
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of an AlI3-doped Li2S positive electrode with superior performance in all-solid-state batteries†
Hirotada
Gamo
a,
Takaki
Maeda
a,
Kazuhiro
Hikima
 a,
Minako
Deguchi
b,
Yushi
Fujita
b,
Yusuke
Kawasaki
b,
Atsushi
Sakuda
b,
Hiroyuki
Muto
ac,
Nguyen Huu Huy
Phuc
*a,
Akitoshi
Hayashi
b,
Masahiro
Tatsumisago
b and
Atsunori
Matsuda
*a
a,
Minako
Deguchi
b,
Yushi
Fujita
b,
Yusuke
Kawasaki
b,
Atsushi
Sakuda
b,
Hiroyuki
Muto
ac,
Nguyen Huu Huy
Phuc
*a,
Akitoshi
Hayashi
b,
Masahiro
Tatsumisago
b and
Atsunori
Matsuda
*a
aDepartment of Electrical and Electronic Information Engineering, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku, Toyohashi, Aichi 441-8580, Japan. E-mail: matsuda@ee.tut.ac.jp; nhhphuc@hcmut.edu.vn
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka Prefecture University, Osaka 599-8531, Japan
cInstitute of Liberal Arts and Science, Toyohashi University of Technology, 1-1 Hibarigaoka, Tempaku, Toyohashi, Aichi 441-8580, Japan
First published on 9th February 2022
Abstract
A (100 − x)Li2S·xAlI3 (0 ≤ x ≤ 30) positive electrode was prepared by the planetary ball-milling method for application in all-solid-state Li–S batteries. X-Ray diffraction results showed that I− in AlI3 dissolved into the Li2S structure in (100 − x)Li2S·xAlI3 with x ≤ 5 to form the solid solution. The change of the electronic structure of Li2S and AlI3 was further proved by UV-Vis spectroscopy and X-ray photoelectron spectroscopy results. The highest conductivity of about 6.0 × 10−5 S cm−1 at room temperature was obtained with 80Li2S·20AlI3 (mol%) and the highest conductivity at 150 °C of about 7.0 × 10−3 S cm−1 was observed in 75Li2S·25AlI3. The maximum capacities of the cells with x = 0, 0.5, 1.5, and 2 were 880, 1059, 1006, and 869 mA h g−1 Li2S, respectively. The capacity retention of the cells with x = 0, 0.5, 1.5, and 2 after 60 cycles was 64.0%, 88.4%, 69.6%, and 71.2%, respectively. This study showed that AlI3 doping could improve not only the conductivities of Li2S but also the cyclic properties of all-solid-state Li–S batteries.
1. Introduction
All-solid-state (ASS) Li–S batteries have attracted attention from a wide research community because of their potential in high theoretical energy density and ease of electrode design.1,2 Research on ASS Li–S batteries has accelerated recently and most of the studies focus on the synthesis of new electrode materials or electrode composite design.3 The active materials of positive electrodes have been developed for years and are mainly divided into three different groups: sulfur based, transition metal chalcogenides, and sulfide solid electrolyte based.4–7Sulfur is the simplest active material for Li–S batteries but its use has faced difficulties arising from its electronic and ionic insulation. Therefore, sulfur must be blended with materials with decent electronic and ionic conductivities. Very often, sulfur is embedded in the pore structures of either carbonaceous or oxide materials to improve its conduction properties.8,9 In another approach, sulfur was embedded into transition metal chalcogenides and employed as an active material in Li–S batteries because there are abundant and simple methods for composite electrode preparation.10 However, the use of either sulfur or transition metal chalcogenides results in the need for Li metal at the negative electrode and then dendritic Li formation becomes a major issue to solve.11
Lithium sulfide (Li2S) is the discharged form of sulfur and, thus, its use in the positive electrode will result in the elimination of Li metal and open the doors for graphite or silicon, the safety of which has been proven for years in conventional Li-ion batteries.12 Li2S could be blended with reduced graphene oxide (electronic conductor) to form a positive electrode composite that provides a capacity of approximately 400 mA h g−1 after 20 cycles.13 In addition, Li2S nanoparticles could form a core–shell structure with the solid electrolyte Li3PS4 to improve interfacial resistivity for better cycle performance and capacity.14,15 Hakari et al. showed that an 80Li2S–20LiI solid solution could improve Li2S utility and cyclic stability due to the electrochemical sites provided by the anion I−.16 The multivalence cations Mg2+ and Al3+ also enhanced not only the ionic conductivities of Li2S and Li3PS4 but also the capacity and cyclic performance of ASS Li–S batteries because of defect formation and reduction in activation energy for the sulfur, lithium ion, and electron combination reactions.6,17,18
In this study AlI3-doped Li2S was prepared by the planetary ball-milling method for further improvement of the Li2S positive electrode performance. The prepared samples were characterized and their battery performances were investigated. Li10P3S12I glass ceramic was used as a solid electrolyte because of its high ionic conductivity, ease of preparation, and stability at low voltage.19,20 X-Ray diffraction (XRD), ultraviolet-visible spectroscopy (UV-Vis), and X-ray photoelectron spectroscopy (XPS) were employed to analyze the structure of prepared (100 − x)Li2S·xAlI3. The capacity of the cell using the sample x = 0.5 remained at 936 mA h g−1 compared with 563 mA h g−1 for x = 0 after 60 cycles.
2. Experimental
Li2S (99.9%) and P2S5 (99%) were purchased from Mitsuwa Chemical and Merck Group, respectively, and used without purification. AlI3 (99.9%) and LiI (99.9%) were received from Kojundo Chemical Laboratory and Aldrich, respectively.(100 − x)Li2S·xAlI3 (0 ≤ x ≤ 30) was prepared via planetary ball milling. Li2S and AlI3 were mixed for 10 min using an agate mortar, and then put into 45 ml zirconia pots with zirconia balls (10 mm, 15 balls). The pots were rotated at 500 rpm for 12 h using a Pulverisette 7 (Fritsch). The obtained samples were recovered and used without any further heat treatment.
Li10P3S12I glass ceramic solid electrolyte was prepared by a solid-state reaction. 1.5 g amounts of Li2S, P2S5, and LiI in a molar ratio of Li2S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P2S5:LiI = 9:3:2 were weighed, mixed well using an agate mortar, and then put into 45 ml zirconia pots with 70 g of zirconia balls (4 mm). The pots were rotated at 510 rpm for 15 h using a Pulverisette 7 (Fritsch). The obtained glassy powder was then sintered at 190 °C for 1 h in an Ar atmosphere to obtain a Li10P3S12I glass ceramic solid electrolyte with an ionic conductivity at RT of approximately 1.0 mS cm−1.
:P2S5:LiI = 9:3:2 were weighed, mixed well using an agate mortar, and then put into 45 ml zirconia pots with 70 g of zirconia balls (4 mm). The pots were rotated at 510 rpm for 15 h using a Pulverisette 7 (Fritsch). The obtained glassy powder was then sintered at 190 °C for 1 h in an Ar atmosphere to obtain a Li10P3S12I glass ceramic solid electrolyte with an ionic conductivity at RT of approximately 1.0 mS cm−1.
A composite cathode composed of (100 − x)Li2S·xAlI3, Li10P3S12I glass ceramic, and Ketjen Black (KB) in a weight ratio of 50:40:10 was prepared via two-step planetary ball milling. An amount of 0.3 g of (100 − x)Li2S·xAlI3 and KB (weight ratio of 5:1) were put into zirconia pots with 30 g of zirconia balls (4 mm) and the pots were rotated at 510 rpm for 10 h. Then 0.2 g of Li10P3S12I glass ceramic was added to the pots and they were rotated at 400 rpm for 2 h to obtain the positive electrode composites.
The structures of the prepared (100 − x)Li2S·xAlI3 powders were characterized via XRD (Ultima IV, Rigaku) and diffuse reflection UV-Vis (V-670, Jasco). For analysis, the samples were sealed in special holders in an Ar-filled glove box to avoid exposure to humidity. The electronic structure was characterized using XPS (K-Alpha, Thermo Fisher Scientific) using a monochromatic Al Kα source (1486.6 eV).21 Calibration of the observed binding energies was performed with respect to the advantageous C 1s peak at 284.7 eV after etching. Ar+ ion-etching was carried out using Ar gas cluster ion beams with an energy of 6 keV for 30 min to suppress damage to the samples. The etching rate was 1 nm min−1. The sample was characterized without exposure to air by using an Ar-filled transfer vessel.
The temperature dependence of the ionic conductivity of the prepared (100 − x)Li2S·xAlI3 samples was investigated using a previously reported procedure.22 The electronic conductivity at RT was measured via the direct current polarization method.5 Prior to the measurements, the samples were pressed into pellets of ∼10 mm in diameter at a pressure of 550 MPa (at RT). The pellets were then placed in a PEEK holder with two stainless steel rods as blocking electrodes. Voltages of 0.1–2.0 V (DC) were then applied to the prepared cells for 60 min and the currents were measured. The experiments were carried out using a potentiostat (SI 1287; Solatron). ASS Li–S cells were fabricated with a structure resembling the one reported elsewhere.5 The current density was ∼0.25 mA cm−2. The cut-off voltages of all batteries were 0.9–3.0 V vs. Li–In for the first cycle and 0.9–2.25 V vs. Li–In for subsequent cycles. The charge–discharge test after the first charge was carried out in CC (Constant Current)–CV (Constant Voltage) mode. The normalized Li2S loading in each cell was 1.8–2.2 mg. ASS Li–S cells were cycled under an applied external pressure of about 36 MPa at 30 °C. All the experiments were conducted in an Ar-filled glove box (water < 0.1 ppm) or an airtight sample holder to avoid direct exposure of the samples to ambient humidity.
3. Results and discussion
Fig. 1 illustrates the XRD patterns of (100 − x)Li2S·xAlI3 samples. Peaks of Li2S were preserved until x = 5 but those of AlI3 disappeared. The peaks of (100 − x)Li2S·xAlI3 (x ≤ 5) were shifted to lower angles compared with x = 0 (pristine Li2S). These facts proved that the lattice volume of the Li2S structure increased until x = 5 because I− within AlI3 was dissolved into the Li2S structure. Calculating the lattice parameters from the diffraction angles corresponding to the (111), (200), (220), and (311) lattice planes, the lattice parameters for the samples with x = 0, 0.5, 3, and 5 were 5.718, 5.741, 5.752, and 5.777 Å, respectively. The lattice parameters of the Li2S structure increased with increasing AlI3 doping levels in Li2S. From x = 7, peaks of LiI were detected and from x = 20 peaks of Li2S disappeared, as shown in Fig. S1 (ESI†). According to the previous study, Li2S·AlS1.5 prepared by high-energy mechanical milling involves an XRD halo pattern and no diffraction peak at a specific composition with high Al-ion concentration.23 Therefore, at higher x values AlI3 can dissolve into the Li2S glass matrix to form a solid solution. It was also proved that Li2S could form a solid solution with LiI at a molar ratio of Li2S:LiI = 80:20.16 The peak-shift to lower angles was also detected in that study. Further increase of the LiI concentration to 25 mol% resulted in LiI remaining in addition to the Li2S–LiI solid solution. This is consistent with the fact that the LiI crystal phase was produced in the sample with x = 7 (in this case, the I concentration was 21 mol%).
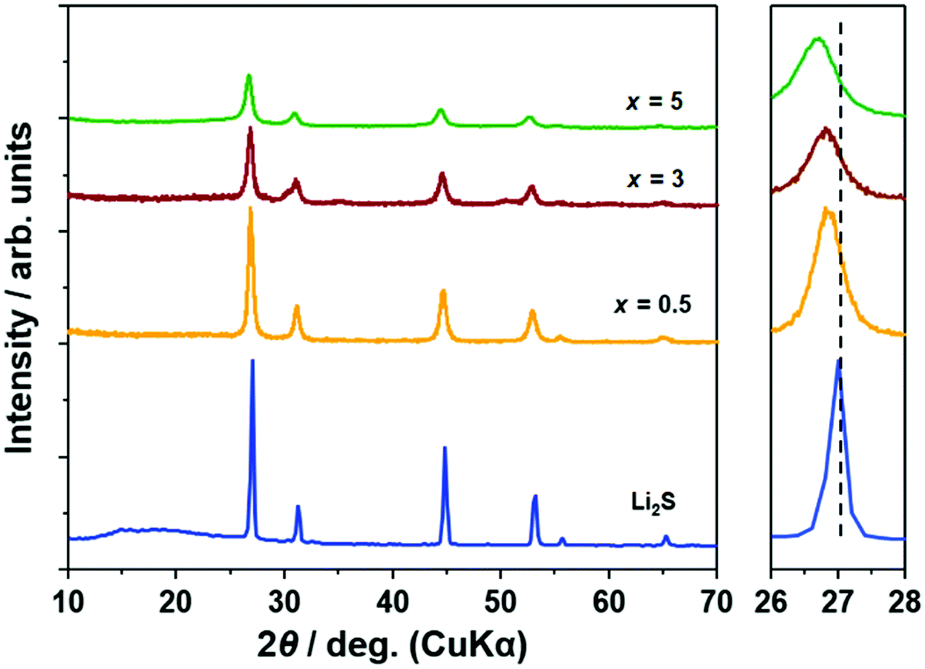 | ||
| Fig. 1 X-Ray diffraction patterns of (100 − x)Li2S·xAlI3 and a magnified view. | ||
Fig. 2 illustrates the UV-Vis spectra of (100 − x)Li2S·xAlI3 (x ≤ 5). Li2S exhibited one small shoulder centered at 270 nm and a large shoulder in the range of 250–190 nm. AlI3 showed a large absorption shoulder ranging from ∼320 nm to 190 nm. Doping a small amount of AlI3 into the Li2S structure (x ≤ 2) resulted in the appearance of a small absorption shoulder centered at approximately 350 nm while the absorption bands centered at 270 nm and 290 nm of Li2S and AlI3 disappeared. At higher doping levels of 2 < x ≤ 5, the band at 350 nm was replaced by a new absorption band centered at 307 nm. This is consistent with the change of the UV-Vis spectra observed in the Li2S·Al2S3 solid solution,6 indicating that Al-ions in AlI3 were well dispersed in the Li2S glass matrix. Therefore, the (100 − x)Li2S·xAlI3 (x ≤ 5) solid solution formation drastically changed the electronic structure.
 | ||
| Fig. 2 UV-Vis spectra of Li2S (x = 0), AlI3 (x = 100), and (100 − x)Li2S·xAlI3. | ||
Fig. 3(a–d) show the XPS spectra (envelope after background subtraction) of S 2p, I 3d, Li 1s, and Al 2p, respectively, for the prepared samples and standard materials Li2S (ball milled), AlI3 (ball milled), LiI, and 80Li2S–20LiI for comparison. The details of the measured data, peak deconvolutions, and envelopes are illustrated in Fig. S2 (ESI†). The binding energy values provided in Fig. 3 were adapted from the deconvolution peaks, the details of which are shown in Fig. S2 (ESI†). The two peaks at 160.1 eV and 161.3 eV of Li2S were assigned to the binding energies of S 2p3/2 and 2p1/2 (Fig. 3(a)).24 Those for 80Li2S·20LiI were 160.6 eV and 161.8 eV. Doping Li2S with a small amount of AlI3 (x = 0.5) resulted in the peaks shifting to higher binding energies, 160.5 eV and 161.6 eV. The peaks were shifted to 160.8 eV and 162.0 eV for both the higher doping levels, x = 3 and x = 5. These results suggested the existence of a bond between S and Al in the Li2S·AlI3; this could be described by high electronegativity of the Al-ion compared with that of the Li-ion. The S 2p spectra and their deconvolution results showed that S in the samples and standard materials were in the single electronic state (Fig. S2, ESI†). Energy separations of approximately 1.20 eV and peak intensity ratios of about 2:1 were observed in all the spectra. The results for S 2p in this study were consistent with the reported values.25,26 The I 3d XPS data is provided in Fig. 3(b). The two peaks located at 619.0 eV and 630.4 eV were assigned to 3d5/2 and 3d3/2 of I−.27,28 Those values for I− in AlI3 were 619.7 eV and 631.2 eV. The 80Li2S–20LiI expressed the peaks of I− at 619.1 eV and 630.5 eV. Peaks of I− in all the prepared samples (x = 0.5, 3, and 5) appeared in the region of I− in 80Li2S–20LiI, 619.1 eV and 630.5 eV, instead of AlI3. This observation indicates that doping I-ion dissolved into the Li2S structure, which is consistent with the formation of a solid solution described by the XRD results. The signal of Li 1s in LiI appeared at 56.3 eV while that of Li2S was at 54.5 eV.27,29 The peak of Li 1s in 80Li2S–20LiI was detected at 55.1 eV, which was in between the values of those in LiI and Li2S. Doping AlI3 into Li2S resulted in the peak-shift of Li 1s from 54.5 eV in Li2S to 54.8 eV, 55.2 eV, and 55.4 eV with x = 0, 3, and 5, respectively. The Al 2p of AlI3 showed a peak at 75.0 eV.30 The addition of AlI3 into Li2S at x = 0.5 led to a drastic peak-shift to 73.6 eV. For x = 3 and x = 5, the signal of Al 2p appeared at 73.9 eV and 74.0 eV. The peak-shift observed in (100 − x)Li2S·xAlI3 (0 < x ≤ 5) compared with those of standard materials demonstrates that the Al3+ is bound to S with low electronegativity in comparison to that of I−. Therefore, these XPS results also confirmed that the Al3+ in AlI3 dissolved into the Li2S matrix. These results were also in good agreement with those obtained from XRD and UV-Vis measurements. The formation of the Li2S·AlI3 solid solution should dramatically change the electronic structures in both Li2S and AlI3. It should be noted that the obtained XPS spectra is reflected the chemical binding state from the surface to a depth of 10 nm in Li2S·AlI3.
 | ||
| Fig. 3 X-Ray photoelectron spectroscopy spectra (envelope) of the prepared samples and standard materials. (a) S 2p; (b) I 3d; (c) Li 1s; and (d) Al 2p. | ||
The temperature dependence of ionic conductivity and polarized I–V correlation (electronic current) of (100 − x)Li2S·xAlI3 (0 ≤ x ≤ 5) samples are shown in Fig. 4. The reported ionic conductivity of Li2S at RT was reported to be 10−9–10−8 S cm−1.16 However, the value obtained in this study was lower than 10−12 S cm−1, which was in agreement with the value reported by Z. Lin et al.14 A 99Li2S·1AlI3 solid solution exhibited ionic conductivity approximately three orders of magnitude higher than that of Li2S. 95Li2S·5AlI3 expressed ionic conductivity at RT of 4.5 × 10−6 S cm−1, which was comparable to that of 80Li2S·20LiI.15 A slight increase in the amount of AlI3 from 5 to 7 resulted in the increase of ionic conductivity from 4.5 × 10−6 S cm−1 to 2.8 × 10−5 S cm−1, as shown in Fig. S3 (ESI†). The highest conductivity at RT was obtained for 80Li2S·20AlI3, which was approximately 6.0 × 10−5 S cm−1; while the highest conductivity at 150 °C was approximately 7.0 × 10−3 S cm−1 and was observed in 75Li2S·25AlI3. The values of ionic conductivity obtained in this study were much higher than that of any Li2S–based substances reported so far.31 Hayashi et al. reported that the 60Li2S·40Al2S3 amorphous solid electrolyte prepared by mechanical milling showed 3.4 × 10−5 S cm−1 of ionic conductivity at room temperature as a pure Li ion conductor.32 Thus, aluminum ions in Li2S matrix spatially localize to occupied sites, and multi-ion concerted migration should not occur in the Li2S–AlI3 system.
 | ||
| Fig. 4 Temperature dependence of ionic conductivity (a) and I–V correlation (polarized electronic current) (b) of (100 − x)Li2S − xAlI3. | ||
The results of the I–V correlation measurements using blocking electrodes for evaluation of electron conductivity are shown in Fig. 4(b). The results confirmed that Li2S was nearly insulating toward electrons at applied voltages up to 1 V. The current in the doped samples clearly exhibited a dependence on the applied voltage. Dissolving AlI3 into Li2S led to the decrease of binding energy for both Al 2p and I 3d orbitals, which meant that the ionization energies of those electrons were reduced (Fig. 3(b and d)). Conversely, the binding energies of both Li 1s and S 2p increased after AlI3 doping (Fig. 3(a and c)). Those increased values meant that the movement of Li+ was enhanced in the vicinity of I−, Al3+, and the vacancies; thus, the ionic conductivity was improved. Hence, the electronic structure of Li2S was altered from being an insulator to a semiconductor with the appearance of new UV-Vis absorption bands and voltage dependence of polarized electronic current.
Fig. 5(a) shows the charge–discharge curves at the 1st, 10th, and 50th cycles of the cell using samples x = 0 and x = 0.5 as active materials. The curves illustrated that the overpotential of the cell was drastically reduced with 0.5 mol% AlI3 doping. Furthermore, it was also pointed out that the overpotentials of both cells were decreasing during the first 10 cycles. Fig. 5(b) plots the cyclic properties of all-solid-state cells employing electrode composites with x = 0, 0.5, 1.5, and 2. The capacity and stability of cells with x = 0.5 and x = 1.5 were better than those of x = 0 and x = 2. The initial coulombic efficiency of x = 0.5 and x = 1.5 were 91.0% and 82.3% but those of x = 0 and x = 2 were 79.1% and 76.8%, respectively. The highest capacities of the cells x = 0, 0.5, 1.5, and 2 were 880, 1059, 1006, and 869 mA h g−1 Li2S, respectively. The capacity retention of the cells x = 0, 0.5, 1.5, and 2 after 60 cycles was 64.0%, 88.4%, 69.6% and 71.2%, respectively. Thus, doping AlI3 into Li2S could improve not only the initial capacity and the initial coulombic efficiency but also the capacity retention. The Li2S cathode to which a trace amount of AlI3 is added (i.e. 99.5Li2S·0.5AlI3) demonstrated the best cell performance among the prepared cathode materials even though the ionic and electronic conductivity of Li2S–AlI3 increased with increasing AlI3 content. The fact indicates no correlation between the electric conductivity and the cell performance in the Li2S–AlI3 system. The enhanced cell performance of 99.5Li2S·0.5AlI3 cannot be described only by the improved electrochemical properties. The reaction kinetics in Li–S batteries depends on the surface reaction of Li2S or S particles. Augustyn et al. revealed that LiI coating on Li2S particles lowers the barrier for grain boundary diffusion between the Li2S and Li6PS5Cl based on NMR analysis.33 AlI3 doping could enhance the grain boundary diffusion between the electrode and the solid electrolyte because of high polarization of I-ions. The experimental results suggest the importance of interface design to establish facile Li-ion transport in ASS Li–S batteries. The capacities of all cells increased during the first 10 cycles, which has been well reported so far but the reason is not yet evident.6,18,34–36 One reason for the increase of capacity during the starting cycles may be the decomposition reactions of the solid electrolyte.37 It is believed that the solid electrolyte in the cathode composite is activated by the high-energy ball milling employed to prepare the positive electrode composite in our study. The composite 80(95Li2S–5AlI3)–20KB (weight ratio) itself could function as a positive electrode composite with a stable charge–discharge cyclic performance over 100 cycles, as shown in Fig. S5 (ESI†). Despite its stability, the low capacity illustrated that only a part of the 95Li2S·5AlI3 participated in the redox reaction during the charge–discharge process. Previous efforts reported that altering the lower cut-off potential influences the capacity retention. Four different cells, which employed the 50(97Li2S–3AlI3)–40Li10P3S12I–10KB composite as the positive electrode, were prepared and cycled with different charge–discharge processes. The results are illustrated in Fig. S6 (ESI†). Among the cycled cells, cell 3 with CC (Constant Current) charge and CV (Constant Voltage) (0.9 V vs. Li–In or 1.52 V vs. Li+/Li) exhibited the most stability. Other cells with a lower cut-off voltage had a faster discharge capacity degradation than cell 3. In fact, cells 1, 2, and 4 were cycled with lower discharge cut-off voltages than that of cell 3 and they exhibited similar cyclic properties, which were different from those of cell 3. Those results proved that an irreversible reaction occurred inside the positive electrode at voltages lower than 0.9 V vs. Li–In or 1.52 V vs. Li+/Li. A comparison of battery performance including cell design and operating conditions is listed in Table 1. Our positive electrode composite is more attractive than the other Li2S-based positive electrode composites reported in the recent literature in terms of a high specific capacity per gram of positive electrode composite, as well as areal Li2S loading and cycling stability.
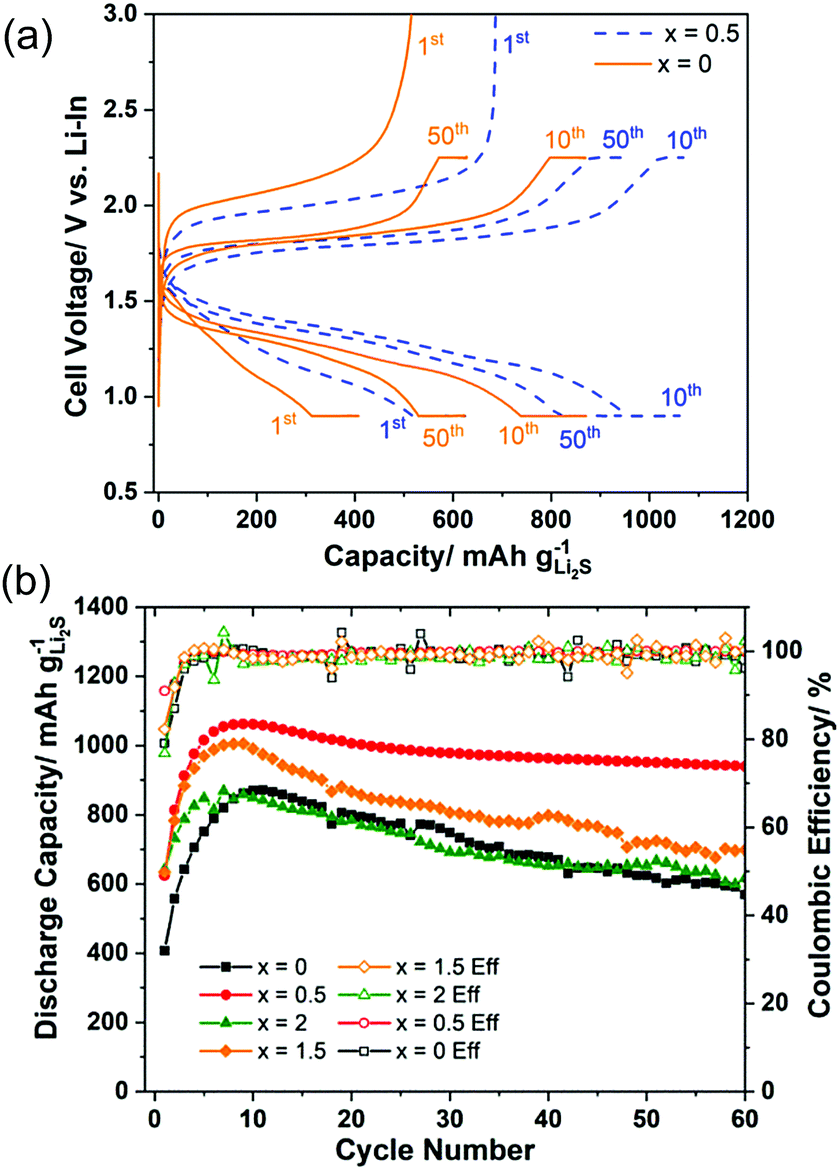 | ||
| Fig. 5 Charge–discharge curves of all-solid-state cells employing electrode composites with x = 0 and 0.5 (a); cyclic properties of all-solid-state cells employing electrode composites with x = 0, 0.5, 1.5, and 2 (b). | ||
| Active material | Content of active materials (%) | Loading [mg cm−2] | Cutoff voltage [V vs. Li+/Li] | Current density [mA cm−2] | Cycle number | Specific capacity [mAh g−1 of active materials] | Specific capacity [mAh g−1 of composite cathode] | Operating temperature [°C] | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Li2S | 30.6 | 0.2–0.5 | 1.5–2.8 | 0.04 | 30 | 720 | 220 | 60 | 14 |
| Li2S | 28.9 | 0.9–1.1 | 1.2–3.6 | 0.58 | 60 | 1100 | 318 | 25 | 16 |
| Li2S | 38.0 | 3.5 | 1.5–3.0 | 0.2 | 10 | 1047 | 398 | RT | 34 |
| Li2S | 50.0 | 3.84 | 1.49–3.5 | 0.2 | 30 | 674 | 337 | 60 | 35 |
| Li2S | 35.8 | 3.6 | 0.9–3.6 | 0.13 | 60 | 830 | 297 | RT | 38 |
| Li2S | 30.0 | 2.24 | 0.6–3.6 | 0.044 | 50 | 525 | 161 | RT | 39 |
| Li2S | 48.0 | 2.45 | 1.52–2.87 | 0.285 | 60 | 936 | 449 | 30 | This work |
4. Conclusions
In summary, this work demonstrated (100 − x)Li2S–xAlI3 (0 ≤ x ≤ 5) solid solutions for ASS Li–S batteries. The solid solutions were prepared by the planetary ball-milling method. It was proved by both XRD and UV-Vis spectra that AlI3 was dissolved in the Li2S matrix during the preparation process. XPS results pointed out that the electronic structure of Li2S was changed due to the solid solution formation. This change led to the formation of new UV-Vis absorption bands, increasing both ionic and electronic conductivities. Better initial capacity, coulombic efficiency, and capacity retention were also obtained with the cells using Li2S–AlI3 solid solution compared with the one employing bare Li2S. This work provided a facile strategy to improve the electrochemical performance of materials for all-solid-state Li–S batteries.Author contributions
H. Gamo: XRD analysis, data curation, visualization, conception, writing – review & editing; T. Maeda: UV-Vis measurement, XRD measurement, synthesis, electrochemical measurement, data curation; K. Hikima: design of the experiment, project administration, writing – review & editing; M. Deguchi: XPS measurements, Y. Fujita: XPS measurements; Y. Kawasaki: XPS measurements; A. Sakuda: XPS analysis, resources, writing – review & editing; H. Muto: funding acquisition, resources; N. H. H. Phuc: XRD measurements, electrochemical measurements, data curation, visualization, designed the study and experiment, conception, writing – original draft; A. Hayashi: resources, writing – review & editing; M. Tatsumisago: discussion and resources; A. Matsuda: supervision, funding acquisition, resources, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Advanced Low Carbon Technology Specially Promoted Research for Innovative Next Generation Batteries program of the Japan Science and Technology Agency (JST-ALCA-SPRING, Grant no. JPMJAL1301). The authors would like to thank Enago (http://www.enago.jp) for the English language review.References
- N. H. H. Phuc, K. Hikima, H. Muto and A. Matsuda, Crit. Rev. Solid State Mater. Sci., 2021, 1–26 CrossRef.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- B. Ding, J. Wang, Z. Fan, S. Chen, Q. Lin, X. Lu, H. Dou, A. Kumar Nanjundan, G. Yushin, X. Zhang and Y. Yamauchi, Mater. Today, 2020, 40, 114–131 CrossRef CAS.
- T. Ando, Y. Sato, T. Matsuyama, A. Sakuda, M. Tatsumisago and A. Hayashi, J. Ceram. Soc. Jpn., 2020, 128, 233–237 CrossRef CAS.
- L. Cai, H. Wan, Q. Zhang, J. P. Mwizerwa, X. Xu and X. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 33810–33816 CrossRef CAS PubMed.
- N. H. H. Phuc, T. Maeda, H. Muto and A. Matsuda, Front. Energy Res., 2021, 8 Search PubMed.
- S. Wang, M. Tang, Q. Zhang, B. Li, S. Ohno, F. Walther, R. Pan, X. Xu, C. Xin, W. Zhang, L. Li, Y. Shen, F. H. Richter, J. Janek and C. W. Nan, Adv. Energy Mater., 2021, 11, 31–40 Search PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- J. Shi, G. Liu, W. Weng, L. Cai, Q. Zhang, J. Wu, X. Xu and X. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 14079–14086 CrossRef CAS PubMed.
- D. Cao, Y. Zhao, X. Sun, A. Natan, Y. Wang, P. Xiang, W. Wang and H. Zhu, ACS Energy Lett., 2020, 5, 3468–3489 CrossRef CAS.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- X. Xu, J. Cheng, Y. Li, X. Nie, L. Dai and L. Ci, J. Solid State Electrochem., 2019, 23, 3145–3151 CrossRef CAS.
- Z. Lin, Z. Liu, N. J. Dudney and C. Liang, ACS Nano, 2013, 7, 2829–2833 CrossRef CAS PubMed.
- Z. Jiao, L. Chen, J. Si, C. Xu, Y. Jiang, Y. Zhu, Y. Yang and B. Zhao, J. Power Sources, 2017, 353, 167–175 CrossRef CAS.
- T. Hakari, A. Hayashi and M. Tatsumisago, Adv. Sustainable Syst., 2017, 1, 1700017 CrossRef.
- N. H. H. Phuc, K. Hikima, H. Muto and A. Matsuda, Solid State Ionics, 2020, 351, 115324–115327 CrossRef CAS.
- N. H. H. Phuc, T. Maeda, K. Hikima, H. Muto and A. Matsuda, SN Appl. Sci., 2020, 2, 1–9 Search PubMed.
- S. Spannenberger, V. Miß, E. Klotz, J. Kettner, M. Cronau, A. Ramanayagam, F. di Capua, M. Elsayed, R. Krause-Rehberg, M. Vogel and B. Roling, Solid State Ionics, 2019, 341, 115040–115046 CrossRef CAS.
- X. Feng, P.-H. Chien, S. Patel, J. Zheng, M. Immediato-Scuotto, Y. Xin, I. Hung, Z. Gan and Y.-Y. Hu, Energy Storage Mater., 2019, 22, 397–401 CrossRef.
- S. Yubuchi, M. Uematsu, M. Deguchi, A. Hayashi and M. Tatsumisago, ACS Appl. Energy Mater., 2018, 1, 3622–3629 CrossRef CAS.
- N. H. H. Phuc, T. Maeda, T. Yamamoto, H. Muto and A. Matsuda, Electron. Mater., 2021, 2, 39–48 CrossRef.
- A. Hayashi, T. Fukuda, S. Hama, H. Yamashita, H. Morimoto, T. Minami and M. Tatsumisago, J. Ceram. Soc. Jpn., 2004, 112, 695–699 Search PubMed.
- Y. Kawasaki, H. Tsukasaki, T. Ayama, S. Mori, M. Deguchi, M. Tatsumisago, A. Sakuda and A. Hayashi, ACS Appl. Energy Mater., 2020, 4, 20–24 CrossRef.
- D. G. Dominique Foix, G. Taillades, A. Pradel and M. Ribes, Solid State Sci., 2001, 3, 235–243 CrossRef.
- M. Yu, Z. Wang, Y. Wang, Y. Dong and J. Qiu, Adv. Energy Mater., 2017, 7, 1700018 CrossRef.
- S. Kim, S. K. Kim, P. Sun, N. Oh and P. V. Braun, Nano Lett., 2017, 17, 6893–6899 CrossRef CAS PubMed.
- Y. Lin, Z. Wen, J. Liu, D. Wu, P. Zhang and J. Zhao, J. Energy Chem., 2021, 55, 129–135 CrossRef.
- Z. W. Seh, H. Wang, N. Liu, G. Zheng, W. Li, H. Yao and Y. Cui, Chem. Sci., 2014, 5, 1396–1400 RSC.
- L. Yu, F.-C. Liu and Z.-W. Fu, Electrochim. Acta, 2009, 54, 2818–2822 CrossRef CAS.
- S. Lorger, R. E. Usiskin and J. Maier, Adv. Funct. Mater., 2018, 29, 1807688 CrossRef.
- A. Hayashi, T. Fukuda, S. Hama, H. Yamashita, H. Morimoto, T. Minami and M. Tatsumisago, J. Ceram. Soc. Jpn., 2004, 5, S695–S699 Search PubMed.
- M. Liu, C. Wang, C. Zhao, E. van der Maas, K. Lin, V. A. Arszelewska, B. Li, S. Ganapathy and M. Wagemaker, Nat. Commun., 2021, 12, 5943–5952 CrossRef CAS PubMed.
- H. Yan, H. Wang, D. Wang, X. Li, Z. Gong and Y. Yang, Nano Lett., 2019, 19, 3280–3287 CrossRef CAS.
- H. Jiang, Y. Han, H. Wang, Y. Zhu, Q. Guo, H. Jiang, C. Zheng and K. Xie, Energy Technol., 2020, 8, 2000023 CrossRef CAS.
- H. Jiang, Y. Han, H. Wang, Y. Zhu, Q. Guo, H. Jiang, C. Zheng and K. Xie, Ionics, 2020, 26, 4257–4265 CrossRef CAS.
- S. Ohno, R. Koerver, G. Dewald, C. Rosenbach, P. Titscher, D. Stechermeier, A. Kwade, J. Janek and W. G. Zeier, Chem. Mater., 2019, 31, 2930–2940 CrossRef CAS.
- J. Liang, Q. Sun, Y. Sun, C. Wang, W. Li, M. Li, D. Wang, X. Li, Y. Liu, K. Adair, R. Li, L. Zhang, R. Yang, S. Lu, H. Huang and X. Sun, J. Mater. Chem. A, 2018, 6, 23712–23719 RSC.
- M. Eom, S. Son, C. Park, S. Noh, W. T. Nichols and D. Shin, Electrochim. Acta, 2017, 230, 279–284 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01228b |
| This journal is © The Royal Society of Chemistry 2022 |