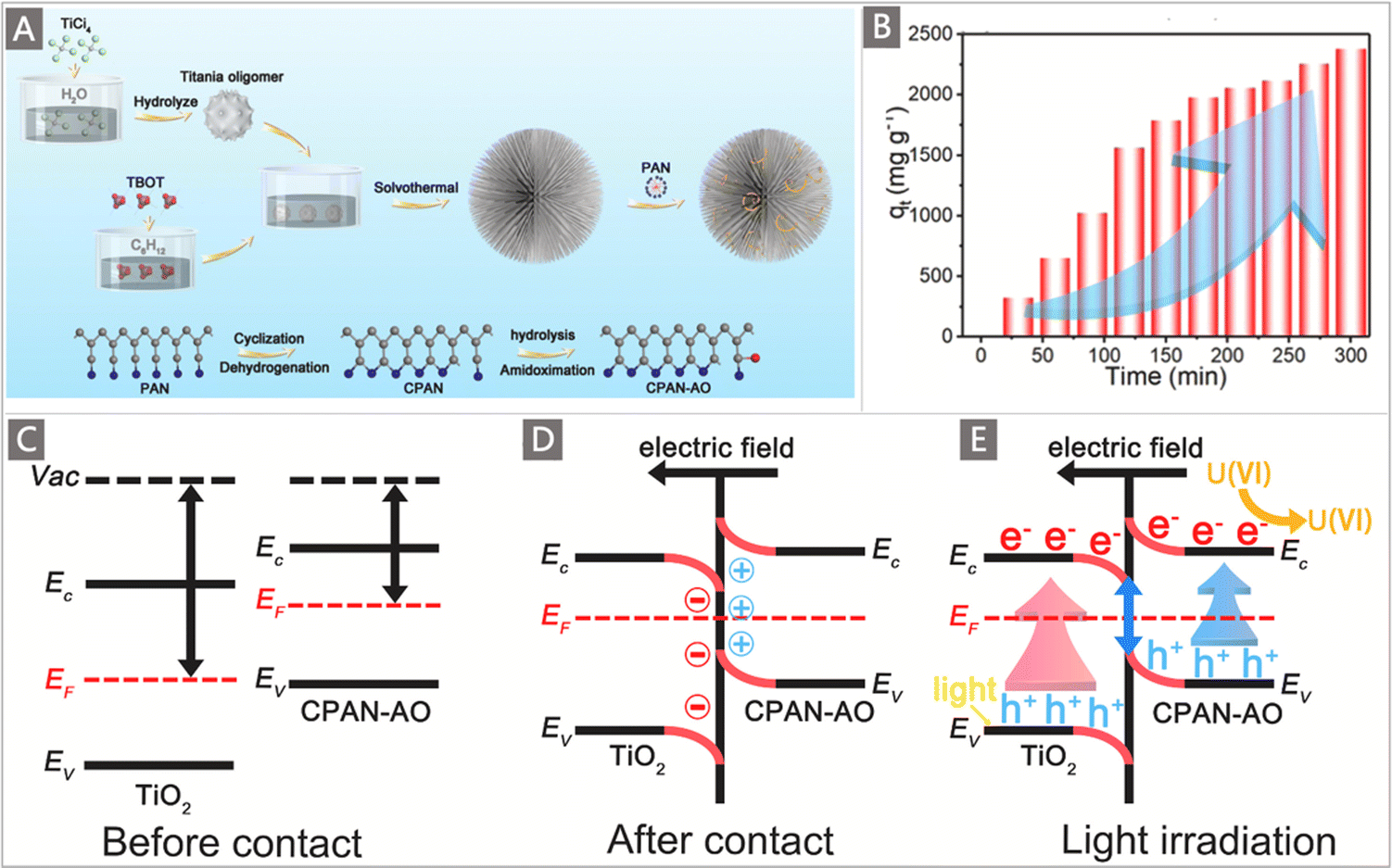 | ||
| Fig. 35 (A) Schematic representation of the formation process of TiO2/CPAN-AO microspheres. (B) The effect of time on the photocatalytic reduction ability of TiO2/CPAN-AO at 0.01 M L−1 uranium concentration. (C–E) The mechanisms of uranium adsorption, coordination and photoexcited electron transfer in TiO2/CPAN-AO. Reproduced with permission.295 Copyright 2021, Elsevier. | ||
The above studies show that TiO2 may be a promising photocatalyst for UES, and through proper modification, the reduction and separation of uranyl ions under visible light could be achieved. To further improve the TiO2 involved photocatalytic system for UES, Li et al. designed and fabricated a novel magnetic core–shell adsorbent, Fe3O4@TiO2-AO.229 The inner core was the magnetic Fe3O4 and the shell layer was TiO2 modified with AO groups. The magnetic Fe3O4@TiO2-AO could be conveniently separated under an external magnetic field, and the regeneration efficiency still reached 78.5% after 10 adsorption–desorption–regeneration cycles. This photocatalytic system achieved a qU of 87.5 μg g−1 within 33 d in natural seawater.
Graphitic carbon nitride (g-C3N4), as a kind of metal-free photocatalyst, attracts widespread attention because of its moderate energy gap, high stability, comparatively facile synthetic method, and low cost.296 Compared with bulk g-C3N4, porous g-C3N4 has a higher specific area, which can provide more active sites for photocatalytic reactions.297 Wang and co-workers prepared tunable mesoporous g-C3N4 (MCN) using a hard template method and tested its photocatalytic performance for uranium.298 Due to the high light utilization and photo-generated carrier separation efficiency, MCN showed excellent photocatalytic performance for the reduction of uranyl under visible light illumination. Among the mesoporous g-C3N4 with different specific surface areas (MCN0.25, MCN0.5, MCN1.0, MCN2.0, MCN4.0), MCN1.0 showed the best uranium separation efficiency, completely enriching the uranium in the U(VI)-spiked real seawater within 20 min.
Previous investigations showed that under weakly alkaline conditions the carbonates in the seawater system seriously inhibited the photocatalytic reduction of U(VI). To overcome the limitation, Li et al. prepared carboxylated g-C3N4 catalysts (CCN-5, CCN-24) with different amounts of carboxyl groups by oxidizing bulk carbon nitride (BCN).299 The introduced carboxylic groups on the surface of carbon nitride enhanced the adsorption ability of U(VI). DFT calculations showed that the modified g-C3N4 had a narrower energy band gap and a lower conduction band position, leading to higher photocatalytic performance for U(VI) reduction. Compared with BCN, the uranium extraction capacity of CCN-24 was increased by 30% in a solution containing 2.0 mmol L−1 NaHCO3. Moreover, the U(VI) reduction rate of CCN-24 was 0.0867 min−1, which was ∼33 times higher than that of BCN (0.0026 min−1). To further improve the photocatalytic activity and adsorption capacity of the carbon nitride photocatalyst, Liu et al. synthesized a new kind of bifunctional carbon nitride material (CN550) by a one-step molten salt method,300 which had more adsorption sites and higher specific surface area (75.6 m2 g−1) (Fig. 36A). The adsorption behaviours of g-C3N4 and CN550 were first studied under dark and light conditions. It was found that CN550 exhibited much higher adsorption and photocatalytic activity than g-C3N4 (Fig. 36Ba–c). Subsequently, the performance of photocatalysis-assisted adsorption of uranium using CN550 as the adsorbent was explored. Compared with the traditional physicochemical adsorption (PA) method, a significantly improved extraction capacity for uranium was observed under light illumination (Fig. 36Bd). The uranium extraction capacity of CN550 could reach 1556 mg g−1 using the sunlight-driven photocatalysis-assisted extraction (SUPER) method with an initial uranium concentration of 500 mg L−1, which was nearly 10 times higher than that of the physicochemical method (149 mg g−1). When tested in uranium-spiked real seawater at a relatively low concentration of 3 mg L−1, a removal rate of 98.7% and an extraction capacity of 17 mg g−1 could be obtained by CN550 using the SUPER method, while the PA method only exhibited a 17.9% removal rate and an extraction capacity of 3 mg g−1 (Fig. 36Be). A high uranium extraction capacity of 1057 mg g−1 by CN550 could be acquired when the initial uranium concentration was increased to 330 mg L−1. Moreover, the SUPER method by utilizing CN550 also showed a higher removal rate for uranium than the PA method under natural sunlight illumination (Fig. 36Bf). These results suggested the potential of CN550 in the application of UES under sunlight by the SUPER method.
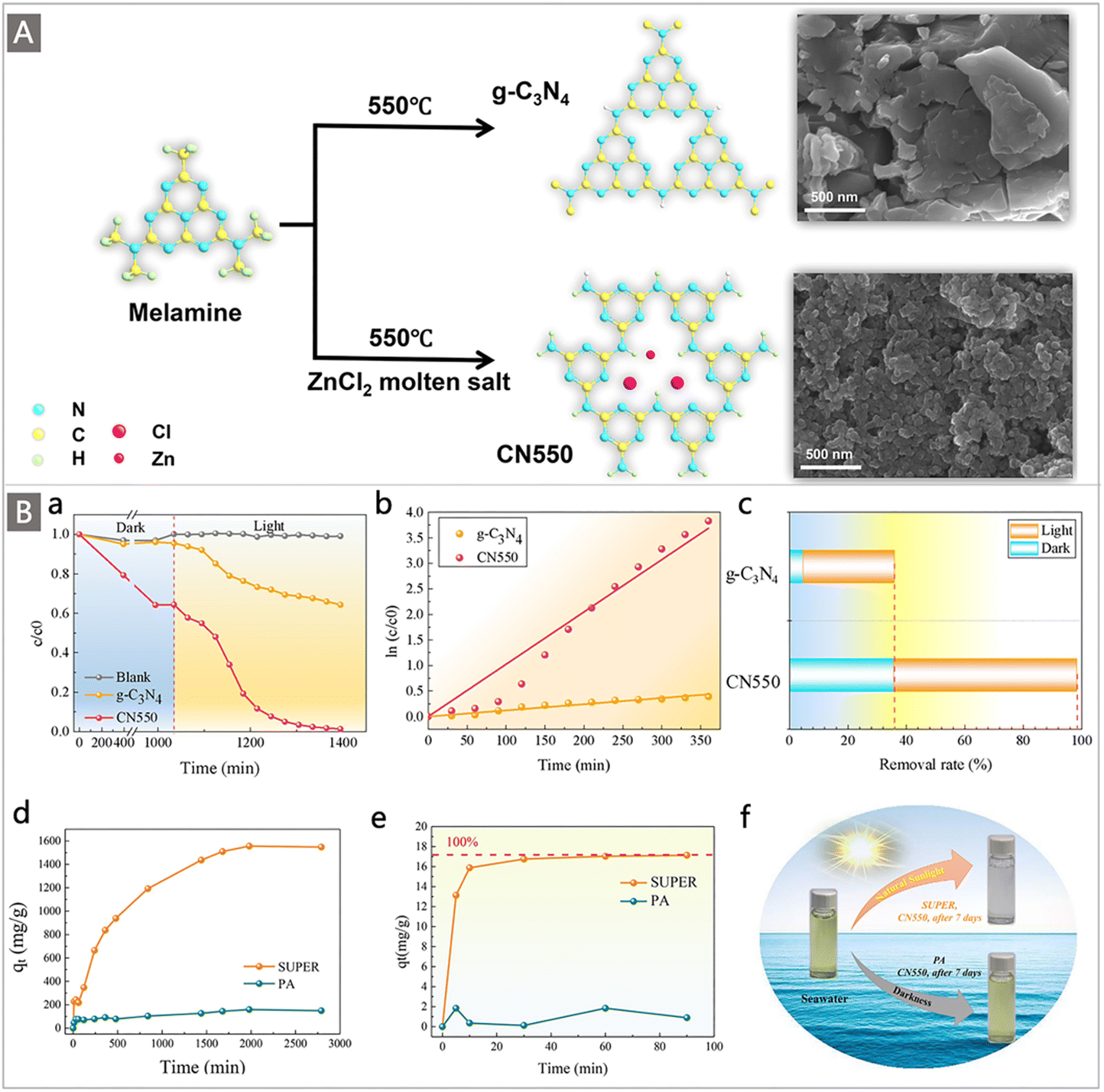 | ||
| Fig. 36 (A) Scheme for the formation of g-C3N4 and CN550 with PTI structure and the SEM images for both materials. (B) (a–c) The photocatalytic and adsorption behaviours of CN550 and g-C3N4 under both dark and light conditions (0.2 g L−1 CN550 and 1 g L−1 g-C3N4 as photocatalysts with initial 200 mg L−1 uranium solution); (d) uranium extraction by using the SUPER and PA methods with an initial uranium concentration of 500 mg L−1 (0.2 g L−1 CN550, pH = 5.0); (e) uranium extraction from spiked seawater using the SUPER and PA methods with an initial uranium concentration of 3 mg L−1 (0.2 g L−1 CN550, pH = 5.0); and (f) photograph of uranium-spiked seawater solutions (around 1500 mg L−1) after a week of extraction under natural sunlight and after a week of adsorption in the dark followed by filtration. Reproduced with permission.300 Copyright 2021, Elsevier. | ||
In recent years, a variety of porous framework materials such as MOFs,301–303 COFs,83,304 CMPs92,305 and HOFs306 have been regarded as new types of photocatalysts for uranium extraction, due to their high specific surface area and multiple reaction sites. Li et al. synthesized a post-synthetically functionalized metal–organic framework (MOF PCN-222) by combining ligand complexation and photocatalytic reduction.302 The MOF PCN-222 was modified with phosphono- and amino groups, which endowed it with the capability of uranyl complexation (Fig. 37A). Upon visible light irradiation, the uranium uptake capacity could be significantly improved, compared with the results obtained under dark conditions (Fig. 37B). In addition, it was observed that U(VI) can almost be completely separated with PCN-222 at high or low concentration over an extremely wide pH range, and a maximum uranium uptake amount of 1289.3 mg g−1 could be achieved at an initial uranyl concentration of 400 ppm and a solid–liquid ratio of 0.25 g L−1. By introducing U(VI)-recognizing ligands that were pre-enriched into the adsorption sites, U(VI) was simultaneously reduced efficiently by the photoinduced electrons from the photoactive MOF host under visible-light irradiation, resulting in neutral U(IV) species that were stored in the open space (Fig. 37C). The active sites for additional uranium binding could also be quickly regenerated, thus affording an ultrahigh uranium-extraction capacity.
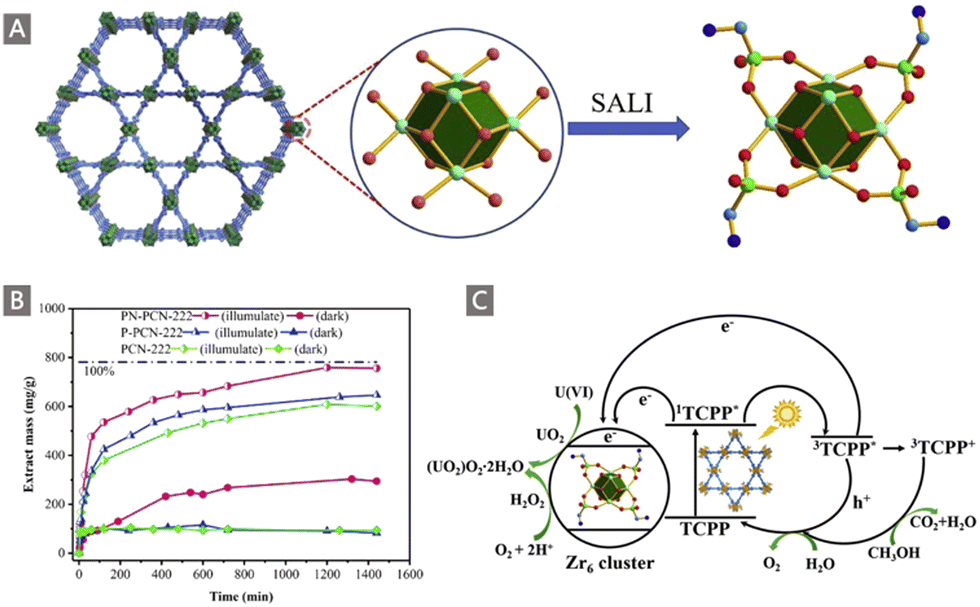 | ||
| Fig. 37 (A) Schematic representation of SALI. (B) Uranium extraction with PCN-222, P-PCN-222 and PN-PCN-222 using the photocatalytic reduction method compared to the adsorption method in the dark with initial uranium concentrations of ∼400 ppm and the solid to liquid ratio of 0.5 g L−1. (C) Schematic illustration of selective enrichment and photocatalytic reduction of U(VI) based on PN-PCN-222. Reproduced with permission.302 Copyright 2019, Elsevier. | ||
Yu et al. presented a molecular engineering strategy to boost the photocatalysis performance of donor–acceptor CMPs (ECUT-SO) for uranium reduction under visible light.305 Significantly, ECUT-SO demonstrated an ultrahigh uranium extraction capacity of 1780 mg g−1 with a nearly 100% extraction efficiency in uranium-spiked real seawater with an initial uranium concentration of 50 mg L−1. Building on this work, the same group synthesized a sulfonated perylene-based CMP (PyB-SO3H), which displayed a higher uranium extraction capacity of 1989 mg g−1.252 The Wang group utilized the photocatalytic and photoelectric effects of black phosphorus,232 anchored UiO-66-NH2/black phosphorus quantum dot (MOF/BPQD) heterojunctions onto the carboxyl cellulose nanofiber (CNF) aerogel, realizing the photocatalytic reduction and efficient separation of uranium.233 For improving the uranium adsorption ability as well as resistance to biofouling, the Qiu group developed a series of COFs by combining selective ligand binding (amidoxime groups or hydroxyl groups), chemical reduction, and a photo-enhanced strategy.85,249,307 These COF photocatalysts exhibited excellent uranium extraction capacity as well as high anti-biofouling activity, due to their multifunctional coupling strategies.
In general, developing photocatalytic strategies for UES has been widely considered to be a promising way for high efficiency and low cost. Continuous effects are still being made to exploit the photocatalytic technology while optimizing the photocatalyst design and synthesis for UES. The principles for the development of achievable photocatalytic systems have been elaborated in several articles.41,308 Researchers in the field of UES could draw inspiration from state-of-the-art photocatalysis. Nonetheless, similar to the electrochemical approaches mentioned above, there remain significant obstacles to the practical application of photocatalytic approaches. A specific challenge that should not be ignored is the use of sacrificial agents, such as methanol and ethanol, in photocatalytic systems. Although sacrificial agents play an important role in facilitating the reduction of uranium on semiconductors with lower reduction potentials, it seems impractical to use these sacrificial agents in real seawater systems. A more environmentally friendly and effective UES process would be established if the issue of sacrificial agents is addressed. Besides, major challenges still lie in the deployment of photocatalytic systems and the stability of photocatalysts under realistic marine conditions.
5. Marine field tests and cost–efficiency analysis
Natural seawater is a complex biogeochemical system with a great amount of coexisting metal ions, high salinity, specific pH conditions, and substantial marine biofouling. The laboratory screening tests with artificial seawater cannot accurately reflect the adsorption behaviours of adsorbents in the natural seawater. Before the evaluation of the commercialization potential of UES, the performance of the diverse uranium adsorbents developed in laboratories needs to be validated in marine field tests. Information from marine field studies is valuable for evaluating the adsorption capacity, selectivity, biofouling resistance and durability of the adsorbents under real seawater conditions. These data are required to optimize the structural parameters of the adsorbents and their deployment conditions to establish an effective uranium recovery system.9 To date, marine tests for uranium extraction have been conducted in countries including Japan, the United States, and China. In this section, the up-to-date achievements in marine tests made by these countries are detailed, which is envisioned to contribute to a more in-depth understanding of UES from an engineering point of view. Since the early UES programs and marine infrastructures in Japan and the United States have been covered in some reviews and reports,9,26,309,310 we highlight the contributions of Chinese researchers in the past five years, especially the breakthroughs in marine tests initiated by the China Academy of Engineering Physics (CAEP) and the Chinese Academy of Sciences (CAS). Moreover, since UES is largely a game of economics and scale, the presentation of the implemented marine field tests will be accompanied by a brief introduction of the cost estimation, and potential approaches to reduce the cost will be proposed at the end of this section.5.1. Marine tests in Japan
Driven by the scarcity of domestic uranium resources and the sharp increase in uranium demand, Japan turned its attention to the ocean as early as the 1960s to exploit marine uranium reserves. Laboratory experiments were then carried out by the National Institute of Advanced Science and Technology (Shikoku), the University of Tokyo, Kyoto University and other research institutes. In 1974, the Research Committee on Rare Resources from Seawater organized by the Ministry of International Trade and Industry (MITI) started the overall study on the recovery of uranium from seawater. In the subsequent year, the Metal Mining Agency of Japan participated in the project, and a coordinating committee was then established.309 These early extensive efforts focused on the screening of various adsorbents where hydrous titanium oxide (HTO) was identified as a promising adsorbent.19–21,311 The first experimental plant for UES was operated by the Agency for Natural Resources and Energy (ANRE), MITI and the Metal Mining Agency of Japan from 1981 to 1988, with HTO as the adsorbent. However, the reported adsorption capacity of the HTO was only 0.1 mg g−1, which was clearly not economically feasible. Moreover, the requirement for mechanical pumping of seawater across the adsorbent bed and the poor mechanical resistance of the HTO limited its practical applications, leading to the cessation of the project.26The focus has been turned to synthetic polymers, which represent the category of adsorbents most capable of large-scale deployment for UES. In particular, AO was found to be a new promising chelating agent for uranium recovery, and in 1979 and 1980, Egawa and co-workers first reported the preparation and application of AO-functionalized polymeric adsorbents for UES.27–29 The development of AO-derived adsorption systems in Japan has aroused a research upsurge for UES, leading to both laboratory studies and marine tests from the 1980s to the early 2000s. From the viewpoint of practical handling in the adsorption process, great efforts on the laboratory scale have been devoted to the development of adsorbents for practical applications to the seawater system over the 1980s and 1990s.26 During this period, three types of AO-based adsorbents were developed successively, including AO-based polymer beads,27,312 AO-based fibers prepared by chemically reacting commercially available AN fibers with hydroxylamine,313,314 and AO-based fibers prepared by the well-known RIGP process as depicted in Fig. 5B.66,68 The adsorbents prepared by RIGP using PE non-woven fabrics as trunk polymers demonstrated favorable mechanical strength and adsorption capacity, which were chosen for subsequent large-scale marine experiments.315
The marine field tests in Japan began in the early 1990s with the work of Takeda and colleagues.266 A fixed bed charged with hollow fibers containing AO groups (labeled AO-H fibers) was fabricated via RIGP and placed on the coast of the Pacific Ocean for UES. After 30 d of contact with the coastal seawater, a qU of 0.97 mg g−1 was obtained by the AO-H fibers. Shortly after, a field test for UES was conducted by Goto and co-workers in Imari Bay in the northern part of the Kyushu Island of Japan.316 A mooring system was established where AO fibers were packed in an adsorption cage (unit), which was suspended with ropes from a buoy moored in the seawater and was allowed to swing with the wave motion (Fig. 38A). After the mooring of 37 d, the stability of AO-based fiber was confirmed, and a qU of 1.30 mg g−1 was achieved (Table 1).
 | ||
| Fig. 38 Schematic images of (A) the mooring system (Reproduced with permission.65 Copyright 2000, American Chemical Society); (B) the stack collection system; and (C) the braid collection system. (B and C) reproduced with permission.320 Copyright 2016, EDP Sciences. | ||
| Time | Place | Adsorbent | Adsorption system | Collected U amount | Time | Ref. |
|---|---|---|---|---|---|---|
| 1991 | The coast of the Pacific Ocean | AO-based fiber | Continuous-flow system | 0.97 mg g−1 | 30 d | 266 |
| 1991 | Imari Bay, Japan | AO-based fiber | Mooring system | 1.30 mg g−1 | 37 d | 316 |
| 1993 | Imari Bay, Japan | AO-based resin | Towing–mooring system | 1.32 mg g−1 | 930 h | 317 |
| 2000 | 6 km offshore at Mutsu Sekine-Hama in Aomori Prefecture, Japan | AO-based fiber | Mooring system | 0.90 mg g−1 | 20 d | 65 |
| 1999–2001 | 7 km offshore from Mustu-Sekine in Aomori prefecture, Japan | AO-based fabric | Stack collection system | 1083 g | 240 d | 315 |
| 2004 | Okinawa, Japan | AO-based fiber | Braid collection system | 1.50 mg g−1 | 30 d | 318 |
The performances of porous AO chelating resins for UES were also evaluated through towing and mooring systems in Imari Bay.317 For both sets of trials, the adsorption beds of the as-prepared AO resins were suspended in the ocean from a buoy. After 930 h exposure, the highest qU of 1.32 mg g−1 was acquired. Besides, several hydrophilic AO-based fibers prepared by RIGP of AN with hydrophilic comonomers of MA or HEMA from PE trunk fibers were tested offshore at Mutsu Sekine-Hama in Aomori Prefecture, using a mooring system.65 After being submerged at 15 m below the surface for 20 days, the fibers with an AO:MA weight ratio of 60:40 exhibited the best performance, affording a maximum qU of 0.90 mg g−1.
Significantly, the largest marine field test led by the Japan Atomic Energy Agency (JAEA) was carried out and a two-year-long effort from 1999 to 2001 was devoted to uranium recovery from the Pacific Ocean. In this project, two different uranium collection systems using adsorbents stacks315,319,320 and braided adsorbents318,321 were designed and prepared, respectively. The detailed design parameters of the two systems were provided by Tamada and co-workers.26 As for the stack collection system (Fig. 38B), the adsorption stacks were prepared from PE nonwoven fabrics grafted with AO ligands, which were then cut into smaller sheets and packed into cages that were lowered from a floating frame into the Pacific Ocean. The adsorption beds were hanged out every 20–40 d and the adsorbed uranium on the adsorbent fabric was fractionally eluted with 0.5 mol L−1 HCl. Over 240 d of contact with seawater, more than 1 kg yellowcake was yielded, affording an average adsorption ability of 0.5 mg g−1 for 30 d soaking. However, one major problem with the stack collection system was the high collection cost. Due to the large weight of the marine equipment for mooring the adsorbents in seawater, the mooring operations of the stack collection system were found to account for more than 70% of the total cost.322
To address this issue, an innovative system using braided adsorbents was designed in 2004, as shown in Fig. 38C. These long kelp-like braids of AO-functionalized PE fibers could be anchored to the seafloor without a floating frame and adsorption beds, and could be cut off from the anchor using wireless operation when collected. Besides, the braid adsorbents that appeared on the sea surface could be recovered with a fishing boat. The braid system was evaluated in the sea of Okinawa area in Japan. Impressively, a breakthrough in UES was achieved in this work where a successful uptake of 1.5 mg g−1 for uranium was acquired by the braided AO-based fibers after 30 d, which was three times higher than that of adsorbent stacks. Compared with the stack collection system, the higher adsorption capacity acquired by the braided-type adsorbents may be attributed to the enhanced contact between the adsorbents and seawater. Moreover, the braid collection system was estimated to achieve a reduction of 40% of the cost for UES compared to the stack collection system, since the construction of the floating frame and the adsorption beds could be avoided.
An extended economic analysis of the braid collection system was performed by Tamada and colleagues in 2006,323 with the consideration of the three basic steps in the uranium production process, including adsorbent production, uranium extraction, and elution and purification. A design for a large-scale UES system was proposed, with an annual output of 1200 tU, adsorption ability of 2 mg g−1 for 60 d soaking and 6 times repeated usage. As a result, the collection cost was estimated to be 90000 yen per kg-U (about $1000 per kg-U, in 2011 dollars). Additionally, the critical cost driver was identified as adsorbent production in this analysis, which accounted for 69% of the total cost. Later, Schneider and Sachde conducted an updated cost estimation for UES through the braid collection system developed by JAEA,324 incorporating the uncertainties in chemical, commodity and capital equipment costs. Using the same reference conditions as JAEA for adsorption capacity and recycles (a 60 d loading of 2 mg g−1 and 6 recycles of use of adsorbent) as a base case, a production cost of $1230 per kg-U with a 95% confidence interval of ($1040 per kg-U, $1440 per kg-U) was estimated in this study. The difference in cost was mostly attributed to their consideration of an experimentally observed 5% degradation of adsorbent capacity per use cycle. Further sensitivity studies highlighted that the major cost drivers in the braid collection system were the adsorbent capacity and the number of recycles of the adsorbents.
5.2. Marine tests in the United States
Although the United States had carried out preliminary research on UES in the 1960s, few experimental efforts have been made before 1980. After the 2005–2007 uranium price bubble, the researchers in the United States were motivated by the need for economically viable uranium sources beyond terrestrial ores. Renewed enthusiasm was thus aroused to speed up the development of UES. The “Fuel Resources Project” was launched with the objective of developing advanced adsorbents with an adsorption capacity at least double that of the best adsorbent pioneered by JAEA (1.5 mg g−1).325 A multidisciplinary team was assembled with scientists from the United States national laboratories including Oak Ridge National Laboratory (ORNL), Lawrence Berkeley National Laboratory (LBNL), Pacific Northwest National Laboratory (PNNL), universities, and research institutes to address this challenging problem in 2011.326 Multifaceted efforts have been made since then, with major contributions achieved such as the development of “HiCap” and “U-Grabber” polymer adsorbents which received the ‘R&D 100’ award in 2012 and 2016,120 respectively, the optimized “AI” and “AF” series adsorbents prepared by RIGP,110,111 and the application of the ATRP approach for synthesis of novel adsorbents.75,76 Herein, we provide an overview of the establishment of marine test activities for performance assessment done in the United States over the period 2012–2016.155,202,310,327 The marine test program was started at the Marine Science Laboratory (MSL) of PNNL in 2012, to determine the adsorption behaviours of the adsorbents produced by program participants towards uranium and other elements (e.g., V, Fe, Cu, Zn, Ni and Cd) under natural seawater conditions. Specifically, three formulations of the ORNL AO-based polymeric adsorbents, 38H, AF1 and AI8, which exhibited high capacity in batch screening experiments with simulated seawater,310 were initially employed in field tests with natural seawater. These adsorbents were produced in a fibrous form (40–100 mg samples) or further prepared in large mass as a braided material (5–10 g samples). Instead of deploying the adsorbents directly in the ocean, two kinds of indoor exposure systems, a flow-through column system and a recirculating flume exposure system (Fig. 39), were designed and utilized for performance assessment. Ambient seawater from Sequim Bay, WA, was used for seawater exposure studies; it was delivered to a “wet laboratory” through a seawater delivery system developed in the MSL. Briefly, for the flow-through column system, the conditioned fibrous adsorbents were dispersed and packed in adsorption columns, and held in place using a combination of glass wool and/or glass beads (3–5 mm). For the flume exposure system, the conditioned braided adsorbent materials were mounted in the flume for seawater exposure. In both seawater exposure systems, the filtered natural seawater was pumped from a large-volume reservoir tank and constantly fed into the columns or flume under controlled temperature and flow-rate conditions, and then recycled back to the tanks. Additional details about the design parameters and concepts for both systems were given by Gill and colleagues.155 | ||
| Fig. 39 (A) Layout and components of the seawater delivery and multi-port manifold system used for exposing uranium adsorbent materials in flow-through columns to filtered ambient seawater under controlled temperature and flow conditions; (B) side view depiction of the recirculating flume system used for exposing braided adsorbent material to filtered or unfiltered natural seawater under controlled temperature and flow-rate (linear velocity) conditions; (C) two, 24 port manifold systems (back to back) with flow-through columns attached; and (D) translucent flumes for unfiltered seawater exposure studies with braided adsorbent materials. Reproduced with permission.155 Copyright 2016, American Chemical Society. | ||
The time series measurements of uranium adsorption capacity with 38H, AF1 and AI8 adsorbents were first performed using the flow-through column exposure system with filtered Sequim Bay seawater, and the results are summarized in Table 2.155,328,329 After 56 d of seawater exposure at 20 °C, all three formulations of adsorbents demonstrated adsorption capacities higher than 3 mg g−1, of which the AF1 formulation exhibited the best performance with a 56 d adsorption capacity of 3.9 mg g−1 and a predicted saturated adsorption capacity of 5.4 mg g−1.310 Notably, the uranium adsorption amounts of the three adsorbents after 56 d exposure to seawater were all 3–4 times higher than the maximum capacity (1.1 mg g−1) achieved by the adsorbents of JAEA under similar conditions.327 Based on the uranium uptake measurements, a cost analysis was also conducted by updating the adsorption capacity in the mooring system proposed by JAEA323 with that of the ORNL adsorbents obtained in this work (3.09 mg g−1 of 60 d uptake). Approximately, the ORNL adsorbent enabled the recovery of uranium from the seawater at a cost of $760 per kg-U using the adsorption system proposed by JAEA, while a cost of $610 per kg-U might be achieved in a modified system using smaller boats working from offshore elution and stronger polymer anchor ropes.327
| Adsorbents | Saturation capacity (mg g−1) | 56 day adsorption capacity (mg g−1) | Half-saturation time (days) |
|---|---|---|---|
| a The data were determined from one-site ligand saturation modelling of time series data and were normalized to a salinity of 35 to allow for a direct comparison of uranium concentrations at a common salinity. Unless otherwise specified, the experiment was conducted at 20 °C, and the flowrate (linear velocity) of the seawater through the column or flume was between 2 and 2.5 cm s−1. All data were taken from the report by Gill et al.328 b The data were determined from one-site ligand saturation modelling of time series data obtained at ambient temperature (26–31 °C) and ambient salinity (35.7 to 37.4). All data were taken from the report by Todd et al.329 | |||
| PNNL, flow-through column testinga | |||
| 38H | 4.29 ± 0.24 | 3.30 ± 0.18 | 16.9 ± 2.8 |
| AI8 | 5.17 ± 0.18 | 3.54 ± 0.12 | 25.8 ± 2.1 |
| AF1 | 5.56 ± 0.15 | 3.91 ± 0.11 | 24.0 ± 1.5 |
| AF1FR2 | 7.05 ± 0.21 | 5.00 ± 0.15 | 22.9 ± 1.7 |
| AN/MA/42kGY-a1 | 8.43 ± 0.72 | 5.13 ± 0.44 | 36.0 ± 6.1 |
| MAN1-AO/DMSO | 6.70 ± 0.22 | 5.04 ± 0.16 | 18.5 ± 1.6 |
| MAN8-AO/DMSO | 7.75 ± 0.37 | 5.09 ± 0.24 | 29.2 ± 3.0 |
| LCW-2 | 6.04 ± 1.04 | 3.78 ± 0.65 | 33.5 ± 12.0 |
| LCW-MSL-10 | 6.34 ± 0.19 | 5.28 ± 0.16 | 11.2 ± 1.3 |
| SB12-8 | 8.90 ± 0.45 | 6.56 ± 0.33 | 20.0 ± 2.7 |
| SB11-295 | 11.1 ± 2.40 | 6.18 ± 1.34 | 44.6 ± 16.4 |
| PNNL, recirculating flume testinga | |||
| AI8 | 6.86 ± 0.68 | 4.13 ± 0.41 | 37.0 ± 7.0 |
| AF1 | 5.93 ± 0.17 | 4.03 ± 0.12 | 26.5 ± 1.6 |
| AF1-13-AO/DMSO | 6.51 ± 0.69 | 4.27 ± 0.15 | 29.3 ± 7.0 |
| AF8 | 7.04 ± 1.42 | 3.62 ± 0.73 | 52.9 ± 19.0 |
| WHOI, flow-through column testinga | |||
| AF1 | 5.97 ± 0.27 | 4.43 ± 0.20 | 19.4 ± 2.4 |
| WHOI, recirculating flume testinga | |||
| AF1 | 9.84 ± 0.48 | 5.96 ± 0.29 | 36.4 ± 3.7 |
| BKI, recirculating flume testingb | |||
| AF1 | — | 6.35 ± 0.10 | 25 ± 1 |
| AI8 | — | 5.96 ± 0.24 | 21 ± 2 |
| AF8 | — | 4.43 ± 0.81 | 51 ± 2 |
| AF1-AO-DMSO | — | 6.77 ± 0.56 | 21 ± 2 |
Other formulations of adsorbents produced by either RIGP or ATRP approaches were also explored. Significantly, the ATRP-adsorbent (referred to as SB12-8) grafted with AN and HEA using PVC-co-CPVC fibers as the backbone achieved the highest qU of 6.56 mg g−1 after 56 d of exposure to natural seawater, nearly a doubling in adsorption capacity over that of the 38H adsorbent. Subsequently, scale-up tests using a recirculating flume were carried out with braided adsorbents under controlled temperature and flow-rate conditions. Multiple cycles of exposure were first conducted with AF1 braided adsorbents, with the final adsorption capacity determined to be 4.03 mg g−1 after 56 d, which was comparable with the data obtained in the flow-through column test. The coincidence of the test results obtained in the two exposure systems validated the performance of the adsorbents. The production of the adsorbents was highly reproducible, and the test procedures were reliable. It is worth mentioning that there has been a continuous improvement in the adsorption capacity of AO-based polymeric adsorbents from 3.30 mg g−1 to 6.56 mg g−1 in 56 d in the marine tests conducted at PNNL.
Building on this work, the Woods Hole Oceanographic Institution (WHOI) and the University of Miami's Broad Key Island (BKI) Research Facility also served as coastal marine sites for additional marine tests using an identical exposure set-up as used at PNNL, to assess the effects of different oceanographic conditions and water quality conditions (e.g., temperature, salinity, the concentration of dissolved organic carbon (DOC), and competing ions) on uranium recovery from seawater. The time series measurements of the ONRL AF1 adsorbents were investigated at the WHOI using both flow-through column and flume exposure systems. The obtained adsorption capacity in the WHOI experiments was 15% and 55% higher than that observed in the PNNL experiments for column and flume tests, respectively. The regional differences in U adsorption capacity were attributed to the variations in competing ions and/or water quality parameters such as DOC concentrations.155
In addition, several formulations of AO-based polymeric adsorbents (AF1, AI8, AF8, and AF1-AO-DMSO) were selected and identical exposure systems were employed for investigations at BKI. The marine tests at BKI provided opportunities to assess the performances of these adsorbents under warmer ambient and higher saline conditions than those existing at Sequim Bay off the Washington coast. Following contact with seawater for a period of 56 d at ambient temperature (26–31 °C) and salinity (35.7 to 37.4), the AF1, AI8 and AF1-AO-DMSO adsorbents all exhibited similar uranium adsorption capacities (6.0 mg g−1 to 6.6 mg g−1) in flume system tests, while the AF8 adsorbent showed a lower uptake of 4.4 mg g−1. Obviously, all adsorbents tested at BKI exhibited higher capacities than those obtained at PNNL, which was mainly ascribed to the higher temperatures. These results acquired from different coastal marine environments highlighted the critical role of temperature, the competing ions and other water quality conditions such as DOC in the process of UES.330
To save energy and costs associated with the deployment, mooring and recovery of adsorbents, a mechanically driven seawater extraction system called the symbiotic system was designed by Picard and co-workers in 2014.331 As illustrated in Fig. 40A, the initial proposed concept of the symbiotic system consists of a continuous belt of adsorbents 4000 m in length, and a platform at the base of the wind tower supports the belt of adsorbent that loops in and out of the water. This belt cycles through the seawater beneath the turbine and through an elution plant located on a platform above the sea surface, thereby allowing for an autonomous elution procedure that can be precisely timed depending on the type of adsorbent used. The proposed system was sized to collect 1.2 tons of uranium per year, which was enough for supplying 5 MW nuclear power. An independent cost-analysis of such a symbiotic system was performed by Byers et al., and the results were compared to a reference kelp-like deployment strategy. It was found that the symbiotic system could reduce the production cost by at least 11% from $450–890 per kg-U (in 2015 dollars) for the reference kelp-like scheme to $400–850 per kg-U,332 and up to 30% with further design optimization.333 The cost reduction was achieved by eliminating the offshore deployment and mooring cost through the symbiotic system, while it also increased the energy output of the wind farm. However, a limitation of the symbiotic system is that it typically requires the adsorbents to be braided into a belt held in tension, which may face difficulties in a marine environment.334
 | ||
| Fig. 40 (A) Three-dimensional view of a continuous uranium recovery system with an adsorbent belt looped around the turbine mast. Reproduced with permission.331 Copyright 2014, American Nuclear Society. Design details of SMORE: illustrations of side (B) and top (C) views of the SMORE system. (D) The design of the outer protective sphere used for encapsulating the polymer adsorbent. (E) 3D model of the 1/10th physical scale model for ocean testing of the SMORE design, where one version is the stationary system and the other is the dynamic system. (F) Photo of both prototypes mounted onto the wooden float and moored to the end of the MMA dock during testing. (G) A 1:10 physical scale adsorbent ball-chain net as used in SMORE. White adsorbent enclosure shells were alternated with orange placeholder shells used for mechanical testing. (B–G) reproduced with permission.340 Copyright 2019, American Chemical Society. | ||
Taking that into account, a two-part system called the Symbiotic Machine for Ocean uRanium Extraction (SMORE) (Fig. 40B and C) was proposed by Haji and colleagues,335–340 to decouple the mechanical and chemical requirements of adsorbents for uranium harvesting from seawater. The SMORE concept involved the structure of an offshore floating wind turbine that was utilized to provide the mooring and structural support for an autonomous, offshore uranium-harvesting platform. Especially, the adsorbents were enclosed in a hard permeable outer shell with sufficient mechanical strength and durability for use in offshore environments and chemical resilience against elution treatments (Fig. 40D).336 In this design, the holes of the shell enclosures allowed the adsorbents to remain exposed to adequate seawater, but at the same time, the adsorbents were also mechanically protected with the spherical enclosures, thus meeting the durability requirements for UES. The shell enclosures were incrementally spaced along high-strength mooring ropes, resembling conventional ball-chain belts. These ball-chains were then strung together to create a net using cross-members which add rigidity and reduce the likelihood of tangling of individual lengths. Accordingly, this design was developed into two versions of prototypes at a 1:10 physical scale, one in which the adsorbent ball-chain net was continuously moving through the water and the other was only subjected to the ocean currents at the test site, known as a dynamic and stationary system, respectively (Fig. 40E and F).337 For both of the prototypes, four lengths of shells were combined to make a single net.
These prototypes were successfully tested in the Massachusetts Maritime Academy (MMA) in Buzzards Bay, MA, in a nine-week ocean trial, with the ORNL AI8 adsorbent fibers as the reference adsorbent.340 Besides, two shell enclosures using slotted holes and circular holes were tested in this ocean trial (Fig. 40G). At the end of a 56 d ocean test, a very little difference in uranium uptake by AI8 adsorbent fibers between the different enclosure or system types was observed, indicating that the system movement and shell designs did not increase the adsorption amount for uranium. The maximum qU of approximately 1.25 mg g−1 by the AI8 adsorbent was achieved using the stationary system and the shell enclosure with slotted holes. This value was much lower than the capacities obtained for AI8 adsorbents in previous ocean tests (Table 2), which were partly attributed to the lower average ambient seawater temperature at MMA (5.8 to 17 °C), compared to other marine testing locations (26 to 31 °C). Temperature has been proven to exert a significant impact on uranium uptake, and there is a positive correlation between seawater temperature and uranium adsorption.341
In addition, the results also demonstrated that the movement of the shells through the water could reduce biofouling if above a certain threshold speed, which provided new ideas for integrating design elements such as bristle brushes and UV lamps to mitigate biofouling in such a uranium harvester. Additionally, the cost analysis of a continuous version of SMORE was carried out, where the uranium production cost using a SMORE deployment strategy was estimated to range from $313 to 593 per kg-U.342 Compared to the results of the original kelp-field design by JAEA ($430–870 per kg-U), in the best-case scenario using a SMORE system (the cost was estimated to be $313 per kg-U) could achieve a uranium cost reduction by 27%.333 The lower recovery cost was partially attributed to the higher number of optimized uses for the SMORE system. Besides, the dramatic decrease (over 43% reduction compared to the kelp-like scheme) in mooring and deployment costs of the SMORE deployment strategy also contributed to reducing the overall uranium production cost. In a nutshell, the symbiotic design of coupling a seawater uranium harvester with an existing offshore wind turbine allows for denser energy recovery per unit ecosystem, as well as lowering the overall uranium production cost.333 Although the full-scale uranium harvesting system of the symbiotic design is only conceptually proposed and has not been verified in practice, it provides ideas for the future development of sustainable UES systems with less capital and operating costs.
5.3. Marine tests in China
According to declassified documents, China launched studies on UES as early as the 1960s. In early 1965, a research group was established by East China Normal University to officially carry out studies on UES. In 1967, scientists from several institutes and universities in Shanghai (including East China University of Science and Technology, Institute of Applied Physics, CAS, and Shanghai University) joined the group, and “Team 671” was then established,343 aiming to explore approaches for the large-scale filed test of UES. In 1970, significant progress was made in China where 30 g of yellowcake was successfully recovered from seawater by “Team 671”.344 It might be the first time that such a considerable amount of uranium was recovered from seawater. From then on, more scientific institutions for research in oceanic sciences or environmental protection in China have joined the project, and several test facilities were set up at different locations. Active programs on UES were conducted in the late 1970s to early 1980s, focusing on testing the performances of adsorbents (e.g., hydrous titanium oxide, basic zine carbonate, aluminium hydroxide and organic resins) and understanding their adsorption kinetics and mechanisms.309,345 However, in the late 1980s, due to the breakthroughs in land-based uranium mining technology, the urgent demand for uranium was alleviated, resulting in the suspension of research studies on UES in China.A renewed interest in UES has been sparked by the surge in uranium price starting in 2005 and peaking in mid-2007. Under such circumstances, the plan for developing UES was restarted in China in 2011. Since then, interest in UES has been continuously increased by supporting fundamental research on the design and creation of adsorbents, developing advanced characterization techniques, and establishing computational models. Significantly, engineering tests for UES have been initiated. Several offshore systems have been successfully constructed in China for marine field tests. Considerable advancements have been made, especially in recent years, by locating adaptable materials and building infrastructures for scale-up marine tests. In 2019, an innovation alliance for UES initiated by the China National Nuclear Corporation (CNNC) has been founded, bringing together more than 20 universities and institutes to promote the development of UES. In what follows, we provide the first comprehensive review of the progress made by the institutions in China in the implementation of marine tests.
In 2011, CAEP pioneered marine field tests in China to verify uranium extraction from real seawater. The AO-functionalized non-woven fabric materials of 10 kg were prepared by a pre-irradiation amplification method, packed into a special mesh stainless steel container, and deployed in the South China Sea using a floating mooring system for the first marine test. After 20 d, however, only a qU of about 0.1 mg g−1 was obtained.345 The second field experiment was then conducted in 2013, using AO-functionalized fibers with a skin–core structure with a high DOG of 300 wt% as the adsorbents. Loaded into a net and deployed in natural seawater over 30 d, the adsorbents exhibited a slight increase in the uranium adsorption capacity (0.3 mg g−1), which was still far from meeting the needs of practical applications.345 In 2019, an offshore platform with an area of 100 m2 was established in the South China Sea by CAEP and the third marine test was carried out at this platform (Fig. 41). Two guanidine-modified AO-fibers, namely, PAO-G-A and PAO-PAPB-A, that exhibited a satisfactory adsorption ability and high antimicrobial activity against marine bacteria in laboratory tests were selected and prepared in the kilogram-grade for the test.216,218 The adsorbent materials were fixed into Nylon mesh pockets, and deployed into the seawater at a depth of approximately 10 m. After 40 d, a qU of 1.14 mg g−1 was acquired by PAO-G-A, whereas an adsorption capacity of 3.19 mg g−1 was obtained by PAO-PAPB-A within 30 d, showing an increase of 178% compared to the PAO-G-A adsorbent. More importantly, no obvious degradation of the adsorbents and no marine microorganism attachment were observed, demonstrating their mechanical strength and antibiofouling properties.
 | ||
| Fig. 41 Images of offshore platforms developed in China for marine engineering of UES. | ||
In order to perform large-scale uranium extraction tests in real seawater, the Shanghai Advanced Research Institute (SARI) of CAS successfully developed a nanofiber-based membrane with high adsorption capacity, a fast uranium adsorption rate, and good selectivity based on electrospinning technology. In the past 10 years, batch preparation from laboratory membranes to industrial membrane modules has been completed. In 2018, a small-scale marine test for uranium extraction in the East China Sea was carried out with kilogram-scale membranes, affording a recovery of nearly 20 g of yellowcake after 30 d in seawater.343 In the next year, an adsorption platform and a matched elution platform for UES were established in the South China Sea by SARI, as shown in Fig. 41. The adsorption performances of the nanofiber-based membrane at the platform are currently being tested.
Since the low concentration of uranium in seawater is a huge obstacle for UES, the concept of uranium extraction from brine which has a significantly higher uranium concentration was born. A ready supply of more concentrated saline solution would potentially allow for greater uranium recovery using the currently available adsorbents.346–348 Accordingly, the Beijing Research Institute of Chemical Engineering and Metallurgy (BRICEM) has developed several uranium extraction polymers with high antimicrobial activity. In 2019, a field test was carried out in Dagze Co Salt Lake, Tibet, for uranium recovery. More than 300 g of yellowcake was recovered, with an average adsorption ability of 7.1 mg g−1. In addition, the performances of the polymers for uranium recovery from seawater were also evaluated. Recently, polymers were anchored and deployed in the South China Sea near Changjiang, Hainan (Fig. 41). Over a three-month marine test, a qU of 4.1 mg g−1 was obtained.343
The Shanghai Institute of Applied Physics (SINAP) of CAS also conducted marine experiments to test the performances of the AO-based fibers they developed in real seawater. In 2015, the performances of AO-UHMWPE fibers that were prepared by pre-irradiation-induced graft copolymerization of AN and AA on UHMWPE fibers, followed by amidoximation,69,71,72 were initially explored. UHMWPE was selected as a substrate polymer to overcome the poor mechanical properties demonstrated by conventional matrix polymers after irradiation. The test site was located at the East China Sea, which was about 100 m from the Chinese coast.71 The fibers were freely placed in a sea stream ∼3 m below the sea surface by a combination of floating buoys and an anchor. After 60 d, a moderate uptake of 0.25 mg g−1 was observed, whereas a higher adsorption amount of 0.48 mg g−1 was obtained when the fibers were sent to the MSL, PNNL in Sequim, WA, for the test. The lower adsorption in the East China Sea was ascribed to the coexistence of competitive ions and marine biofouling.
In a follow-up study, several AO-UHMWPE fibers were synthesized by varying preparation conditions (e.g., irradiation dose, AN:AA ratio, grafting temperature and amidoximation conditions), resulting in adsorbents with different AO densities.72 These fibers were subsequently tested at three different adsorption platforms developed in Xiamen, Daishan and Raoping, respectively, to examine their adsorption performances. When tested in the Xiamen and Daishan adsorption platforms, the adsorbents were strapped and directly immersed in seawater, about 3 m or 1 m below the water surface, respectively. On the other hand, during the tests at the Raoping adsorption platform, seawater was pumped into an underground impounding reservoir where adsorbents were immersed. In such a manner, the seawater in the reservoir was almost static with a very low flow velocity of less than 0.01 m s−1. As indicated in Table 3, the uranium adsorption capacities obtained from the marine adsorption test varied with the adsorbent, adsorption platform and adsorption time, affording 0.04–1.41 mg g−1 of the seven fiber samples (AO-UHMWPE-1-7). These results suggested that in addition to the characteristics of the adsorbents, marine hydrological conditions, such as temperature, ion concentrations, water quality and biofouling, are all important for uranium extraction in real marine experiments. Regarding uranium uptake performance in this test, the AO-UHMWPE-7 adsorbent acquired a maximum capacity of 1.41 mg g−1 after 15 d in the Daishan adsorption platform, which may be attributed to the high AO density of AO-UHMWPE-7 and the appropriate marine hydrological conditions of the Daishan adsorption platform.
| Time | Place | Adsorbents | Collected U amount | Time (d) | Ref. |
|---|---|---|---|---|---|
| CAEP, soaking in oceans directly | |||||
| 2011 | South China Sea | AO-based non-woven fabrics | 0.10 mg g−1 | 20 | 345 |
| 2013 | South China Sea | AO-based fibers | 0.10 mg g−1 | 20 | 345 |
| 2019 | South China Sea | Guanidine-modified AO-fibers (PAO-G-A) | 1.144 mg g−1 | 40 | 216 |
| 2019 | South China Sea | Guanidine-modified AO-fibers (PAO-PAPB-A) | 3.19 mg g−1 | 30 | 218 |
| SARI, soaking in oceans directly | |||||
| 2018 | East China Sea | Nanofiber-based membranes | 20 g | 30 | 343 |
| BRICEM, soaking in oceans directly | |||||
| 2019 | Dagze Co Salt Lake, Tibet | Polymers | 300 g | — | 343 |
| 2021 | Changjiang, Hainan | Polymers | 4.10 mg g−1 | 90 | 343 |
| SINAP, soaking in oceans directly | |||||
| 2015 | Xiamen | AO-UHMWPE | 0.25 mg g−1 | 60 | 71 |
| MSL, PNNL | AO-UHMWPE | 0.48 mg g−1 | 42 | ||
| 2015 | Xiamen | AO-UHMWPE-1 | 0.25 mg g−1 | 68 | 72 |
| Xiamen | AO-UHMWPE-2 | 0.04 mg g−1 | 68 | ||
| Xiamen | AO-UHMWPE-3 | 0.50 mg g−1 | 110 | ||
| Xiamen | AO-UHMWPE-4 | 0.77 mg g−1 | 110 | ||
| Xiamen | AO-UHMWPE-5 | 0.33 mg g−1 | 73 | ||
| Daishan | AO-UHMWPE-6 | 0.05 mg g−1 | 15 | ||
| Daishan | AO-UHMWPE-7 | 1.41 mg g−1 | 15 | ||
| Raoping | AO-UHMWPE-5 | 0.44 mg g−1 | 101 | ||
| Raoping | AO-UHMWPE-7 | 0.18 mg g−1 | 72 | ||
| SINAP, flow-through flume system | |||||
| 2019 | Raoping | Zn@AO-UHMWPE | 0.29 mg g−1 | 30 | 231 |
| 2019 | Raoping | H-ABP fibers | 11.50 mg g−1 | 90 | 132 |
| 2020 | Raoping | AO-OpNpNc fibers | 15.42 mg g−1 | 90 | 133 |
| Hainan University, flow-through column system with parallel multi-channels | |||||
| 2018 | Boundary Island of the South China Sea | Poly(imide dioxime) nanofibers (PIDO NFs) | 8.70 mg g−1 | 56 | 125 |
| 2019 | Boundary Island in the South China Sea | SMON–PAO | 9.59 mg g−1 | 56 | 124 |
| 2019 | Coastal water near the Boundary Island of the South China Sea | MS@PIDO/Alg sponge | 1.87 mg g−1 | 56 | 134 |
| 2019 | Not given | PAO hydrogel membrane | 4.87 mg g−1 | 28 | 137 |
| 2020 | Boundary Island in the South China Sea | PAO/Alg NFs | 8.42 mg g−1 | 56 | 126 |
| 2020 | Qiongzhou strait nearby the Haikou city of Hainan province | Zn2+-PAO hydrogel membrane | 9.23 mg g−1 | 28 | 138 |
| 2020 | The Chunyuan Sea nearby the Ganze Island in Wanning city of Hainan province | CP–PAO composite hydrogel | 6.21 mg g−1 | 42 | 140 |
| 2020 | South China Sea close to Hainan province | PAO PNMs | 9.35 mg g−1 | 35 | 143 |
| 2021 | South China Sea close to Hainan province | AUPM | 8.78 mg g−1 | 25 | 221 |
In addition, a flow-through flume test platform was also developed in the coastal area of Raoping, Guangdong Province, by SINAP and used for the marine test (Fig. 42). The natural seawater was pumped into an underground impounded reservoir. After filtering out the sediment with a polyester non-woven fabric, the seawater was pumped into a flume, where the adsorbent materials were freely dispersed for seawater exposure. The flow rate of the flume could be controlled with a motor controller. A representative test was performed using the flow-through flume test platform with the H-ABP fibers prepared through a two-step graft polymerization (RIGP-CIGP) in 2019.132 As shown in Fig. 42C, the H-ABP fibrous adsorbents were immersed in the flume at the controlled flow rate. After a duration of 90 d, the well-designed adsorbent achieved a high adsorption capacity of 11.5 mg g−1. Meanwhile, the H-ABP fibers also exhibited advantages in mechanical strength, selectivity, and especially service life (of at least ten adsorption–desorption cycles). In a follow-up study, the marine adsorption test of AO-OpNpNc fibers was performed with the same experimental setup.133 Encouragingly, an exceptional qU of 15.42 mg g−1 was obtained, while the contamination of marine microorganisms was noted on the adsorbents’ surfaces after 90 d. These results represented a significant breakthrough in the field of UES in China.
 | ||
| Fig. 42 Recovery of uranium from natural seawater. (A) Seawater reservoir. (B) Adsorption flume. (C) H-ABP fibrous adsorbent immersed in the flume at the beginning of adsorption. Reproduced with permission.132 Copyright 2019, Royal Society of Chemistry. | ||
Scientists from Hainan University have devoted their efforts towards the research on UES for developing novel adsorbent materials that can extract uranium from seawater with good performances. In recent years, multiple novel nanomaterial adsorbents have been synthesized and evaluated for uranium recovery in laboratory studies. Meanwhile, a flow-through column system with parallel multi-channels was designed and used for natural seawater exposure studies by Hainan University (Fig. 43).124–126 Similar to the design of the flow-through column system developed in PNNL, the natural seawater was collected and stored in a 5-T volume water tank, and was filtered through a microfiltration membrane (0.22 μm) system before being used for adsorption. The filtered natural seawater was stored in a 1-T tank and constantly fed into the columns at controlled speed, which were packed with the conditioned adsorbents. The filtered natural seawater was circulated in 12 parallel adsorption columns for each phase of test, and was regularly refreshed with new filtered natural seawater. The field column experiments of various kinds of adsorbents (e.g., fibers, membranes and hydrogels) developed by Hainan University were performed at the State Key Laboratory of Marine Resource Utilization in the South China Sea, Hainan, and the results are compiled in Table 3. The detailed design strategies and preparation methods of these adsorbents are covered in Sections 3 and 4. It was demonstrated that the adsorption performances could be improved by controlling the structural features of the adsorbent materials, as discussed in Section 3.1.3. It is worthy of note that most of the tested adsorbents such as PIDO NFs,125 SMON–PAO,124 PAO/Alg NFs,126 Zn2+-PAO hydrogel membranes,138 PAO PNMs143 and AUPM221 achieved higher adsorption capacities (8.42–9.59 mg g−1) than those of adsorbents developed by ORNL of the U.S. (3.30 to 6.56 mg g−1). Besides, the cost estimation was also performed by the authors with the developed PAO PNMs143 and AUPM221 adsorbent materials that had membrane structures, based on the assumption that the materials were directly immersed into the ocean for UES.
 | ||
| Fig. 43 (A) Design of a continuous flow-through test system with a parallel configuration. The flow rate for each channel can be controlled from 0 to 10 L min−1 individually using a motor controller. (B) The components of an adsorption study in natural seawater via a continuous flow-through test system. Reproduced with permission.125 Copyright 2018, Wiley-VCH. | ||
As described above, substantial efforts have been devoted by different countries for conducting marine experiments at selected locations. The information obtained from these studies is extremely valuable in evaluating the practical performances of adsorbents and verifying the technical feasibility of UES. The well-developed AO-functionalized polymeric adsorbents have demonstrated their potential in marine tests due to the high binding affinity to uranium and the chemical stability in ocean environments. Continuous progress has been made to improve the uranium adsorption capacity for AO-based polymeric adsorbents during marine tests. However, there is still a huge gap between the data obtained in marine tests and those in laboratories, suggesting the matrix complexity of the real seawater. Since uranium adsorption is highly dependent on the environmental conditions of the ocean, the fundamental understanding of the effect of environmental parameters (e.g., temperature and flow rate of seawater) on the extraction process is of vital importance for the scalability of UES. On the other hand, regarding the deployment of adsorbents, several systems such as floating platforms or stacks, flow-through or recirculating flume exposure systems, tethered braids and SMORE systems have been established. Meanwhile, various contact manners between adsorbents and seawater including mooring, soaking, pumping and towing have been evaluated with different adsorption bed configurations. From a practical point of view, further systematic studies on scaling up these technologies and evaluating their performances under realistic operation conditions are required.
In addition, the cost is another major obstacle facing the existing technology for UES. As mentioned above, several studies have conducted the cost–efficiency analysis based on the established systems or conceptual protocols for UES.144,323,324,327,332,342,349 Representative passive, current-driven systems and mechanically driven symbiotic systems were evaluated, highlighting three major components in the cost of uranium recovery from the oceans (i.e., adsorbent production, mooring and deployment, and elution and regeneration). It is generally concluded from these examinations that adsorbent production is the major cost in the process of UES. Therefore, minimizing the cost of adsorbent production is regarded as a key objective for UES. Efforts have been also devoted to improving the adsorption capacity and number of recycles of the adsorbents, which are identified as two pathways that could reduce the cost.327 Clearly, dramatically improving the adsorption capacity, which would perhaps allow a single use of the adsorbents, could significantly reduce the cost of UES.84,133 On the other hand, the adsorbents that can be effectively reused for multiple cycles without loss of adsorption capacity can also significantly reduce the overall cost of uranium production, but this simultaneously leads to increased mooring and collection cost, which is also a major cost driver for UES.
Currently, none of the established systems for UES can compete economically with terrestrial mining, and the estimated cost of uranium is still higher than the price of uranium from conventional sources. For improving a UES system to be economically viable, a priority in future work lies in continuously promoting the adsorption capacity of adsorbents. With the reduction in the cost of raw materials and the development of synthetic techniques, the production cost of adsorbents is expected to be gradually decreased. In the meantime, the mooring and deployment costs will become the greater contributor to the final cost. Linking the UES system to a local power source appears to be effective in reducing the cost by eliminating the offshore deployment and mooring cost.144,331 Equally important, efforts should also be put into reducing the degradation arising due to elution, which will lower the overall production cost.152 Since adsorbent regeneration is regarded as a necessity for economic viability, an effective, non-destructive elution/regeneration process to enable the maximum reuse of adsorbents is required. Overall, extensive R&D is still needed before economically viable UES can be achieved.
6. Current challenges and future perspectives
As hereinbefore described, the sustainable development of nuclear energy is crucial for the global low-carbon energy transition. Due to the dwindling availability of terrestrial uranium reserves, UES has been considered to be a potential approach for unconventional uranium harvesting to secure the expansion of nuclear energy. Starting from the “Project Oyster”, UES has attracted extensive academic and industrial attention over the past decades, evoking steady research and development efforts in multiple countries. In particular, the past five years have witnessed great progress in the field of UES. Salient improvements have been achieved in adsorption performances (i.e., adsorption capacity, selectivity, biofouling resistance, durability) by using various novel strategies for the function-oriented development of adsorbents. More mechanistic insights into uranyl–ligand interactions have been obtained, inspiring innovative design and preorganization of the ligands to promote the binding ability and selectivity to uranium. The improvement of polymerization methods for the preparation of polymeric materials endows adsorbents with a maximum number of binding sites. Meanwhile, based on a better understanding of the structure–property relationship of polymers from the molecular, mesoscopic and macroscopic levels, by tuning the physicochemical properties (e.g., ligand density, spatial conformation, surface area, pore size) of adsorbents, constantly increased adsorption capacities have been achieved. In addition, a deeper understanding of the effects of marine biofouling on uranium extraction has been obtained, and a variety of biofouling-proof adsorbents have been developed, which have demonstrated high inhibitory activity to marine bacteria. Especially, a noteworthy trend is the introduction of electrochemical and photochemical approaches to UES systems, which displayed faster kinetics and enhanced uranium extraction compared to conventional physicochemical adsorption. These appealing methods are believed to hold promise for developing new UES systems. Meanwhile, progress has also been realized in the engineering aspects of UES, evidenced by the implementation of scale-up marine tests, the building of offshore adsorption platforms, and the successful acquisition of yellowcake from the ocean. Overall, these multi-faceted efforts have propelled UES towards the industrialization goal of marine uranium recovery.Although great progress has been made in the field of UES, there are still significant challenges and obstacles that hinder the commercialization of UES. To develop technically and economically viable UES systems, additional breakthroughs are required. Herein, we will discuss the current challenges and describe future research directions for UES in terms of fundamental, technical and engineering aspects, respectively.
(1) Fundamental aspects of uranium coordination chemistry and adsorption science
Uranium harvesting from seawater is essentially chemical separation associated with the adsorption–desorption equilibrium of uranyl ions on the interfaces between adsorbents and seawater. In principle, achieving efficient chemical separation depends on the simultaneous tuning of the thermodynamic and kinetic factors. First, the uranyl–ligand interaction plays a pivotal role which determines the thermodynamic basis of the uranium adsorption–desorption equilibrium. Developing adsorbents with a sufficient binding ability and selectivity to uranium in seawater fundamentally relies on the engineering of uranyl–ligand interactions at the molecular level. With that in mind, understanding the basic principles for uranium coordination will accelerate the design and screening of functional ligands. Recently, while AO moieties have been demonstrated to be a category of most effective ligands for UES, the underlying coordination chemistry between AO ligands and uranyl in the seawater, as well as the nature of the complexes, has not been completely understood. More mechanistic insights into the uranyl–AO binding interactions in ocean environments are required, which could serve as guidance for the design of alternative or next-generation ligands with an enhanced binding ability.Learning from biological systems with a high affinity to uranium offers an intriguing way to screen favourable ligands. By identifying the specific sites of biomacromolecules, such as peptides, proteins and DNA, responsible for uranium enrichment, biological ligands with a superb binding ability to uranyl ions have been located. These state-of-the-art ligands efficiently coordinate with uranium by reaching a more thermodynamically stable state. Novel adsorbents functionalized with these biological ligands exhibit exceptional selectivity to uranyl ions in the seawater. However, there remain severe challenges for the practical use of these biocomponent-containing adsorbents in the complex ecological environment of the oceans. In the meantime, the research in this field inspired the idea to develop artificial uranyl nanotraps by tuning the preorganization and/or synergistic effect of the binding sites. These nascent concepts are encouraging, but more research concerning the geometry of uranyl coordination complexes is needed before the principles for regulating the spatial arrangements of the ligands while eliminating the steric hindrance could be established. Technically, more advanced spectroscopic analysis and the methods of computational chemistry would be supportive to this end.
Aside from the thermodynamic factors controlling the uranyl–ligand interactions, the kinetic aspects of the uranium adsorption equilibrium involving the mass transfer efficiency are of equal importance. A critical issue that must be highlighted is the effectiveness of the ligands or, in other words, their accessibility to the uranyl ions, which is significantly influenced by the chain conformation of polymeric adsorbents. Currently, established approaches to regulate the polymer chain conformation mainly rely on the introduction of hydrophilic copolymer chains through random or block copolymerization. However, most of these polymer architectures still cannot afford favourable conformations in high-salinity seawater due to the polyelectrolyte effect, resulting in an insufficient utilization of the ligands, and thus an inadequate adsorption capacity much lower than the theoretical value. More interactions to regulate the chain conformation of polymers, such as the electrostatic interaction, should be exploited to maximize the ligand availability by reducing the internal diffusion barriers of the uranyl ions. On the other hand, the relationship between the structure, chain conformation and adsorption behaviour of polymer adsorbents remains an area that is largely unexplored. In this regard, the vigorous development of computer technology, combined with constantly improved computational chemistry theories and simulation models, offers an avenue for the multi-scale investigation of the underlying scientific issues associated with uranium adsorption in aqueous solutions. Meanwhile, pivotal information about the structural and conformational properties of polymeric adsorbents could be experimentally obtained by using the neutron reflectometry (NR) method. These fundamental research studies would establish a more comprehensive understanding of the structure–property relationship, inspiring the design of high-performance polymeric adsorbents for UES.
(2) Technical aspects for developing effective adsorbents and active UES systems
Based on the fundamental insights into uranyl–ligand interactions, chemists have employed miscellaneous synthetic methods to introduce the identified ligands (e.g., AO) to specific substrates for developing uranium adsorbents. A majority of previous research tended to emphasize a great number or a high density of carefully-designed ligands introduced into adsorbents. However, as described above, most of the reported adsorbents were limited by the poor utilization of the functional ligands. Thus, a primary technical challenge lies in how to preserve the effectiveness and accessibility of the functional ligands by using more controllable synthetic methods for preparing uranium adsorbents. Taking the well-developed polymeric adsorbents bearing AO ligands as an example, since the AO ligands are tethered to the polymer backbones, the role of local chemistry and steric factors need to be examined. On this basis, rational design and synthesis of polymeric adsorbents is required to create a favourable coordination environment to promote the effective formation of uranyl–ligand complex species. Additionally, for the bio-inspired binding motifs involving the preorganization and/or synergistic effect of ligands, more advanced synthesis is needed to tune the distribution and arrangement of the ligands for achieving collaborative binding. On the other hand, the accessibility of the ligands can be improved by regulating the polymer chain conformation to eliminate the mass transport barriers. But how to precisely control the chain structure and topology of polymers remains a technical challenge when synthesizing specific polymeric adsorbents. More efforts are needed to develop controlled polymerization techniques to achieve the precise synthesis of polymers with conformational benefits to address the kinetic obstacles for uranium adsorption.The structural robustness of adsorbents is also a crucial point to consider. It has been demonstrated that increasing the number of recycles of adsorbents will significantly lower the overall cost of uranium production. However, the current fact is that the adsorption capacity of adsorbents degrades rapidly during cycle use. To enhance the durability of uranium adsorbents, the following issues should be considered. The first is the improvement of structural stability of adsorbents in the preparation process. Studies have shown that during the synthesis process of adsorbents, various steps including radiation graft polymerization, hydroxylamine treatment, and hot alkali treatment will cause certain damage to the mechanical strength of the adsorbents. In this way, hosts with high mechanical strength are preferred, and effective but mild synthesis methods are needed to reduce damage to the materials. Besides, the structural stability in seawater environments is also essential for any adsorbents since an unstable adsorbent suffers from structural degradation over prolonged seawater exposure. From this perspective, polymer adsorbents have more potential for practical application than most of the emerging porous framework adsorbents in realistic marine tests due to their excellent structural stability. The second issue is the durability of adsorbents in the elution process. The loss and deterioration of adsorbents in the elution process (especially using a strong acid leaching method) is a significant challenge that limits the reusability of the adsorbents. Improving the elution/regeneration process by using a non-acidic solution seems to be an effective method to avoid the deterioration of the adsorbent performance, and more eluents need to be examined in this respect. Besides, it should be noted that, for the various popular porous materials explored for uranium adsorption in recent years, an appropriate form for deployment in the seawater is the prerequisite for considering them as promising candidates for UES. So, there remains significant room to develop versatile synthetic and processing techniques for shaping these potential materials as deployable adsorbents.
Improving biofouling-resistant properties is another technical issue for developing adaptable adsorbents for UES. As described previously, the uranium adsorbents deployed in the ocean are inevitably subjected to biofouling. The attachment of marine microorganisms inhibits the contact between uranyl ions and the adsorbent surfaces, resulting in a significant reduction in uranium recovery. Additionally, biofouling also affects the reusability of the adsorbent due to the harsh treatments required to remove the foulants. In recent years, achievable strategies have been proposed to develop adsorbents with anti-biofouling properties, such as incorporating antibacterial components that can actively kill bacteria or introducing anti-adhesion coatings to prevent the attachment of marine organisms on the adsorbent surfaces. Several biofouling-proof adsorbents have been successfully developed, demonstrating effective biofouling resistance for specific bacteria. However, current antimicrobial evaluation experiments are basically carried out in laboratory tests. The complexity of biological species in real seawater and the fluidity of seawater bring more uncertainties and challenges to the practical antifouling performance of these adsorbents in the ocean. Future research on antimicrobial assessments should be carried out in the marine environment to verify the realistic performance of adsorbents.
(3) Engineering aspects for expanding marine field tests
The ultimate goal of UES is to realize scale-up marine engineering for stable and economically viable harvesting of uranium. Up to now, although several marine field tests have been implemented and multiple offshore platforms have been established, there are still many bottlenecks associated with marine engineering. Substantial efforts still are required to address the challenges. The first challenge comes from the complex ocean environment. Prior to marine engineering, an overall understanding of oceanographic conditions is of great importance, because there remain huge gaps between the data obtained in marine field tests and those obtained in simulated seawater. These gaps suggest that the performance of adsorbents developed in laboratories needs to be validated under real seawater conditions. Therefore, the solvation structures of dominant uranium species and the uranyl–ligand interactions need to be examined in natural seawater environments with specific pH values and high salinity. Meanwhile, the influence of the temperature and velocity of seawater should be evaluated to determine the optimal locations and conditions for marine field tests. Specifically, as discussed above, marine biofouling is a serious and inevitable issue related to the deployment of adsorbents in real seawater. The attachment of marine organisms to the adsorbent surfaces severely limits the uptake of uranium and the reusability of the adsorbents. In this sense, it is quite necessary to identify the dominant marine organisms affecting uranium harvesting. Antibiofouling adsorbents targeting specific biological species can thus be developed. Conversely, the deployment of UES adsorbents, especially for those with bactericidal activity, and facilities in seawater also affects local marine ecosystems. The research on these impacts would be necessary if scale-up marine field tests for UES are implemented.Another major challenge is to develop an effective and economically competitive system for the deployment of adsorbents. In this regard, both actively pumped systems and passive, current-driven systems have been evaluated. The latter outperform due to their simplicity and lower cost. For instance, the braid collection system developed by Japan, with the braided adsorbents anchored to the seafloor, has been acknowledged as a promising deployment mode, enabling efficient contact between uranyl ions and adsorbents with the aid of wave or current forces. From an economic perspective, the passive extraction processes that avoid the energy consumption required for seawater pumping have advantages. However, the extremely low concentration of uranium in seawater leads to slow adsorption kinetics in the passive extraction processes, influencing the uranium extraction efficiency. In the meantime, the prolonged exposure of the adsorbents in seawater results in more serious biofouling, which could compromise the capacity and reusability of the adsorbents. These aspects are not evaluated in the previously reported cost–efficiency analysis.
The emerging electrochemical and photochemical approaches for uranium extraction seem to provide solutions to this problem. Compared to the passive extraction processes, the application of an extra electrical field or light energy to the adsorption system improves the adsorption rate and capacity, thereby achieving a more efficient uranium extraction. As no pilot operations of these strategies have been performed, and any power requirements are based on lab-scale assessments, the scale-up deployment of these scenarios and the corresponding cost assessments require further efforts. On the other hand, it is generally believed that linking the UES to coastal nuclear power stations, desalination plants, or offshore energy technologies such as wind and tidal power could substantially promote the extraction process and reduce the cost. As claimed by the CNNC, a project to integrate the UES components with the warm water discharge system of a coastal nuclear power station has been launched. While the existing techniques for UES are mostly in the R&D stage, substantial efforts are still needed to demonstrate the feasibility and economic efficiency of the integrative systems.
Of particular note, standard protocols for validating the adsorption capacity or extraction efficiency of different UES systems, either in laboratory studies or in marine field tests, should be proposed. Many data sets reported in the current literature were obtained under experimental conditions of very little relevance to realistic marine environments in terms of uranium concentration, pH value, competing ions and microorganisms. A head-to-head comparison of potential candidates cannot be made when screening the most promising uranium adsorbents. Besides, general facilities or platforms, either coastal or offshore, to demonstrate the deployments of UES systems should be established, and open opportunities should be offered to researchers in universities or institutes to access these resources. Additionally, an area that has not been explored yet is the evaluation of the impact of coastal or offshore UES systems, in terms of both infrastructures and adsorbent components, on marine ecological systems. The results would be valuable for the governments to make policies for scale-up marine tests of UES, thereby preventing negative impacts on the marine ecological systems. Before the commercialization of UES, the corresponding regulations or even laws should be established to protect the marine environment and maintain the ecological balance.
Ultimately, the commercialization of UES depends on its economic viability. While several cost estimations have been made for some of the currently reported adsorbents and extraction systems, most of them are based on assumptions regarding adsorption capacity, recycle numbers and equipment requirements. However, an accurate economic assessment system should rest on the practical marine field tests of UES, with real and repeatable measured data as a reference, so as to comprehensively assess the contribution of each factor (i.e., adsorbent production, mooring and deployment, elution and regeneration, etc.) in the final uranium production cost. To this end, a reliable economic assessment system for the marine engineering of UES should be established.
In sum, future research on UES should focus on addressing the above challenges involving fundamental, technical and engineering issues. It is highly believed by the authors that a more comprehensive understanding of the coordination chemistry of uranium is of fundamental significance. This would inspire the exploration of new effective binding motifs by regulating the uranyl–ligand interactions at the molecular level. Meanwhile, the thermodynamic and kinetic factors influencing the uranium adsorption equilibrium should be simultaneously tuned to achieve an efficient uranium extraction. Particular attention should be given to the kinetic aspects involving the accessibility of the binding sites and the mass transfer efficiency of uranyl ions. Technically, advanced synthesis and polymerization methods are required for the controlled preparation of uranium adsorbents, allowing the tuning of the arrangement of binding sites while promoting their accessibility. Innovative ideas are encouraged to explore bio-inspired U-specific ligands for improving the selectivity of adsorbents, as well as emerging electrochemical and photochemical approaches for increasing the uranium extraction efficiency. Besides, from an engineering perspective, additional efforts should be made to develop infrastructures for marine field tests while optimizing individual or integrated UES systems. On this basis, a comprehensive cost–efficiency analysis of UES could be made, and eventually, a realistic roadmap for the commercialization of UES could be established.
Conclusively, UES is one of the remarkable activities for human beings to exploit strategic resources from the ocean. We provided here a comprehensive review of the state-of-the-art in the UES field and the efforts that are needed to make it technically and economically viable. Such an interdisciplinary field requires close cooperation between scientists and industrial engineers in different disciplines. Despite the critical challenges, encouraging achievements have been made in the past five years. New opportunities have been opened up for the development of more competitive UES technologies for commercial applications in the future. More significantly, elaborating the fundamental, technical and engineering issues of UES would provide concrete guidance for more general research on metal resource extraction from dilute media, such as lithium extraction from oceans or salt lakes.
Author contributions
Y. Xie, Z. Y. Liu and G. Ye conceived and prepared the outline of this review. X. L. Wang, S. Q. Ma and G. Ye supervised this review. Y. Xie drafted the Abstract, Introduction, Sections 3, 5 and 6 with the advice from J. C. Wang and J. Chen. Y. Xie and Z. Y. Liu co-wrote Sections 2 and 4. N. Wang revised Section 3. X. L. Wang, Y. P. Song and S. Q. Ma revised Section 6. G. Ye carefully revised the whole manuscript. Y. Y. Geng and H. Li prepared the graphic design and compiled the tables. All the authors contributed to the discussion of the content and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the financial support provided by the National Natural Science Fund for Excellent Young Scholars (No. 21922604, G. Ye), the National Natural Science Foundation of China (No. 22206104, Y. Y. Geng), the Robert A. Welch Foundation (B-0027 to S. Q. Ma), the China National Postdoctoral Program for Innovative Talents (No. BX2021142, H. Li) and the Shuimu Tsinghua Scholar Program (No. 2020SM139, H. Li).References
- J. Chilvers, R. Bellamy, H. Pallett and T. Hargreaves, Nat. Energy, 2021, 6, 250–259 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981–987 CrossRef CAS PubMed.
- N. Kaltsoyannis and S. T. Liddle, Chem, 2016, 1, 659–662 CAS.
- R. Taylor, Chem, 2016, 1, 662–663 CAS.
- L. Grancea, M. Mihalasky and M. Fairclough, Uranium 2020: Resources, Production and Demand, Nuclear Energy Agency and International Atomic Energy Agency, 2020 Search PubMed.
- K. Dungan, G. Butler, F. R. Livens and L. M. Warren, Prog. Nucl. Energy, 2017, 99, 81–85 CrossRef.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef.
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Sep. Sci. Technol., 2013, 48, 367–387 CrossRef CAS.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed.
- M. M. Aly and M. F. Hamza, J. Disper. Sci. Technol., 2013, 34, 182–213 CrossRef CAS.
- S. Kushwaha and K. Patel, Chem, 2021, 7, 271–274 CAS.
- B. C. Melot and J. M. Tarascon, Acc. Chem. Res., 2013, 46, 1226–1238 CrossRef CAS PubMed.
- S. Dai, Chem, 2021, 7, 537–539 CAS.
- M. Flicker Byers and E. Schneider, Ind. Eng. Chem. Res., 2016, 55, 4351–4361 CrossRef CAS.
- R. V. Davies, J. Kennedy, R. W. Mcilroy, R. Spence and K. M. Hill, Nature, 1964, 203, 1110–1115 CrossRef.
- M. Kanno, J. Nucl. Sci. Technol., 1984, 21, 1–9 CrossRef CAS.
- C. K. Nitta, F. R. Best and M. J. Driscoll, Delayed neutron assay to test sorbers for uranium-from-seawater applications, Report MIT-EL 82-008, United States, 1982.
- Y. Hisao, O. Yoshihiro, F. Nakajima and M. Toshifumi, Bull. Chem. Soc. Jpn., 1980, 53, 1–5 CrossRef.
- Y. Hisao, O. Yoshihiro, N. Fumito and M. Toshifumi, Bull. Chem. Soc. Jpn., 1980, 53, 1331–1334 CrossRef.
- Y. Hisao, O. Yoshihiro, N. Fumito and M. Toshifumi, Bull. Chem. Soc. Jpn., 1980, 53, 3050–3053 CrossRef.
- I.-H. Park and J.-M. Suh, Angew. Makromol. Chem., 1996, 239, 121–132 CrossRef CAS.
- K. Sugasaka, S. Katoh, N. Takai, H. Takahashi and Y. Umezawa, Sep. Sci. Technol., 1981, 16, 971–985 CrossRef CAS.
- Y. Kobuke, I. Tabushi, T. Aoki, T. Kamaishi and I. Hagiwara, Ind. Eng. Chem. Res., 1988, 27, 1461–1466 CrossRef CAS.
- N. Tang, J. Liang, C. G. Niu, H. Wang, Y. Luo, W. L. Xing, S. J. Ye, C. Liang, H. Guo, J. Y. Guo, Y. F. Zhang and G. M. Zeng, J. Mater. Chem. A, 2020, 8, 7588–7625 RSC.
- M. Tamada, International Seminar on Nuclear War and Planetary Emergencies – 42nd Session, World Scientific, 2010, pp. 243–252 Search PubMed.
- H. Egawa and H. Harada, Nippon Kagaku Kaishi, 1979, 7, 958–959 CrossRef.
- H. Egawa, H. Harada and T. Shuto, Nippon Kagaku Kaishi, 1980, 1980, 1773–1776 CrossRef.
- H. Egawa, H. Harada and T. Nonaka, Nippon Kagaku Kaishi, 1980, 11, 1767–1772 CrossRef.
- J. B. Xiong, Y. L. Fan and F. Luo, Dalton Trans., 2020, 49, 12536–12545 RSC.
- H. Guo, P. Mei, J. T. Xiao, X. S. Huang, A. Ishag and Y. B. Sun, Chemosphere, 2021, 278, 130411 CrossRef CAS.
- M. Carboni, C. W. Abney, S. B. Liu and W. B. Lin, Chem. Sci., 2013, 4, 2396–2402 RSC.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2013, 1, 3016–3026 RSC.
- L. L. Wang, F. Luo, L. L. Dang, J. Q. Li, X. L. Wu, S. J. Liu and M. B. Luo, J. Mater. Chem. A, 2015, 3, 17880 RSC.
- W. Chouyyok, J. W. Pittman, M. G. Warner, K. M. Nell, D. C. Clubb, G. A. Gill and R. S. Addleman, Dalton Trans., 2016, 45, 11312–11325 RSC.
- C. T. Yavuz, Chem, 2021, 7, 276–277 CAS.
- G. S. Zhu, Chem, 2021, 7, 277–278 CAS.
- F. Sun and C. He, Chem, 2021, 7, 274–275 CAS.
- F. C. Wu, N. Pu, G. Ye, T. X. Sun, Z. Wang, Y. Song, W. Q. Wang, X. M. Huo, Y. X. Lu and J. Chen, Environ. Sci. Technol., 2017, 51, 4606–4614 CrossRef CAS PubMed.
- P. Singhal, B. G. Vats and V. Pulhani, J. Ind. Eng. Chem., 2020, 90, 17–35 CrossRef CAS.
- B. F. Parker, Z. Zhang, L. Rao and J. Arnold, Dalton Trans., 2018, 47, 639–644 RSC.
- F. Endrizzi, C. J. Leggett and L. F. Rao, Ind. Eng. Chem. Res., 2016, 55, 4249–4256 CrossRef CAS.
- F. Endrizzi and L. F. Rao, Chem. – Eur. J., 2014, 20, 14499–14506 CrossRef CAS.
- C. J. Leggett, F. Endrizzi and L. F. Rao, Ind. Eng. Chem. Res., 2016, 55, 4257–4263 CrossRef CAS.
- Z. Szabo, T. Toraishi, V. Vallet and I. Grenthe, Coordin. Chem. Rev., 2006, 250, 784–815 CrossRef CAS.
- C. Z. Wang, J. H. Lan, Q. Y. Wu, Q. Luo, Y. L. Zhao, X. K. Wang, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2014, 53, 9466–9476 CrossRef CAS PubMed.
- M. A. Lashley, N. Mehio, J. W. Nugent, E. Holguin, C.-L. Do-Thanh, V. S. Bryantsev, S. Dai and R. D. Hancock, Polyhedron, 2016, 109, 81–91 CrossRef CAS.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS.
- J. P. Dognon, Coordin. Chem. Rev., 2014, 266, 110–122 CrossRef.
- S. Fortier and T. W. Hayton, Coordin. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- Z. Qin, Y. M. Ren, S. W. Shi, C. T. Yang, J. Yu, S. F. Wang, J. P. Jia, H. Z. Yu and X. L. Wang, RSC Adv., 2017, 7, 18639–18642 RSC.
- C. W. Abney, S. B. Liu and W. B. Lin, J. Phys. Chem. A, 2013, 117, 11558–11565 CrossRef CAS.
- S. C. Liu, M. B. Wu, H. Ye, L. Liu, L. L. Ma and J. M. Yao, Chem. Eng. J., 2021, 426, 131378 CrossRef CAS.
- S. Das, S. Brown, R. T. Mayes, C. J. Janke, C. Tsouris, L. J. Kuo, G. Gill and S. Dai, Chem. Eng. J., 2016, 298, 125–135 CrossRef CAS.
- H. Omichi, A. Katakai, T. Sugo and J. Okamoto, Sep. Sci. Technol., 1985, 20, 163–178 CrossRef CAS.
- H. Omichi, A. Katakai, T. Sugo and J. Okamoto, Sep. Sci. Technol., 1986, 21, 299–313 CrossRef CAS.
- X. Y. Liu, H. Z. Liu, H. J. Ma, C. Q. Cao, M. Yu, Z. Q. Wang, B. Deng, M. Wang and J. Y. Li, Ind. Eng. Chem. Res., 2012, 51, 15089–15095 CrossRef CAS.
- T. L. Prasad, P. K. Tewari and D. Sathiyamoorthy, Ind. Eng. Chem. Res., 2010, 49, 6559–6565 CrossRef CAS.
- S. Das, A. K. Pandey, A. Athawale, V. Kumar, Y. K. Bhardwaj, S. Sabharwal and V. K. Manchanda, Desalination, 2008, 232, 243–253 CrossRef CAS.
- T. L. Prasad, A. K. Saxena, P. K. Tewari and D. Sathiyamoorthy, Nucl. Eng. Technol., 2009, 41, 1101–1108 CrossRef CAS.
- S. H. Choi and Y. C. Nho, Radiat. Phys. Chem., 2000, 57, 187–193 CrossRef CAS.
- S. Rattan, J. Maitra, B. N. Misra and I. Kaur, J. Appl. Polym. Sci., 2008, 108, 3104–3113 CrossRef CAS.
- R. Li, H. J. Ma, Z. Xing and G. Z. Wu, J. Radioanal. Nucl. Chem., 2018, 315, 111–117 CrossRef CAS.
- S. H. Choi and Y. C. Nho, Radiat. Phys. Chem., 2000, 58, 157–168 CrossRef CAS.
- T. Kawai, K. Saito, K. Sugita, A. Katakai, N. Seko, T. Sugo, J. Kanno and T. Kawakami, Ind. Eng. Chem. Res., 2000, 39, 2910–2915 CrossRef CAS.
- T. Kawai, K. Saito, K. Sugita, T. Kawakami, J.-i Kanno, A. Katakai, N. Seko and T. Sugo, Radiat. Phys. Chem., 2000, 59, 405–411 CrossRef CAS.
- R. Li, X. X. Feng, M. X. Zhang, Z. Xing and G. Z. Wu, ACS Omega, 2021, 6, 1894–1900 CrossRef CAS PubMed.
- N. Seko, A. Katakai, M. Tamada, T. Sugo and F. Yoshii, Sep. Sci. Technol., 2004, 39, 3753–3767 CrossRef CAS.
- R. Li, L. J. Pang, H. J. Ma, X. Y. Liu, M. X. Zhang, Q. H. Gao, H. L. Wang, Z. Xing, M. H. Wang and G. Z. Wu, J. Radioanal. Nucl. Chem., 2017, 311, 1771–1779 CrossRef CAS.
- Z. Xing, J. T. Hu, M. H. Wang, W. L. Zhang, S. N. Li, Q. H. Gao and G. Z. Wu, Sci. China Chem., 2013, 56, 1504–1509 CrossRef CAS.
- J. T. Hu, H. J. Ma, Z. Xing, X. Y. Liu, L. Xu, R. Li, C. J. Lin, M. H. Wang, J. Y. Li and G. Z. Wu, Ind. Eng. Chem. Res., 2016, 55, 4118–4124 CrossRef CAS.
- C. J. Ling, X. Y. Liu, X. J. Yang, J. T. Hu, R. Li, L. J. Pang, H. J. Ma, J. Y. Li, G. Z. Wu, S. M. Lu and D. L. Wang, Ind. Eng. Chem. Res., 2017, 56, 1103–1111 CrossRef CAS.
- Q. H. Gao, J. T. Hu, R. Li, Z. Xing, L. Xu, M. H. Wang, X. J. Guo and G. Z. Wu, Radiat. Phys. Chem., 2016, 122, 1–8 CrossRef.
- Y. N. Zhao, M. H. Wang, Z. F. Tang and G. Z. Wu, Radiat. Phys. Chem., 2010, 79, 429–433 CrossRef CAS.
- T. Saito, S. Brown, S. Chatterjee, J. Kim, C. Tsouris, R. T. Mayes, L. J. Kuo, G. Gill, Y. Oyola, C. J. Janke and S. Dai, J. Mater. Chem. A, 2014, 2, 14674–14681 RSC.
- S. Brown, S. Chatterjee, M. J. Li, Y. F. Yue, C. Tsouris, C. J. Janke, T. Saito and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4130–4138 CrossRef CAS.
- J.-S. Wang and M. Krzysztof, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- V. S. Neti, S. Das, S. Brown, C. J. Janke, L.-J. Kuo, G. A. Gill, S. Dai and R. T. Mayes, Ind. Eng. Chem. Res., 2017, 56, 10826–10832 CrossRef CAS.
- Y. F. Yue, R. T. Mayes, J. Kim, P. F. Fulvio, X. G. Sun, C. Tsouris, J. H. Chen, S. Brown and S. Dai, Angew. Chem., Int. Ed., 2013, 52, 13458–13462 CrossRef CAS.
- S. Brown, Y. F. Yue, L. J. Kuo, N. Mehio, M. J. Li, G. Gill, C. Tsouris, R. T. Mayes, T. Saito and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4139–4148 CrossRef CAS.
- F. Chi, J. Wen, J. Xiong, H. Sheng, Z. Gong, T. Qiu, G. Wei, F. Yi and X. Wang, J. Radioanal. Nucl. Chem., 2016, 309, 787–796 CAS.
- L. Chen, Z. L. Bai, L. Zhu, L. J. Zhang, Y. W. Cai, Y. X. Li, W. Liu, Y. L. Wang, L. H. Chen, D. W. Juan, J. Q. Wang, Z. F. Chai and S. A. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32446–32451 CrossRef CAS.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Q. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef.
- S. L. Zhao, Y. H. Yuan, Q. H. Yu, B. Y. Niu, J. H. Liao, Z. H. Guo and N. Wang, Angew. Chem., Int. Ed., 2019, 58, 14979–14985 CrossRef CAS.
- W. R. Cui, F. F. Li, R. H. Xu, C. R. Zhang, X. R. Chen, R. H. Yan, R. P. Liang and J. D. Qiu, Angew. Chem., Int. Ed., 2020, 59, 17684–17690 CrossRef CAS.
- G. Cheng, A. R. Zhang, Z. W. Zhao, Z. M. Chai, B. W. Hu, B. Han, Y. J. Ai and X. K. Wang, Sci. Bull., 2021, 66, 1994–2001 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, A. S. Ivanov, V. S. Bryantsev, L. D. Earl, C. W. Abney, L. Wojtas and S. Q. Ma, Nat. Commun., 2018, 9, 1644 CrossRef.
- Z. N. Li, Q. H. Meng, Y. J. Yang, X. Q. Zou, Y. Yuan and G. S. Zhu, Chem. Sci., 2020, 11, 4747–4752 RSC.
- J. W. Wang, Y. Sun, X. M. Zhao, L. Chen, S. Y. Peng, C. X. Ma, G. G. Duan, Z. Z. Liu, H. Wang, Y. H. Yuan and N. Wang, e-Polymers, 2022, 22, 399–410 CrossRef CAS.
- L. Ma, J. Gao, C. Huang, X. Xu, L. Xu, R. H. Ding, H. L. Bao, Z. Q. Wang, G. Xu, Q. N. Li, P. Y. Deng and H. J. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 57831–57840 CrossRef CAS PubMed.
- B. X. Yu, L. Zhang, G. Ye, Q. Z. Liu, J. L. Li, X. D. Wang, J. Chen, S. M. Xu and S. Q. Ma, Nano Res., 2021, 14, 788–796 CrossRef CAS.
- L. Zhang, N. Pu, B. X. Yu, G. Ye, J. Chen, S. M. Xu and S. Q. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 3688–3696 CrossRef CAS.
- Z. Y. Wang, Q. H. Meng, R. C. Ma, Z. K. Wang, Y. J. Yang, H. Y. Sha, X. J. Ma, X. H. Ruan, X. Q. Zou, Y. Yuan and G. S. Zhu, Chem, 2020, 6, 1683–1691 CAS.
- S. Zhang, H. Li and S. A. Wang, Chem, 2020, 6, 1504–1505 CAS.
- Y. F. Yue, C. X. Zhang, Q. Tang, R. T. Mayes, W. P. Liao, C. Liao, C. Tsouris, J. J. Stankovich, J. H. Chen, D. K. Hensley, C. W. Abney, D. E. Jiang, S. Brown and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4125–4129 CrossRef CAS.
- S. D. Alexandratos, X. P. Zhu, M. Florent and R. Sellin, Ind. Eng. Chem. Res., 2016, 55, 4208–4216 CrossRef CAS.
- S. H. Choi, M. S. Choi, Y. T. Park, K. P. Lee and H. D. Kang, Radiat. Phys. Chem., 2003, 67, 387–390 CrossRef CAS.
- D. D. Shao, X. L. Wang, X. M. Ren, S. Hu, J. Wen, Z. Y. Tan, J. Xiong, A. M. Asiri and H. M. Marwani, J. Ind. Eng. Chem., 2018, 67, 380–387 CrossRef CAS.
- Z. Ahmad, Y. Li, J. J. Yang, N. B. Geng, Y. Fan, X. Y. Gou, Q. Y. Sun and J. P. Chen, J. Hazard. Mater., 2022, 425, 127995 CrossRef CAS.
- H.-B. Pan, C. M. Wai, L.-J. Kuo, G. A. Gill, J. S. Wang, R. Joshi and C. J. Janke, Dalton Trans., 2020, 49, 2803–2810 RSC.
- P. A. Kavaklı, N. Seko, M. Tamada and O. Güven, Sep. Sci. Technol., 2005, 39, 1631–1643 CrossRef.
- N. Seko, L. T. Bang and M. Tamada, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 265, 146–149 CrossRef CAS.
- C. Huang, L. Xu, X. Xu, L. Ma, H. L. Bao, J. Liao, J. J. Wang, J. G. Han, G. Xu, D. M. Huang, B. J. Ye, H. J. Zhang, M. H. Wu, X. Y. Zhao and H. J. Ma, Chem. Eng. J., 2022, 443, 136312 CrossRef CAS.
- X. M. Wang, S. Q. Wu, J. W. Guan, L. H. Chen, C. Shi, J. M. Wan, Y. Liu, J. Diwu, J. Q. Wang and S. A. Wang, Inorg. Chem., 2019, 58, 3349–3354 CrossRef CAS PubMed.
- X. M. Wang, X. Dai, C. Shi, J. M. Wan, M. A. Silver, L. J. Zhang, L. H. Chen, X. Yi, B. Z. Chen, D. Zhang, K. Yang, J. Diwu, J. Q. Wang, Y. J. Xu, R. H. Zhou, Z. F. Chai and S. A. Wang, Nat. Commun., 2019, 10, 2570 CrossRef PubMed.
- J. W. Guan, X. M. Wang, P. H. Shi, L. H. Chen, B. Chen, Y. G. Zhang, Y. M. Chen, Y. G. Xu, Z. F. Chai, S. A. Wang and J. Diwu, Inorg. Chem., 2022, 61, 3886–3892 CrossRef CAS.
- B. Chen, S. Hong, X. Dai, X. M. Li, Q. Huang, T. F. Sun, D. H. Cao, H. L. Zhang, Z. F. Chai, J. Diwu and S. A. Wang, J. Am. Chem. Soc., 2022, 144, 11054–11058 CrossRef CAS PubMed.
- Z. F. Sun, F. C. Lv, L. J. Cao, L. Liu, Y. Zhang and Z. G. Lu, Angew. Chem., Int. Ed., 2015, 54, 7944–7948 CrossRef CAS.
- Y. Oyola and S. Dai, Dalton Trans., 2016, 45, 8824–8834 RSC.
- S. Das, Y. Oyola, R. T. Mayes, C. J. Janke, L. J. Kuo, G. Gill, J. R. Wood and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4110–4117 CrossRef CAS.
- S. Das, Y. Oyola, R. T. Mayes, C. J. Janke, L. J. Kuo, G. Gill, J. R. Wood and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4103–4109 CrossRef CAS.
- A. I. Wiechert, W. P. Liao, E. Hong, C. E. Halbert, S. Yiacoumi, T. Saito and C. Tsouris, J. Colloid. Interf. Sci, 2018, 524, 399–408 CrossRef CAS.
- L. Xie, Y. L. Wang, Y. Wang, X. X. Li, Q. Tian, D. Liu, G. G. Sun and X. L. Wang, Mater. Lett., 2018, 220, 47–49 CrossRef CAS.
- P. H. Ju, Q. Liu, H. S. Zhang, R. R. Chen, J. Y. Liu, J. Yu, P. L. Liu, M. L. Zhang and J. Wang, Chem. Eng. J., 2019, 374, 1204–1213 CrossRef CAS.
- P. H. Ju, H. Guo, J. W. Bai, Q. Liu, H. S. Zhang, J. Y. Liu, J. Yu, R. R. Chen and J. Wang, J. Colloid. Interf. Sci, 2020, 576, 109–118 CrossRef CAS PubMed.
- Z. Y. Liu, Y. S. Lan, J. F. Jia, Y. Y. Geng, X. B. Dai, L. T. Yan, T. Y. Hu, J. Chen, K. Matyjaszewski and G. Ye, Nat. Commun., 2022, 13, 3918 CrossRef CAS PubMed.
- J. J. Jiang, G. Ye, Z. Wang, Y. X. Lu, J. Chen and K. Matyjaszewski, Angew. Chem., Int. Ed., 2018, 57, 12037–12042 CrossRef CAS.
- J. J. Jiang, G. Ye, F. Lorandi, Z. Y. Liu, Y. Q. Liu, T. Y. Hu, J. Chen, Y. X. Lu and K. Matyjaszewski, Angew. Chem., Int. Ed., 2019, 58, 12096–12101 CrossRef CAS.
- H. Omichi, A. Katakai, T. Sugo, J. Okamoto, S. Katoh, K. Sakane, K. Sugasaka and T. Itagaki, Sep. Sci. Technol., 1987, 22, 1313–1325 CrossRef CAS.
- Y. Oyola, C. J. Janke and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4149–4160 CrossRef CAS.
- S. Y. Xie, X. Y. Liu, B. W. Zhang, H. J. Ma, C. J. Ling, M. Yu, L. F. Li and J. Y. Li, J. Mater. Chem. A, 2015, 3, 2552–2558 RSC.
- B. W. Zhang, X. J. Guo, S. Y. Xie, X. Y. Liu, C. J. Ling, H. J. Ma, M. Yu and J. Y. Li, RSC Adv., 2016, 6, 81995–82005 RSC.
- F. Ashrafi, M. Firouzzare, S. J. Ahmadi, M. R. Sohrabi and M. Khosravi, Ecotoxicol. Environ. Saf., 2019, 186, 109746 CrossRef CAS.
- Y. H. Yuan, S. L. Zhao, J. Wen, D. Wang, X. W. Gu, L. L. Xu, X. L. Wang and N. Wang, Adv. Funct. Mater., 2019, 29, 1805380 CrossRef.
- D. Wang, J. A. Song, J. Wen, Y. H. Yuan, Z. L. Liu, S. Lin, H. Y. Wang, H. L. Wang, S. L. Zhao, X. M. Zhao, M. H. Fang, M. Lei, B. Li, N. Wang, X. L. Wang and H. Wu, Adv. Energy Mater., 2018, 8, 1802607 CrossRef.
- X. Xu, Y. R. Yue, D. Cai, J. N. Song, C. N. Han, Z. J. Liu, D. Wang, J. X. Xiao and H. Wu, Small Methods, 2020, 4, 2000558 CrossRef CAS.
- Z. Li, Z. Q. Yu, Y. D. Wu, X. L. Wu, Y. Wan, Y. H. Yuan and N. Wang, Chem. Eng. J., 2020, 390, 124648 CrossRef CAS.
- D. Wang, Z. Liu, Y. Yue, X. Xu, D. Cai, C. Han, J. Song, J. Xiao and H. Wu, Mater. Today Energy, 2021, 21, 100735 CrossRef CAS.
- H. L. Wang, X. Zhang, N. Wang, Y. Li, X. Feng, Y. Huang, C. S. Zhao, Z. L. Liu, M. H. Fang, G. Ou, H. J. Gao, X. Y. Li and H. Wu, Sci. Adv., 2017, 3, e1603170 CrossRef.
- J. J. Xue, J. W. Xie, W. Y. Liu and Y. N. Xia, Acc. Chem. Res., 2017, 50, 1976–1987 CrossRef CAS.
- V. Thavasi, G. Singh and S. Ramakrishna, Energ. Environ. Sci., 2008, 1, 205–221 RSC.
- X. Xu, H. J. Zhang, J. X. Ao, L. Xu, X. Y. Liu, X. J. Guo, J. Y. Li, L. Zhang, Q. N. Li, X. Y. Zhao, B. J. Ye, D. L. Wang, F. Shen and H. J. Ma, Energ. Environ. Sci., 2019, 12, 1979–1988 RSC.
- X. Xu, L. Xu, J. X. Ao, Y. L. Liang, C. Li, Y. J. Wang, C. Huang, F. Ye, Q. N. Li, X. J. Guo, J. Y. Li, H. T. Wang, S. Q. Ma and H. J. Ma, J. Mater. Chem. A, 2020, 8, 22032–22044 RSC.
- D. Wang, J. N. Song, S. Lin, J. Wen, C. X. Ma, Y. H. Yuan, M. Lei, X. L. Wang, N. Wang and H. Wu, Adv. Funct. Mater., 2019, 29, 1901009 CrossRef.
- X. Wang, Q. Liu, J. Y. Liu, R. R. Chen, H. S. Zhang, R. M. Li, Z. S. Li and J. Wang, Appl. Surf. Sci., 2017, 426, 1063–1074 CrossRef CAS.
- F. H. Wang, H. P. Li, Q. Liu, Z. S. Li, R. M. Li, H. S. Zhang, L. H. Liu, G. A. Emelchenko and J. Wang, Sci. Rep., 2016, 6, 19367 CrossRef CAS.
- C. X. Ma, J. X. Gao, D. Wang, Y. H. Yuan, J. Wen, B. J. Yan, S. L. Zhao, X. M. Zhao, Y. Sun, X. L. Wang and N. Wang, Adv. Sci., 2019, 6, 1900085 CrossRef PubMed.
- B. J. Yan, C. X. Ma, J. X. Gao, Y. H. Yuan and N. Wang, Adv. Mater., 2020, 32, 1906615 CrossRef CAS.
- N. Wang, X. M. Zhao, J. W. Wang, B. J. Yan, S. X. Wen, J. C. Zhang, K. Lin, H. Wang, T. Liu, Z. Z. Liu, C. X. Ma, J. B. Li and Y. H. Yuan, Adv. Sci., 2021, 8, 2102250 CrossRef CAS.
- J. X. Gao, Y. H. Yuan, Q. H. Yu, B. J. Yan, Y. X. Qian, J. Wen, C. X. Ma, S. H. Jiang, X. L. Wang and N. Wang, Chem. Commun., 2020, 56, 3935–3938 RSC.
- Q. H. Yu, Y. H. Yuan, L. J. Feng, T. T. Feng, W. Y. Sun and N. Wang, Angew. Chem., Int. Ed., 2020, 59, 15997–16001 CrossRef CAS.
- Y. H. Yuan, T. T. Liu, J. X. Xiao, Q. H. Yu, L. J. Feng, B. Y. Niu, S. W. Feng, J. C. Zhang and N. Wang, Nat. Commun., 2020, 11, 5708 CrossRef CAS.
- S. Shi, Y. X. Qian, P. P. Mei, Y. H. Yuan, N. Jia, M. Y. Dong, J. C. Fan, Z. H. Guo and N. Wang, Nano Energy, 2020, 71, 104629 CrossRef CAS.
- W. Luo, G. Xiao, F. Tian, J. J. Richardson, Y. P. Wang, J. F. Zhou, J. L. Guo, X. P. Liao and B. Shi, Energy. Environ. Sci., 2019, 12, 607–614 RSC.
- L. S. Yang, H. Y. Xiao, Y. C. Qian, X. L. Zhao, X. Y. Kong, P. Liu, W. W. Xin, L. Fu, L. Jiang and L. P. Wen, Nat. Sustainability, 2022, 5, 71–80 CrossRef.
- Y. H. Sihn, J. Byun, H. A. Patel, W. Lee and C. T. Yavuz, RSC Adv., 2016, 6, 45968–45976 RSC.
- A. I. Wiechert, S. Yiacoumi and C. Tsouris, Nat. Sustainability, 2022, 5, 13–14 CrossRef.
- F. Q. Ma, Y. Y. Gui, P. Liu, Y. Xue and W. Song, Chem. Eng. J., 2020, 390, 124597 CrossRef CAS.
- X. Chen, C. X. Wan, R. Yu, L. P. Meng, D. L. Wang, T. Duan and L. B. Li, Desalination, 2020, 486, 114447 CrossRef CAS.
- H. Wang, B. H. Zheng, T. H. Xu, M. Cao, F. Gao, G. B. Zhou, C. Ma, J. Dang, W. K. Yao and K. C. Wu, Sep. Purif. Technol., 2022, 289, 120823 CrossRef CAS.
- Y. Wang, Y. P. Zhang, Q. Li, Y. X. Li, L. X. Cao and W. L. Li, Carbohyd. Polym., 2020, 245, 116627 CrossRef CAS.
- N. Li, L. Yang, D. Wang, C. Y. Tang, W. Q. Deng and Z. N. Wang, Environ. Sci. Technol., 2021, 55, 9181–9188 CrossRef CAS PubMed.
- W. H. Zhang, C. L. Xu, X. P. Che, T. Wang, S. Willför, M. J. Li and C. X. Li, ACS Nano, 2022, 16, 13144–13151 CrossRef CAS PubMed.
- A. P. Ladshaw, S. Das, W. P. Liao, S. Yiacoumi, C. J. Janke, R. T. Mayes, S. Dai and C. Tsouris, Ind. Eng. Chem. Res., 2016, 55, 4241–4248 CrossRef CAS.
- G. A. Gill, L.-J. Kuo, C. J. Janke, J. Park, R. T. Jeters, G. T. Bonheyo, H.-B. Pan, C. Wai, T. Khangaonkar, L. Bianucci, J. R. Wood, M. G. Warner, S. Peterson, D. G. Abrecht, R. T. Mayes, C. Tsouris, Y. Oyola, J. E. Strivens, N. J. Schlafer, R. S. Addleman, W. Chouyyok, S. Das, J. Kim, K. Buesseler, C. Breier and E. D’Alessandro, Ind. Eng. Chem. Res., 2016, 55, 4264–4277 CrossRef CAS.
- T. P. Rao, R. Kala and S. Daniel, Anal. Chim. Acta, 2006, 578, 105–116 CrossRef CAS.
- L. X. Chen, X. Y. Wang, W. H. Lu, X. Q. Wu and J. H. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- C. Branger, W. Meouche and A. Margaillan, React. Funct. Polym., 2013, 73, 859–875 CrossRef CAS.
- Z. Yi, L. Jun Wen, W. Si Jin and C. Hai Ming, RSC Adv., 2022, 12, 15470–15478 RSC.
- J. Q. Fu, L. X. Chen, J. H. Li and Z. Zhang, J. Mater. Chem. A, 2015, 3, 13598–13627 RSC.
- P. Metilda, J. M. Gladis, G. Venkateswaran and T. P. Rao, Anal. Chim. Acta, 2007, 587, 263–271 CrossRef CAS.
- H. J. Zhang, H. L. Liang, Q. D. Chen and X. H. Shen, J. Radioanal. Nucl. Chem., 2013, 298, 1705–1712 CrossRef CAS.
- D. K. Singh and S. Mishra, Anal. Chim. Acta, 2009, 644, 42–47 CrossRef CAS PubMed.
- M. Shamsipur, J. Fasihi and K. Ashtari, Anal. Chem., 2007, 79, 7116–7123 CrossRef CAS.
- J. X. Ao, H. J. Zhang, X. Xu, F. J. Yao, L. Ma, L. Zhang, B. J. Ye, Q. N. Li, L. Xu and H. J. Ma, RSC Adv., 2019, 9, 28588–28597 RSC.
- L. X. Zhang, S. Yang, J. Qian and D. B. Hua, Ind. Eng. Chem. Res., 2017, 56, 1860–1867 CrossRef CAS.
- Y. Yuan and G. S. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS.
- B. Y. Li, Q. Sun, Y. M. Zhang, C. W. Abney, B. Aguila, W. B. Lin and S. Q. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 12511–12517 CrossRef CAS PubMed.
- Y. Yuan, Y. J. Yang and G. S. Zhu, ACS Cent. Sci., 2020, 6, 1082–1094 CrossRef CAS PubMed.
- Y. Y. Tian and G. S. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS.
- Y. Yuan, Y. J. Yang, X. J. Ma, Q. H. Meng, L. L. Wang, S. Zhao and G. S. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef.
- Y. Yuan, Q. H. Meng, M. Faheem, Y. J. Yang, Z. N. Li, Z. Y. Wang, D. Deng, F. X. Sun, H. M. He, Y. H. Huang, H. Y. Sha and G. S. Zhu, ACS Cent. Sci., 2019, 5, 1432–1439 CrossRef CAS PubMed.
- D. T. Sun and W. L. Queen, ACS Cent. Sci., 2019, 5, 1307–1309 CrossRef CAS PubMed.
- L. J. Feng, H. Wang, T. T. Feng, B. J. Yan, Q. H. Yu, J. C. Zhang, Z. H. Guo, Y. H. Yuan, C. X. Ma, T. Liu and N. Wang, Angew. Chem., Int. Ed., 2022, 61, 82–86 Search PubMed.
- A. S. Ivanov, B. F. Parker, Z. C. Zhang, B. Aguila, Q. Sun, S. Q. Ma, S. Jansone-Popova, J. Arnold, R. T. Mayes, S. Dai, V. S. Bryantsev, L. F. Rao and I. Popovs, Nat. Commun., 2019, 10, 819 CrossRef CAS.
- S. V. Wegner, H. Boyaci, H. Chen, M. P. Jensen and C. He, Angew. Chem., Int. Ed., 2009, 48, 2339–2341 CrossRef CAS.
- E. A. Heide, K. Wagener, M. Paschke and M. Wald, Naturwissenschaften, 1973, 60, 431 CrossRef CAS.
- L. Zhou, M. Bosscher, C. S. Zhang, S. Ozcubukcu, L. Zhang, W. Zhang, C. J. Li, J. Z. Liu, M. P. Jensen, L. H. Lai and C. He, Nat. Chem., 2014, 6, 236–241 CrossRef CAS.
- Y. W. Lin, Biomolecules, 2020, 10, 457 CrossRef CAS.
- S. O. Odoh, G. D. Bondarevsky, J. Karpus, Q. Cui, C. He, R. Spezia and L. Gagliardi, J. Am. Chem. Soc., 2014, 136, 17484–17494 CrossRef CAS PubMed.
- S. Z. Kou, Z. G. Yang and F. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 2035–2039 CrossRef CAS PubMed.
- X. Y. Yang, J. Y. Wei, Y. Q. Wang, C. R. Yang, S. J. Zhao, C. Li, Y. M. Dong, K. Bai, Y. X. Li, H. Y. Teng, D. Y. Wang, N. Y. Lyu, J. M. Li, X. Y. Chang, X. Ning, Q. Ouyang, Y. H. Zhang and L. Qian, ACS Synth. Biol., 2018, 7, 2331–2339 CrossRef CAS.
- Y. H. Yuan, Q. H. Yu, J. Wen, C. Y. Li, Z. H. Guo, X. L. Wang and N. Wang, Angew. Chem., Int. Ed., 2019, 58, 11785–11790 CrossRef CAS PubMed.
- Q. Y. Hu, S. Q. Huang, T. B. Wei, J. Wang, Z. Z. Huo, T. Y. Zhu, C. Wu and H. Chen, ACS Appl. Polym. Mater., 2022, 4, 2189–2196 CrossRef CAS.
- F. Sun and W. B. Zhang, Chinese Chem. Lett., 2017, 28, 2078–2084 CrossRef CAS.
- B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy and M. Howarth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E690–E697 CrossRef CAS.
- S. Z. Kou, Z. G. Yang, J. R. Luo and F. Sun, Polym. Chem., 2017, 8, 6158–6164 RSC.
- X. J. Zhang, X. W. Wang, X. D. Da, Y. L. Shi, C. L. Liu, F. Sun, S. G. Yang and W. B. Zhang, Biomacromolecules, 2018, 19, 1065–1073 CrossRef CAS.
- X. F. Luan, C. Z. Wang, Q. Y. Wu, J. H. Lan, Z. F. Chai, L. S. Xia and W. Q. Shi, J. Phys. Chem. A, 2022, 126, 406–415 CrossRef CAS.
- A. S. Ivanov, C. J. Leggett, B. F. Parker, Z. C. Zhang, J. Arnold, S. Dai, C. W. Abney, V. S. Bryantsev and L. F. Rao, Nat. Commun., 2017, 8, 1560 CrossRef.
- S. P. Kelley, P. S. Barber, P. H. K. Mullins and R. D. Rogers, Chem. Commun., 2014, 50, 12504–12507 RSC.
- A. S. Ivanov and V. S. Bryantsev, Dalton Trans., 2016, 45, 10744–10751 RSC.
- Q. Sun, Y. P. Song, B. Aguila, A. S. Ivanov, V. S. Bryantsev and S. Q. Ma, Adv. Sci., 2021, 8, 2001573 CrossRef CAS.
- Y. P. Song, C. J. Zhu, Q. Sun, B. Aguila, C. W. Abney, L. Wojtas and S. Q. Ma, ACS Cent. Sci., 2021, 7, 1650–1656 CrossRef CAS.
- Y. H. Yuan, S. W. Feng, L. J. Feng, Q. H. Yu, T. T. Liu and N. Wang, Angew. Chem., Int. Ed., 2020, 59, 4262–4268 CrossRef CAS PubMed.
- Q. Sun, B. Aguila and S. Q. Ma, Trends Chem., 2019, 1, 292–303 CrossRef CAS.
- C. Z. Wang, Z. F. Chai and W. Q. Shi, ACS Cent. Sci., 2021, 7, 1602–1604 CrossRef CAS PubMed.
- M. Lejars, A. Margaillan and C. Bressy, Chem. Rev., 2012, 112, 4347–4390 CrossRef CAS PubMed.
- Q. Y. Xie, J. S. Pan, C. F. Ma and G. Z. Zhang, Soft Matter, 2019, 15, 1087–1107 RSC.
- L. D. Chambers, K. R. Stokes, F. C. Walsh and R. J. K. Wood, Surf. Coat. Tech., 2006, 201, 3642–3652 CrossRef CAS.
- M. S. Selim, M. A. Shenashen, S. A. El-Safty, S. A. Higazy, M. M. Selim, H. Isago and A. Elmarakbi, Prog. Mater. Sci., 2017, 87, 1–32 CrossRef CAS.
- J. Park, G. A. Gill, J. E. Strivens, L. J. Kuo, R. T. Jeters, A. Avila, J. R. Wood, N. J. Schlafer, C. J. Janke, E. A. Miller, M. Thomas, R. S. Addleman and G. T. Bonheyo, Ind. Eng. Chem. Res., 2016, 55, 4328–4338 CrossRef CAS.
- Z. R. Xin, S. S. Du, C. Y. Zhao, H. Chen, M. Sun, S. J. Yan, S. F. Luan and J. H. Yin, Appl. Surf. Sci., 2016, 365, 99–107 CrossRef CAS.
- S. Das, A. K. Pandey, A. A. Athawale, M. Subramanian, T. K. Seshagiri, P. K. Khanna and V. K. Manchanda, J. Hazard. Mater., 2011, 186, 2051–2059 CrossRef CAS PubMed.
- M. F. Byers, S. Landsberger and E. Schneider, Sustainable Energy Fuels, 2018, 2, 2303–2313 RSC.
- X. J. Guo, R. R. Chen, Q. Liu, J. Y. Liu, H. S. Zhang, J. Yu, R. M. Li, M. L. Zhang and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 6185–6195 CrossRef CAS.
- J. H. Zhu, H. S. Zhang, R. R. Chen, Q. Liu, J. Y. Liu, J. Yu, R. M. Li, M. L. Zhang and J. Wang, J. Colloid. Interf. Sci, 2019, 543, 192–200 CrossRef CAS.
- F. F. Zhang, H. S. Zhang, R. R. Chen, Q. Liu, J. Y. Liu, C. Wang, Z. Y. Sun and J. Wang, J. Colloid. Interf. Sci, 2019, 534, 172–182 CrossRef CAS PubMed.
- X. J. Guo, H. C. Yang, Q. Liu, J. Y. Liu, R. R. Chen, H. S. Zhang, J. Yu, M. L. Zhang, R. M. Li and J. Wang, Chem. Eng. J., 2020, 382, 122850 CrossRef CAS.
- Q. H. Yu, Y. H. Yuan, J. Wen, X. M. Zhao, S. L. Zhao, D. Wang, C. Y. Li, X. L. Wang and N. Wang, Adv. Sci., 2019, 6, 1900002 CrossRef.
- S. Shi, B. C. Li, Y. X. Qian, P. P. Mei and N. Wang, Chem. Eng. J., 2020, 397, 125337 CrossRef CAS.
- X. J. Guo, H. C. Yang and J. Wang, Inorg. Chem. Front., 2022, 9, 155–164 RSC.
- X. J. Guo, R. R. Chen, Q. Liu, J. Y. Liu, H. S. Zhang, J. Yu, R. M. Li, M. L. Zhang and J. Wang, Environ. Sci.: Nano, 2018, 5, 2346–2356 RSC.
- Y. Wang, Z. W. Lin, H. S. Zhang, Q. Liu, J. Yu, J. Y. Liu, R. R. Chen, J. H. Zhu and J. Wang, J. Colloid. Interf. Sci., 2021, 598, 455–463 CrossRef CAS PubMed.
- H. J. Zhang, L. X. Zhang, X. L. Han, L. J. Kuang and D. B. Hua, Ind. Eng. Chem. Res., 2018, 57, 1662–1670 CrossRef CAS.
- H. Li, N. N. He, C. Cheng, H. Dong, J. Wen and X. L. Wang, Chem. Eng. J., 2020, 388, 124273 CrossRef CAS.
- Y. Liang, M. Xia, Q. H. Yu, Y. P. Li, Z. Y. Sui, Y. H. Yuan, X. M. Hu, Q. Chen and N. Wang, Adv. Compos. Hybrid Mater., 2022, 5, 184–194 CrossRef CAS.
- N. N. He, H. Li, L. Y. Li, C. Cheng, X. R. Lu, J. Wen and X. L. Wang, J. Hazard. Mater., 2021, 416, 126192 CrossRef CAS PubMed.
- S. Yang, G. X. Ji, S. Y. Cai, M. Y. Xu and D. B. Hua, J. Radioanal. Nucl. Chem., 2019, 321, 323–332 CrossRef CAS.
- N. N. He, H. Li, C. Cheng, H. Dong, X. R. Lu, J. Wen and X. L. Wang, Chem. Eng. J., 2020, 395, 125162 CrossRef CAS.
- Y. Sun, R. R. Liu, S. X. Wen, J. W. Wang, L. Chen, B. J. Yan, S. Y. Peng, C. Ma, X. Y. Cao, C. X. Ma, G. G. Duan, H. Wang, S. Shi, Y. H. Yuan and N. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 21272–21285 CrossRef CAS PubMed.
- S. Shi, R. Wu, S. L. Meng, G. P. Xiao, C. X. Ma, G. C. Yang and N. Wang, J. Hazard. Mater., 2022, 436, 128983 CrossRef CAS PubMed.
- Y. H. Yuan, Q. H. Yu, M. Cao, L. J. Feng, S. W. Feng, T. T. Liu, T. T. Feng, B. J. Yan, Z. H. Guo and N. Wang, Nat. Sustainability, 2021, 4, 708–714 CrossRef.
- K. Lin, W. Y. Sun, L. J. Feng, H. Wang, T. T. Feng, J. C. Zhang, M. Cao, S. L. Zhao, Y. H. Yuan and N. Wang, Chem. Eng. J., 2022, 430, 133121 CrossRef CAS.
- Q. H. Yu, Y. H. Yuan, L. J. Feng, W. Y. Sun, K. Lin, J. C. Zhang, Y. B. Zhang, H. Wang, N. Wang and Q. Peng, J. Hazard. Mater., 2022, 424, 127758 CrossRef CAS.
- H. Ye, C. Liu, M. B. Wu, L. L. Ma, S. C. Liu, Y. Zhong and J. Yao, J. Mater. Chem. A, 2022, 10, 2987–2994 RSC.
- W. Wang, Q. Luo, J. Y. Li, Y. H. Li, R. G. Wu, Y. L. Li, X. B. Huo and N. Wang, Chem. Eng. J., 2022, 431, 133483 CrossRef CAS.
- J. Wen, Q. Y. Li, H. Li, M. Chen, S. Hu and H. M. Cheng, Ind. Eng. Chem. Res., 2018, 57, 1826–1833 CrossRef CAS.
- N. Li, P. Gao, H. W. Chen, F. L. Li and Z. N. Wang, Chemosphere, 2022, 287, 132137 CrossRef CAS PubMed.
- H. C. Ma, F. Zhang, Q. Y. Li, G. B. Chen, S. Hu and H. M. Cheng, RSC Adv., 2019, 9, 18406–18414 RSC.
- J. X. Ao, Y. H. Yuan, X. Xu, L. Xu, Z. Xing, R. Li, G. Z. Wu, X. J. Guo, H. J. Ma and Q. N. Li, Ind. Eng. Chem. Res., 2019, 58, 8026–8034 CrossRef CAS.
- Y. H. Yuan, B. Y. Niu, Q. H. Yu, X. Guo, Z. H. Guo, J. Wen, T. Liu, H. Q. Zhang and N. Wang, Angew. Chem., Int. Ed., 2020, 59, 1220–1227 CrossRef CAS.
- M. W. Chen, T. Liu, X. B. Zhang, R. Q. Zhang, S. Tang, Y. H. Yuan, Z. J. Xie, Y. J. Liu, H. Wang, K. V. Fedorovich and N. Wang, Adv. Funct. Mater., 2021, 31, 2100106 CrossRef CAS.
- H. Wang, T. H. Xu, B. H. Zheng, M. Cao, F. Gao, G. B. Zhou, C. Ma, J. Dang, W. K. Yao, K. C. Wu, T. Liu, Y. H. Yuan, Q. Y. Fu and N. Wang, J. Hazard. Mater., 2022, 433, 128789 CrossRef CAS PubMed.
- Z. Q. Yu, D. P. Ye, J. Zhao, X. L. Wu and Y. D. Wu, Chem. Eng. J., 2021, 420, 129691 CrossRef CAS.
- A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- K.-S. Huang, C.-H. Yang, S.-L. Huang, C.-Y. Chen, Y.-Y. Lu and Y.-S. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef PubMed.
- N. F. Kamaruzzaman, L. P. Tan, R. H. Hamdan, S. S. Choong, W. K. Wong, A. J. Gibson, A. Chivu and M. d F. Pina, Int. J. Mol. Sci., 2019, 20, 2747 CrossRef CAS.
- E. I. Rabea, M. E. T. Badawy, C. V. Stevens, G. Smagghe and W. Steurbaut, Biomacromolecules, 2003, 4, 1457–1465 CrossRef CAS PubMed.
- A. Gobbi and G. Frenking, J. Am. Chem. Soc., 1993, 115, 2362–2372 CrossRef CAS.
- Y. H. Yuan, Q. H. Yu, S. Yang, J. Wen, Z. H. Guo, X. L. Wang and N. Wang, Adv. Sci., 2019, 6, 1900961 CrossRef CAS.
- Y. W. Ren, H. P. Liu, X. M. Liu, Y. F. Zheng, Z. Y. Li, C. Y. Li, K. W. K. Yeung, S. L. Zhu, Y. Q. Liang, Z. D. Cui and S. L. Wu, Cell Rep. Phys. Sci., 2020, 1, 100245 CrossRef.
- T. Wei, Q. Yu and H. Chen, Adv. Healthcare Mater., 2019, 8, e1801381 CrossRef PubMed.
- J. W. Liou and H. H. Chang, Arch. Immunol. Ther. Exp., 2012, 60, 267–275 CrossRef CAS PubMed.
- W. Y. Sun, L. J. Feng, J. C. Zhang, K. Lin, H. Wang, B. J. Yan, T. T. Feng, M. Cao, T. Liu, Y. H. Yuan and N. Wang, Adv. Sci., 2022, 9, 2105008 CrossRef CAS.
- W. R. Cui, C. R. Zhang, R. P. Liang, J. W. Liu and J. D. Qiu, ACS Appl. Mater. Interfaces, 2021, 13, 31561–31568 CrossRef CAS PubMed.
- C. R. Zhang, W. R. Cui, R. H. Xu, X. R. Chen, W. Jiang, Y. D. Wu, R. H. Yan, R. P. Liang and J. D. Qiu, CCS Chem., 2021, 3, 168–179 CrossRef CAS.
- Y. D. Wu, W. R. Cui, C. R. Zhang, R. P. Liang and J. D. Qiu, Environ. Chem. Lett., 2021, 19, 1847–1856 CrossRef CAS.
- W. R. Cui, C. R. Zhang, R. H. Xu, X. R. Chen, R. H. Yan, W. Jiang, R. P. Liang and J. D. Qiu, Small, 2021, 17, 2006882 CrossRef CAS.
- C. R. Zhang, W. R. Cui, C. P. Niu, S. M. Yi, R. P. Liang, J. X. Qi, X. J. Chen, W. Jiang, L. Zhang and J. D. Qiu, Chem. Eng. J., 2022, 428, 131178 CrossRef CAS.
- W. R. Cui, C. R. Zhang, R. P. Liang and J. D. Qiu, J. Mater. Chem. A, 2021, 9, 25611–25620 RSC.
- F. T. Yu, F. R. Song, R. Z. Wang, M. Xu and F. Luo, Polym. Chem., 2021, 12, 867–875 RSC.
- Z. Y. Bai, Q. Liu, H. S. Zhang, J. Y. Liu, R. R. Chen, J. Yu, R. M. Li, P. L. Liu and J. Wang, J. Hazard. Mater., 2020, 381, 120984 CrossRef CAS.
- Z. H. Bai, Q. Liu, H. S. Zhang, J. Y. Liu, J. Yu and J. Wang, Chem. Eng. J., 2020, 382, 122555 CrossRef CAS.
- Z. Y. Bai, Q. Liu, H. S. Zhang, J. Yu, R. R. Chen, J. Y. Liu, D. L. Song, R. M. Li and J. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 18012–18022 CrossRef CAS.
- Y. H. Yuan, X. Guo, L. J. Feng, Q. H. Yu, K. Lin, T. T. Feng, B. J. Yan, K. V. Fedorovich and N. Wang, Chem. Eng. J., 2021, 421, 127878 CrossRef CAS.
- S. Yang, Y. Cao, T. Wang, S. Y. Cai, M. Y. Xu, W. H. Lu and D. B. Hua, Environ. Res., 2020, 183, 109214 CrossRef CAS PubMed.
- Z. Huang, H. Dong, N. Yang, H. Li, N. N. He, X. R. Lu, J. Wen and X. L. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 16959–16968 CrossRef CAS.
- P. P. Mei, R. Wu, S. Shi, B. C. Li, C. X. Ma, B. W. Hu, Y. H. Yuan, H. Wang, T. Liu and N. Wang, Chem. Eng. J., 2021, 420, 130382 CrossRef CAS.
- M. Debayle, E. Balloul, F. Dembele, X. Z. Xu, M. Hanafi, F. Ribot, C. Monzel, M. Coppey, A. Fragola, M. Dahan, T. Pons and N. Lequeux, Biomaterials, 2019, 219, 119357 CrossRef CAS.
- L. Mi and S. Y. Jiang, Angew. Chem., Int. Ed., 2014, 53, 1746–1754 CrossRef CAS.
- D. J. Vanderah, H. La, J. Naff, V. Silin and K. A. Rubinson, J. Am. Chem. Soc., 2004, 126, 13639–13641 CrossRef CAS PubMed.
- H. B. Pang, L. J. Kuo, C. M. Wai, N. Miyamoto, R. Joshi, J. R. Wood, J. E. Strivens, C. J. Janke, Y. Oyola, S. Das, R. T. Mayes and G. A. Gill, Ind. Eng. Chem. Res., 2016, 55, 4313–4320 CrossRef.
- T. Suzuki, K. Saito, T. Sugo, H. Ogura and K. Oguma, Anal. Sci., 2000, 16, 429–432 CrossRef CAS.
- H. Egawa, T. Nonaka and M. Nakayama, Ind. Eng. Chem. Res., 1990, 29, 2273–2277 CrossRef CAS.
- T. Takeda, K. Saito, K. Uezu, S. Furusaki, T. Sugo and J. Okamoto, Ind. Eng. Chem. Res., 1991, 30, 185–190 CrossRef CAS.
- L. Astheimer, H. J. Schenk, E. G. Witte and K. Schwochau, Sep. Sci. Technol., 1983, 18, 307–339 CrossRef CAS.
- S. O. Kang, S. Vukovic, R. Custelcean and B. P. Hay, Ind. Eng. Chem. Res., 2012, 51, 6619–6624 CrossRef CAS.
- H. Omichi, A. Katakai, T. Sugo and J. Okamoto, Sep. Sci. Technol., 1986, 21, 563–574 CrossRef CAS.
- S. Das, W. P. Liao, M. Flicker Byers, C. Tsouris, C. J. Janke, R. T. Mayes, E. Schneider, L. J. Kuo, J. R. Wood, G. A. Gill and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4303–4312 CrossRef CAS.
- S. Das, C. Tsouris, C. Zhang, J. Kim, S. Brown, Y. Oyola, C. J. Janke, R. T. Mayes, L. J. Kuo, J. R. Wood, G. A. Gill and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4294–4302 CrossRef CAS.
- H. B. Pan, L. J. Kuo, J. Wood, J. Strivens, G. A. Gill, C. J. Janke and C. M. Wai, RSC Adv., 2015, 5, 100715–100721 RSC.
- H.-B. Pan, W. S. Liao, C. M. Wai, Y. Oyola, C. J. Janke, G. X. Tian and L. F. Rao, Dalton Trans., 2014, 43, 10713–10718 RSC.
- H.-B. Pan, C. M. Wai, L.-J. Kuo, G. Gill, G. X. Tian, L. F. Rao, S. Das, R. T. Mayes and C. J. Janke, ChemistrySelect, 2017, 2, 3769–3774 CrossRef CAS.
- L. J. Kuo, H. B. Pan, C. M. Wai, M. F. Byers, E. Schneider, J. E. Strivens, C. J. Janke, S. Das, R. T. Mayes, J. R. Wood, N. Schlafer and G. A. Gill, Ind. Eng. Chem. Res., 2017, 56, 11603–11611 CrossRef CAS.
- C. Liu, P. C. Hsu, J. Xie, J. Zhao, T. Wu, H. T. Wang, W. Liu, J. S. Zhang, S. Chu and Y. Cui, Nat. Energy, 2017, 2, 17007 CrossRef CAS.
- C. Tsouris, Nat. Energy, 2017, 2, 17022 CrossRef.
- F. T. Chi, S. Zhang, J. Wen, J. Xiong and S. Hu, Ind. Eng. Chem. Res., 2018, 57, 8078–8084 CrossRef CAS.
- X. R. Tang, Y. Liu, M. Liu, H. M. Chen, P. L. Huang, H. M. Ruan, Y. M. Zheng, F. Yang, R. He and W. K. Zhu, Nanoscale, 2022, 14, 6285–6290 RSC.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- A. F. Ismail and M. S. Yim, Nucl. Eng. Technol., 2015, 47, 579–587 CrossRef.
- Y. Liao, M. Wang and D. J. Chen, Appl. Surf. Sci., 2019, 484, 83–96 CrossRef CAS.
- L. Chen and D. G. Tong, Sep. Purif. Technol., 2020, 250, 117175 CrossRef CAS.
- J. Zhou, H. J. Zhou, Y. Z. Zhang, J. Wu, H. M. Zhang, G. Z. Wang and J. X. Li, Chem. Eng. J., 2020, 398, 125460 CrossRef CAS.
- X. H. Tang, L. M. Zhou, J. Xi, J. B. Ouyang, Z. R. Liu, Z. S. Chen and A. A. Adesina, Sep. Purif. Technol., 2021, 274, 119005 CrossRef CAS.
- Y. Z. Zhang, J. Zhou, D. Wang, R. Y. Cao and J. X. Li, Chem. Eng. J., 2022, 430, 132702 CrossRef CAS.
- H. Yang, X. L. Liu, M. J. Hao, Y. H. Xie, X. K. Wang, H. Tian, G. I. N. Waterhouse, P. E. Kruger, S. G. Telfer and S. Q. Ma, Adv. Mater., 2021, 33, 2106621 CrossRef CAS.
- X. L. Liu, Y. H. Xie, M. J. Hao, Z. S. Chen, H. Yang, G. I. N. Waterhouse, S. Q. Ma and X. K. Wang, Adv. Sci., 2022, 9, 2201735 CrossRef CAS.
- C. Wang, A. S. Helal, Z. Q. Wang, J. Zhou, X. H. Yao, Z. Shi, Y. Ren, J. H. Lee, J. K. Chang, B. Fugetsu and J. Li, Adv. Mater., 2021, 33, 2102633 CrossRef CAS.
- J. Chen, D. F. Ollis, W. H. Rulkens and H. Bruning, Colloids Surf., A, 1999, 151, 339–349 CrossRef CAS.
- C. H. Lu, P. Zhang, S. J. Jiang, X. Wu, S. Q. Song, M. S. Zhu, Z. Z. Lou, Z. Li, F. Liu, Y. H. Liu, Y. Wang and Z. G. Le, Appl. Catal. B: Environ., 2017, 200, 378–385 CrossRef CAS.
- P. Li, J. J. Wang, Y. Wang, J. J. Liang, B. H. He, D. Q. Pan, Q. H. Fan and X. K. Wang, Chem. Eng. J., 2019, 365, 231–241 CrossRef CAS.
- Y. Wang, J. J. Wang, J. Wang, J. J. Liang, D. Q. Pan, P. Li and Q. H. Fan, J. Mol. Liq., 2020, 308, 113007 CrossRef CAS.
- J. J. Wang, Y. Wang, W. Wang, T. Peng, J. J. Liang, P. Li, D. Q. Pan, Q. H. Fan and W. S. Wu, Environ. Pollut., 2020, 262, 114373 CrossRef CAS PubMed.
- Y. C. Xu, H. S. Zhang, Q. Liu, J. Y. Liu, R. R. Chen, J. Yu, J. H. Zhu, R. M. Li and J. Wang, J. Hazard. Mater., 2021, 416, 125812 CrossRef CAS.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Li, J. G. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- J. J. Wang, Y. Wang, W. Wang, Z. Ding, R. Y. Geng, P. Li, D. Q. Pan, J. J. Liang, H. B. Qin and Q. H. Fan, Chem. Eng. J., 2020, 383, 123193 CrossRef CAS.
- P. Li, Y. Wang, J. J. Wang, L. Dong, W. T. Zhang, Z. H. Lu, J. J. Liang, D. Q. Pan and Q. H. Fan, Chem. Eng. J., 2021, 414, 128810 CrossRef CAS.
- S. Liu, Z. Wang, Y. X. Lu, H. P. Li, X. J. Chen, G. Y. Wei, T. Wu, D. J. Maguire, G. Ye and J. Chen, Appl. Catal., B, 2021, 282, 119523 CrossRef CAS.
- H. Li and S. A. Wang, Chem, 2021, 7, 279–280 CAS.
- H. Li, F. W. Zhai, D. X. Gui, X. X. Wang, C. F. Wu, D. Zhang, X. Dai, H. Deng, X. T. Su, J. Diwu, L. Zhang, Z. F. Chai and S. A. Wang, Appl. Catal., B, 2019, 254, 47–54 CrossRef CAS.
- H. L. Zhang, W. Liu, A. Li, D. Zhang, X. Y. Li, F. W. Zhai, L. H. Chen, L. Chen, Y. L. Wang and S. A. Wang, Angew. Chem., Int. Ed., 2019, 58, 16110–16114 CrossRef CAS.
- M. J. Hao, Z. S. Chen, X. L. Liu, X. H. Liu, J. Y. Zhang, H. Yang, G. I. N. Waterhouse, X. K. Wang and S. Q. Ma, CCS Chem., 2022, 4, 2294–2307 CrossRef CAS.
- F. T. Yu, Z. Q. Zhu, S. P. Wang, Y. K. Peng, Z. Z. Xu, Y. A. Tao, J. B. Xiong, Q. W. Fan and F. Luo, Chem. Eng. J., 2021, 412, 127558 CrossRef CAS.
- L. J. Feng, Y. H. Yuan, B. J. Yan, T. T. Feng, Y. P. Jian, J. C. Zhang, W. Y. Sun, K. Lin, G. S. Luo and N. Wang, Nat. Commun., 2022, 13, 1389 CrossRef CAS.
- W. R. Cui, C. R. Zhang, R. H. Xu, X. R. Chen, R. H. Yan, W. Jiang, R. P. Liang and J. D. Qiu, ACS EST Water, 2021, 1, 440–448 CrossRef CAS.
- T. Chen, K. F. Yu, C. X. Dong, X. Yuan, X. Gong, J. Lian, X. Cao, M. Z. Li, L. Zhou, B. W. Hu, R. He, W. K. Zhu and X. K. Wang, Coordin. Chem. Rev., 2022, 467, 214615 CrossRef CAS.
- L. Rao, Recent international R&D activities in the extraction of uranium from seawater, Report LBNL–4034E, United States, 2010.
- L.-J. Kuo, C. J. Janke, J. R. Wood, J. E. Strivens, S. Das, Y. Oyola, R. T. Mayes and G. A. Gill, Ind. Eng. Chem. Res., 2016, 55, 4285–4293 CrossRef CAS.
- H. Yamashita, Y. Ozawa, F. Nakajima and T. Murata, Nippon Kagaku Kaishi, 1978, 1978, 1057–1061 CrossRef.
- H. J. Schenk, L. Astheimer, E. G. Witte and K. S. Schwochau, Sep. Sci. Technol., 1982, 17, 1293–1308 CrossRef CAS.
- H. Nobukawa, M. Tamehiro, M. Kobayashi, H. Nakagawa, J. Sakakibara and N. Takagi, J. Shipbuild. Soc. Japan, 1989, 165, 281–292 Search PubMed.
- H. Nobukawa, M. Kitamura, M. Kobayashi, H. Nakagawa, N. Takagi and M. Tamehiro, J. Shipbuild. Soc. Japan, 1992, 172, 519–528 Search PubMed.
- N. Seko, A. Katakai, S. Hasegawa, M. Tamada, N. Kasai, H. Takeda, T. Sugo and K. Saito, Nucl. Technol., 2003, 144, 274–278 CrossRef CAS.
- A. Goto, K. Kusakabe, S. Morooka and T. Kago, Sep. Sci. Technol., 1993, 28, 1273–1285 CrossRef.
- H. Egawa, N. Kabay, T. Shuto and A. Jyo, Ind. Eng. Chem. Res., 1993, 32, 709–715 CrossRef CAS.
- T. Shimizu and M. Tamada, Proc. Civ. Eng. Ocean, 2004, 20, 617–622 CrossRef.
- M. Tamada, N. Seko and F. Yoshii, Radiat. Phys. Chem., 2004, 71, 221–225 CrossRef.
- J. Guidez and S. Gabriel, EPJ Nucl. Sci. Technol., 2016, 2, 10 CrossRef.
- N. Seko, M. Tamada, N. Kasai, F. Yoshii and T. Shimizu, Proc. Civ. Eng. Ocean, 2002, 18, 737–742 CrossRef.
- T. Sugo, M. Tamada, T. Seguchi, T. Shimizu, M. Uotani and R. Kashima, J. Atom. Energy Soc. Japan, 2001, 43, 1010–1016 CrossRef CAS.
- M. Tamada, N. Seko, N. Kasai and T. Shimizu, Trans. At. Energy Soc. Jpn., 2006, 5, 358–363 CrossRef CAS.
- E. Schneider and D. Sachde, Sci. Global Secur., 2013, 21, 134–163 CrossRef.
- Uranium from seawater program review; Fuel resources uranium from seawater program DOE Office of Nuclear Energy, Report ORNL/TM-2013/295, United States, 2013.
- S. D. Alexandratos and S. Kung, Ind. Eng. Chem. Res., 2016, 55, 4101–4102 CrossRef CAS.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076–6083 CrossRef CAS.
- G. A. Gill, L.-J. Kuo, J. E. Strivens, J. R. Wood, N. J. Schlafer, C. J. Janke, S. Das, R. Mayes, T. Saito, S. S. Brown, C. Tsouris, C. Tsouris, C. M. Wai and H.-B. Pan, Summary of adsorption capacity and adsorption kinetics of uranium and other elements on amidoxime-based adsorbents from time series marine testing at the Pacific Northwest National Laboratory, Report PNNL–25899, United States, 2016.
- T. A. Todd, J. Vienna and P. Paviet, Material recover and waste form development--2016 Accomplishments, Report INL/EXT-16-40677, United States, 2016.
- J. A. Drysdale and K. O. Buesseler, Prog. Nucl. Energy, 2019, 119, 103170 CrossRef.
- M. Picard, C. Baelden, Y. Wu, L. Chang and A. H. Slocum, Nucl. Technol., 2014, 188, 200–217 CrossRef CAS.
- M. Byers, E. Schneider, M. N. Haji and A. H. Slocum, Trans. Am. Nucl. Soc., 2016, 115, 271–274 Search PubMed.
- M. F. Byers, M. N. Haji, A. H. Slocum and E. Schneider, Ocean Eng., 2018, 169, 227–241 CrossRef.
- M. N. Haji, J. M. Kluger, T. P. Sapsis and A. H. Slocum, Ocean Eng., 2018, 169, 673–681 CrossRef.
- M. N. Haji, C. Vitry and A. H. Slocum, Trans. Am. Nucl. Soc., 2015, 113, 158–161 Search PubMed.
- M. N. Haji and A. H. Slocum, Trans. Am. Nucl. Soc., 2016, 115, 153–156 Search PubMed.
- M. N. Haji, J. Drysdale, K. Buesseler and A. H. Slocum, presented in part at The 27th International Ocean and Polar Engineering Conference, 2017.
- M. N. Haji, M. Byers, E. Schneider and A. H. Slocum, Trans. Am. Nucl. Soc., 2017, 116, 89–92 Search PubMed.
- M. N. Haji, J. Gonzalez, J. A. Drysdale, K. O. Buesseler and A. H. Slocum, Ind. Eng. Chem. Res., 2018, 57, 15534–15541 CAS.
- M. N. Haji, J. A. Drysdale, K. O. Buesseler and A. H. Slocum, Environ. Sci. Technol., 2019, 53, 2229–2237 CrossRef CAS.
- L.-J. Kuo, G. A. Gill, C. Tsouris, L. F. Rao, H.-B. Pan, C. M. Wai, C. J. Janke, J. E. Strivens, J. R. Wood, N. Schlafer and E. K. D’Alessandro, ChemistrySelect, 2018, 3, 843–848 CrossRef CAS.
- A. Slocum, Extraction of uranium from seawater: Design and testing of a symbiotic system, Report 14-6557, United States, 2018.
- Z. M. Li, Y. Q. Niu, Y. T. Su, Y. Song, F. j Wang, Y. F. Gou, H. Z. Wang and S. S. Chen, At. Energy Sci. Technol., 2022, 44, 233–245 CAS.
- Z. Y. Liu, Y. Xie, Y. F. Wang, T. Y. Hu, G. Ye and J. Chen, Tsinghua Sci. Technol., 2021, 61, 279–301 Search PubMed.
- H. Li, J. Wen and X. L. Wang, Chin. Sci. Bull., 2018, 63, 481–494 CrossRef.
- A. I. Wiechert, A. P. Ladshaw, G. A. Gill, J. R. Wood, S. Yiacoumi and C. Tsouris, Ind. Eng. Chem. Res., 2018, 57, 17237–17244 CrossRef CAS.
- D. Wongsawaeng, W. Wongjaikham, P. Hosemann, D. Swantomo and K. T. Basuki, Int. J. Energy. Res., 2021, 45, 1748–1760 CrossRef CAS.
- M. B. Altay, C. Kalipcioglu and Z. Kurt, Resour. Conserv. Recy., 2022, 181, 106237 CrossRef CAS.
- H. Lindner and E. Schneider, Energy. Econ., 2015, 49, 9–22 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |