
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous and scalable synthesis of a porous organic cage by twin screw extrusion (TSE)†
Benjamin D.
Egleston
 a,
Michael C.
Brand
a,
Francesca
Greenwell
a,
Michael E.
Briggs
a,
Stuart L.
James
b,
Andrew I.
Cooper
a,
Deborah E.
Crawford
*c and
Rebecca L.
Greenaway
*ad
a,
Michael C.
Brand
a,
Francesca
Greenwell
a,
Michael E.
Briggs
a,
Stuart L.
James
b,
Andrew I.
Cooper
a,
Deborah E.
Crawford
*c and
Rebecca L.
Greenaway
*ad
aDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool, L7 3NY, UK
bSchool of Chemistry and Chemical Engineering, Queen's University Belfast, 39-123 Stranmillis Road, Belfast, Northern Ireland BT9 5AG, UK
cSchool of Chemistry and Biosciences, University of Bradford, Richmond Road, Bradford, BD7 1DP, UK. E-mail: d.crawford@bradford.ac.uk
dDepartment of Chemistry, Imperial College London, White City Campus, Wood Lane, London, W12 0BZ, UK. E-mail: r.greenaway@imperial.ac.uk
First published on 11th May 2020
Abstract
The continuous and scalable synthesis of a porous organic cage (CC3), obtained through a 10-component imine polycondensation between triformylbenzene and a vicinal diamine, was achieved using twin screw extrusion (TSE). Compared to both batch and flow syntheses, the use of TSE enabled the large scale synthesis of CC3 using minimal solvent and in short reaction times, with liquid-assisted grinding (LAG) also promoting window-to-window crystal packing to form a 3-D diamondoid pore network in the solid state. A new kinetically trapped [3+5] product was also observed alongside the formation of the targeted [4+6] cage species. Post-synthetic purification by Soxhlet extraction of the as-extruded ‘technical grade’ mixture of CC3 and [3+5] species rendered the material porous.
Introduction
The use of mechanochemistry in the formation of supramolecular assemblies1 and porous materials2 has emerged recently as an alternative to conventional solution-based synthetic procedures. Mechanochemistry can remove or greatly reduce the use of solvent compared to conventional solution methods, leading to more efficient and greener synthetic routes.3 Recent examples include the use of ball-milling in the synthesis of macrocycles,4,5 metal–organic cages,6 and rotaxanes,7 as well the formation of co-crystals.8 Ball-milling has also been used to make both crystalline and amorphous porous materials such as metal–organic frameworks (MOFs),9 covalent organic frameworks (COFs),10 and covalent triazine-based frameworks (CTFs).11 The use of solvent-free mechanochemical synthesis for porous materials is particularly attractive since it offers the potential to avoid additional desolvation steps, which can negatively affect porosity. However, drawbacks of using ball-mills to carry out mechanochemical synthesis include the limited ability to control temperature and the difficulty in scaling up the technique. To overcome these limitations, researchers have turned to using twin screw extrusion (TSE) for the continuous and scalable synthesis of MOFs,12 and discrete organic compounds,13 with little or no solvent. TSE permits mechanochemical synthesis, typically carried out on gram scale by ball-milling, to be scaled up to kg h−1 quantities.Porous organic cages (POCs) are discrete molecules that contain a permanent internal cavity accessible through windows, which can pack together into 3-dimensional structures with interconnected pores. POCs are a relatively new class of porous material, but they have already shown potential in a number of applications,14,15 most recently in the formation of porous liquids.16,17 In particular, the imine-based POC, CC318 (Scheme 1), has found a wide range of applications,19 and is surprisingly hydrolytically stable, expanding its potential in a range of applications.20CC3 has been used for molecular shape-sorting,21 chiral separations,22,23 and for the separation of rare gases.24 It was also processed into composite membranes,25 thin films for molecular sieving,26 and used to form organic alloys.27 Simple chemical modification of CC3 has led to new proton conductors28 and porous molecular crystals that are exceptionally stable under both acidic and basic conditions.29 Given the wide range of potential applications, it was desirable to determine a scalable and efficient route to CC3 to improve its commercial viability and, by extension, to allow the scale up of other POCs. CC3 can be synthesised in batch30 or in flow,31 but this requires long reactions times or the use of a large amount of solvent, respectively. We therefore investigated mechanochemical synthesis, specifically TSE, to establish a continuous and scalable synthetic route to CC3, employing little or no solvent.
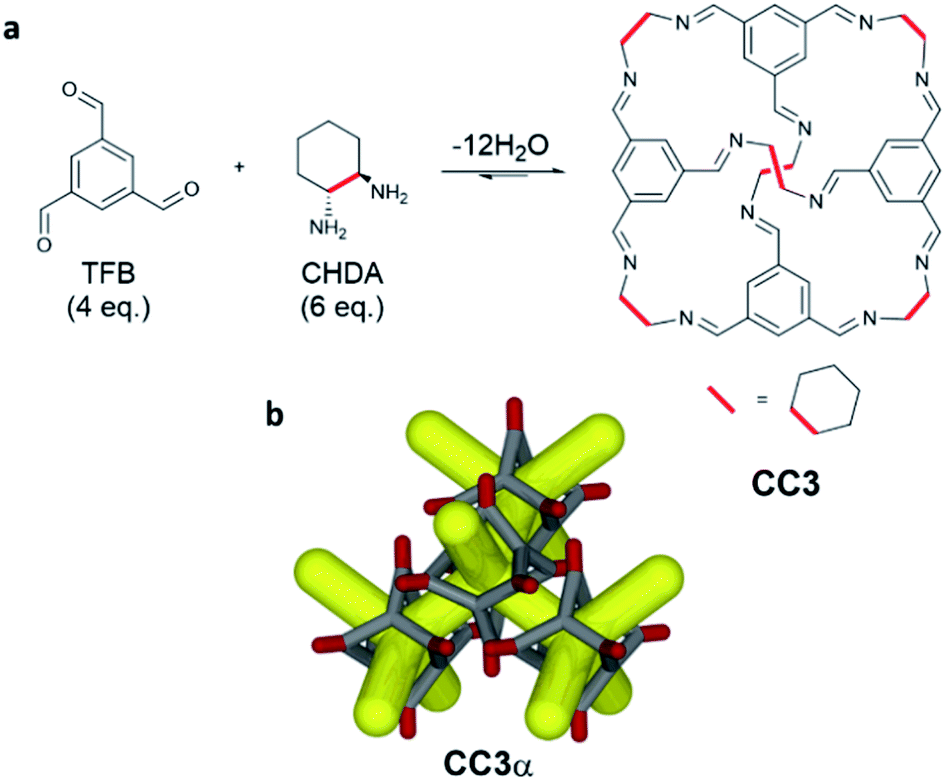 | ||
| Scheme 1 (a) Synthesis of CC3 – a [4+6] porous organic cage formed through the dynamic imine condensations of 4 equivalents of 1,3,5-triformylbenzene (TFB) with 6 equivalents of cyclohexanediamine (CHDA). Two cage enantiomers can be formed, CC3-R and CC3-S, by using the different chiralities of CHDA, (R,R)-CHDA and (S,S)-CHDA, respectively. (b) The cages can pack together window-to-window to form a 3D diamondoid pore network in the solid state (CC3α). | ||
To date, there is only a single example of the formation of orthogonal imine and boronate ester organic cages using mechanochemical grinding in a ball-mill, and those cages were non-porous.32 Here, we report the successful and scalable synthesis of CC3 by TSE, with minimal added solvent (8 equiv. relative to the formed cage). To our knowledge, this is the first example of the formation of a permanently porous organic cage using mechanochemistry, and the first example of using TSE for these types of materials. By using TSE, we also discovered the formation of a new kinetically trapped [3+5] oligomeric species that had not been observed before in solvent-based syntheses. This highlights the potential of mechanochemistry to give access to compounds that would not otherwise be possible.33
Results and discussion
The synthesis of CC3-S involves the reaction of four molecules of 1,3,5-triformylbenzene (TFB) with six molecules of (S,S)-1,2-cyclohexanediamine ((S,S)-CHDA) via 12 reversible imine condensations (Scheme 1a). Typically, high-dilution is required for the formation of POCs to ensure that any kinetically formed oligomers or products can equilibrate to the thermodynamic cage product within the reaction timeframe.34 Once synthesised, discrete cages can then be directed to pack together using solvent to form 3-D structures, which after desolvation can be porous. Alternatively, POCs can be rendered amorphous by disrupting their ability to crystallise.35 In the batch, solution-phase synthesis of CC3, the cages favour packing together in a window-to-window fashion to form a 3-D diamondoid pore network in the solid state (CC3α, Scheme 1b).18 Due to the nature of mechanochemical synthesis, involving removal of solvent and application of mixing, compression, and shearing forces in TSE, it was difficult to predict how the synthesis of CC3 would proceed and whether it would successfully undergo subsequent assembly into a 3-dimensional network. Furthermore, based on previous experience with attempted batch syntheses at high-concentrations, we expected that undesired kinetic products were likely to form, such as insoluble polymers, or amorphous rather than crystalline materials.30Optimisation of cage formation using TSE
To optimise the formation of CC3-S via TSE (Fig. 1), we investigated a range of conditions including temperature, reaction stoichiometry, extruder screw speed, liquid-assisted grinding (LAG) with different solvents, screw configuration, and the use of different additives (Tables 1 and S1, ESI†). For each set of conditions, the extruded samples were analysed using 1H NMR spectroscopy in the first instance, followed by HPLC, HRMS, and PXRD. Care was taken with the solution-based analyses to run the samples immediately after sample preparation to avoid equilibration to CC3; that is, to capture the identity of the product that came directly out of the extruder, rather than allowing the mixtures to further equilibrate once fully dissolved (see Fig. S2, ESI†). | ||
| Fig. 1 Twin screw extruder used in this study to investigate the synthesis of POCs. | ||
TFB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHDA :CHDA |
Temp. (°C) | Speed (rpm) | LAG | Sol.:CHDA |
Screw config. | |
|---|---|---|---|---|---|---|
| a TFA used as an additive. b NaHCO3 used as an additive. | ||||||
| 1 | 4:7 |
RT | 55 | ✗ | — | Standard |
| 2 | 4:7 |
60 | 55 | ✗ | — | Standard |
| 3 | 4:7 |
100 | 55 | ✗ | — | Standard |
| 100 | ✗ | — | Standard | |||
| 4 | 4:7 |
160 | 75 | ✗ | — | Standard |
| 5 | 4:7 |
60–100 | 75–100 | ✗ | — | Standard |
| 6 | 4:6 |
60 | 55 | ✗ | — | Standard |
| 7 | 4:8 |
60 | 55 | ✗ | — | Standard |
| 8 | 4:8 |
60–80 | 200 | ✗ | — | Reverse |
| 9 | 4:8 |
60–80 | 200 | CHCl3 | 1:1 |
Reverse |
| 10 | 4:8 |
60–80 | 200 | H2O | 1:1 |
Reverse |
| 11 | 4:8 |
60–80 | 100 | CHCl3 | 1:1 |
Reverse |
| 12a | 4:8 |
60–80 | 200 | CHCl3 | 1:1 |
Reverse |
| 13b | 4:8 |
60–80 | 200 | CHCl3 | 1:1 |
Reverse |
| 14 | 4:8 |
60–80 | 200 | CHCl3 | 1:4 |
Reverse |
| 15 | 4:8 |
60–80 | 200 | CHCl3 | 1:8 |
Reverse |
| 16 | 4:6.2 |
60–80 | 200 | CHCl3 | 1:1 |
Reverse |
| 17 | 4:10 |
60–80 | 200 | CHCl3 | 1:1 |
Reverse |
Initially, the reactants were briefly mixed together and then manually fed into the extruder with a standard screw configuration (Fig. 2a, Table 1, entries 1–7). This consisted of conveying and kneading sections at angles of 30° and 60°, at a speed of 55 rpm, which was previously reported to provide efficient mixing with a reasonable residence time of less than 2 minutes.13
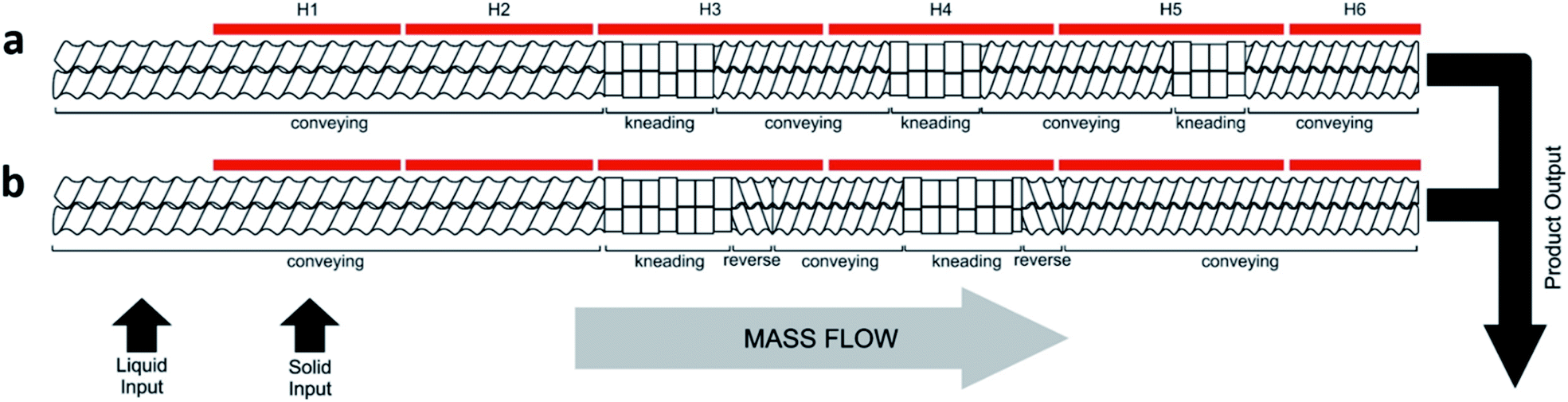 | ||
| Fig. 2 (a) Schematic illustration of the standard screw configuration, and (b) the reverse screw configuration, used during the investigations and optimisation of the synthesis for CC3. The six heating zones down the length of the barrel are labelled (H1–H6). | ||
A range of temperatures both above and below the reactant melting points were investigated (ambient, 60, 100, and 160 °C; entries 1–4) for the reaction between TFB (4 equiv., melting point 156–158 °C) and S,S-CHDA (7 equiv., melting point 40–43 °C). Initially, an excess of CHDA was used since this had been previously reported to be well tolerated and improve conversion in the formation of CC3 by solvent-based methods.31 No conversion to CC3 was observed at ambient temperature. Partial conversion to CC3 was observed at 60 °C, but no further conversion was apparent upon passing the same mixture through the extruder a further two times under the same conditions (Fig. S8, ESI†). At the higher temperatures of 100 and 160 °C, all of the TFB was consumed and an increased torque (exceeding the extruder's built-in limit of 12.5 Nm) was observed, but this was associated with the formation of an insoluble material, assumed to be polymeric in nature. In an attempt to increase conversion to CC3 and to avoid insoluble polymer formation, a temperature gradient was introduced along the barrel from room temperature to 100 °C (entry 5). However, again the torque limit was exceeded, which coincided with the formation of insoluble material, possibly due to the loss of water vapour that might reduce the reversibility of the system. An alternative approach was therefore taken, whereby the reagent stoichiometry was varied, ranging from a stoichiometric balance (4:6 TFB:CHDA, entry 6) to a large excess of CHDA (4:8, entry 7), while maintaining the temperature at 60 °C. The latter afforded better conversion to CC3, but residual TFB was still present.
We next employed an alternative screw configuration that contained a number of reverse conveying segments (Fig. 2b) which retards the flow of material along the extruder (Table 1, entries 8–17). Our aim was to drive the reaction to completion by increasing the amount of time the reagents would be kneaded.13 The reaction was carried out at 200 rpm because a higher speed was needed to reduce the increased torque resulting from the reverse screw configuration (entry 8). Also, by using a slight temperature gradient along the barrel from 60 to 80 °C, complete consumption of TFB could be achieved (confirmed by 1H NMR spectroscopy), although other oligomers were still observed along with the desired CC3 product (Fig. S3, ESI†).
In all extruded mixtures, analysis of relative peak areas by HPLC indicated the formation of one additional main species along with CC3 in an approximate 30:70 ratio (Table S2, ESI†). HRMS indicated two main mass ions corresponding to the formation of a [3+5] ‘partial cage’ and the targeted [4+6] CC3 (Fig. 3, S5 and S7, ESI†). This [3+5] species was not isolated with CC3 previously when using solvent-based synthesis methods. However, it has been reported by Lively and co-workers to be an observable intermediate in the formation of CC3 using mass spectrometry, although based on computational modelling the formation energy of a [3+5] species was reported to be ∼154 kJ mol−1 higher than for the thermodynamically favoured [4+6] cage.36 It is likely, therefore, that the consistent formation of a mixture of [3+5] and [4+6] species in an approximate 30:70 ratio respectively is due to the kinetic trapping of this intermediate species, which does not occur in solution (Fig. 3).
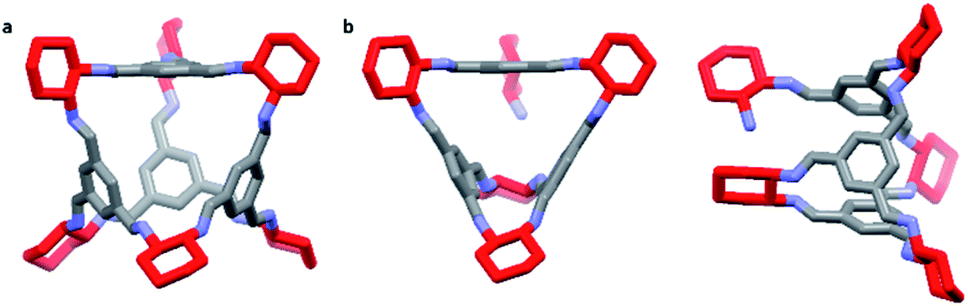 | ||
| Fig. 3 Comparison of the formed [4+6] and [3+5] structures by TSE: (a) targeted [4+6] (CC3) cage viewed through one of the cage windows; (b) proposed structure of the [3+5] species (window view (left) and side view (right)) showing the free NH2 group available to interact with the pore. Hydrogens are omitted for clarity, with CHDA vertices highlighted in red. | ||
With complete consumption of TFB observed with the alternative screw configuration (200 rpm, temperature gradient: 60–80 °C, 4:8 TFB:CHDA), we believe that CHDA ‘wets’ the reaction mixture because its melting point is lower than the 60 °C reaction temperature. This, combined with the formation of water as a reaction side-product, provides some level of reversibility in the extruder, affording the mixture of these two species, but it does not allow complete conversion to the desired cage product. Therefore, using the same screw speed, temperature gradient, and ratio of precursors, we next investigated the use of liquid-assisted grinding (LAG) in an attempt to increase reversibility and to favour the formation of CC3. The use of both chloroform (entry 9) and water (entry 10) was investigated; chloroform is a typical solvent used in the formation of these types of organic cages in solution based methods,34 and whilst imine bonds are not typically stable in water, CC3 is surprisingly hydrolytically stable.20 Additionally, we were interested in what effect water might have on the formation of the kinetically trapped [3+5] species, with the potential for hydrogen-bonding with the free amine. For ease of reagent addition to the extruder, CHDA was dissolved in each of the solvents (1:1, based on molar equiv.) and added via a syringe pump (0.25 mL min−1) prior to the solid hopper, where solid TFB was manually fed in at ∼100 mg min−1. Full consumption of TFB was observed with both solvents. While a similar mixture of 30:70 species was formed in the presence of water, the use of chloroform favoured formation of CC3, with a 15:85 ratio of [3+5]:[4+6] apparent, as estimated by comparison of the relative peak areas in the HPLC traces. Further investigations into reducing the screw speed (200 to 100 rpm, entry 11), the use of additives (trifluoroacetic acid added to the CHDA/CHCl3 mixture to catalyse imine formation, entry 12; NaHCO3 pre-mixed with the TFB to remove some of the formed water during cage formation, entry 13), increased quantities of chloroform (1:4 and 1:8 CHDA:CHCl3 based on molar equiv., entries 14 and 15), and alternative equivalents of the diamine (from 6.2 equiv. CHDA, up to an excess of 10 equiv. relative to 4 equiv. TFB, entries 16 and 17) did not improve the conversion to CC3. For a summary of all the reaction conditions attempted, and their subsequent analyses, see Tables S1–S3 and Fig. S3–S7 in the ESI.†
Due to the inherent reversibility of the imine bonds, to confirm that the mechanical grinding was not causing decomposition of the cage to the [3+5] species, a sample of pure CC3, pre-formed using standard solution-based batch conditions, was processed in the extruder using the alternative screw configuration. No conversion to any side-products was observed (Fig. S9, ESI†). To further demonstrate that the mechanical grinding was responsible for the formation of CC3, a series of studies were carried out using dynamic scanning calorimetry (DSC), followed by analysis using 1H NMR spectroscopy, to rule out product formation solely due to heating. By simply mixing the two precursors at room temperature, no formation of cage was observed (Fig. S10, ESI†). Generally, polymer formation was observed when the reactions were heated without mixing, with the formed material being insoluble. This was particularly evident when the reactions were heated above 60 °C, an exotherm is observed between 60–75 °C when the reaction mixture is analysed by DSC (Fig. S11, ESI†). This probably explains the observed torque increase and insoluble polymer formation during the initial extrusion studies.
Scale-up and continuous processing
With the successful formation of CC3 by TSE, albeit alongside a [3+5] side-product, we next investigated scale-up using the optimised conditions, but now using a solid addition unit for the controlled addition of TFB. Initially, the solid addition hopper was calibrated with TFB to determine an average dispensing rate of 86 ± 3.5 mg min−1 (Table S4, ESI†), and a stock solution of CHDA:CHCl3 (∼53 mL, 1:1 molar equiv.) was prepared. It was found that maintaining a 4:8 TFB:CHDA molar ratio was an important factor in obtaining consistent formation of the products, and the addition rate of the CHDA solution was adjusted accordingly (i.e., 0.22 mL min−1 stock solution, equating to 1.10 mmol min−1 of CHDA, for 0.55 mmol min−1 of TFB, see Tables S5 and S6, ESI†).
The stock solution was introduced into the extruder containing the reverse screws via a syringe pump, prior to the solid, using the above addition rates, with a screw speed of 200 rpm and a temperature gradient of 60 to 80 °C along the barrel (Fig. 5a). Overall, it took 38 minutes of reagent addition (total addition rate = 20.6 g h−1) before material first began to be extruded (this equates to ∼13 g of material in the barrel), at which point, the product mixture was steadily extruded at ∼16.3 g h−1 with a residence time of 11 minutes. By comparing samples of the material taken from the 1st and 2nd kneading sections along the barrel length to the extrudate, the reaction was actually found to be complete at the 1st kneading section, and no further conversion was observed down the remaining length (Fig. S22, ESI†). This means that a shorter barrel could in principle be used, and the 2nd reverse kneading section removed, which would shorten the residence time considerably and increase throughput rate.
Once the extrusion process reached a ‘steady state’, starting materials were continuously fed into the extruder for 3 hours to yield 49 g of material with no major issues being encountered such as increased temperature or torque (Fig. S16 and S17, ESI†). Analysis of aliquots throughout the process at 30 min intervals (using 1H NMR spectroscopy and PXRD) confirmed a similar purity, product distribution, and crystallinity throughout the whole reaction process, with full consumption of the TFB (see Fig. S20 and S21, ESI†). The as-made extruded solid was fully soluble in both CDCl3 (NMR) and a 1:1 mixture of DCM:MeOH (HPLC), suggesting that no polymer had been formed as a side-product, and the product contained a 12:88 ratio of [3+5]:CC3 with an overall 95% purity, as determined by HPLC and LCMS (Fig. S14 and S15, ESI†). Comparison of the average mass of materials being fed into the extruder (20.6 g h−1), the mass being extruded (∼16.3 g h−1), and the maximum theoretical extruded mass of CC3 assuming complete conversion (8.89 g h−1), indicates that while chloroform or water was lost by evaporation during the extrusion process, some also remains in the extruded material (Table S7 and Fig. S18, ESI†).
In comparison with both batch and flow methods, the use of TSE reduces the volume of solvent used during the reaction vastly (by 98–99.5%), while also drastically lowering the reaction time (Table 2, Fig. 4).
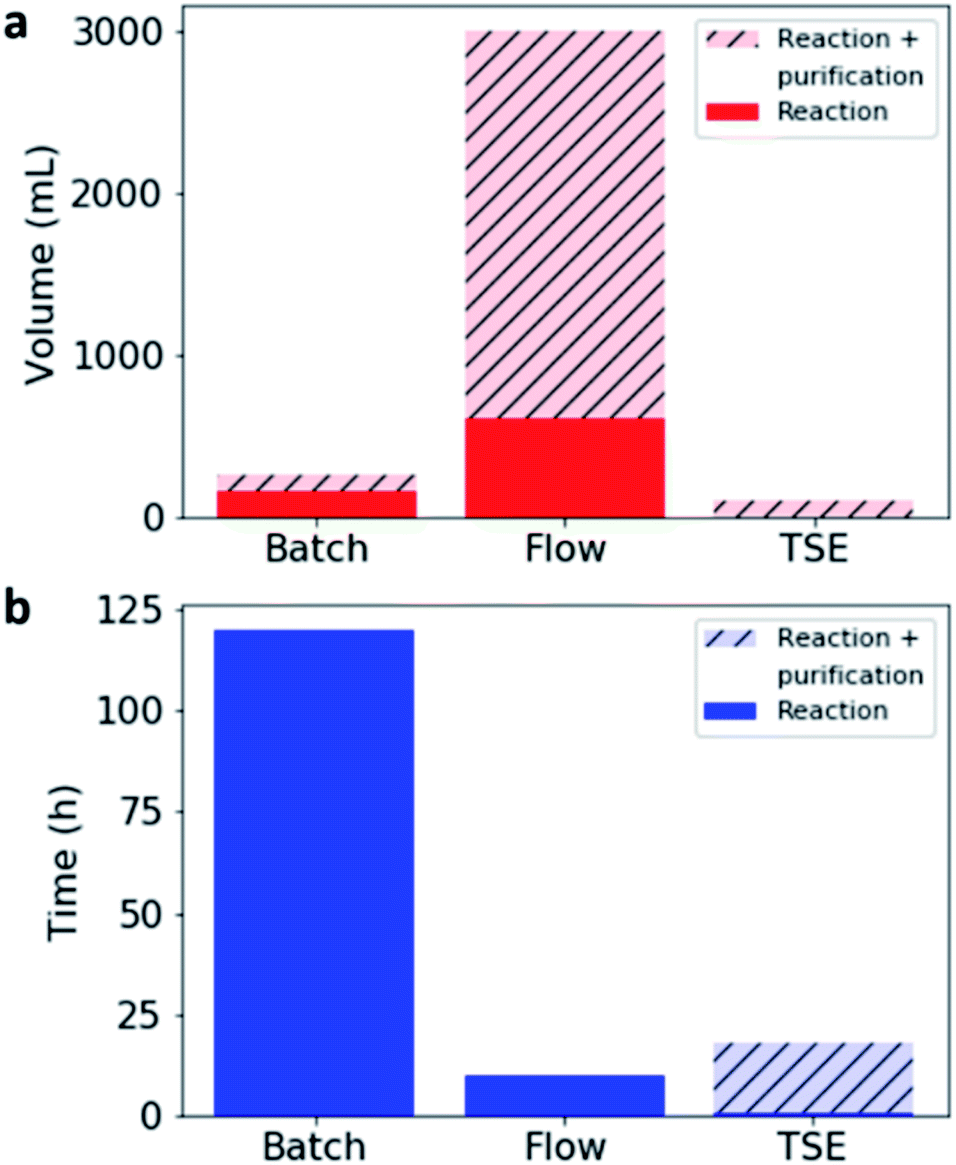 | ||
| Fig. 4 (a) Comparison of solvent volume used to form 5 g of CC3 (red) by each synthetic method, compared to the overall solvent used for both the reaction and subsequent purification; (b) comparison of the time required to synthesise and purify 5 g of CC3. Solid colour bars for TSE are plotted, but due to the low solvent volume used and the short reaction time compared to both batch and flow, on the plotted scale they are negligible. | ||
Cage packing
Analysis by PXRD of the extruded solids suggested that the products prepared in the complete absence of solvent were all amorphous. Crystalline TFB was observed in the reactions when incomplete consumption had occurred (Fig. S6, ESI†). However, upon introducing LAG using chloroform, the presence of some crystalline CC3α was apparent by PXRD, albeit alongside a large amount of amorphous material that could be observed by SEM analysis (Fig. S6 and S28, ESI†). Unfortunately, while previous reports suggest that defect engineered CC3 can show interesting sorption properties,36 and amorphous CC3 demonstrates higher porosity than crystalline CC3α,30 the as-extruded mixtures of [3+5]:[4+6] (70:30, amorphous and 80:20, semi-crystalline) were found to be non-porous after desolvation (Fig. 5c), possibly because of the unreacted excess diamine used during the synthesis occupying the cage cavities. Unfortunately, when a stoichiometric quantity of diamine was employed under the optimised conditions, instead of an excess, whilst all of the TFB was consumed and a similar ratio of [3+5]:[4+6] species was formed, the reaction mixture contained a larger number of impurities (see Tables S1–S3 and Fig. S4, S5, ESI†).
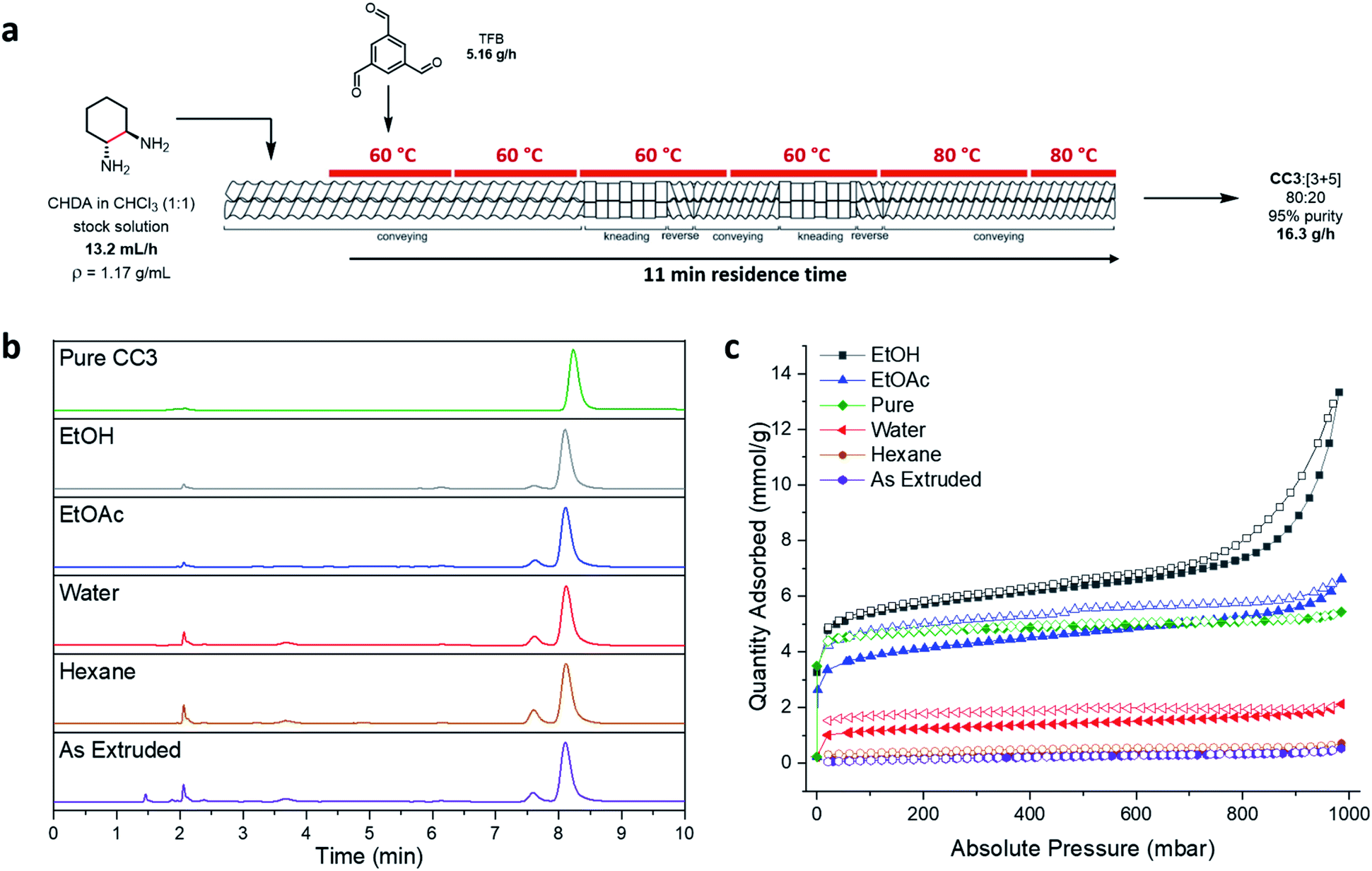 | ||
| Fig. 5 (a) Scale-up procedure for the formation of CC3 by TSE (screw speed = 200 rpm); (b) HPLC traces of pure CC3 formed using batch synthesis and post-processed by TSE under the same conditions (reverse screw configuration, 200 rpm, temperature gradient: 60–80 °C), compared to the as-extruded mixture from the scale-up (88:12 CC3:[3+5], 95% purity), and the purified solids post-Soxhlet extraction with a range of solvents (CC3 ∼8.2 min, [3+5] ∼7.6 min); (c) nitrogen adsorption (filled) and desorption (empty) isotherms for pure CC3, the as-extruded material, and the samples purified by Soxhlet extraction. | ||
Purification of bulk CC3
Purification of the scaled-up reaction mixture was next investigated because the as-extruded mixtures were non-porous. Previously, purification of these cages has typically involved fully dissolving the material in dichloromethane followed by the removal of any insoluble precipitate by filtration. An anti-solvent, such as hexane, is then added to precipitate out the cage material, which can be collected by filtration.34 This method could be used to purify extruded CC3, but it would require a large amount of solvent. Clearly, this would negate the solvent savings achieved by using TSE. We therefore looked to exploit the low solubility of CC3 in many solvents and investigated purification procedures where the produced material was simply washed in a minimal volume of solvent in which the [3+5] species and other impurities would dissolve.A range of solvents were investigated and Soxhlet extraction was utilised to minimize the volume of solvent used (Table S8, ESI†). Complete removal of the [3+5] species was not achieved, even with prolonged Soxhlet extraction, although the proportion in the mixture could be reduced, and the overall purity and CC3 percentage could be improved by washing with ethyl acetate or ethanol (9:91 and 5:95 [3+5]:CC3, respectively, Fig. 5b and S24†). However, despite this small amount of residual [3+5] product, the washed products were found to be porous to nitrogen (Fig. 5c) and carbon dioxide (Fig. S29, ESI†). In particular, washing with ethyl acetate and ethanol formed ‘technical grade’ CC3 that was as porous, or even more porous, than pure CC3 samples (SABET (N2) = 339 and 474 m2 g−1 respectively for EtOAc and EtOH washed samples; SABET (N2) = 420 m2 g−1 for pure CC3).
It was found that the solvent-produced, 100% pure CC3, after post-synthetic processing in the extruder, appeared to be somewhat less crystalline by SEM, although crystalline CC3α was still apparent by PXRD and the material was still porous (Fig. S27 and S28, ESI†). This suggests that whilst mechanical grinding does not cause decomposition of the cage (as discussed earlier), it can lead to some loss in crystallinity. By comparison, the as-extruded material, which was largely amorphous by SEM, became much more porous after washing with ethyl acetate, and in particular with ethanol. These products were as porous as the pure batch-made CC3 that had been post-synthetically processed in the extruder. This suggests that the use of a Soxhlet extraction not only removes impurities but also improves crystallinity, which has a direct impact on the porosity of the resulting material. Additionally, even taking into account the solvent used and the time taken to purify the as-extruded material by Soxhlet extraction, the solvent saving is large compared to flow methods (96.5% reduction, Fig. 4a), and the reaction time is greatly improved compared to the batch synthesis (85% reduction, Fig. 4b).
Conclusions
In conclusion, the use of TSE has enabled the large-scale synthesis of a porous organic cage, CC3. During optimisation, increased temperature and residence time, through use of reverse screws to specifically prolong the kneading time, were required to promote conversion. Inclusion of LAG improved the preferential formation of the desired cage species in the product mixture over the kinetically trapped [3+5] side product. Whilst clean conversion to solely CC3 was not achieved after optimisation, the mixtures could be simply washed by Soxhlet extraction to obtain ‘technical grade’ CC3 of improved purity, crystallinity, and containing a smaller proportion of the [3+5] species, resulting in porous material that is comparable to pure CC3 in terms of gas uptake. Overall, the formation of CC3 by TSE uses minimal solvent and has a greatly shortened reaction time, and is more favourable than both the batch and flow syntheses previously employed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) under the Grants EP/R005710/1 (AIC) and EP/R005540/1 (SLJ), and for an EPSRC Summer Vacation Bursary at the University of Liverpool (FG, RLG). We also thank the European Research Council under FP7, RobOT, ERC Grant Agreement No. 321156 (AIC), for financial support. RLG thanks the Royal Society for a University Research Fellowship. We also thank Dr Kim Jelfs for helpful comments and discussion. We acknowledge the MicroBioRefinery for assistance with QTOF-MS measurements.Notes and references
- T. Friščić, Supramolecular concepts and new techniques in mechanochemistry: cocrystals, cages, rotaxanes, open metal–organic frameworks, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- P. Zhang and S. Dai, Mechanochemical synthesis of porous organic materials, J. Mater. Chem. A, 2017, 5, 16118–16127 RSC.
- D. Tan and T. Friščić, Mechanochemistry for Organic Chemists: An Update, Eur. J. Org. Chem., 2018, 18–33 CrossRef CAS.
- M. Pascu, A. Ruggi, R. Scopelliti and K. Severin, Synthesis of borasiloxane-based macrocycles by multicomponent condensation reactions in solution or in a ball mill, Chem. Commun., 2013, 49, 45–47 RSC.
- S. Kaabel, R. S. Stein, M. Fomitšenko, I. Järving, T. Friščić and R. Aav, Size-Control by Anion Templating in Mechanochemical Synthesis of Hemicucurbiturils in the Solid State, Angew. Chem., Int. Ed., 2019, 58, 6230–6234 CrossRef CAS PubMed.
- C. Giri, P. K. Sahoo, R. Puttreddy, K. Rissanen and P. Mal, Solvent-Free Ball-Milling Subcomponent Synthesis of Metallosupramolecular Complexes, Chem.–Eur. J., 2015, 21, 6390–6393 CrossRef CAS PubMed.
- C.-C. Hsu, N.-C. Chen, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Solvent-Free Synthesis of the Smallest Rotaxane Prepared to Date, Angew. Chem., Int. Ed., 2008, 47, 7475–7478 CrossRef CAS PubMed.
- F. Fischer, D. Lubjuhn, S. Greiser, K. Rademann and F. Emmerling, Supply and Demand in the Ball Mill: Competitive Cocrystal Reactions, Cryst. Growth Des., 2016, 16, 5843–5851 CrossRef CAS.
- A. Pichon, A. Lazuen-Garay and S. L. James, Solvent-free synthesis of a microporous metal–organic framework, CrystEngComm, 2006, 8, 211–214 RSC.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, Mechanochemical synthesis of chemically stable isoreticular covalent organic frameworks, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- E. Troschke, S. Grätz, T. Lübken and L. Borchardt, Mechanochemical Friedel-Crafts Alkylation-A Sustainable Pathway Towards Porous Organic Polymers, Angew. Chem., Int. Ed., 2017, 56, 6859–6863 CrossRef CAS PubMed.
- D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Synthesis by extrusion: continuous, large-scale preparation of MOFs using little or no solvent, Chem. Sci., 2015, 6, 1645–1649 RSC.
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Organic synthesis by Twin Screw Extrusion (TSE): continuous, scalable and solvent-free, Green Chem., 2017, 19, 1507–1518 RSC.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Synthesis and Applications of Porous Organic Cages, Chem. Lett., 2015, 44, 582–588 CrossRef CAS.
- T. Hasell and A. I. Cooper, Porous organic cages: soluble, modular and molecular pores, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- N. Giri, M. G. Del Popolo, G. Melaugh, R. L. Greenaway, K. Ratzke, T. Koschine, L. Pison, M. F. Gomes, A. I. Cooper and S. L. James, Liquids with permanent porosity, Nature, 2015, 527, 216–220 CrossRef CAS PubMed.
- R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Understanding gas capacity, guest selectivity, and diffusion in porous liquids, Chem. Sci., 2017, 8, 2640–2651 RSC.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Porous organic cages, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- F. Beuerle and B. Gole, Covalent Organic Frameworks and Cage Compounds: Design and Applications of Polymeric and Discrete Organic Scaffolds, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- T. Hasell, M. Schmidtmann, C. A. Stone, M. W. Smith and A. I. Cooper, Reversible water uptake by a stable imine-based porous organic cage, Chem. Commun., 2012, 48, 4689–4691 RSC.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Molecular shape sorting using molecular organic cages, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- A. Kewley, A. Stephenson, L. Chen, M. E. Briggs, T. Hasell and A. I. Cooper, Porous Organic Cages for Gas Chromatography Separations, Chem. Mater., 2015, 27, 3207–3210 CrossRef CAS.
- J.-H. Zhang, S.-M. Xie, L. Chen, B.-J. Wang, P.-G. He and L.-M. Yuan, Homochiral Porous Organic Cage with High Selectivity for the Separation of Racemates in Gas Chromatography, Anal. Chem., 2015, 87, 7817–7824 CrossRef CAS PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Separation of rare gases and chiral molecules by selective binding in porous organic cages, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Nanoporous Organic Polymer/Cage Composite Membranes, Angew. Chem., Int. Ed., 2013, 52, 1253–1256 CrossRef CAS PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Porous Organic Cage Thin Films and Molecular-Sieving Membranes, Adv. Mater., 2016, 28, 2629–2637 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Porous Organic Alloys, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed.
- M. Liu, L. Chen, S. Lewis, S. Y. Chong, M. A. Little, T. Hasell, I. M. Aldous, C. M. Brown, M. W. Smith, C. A. Morrison, L. J. Hardwick and A. I. Cooper, Three-dimensional protonic conductivity in porous organic cage solids, Nat. Commun., 2016, 7, 12750 CrossRef CAS PubMed.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, Acid- and Base-Stable Porous Organic Cages: Shape Persistence and pH Stability via Post-synthetic “Tying” of a Flexible Amine Cage, J. Am. Chem. Soc., 2014, 136, 7583–7586 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, Porous Organic Cage Nanocrystals by Solution Mixing, J. Am. Chem. Soc., 2012, 134, 588–598 CrossRef CAS PubMed.
- M. E. Briggs, A. G. Slater, N. Lunt, S. Jiang, M. A. Little, R. L. Greenaway, T. Hasell, C. Battilocchio, S. V. Ley and A. I. Cooper, Dynamic flow synthesis of porous organic cages, Chem. Commun., 2015, 51, 17390–17393 RSC.
- B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, Synthesis of Molecular Nanostructures by Multicomponent Condensation Reactions in a Ball Mill, J. Am. Chem. Soc., 2009, 131, 3154–3155 CrossRef PubMed.
- J. L. Howard, M. C. Brand and D. L. Browne, Switching Chemoselectivity: Using Mechanochemistry to Alter Reaction Kinetics, Angew. Chem., Int. Ed., 2018, 57, 16104–16108 CrossRef CAS PubMed.
- M. E. Briggs and A. I. Cooper, A perspective on the synthesis, purification, and characterisation of porous organic cages, Chem. Mater., 2017, 29, 149–157 CrossRef CAS PubMed.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, Molecular Dynamics Simulations of Gas Selectivity in Amorphous Porous Molecular Solids, J. Am. Chem. Soc., 2013, 135, 17818–17830 CrossRef CAS PubMed.
- G. Zhu, Y. Liu, L. Flores, Z. R. Lee, C. W. Jones, D. A. Dixon, D. S. Sholl and R. P. Lively, Formation mechanisms and defect engineering of imine-based porous organic cages, Chem. Mater., 2017, 30, 262–272 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: full experimental and characterisation details. See DOI: 10.1039/d0sc01858a |
| This journal is © The Royal Society of Chemistry 2020 |