
Regulating the *OCCHO intermediate pathway towards highly selective photocatalytic CO2 reduction to CH3CHO over locally crystallized carbon nitride†
Qiong
Liu‡
a,
Hui
Cheng‡
 a,
Tianxiang
Chen
b,
Tsz Woon Benedict
Lo
b,
Zhangmin
Xiang
a and
Fuxian
Wang
*a
a,
Tianxiang
Chen
b,
Tsz Woon Benedict
Lo
b,
Zhangmin
Xiang
a and
Fuxian
Wang
*a
aInstitute of Analysis, Guangdong Academy of Sciences (China National Analytical Center, Guangzhou), Guangzhou, Guangdong 510070, China. E-mail: wangfuxian@fenxi.com.cn
bState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong, China
First published on 16th November 2021
Abstract
Photocatalytic conversion of CO2 to CH3CHO is of increasing interest but confronts the significant challenges of forming C–C bonds and keeping the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond intact throughout the process. Here, we report the selective photocatalytic hydrogenation of CO2 to CH3CHO using a modified polymeric carbon nitride (PCN) under mild conditions. The locally crystallized PCN offers a photocatalytic activity of 1814.7 μmol h−1 g−1 with a high selectivity of 98.3% for CH3CHO production and a quantum efficiency of 22.4% at 385 nm, outperforming all the state-of-art CO2 photocatalysts. The promoted formation of the *OCCHO intermediate on the locally crystallized PCN is disclosed as the key factor leading to the highly selective CH3CHO generation. The locally crystallized PCN favors spontaneous C–C coupling towards *OCCHO formation rather than *CHO protonation, thus preventing HCHO formation. This work provides a new strategy for designing carbon nitrides for highly selective and sustainable conversion of CO2 to CH3CHO.
O bond intact throughout the process. Here, we report the selective photocatalytic hydrogenation of CO2 to CH3CHO using a modified polymeric carbon nitride (PCN) under mild conditions. The locally crystallized PCN offers a photocatalytic activity of 1814.7 μmol h−1 g−1 with a high selectivity of 98.3% for CH3CHO production and a quantum efficiency of 22.4% at 385 nm, outperforming all the state-of-art CO2 photocatalysts. The promoted formation of the *OCCHO intermediate on the locally crystallized PCN is disclosed as the key factor leading to the highly selective CH3CHO generation. The locally crystallized PCN favors spontaneous C–C coupling towards *OCCHO formation rather than *CHO protonation, thus preventing HCHO formation. This work provides a new strategy for designing carbon nitrides for highly selective and sustainable conversion of CO2 to CH3CHO.
Broader contextAcetaldehyde is an indispensable chemical in the synthesis of pharmaceuticals, agrochemicals and fragrances. Photocatalytic conversion of CO2 to acetaldehyde has emerged as a promising method for sustainable acetaldehyde production. However, the current carbon-based products of photocatalytic CO2 conversion are mostly dominated by CO, HCOOH and CH4. Specifically, a ten-electron transfer process and multiple hydrogenation steps are involved in the conversion of CO2 to acetaldehyde, resulting in unexpected products (e.g., CO, HCHO, HCOOH) and limited conversion efficiency. This calls for a systematic mechanism investigation on the whole reaction at the atomic level and rational design of highly selective photocatalysts. Herein, we design a high-performance polymeric carbon nitride (PCN) catalyst with a locally crystallized structure through an amino-2-propanol (AP)-assisted hydrothermal pretreatment. The energy barrier from *CHO to the *OCCHO intermediate is significantly reduced on the locally crystallized PCN, whereas the hydrogenation of *CHO to *CH2O is efficiently suppressed. The formation of the *OCCHO intermediate is beneficial to the subsequent proton-coupled electron transfer process to form CH3CHO rather than HCHO. Eventually, a high CH3CHO generation rate and high selectivity are achieved on the elaborately crafted locally crystallized PCN. |
Introduction
Acetaldehyde, as an indispensable intermediate in the manufacture of high-value-added chemicals, is widely applied in the fields of pharmaceuticals, agrochemicals and fragrances.1 Ethylene oxidation catalyzed by palladium(II) and copper chloride in strong acidic solutions via the Wacker process is a common method for acetaldehyde production; however, it involves intensive energy consumption and acid contamination, and it is restricted by the petroleum-based ethylene resource.2,3 Partial oxidation of ethanol at high temperature via an endothermic reaction or partial dehydrogenation of ethanol is an alternative route.4,5 However, this method possesses the drawback of massive consumption of agriculture crops. Thus, there is substantial interest in developing sustainable and atomic economic approaches for acetaldehyde production.Using solar energy radiation instead of thermal activation to photo-reduce CO2 to C2 hydrocarbon-based products under mild operating conditions appears to be a promising strategy for the sustainable production of acetaldehyde, accompanied by the alleviation of ever-increasing CO2 emission.6,7 The current carbon-based products of photocatalytic CO2 conversion are dominated by CO8–10 and HCOOH11via a two-electron reduction reaction. Other typical C1 products include HCHO,12,13 CH4,14–16 and CH3OH,17–19 whereas liquid multi-carbon (C2+) compounds,6,20–22 specifically CH3CHO as a product, have been rarely reported.23,24 Recently, several works have reported the photocatalytic conversion of CO2 to CH3CHO by using transition-metal semiconductors with a d0 or d10 electronic configuration, including TiO2,25–28 lnTaO429 or SnS2.30 The ternary metal materials of ZnxCd1−xS31 and ZnFe2O432 and copper-containing species24,33 have also been demonstrated to yield CH3CHO. However, they still suffer from low productivity (10–500 μmol h−1 g−1) and uncontrollable selectivity.
The remaining great challenge for photocatalytic reduction of CO2 to CH3CHO is the trade-off between catalytic activity and selectivity.34 This is because the complicated ten-electron transfer process involves multiple hydrogenation steps from CO2 to CH3CHO, during which various competitive unexpected products (e.g., CO, HCHO, HCOOH) can be generated through different pathways.7,35,36 Moreover, currently, the mechanism for this complicated reaction is yet to be disclosed; to date, only the CO2 adsorption and the initiate CO2 activation process have been investigated.30 This calls for a systematic mechanism investigation on the whole reaction at an atomic level and rational design of highly selective photocatalysts.
Here, we elaborately craft a polymeric carbon nitride (PCN) through an amino-2-propanol (AP)-assisted hydrothermal pretreatment. The modified PCN presents a unique bubble-like structure with amorphous domains and well-crystallized lattice matrices that appear alternately in the framework. It is found that the energy barrier from *CHO to the *OCCHO intermediate is significantly reduced on the locally crystallized PCN, whereas the hydrogenation of *CHO to *CH2O is suppressed. Thus, the formation of the *OCCHO intermediate is beneficial to the subsequent proton-coupled electron transfer process to form CH3CHO rather than HCHO. Moreover, the optimized PCN can prevent the excessive protonation of CH3CHO to form CH3CH2OH, eventually affording highly selective CH3CHO generation. Furthermore, isotopic tests prove that the components of carbon and oxygen in CH3CHO originate from CO2, and hydrogen in CH3CHO is extracted from H2O. For the first time, the reaction mechanism for the whole reaction of CO2 reduction to CH3CHO is systematically investigated and our strategy for fabricating dyadic PCN could be easily applied for the rational design of conjugated polymer semiconductors for a variety of energy conversion reactions.
Results and discussion
Usually, dicyandiamide (DCDA) can form a supramolecular precursor for polymeric carbon nitride (PCN) under the hydrothermal pretreatment strategy (Fig. 1a).37 Previously, the addition of amino-2-propanol (AP) was found to improve the polymerization degree of the monomers and polymers for the transformation of oleochemicals.38 Here, we attempted to improve the crystallinity and photocatalytic performance of PCN by introducing AP into the precursor during the hydrothermal pretreatment. The precursor with different amounts of AP addition was calcined at 550 °C in air to fabricate carbon the nitride products, denoted as HCN-Ax. The photocatalytic tests (vide infra) show that HCN-A3 had the best performance; therefore, we used HCN-A to denote HCN-A3 for the following description, unless otherwise specified. For comparison, CN and HCN were synthesized from the direct calcination of DCDA with and without hydrothermal pretreatment, respectively. The SEM image (Fig. 1b) reflects that the bubble-shaped structure of HCN-A is obviously different from that of the bulk particles of CN and shows the two-dimensional layered framework of HCN (ESI,† Fig. S1). The high-magnification SEM image (Fig. 1c) and transmission electron microscopy (TEM) image (Fig. 1d) manifest the formation of a hollow framework in the bubble-shaped HCN-A. The entity size was enlarged as the AP content increased (Fig. S2 and S3, ESI†). The selected area electron diffraction (SAED) image (inset in Fig. 1d) of HCN-A presents a bright diffraction ring indexed to the (002) plane, verifying a certain degree of highly crystalline configuration (Fig. S4, ESI†). In contrast, the HCN shows an amorphous structure (Fig. S5, ESI†), confirming that the crystallization in HCN-A is caused by the addition of AP. A series of characterizations were conducted (as shown in Fig. S6–S12, ESI†) to reveal the role of AP in the morphology and structure of the HCN-A. Briefly, the AP-modified precursor presents a higher thermal decomposition temperature, implying stronger thermal stability. The NMR peaks of HCN-A shift towards a high field when AP is added, suggesting that increased electron cloud density surrounds the carbon atom near the H-bonds and thus improves the degree of crystallinity.39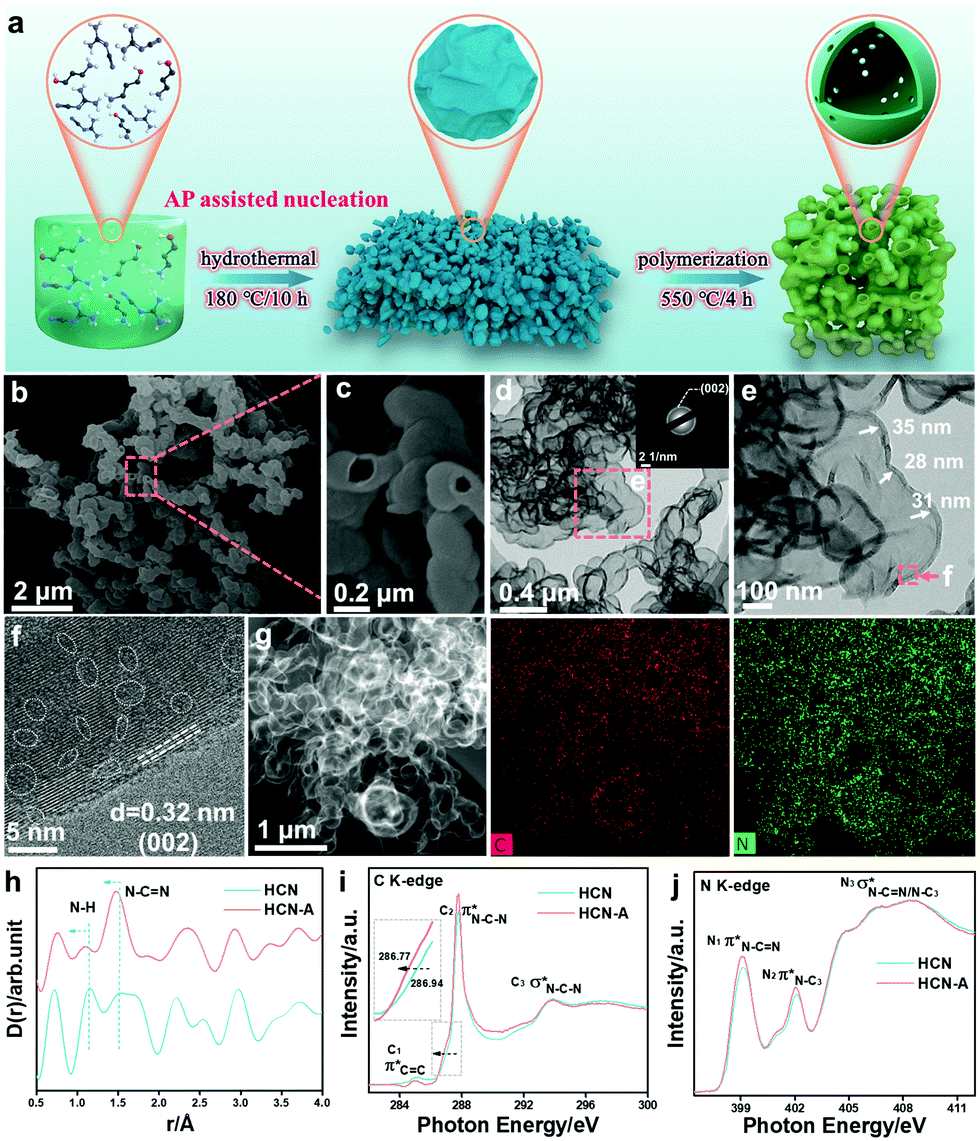 | ||
| Fig. 1 (a) Schematic of the synthesis of the HCN-A photocatalysts. (b and c) SEM images of HCN-A. (d and e) TEM images; inset is the corresponding selected area electron diffraction (SAED) pattern. (f) HR-TEM image of FCN-V. (g) High-angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) images and C, N element mappings; (h) high-resolution X-ray total scattering spectra with the PDFgui refinement, with a comparison of the differential distance within the structures of HCN and HCN-A via the differential correlation function of D(r). (i) C K-edge and (j) N K-edge XANES spectra of HCN and HCN-A. | ||
The hollow bubble-shaped HCN-A (Fig. 1e) presents a well-defined wall with an average thickness of 32 nm. The HCN-A exhibits a clear lattice fringe of 0.32 nm that corresponds to the (002) plane of carbon nitride (Fig. 1f and Fig. S13, ESI†), further demonstrating the well-crystallized lattice matrices.40 Interestingly, amorphous domains are observed between the well-crystallized lattice matrices (marked by white dotted lines). These results reveal the formation of an interface dyadic homo-junction that features alternately appearing amorphous domains and well-crystallized lattice matrices, which may rearrange the reaction pathway and the product selectivity.41,42 High-angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) further discloses the hollow bubble structures (Fig. 1g), and the corresponding energy dispersive X-ray spectroscopy (EDS) mapping images (Fig. 1g) reveal the uniform distributions of C and N elements in HCN-A. The specific surface area of HCN-A is determined to be 81.6 m2 g−1 and the pore volume is 0.28 m3 g−1, which is slightly smaller than that of HCN (Fig. S14 and Table S1, ESI†).
As shown in Fig. S15 and S16 (ESI†), the (002) peak of HCN-A shifts towards a lower angle compared with that of HCN, suggesting enhanced distance of the interplanar space.43 The FTIR spectra prove that the HCN-A maintains the backbone unit of the heptazine structure of CN (Fig. S17, ESI†). The chemical environment variation was further investigated by high-resolution X-ray total scattering spectra with the PDFgui refinement (Fig. 1i and Fig. S18, ESI†).
The partial double bond length of CN–C is reduced from 1.52 Å (HCN) to 1.47 Å (HCN-A). The N–H bond length is 1.11 Å for HCN-A, which is shorter than that of HCN (1.15 Å). The reduced bond lengths of both C–N and N–H manifest the promoted crystallinity.44 The detailed N1s XPS spectrum is shown in Fig. S19a (ESI†), from which the C–NC, N–(C)3 and C–N–H groups are found in HCN-A (Tables S2–S4, ESI†). The solid 13C NMR spectra (Fig. S19b, ESI†) reveal the presence of C–(N)3 and NH2–C(N)2 in the tri-s-triazine motifs. Moreover, the C K-edge synchrotron-based X-ray absorption near-edge structure spectroscopy (XANES) results (Fig. 1i) demonstrate two characteristic dipole transition 2p π* resonance signals of CC bonds at the defect sites, and signals of C–N–C bonds at 284.9 and 287.8 eV are observed for HCN and HCN-A. In the N K-edge region (Fig. 1j), two 2p π*characteristic resonances are observed at 399.1/399.2 eV and 402.0/402.1 eV in HCN-A and HCN, respectively, which correspond to the N–CN (N1) coordination structure in a heptazine unit and graphitic-type N–(C)3 (N2) bridging among the three heptazine units. Notably, a slight negative shift (about 0.17 eV) in the absorption edge is observed in HCN-A (Fig. 1i), suggesting the existence of a strong interaction of C–N–C and providing evidence of the elevated crystalline structure.45 The intensity of both the N1 and N2 peaks in HCN-A is higher than that of the peaks in HCN, further signifying the well-ordered structure.46
Collectively, based on the abovementioned results, we demonstrate that a unique hollow bubble-shape carbon nitride catalyst with alternately appeared amorphous domains and well-crystallized lattice matrices was successfully prepared. To unveil the electronic structure, the densities of states (DOS) by virtue of first principles calculations based on density functional theory (DFT) were determined (Fig. S20 and S21, ESI†). For HCN, the CB is mainly composed of the C 2p and N 2p orbitals, while the N 2p orbitals contribute to the VB; this is in agreement with the literature.46 The calculated band gap of HCN-A is smaller than that of HCN. An additional midgap state is presented in HCN-A, which could facilitate carrier separation and lead to charge enrichment on the active sites of pyridinic N of the tri-s-triazine structure units.14 In addition, the optical absorption properties and band alignment positions are demonstrated in Fig. S22–S27 (ESI†), signifying that the conduction band (CB) of HCN-A is negative enough for efficient CH3CHO generation.23 The charge dynamics were investigated by femtosecond transient absorption measurements (fs-TA, Fig. S28 and S29, ESI†). The longer relaxation time in HCN-A indicates more efficient charge separation and a higher concentration of charge carriers to participate in the catalytic reaction.47
The photocatalytic CO2 conversion performance of CN, HCN and HCN-A was investigated (for experimental details, see Fig. S30–S33, ESI†). As shown in Fig. 2a, HCN-A presents a CH3CHO generation rate of 1083.5 μmol h−1 g−1 for the first 2 h, which was 45.6 fold higher than that of CN (22.3 μmol h−1 g−1) and 7.4 fold higher than that of HCN (145.3 μmol h−1 g−1) (the gas chromatograph (GC) signals for the products are shown in Fig. S34, ESI†). The inset image in Fig. 2a presents that the HCN exhibits a HCHO evolution rate of 125.5 μmol h−1 g−1. In contrast, HCN-A exhibits a much lower HCHO production rate of 34.7 μmol h−1 g−1 (Fig. S35, ESI†). As shown in Fig. 2b and Table S5 (ESI†), the photocatalytic activity and selectivity of HCN-A could be regulated by varying the amount of AP added. Specifically, the selectivity for CH3CHO increases from 51.6% in HCN to 95.5% in HCN-A3 (1083.5 μmol h−1 g−1). As such, the HCN-A with locally crystallized domains achieves high selectivity for CH3CHO production, which can be ascribed to the AP-assisted nucleation during the hydrothermal treatment. Notably, the typical gas products for the CO2 reduction, such as CO and CH4, were not detected in this reaction system. The addition of recently routinely exploited co-catalysts, including the Fe, Co, Ni, Ru and Cu species, to HCN-A did not lead to any improvement in the photocatalytic activity or selectivity (Fig. S36, ESI†). Specifically, the addition of Co species in HCN-A results in a main product of gaseous CO (Fig. S37, ESI†). Control experiments in Fig. 2c revealed that the HCN-A catalyst, light source, CO2 gas, and H2O were indispensable for this reaction.
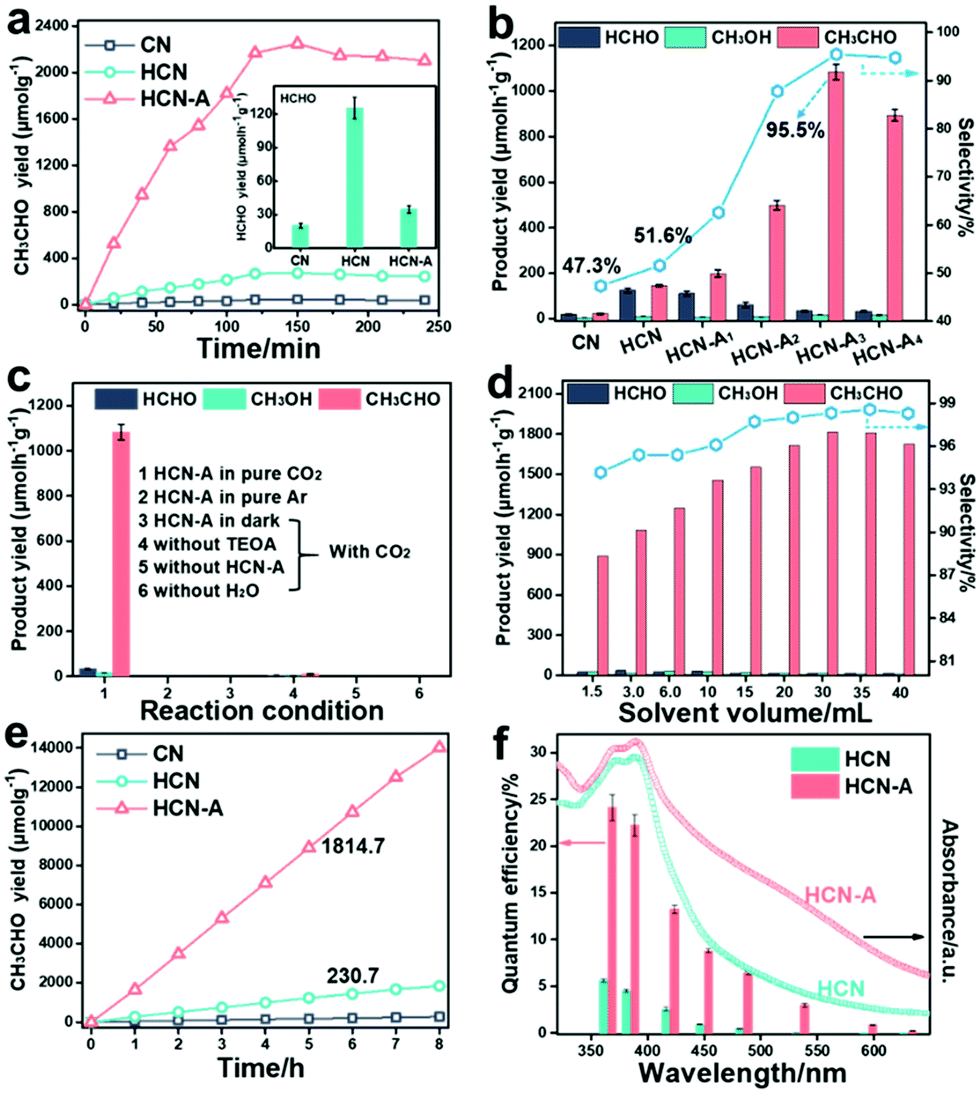 | ||
| Fig. 2 (a) Time dependence of the CH3CHO product evolution rates of CN, HCN, and HCN-A with 3 mL solvent; inset shows the HCHO generation rates over CN, HCN, and HCN-A. (b) The average generation rates of the CO2 reduction products and the selectivities of CH3CHO using CN, HCN, HCN-A1, HCN-A2, HCN-A3, and HCN-A4 with 3 mL solvent. (c) The average generation rates of CO2 reduction products under multiple experiments of control conditions by using HCN-A3 with 3 mL solvent. (d) The generation rates of CO2 reduction products upon altering the volume of the reaction solution in this system. (e) Time dependence of the CO2 reduction product evolution rates of CN, HCN, and HCN-A with 30 mL solvent. (f) Quantum efficiencies of HCN and HCN-A for the CH3CHO product (irradiated by LED light sources with different monochromatic wavelengths of 365 nm, 385 nm, 420 nm, 450 nm, 485 nm, 535 nm, 595 nm and 600 nm), together with the DRS spectra of HCN and HCN-A with 30 mL solvent. | ||
As shown in Fig. 2a, the product yield reaches a plateau after 2 h and stops increasing.29,48 To address this problem, we further investigated the effect of the reaction solution volume on the reaction activity and selectivity, as shown in Fig. 2d. Clearly, as the volume increased to 30 mL, the generation yield of CH3CHO on HCN-A further increased to 1814.7 μmol h−1 g−1 with a selectivity of 98.3%, compared to 230.7 μmol h−1 g−1 on HCN with a selectivity of 54.7%. The increase in the CH3CHO product yield is due to the dilution of the initially formed CH3CHO, the accumulation of which was found to inhibit the right-shifting of the reaction equilibrium (Fig. S38, ESI†). Further increasing the volume to 40 mL resulted in a slight decrease in the CH3CHO yield, which is due to the decreased mass concentration of the catalyst, as demonstrated in Fig. S39 (ESI†).
Furthermore, when the volume is 30 mL, the CH3CHO product yield progressively increases during the measurement (8 h), as shown in Fig. 2e. In this case, increasing the solvent volume is an efficient strategy for boosting both the selectivity and yield for CH3CHO production. Actually, we have screened a series of other semiconductor photocatalysts under identical reaction conditions for comparison (Fig. S40, ESI†). Most of the widely used catalysts were almost inactive for the conversion of CO2 to CH3CHO, and only trace amounts of CH3CHO were found in the cases of Znln2S4 and CdS.
Increasing the light intensity from 50 to 150 mW cm−2 led to enhancement of the product yield (Fig. S41, ESI†), demonstrating that the successful conversion of CO2 to CH3CHO is driven by the light source. Along with its outstanding activity and high selectivity, HCN-A exhibited prominent quantum efficiencies (QEs) of 22.4% and 13.3% at 385 nm and 420 nm, respectively (the calculation process is listed in Note S1, Tables S6 and S7, ESI†); these are clearly higher than those for HCN of 4.5% and 2.5%, respectively, as shown in Fig. 2f (the relationship between the catalyst dosage and performance activity is shown in Fig. S42, ESI†). The QE value of HCN-A decreases as the light wavelength increases, which coincides well with the trend in the diffuse reflection spectra. Compared to HCN, the CH3CHO generation by HCN-A could be extended to longer wavelengths (595–630 nm) than that by HCN (Table S7, ESI†). The QE of HCN-A was determined to be 0.86% at λ = 595 nm and 0.19% at λ = 630 nm, signifying improved light-responsive activity over a broadened visible light region. More importantly, the HCN-A catalyst showed high stability after 10 cycle tests, as shown in Fig. S43 (ESI†). No obvious change was found in the structure or morphology after the stability tests, as confirmed by the XRD patterns, FTIR spectra (Fig. S44, ESI†) and TEM images (Fig. S45, ESI†). HCN-A exhibits excellent photocatalytic activity and selectivity towards CH3CHO generation, outperforming the state-of-art photocatalysts, as summarized in Table S8 (ESI†).
To further confirm the CH3CHO formation, GC-MS and 13CO2 isotope labeling experiments were first conducted. The photographs of the employed devices for the measurements are shown in Fig. S46 and S47 (ESI†). The GC spectra confirm the dissolved CO2 molecule and the CH3CHO product (Fig. 3c) with either CO2 or 13CO2 as the feedstock. The dissolved CO2 molecule weight increased when CO2 was replaced with 13CO2 isotope, as shown in Fig. 3d. The molecular weight of the generated CH3CHO product was measured to be m/z = 43 (the molecular weight for the anion should minus 1), corresponding to the formation of CH3CHO (Fig. 3e).30,49 The 13CH3 and 13CHO peaks located at m/z = 46 and 47 evidently prove that the carbon source of CH3CHO is derived from the CO2 feedstock, which is in accord with previous results.50 We further employed deuterium oxide (D2O) and 18O-labeled CO2 in the reaction to trace the origin of the H and O elements in the produced CH3CHO. The product peak at m/z = 47 was indexed to CD3CDO (Fig. 3f) when using D2O in the reaction, indicating that the proton in CH3CHO comes from H2O. This result further manifests that the formation of CH3CHO is a multi-step hydrogenation process and that the proton is provided by H2O.51–53 Meanwhile, the molecule weight increased to m/z = 45 when C18O2 was used, suggesting that the oxygen atom in CH3CHO is derived from the CO2 feedstock. The applied TEOA was oxidized to N-ethyl-2-methoxy-N-(2-methoxyethyl)-ethanamine, as verified by GC-MS (Fig. S48 and S49, ESI†).40 All these results explicitly reveal that HCN-A photocatalysts can catalyze the stepwise conversion of CO2 into CH3CHO products via C–C coupling and a multiple proton-coupled electron transfer process. We note that this the first time that the origin of CH3CHO through photocatalytic CO2 conversion has been revealed with the assistance of isotope labeling experiments by using 13CO2, CO2, D2O and C18O2 as the feedstocks. Furthermore, the 1H NMR spectra of the product confirm the formation of CH3CHO (Fig. S50, ESI†). To the best of our knowledge, this is also the first time that a PCN-based catalyst was designed and applied for highly selective and efficient conversion of CO2 to CH3CHO.
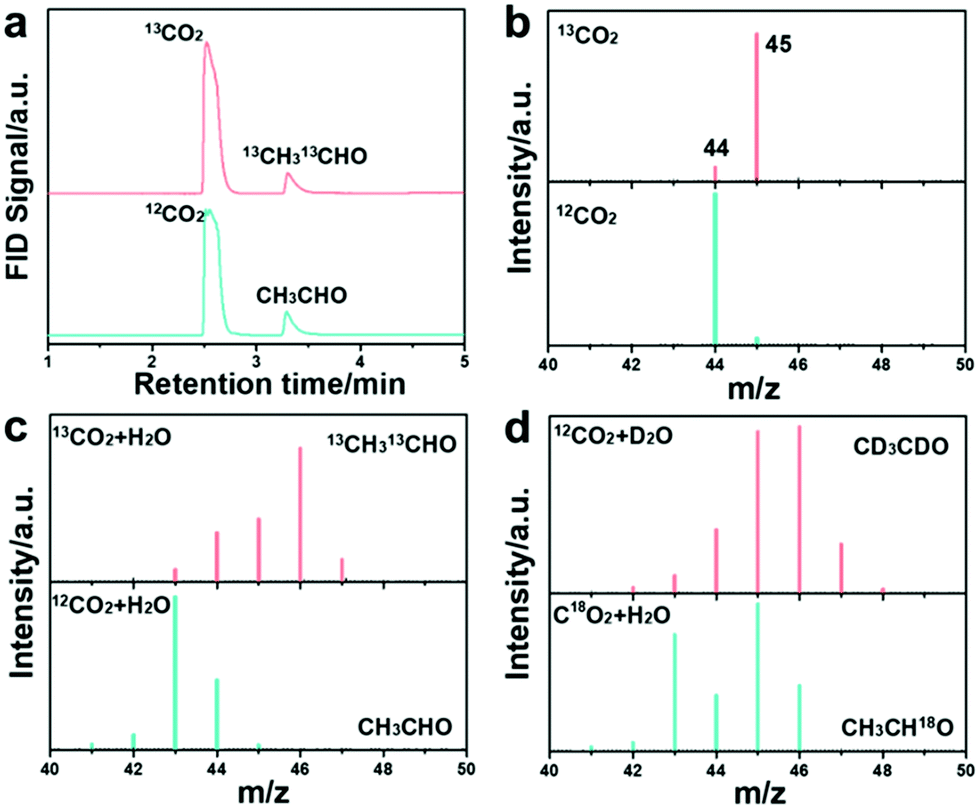 | ||
| Fig. 3 GC-MS spectra of CH3CHO generated over HCN-A in the photocatalytic CO2 conversion. (a) GC spectra of dissolved CO2 molecules and the CH3CHO product with CO2 and 13CO2. (b) MS spectra of CO2 and 13CO2. (c) MS spectra of CH3CHO with the use of CO2 + H2O, 13CO2 + H2O. (d) MS spectra of CH3CHO with the use of 13CO2 + D2O, C18O2 + H2O. | ||
To understand the underlying reasons for the enhanced photocatalytic performance, we investigated the fundamental steps of the CO2 conversion. The surface adsorption property of CO2 is a prerequisite for conversion of CO2 molecules. We therefore first measured the CO2 adsorption capacity of the samples. As shown in Fig. S51 (ESI†), both HCN-A and HCN present fundamentally higher CO2 adsorption than the pristine CN, which is due to the enhanced surface area. No significant change in the CO2 adsorption capacity was found between HCN-A and HCN; therefore, the advanced photocatalytic performance of HCN-A compared to HCN cannot be ascribed to the CO2 adsorption property.
It is worth noting that currently, the mechanism for the conversion of CO2 to CH3CHO has been seldom disclosed. To date, only the CO2 adsorption and the initiate CO2 activation process have been investigated.30 In this work, we aimed to disclose the whole reaction mechanism. The photocatalysis intermediate on HCN-A was verified by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS, Fig. 4a–c) and compared with HCN (Fig. 4d–f) under visible light illumination. The peaks attributed to the monodentate bicarbonates (1532, 1513 cm−1), bidentate bicarbonates (1420, 1375, 1320 cm−1) and hydroxyl groups of carboxylic acid (1154, 1465 cm−1) were recorded with increasing irradiation time (Fig. 4a).54 The characteristic peak of *CO at 2078 cm−1 obviously appears under illumination (Fig. 4b).54 The peak intensities of HCN-A are notably higher than those of HCN (Fig. 4d), signifying the accelerated CO2 transformation process to *CO in HCN-A. The asymmetric C–H stretching vibration (2942 and 2968 cm−1), symmetric C–H stretching vibration (2878 cm−1), and CH3 bending vibration (1380–1408 cm−1) were observed in the HCN-A reaction system, which can be assigned to the –CH3 group of CH3CHO.55 More importantly, the vibration of the *CHO group at 1097 cm−1 and the asymmetric stretching vibrations of C–C–O at 1038 cm−1 and for *OCCHO at 917 cm−1 were investigated. The *CHO formed from the hydrogenation of the *CO group is the prerequisite to generate the *OCCHO intermediate (eqn (1)), and the *OCCHO intermediate is generally regarded as the crucial intermediate for the conversion of CO2 to the multicarbon (C2+) products.14,53 The formation of *OCCHO through the C–C coupling of *CHO with *CO56 is depicted in eqn (2).
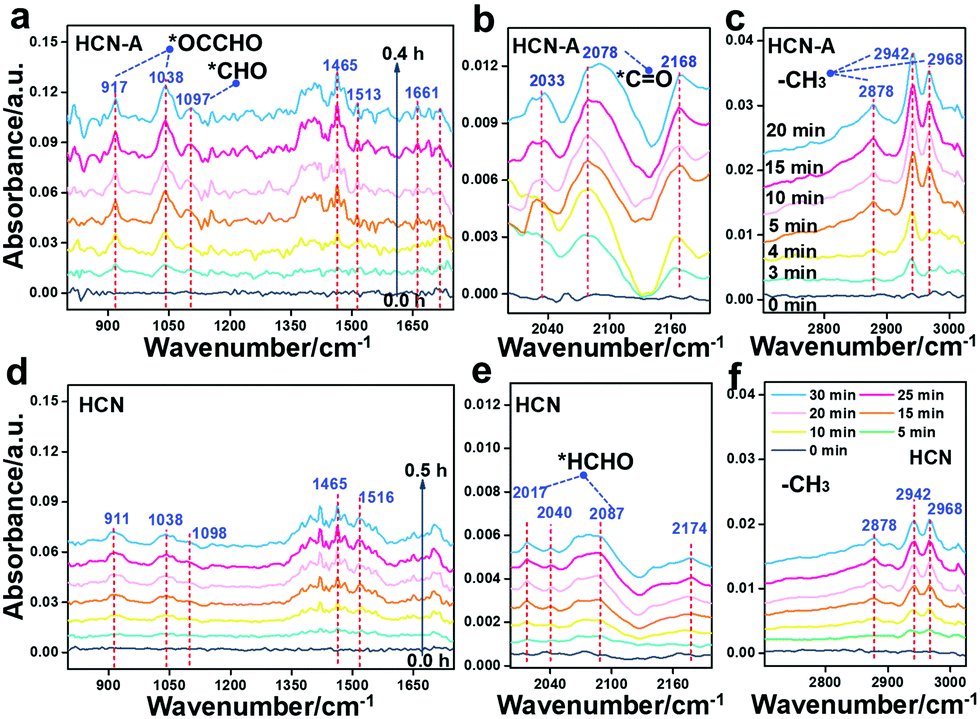 | ||
| Fig. 4 In situ diffuse reflectance infrared Fourier transform spectroscopy of HCN-A (a–c) and HCN (d–f). The IR spectrum was collected with interval times of 0 min, 3 min, 4 min, 5 min, 10 min, 15 min and 20 min for HCN-A and at 5 min intervals for HCN under illumination. | ||
The possible reduction paths are as follows (for details, see Experimental section 1.6 in the ESI†):
| *CO + H+ + e− → *CHO | (1) |
| *CHO + *CO + e− → *OCCHO | (2) |
| *CHO + H+ + e− → *CH2O → HCHO | (3) |
Additionally, the CO stretching vibration (1717, 1661, 1630 cm−1) and C–H out-of-plane bending vibration (1102 cm−1) are present in the spectrum of HCN-A, together verifying the formation of CH3CHO.57 These peaks were also observed when using HCN, but the intensity was obviously lower (Fig. 4d–f); this suggests that CH3CHO can also be generated by HCN, although the activity is substantially limited. This is in agreement with the photocatalytic performance observed experimentally. For HCN, two new peaks at 2017 cm−1 and 2087 cm−1 appear in addition to the *CHO band (1098 cm−1), which can be attributed to the stretching vibration of the surface-absorbed *CH2O58 (eqn (3)), as shown in Fig. 4e. This indicates that the HCHO intermediate can be easily generated on HCN, leading to HCHO formation.49,51–53 By contrast, this was not found in HCN-A; this may be due to the fact that *CHO in HCN-A is capable of rapid C–C coupling to form *OCCHO (eqn (2)) rather than being competitively protonated to form *CH2O (eqn (3)). The successful formation of *OCCHO is the crucial step that results in high selectivity for the CH3CHO product. These results conclusively demonstrate that the PCN-based samples with AP modification exhibit suppressed *CHO protonation while simultaneously boosting the C–C coupling and multiple proton-coupled electron transfer of CO2 to form the CH3CHO species. In situ EPR measurements were further applied to detect the radical active species during the CO2 reduction reaction (Fig. S52, ESI†). Under illumination, the EPR signal of the characteristic DMPO–CO2˙− is clearly present in the reaction system with HCN-A as the photocatalyst, indicating that the CO2 molecules can be activated to CO2˙− radicals. The CO2 to CO2˙− process is then followed by the generation of the important *CO intermediate, which is the prerequisite for the conversion of CO2 to the final CH3CHO product. After bubbling CO2 into the HCN-A catalyst suspension, the charge dynamics (fs-TA) were investigated (Fig. S53, ESI†). Notable decay of the trapped electrons in HCN-A was observed. This indicates that the reactant CO2 would preferentially react with the trapped electrons and then be activated to CO2˙− radical.
To further reveal the reaction mechanisms, Gibbs free energy calculations (Fig. S54, ESI†) based on density functional theory (DFT) were performed; as illustrated in Fig. 5a, the energy barriers for the *CHO intermediate formation are 0.28 eV and −0.46 eV on HCN and HCN-A, respectively, suggesting easier formation of *CHO on HCN-A. The Gibbs free energy of CHO* formation is obviously smaller than the desorption energy of the CO molecules (Fig. 5b) on HCN-A. This means that the formation of the CHO* intermediate is an exothermic and spontaneous process (eqn (1)), while the desorption of CO* is endothermic and has a large activation energy barrier on HCN-A. This signifies that HCN-A favors the hydrogenation of CO* to generate CHO* rather than the desorption of CO* molecules from the catalyst surface. Therefore, there is no CO evolution on HCN-A. Meanwhile, a likely intermediate of *OCCHO in the C–C coupling pathway via a favorable thermodynamic process could be spontaneously achieved on HCN-A, which serves as a crucial intermediate for the formation of C2 species56 (eqn (2)). For the C–C coupling on the HCN-A surface, the formation energy of *OCCHO when *CO species reacts with the carbon atom of *CHO is −0.18 eV. In contrast, the *CHO is difficult to protonate on HCN-A to form *CH2O (formaldehyde), which accounts for the high selectivity of the visible light-driven CO2 conversion to CH3CHO. Contrastingly, the formation of *OCCHO on HCN (Fig. 5a) still needs to overcome a certain energy barrier, whereas the Gibbs energy of hydrogenation of *CHO to form *CH2O is close to the energy barrier for the C–C coupling to form *OCCHO, leading to a high possibility of HCHO formation. As such, the HCN could simultaneously yield HCHO and CH3CHO with lower product selectivity, which is in accord with the experimental results (Fig. 2b).
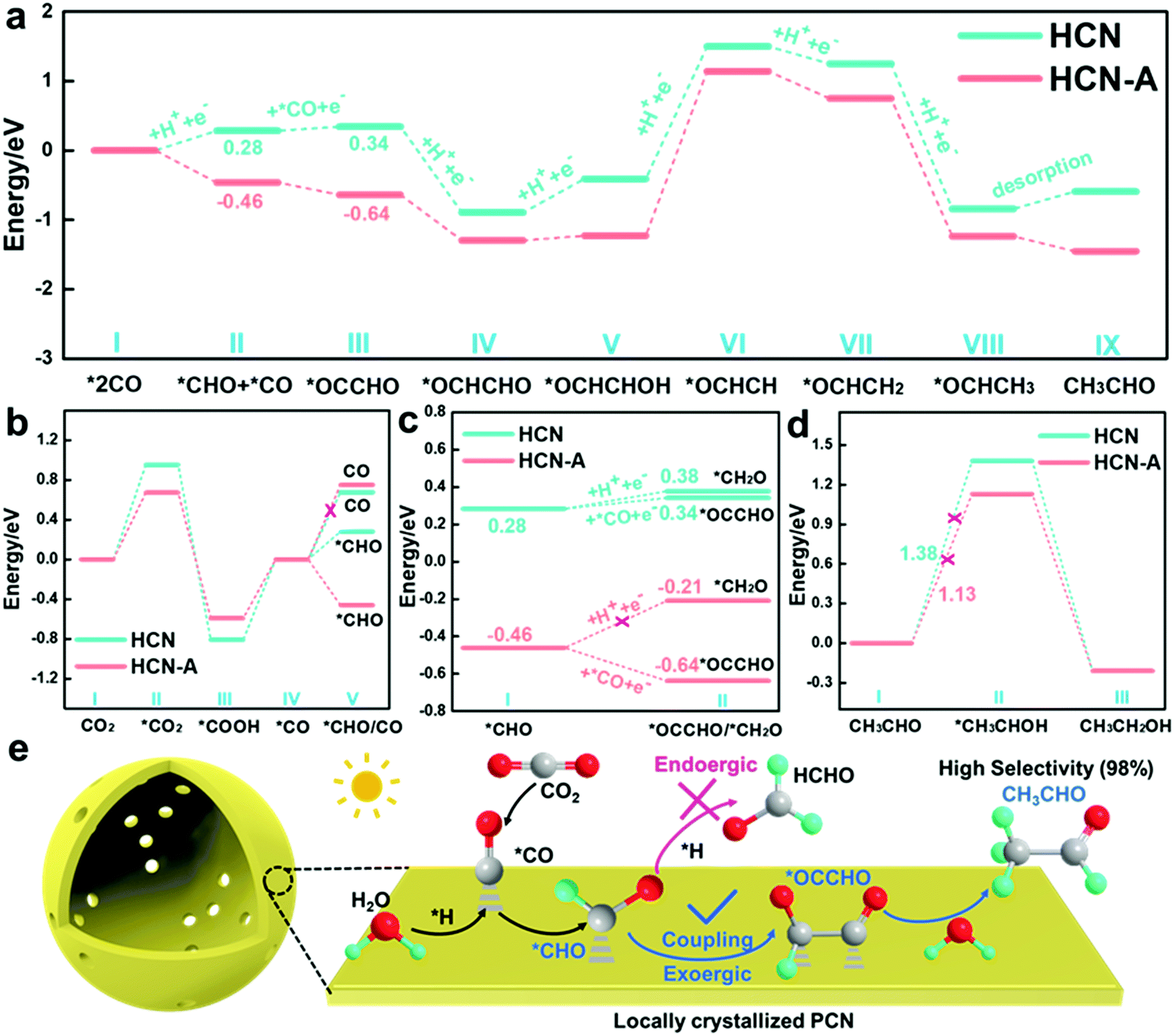 | ||
| Fig. 5 Calculated free energy diagram for the reduction of CO2 to CH3CHO on the HCN and HCN-A catalysts (a–d). (e) The proposed reaction mechanism for the photocatalytic CO2 reduction conversion to CH3CHO. The grey, red, and green color spheres denote carbon, oxygen, and hydrogen atoms, respectively. | ||
It is worth noting that the formed CH3CHO may also be further hydrogenated into CH3CH2OH via the formation of an *OCH2CH3 intermediate.49 The formation of *OCH2CH3 is an uphill process, with an energy of 1.13 V vs. RHE in HCN-A; this is in accordance with previously reported results of CO2 reduction.49,52 This endothermic nature of the process limits further hydrogenation of CH3CHO to CH3CH2OH (eqn (4) and (5)), which is supported by the experimental observation that no CH3CH2OH is released from the reaction system of HCN-A (Fig. 5d).
| CH3CHO + H+ + e− → *OCH2CH3 | (4) |
| *OCH2CH3 + H+ + e− → CH3CH2OH | (5) |
The primary product CH3CHO is eventually generated through the multi-hydrogenation-coupled electron transfer process with 98.3% product selectivity under visible-light irradiation on HCN-A. This result substantially indicates that the locally crystallized HCN-A catalyst favors *OCCHO formation via C–C coupling rather than the hydrogenation of *CHO to form *CH2O. Meanwhile, HCN-A also possesses a high CH3CH2OH thermodynamic energy barrier, thus prohibiting CH3CH2OH formation and accounting for the excellent selectivity and activity of the conversion of CO2 to CH3CHO.
Thus, a reasonable mechanism for the reduction of CO2 on HCN-A is proposed (Fig. 5e). Under visible light illumination, the AP-induced locally crystallized HCN-A enables the facile formation of C2 intermediates and exhibits enhanced photocatalytic activity for the conversion of CO2 to CH3CHO. HCN-A could efficiently activate the CO2 molecules to CO2˙− radicals and generate the *CO intermediate, which is further hydrogenated to form the *CHO intermediate. Compared to the amorphous HCN, the locally crystallized HCN-A is prone to have stronger bonding with the *OCCHO group, which regulates the endoergic C–C coupling step to a spontaneous process and changes the reaction pathway to form CH3CHO instead of HCHO. The proton is rapidly extracted from the dissociation of H2O. Eventually, CH3CHO can be obtained via the C–C coupling and multi-step hydrogenation process with high selectivity and activity.
Conclusions
In summary, we have developed a locally crystallized HCN-A catalyst via hydrothermal pre-treatment with an amine-assisted nucleation strategy to enable the formation of alternately appearing amorphous domains and a well-crystallized lattice matrix framework. The HCN-A breaks the trade-off between the efficiency and the selectivity for photocatalytic CO2 reduction to CH3CHO. It furnishes CH3CHO product with a high-yielding rate of 1814.7 μmol h−1 g−1 under mild conditions, accompanied with a QE of 13.3% at λ = 420 nm. The CH3CHO selectivity on HCN-A reaches 98.3%, which is significantly higher than the value of 54.7% on HCN. This locally crystallized HCN-A exhibits significantly improved selectivity and activity compared to reported CO2 reduction photocatalysts. We reveal the C–C coupling and multi-step hydrogenation mechanisms for the photocatalytic conversion of CO2 to CH3CHO on the locally crystallized HCN-A, and *OCCHO is determined to be the key reaction intermediate. Compared to the endoergic C–C coupling step on HCN, the locally crystallized HCN-A enables a spontaneous exoergic reaction to form the *OCCHO intermediates. The HCN-A shows suppressed hydrogenation of *CHO to *CH2O, leading the reaction pathway towards CH3CHO instead of HCHO. Our work not only demonstrates an efficient strategy for grafting locally crystallized PCN for highly selective CH3CHO photosynthesis in a sustainable way, but also proposes a plausible mechanism for the whole reaction. The research findings shed light on the highly selective photosynthesis of liquid multi-carbon products by CO2 reduction.Author contributions
Q. L. Conceptualization, methodology, investigation, writing-original draft, visualization; H. C. Methodology, investigation, visualization; T. C. and T. B. L. Performed high-resolution X-ray total scattering spectra, PDFgui refinement, synchrotron-based X-ray absorption structure spectroscopy; M. X. Analysed GC-MS. F. W. Project administration; conceptualization and supervision; writing – review & editing; funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (61904167), the Natural Science Foundation of Guangdong Province (2019A1515012081), the Science and Technology Program of Guangzhou (202002030017), the GDAS' Project of Science and Technology Development, and the China Postdoctoral Science Foundation (2020M672638).Notes and references
- M. Eckert, G. Fleischmann, R. Jira, H. M. Bolt and K. Golka, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034–9037 CrossRef CAS PubMed.
- J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038–9049 CrossRef CAS PubMed.
- P. Liu and E. J. M. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS PubMed.
- J. Mielby, J. O. Abildstrøm, F. Wang, T. Kasama, C. Weidenthaler and S. Kegnæs, Angew. Chem., Int. Ed., 2014, 126, 12721–12724 CrossRef.
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- W. Yang, H.-J. Wang, R.-R. Liu, J.-W. Wang, C. Zhang, C. Li, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2021, 60, 409–414 CrossRef CAS.
- Q. Liu, L. Wei, Q. Xi, Y. Lei and F. Wang, Chem. Eng. J., 2020, 383, 123792 CrossRef CAS.
- Y. Li, M. Wen, Y. Wang, G. Tian, C. Wang and J. Zhao, Angew. Chem., Int. Ed., 2021, 60, 910–916 CrossRef CAS.
- Q. Wang, J. Warnan, S. Rodríguez-Jiménez, J. J. Leung, S. Kalathil, V. Andrei, K. Domen and E. Reisner, Nat. Energy, 2020, 5, 703–710 CrossRef CAS.
- G. Qin, Y. Zhang, X. Ke, X. Tong, Z. Sun, M. Liang and S. Xue, Appl. Catal., B, 2013, 129, 599–605 CrossRef CAS.
- Y. Wang, Y. Tian, L. Yan and Z. Su, J. Phys. Chem. C, 2018, 122, 7712–7719 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- H. Wu, X. Y. Kong, X. Wen, S.-P. Chai, E. C. Lovell, J. Tang and Y. H. Ng, Angew. Chem., Int. Ed., 2021, 60, 8455–8459 CrossRef CAS PubMed.
- J. Xu, Z. Ju, W. Zhang, Y. Pan, J. Zhu, J. Mao, X. Zheng, H. Fu, M. Yuan and H. Chen, Angew. Chem., Int. Ed., 2021, 133, 8787–8791 CrossRef.
- Y. Wang, X. Liu, X. Han, R. Godin, J. Chen, W. Zhou, C. Jiang, J. F. Thompson, K. B. Mustafa, S. A. Shevlin, J. R. Durrant, Z. Guo and J. Tang, Nat. Commun., 2020, 11, 2531 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- J. Li, W. Pan, Q. Liu, Z. Chen, Z. Chen, X. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS.
- Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang and M.-H. Whangbo, Catal. Commun., 2009, 11, 210–213 CrossRef CAS.
- S. Zhu, X. Li, X. Jiao, W. Shao, L. Li, X. Zu, J. Hu, J. Zhu, W. Yan, C. Wang, Y. Sun and Y. Xie, Nano Lett., 2021, 21, 2324–2331 CrossRef CAS PubMed.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- X. Zhao, L. Du, B. You and Y. Sun, Catal. Sci. Technol., 2020, 10, 2711–2720 RSC.
- I. Shown, H.-C. Hsu, Y.-C. Chang, C.-H. Lin, P. K. Roy, A. Ganguly, C.-H. Wang, J.-K. Chang, C.-I. Wu, L.-C. Chen and K.-H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- X. Qian, W. Yang, S. Gao, J. Xiao, S. Basu, A. Yoshimura, Y. Shi, V. Meunier and Q. Li, ACS Appl. Mater. Interfaces, 2020, 12, 55982–55993 CrossRef CAS PubMed.
- O. Ola, M. Maroto-Valer, D. Liu, S. Mackintosh, C.-W. Lee and J. C. S. Wu, Appl. Catal., B, 2012, 126, 172–179 CrossRef CAS.
- O. Ola and M. M. Maroto-Valer, Chem. Eng. J., 2016, 283, 1244–1253 CrossRef CAS.
- C.-W. Lee, R. Antoniou Kourounioti, J. C. S. Wu, E. Murchie, M. Maroto-Valer, O. E. Jensen, C.-W. Huang and A. Ruban, J. CO2 Util., 2014, 5, 33–40 CrossRef CAS.
- P.-Y. Liou, S.-C. Chen, J. C. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer and R. Linforth, Energy Environ. Sci., 2011, 4, 1487–1494 RSC.
- I. Shown, S. Samireddi, Y.-C. Chang, R. Putikam, P.-H. Chang, A. Sabbah, F.-Y. Fu, W.-F. Chen, C.-I. Wu, T.-Y. Yu, P.-W. Chung, M. C. Lin, L.-C. Chen and K.-H. Chen, Nat. Commun., 2018, 9, 169 CrossRef.
- L. Tang, L. Kuai, Y. Li, H. Li, Y. Zhou and Z. Zou, Nanotechnology, 2018, 29, 064003 CrossRef PubMed.
- J. Xiao, W. Yang, S. Gao, C. Sun and Q. Li, J. Mater. Sci. Technol., 2018, 34, 2331–2336 CrossRef.
- M. L. Ovcharov, A. M. Mishura, N. D. Shcherban, S. M. Filonenko and V. M. Granchak, Sol. Energy, 2016, 139, 452–457 CrossRef CAS.
- X. Zhi, A. Vasileff, Y. Zheng, Y. Jiao and S.-Z. Qiao, Energy Environ. Sci., 2021, 14, 3912–3930 RSC.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- Q. Liu, Z. Chen, W. Tao, H. Zhu, L. Zhong, F. Wang, R. Zou, Y. Lei, C. Liu and X. Peng, J. Mater. Chem. A, 2020, 8, 11761–11772 RSC.
- G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2111–2124 CrossRef CAS.
- X. Xiao, Y. Gao, L. Zhang, J. Zhang, Q. Zhang, Q. Li, H. Bao, J. Zhou, S. Miao, N. Chen, J. Wang, B. Jiang, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2003082 CrossRef CAS.
- Q. Liu, H. Cheng, T. Chen, T. W. B. Lo, J. Ma, A. Ling and F. Wang, Appl. Catal., B, 2021, 295, 120289 CrossRef CAS.
- W.-J. Jiang, S. Niu, T. Tang, Q.-H. Zhang, X.-Z. Liu, Y. Zhang, Y.-Y. Chen, J.-H. Li, L. Gu, L.-J. Wan and J.-S. Hu, Angew. Chem., Int. Ed., 2017, 56, 6572–6577 CrossRef CAS.
- S. Kusama, T. Saito, H. Hashiba, A. Sakai and S. Yotsuhashi, ACS Catal., 2017, 7, 8382–8385 CrossRef CAS.
- C. Hu, F. Chen, Y. Wang, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Mater., 2021, 33, 2101751 CrossRef CAS.
- R. R. Lieten, C. Fleischmann, S. Peters, N. M. Santos, L. M. Amorim, Y. Shimura, N. Uchida, T. Maeda, S. Nikitenko, T. Conard, J.-P. Locquet, K. Temst and A. Vantomme, ECS J. Solid State Sci. Technol., 2014, 3, P403–P408 CrossRef CAS.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS PubMed.
- D. Zhao, C.-L. Dong, B. Wang, C. Chen, Y.-C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, 1903545 CrossRef CAS PubMed.
- Q. Liu, C. Chen, K. Yuan, C. D. Sewell, Z. Zhang, X. Fang and Z. Lin, Nano Energy, 2020, 77, 105104 CrossRef CAS.
- Z. Jin, N. Murakami, T. Tsubota and T. Ohno, Appl. Catal., B, 2014, 150-151, 479–485 CrossRef CAS.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio, J. H. Montoya, D. B. Trimarco, C. Roy, S. Meier, J. Wendland, J. K. Nørskov, I. E. L. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2016, 55, 1450–1454 CrossRef CAS.
- A. Ott, J.-E. Germond and A. Chaintreau, J. Agric. Food Chem., 2000, 48, 1512–1517 CrossRef CAS PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahn and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS PubMed.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS PubMed.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- Z. Yu and S. S. C. Chuang, J. Catal., 2007, 246, 118–126 CrossRef CAS.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- X.-Y. Ma, C. Ding, H. Li, K. Jiang, S. Duan and W.-B. Cai, J. Phys. Chem. Lett., 2020, 11, 8727–8734 CrossRef CAS.
- J. T. Yates and R. R. Cavanagh, J. Catal., 1982, 74, 97–109 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee02073k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |