
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpider silk-inspired peptide multiblock hybrid copolymers for self-healable thin film materials†
Tomoyuki
Koga
 *a,
Tomotaka
Morishita
a,
Yushi
Harumoto
a,
Shin-nosuke
Nishimura
b and
Nobuyuki
Higashi
*a
*a,
Tomotaka
Morishita
a,
Yushi
Harumoto
a,
Shin-nosuke
Nishimura
b and
Nobuyuki
Higashi
*a
aDepartment of Molecular Chemistry and Biochemistry, Faculty of Science and Engineering, Doshisha University, Kyotanabe, Kyoto 610-0321, Japan. E-mail: tkoga@mail.doshisha.ac.jp; nhigashi@mail.doshisha.ac.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, Fukuoka 819-0395, Japan
First published on 28th October 2021
Abstract
Natural biopolymers exhibit smart functions and extraordinary physical properties because of their optimized and rational molecular structures. Understanding and mimicking such architectural principles on a nanoscale offer promising opportunities for designing novel functional polymer materials. Herein, we report unique nano/microfilm materials fabricated using spider silk-inspired peptide multiblock hybrid copolymers. The multiblock copolymers are composed of self-assembling oligopeptides (Ala, Gly, Val, and Leu-based peptides) and non-crystalline flexible polypropylene glycol. Self-supporting microfilms are easily obtained by casting from polymer solutions via β-sheet network formation, and their structural and mechanical properties are characterized comprehensively. The resulting spider silk-like specific nanostructure comprises balanced crystalline and non-crystalline regions; additionally, it exhibits reversible interactions among peptide blocks based on multiple hydrogen bonds. This enables mechanical toughness and self-healing characteristics in the thin films. Moreover, such film characteristics, including healing efficiency, can be modulated by manipulating the sequence and length of the peptide blocks. Additionally, these hybrid multiblock copolymers are used to prepare free-standing and ultraflexible nanofilms, which demonstrate excellent followability to nanoscale roughness based on film thickness and serve as a nano-coating with ion barrier function. The biomimetic approach presented herein is a facile and effective method for the development of high-performance polymer films and is promising in various fields, such as industrial, nanotechnological, and biomedical material fields.
Introduction
Molecular biomimetics has recently garnered significant attention as an effective approach for designing novel functional and practical materials with wide-ranging applications, from nanotechnology to biomedical fields.1–4 The understanding of natural functional polymers can facilitate the development of novel polymer materials. In particular, proteins and peptides are promising models because of their intelligent functions, biocompatibility, and exceptional physical properties, i.e., they fold5 or assemble appropriately into precisely defined three-dimensional nanoarchitectures based on their optimized and perfectly controlled amino acid sequences.6–11 Therefore, various de novo designed proteins and peptides have been developed and used as nanobiomaterials for tissue engineering, controlled drug delivery, catalysis, nano-templates, and bioimaging.12–21 In nature, such self-structuring of proteins and peptides, including their high-order structuring, is achieved by exploiting non-covalent interactions such as hydrogen bonds, electrostatic interactions, van der Waals interactions, and hydrophobic interactions, resulting in sophisticated biofunctions and extraordinary material properties. For example, major ampullate silk produced by a spider (Nephila clavipes), known as dragline silk, contains two major proteins in the core (major ampullate spidroins 1 and 2 [MaSp1, MaSp2]) that possess repetitive modules flanked by conserved non-repetitive domains.22,23 The repetitive module possesses significant amounts of non-polar amino acids such as alanine (Ala) or glycine (Gly) for β-sheet formation, and the non-repetitive domains serve as control units for shear force- and ion/pH change-induced self-assembly during spinning.24,25 The resultant dragline silk exhibits outstanding mechanical properties that are superior to those of most synthetic fibers, such as Nylon (comparable to Kevlar), owing to its unique self-assembled nanostructure.22,23 Interestingly, spiders produce various types of silks with diverse mechanical properties for different purposes to accommodate specific tasks. Dragline silk used as a scaffold, which is generated from the major ampullate gland, exhibits extremely high strength and moderate elasticity. By contrast, silk fibers produced in the flagelliform gland are highly extensible and are used to produce the capture spiral of orb webs. From a structural perspective, the self-assembled structure of spider silks can be regarded as nanoscale networks composed of crystalline and non-crystalline regions.26 Rigid crystalline regions are composed of β-sheet structures derived from oligo(Ala) and exhibit high tensile strength.22,23,27 By contrast, flexible non-crystalline regions are composed of amorphous random coil, 310-helix, and β-turn spiral structures derived from random amino acid arrays including Gly and Pro-rich sequences, which endow the spider silk with high elasticity. Hence, the spider silk allows the mechanical properties of fibers to be adjusted by appropriately controlling the balance of crystalline and non-crystalline regions via self-assembly.Recent advances in the study of such structure–property relationships and self-assembling mechanisms provide useful information for designing novel and high-performance protein/peptide-based materials,28–30 such as those that exhibit the outstanding toughness of spider silk. It is well known that solid phase peptide synthesis (SPPS) and recombinant protein expression methods allow the preparation of artificial peptides/proteins with perfectly controlled monomer sequences and chain lengths. However, SPPS is difficult to scale up and does not facilitate the easy synthesis of high-molecular-weight samples; additionally, the recombinant DNA method occasionally requires complicated and specialized operations and is often difficult to scale up. A promising strategy is to hybridize sequence-controlled peptides with conventional synthetic polymers. This chemical approach enables the precise control of both structural and functional properties by manipulating the element type (e.g., peptides and synthetic polymers) and chain length; furthermore, it can overcome issues such as the large-scale production, processability, and preparation of high-molecular-weight samples. Various peptide–polymer hybrids with different conformations (e.g., α-helix, β-sheet, and coiled-coil motifs) and architectures (e.g., diblock, triblock, multiblock, and graft-types) have been designed and synthesized by employing suitable synthetic polymers for nanotechnological and biomedical applications.31–40
An additional advantage of peptide-based hybrids is that they enable self-healing in materials. Self-healable materials that recover their original characteristics after damage improve the longevity, durability, and reliability of materials, thereby being beneficial to both industry and biomedicine.41 To date, several self-healable polymeric materials have been developed using various approaches, such as monomer release and in situ polymerization,42,43 irreversible chemical reactions,44,45 and reversible dynamic bond/interactions40,46–52 including hydrogen bonds,40 electrostatic interactions,48 disulfide bonds,50 and Diels–Alder reactions.51,52 The β-sheet peptide is useful not only as a nanostructuring unit, but also as a self-repairing unit, afforded by multiple reversible hydrogen bonds occurring intermolecularly among the peptide strands. Some β-sheet peptide–polymer hybrids have been synthesized and characterized previously.53–58 However, in many cases, structural characterization is prioritized, and the structure–property and structure–function relationships are yet to be understood comprehensively, particularly in multiblock polymer systems. The creation of a new β-sheet peptide-based hybrid multiblock polymer system that can serve as a self-healable thin film with controllable mechanical properties via a biomimetic approach remains challenging.
Herein, we report novel self-healable film materials with thicknesses ranging from the nanoscale to microscale fabricated from spider silk-inspired peptide multiblock hybrids composed of β-sheet-formable hydrophobic oligopeptides (Ala, Val, Gly, and Leu-based sequences) and non-crystalline flexible polypropylene glycol (PPG) (Fig. 1). Detailed analyses of the structural, physical, mechanical, and self-healing properties of the resulting films were conducted, particularly with respect to the effect of the peptide structure. The reversible and controlled self-assembly of peptide multiblocks allows finely tunable control over the mechanical strength of the film, as well as affords high self-healing efficiency. Furthermore, the mechanical toughness of these multiblock hybrids induced by the formation of spider silk-like β-sheet nano-networks enables the successful preparation of ultraflexible and self-supporting nanofilms. These results emphasize the great potential of molecular biomimetics via a peptide–polymer hybrid strategy for advancing material, nanotechnology and biomedical fields and also provide essential insights for understanding the structure–property correlation of peptide-based materials.
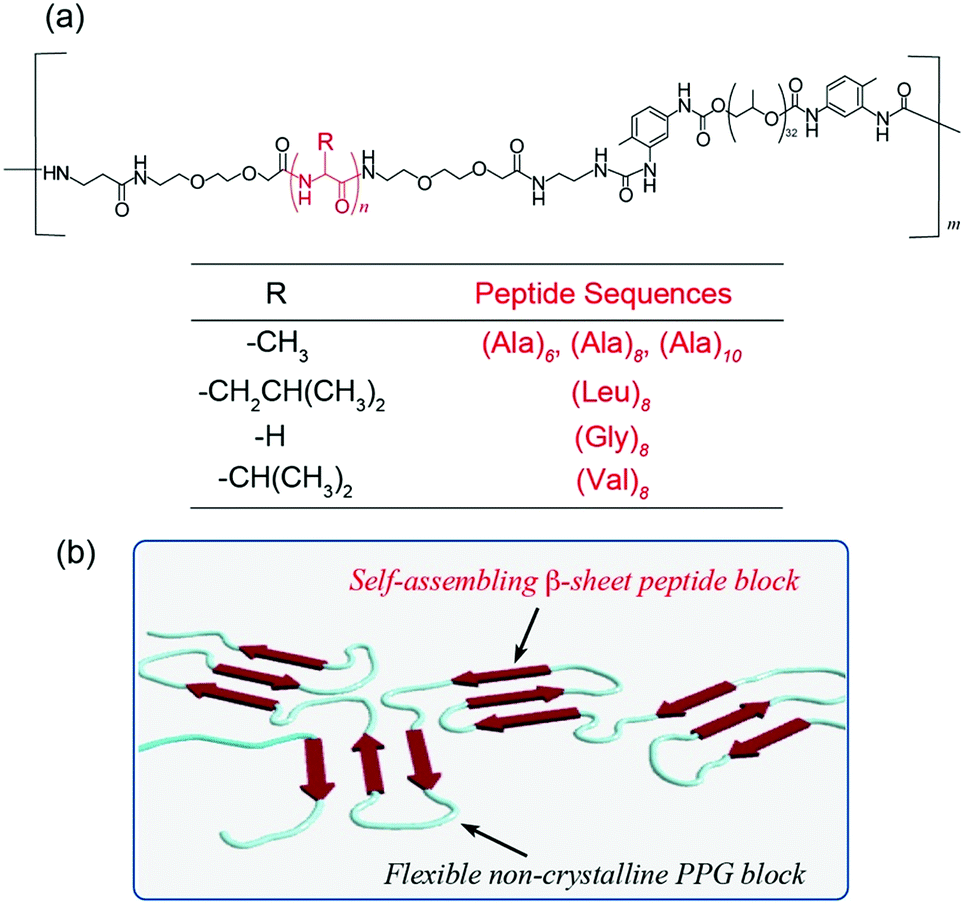 | ||
| Fig. 1 (a) Chemical structure of multiblock hybrid copolymers composed of self-assembling peptides and flexible polypropylene glycol. (b) Schematic illustration of the self-assembling model at the film state. | ||
Experimental
Materials
N,N′-Diisopropyl carbodiimide (DIPC), triethylamine, N,N-dimethylformamide (DMF), dichloromethane (DCM), 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), urea, and D2O were purchased from Wako Pure Chemical. Chloroform, diethyl ether, piperidine and KBr were purchased from Nacalai Tesque. H2N–(CH2)2–NH–Trt–resin, Fmoc-L-Ala–OH, Fmoc-Gly–OH, Fmoc-L-Leu–OH, Fmoc-L-Val–OH, Fmoc-β-Ala–OH, 1-hydroxybenzotriazole anhydrous (HOBt), and trifluoroacetic acid (TFA) were purchased from Watanabe Chemical Industries. Fmoc-8-amino-3,6-dioxaoctanoic acid (Fmoc-deg-COOH) was purchased from Peptide Institute. Triisopropylsilane (TIPS), tolylene-2,4-diisocyanate-terminated poly(propylene glycol) (MW: ∼2300), poly(propylene glycol) (MW: 1000, 2700, and 4000), and cyano-4-hydroxy cinnamic acid (CHCA) were purchased from Sigma-Aldrich. All reagents were used as received. DMF was used for the polyaddition reaction after purification by distillation.Measurements
1H NMR spectra were measured using a JEOL FT-NMR AL400 (JEOL Resonance) spectrometer (400 MHz). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analyses were performed on an Autoflex Speed (Bluker Daltonics) using CHCA as the matrix. Circular dichroism (CD) spectra were recorded on a J-820 spectropolarimeter (JASCO Ltd) equipped with a Peltier-type thermostatic cell holder coupled with a PTC-423L controller under inert N2 gas at 25 °C. Experiments were performed in a quartz cell with a path length of 1 mm or on a quartz plate (for the film state). Transmission (TM)- and reflection absorption (RA)-Fourier-transform infrared spectroscopy (FTIR) spectra of the multiblock polymer films on CaF2 and Au-deposited glass plates were measured using a Nexus 470 spectrometer (Thermo Nicolet Co.) equipped with a mercury–cadmium–tellurium detector (resolution: 4 cm−1, number of scans: 256 or 512). The mechanical properties of the microfilms (∼40 μm thick) were evaluated via tensile tests in air at crosshead speeds of 60 and 180 mm min−1 at room temperature (∼25 °C) using an EZ Graph tester (Shimadzu Co.) equipped with a 50 N load cell. Note that the difference in the crosshead speed did not significantly affect the stress–strain curves in this study. The microfilms were punched into dumbbell-shaped specimens (JIS K6251-7 (ISO37-4)) using a punching blade (Kobunshi Kenki Co. Ltd), and the film thicknesses were precisely measured using a digimatic micrometer (MDC-25MJ, Mitutoyo Co.). At least three independent tensile tests were conducted. AFM images were obtained at room temperature on an SPM9700 (Shimadzu Co.) in the tapping mode (MPP-11100, tip radius < 12 nm) to evaluate the thickness and followability of the nanofilms to nanostructured surfaces. Scanning electron microscopy (SEM) was conducted to evaluate the thickness of the nanofilms on a JEOL JSM7001FD (JEOL Resonance). Prior to SEM analysis, the cross section of the nanofilm on the silicon wafer was sputtered with Pt (6 nm). Laser microscope images were obtained at room temperature using a VK-X210 microscope (Keyence Co.). The water contact angles (CAs) of the multiblock polymer films were evaluated at room temperature using a DropMaster-501 (Kyowa Interface Science Co., Ltd). Thermogravimetric analyses were conducted using a DTG60/60H (Shimadzu Co.) at 30 °C–280 °C (10 °C min−1), and α-Al2O3 was used as a reference.Synthesis of peptide–PPG multiblock copolymers via polyaddition
Block copolymerization of the amine-terminated peptides (see Fig. S1, ESI†) with tolylene-2,4-diisocyanate-terminated poly(propylene glycol) (PPG–(NCO)2) via a polyaddition reaction was performed in distilled DMF at room temperature in the presence of triethylamine. In a typical polyaddition reaction, amine-terminated (Ala)8 (256 μmol) and triethylamine (767 μmol) were initially dissolved in DMF (20 mL), and the solutions were stirred for 1 h. Subsequently, the DMF solution (5 mL) of PPG–(NCO)2 (256 μmol) was poured into the peptide solution. After polyaddition at room temperature, the resulting multiblock polymer was purified via reprecipitation using DMF/diethyl ether and HFIP/water. The chemical structures were determined via FTIR and 1H NMR spectroscopy (Fig. S2, ESI†). The preparation of other multiblock copolymers was conducted in a similar manner. The isolated multiblock copolymers were insoluble in general organic solvents because of their prominent self-assembling nature once they were extracted as a dried solid, and they were only soluble in highly polar solvents such as HFIP and TFA. Therefore, inherent viscosity analyses of the polymers in HFIP were conducted at 30 °C using an Ostwald-type viscometer to evaluate the viscosity-average molecular weight (Mv) of the multiblock copolymers using the Mark–Houwink–Sakurada equation, as follows: [η] = KMvα. Here, the K and α values estimated from pure PPG standards (MW 1000, 2700, and 4000) in HFIP (30 °C) were used to calculate Mv.Preparation of nanofilms
Two-dimensional nanofilms of peptide–PPG multiblock hybrids were prepared as follows: HFIP solutions (200 μL) of [(Ala)n–PPG]m (n = 6, 8, and 10) (0.5, 1, and 2 wt%) were spin coated using a K-3591S spin coater (Kyowariken Co. Ltd) at 1000, 2000, and 4000 rpm onto a polystyrene substrate (25 mm × 25 mm × 0.8 mm), which was used as a sacrificial layer. Subsequently, the substrate was treated with chloroform to dissolve the polystyrene sacrificial layer. Self-supporting nanofilms were formed successfully on the surface of chloroform and were carefully scooped up subsequently. The thicknesses of the obtained nanofilms were evaluated using SEM and AFM analyses.Characterization of [(Ala)10–PPG]m nanofilms as coating materials
The followability of the nanofilms over nanoscale structures was evaluated to assess the ultraflexibility of the [(Ala)n–PPG]m nanofilm. Line patterns of commercially available CD-R and DVD-R were used as nanostructured surfaces. CD-R and DVD-R were used after peeling off the polycarbonate layer and removing the adhesive by washing with water. Nanofilms with different thicknesses were coated onto the nanostructured materials, and the surface topographies were evaluated by comparing them to the uncoated bare surfaces via AFM analyses. The ion barrier ability of the nanofilms was evaluated via permeation experiments using pure water (45 mL) and aqueous solutions (6 mL) of sodium chloride (1.67 M) or sodium p-toluenesulfonate (1.67 × 10–2 M) using a conventional, thermostatically controlled H-shaped cell. First, the [(Ala)10–PPG]m nanofilm with a thickness of 300 nm was prepared by a spin-coating method using a flat PSt sacrificial substrate as described above. The nanofilm was then sandwiched between glass filter papers (thickness, 440 μm; retention particle size, 1 μm) and set in the cell using a silicon sheet. The permeation nanofilm area between the cells was 0.385 cm2, and both sides of the cell were stirred at a constant speed. The permeation of the salts was monitored by increasing the electrical conductance on the water side at room temperature (25 °C). The electrical conductance was measured using an electrical conductivity meter CM-7B (DKK-TOA Co.). The experiments were performed thrice for each sample.Results and discussion
Design and synthesis of peptide-based multiblock copolymers
Two distinct alternating blocks, i.e., (i) flexible and elastic blocks and (ii) hard crystalline β-sheet blocks, which serve as cross-links for the network structure, were used. The balance of the blocks is important for controlling the physical characteristics of natural spider silk materials, including their mechanical strength. In the spiders’ draglines and frame lines, the oligo(Ala) sequence with four to nine residues are typically employed as such β-sheet nano-crystal blocks.27 Based on the biomimetic design principle, we synthesized various spider silk-mimetic peptide–synthetic polymer multiblock hybrids composed of self-assembling hydrophobic oligopeptides (Ala, Gly, Val, and Leu-based peptides) and a non-crystalline and flexible PPG, i.e., [peptide–PPG]m, via step-growth polymerization, as shown in Fig. 1.Amine-terminated hydrophobic oligopeptides ((Ala)6, (Ala)8, (Ala)10, (Gly)8, (Val)8, and (Leu)8) with diethylene glycol (deg) spacers were employed as self-assembling units and synthesized via the conventional SPPS method using Fmoc chemistry. The introduction of flexible deg spacers at both the N- and C-termini of the peptide unit enables the molecular motility of peptide blocks in the polymer chain and enhances the solubility in organic solvents. The six target peptides were successfully prepared and identified via1H NMR and MALDI-TOF MS analyses (Fig. S1, ESI†). Subsequently, the polyaddition reactions of the obtained peptides with commercially available isocyanate-functionalized PPG (MW: 2300) were performed in DMF, and the progress of the reactions was confirmed by the disappearance of the peak of the isocyanate group at 2280 cm−1via FTIR analyses. 1H NMR studies of the purified polymers confirmed the presence of the peptide block in the PPG-based polymer, which demonstrated the formation of a multiblock structure (Fig. S2, ESI†). It is noteworthy that all isolated block copolymers were relatively insoluble owing to their prominent self-assembling nature, and they were only soluble in highly polar solvents such as HFIP and TFA. Therefore, inherent viscosity [η] analyses of these block copolymers in HFIP were conducted to characterize their molecular weights. The [η] values of these block polymers ([η] = 0.47–0.89 (dL g−1)) were much higher than that of the starting PPG (MW = 2300, [η] = 0.18 (dL g−1)) (see the Experimental section), demonstrating the increase in molecular weight by the polyaddition reaction. The Mv values of multiblock [peptide–PPG]m estimated from the abovementioned [η] values using the Mark–Houwink–Sakurada equation were in the range of 6000–12000 for pure PPG standards. These Mv values correspond to approximately two to four repeat numbers (m). Hence, six types of multiblock hybrid polymers with systematically different peptide lengths and sequences were successfully synthesized.
Characterization of the cast film properties of peptide/PPG hybrid multiblock copolymers
Self-supporting films (approximately 40 μm thick) were prepared by casting 4 wt% HFIP solutions of [peptide–PPG]m block copolymers on Teflon plates under ambient conditions. Initially, we characterized the structural and mechanical properties of these microfilms, particularly with respect to the effects of peptide structures, such as the sequence and chain length. Fig. 2a shows the FTIR spectra of the multiblock polymer films. We focused on the amide I band, which reflects the secondary structure of the peptide blocks.35,40,59 In all cases, the characteristic absorption of the amide I band in the β-sheet structure was observed at approximately 1625 cm−1. In addition, a weak peak appeared at 1695 cm−1 because of the antiparallel β-sheet structure. It is noteworthy that the shoulders at 1660 cm−1 observed in these spectra, which were considerably weaker than the β-sheet amide I band, were attributed to amide I in the random coil form and/or the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the urea linkage. The β-sheet content of peptide blocks in the film states was evaluated to be approximately 62% ((Ala)6), 70% ((Ala)8), 74% ((Ala)10), 70% ((Gly)8), 77% ((Val)8), and 81% ((Leu)8), based on the ratio of integrated peak intensities assigned to a β-sheet conformation to that of non-sheet conformations, which were obtained by the peak deconvolution of the amide I bands (Fig. S3, ESI†). The data revealed that the β-sheet content depended slightly on the peptide sequence and length, and that the shorter peptide ((Ala)6)-hybrid possessed a slightly lower β-sheet content but remained higher than 60%. In the HFIP solutions, these polymers appeared as random coil conformations with a negative maximum at 198 nm, as confirmed via CD analysis. However, in the film state, the spectrum showed a typical pattern of the β-sheet structure with a negative maximum at 216 nm (Fig. 2b). Hence, the peptide multiblocks self-assembled into a predominantly antiparallel β-sheet structure during casting, as observed in oligo(Ala) domains in MaSp1 and MaSp2 of the dragline fiber during the spinning process from the gland of the spider.24–26 It is noteworthy that the peptide-free starting PPG (MW 2300) failed to form a film; therefore, it is assumed that multipoint cross-links by β-sheet self-assembly are vital to film formation.
O stretching of the urea linkage. The β-sheet content of peptide blocks in the film states was evaluated to be approximately 62% ((Ala)6), 70% ((Ala)8), 74% ((Ala)10), 70% ((Gly)8), 77% ((Val)8), and 81% ((Leu)8), based on the ratio of integrated peak intensities assigned to a β-sheet conformation to that of non-sheet conformations, which were obtained by the peak deconvolution of the amide I bands (Fig. S3, ESI†). The data revealed that the β-sheet content depended slightly on the peptide sequence and length, and that the shorter peptide ((Ala)6)-hybrid possessed a slightly lower β-sheet content but remained higher than 60%. In the HFIP solutions, these polymers appeared as random coil conformations with a negative maximum at 198 nm, as confirmed via CD analysis. However, in the film state, the spectrum showed a typical pattern of the β-sheet structure with a negative maximum at 216 nm (Fig. 2b). Hence, the peptide multiblocks self-assembled into a predominantly antiparallel β-sheet structure during casting, as observed in oligo(Ala) domains in MaSp1 and MaSp2 of the dragline fiber during the spinning process from the gland of the spider.24–26 It is noteworthy that the peptide-free starting PPG (MW 2300) failed to form a film; therefore, it is assumed that multipoint cross-links by β-sheet self-assembly are vital to film formation.
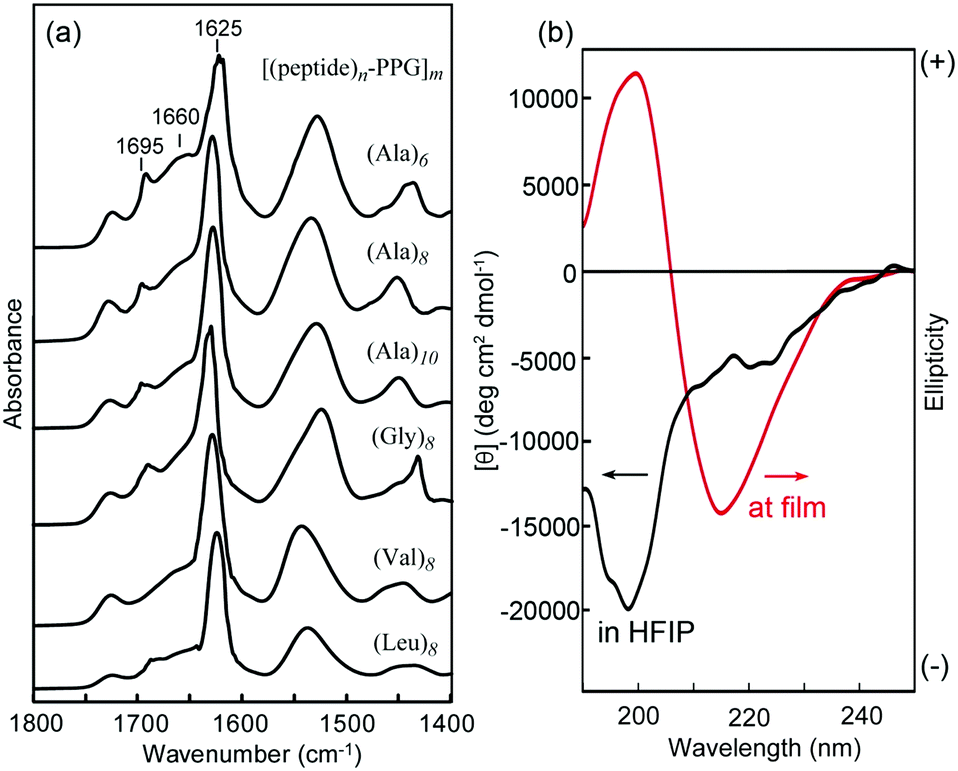 | ||
| Fig. 2 (a) Transmission FTIR spectra of various [(peptide)n–PPG]m microfilms. (b) CD spectra of [(Ala)8–PPG]m in HFIP (2 mg mL−1) and at the film state on a quartz plate. | ||
The surface wettability and thermal stability of various [peptide–PPG]m films were characterized by water CA and thermogravimetric analyses, respectively. Most of the films, except the [(Gly)8–PPG]m film, afforded relatively high CAs of 90°–105°, which correspond to hydrophobic surfaces (CA > 90°) (Fig. S4a–e, ESI†). By contrast, the CA of the [(Gly)8–PPG]m film was lower (85°) compared with those of the other films; this is likely due to the lower hydrophobicity of the constituent amino acid residue compared with those of the other residues (Fig. S4f, ESI†). TGA analysis showed that the weights of these films began to decrease at 230–240 °C (Fig. S5, ESI†). The decomposition temperatures of these films were lower than those of previously reported native spider silk materials (Td = 250–300 °C)60 but were sufficient for practical use.
The mechanical properties of the multiblock polymer films were subsequently evaluated via tensile tests. Fig. 3 shows the representative stress–strain curves for various films with different peptide sequences (Fig. 3a) and chain lengths (Fig. 3b). All films indicated good mechanical properties with relatively high strength (6–10 MPa) and high extensibility (elongation at break: 300–700%). Fig. 3c and d summarize the tensile strength, elongation at break, Young's modulus, and breaking energy evaluated from the stress–strain curves. The observed high breaking energies of these films were attributable to the spider silk-like nano-separation, in which the rigid β-sheet multiblocks serving as non-covalent cross-links were dispersed in the non-crystalline elastic PPG matrix. However, their tensile behaviors on the peptide sequence showed slight but clear differences, despite the same peptide length (8 mer). Specifically, the Young's moduli of the Ala-based films (15–25 MPa) were significantly higher than those of the Gly-, Val-, and Leu-based films (5–8 MPa). Tirrell et al. comprehensively evaluated the molecular architecture of artificial extracellular matrix (aECM) protein films, which comprise an elastin-based sequence cross-linked by hexamethylene diisocyanate, based on their stress–strain curves.61 They evaluated the shear modulus, G, from the slope of a plot of σ vs. (λ − 1/λ2) up to a low strain of 0.25 (λ = 1.25), where σ is the stress, and λ is the elongation ratio. The obtained G value is inversely proportional to the average molecular weight between cross-links (Mc), expressed as G = ρRT/Mc, where ρ is the effective polymer density, R is the gas constant, and T is the temperature (25° C). Using this equation, the apparent Mc values for all multiblock polymer films were calculated using their G values estimated from the stress–strain curves (Table 1, Fig. S6, ESI†). To calculate the ρ values for each film, the polymer densities of β-sheet peptides (1.34 (A), 1.30 (G), 1.05 (V), and 0.97 (L) g cm−3) and PPG (1.0 g cm−3) were used considering the ratio of their molecular weights (∼1/2 (peptide/PPG)).62 It was discovered that the calculated Mc values for the Ala-based films (∼300–400 g mol−1) were significantly lower than those for the other peptide films (∼1000–2000 g mol−1) (Table 1). This indicates a high cross-linking density of the Ala-based films, reflecting the high aggregation propensity of oligo(Ala) blocks owing to their residual structure with moderate hydrophobicity and sterically non-bulky side chains, which allow tight packing between β-strands. It is noteworthy that this may have resulted in the adoption of short and repetitive Ala sequences in the self-assembling β-sheet crystalline domain in many natural silk proteins, including spider silks. Moreover, the tensile data revealed that an increase in the chain length of the oligo(Ala) block increased the breaking energy of the film, which was likely due to the strengthening of hydrogen bonding.
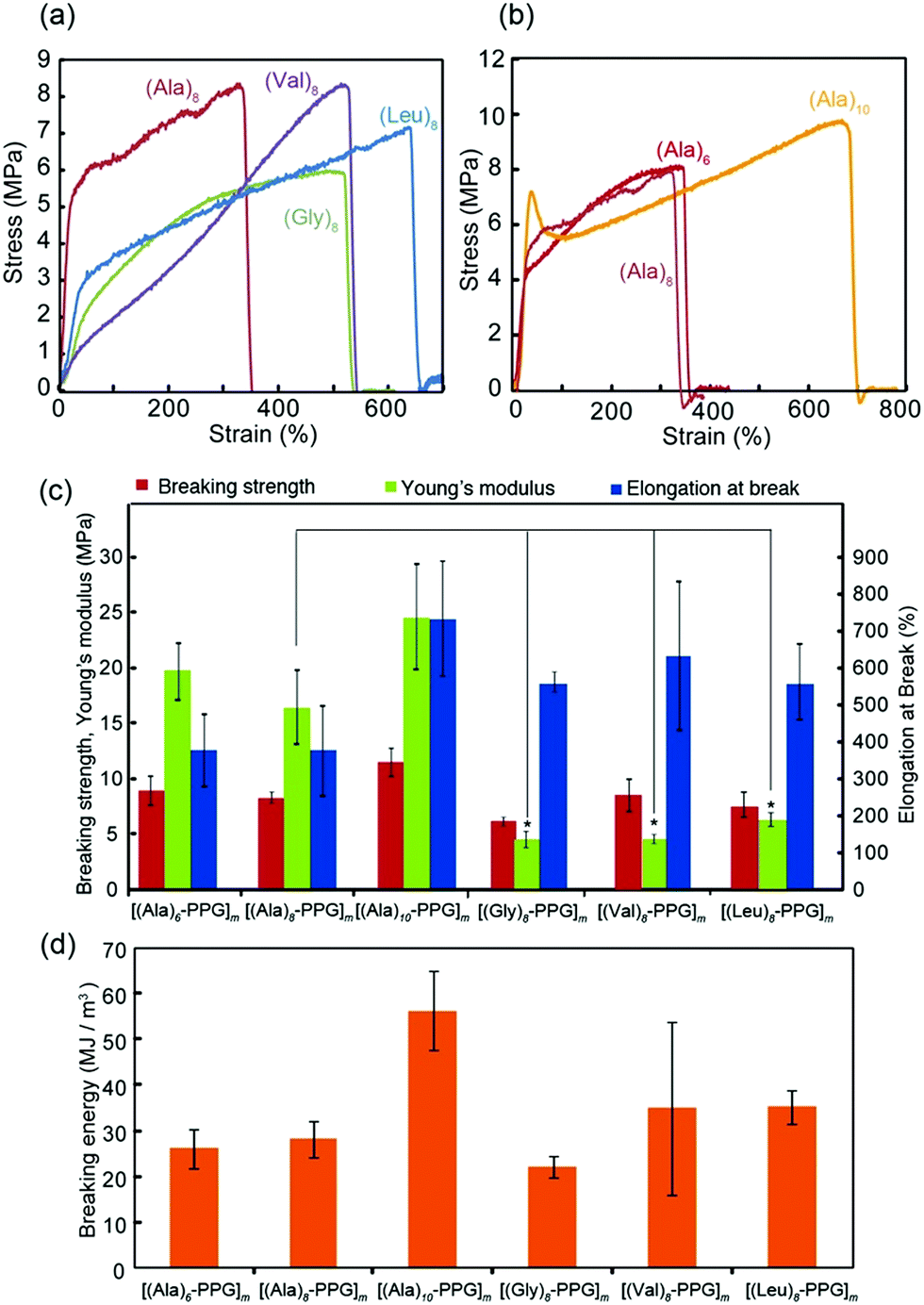 | ||
| Fig. 3 Mechanical properties of [(peptide)n–PPG]m hybrid microfilms. (a and b) Representative stress–strain curves of various [(peptide)n–PPG]m films with different peptide sequences (a) and chain lengths (b). (c) Summary of tensile tests. Statistical analyses performed using Student's t-test. *p < 0.01. (d) Comparison of breaking energy for [(peptide)n–PPG]m films. Error bars represent standard deviation. | ||
| Film | G (MPa) | M c (g mol−1) |
|---|---|---|
| [(Ala)6–PPG]m | 7.6 ± 0.1 | 365 ± 45 |
| [(Ala)8–PPG]m | 6.8 ± 2.3 | 447 ± 183 |
| [(Ala)10–PPG]m | 8.7 ± 2.1 | 330 ± 79 |
| [(Gly)8–PPG]m | 1.5 ± 0.3 | 1894 ± 505 |
| [(Val)8–PPG]m | 1.4 ± 0.2 | 1816 ± 210 |
| [(Leu)8–PPG]m | 2.5 ± 0.3 | 986 ± 110 |
Self-healing behavior of the peptide–PPG multiblock hybrid films
We anticipate the β-sheet-forming ability of peptide multiblocks to facilitate the self-healing function of the polymer films through both multiple/reversible hydrogen bonds among the self-assembling oligopeptide blocks and non-crystalline flexible PPG blocks. Hence, the self-healing behavior of multiblock polymer films was investigated. Fig. 4 shows the healing of the mechanical damage on the [(Ala)10–PPG]m film at room temperature. The multiblock polymer film was scratched using tweezers with fine tips, and healing was observed via laser microscopy. The damaged line (white circling in Fig. 4a) observed immediately after scratching was almost undetectable after 5 d (Fig. 4b). The self-healing properties of the films were characterized more directly, particularly with respect to the effect of the peptide sequence. The polymer films (dumbbell-shaped, ∼40 μm thick) were cut into two pieces using a razor blade, and the cutting surfaces were soaked in pure water for 3 s. The cut pieces were immediately placed in contact for 24 h at room temperature. All multiblock polymer films exhibited self-healing behavior, which was not broken by their own weight. When the cutting surfaces of the film were soaked in a urea solution (8 M) instead of pure water (urea solution is known to be a hydrogen bond breaker), no efficient self-healing occurred, and the film broke easily when lifted with tweezers. These results indicate that the healing ability of these films was primarily attributed to the reversible hydrogen bonds among the peptide multiblocks accompanied by β-sheet formation. For a more comprehensive understanding, tensile tests were conducted on various self-healed films. Fig. 4c shows representative stress–strain curves for the original and self-healed [(Ala)6–PPG]m films. The stress–strain curve of the self-healed film almost overlapped with that of the original one in the low stretching region; namely, the mechanical properties of the film were well repaired, although the film broke at the cut position with a lower breaking elongation than the original one. The healing efficiency, which is defined herein as the ratio of the breaking energy of the original (BEo) film to that of the self-healed (BEh) film, i.e., BEh/BEo × 100, was approximately 35%. Hence, it can be concluded that the peptide-based multiblock hybrid exhibits intrinsic self-healing. Additionally, we performed self-healing tests for five additional multiblock polymer films using the same procedure, and the results are summarized in Fig. 4d. The [(Ala)n–PPG]m films with different peptide lengths (n = 8 and 10) showed similar healing efficiencies, whereas the other films (Gly, Val, and Leu-based peptides) indicated a much lower healing efficiency (∼10%) compared with the Ala-based films. This observation reflected the relatively high self-assembling capability of the oligo(Ala) units, as demonstrated in the tensile analysis. In conclusion, a novel peptide–PPG multiblock hybrid system exhibiting various types of mechanical strength and self-healing capability was successfully developed by manipulating the peptide sequence and chain length.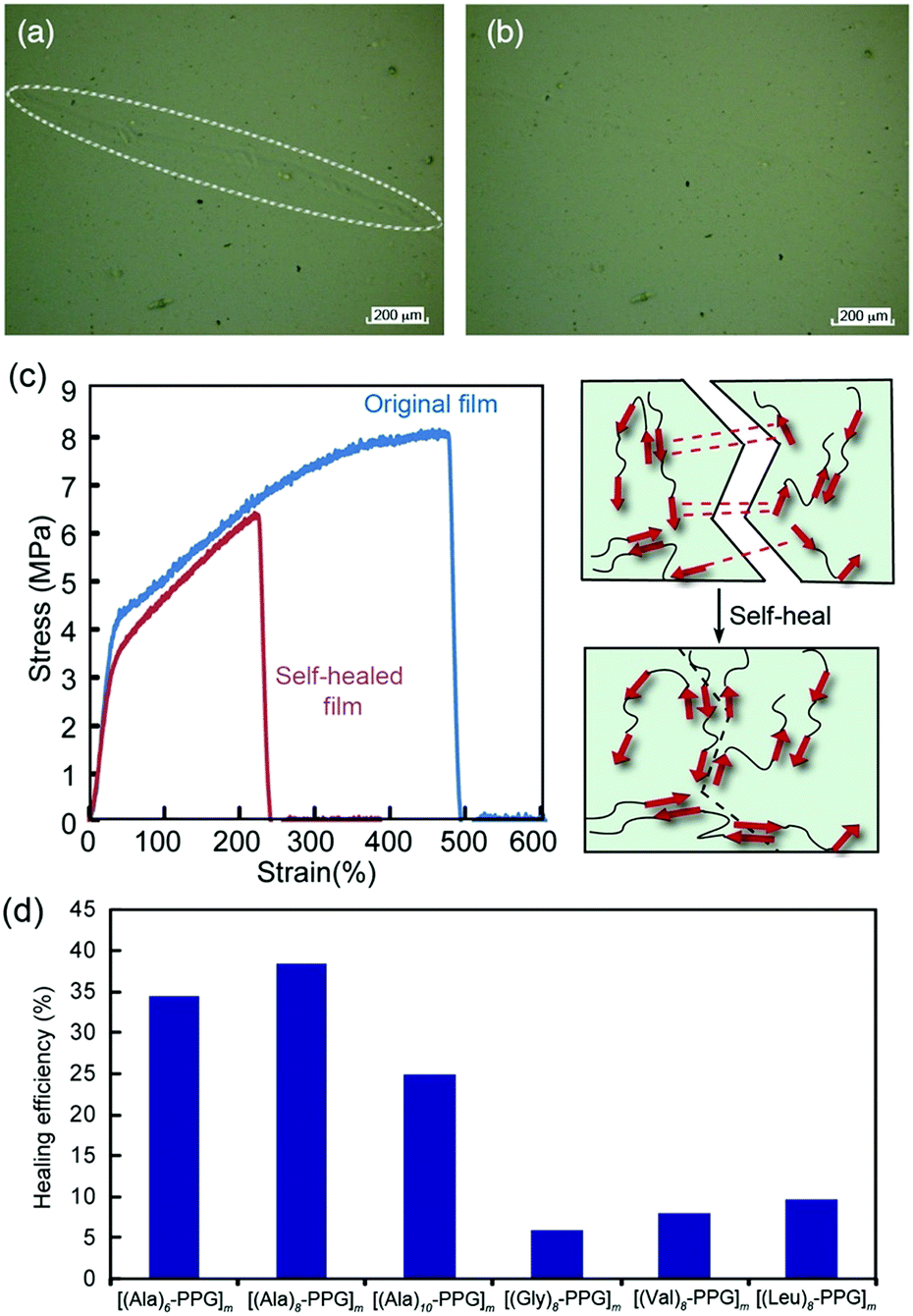 | ||
| Fig. 4 Self-healing behavior of [(peptide)n–PPG]m microfilms. (a) Laser microscope images of the [(Ala)10–PPG]m film immediately after scratching by tweezers with fine tips and after 5 d (b) at room temperature. Scale bar: 200 μm. (c) Comparison of stress–strain curves of the original [(Ala)6–PPG]m film and self-healed film cut into two pieces (left), and the schematic illustration of self-healing by multiple hydrogen bonds among β-sheet peptide blocks (right). (d) Self-healing efficiencies of various multiblock films. | ||
Preparation and characterization of peptide multiblock hybrid nanofilms
Because the multiblock hybrids possessed self-healing capability and sufficient mechanical strength, we subsequently fabricated self-supporting nanofilms using these hybrids and assessed their structural and functional characteristics. In industrial and biomedical fields, polymer thin films of nanoscale thickness have recently garnered significant attention owing to their high flexibility, adhesiveness, and transparency.63 Several fascinating nanofilms have been developed using polymers and polymer/inorganic hybrids via layer-by-layer assembly,64,65 plasma polymerization of nonionic amphiphile (tween20),66 cross-linked Langmuir–Blodgett films,67 and spin coating on a sacrificial underlayer.68–70 Subsequently, they were used as nano-wrapping and coating materials in analytical and biomedical fields, such as for tissue and organism imaging. However, as the film thickness decreases to the nanoscale, the mechanical strength generally tends to decrease. Therefore, although challenging, new functional nanofilm materials to overcome such tradeoff relationships must be identified.The nanofilms were prepared as follows: [(Ala)n–PPG]m hybrids were used as the nanofilm material owing to their high mechanical toughness, including stretchability, and their self-healing capability. First, an HFIP solution of [(Ala)n–PPG]m was spin coated at 2000 rpm onto a PSt sacrificial substrate (2.5 cm × 2.5 cm). Next, the substrate was immersed in chloroform, where the sacrificial PSt was dissolved; consequently, a self-supporting [(Ala)n–PPG]m film buoyant in chloroform was achieved, as shown in Fig. 5a. To control the film thickness, we prepared nanofilms by varying the polymer concentration (0.5–2 wt%) used for spin coating. These nanofilms were transparent, ultraflexible, and did not break when lifted with tweezers (Fig. 5b). To characterize them, the floating nanofilms were transferred onto a silicon wafer and dried at room temperature. Fig. 5c and d show the SEM (cross-sectional view) and AFM images of the [(Ala)8–PPG]m nanofilm (0.5 wt%) as an example. The film thicknesses were estimated to be 50 nm (AFM) and 70 nm (SEM). As shown in Table S1 (ESI†), the film thickness can be controlled easily to 50–300 nm by adjusting the polymer concentration for all [(Ala)n–PPG]m hybrids. It is noteworthy that the rotation speed during spin coating, i.e., 1000–4000 rpm, did not significantly affect the film thickness in this polymer system.
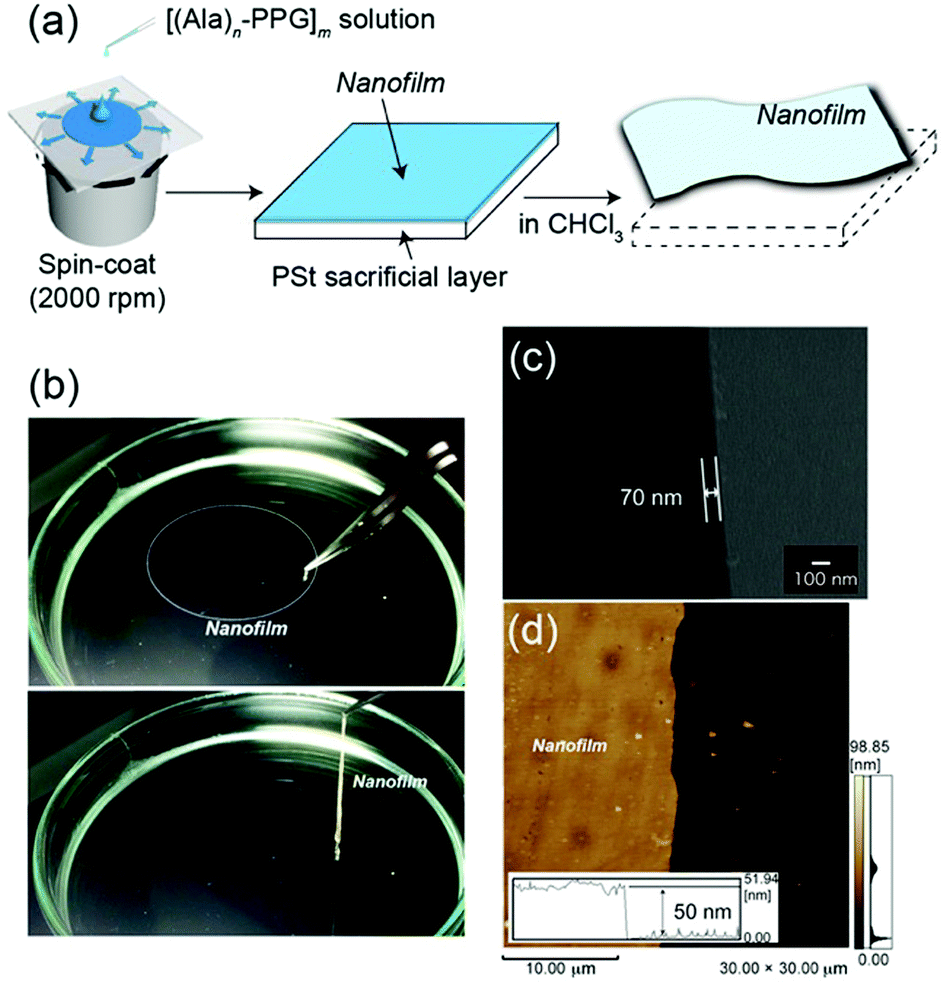 | ||
| Fig. 5 (a) Preparation of the self-supporting [(Ala)n–PPG]m nanofilm via spin coating using a sacrificial under layer. (b) Photographs of the [(Ala)n–PPG]m nanofilm in CHCl3 (top) and that grasped using tweezers (bottom). (c) Cross-sectional SEM and (d) AFM images of the [(Ala)8–PPG]m nanofilm obtained from 0.5 wt% solution. The inset in (d) shows the height profile at the edge of the nanofilm. | ||
The conformational properties of the [(Ala)n–PPG]m nanofilms were investigated via CD and FTIR analyses. Similar to the behavior of the cast microfilm as described above, the oligo(Ala) multiblocks in the nanofilm rapidly self-assembled into an antiparallel β-sheet structure during spin coating, as indicated by the CD pattern with a negative maximum at 216 nm (Fig. S7, ESI†) and the amide I peaks at 1690 and 1627 cm−1 in the FTIR spectrum. However, the orientation of the β-sheet domains in the nanofilm differed from that in the microfilm. Fig. 6 shows the TM and RA-FTIR spectra of the [(Ala)10–PPG]m nanofilm (300 nm thick), which were normalized to the absorbance of the CH2 stretching band of the PPG blocks (2970 cm−1). The intensity of the amide I band at 1627 cm−1 based on the β-sheet structure of the TM spectrum (ITM) was significantly higher than that of the RA spectrum (IRAS). The TM method emphasizes vibrational modes parallel to the film plane; conversely, the RA method emphasizes those perpendicular to the film plane. The ITM/IRAS values were 3.78 (Fig. 6) and 1.96 (Fig. S8, ESI†) for the nanofilm and cast microfilm (∼40 μm), respectively. This indicates that the β-sheet domains in the nanofilm (i.e., amide bonds in the β-strand) were oriented more parallel to the film plane than those in the thick cast film (Fig. 6, inset), likely owing to the centrifugal force generated during spin coating.
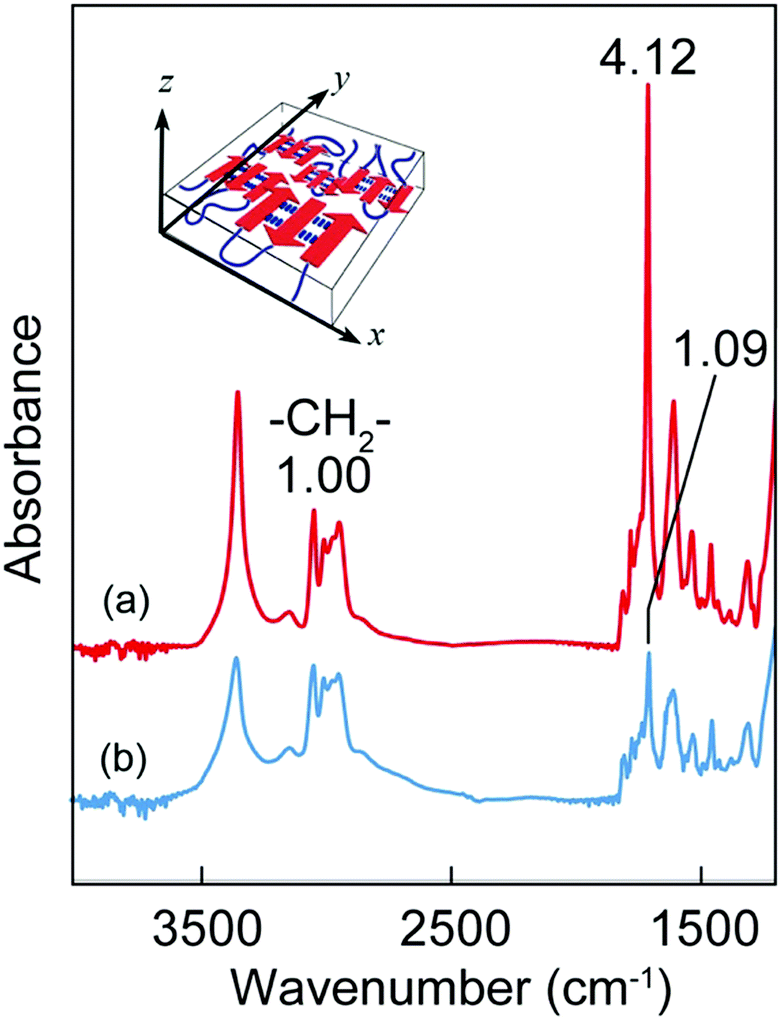 | ||
| Fig. 6 (a) Transmission- and (b) reflection absorption-FTIR spectra of the [(Ala)10-PPG]m nanofilm (300 nm thickness) on a CaF2 plate and Au plate, respectively. The inset shows a schematic illustration of the orientation of β-sheet multiblocks in the nanofilm. | ||
The flexibility and adhesiveness of the [(Ala)10–PPG]m nanofilm were subsequently investigated based on the effect of the film thickness via a followability test performed on the nanostructured surfaces. Two types of line-patterned nanosurfaces derived from CD-R (line pitch: 1600 nm, height: 170 nm) and DVD-R (line pitch: 320 nm, height: 43 nm) were used for this test, and the followability of the nanofilms transferred onto the nanosurfaces was assessed via AFM and laser microscopy. Fig. 7a shows a laser microscopy image of the edge of the [(Ala)10–PPG]m nanofilm with a 300 nm thickness placed on the CD-R surface. A line pattern based on the underlying structure was clearly observed on the nanofilm surface, indicating that the film reflected the nanoscale uneven structure well. Fig. 7b shows the AFM images and cross-sectional height profiles of bare and nanofilm (150 and 300 nm thickness)-covered surfaces. All films demonstrated favorable shape-following and adhesion to the nanostructure without tearing, although the followability to the surface structure, which is defined herein as the ratio of the height of the bare line pattern to that of the nanofilm-covered surface, depended significantly on the film thickness. The decrease in the film thickness resulted in an increase in flexibility, and the followability of the nanofilm with a thickness of 150 nm reached 86% (77% for 200 nm thickness, 53% for 300 nm thickness) over the micro/nanoscale structure. More interestingly, this ultraflexible film (150 nm thick) reflected a finer nanoscale structure (DVD-R surface), as shown in Fig. 7c. The followability of the 150 nm-thick film over the nanoscale roughness maintained at 88%, although for the 200 nm- and 300 nm-thick films, it decreased to 51% and 12%, respectively.
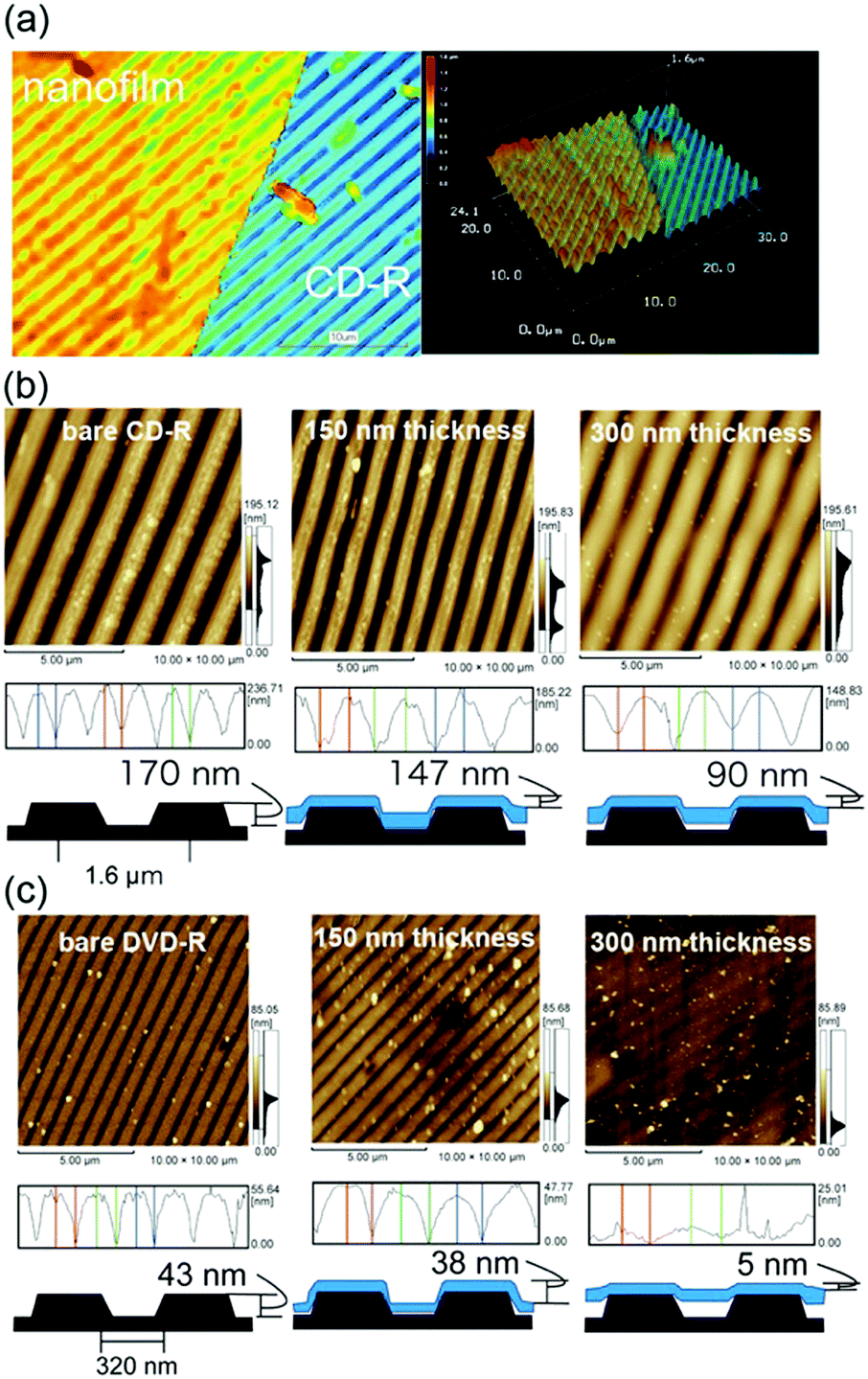 | ||
| Fig. 7 (a) Laser microscope images of the edge of the [(Ala)10–PPG]m nanofilm (300 nm thick) on a CD-R surface. (b) and (c) AFM images of bare and [(Ala)10–PPG]m nanofilms (150 nm and 300 nm thick)-coated CD-R (b) and DVD-R (c) surfaces and their corresponding height profiles. | ||
Because these nanofilms possessed robustness, good shape-following capability, and adhesiveness, we further investigated their ability as coating nanomaterials. Permeation experiments using the [(Ala)10–PPG]m nanofilm (300 nm thick) were performed using pure water and aqueous solutions of sodium chloride (NaCl) and sodium p-toluenesulfonate (pTSNa). Fig. 8 shows the typical time courses of the electrical conductance. As shown in Fig. 8, the conductance increased rapidly and reached stabilized within 3 min in the absence of the nanofilm. By contrast, the conductance change across the nanofilm was remarkably small, and the permeation of both NaCl and the relatively hydrophobic pTSNa (Fig. S9, ESI†) was completely suppressed by the nanofilm for more than 3 h. Hence, the unique nanofilm structure with an oriented β-sheet network, in addition to its stretchability, ultraflexibility, and adhesiveness, is favorable for surface protection, sealing, and wrapping applications, with significant potential in nanotechnology and biomedicine.
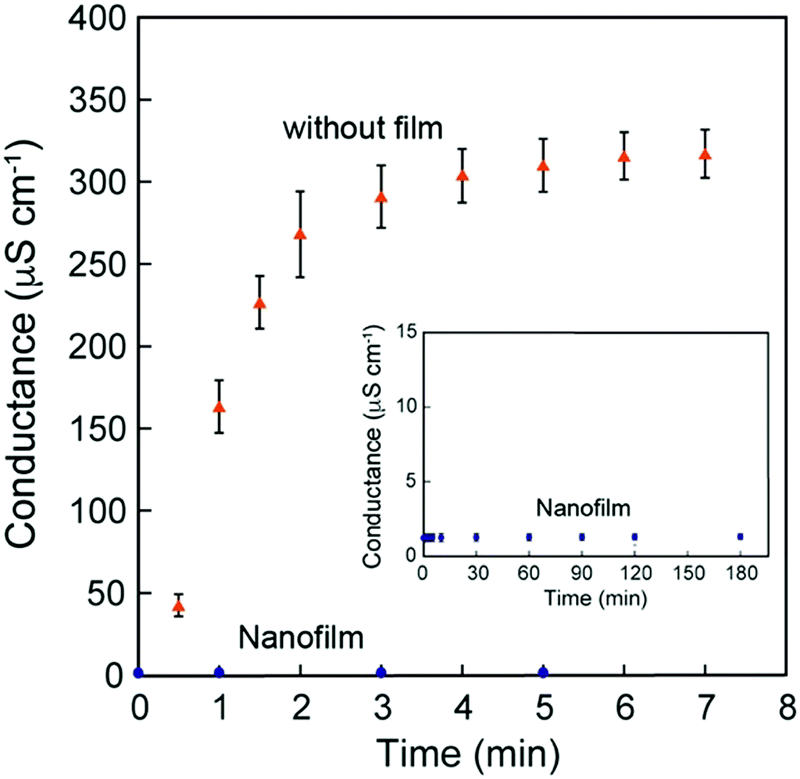 | ||
| Fig. 8 Time courses of ion permeation (NaCl) through the [(Ala)10–PPG]m nanofilm (300 nm thickness). The inset shows the time course of electrical conductance over a long duration (180 min). Errors indicate standard deviation. | ||
Conclusions
In summary, we comprehensively described the synthesis of spider silk-inspired peptide–PPG hybrid multiblock copolymers and their film properties. Six types of hybrids with different peptide chain lengths and sequences were successfully synthesized using a simple SPPS and a subsequent polyaddition reaction. These multiblock hybrids formed stable and reversible β-sheet network structures via self-assembly through casting and yielded unique thin films with mechanical toughness and self-healing capability. Both the type and length of the peptide block affected not only the mechanical properties, but also the self-healing efficiency of the resultant microfilms. The oligo(Ala)-based microfilms indicated significantly lower Mc values and higher self-healing efficiency compared with the other peptide films (i.e., Gly, Val, and Leu-based films), thereby suggesting the high self-assembling propensity of oligo(Ala) blocks in the film. Owing to such unique film characteristics, self-supporting and ultraflexible nanofilms can be prepared as functional coatings for nanostructured surfaces. We believe that this peptide–polymer hybrid strategy will enable the creation of structurally and functionally diverse multiblock polymers that will be beneficial to nanotechnology and biomedicine, as well as provide new directions for the design of smart self-assembling nanomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by Grants-in-Aid for Scientific Research (KAKENHI) (No. 17K04994 and 20H02767) from the Japan Society for the Promotion of Science (JSPS), and a MEXT-Supported Program for the Strategic Research Foundation at Private University.Notes and references
- M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577–585 CrossRef CAS PubMed.
- C. Tamerler and M. Sarikaya, Acta Biomater., 2007, 3, 289–299 CrossRef CAS PubMed.
- A. Miserz, J. C. Weaver and O. Chaudhuri, J. Mater. Chem. B, 2015, 3, 13–24 RSC.
- L. Rodríguez-Arco, A. Poma, L. Ruiz-Pérez, E. Scarpa, K. Ngamkham and G. Battaglia, Biomaterials, 2019, 192, 26–50 CrossRef PubMed.
- C. B. Anfinsen, Science, 1973, 181, 223–230 CrossRef CAS PubMed.
- J. C. M. van Hest and D. A. Tirrell, Chem. Commun., 2001, 1897–1904 RSC.
- H. Yokoi, T. Kinoshita and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS PubMed.
- T. Koga, M. Matsuoka and N. Higashi, J. Am. Chem. Soc., 2005, 127, 17596–17597 CrossRef CAS PubMed.
- M. G. Ryadnov and D. N. Woolfson, Nat. Mater., 2013, 2, 329–332 CrossRef PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- N. Higashi, R. Yoshikawa and T. Koga, RSC Adv., 2020, 10, 15947–15954 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2002, 294, 1684–1688 CrossRef PubMed.
- K. Rajagopal and J. P. Schneider, Curr. Opin. Struct. Biol., 2004, 14, 480–486 CrossRef CAS PubMed.
- Y. Hirano and D. J. Mooney, Adv. Mater., 2004, 16, 17–25 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- D. N. Woolfson and Z. N. Mahmoud, Chem. Soc. Rev., 2010, 39, 3464–3479 RSC.
- J. A. Fallas, L. E. R. O’Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC.
- K. Murai, T. Kinoshita, K. Nagata and M. Higuchi, Langmuir, 2016, 32, 9351–9359 CrossRef CAS PubMed.
- P. Katyal, M. Meleties and J. K. Montclare, ACS Biomater. Sci. Eng., 2019, 5, 4132–4147 CrossRef CAS PubMed.
- H. Inaba and K. Matsuura, Chem. Rec., 2019, 19, 843–853 CrossRef CAS PubMed.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- C. Vepari and D. L. Kaplan, Prog. Polym. Sci., 2007, 32, 991–1007 CrossRef CAS PubMed.
- J. G. Hardy, L. M. Romer and T. R. Scheibel, Polymer, 2008, 49, 4309–4327 CrossRef CAS.
- G. Askarieh, M. Hedhammar, K. Nordling, A. Saenz, C. Casals, A. Rising, J. Johansson and S. D. Knight, Nature, 2010, 465, 236–239 CrossRef CAS PubMed.
- F. Hagn, L. Eisoldt, J. G. Hardy, C. Vendrely, M. Coles, T. Sheibel and H. A. Kessler, Nature, 2010, 465, 239–244 CrossRef CAS PubMed.
- A. H. Simmons, C. A. Michal and L. W. Jelinski, Science, 1996, 271, 84–87 CrossRef CAS PubMed.
- J. Gatesy, C. Hayashi, D. Motriuk, J. Woods and R. Lewis, Science, 2001, 291, 2603–2605 CrossRef CAS PubMed.
- S. Rammensee, U. Slotta, T. Scheibel and A. R. Bausch, Angew. Chem., Int. Ed., 2009, 48, 3584–3596 CrossRef PubMed.
- A. Rising, Acta Biomater., 2014, 10, 1627–1631 CrossRef CAS PubMed.
- A. D. Malay, T. Suzuki, T. Katashima, N. Kono, K. Arakawa and K. Numata, Sci. Adv., 2020, 6, eabb6030 CrossRef CAS PubMed.
- Y. Mei, K. L. Beers, H. C. MichellesByrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS PubMed.
- J. Hentschel and H. G. Börner, J. Am. Chem. Soc., 2006, 128, 14142–14149 CrossRef CAS PubMed.
- H. R. Marsden, A. V. Korobko, E. N. M. van Leeuwen, E. M. Pouget, S. J. Veen, N. A. J. M. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386–9393 CrossRef PubMed.
- J. Kopeček and J. Yang, Angew. Chem., Int. Ed., 2012, 51, 7396–7417 CrossRef PubMed.
- T. Koga, S. Kamiwatari and N. Higashi, Langmuir, 2013, 29, 15477–15484 CrossRef CAS PubMed.
- S. E. Grieshaber, B. A. Paik, S. Bai, K. L. Kiick and X. Jia, Soft Matter, 2013, 9, 1589–1599 RSC.
- S. Nishimura, N. Higashi and T. Koga, Chem. – Eur. J., 2017, 23, 15050–15058 CrossRef CAS PubMed.
- S. Nishimura, N. Hokazono, Y. Taki, H. Motoda, Y. Morita, K. Yamamoto, N. Higashi and T. Koga, ACS Appl. Mater. Interfaces, 2019, 11, 24577–24587 CrossRef CAS PubMed.
- N. Higashi, K. Narimatsu, M. Okumura, S. Nishimura and T. Koga, ACS Omega, 2019, 4, 8104–8111 CrossRef CAS PubMed.
- T. Koga, T. Matsuoka, Y. Morita and N. Higashi, Mater. Adv., 2021, 2, 4068–4074 RSC.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- A. C. Jackson, J. A. Bartelt and P. V. Braun, Adv. Funct. Mater., 2011, 21, 4705–4711 CrossRef CAS.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- O. S. Taskin, B. Kiskan and Y. Yagci, Macromolecules, 2013, 46, 8773–8778 CrossRef CAS.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Y. Sun, A. L. Wollenberg, T. M. O’Shea, Y. Cui, Z. H. Zhou, M. V. Sofroniew and T. J. Deming, J. Am. Chem. Soc., 2017, 139, 15114–15121 CrossRef CAS PubMed.
- K. Imato, A. Takahara and H. Otsuka, Macromolecules, 2015, 48, 5632–5639 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- C. Zeng, H. Seino, J. Ren, K. Hatanaka and N. Yoshie, Macromolecules, 2013, 46, 1794–1802 CrossRef CAS.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231–5239 CrossRef CAS PubMed.
- C. Zhou, B. Leng, J. Yao, J. Qian, X. Chen, P. Zhou, D. P. Knight and Z. Shao, Biomacromolecules, 2006, 7, 2415–2419 CrossRef CAS PubMed.
- L. R. Wu, J. Yang, K. Wu and J. Kopeček, Biomacromolecules, 2009, 10, 2319–2327 CrossRef PubMed.
- E. Croisier, S. Liang, T. Schweizer, S. Balog, M. Mionic, R. Snellings, J. Cugnoni, V. Michaud and H. Frauenrath, Nat. Commun., 2014, 5, 4728 CrossRef CAS PubMed.
- S. Nishimura, N. Higash and T. Koga, Polym. Chem., 2019, 10, 71–76 RSC.
- S. Nishimura, N. Higashi and T. Koga, Chem. Commun., 2019, 55, 1498–1501 RSC.
- T. Miyazawa and E. R. Blout, J. Am. Chem. Soc., 1961, 83, 712–719 CrossRef CAS.
- F. G. Torres, O. P. Troncoso, C. Torres and W. Cabrejos, Mater. Sci. Eng., C, 2013, 33, 1432–1437 CrossRef CAS PubMed.
- P. J. Nowatzki and D. A. Tirrell, Biomaterials, 2004, 25, 1261–1267 CrossRef CAS PubMed.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, Wiley-Interscience Publication, 4th edn, 1999 Search PubMed.
- Z. Jakšić and O. Jakšić, Biomimetics, 2020, 5, 24 CrossRef PubMed.
- A. A. Mamedov and N. A. Kotov, Langmuir, 2000, 16, 5530–5533 CrossRef CAS.
- F. Mallwitz and A. Laschewsky, Adv. Mater., 2005, 17, 1296–1299 CrossRef CAS.
- Y. Takaku, H. Suzuki, I. Ohta, D. Ishii, Y. Muranaka, M. Shimomura and T. Hariyama, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7631–7635 CrossRef CAS PubMed.
- E. Endo, M. Mitsuishi and T. Miyashita, J. Mater. Chem., 2008, 18, 1302–1308 RSC.
- R. Vendamme, S. Onoue, A. Nakao and T. Kunitake, Nat. Mater., 2006, 5, 494–501 CrossRef CAS PubMed.
- Y. Okamura, K. Kabata, M. Kinoshita, D. Saitoh and S. Takeoka, Adv. Mater., 2009, 21, 4388–4392 CrossRef CAS PubMed.
- H. Zhang, A. Masuda, R. Kawakami, K. Yarinome, R. Saito, Y. Nagase, T. Nemoto and Y. Okamura, Adv. Mater., 2017, 29, 1703139 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure for peptide synthesis, Fig. S1–Fig. S9, and Table S1. See DOI: 10.1039/d1ma00823d |
| This journal is © The Royal Society of Chemistry 2021 |