
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced proton conductivity in a flexible metal–organic framework promoted by single-crystal-to-single-crystal transformation†
Xi
Chen
a,
Zhongyue
Zhang
*b,
Jin
Chen
a,
Sergei
Sapchenko
a,
Xue
Han
a,
Ivan
da-Silva
 c,
Ming
Li
d,
Inigo J.
Vitorica-Yrezabal
a,
George
Whitehead
a,
Chiu C.
Tang
e,
Kunio
Awaga
b,
Sihai
Yang
*a and
Martin
Schröder
*a
c,
Ming
Li
d,
Inigo J.
Vitorica-Yrezabal
a,
George
Whitehead
a,
Chiu C.
Tang
e,
Kunio
Awaga
b,
Sihai
Yang
*a and
Martin
Schröder
*a
aDepartment of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Sihai.Yang@manchester.ac.uk; M.Schroder@manchester.ac.uk
bDepartment of Chemistry, Nagoya University, Furocho, Chikusa, 464-8602, Nagoya, Japan. E-mail: zhang.zhongyue@i.mbox.nagoya-u.ac.jp
cISIS Pulsed Neutron and Muon Source, Rutherford Appleton Laboratory, Oxfordshire OX11 0QX, UK
dFaculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK
eDiamond Light Source, Harwell Science Campus, Oxfordshire, OX11 0DE, UK
First published on 17th November 2020
Abstract
MFM-722(Pb)-DMA undergoes a single-crystal-to-single-crystal (SCSC) transformation to give MFM-722(Pb)-H2O via ligand substitution upon exposure to water vapour. In situ single crystal impedance spectroscopy reveals an increase in proton conductivity due to this structural transition, with MFM-722(Pb)-H2O showing a proton conductivity of 6.61 × 10−4 S cm−1 at 50 °C and 98% RH. The low activation energy (Ea = 0.21 eV) indicates that the proton conduction follows a Grotthuss mechanism.
Metal–organic framework (MOF) materials are highly crystalline with rich structural diversity,1 and are excellent candidates for gas storage,2 substrate binding and delivery3 and catalysis.4 Recently, MOFs showing high proton conductivities (>10−2 S cm−1) are considered as emerging candidates for applications in proton exchange membrane fuel cells.5–11 Two approaches have been developed to enhance the proton conductivity in porous MOFs: (i) loading the pores of MOFs with guest molecules of intrinsic proton conductivity, such as H2SO4,5 H3PO4,6 urea7 and an ionic liquid,12 and (ii) decorating the bridging organic ligands with acidic groups (e.g., –SO3H,8 –PO3H2,10 –COOH13,14) to assemble hydrogen-bonding networks within the pore to mediate proton transport. For example, H2SO4@(NH2)2-MIL-125 shows a high proton conductivity of 2.20 × 10−2 S cm−1 at 80 °C under 98% RH.5 Recently, a flexible BUT-8(Cr)A with sulfonic acid (–SO3H) groups within the pore has been found to exhibit a conductivity of 1.27 × 10−1 S cm−1 at 80 °C under 100% RH.8 In contrast, improving the proton conductivity of non-porous MOFs can be highly challenging due to limitations in their design to enable flexible chemical modification or doping.15,16 Phase transition has been reported to be an effective approach to tune the property of flexible MOFs,17 and this can be triggered by ligand substitution,18 guest uptake,19 or by changes in temperature20 and/or pressure.21 However, studies on the impact of single-crystal-to-single-crystal (SCSC) transformations on proton conductivity in MOFs have been reported rarely.22,23 We report herein the SCSC transformation via ligand substitution in a nonporous Pb(II)-based MOF, MFM-722(Pb)-DMA, and the enhancement of proton conductivity in the resultant MFM-722(Pb)-H2O. The diverse coordination environment of Pb(II) ions naturally endows Pb(II)-based MOFs with abundant structural diversity and flexibility. Single crystal impedance spectroscopy can reduce the impact of grain boundaries that are inherent in bulk-pellet measurements,24 and has been employed here to evaluate the change of proton conductivity of MFM-722(Pb)-DMA during the SCSC transformation on exposure to water vapour.
MFM-722(Pb)-DMA was synthesised by solvothermal reaction of Pb(NO3)2 and biphenyl-3,3′,5,5′ tetracarboxylic acid (H4L)25 in DMA at 90 °C for 3 days and isolated as colourless rod-shaped single crystals. The single crystals were collected on cooling by filtration and dried in air. Single crystal X-ray diffraction revealed that MFM-722(Pb)-DMA, [Pb2(L)(DMA)2], crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , featuring a three dimensional framework comprised of extended Pb(II) oxide chains [Pb2O10]∞ bridged by two crystallographically-independent ligands L4− (Fig. 1d and Fig. S1, Table S1, ESI†). This yields a narrow channel along the c axis, which is filled with two coordinated DMA molecules, resulting in a nonporous structure (Fig. 1d). There are two crystallographically-independent Pb atoms, both of which are 7 coordinated by oxygen donors (Fig. 1c). Pb(1) is coordinated to six oxygen atoms from the carboxylate groups (O1, O1′, O4, O5, O6, O7) as well as another oxygen atom (O10) from a terminal DMA molecule. Four Pb–O bonds range from 2.346(6) to 2.648(7) Å, and the other three are longer [2.754(6), 2.776(6) and 2.903(7) Å], consistent with the reported values for Pb–O bonds (Table S2, ESI†). Pb(2) is surrounded by six oxygen atoms (O2, O3, O3′, O4, O8, O9) from the carboxylate groups of five different ligands and another oxygen atom (O9) from the DMA molecule. Similarly, Pb(2) has four short Pb–O bonds and three long secondary Pb–O bonds (Table S2 and Fig. S4, ESI†). According to the VSEPR model,23 the geometry of both [PbO7] polyhedron can be described as a distorted ψ-trigonal bipyramid (ψ-TBP) with a vacant vertex in the equatorial plane. The powder X-ray diffraction (PXRD) patterns of MFM-722(Pb)-DMA confirm its phase purity (Fig. S13, ESI†). TGA shows that the coordinated DMA molecules can be removed at 125–220 °C (weight loss of 19%, calc. 19%), followed by a framework decomposition at ∼400 °C (Fig. S15, ESI†).
, featuring a three dimensional framework comprised of extended Pb(II) oxide chains [Pb2O10]∞ bridged by two crystallographically-independent ligands L4− (Fig. 1d and Fig. S1, Table S1, ESI†). This yields a narrow channel along the c axis, which is filled with two coordinated DMA molecules, resulting in a nonporous structure (Fig. 1d). There are two crystallographically-independent Pb atoms, both of which are 7 coordinated by oxygen donors (Fig. 1c). Pb(1) is coordinated to six oxygen atoms from the carboxylate groups (O1, O1′, O4, O5, O6, O7) as well as another oxygen atom (O10) from a terminal DMA molecule. Four Pb–O bonds range from 2.346(6) to 2.648(7) Å, and the other three are longer [2.754(6), 2.776(6) and 2.903(7) Å], consistent with the reported values for Pb–O bonds (Table S2, ESI†). Pb(2) is surrounded by six oxygen atoms (O2, O3, O3′, O4, O8, O9) from the carboxylate groups of five different ligands and another oxygen atom (O9) from the DMA molecule. Similarly, Pb(2) has four short Pb–O bonds and three long secondary Pb–O bonds (Table S2 and Fig. S4, ESI†). According to the VSEPR model,23 the geometry of both [PbO7] polyhedron can be described as a distorted ψ-trigonal bipyramid (ψ-TBP) with a vacant vertex in the equatorial plane. The powder X-ray diffraction (PXRD) patterns of MFM-722(Pb)-DMA confirm its phase purity (Fig. S13, ESI†). TGA shows that the coordinated DMA molecules can be removed at 125–220 °C (weight loss of 19%, calc. 19%), followed by a framework decomposition at ∼400 °C (Fig. S15, ESI†).
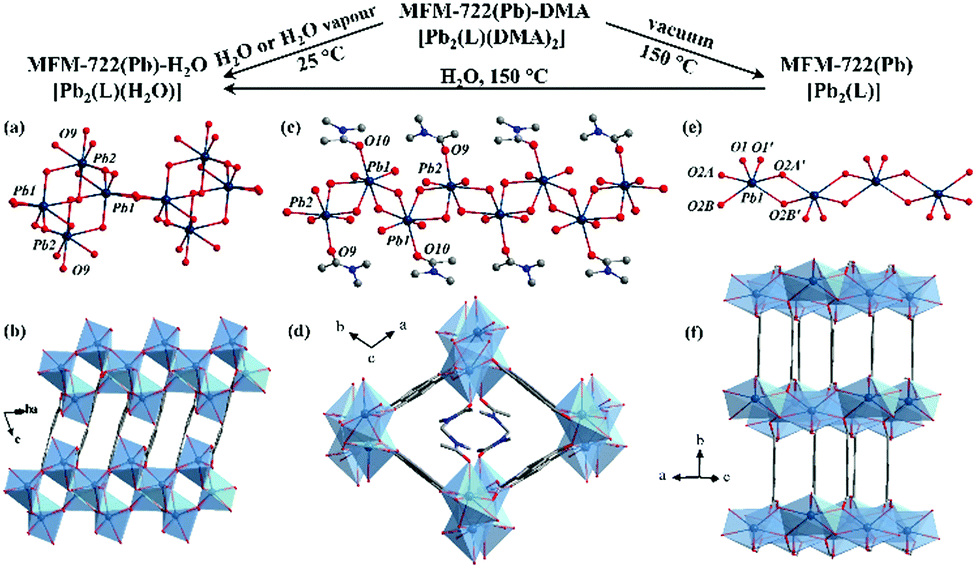 | ||
| Fig. 1 Coordination environment of Pb(II) centres and crystal structures of (a), (b) MFM-722(Pb)-H2O, (c), (d) MFM-722(Pb)-DMA, and (e), (f) MFM-722(Pb) (Pb: dark blue; O: red; C: grey; N: blue; Pb–O polyhedra: light blue). Hydrogen atoms are omitted for clarity. The projected views in (b), (d), (f) are along the ab plane, c axis, and ac plane, respectively. | ||
By immersing the single crystals of MFM-722(Pb)-DMA in water for 3 h or exposing to water vapour for 10 h at 25 °C, we observed a SCSC transformation via ligand substitution to give MFM-722(Pb)-H2O, [Pb2(L)(H2O)]. The space group of MFM-722(Pb)-H2O is also P. On ligand substitution, adjacent chains of [Pb2O10]∞ inter-connect via carboxylate oxygen centres to assemble into a network of layers of [Pb4O18]∞ along the c axis (Fig. 1a and Fig. S7, ESI†). These layers are further bridged by L4− in the ab plane to afford a nonporous three-dimensional structure (window size of 1 × 2 Å; Fig. 1b and Fig. S2, ESI†). The lead oxide layer consists of tetra-nuclear [Pb4O18] clusters, with each cluster containing four Pb atoms from two crystallographically-independent Pb(II) sites connected via corner-sharing and edge-sharing oxygen centres (Fig. S7a, ESI†). Pb(1#) is coordinated to seven oxygen atoms belonging to monodentate carboxylates (O5, O6, O8) and the bidentate-chelate carboxylates (O3, O4, O5′, O6) from five different ligands. Pb(2#) is surrounded by seven oxygen atoms from five carboxylates and one oxygen atom (O9) from a terminal coordinated H2O molecule. The geometry of both [Pb(1#)O7] and [Pb(2#)O8] polyhedra can be described as ψ-TBP (Fig. S5, ESI†). A close examination reveals that the layers of [Pb4O18]∞ are also stabilised by two-fold hydrogen bonding in the ab plane (Fig. 3b), O9–H⋯O2* and O9*–H⋯O2 [O9⋯O2 = 2.837(34) Å, ∠O9HO2 = 143.8(17)°]. This is supplemented by additional hydrogen bonding interactions between O9–H⋯O9* and O9*–H⋯O9 [O9⋯O9 = 3.504(43) Å, ∠O9HO9 = 140.2 (19)°, Fig. 3a and Table S4, ESI†]. The phase transition of bulk materials was confirmed by PXRD (Fig. S13, ESI†). ATR-IR spectroscopy confirms that the peak at 1644 cm−1 attributed to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching vibration of DMA in MFM-722(Pb)-DMA is not present in MFM-722(Pb)-H2O (Fig. S14, ESI†). TGA of MFM-722(Pb)-H2O (Fig. S16, ESI†) shows an initial weight loss of 1.5% at 75 °C owing to the volatilisation of surface and interstitial water molecules, the presence of which has been confirmed by FTIR spectroscopy (Fig. S14, ESI†). This is followed by another weight loss of 2.4% between 75 and 200 °C, corresponding to the loss of the coordinated H2O molecules on Pb(2#) (calc. 2.4%). Framework decomposition occurs at ∼400 °C.
O) stretching vibration of DMA in MFM-722(Pb)-DMA is not present in MFM-722(Pb)-H2O (Fig. S14, ESI†). TGA of MFM-722(Pb)-H2O (Fig. S16, ESI†) shows an initial weight loss of 1.5% at 75 °C owing to the volatilisation of surface and interstitial water molecules, the presence of which has been confirmed by FTIR spectroscopy (Fig. S14, ESI†). This is followed by another weight loss of 2.4% between 75 and 200 °C, corresponding to the loss of the coordinated H2O molecules on Pb(2#) (calc. 2.4%). Framework decomposition occurs at ∼400 °C.
We sought to monitor the change of proton conductivity of MFM-722(Pb)-DMA in situ during the SCSC transformation using single crystal AC impedance spectroscopy (Fig. 2). The measurements were carried out on two single crystals of MFM-722(Pb)-DMA (sizes of 468 × 64 × 54 μm, Fig. 2a, and 325 × 41 × 31 μm, Fig. 2b–d) using a conventional two-contact wire-paste method.24 The single crystals were rested on an insulating glass substrate in a humidity chamber. Soft gold wires (Φ = 25 μm) connected to Pt foil electrodes were contacted with single crystals using gold paste to enable measurement of the proton conductivity (Fig. S18a, ESI†). Analysis of the face index confirms that the proton conductivity was measured along the crystallographic c axis in MFM-722(Pb)-DMA, which remained the same for MFM-722(Pb)-H2O on SCSC transformation (Fig. S18b and c, ESI†). Nyquist plots show a typical semi-circle in the high frequency region indicative of the intrinsic conductivity of the material, with the tail at low frequency representing the blocking of protons at the electrode interface.26 At 25 °C and 98% RH, the proton conductivity of the single crystals increased gradually from 3.64 × 10−5 S cm−1 (at 0 h) to 8.09 × 10−5 S cm−1 (at 9 h), which then stabilised to 1.33 × 10−4 S cm−1 over 30 h (Fig. 2a). The increase in conductivity originates from the phase transition from MFM-722(Pb)-DMA to MFM-722(Pb)-H2O (Fig. S19, ESI†), and Fig. 2b, c show the temperature dependence of the single crystal proton conductivity of MFM-722(Pb)-H2O. The conductivity increases with increasing temperature and reaches 6.61 × 10−4 S cm−1 at 50 °C at 98% RH. This value is comparable to a Cu-MOF system with the proton conductivity of 5.48 × 10−3 S cm−1 at 60 °C and 95% RH after the phase transition (Table S8, ESI†).22 At temperatures above 50 °C, the proton conductivity of MFM-722(Pb)-H2O decreases slightly to 3.55 × 10−4 S cm−1 at 75 °C, most likely due to the partial loss of surface/interstitial water molecules. No apparent structural change is observed for MFM-722(Pb)-H2O between 25 and 75 °C at 98% RH (Fig. S20, ESI†), and the proton conductivity returns to 1.16 × 10−4 S cm−1 on cooling to 25 °C (Fig. S21, ESI†). The activation energy (Ea) for MFM-722(Pb)-H2O was calculated from the variable temperature impedance spectra to be 0.21 eV (Fig. 2d), suggesting that the proton diffusion is governed by the Grotthuss mechanism, where protons hop along the hydrogen bonding networks.
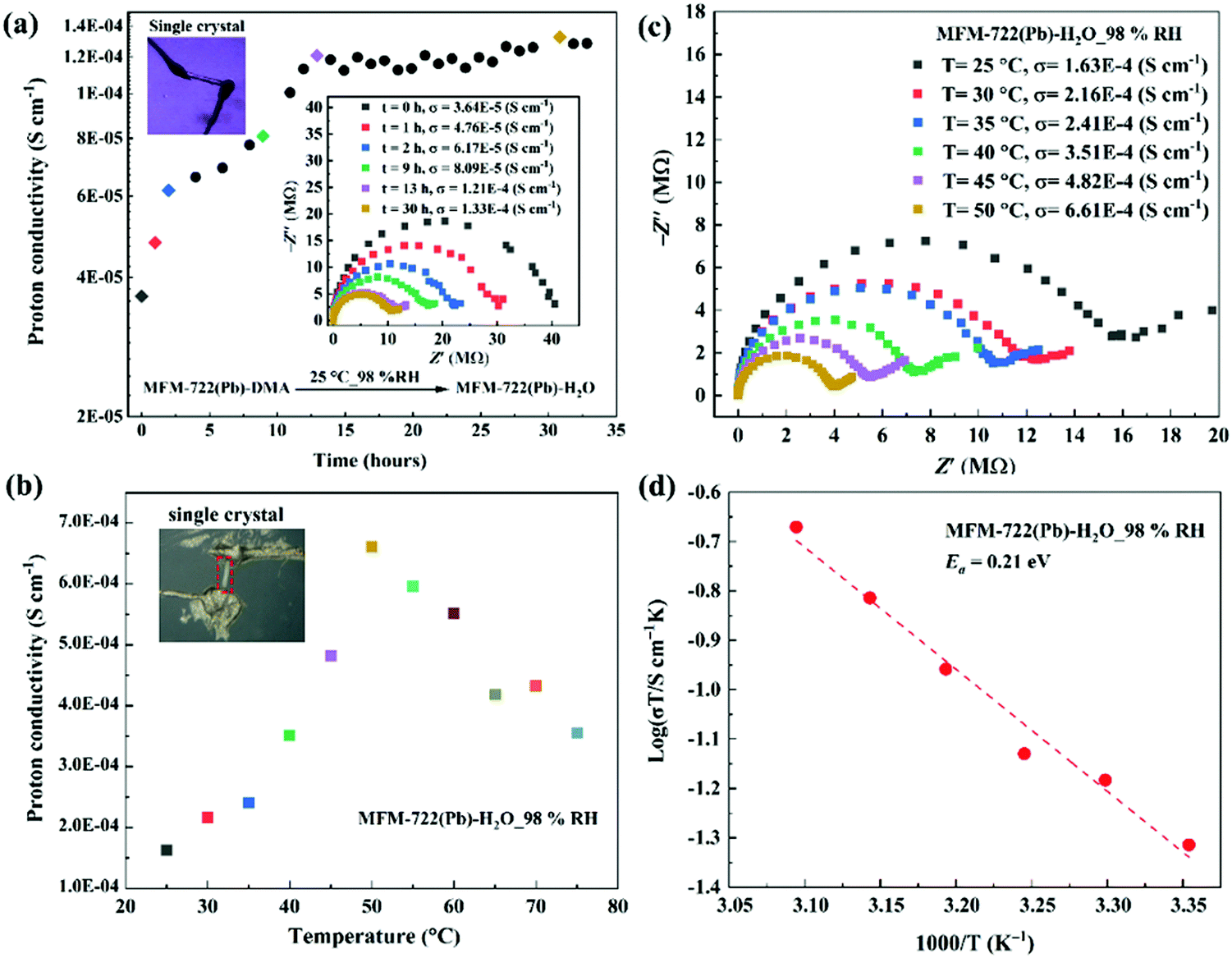 | ||
| Fig. 2 Single crystal impedance spectra and proton conductivity of MFM-722(Pb)-DMA and MFM-722(Pb)-H2O. (a) Time dependence of the single crystal proton conductivity during the phase transition from MFM-722(Pb)-DMA to MFM-722(Pb)-H2O at 25 °C and 98% RH; the insert figure shows selected Nyquist plots. (b) Temperature dependence of the single crystal proton conductivity of MFM-722(Pb)-H2O at 98% RH at 25–75 °C; the corresponding Nyquist plots are shown in (c). (d) Arrhenius plot of the proton conductivity of MFM-722(Pb)-H2O under 98%RH. The proton conductivity of bulk MFM-722(Pb)-DMA has also been measured using compressed pellets at 21 °C and 98% RH (Fig. S22, ESI†). | ||
The proton conductivity of bulk MFM-722(Pb)-DMA has also been measured using compressed pellets at 21 °C at 98% RH (Fig. S22, ESI†). The proton conductivity of the pellet of MFM-722(Pb)-DMA (3.47 × 10−8 S cm−1, Fig. S23, ESI†) increases slowly over time, and reaches 1.20 × 10−4 S cm−1 at 21 °C and 98% RH over ∼7 days linked to the phase transition and formation of MFM-722(Pb)-H2O (Fig. S22, ESI†). Given the nonporous structure of this material, additional time is required to allow diffusion of water through the compressed pellet to drive the phase transition compared to the single crystal. Distinct to systems reported in literature9 where the single crystal impedance measurements often yield a ∼100-fold increase in proton conductivity compared to that obtained from measurements on bulk sample (due to the elimination of resistance from the crystallite boundaries and the judicious utilisation of the anisotropic conductivity), single crystals and pellets of MFM-722(Pb)-H2O exhibit similar values of conductivity.
A detailed examination of the hydrogen bonding network in MFM-722(Pb)-H2O identified the crystallographic a axis as the potential pathway for proton hopping (dashed red lines in Fig. 3c). Six adjacent oxygen centres (O2, O7, O9, O2*, O7*, O9*) from two neighbouring [Pb]4 clusters form a supra-octahedron [O⋯O = 2.837(34)–3.504(43) Å, Table S3, ESI†] that enables proton hopping (Fig. 3d). A continuous hydrogen bonding pathway is established by packing these hexa-oxygen supra-octahedra along the a axis with a gap of ∼4.5 Å, which can be bridged by interstitial water molecules (Fig. 3e). Indeed, the water adsorption isotherm of MFM-722(Pb)-H2O at 25 °C shows a type-II profile with a total uptake of 3.32 mmol g−1 (Fig. S12, ESI†), attributed to the adsorption of surface/interstitial water. Thus, the a axis is predicted to be the conducting axis of MFM-722(Pb)-H2O. However, due to the rod-shaped single crystal, accurate proton conductivities can only be measured along the c axis, which has a much greater barrier for proton hopping due to the ligands (Fig. S8, ESI†). This explains the similar conductivity observed in the single crystal and powder impedance measurements.
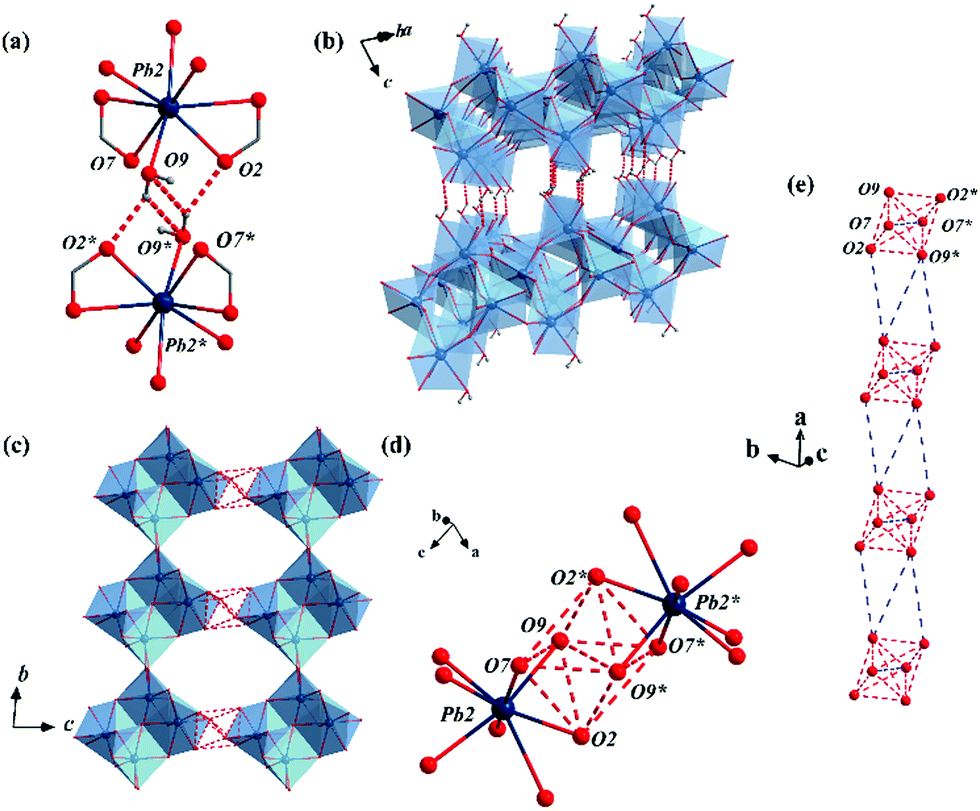 | ||
| Fig. 3 (a) View of the hydrogen bonds of O9–H⋯O2 and O9–H⋯O9 in MFM-722(Pb)-H2O. (b) View of the layers of [Pb4O18]∞ stabilised via inter-layer hydrogen bonding in MFM-722(Pb)-H2O. The linker is omitted for clarity. (c) Potential pathways for proton transfer in MFM-722(Pb)-H2O along the a axis. The linker is omitted for clarity. (d) View of the [O6] supra-octahedron. (e) Continuous hydrogen bonding pathway consisting of the [O6] supra-octahedra viewed along the a axis. Pb: dark blue; O: red; C: grey; N: blue; Pb–O polyhedra: light blue; hydrogen bond: dash red lines. | ||
Desolvation of MFM-722(Pb)-DMA was achieved by heating at 150 °C for 12 h under vacuum to give a fully desolvated phase. The crystal structure of desolvated MFM-722(Pb) was determined by Rietveld refinement of high-resolution synchrotron X-ray powder diffraction data (Fig. S10, ESI†). The space group of MFM-722(Pb), [Pb2(L)], is C2/m with only one crystallographically-independent six co-ordinate Pb(II) centre bound to carboxylate oxygen centres (Fig. 1e and f). The geometry of the [PbO6] polyhedron is distorted ψ-TBP (Fig. S6 and Table S2, ESI†), with extended [Pb2O8]∞ chains inter-connected by the asymmetric [Pb2O2] unit from two different carboxylate groups. At 25 °C and 0% RH, MFM-722(Pb) exhibits negligible proton conductivity but an enhanced dielectric constant of 8.99 at 100 kHz compared with that of 5.58 for MFM-722(Pb)-DMA (Fig. S24, ESI†). The dielectric constant of MFM-722(Pb) is higher than that of HKUST-1 (1.74 at 100 kHz),27 but is lower than that of an In-btc (btc3− = 1,2,3-benzenetricarboxylate) based MOF (28.0 at 100 kHz).28 Thus, the phase transition in MFM-722(Pb) has a positive impact on the dielectric properties. MFM-722(Pb) can be rehydrated to MFM-722(Pb)-H2O by heating in water at 150 °C for 1.5 days (Fig. S13, ESI†).
In summary, a new flexible Pb(II)-based nonporous MOF has been synthesised and its framework flexibility characterised. On exposure to water, MFM-722(Pb)-DMA undergoes a SCSC transformation to MFM-722(Pb)-H2O, induced by the substitution of coordinated DMA by water ligands at room temperature. The single crystal proton conductivity of MFM-722(Pb)-H2O reaches 6.61 × 10−4 S cm−1 at 50 °C under 98% RH, a four-fold enhancement compared with MFM-722(Pb)-DMA. The low activation energy (Ea = 0.21 eV) is consistent with the Grotthuss mechanism with the protons hopping along the hydrogen bonding network. This study will promote future design of flexible nonporous MOFs showing enhanced proton conductivity on phase transition.
We thank EPSRC (EP/I011870, EP/P001386), China Scholarship Council (CSC) and the University of Manchester for funding. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 742401, NANOCHEM). We are grateful to Diamond Light Source for access to the Beamline I11.
Conflicts of interest
The authors declare no competing financial interests.Notes and references
- D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O’Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
- X. Han, S. Yang and M. Schröder, Nat. Rev. Chem., 2019, 3, 108 CrossRef CAS.
- L. Wang, M. Zheng and Z. Xie, J. Mater. Chem. B, 2018, 6, 707–717 RSC.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604 CrossRef CAS.
- W. Deng, P. Naresh Kumar, W. Li, C. Kashi, M. Yao, G. Wu and G. Xu, Inorg. Chim. Acta, 2020, 502, 119317 CrossRef CAS.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640 CrossRef CAS.
- M. K. Sarango-Ramírez, D. Lim, D. I. Kolokolov, A. E. Khudozhitkov, A. G. Stepanov and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 6861 CrossRef.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J. Li and B. Chen, Nat. Energy, 2017, 2, 877 CrossRef CAS.
- K. Otake, K. Otsubo, T. Komatsu, S. Dekura, J. M. Taylor, R. Ikeda, K. Sugimoto, A. Fujiwara, C. Chou, A. W. Sakti, Y. Nishimura, H. Nakai and H. Kitagawa, Nat. Commun., 2020, 11, 843 CrossRef CAS.
- S. Kim, B. Joarder, J. A. Hurd, J. Zhang, K. W. Dawson, B. S. Gelfand, N. E. Wong and G. K. H. Shimizu, J. Am. Chem. Soc., 2018, 140, 1077 CrossRef CAS.
- S. Horike, Y. Kamitsubo, M. Inukai, T. Fukushima, D. Umeyama, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 4612 CrossRef CAS.
- X. Sun, W. Deng, H. Chen, H. Han, J. M. Taylor, C. Wan and G. Xu, Chem. – Eur. J., 2017, 23, 1248–1252 CrossRef CAS.
- P. Rought, C. Marsh, S. Pili, I. P. Silverwood, V. G. Sakai, M. Li, M. S. Brown, S. P. Argent, I. Vitorica-Yrezabal, G. Whitehead, M. R. Warren, S. Yang and M. Schröder, Chem. Sci., 2019, 10, 1492 RSC.
- Y. Qin, M. Xue, B. Dou, Z. Sun and G. Li, New J. Chem., 2020, 44, 2741 RSC.
- S. Pili, S. P. Argent, C. G. Morris, P. Rought, V. García-Sakai, I. P. Silverwood, T. L. Easun, M. Li, M. R. Warren, C. A. Murray, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 6352 CrossRef CAS.
- S. Pili, P. Rought, D. I. Kolokolov, I. da-Silva, Y. Cheng, C. Marsh, I. P. Silverwood, V. García-Sakai, M. Li, J. J. Titman, L. Knight, L. L. Daemen, A. J. Ramirez-Cuesta, C. C. Tang, A. G. Stepanov, S. Yang and M. Schröder, Chem. Mater., 2018, 30, 7593 CrossRef CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062 RSC.
- R. Li, H. Liu, C. Zhou, Z. Chu, J. Lu, S. Wang, J. Jin and W. Yan, Inorg. Chem. Front., 2020, 7, 1880 RSC.
- S.-H. Lo, L. Feng, K. Tan, Z. Huang, S. Yuan, K.-Y. Wang, B.-H. Li, W.-L. Liu, G. S. Day, S. Tao, C.-C. Yang, T.-T. Luo, C.-H. Lin, S.-L. Wang, S. J. L. Billinge, K.-L. Lu, Y. J. Chabal, X. Zou and H.-C. Zhou, Nat. Chem., 2020, 12, 90 CrossRef.
- R. F. Mendes, P. Barbosa, E. M. Domingues, P. Silva, F. Figueiredo and F. A. Almeida Paz, Chem. Sci., 2020, 11, 6305 RSC.
- A. Sussardi, C. L. Hobday, R. J. Marshall, R. S. Forgan, A. C. Jones and S. A. Moggach, Angew. Chem., Int. Ed., 2020, 132, 8195 CrossRef.
- S. Nakatsuka, Y. Watanabe, Y. Kamakura, S. Horike, D. Tanaka and T. Hatakeyama, Angew. Chem., Int. Ed., 2020, 59, 1435 CrossRef CAS.
- Y. Wei, X. Hu, Z. Han, X. Dong, S. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2017, 139, 3505 CrossRef CAS.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, J. Am. Chem. Soc., 2016, 138, 14772 CrossRef CAS.
- X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef CAS.
- J. T. S. Irvine, D. C. Sinclair and A. R. West, Adv. Mater., 1990, 2, 132 CrossRef CAS.
- R. Scatena, Y. T. Guntern and P. Macchi, J. Am. Chem. Soc., 2019, 141, 9382 CrossRef CAS.
- S. Kamal, K. R. Chiou, B. Sainbileg, A. I. Inamdar, M. Usman, A. Pathak, T.-T. Luo, J.-W. Chen, M. Hayashi, C.-H. Hung and K.-L. Lu, J. Mater. Chem. C, 2020, 8, 9724 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre under deposition numbers CCDC 1995412, 1995413 and 2018703. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05270a |
| This journal is © The Royal Society of Chemistry 2021 |