
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA zeolite-based ship-in-a-bottle route to ultrasmall carbon dots for live cell labeling and bioimaging†
Lei
Dong‡
a,
Dehong
Hu‡
b,
Yanding
Wang
a,
Zonghai
Sheng
 b,
Mei
Hong
*a and
Shihe
Yang
*a
b,
Mei
Hong
*a and
Shihe
Yang
*a
aState Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Nano-Micro Materials Research, School of Chemical Biology & Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: hongmei@pku.edu.cn; chsyang@pku.edu.cn
bPaul C. Lauterbur Research Center for Biomedical Imaging, Shenzhen Key Laboratory of Ultrasound Imaging and Therapy, CAS Key Laboratory of Health Informatics, Institute of Biomedical and Health Engineering, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, P. R. China
First published on 14th October 2020
Abstract
Fluorescent carbon quantum dots (CQDs) are a new class of carbon nanomaterials that have excellent biocompatibility and low toxicity, useful for cell labeling and bioimaging. However, it is challenging to reproducibly create CQDs that are uniform and small in size with controlled toxicity and fluorescence properties, and this status quo hinders the practical application of CQDs, especially in vivo. In this report, CQDs with a uniform size of ∼1.53 ± 0.42 nm are synthesized via in situ pyrolysis of organics confined in the smallest-pore zeolite SAPO-20 with a micropore aperture of 0.28 nm. The ultrasmall fluorescent CQDs show a bright green fluorescence with a quantum yield (QY) of 16%, higher than that of most of the bare CQDs without any functionalization and maximum emission at 523 nm under the irradiation of xenon-light at an excitation wavelength of 470 nm. X-ray photoelectron spectroscopy (XPS) characterization confirms the presence of rich carboxyl and hydroxyl groups on the CQD surface containing N- and O-based functional groups. The synthesized CQDs with high QY and low cytotoxicity have been successfully used for cellular imaging with excellent biocompatibility.
Introduction
Carbon quantum dots (CQDs) with a size of less than 10 nm are a new member of the carbon nanomaterial family and usually possess a spherical shape with an obvious crystal lattice.1 The distinguished advantages of CQDs include low toxicity, biocompatibility, low cost, and chemical inertness, which endow CQDs with applicability to a wide range of fields such as bioimaging, chemical sensing, drug delivery, photocatalysis and multicolor patterning.2 Various synthetic methods have been exploited to prepare CQDs from corresponding carbon sources or molecular precursors, such as arc discharge,3 chemical oxidation,4,5 laser ablation,6 ultrasound7 or microwave-assisted synthesis,8,9 combustion10,11 and hydrothermal methods.12 Although significant advances have been achieved, the preparation of CQDs with a compact particle size distribution, high biocompatibility, and bright luminescence for biolabeling and intravital bioimaging remains a significant challenge.13Generally, CQDs processed in the homogeneous liquid phase are difficult to regulate, especially the dimensions of the CQDs due to free space for CQD formation. The only possible method to achieve uniform CQDs is to optimize the experimental conditions, making the synthesis method more complicated. Recently, zeolite-based templates for the preparation of CQDs have attracted wide attention, due to their unique advantages of uniform micropore size, unique rigid crystalline matrix, and confined space. Organic molecular precursors confined in the micropore space of zeolitic frameworks ensured the controlled production of CQDs with multicolor luminescence.14 Up to now, the pore size of the utilized template zeolite has varied from 0.38 nm to 0.73 nm, according to different zeolite framework matrices. For example, Mu et al. prepared N-doped CQDs with a size of 2.5–5.8 nm by pyrolysis of N-methylpiperazine (NMP) confined in zeolite MgAPO-44 possessing a pore aperture of 0.38 nm.15 The modulated full-color photoluminescence can be tuned by altering the concentration and pH value of aqueous dispersions of the CQDs. Baldovi et al. employed pure-silica ITQ-29 zeolite that belongs to LTA zeotype topology with an aperture of 0.42 nm to synthesize carbon dots with a size of 5–12 nm and this composite material can act as a fluorescent oxygen sensor.16 It can be easily envisioned that the design and preparation of a small-pore zeolite would benefit the control of the dimensions, yield, and optical properties of the as-prepared CQDs.
Herein, we report SAPO-20 zeolites as efficient templates to prepare CQDs with a miniature size and high luminescence. As a kind of aluminosilicophosphate (SAPO) molecular sieve, the SAPO-20 zeolite has only one type of pore and the smallest pore size with apertures formed by 6-ring windows of 0.28 × 0.28 nm.17 Due to the comparable size of pyrrolidine and tetraethylammonium hydroxide (TEAOH) as entrapped organic molecules within the apertures, SAPO-20 can significantly boost the confinement effect promoted by the interaction between pyrrolidine, TEAOH and the zeolite framework, which endows the as-synthesized CQDs with a uniform size distribution and excellent optical properties. We have successfully adopted the as-prepared CQDs to achieve efficient labeling and tracking of living cells in vitro and in vivo (Fig. 1), exhibiting the huge potential of CQDs in the field of biomedicine.
 | ||
| Fig. 1 Schematic illustration for the preparation process of CQDs and their application in living cell labeling and bioimaging in vivo. | ||
Results and discussion
Synthesis and modulation of the SAPO-20 zeolite
The SAPO-20 zeolite was synthesized with the additives of pyrrolidine and tetraethylammonium (TEAOH) by a hydrothermal synthesis method. The structure, morphology, and photophysical properties of the obtained products were characterized. The powder X-ray diffraction (PXRD) patterns of zeolite particles exhibited the characteristic sharp peaks of SOD topology, demonstrating a pure SAPO-20 phase with high crystallinity (Fig. 2a). Calcination at 600 °C hardly changed the crystallinity of the zeolite but altered its relative peak intensity. The peak of the (200) crystal planes was greatly suppressed upon calcination, indicating less exposure of the {100} planes by calcination probably through the shielding effect of the organics through specific binding to select crystal faces.18 The SEM image (Fig. 2b) of SAPO-20 showed a well-faceted cubic morphology and a uniform size of 2.65 ± 0.08 μm. It is indicated that the treatments at 600 °C did not alter the morphology of the crystals (Fig. S1†). | ||
| Fig. 2 Characterization of SAPO-20. (a) Powder XRD patterns of the washed and calcined SAPO-20 synthesized in the presence of pyrrolidine and TEAOH as referenced to the standard (lower) pattern. (b) SEM image of the as-synthesized SAPO-20 before calcination (inset, the size distribution of the as-synthesized SAPO-20). (c) Infrared spectra and (d) Raman spectra of the as-synthesized SAPO-20. (e) Digital photos of SAPO-20 zeolites before and after calcination. (f) Elemental analysis of the as-synthesized SAPO-20 before and after calcination. | ||
The strong confinement of organics in the small-pore SAPO-20 which leads to N-containing CQDs upon controlled pyrolysis has been verified through an array of analytical techniques. The IR spectra (Fig. 2c) of the SAPO-20 sample before and after calcination revealed the entrapment of organic species in the zeolite framework. The intense bands, which appear at 1115 cm−1, 720 cm−1, 660 cm−1 and 451 cm−1, derived from the vibrational modes of the framework of the as-obtained SAPO-20 zeolite19 remained after calcination. The characteristic adsorptions of amine N–H stretch at ∼3260 cm−1 were visible in the as-synthesized SAPO-20. The bands at 3058 cm−1 and 1470 cm−1 can be assigned to the asymmetric stretching vibration and symmetric bending vibration of the –CH3 group respectively.
These peaks disappeared after calcination, demonstrating the confinement of organics in the silicoaluminophosphate framework that cannot be removed by water washing. The Raman spectra (Fig. 2d) of the washed SAPO-20 zeolite before calcination provide more support for this conclusion. The Raman peaks occurring at 379 cm−1, 418 cm−1, 875 cm−1, 918 cm−1, 1461 cm−1, and 2800–3000 cm−1 can be attributed to the vibrational modes of the tetraethylammonium cation (TEA+).20,21 Combining with the SEM images and the XRD patterns showing that the most intense peak is the (200) lattice plane, it is likely that the TEA+ helped expose more {100} lattice planes to modulate towards the cubic morphology by selective binding to certain crystallographic faces. Pyrrolidine is a known organic structure-directing agent (OSDA) for SAPO-20.22 However, uncalcined SAPO-20 synthesized only with pyrrolidine is not uniform, showing greater size variation (Fig. S2†). Therefore, TEAOH molecules modified the crystal morphology from nonuniform spheres to uniform cubes. Due to the smallest pore size of SAPO-20, the entrapped molecules form CQDs after calcination at 600 °C, leading to a color change from white to black upon calcination (Fig. 2e). Because of the formation of CQDs, the thermogravimetric analysis (TGA) profile (Fig. S3†) of the as-synthesized SAPO-20 only showed a total weight loss of 7% even by calcination in air for 4 hours at 600 °C, which involves desorption of adsorbed water molecules and incomplete decomposition of organic molecules. Organic elemental analysis (Fig. 2f) quantitatively determined the carbon contents of 3.358% and 2.692% before and after calcination, respectively. The contents of nitrogen originating from TEAOH and pyrrolidine were 1.751 and 0.461% in SAPO-20 before and after calcination. These results further illustrated the power of entrapping the organics by the SAPO-20 framework and the high productivity of N-containing CQDs.
Characterization of the as-prepared CQDs
The CQDs were prepared by pyrolysis of organic small molecules entrapped in the SAPO-20 cages and micropore cavities (Fig. 1). The XRD pattern (Fig. 3a) shows a broad peak centred at 24°, which verifies the graphene-like structures of these CQDs. They are monodispersed and embedded in the zeolite framework with an average diameter of less than 2 nm (Fig. S4†). The high-resolution TEM (HRTEM) images show that these CQDs possess a crystalline structure with a lattice spacing of 0.21 nm which is consistent with that of the (100) plane of graphene (Fig. 3b).23 After extraction from the SAPO-20 matrix by post-dissolution of the SAPO-20 host, the collected CQDs were analyzed by dynamic light scattering (DLS) which showed an average size of 1.53 ± 0.42 nm and a narrow distribution with a maximum size not exceeding 3 nm, confirming the ultra-small size and uniformity of the as-prepared CQDs. It is still larger than the largest sodalite cavity diameter of the cage ∼0.5 nm,24 similar to the results observed using other zeolite frameworks (Table S1†), probably due to expansion of the zeolite host25 upon guest carbon dot formation, and slight expansion of the carbon dots after extraction from the zeolite matrix. Because of the smallest pore size of SAPO-20 compared to other zeolite framework types, the obtained CQDs were also smaller than those reported in the literature (Table S1†). The zeta potential of CQDs dispersed in water is −39 mV (Fig. 3d), showing that they possess a negative charge on the surface, which ensured their good monodispersity and stability for more than 3 months of storage. It provides a prerequisite for living cell labeling and imaging in vivo. The CQD solution exhibited excitation-dependent luminescence (Fig. 3e and f). The optimal excitation and maximum emissions were 470 nm and 523 nm, respectively. The absolute fluorescence quantum yield (QE) of CQD solutions was measured to be 16% without any functionalization (Fig. 3g), which is much higher than that of most of the bare CQDs with typical QE below 10%.15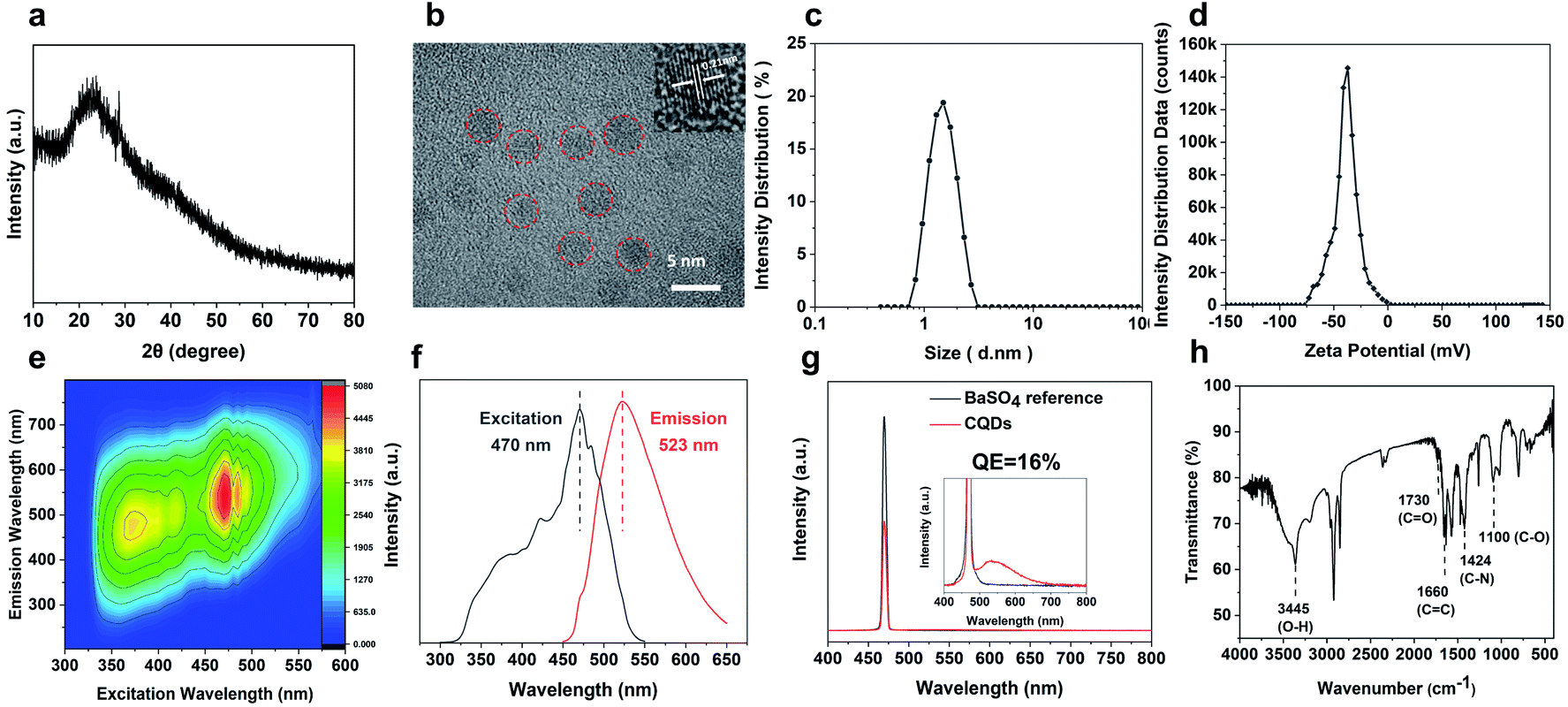 | ||
| Fig. 3 Physical and optical properties of the as-synthesized CQDs. (a) XRD pattern, and (b) TEM images of the CQDs (marked in red circles) after extraction. The inset shows the lattice fringes of the CQDs. (c) Size distribution, (d) zeta potential, (e) excitation–emission matrix spectra, (f) photoluminescence excitation and emission spectra of the CQD solutions, and (g) fluorescence QE measurements of the as-prepared CQDs using BaSO4 as the reference. The inset shows the zoomed-in spectra in the lower intensity range. (h) FTIR spectrum of CQDs. | ||
Owing to the high nitrogen content organics entrapped in the SAPO-20 framework, the CQDs from the confined synthesis in SAPO-20 are rich in carboxyl and hydroxyl groups on the surface containing N- and O-based functional groups, which avoids the requirement for post-synthetic surface functionalization. From the Fourier transform infrared (FT-IR) spectra of CQDs (Fig. 3g), it can be observed that the characteristic peaks at 3445 cm−1, 1730 cm−1 and 1100 cm−1 are due to the vibrations of O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, thus verifying the abundant hydrophilic groups on the surface originating from the pyrrolidine and TEAOH (Fig. S5†) leading to their excellent solubility and stability in water. Besides, the stretching vibrations of CC (1660 cm−1) and C–N (1424 cm−1) could also be observed in the spectrum, showing the highly graphitized structure of CQDs doped with the N element. The X-ray photoelectron (XPS) spectra of the obtained CQDs are shown in Fig. 4. The full XPS survey demonstrated the C 1s peak at 285.3 eV, N 1s peak at 400.5 eV, O 1s at 531.7 eV, and Na KLL peak at 496.5 eV, which confirmed the composition of the CQDs. The presence of Na might be from the post-extraction treatment using NaOH solution to dissolve the SAPO-20 framework, which further supports the negative charge of the obtained CQDs (Fig. 3c). The high-resolution C 1s spectrum of the CQDs can be deconvoluted into four main peaks of C–C (284.8 eV), CC (284.0 eV), CO (288.0 eV), and C–O/C–N (285.8 eV), respectively. The corresponding deconvolution results for investigating the O 1s and N 1s status are displayed in Fig. 4c and d. The O 1s signal might originate from three different oxygen environments: C–O bonds (531.7 eV), CO bonds (533.1 eV), and water (535.5 eV), illustrating the hydrophilicity of the as-made CQDs. The high-resolution N 1s spectrum shows only one peak of graphitic type N atoms at 399.5 eV.
O and C–O, thus verifying the abundant hydrophilic groups on the surface originating from the pyrrolidine and TEAOH (Fig. S5†) leading to their excellent solubility and stability in water. Besides, the stretching vibrations of CC (1660 cm−1) and C–N (1424 cm−1) could also be observed in the spectrum, showing the highly graphitized structure of CQDs doped with the N element. The X-ray photoelectron (XPS) spectra of the obtained CQDs are shown in Fig. 4. The full XPS survey demonstrated the C 1s peak at 285.3 eV, N 1s peak at 400.5 eV, O 1s at 531.7 eV, and Na KLL peak at 496.5 eV, which confirmed the composition of the CQDs. The presence of Na might be from the post-extraction treatment using NaOH solution to dissolve the SAPO-20 framework, which further supports the negative charge of the obtained CQDs (Fig. 3c). The high-resolution C 1s spectrum of the CQDs can be deconvoluted into four main peaks of C–C (284.8 eV), CC (284.0 eV), CO (288.0 eV), and C–O/C–N (285.8 eV), respectively. The corresponding deconvolution results for investigating the O 1s and N 1s status are displayed in Fig. 4c and d. The O 1s signal might originate from three different oxygen environments: C–O bonds (531.7 eV), CO bonds (533.1 eV), and water (535.5 eV), illustrating the hydrophilicity of the as-made CQDs. The high-resolution N 1s spectrum shows only one peak of graphitic type N atoms at 399.5 eV.
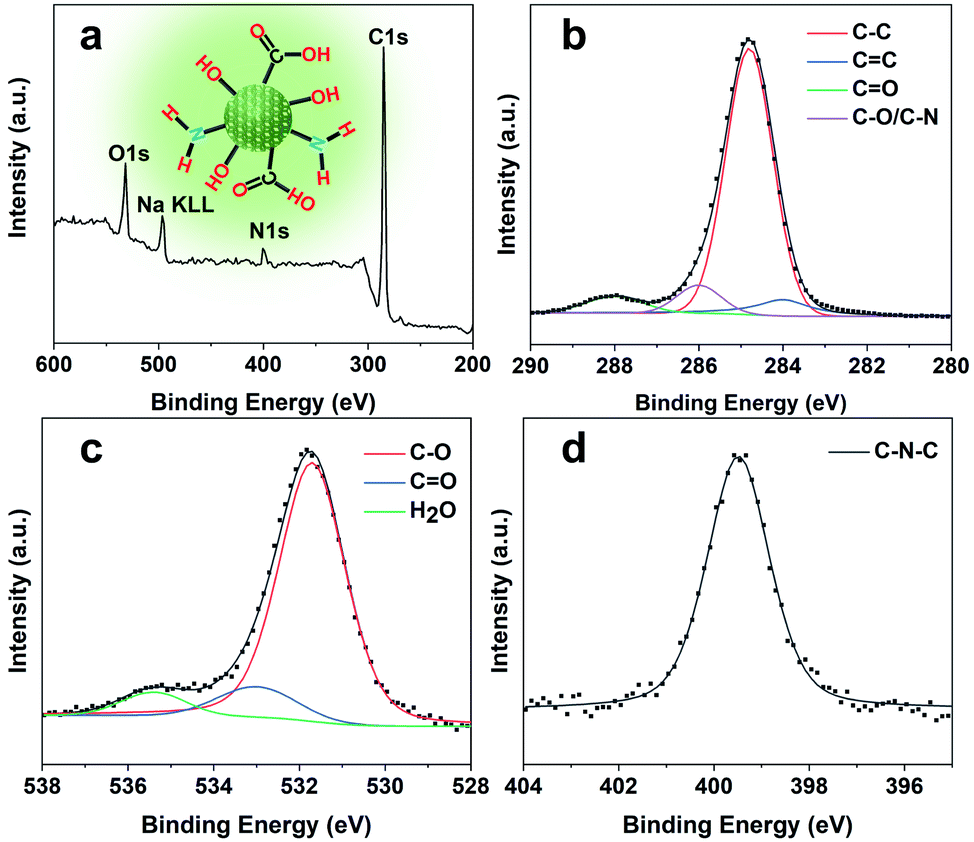 | ||
| Fig. 4 Surface analysis of CQDs. (a) Wide scan XPS survey spectrum (inset shows the chemical structure of CQDs), and (b) high-resolution C 1s, (c) O 1s, and (d) N 1s spectra of the as-prepared CQDs. | ||
Cell uptake of CQDs for bioimaging
Long-term cellular labeling and tracking are of great importance in monitoring biological processes, pathological mechanisms, therapeutic responses, etc.26 To explore the potential applications of the as-prepared CQDs, we attempted to label and trace glioma cells in vitro and in vivo. Before performing this experiment, we firstly evaluated the stability of CQDs in PBS solution simulating the physiological environment and cytotoxicity of CQDs. We dispersed the CQDs into PBS solution and monitored the change of size by DLS. From the DLS results (Fig. S6†), it can be observed that the size of CQDs shows little change within 7 consecutive days, confirming their great stability in a physiological environment. Furthermore after incubating CQDs of different concentrations with C6 glioma cells and bEnd.3 endothelial cells for 24 hours, respectively, the cell viability was determined by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method. The results showed that the cell viability maintained at more than 95% when the concentration of CQDs increased to 400 μg mL−1, indicating very low cytotoxicity of the as-prepared CQDs (Fig. 5a). Then, we further evaluated the blood compatibility of CQDs by incubating CQDs (0–400 μg mL−1) with red blood cells from healthy mice. It indicated that the hemolysis rate of red blood cells was less than 5%, showing good blood compatibility (Fig. 5b). Moreover, after the mice were treated with CQDs for 7 days (intravenous dose = 5 mg kg−1), the major organs, including the heart, liver, spleen, lungs, and kidneys showed no obvious damage or inflammation using hematoxylin and eosin (H&E) staining analysis (Fig. 5c), demonstrating that CQDs have excellent histocompatibility at our experimental dose and period.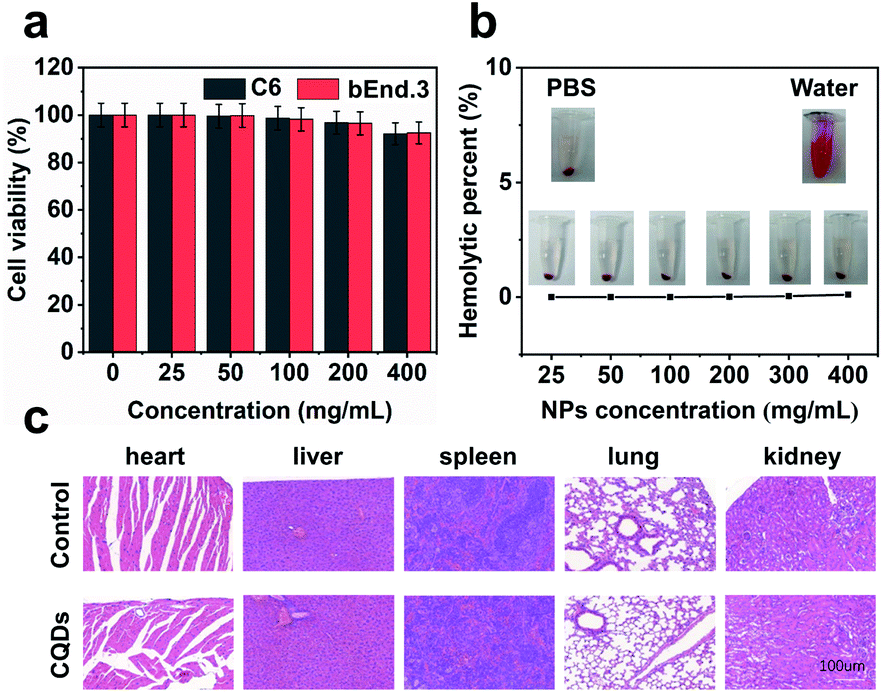 | ||
| Fig. 5 Biocompatible CQDs for living cell labeling and imaging in vitro. (a) Cytotoxicity of CQDs on C6 cells and bEnd.3 cells using MTT assay. (b) Hemolytic percentage of CQD-treated red blood cells after 3 h of incubation. Deionized water (+) and phosphate buffer saline (−) were used as positive and negative controls, respectively. The inset shows the digital photos of CQD-treated red blood cells. (c) Representative H&E stained images of the major organs after injection of CQDs with an injection dose of 5 mg kg−1. | ||
CQD labeled glioma cells for in vivo imaging
Our results preliminarily demonstrate that CQDs have good biocompatibility. Next, the bioimaging performance was studied. We incubated the CQDs with C6 glioma cells for 3, 6, and 12 h, respectively. Confocal fluorescence images showed that the CQDs could enter C6 glioma cells through cell endocytosis, locate in the cytoplasm, and generate strong fluorescence signals (Fig. 6a). Since CQDs could be internalized efficiently by glioma cells, we attempted to investigate the potential of CQD labeled glioma cells for in vivo fluorescence imaging. After treating glioma cells with CQDs for 3 h, we collected these cells and mixed with a matrigel matrix (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and subcutaneously injected into the subcutaneous tissue of Balb/c nude mice. As shown in Fig. 6b and c, with a decrease in the number of glioma cells, the fluorescence intensity of the injection region decreased gradually. Surprisingly, when the number of cells decreased to 1000, we could still detect the fluorescence signal in the injection site. This suggests that the as-prepared CQDs with high QE and good biocompatibility could efficiently label glioma cells for dynamic visualization of cell activity in the mouse model. In the future, CQDs with a longer wavelength and higher QE can be prepared to realize the long-term tracing of living cells in the body, providing efficient methods and technologies for the diagnosis and treatment of diseases.
:1) and subcutaneously injected into the subcutaneous tissue of Balb/c nude mice. As shown in Fig. 6b and c, with a decrease in the number of glioma cells, the fluorescence intensity of the injection region decreased gradually. Surprisingly, when the number of cells decreased to 1000, we could still detect the fluorescence signal in the injection site. This suggests that the as-prepared CQDs with high QE and good biocompatibility could efficiently label glioma cells for dynamic visualization of cell activity in the mouse model. In the future, CQDs with a longer wavelength and higher QE can be prepared to realize the long-term tracing of living cells in the body, providing efficient methods and technologies for the diagnosis and treatment of diseases.
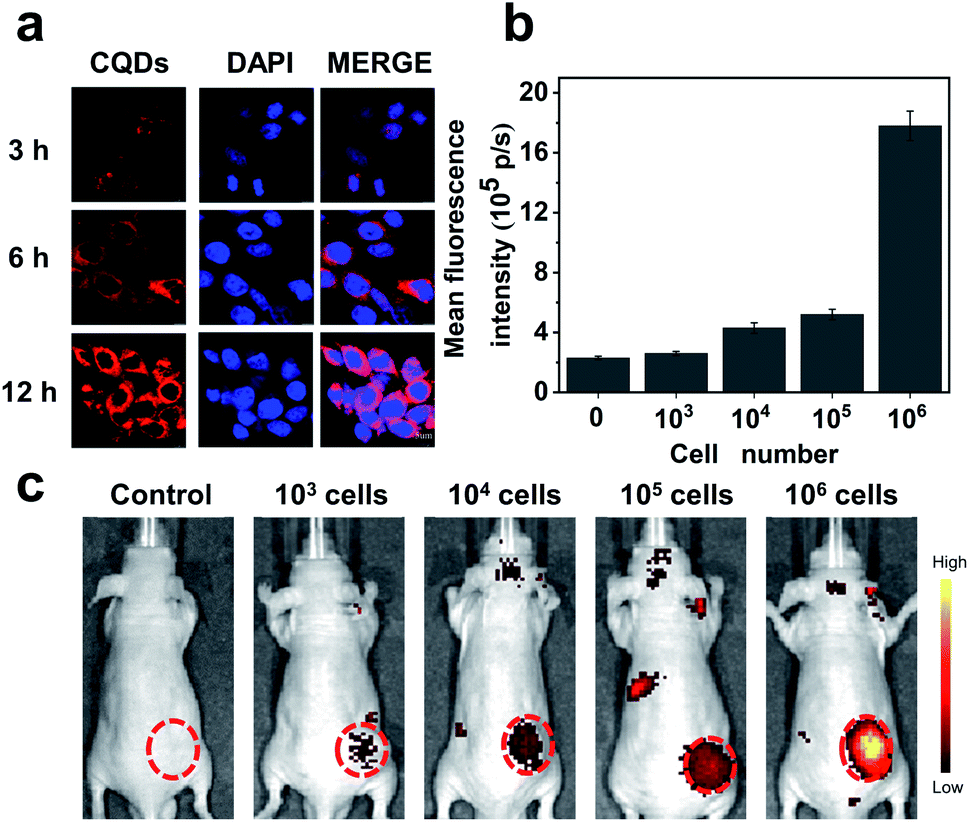 | ||
| Fig. 6 CQD labeled glioma cells for in vitro/in vivo imaging. (a) Confocal fluorescence images of C6 subcellular localization of CQDs after 3 h, 6 h and 12 h incubation. Red and blue represent the fluorescence of CQDs in the cytoplasm and 4′,6-diamidino-2-phenylindole (DAPI) fluorescence in the nucleus, respectively. (b) The mean FL intensity of CQD labeling of living glioma cells with different numbers based on in vivo fluorescence images indicated in (c). The data are shown as mean ± SD (n = 3). (c) CQD labeling of living glioma cells with different numbers for in vivo fluorescence imaging. The red circles point to the injection sites of CQD labeling of living glioma cells. | ||
Experimental
Reagents
All the reagents and solvents used in the experiment were commercially available and directly used without further purification. Pseudoboehmite and H3PO4 [85 wt% in water] were purchased from Guizhou or the Tianjin Chemical Reagent Factory. TEAOH [25 wt% in water] and pyrrolidine were obtained from Adamas Reagent Co., Ltd. Ludox (25% aqueous solution) was supplied from Qingdao Ocean Co., Ltd. Penicillin–streptomycin and high-glucose DMEM were obtained from Hyclone (USA). Trypsin EDTA and fetal bovine serum (FBS) were purchased from Gibco Life Technologies (AG, USA). A cell counting kit-8 (CCK-8) was purchased from Dojindo Laboratories (Shanghai, China).Synthesis of the SAPO-20 zeolite
The SAPO-20 zeolite samples were synthesized by a one-step hydrothermal synthesis. First, 2 g pseudoboehmite, 18 g TEAOH (25%), and 4 g H2O were mixed under stirring for 1 hour. Then 3.5 g phosphoric acid (85%) was added into the above solution. After stirring for 1 hour, 1.08 g pyrrolidine was added into the mixed solution, followed by the addition of 2.1 g SiO2 (25%) gel. The resultant homogeneous gel was further stirred using a magnetic stirrer for 4 hours. The molar ratio of the gel mixture is 1.0Al2O3:1.0P2O5:0.6SiO2:2.0TEAOH:1.0C4H9N:70.0H2O. Then the gel mixture was transferred into a 100 mL Teflon-lined stainless steel autoclave and heated at 200 °C for 40 hours at a controlled heating rate of 1 °C min−1. After crystallization, the obtained solid product was isolated by centrifugation at 6000 rpm and washed several times with distilled water. The collected white crystals were dried at 60 °C overnight.
A similar procedure was adopted for the comparative zeolite samples synthesized without TEAOH. Typically, 2 g pseudoboehmite and 17.5 g H2O were mixed under stirring for 1 hour. Then 3.5 g phosphoric acid (85%) was added into the above solution. After stirring for 1 hour, 1.08 g pyrrolidine were added into the mixed solution, followed by the addition of 2.1 g SiO2 (25%) gel. The resultant homogeneous gel was further stirred using a magnetic stirrer for 4 hours. The molar ratio of the gel mixture is 1.0Al2O3:1.0P2O5:0.6SiO2:1.0C4H9N:70.0H2O. Then the gel mixture was transferred into a 100 mL Teflon-lined stainless steel autoclave and heated at 200 °C for 40 hours at a controlled heating rate of 1 °C min−1. After crystallization, the obtained solid product was isolated by centrifugation at 6000 rpm and washed several times with distilled water. The collected brown crystals were dried at 60 °C overnight.
Preparation of CQDs
The as-synthesized TEAOH/pyrrolidine@SAPO-20 zeolite product was calcined in a furnace (KSL-1100X, JingKe) under an air atmosphere. The oven temperature was ramped to 600 °C at a rate of 2 °C min−1, followed by a 4 h isothermal treatment at this temperature. During this time, the entrapped organic species was carbonized, resulting in a composite material. 100 mg of the CQDs@SAPO-20 composite sample was dissolved in 30 mL of 2.5 M NaOH aqueous solution at room temperature. Then the mixture was ultrasonically treated for 10 min to accelerate the dissolution of the framework of SAPO-20. The dark color solution was neutralized with HCl solution to a pH of about 7. At this moment, a solid precipitate appeared which was separated from the solution by centrifugation. The supernatant containing CQDs was removed by freeze drying. Finally, the CQDs were obtained through extraction with alcohol to remove NaCl produced in the neutralization process and redispersed in water with a concentration of around 14.4 mg mL−1.Cell culture
C6 glioma cells and bEnd.3 brain microvascular endothelial cells were cultured in DMEM containing 10% FBS, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 2 mM L-glutamine. The cells were incubated at 37 °C in an incubator with a humidified atmosphere and 5% CO2.Confocal fluorescence imaging
105 C6 cells in eight-well glass-bottomed culture dishes were incubated with CQDs (50 μg mL−1) for 3, 6 and 12 h, respectively, washed with PBS, fixed with 4% paraformaldehyde, stained with DAPI, and finally imaged using a Laser confocal fluorescence microscope (Leica-TCS-SP5).Cytotoxicity assessment
To evaluate the toxicity of CQDs, normal bEnd.3 cells and glioma C6 cells were seeded in a 96-well plate (1 × 104 cells in 100 μL of medium for each well). After 12 h of incubation, cells were treated with CQDs at various concentrations from 25 μg mL−1 to 400 μg mL−1. The cell viability was measured by MTT assay.Hemolytic test
The whole blood of Balb/c mice was collected, centrifuged (1500 rpm, 3 min) to separate the red blood cells (RBC) and washed with PBS three times. 10% RBC (v/v, in PBS) was incubated with CQDs at final concentrations of 25, 50, 100, 200, 300, and 400 μg mL−1 in PBS at 37 °C for 3 h, respectively. After centrifugation (1500 rpm, 3 min), the supernatant of the suspensions was collected and analyzed using a UV-vis spectrometer at 541 nm.Histology
After the mice were treated with CQDs for 7 days (intravenous dose = 5 mg kg−1), the major organs, including the heart, liver, spleen, lungs, and kidneys showed no obvious damage or inflammation using hematoxylin and eosin (H&E) staining analysis. H&E staining was performed according to a protocol provided by the vendor (BBC Biochemical). The images were acquired on a Nikon Eclipse 90i microscope.In vivo imaging
After treating glioma cells with CQDs for 3 h, we collected these cells and mixed with a matrigel matrix (v/v = 1:1) and subcutaneously injected into the subcutaneous tissue of Balb/c nude mice, and acquired fluorescence images on an IVIS fluorescence imaging system using a 480 nm excitation wavelength and a 700 nm filter at different time points. The images were unmixed using the Maestro software. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, and approved by the Animal Ethics Committee of Shenzhen Institutes of Advanced Technology.
Characterization
The powder X-ray diffraction (XRD) patterns of all samples were recorded using a D8 Advance X-ray diffractometer with a diffraction angle between 4 and 50° equipped with Cu Kα radiation (λ = 1.5418 Å) at 40 kV, 40 mA. The scanning electron microscopy (SEM) image was obtained on a JEOL JSM-7800F electron microscope operated at 5.0 kV with platinum coating. Transmission electron microscopy (TEM) was performed on a Tecnai G2 F30 field emission source transmission electron microscope operated at 300 kV. X-ray photoelectron spectroscopy (XPS) measurements were performed on a PHI 5000 VersaProbe II spectrometer with monochromatized Al Kα excitation. Thermogravimetric analyses (TGA) were performed using a Shimadzu TGA-50 analyzer and Fourier transform infrared spectroscopy (FTIR) spectra were recorded in KBr pellets using a Shimadzu IR-Prestige 21 Spectrophotometer. Raman spectra were obtained using a Mettler Toledo ReactRaman 785. The contents of C and N were measured using an Organic Element Analyzer (Vario PYRO cube, Elementar, Germany). The particle size and zeta potential measurements of the CQDs were performed on a Zetasizer Nano analyzer (Zetasizer Nano ZS90, Malvern). The photoluminescence excitation and emission spectra were recorded using a spectrofluorometer (Lumina, Thermo Scientific). The absolute fluorescence quantum yields were measured on an Edinburgh FLS980 fluorimeter.Conclusions
We have developed a simple and facile strategy to prepare ultrasmall-sized CQDs with tunable fluorescence properties. By choosing a small-pore SAPO-20 with SOD topology, through the joint action of structure-directing agent pyrrolidine and growth modulator TEAOH, monodisperse cubic crystals with a micropore aperture of 0.28 nm were obtained. Pyrolysis of the entrapped organics afforded monodisperse ultrasmall CQDs with a surface rich in carboxyl and hydroxyl groups with an average size of 1.53 nm, smaller than those obtained from larger-pore zeolites. These CQDs exhibited excellent stability and were easily dispersible in solution, showing a high fluorescence QE of 16% in the non-functionalized form. The as-synthesized CQDs have been successfully adopted as an effective fluorescent probe for efficient labeling of living cells and in vivo biological imaging due to their high fluorescence QE, low cytotoxicity, and excellent biocompatibility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the financial support from the National Natural Science Foundation of China (21671010, 91859117, and 81771906), the Guangdong Science and Technology Program (2020A1515011260), the Shenzhen Science and Technology Program (GJHZ20180928162402322, JCYJ20170818085754055, JCYJ20190812163614809, and JCYJ20170818161918918), the Shenzhen Peacock Plan (KQTD2016053015544057), the CAS Key Laboratory of Health Informatics (2011DP173015), and the Shenzhen Key Laboratory of Ultrasound Imaging and Therapy (ZDSYS201802061806314).Notes and references
- Y. F. Wang and A. G. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS.
- L. Bao, Z.-L. Zhang, Z.-Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D.-W. Pang, Adv. Mater., 2011, 23, 5801–5806 CrossRef CAS.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS.
- H. Li, X. He, Y. Liu, H. Huang, S. Lian, S. T. Lee and Z. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 1, 5118–5120 RSC.
- Q. Wang, X. Liu, L. Zhang and Y. Lv, Analyst, 2012, 137, 5392–5397 RSC.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS.
- Z. Jiang, A. Nolan, J. G. Walton, A. Lilienkampf, R. Zhang and M. Bradley, Chem.–Eur. J., 2014, 20, 10926–10931 CrossRef CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS.
- K. O. Boakye-Yiadom, S. Kesse, Y. Opoku-Damoah, M. S. Filli, M. Aquib, M. M. B. Joelle, M. A. Farooq, R. Mavlyanova, F. Raza, R. Bavi and B. Wang, Int. J. Pharm., 2019, 564, 308–317 CrossRef CAS.
- B. Wang, Y. Mu, H. Yin, Z. Yang, Y. Shi and J. Li, Nanoscale, 2018, 10, 10650–10656 RSC.
- Y. Mu, N. Wang, Z. Sun, J. Wang, J. Li and J. Yu, Chem. Sci., 2016, 7, 3564–3568 RSC.
- H. G. Baldovi, S. Valencia, M. Alvaro, A. M. Asiri and H. Garcia, Nanoscale, 2015, 7, 1744–1752 RSC.
- M. Bandyopadhyay, R. Bandyopadhyay, S. Tawada, Y. Kubota and Y. Sugi, Appl. Catal., A, 2002, 225, 51–62 CrossRef CAS.
- G. Zheng, Z. Chen, K. Sentosun, I. Perez-Juste, S. Bals, L. M. Liz-Marzan, I. Pastoriza-Santos, J. Perez-Juste and M. Hong, Nanoscale, 2017, 9, 16645–16651 RSC.
- A. M. Mofrad, C. Peixoto, J. Blumeyer, J. Liu, H. K. Hunt and K. D. Hammond, J. Phys. Chem. C, 2018, 122, 24765–24779 CrossRef CAS.
- T. Ikuno, W. Chaikittisilp, Z. Liu, T. Iida, Y. Yanaba, T. Yoshikawa, S. Kohara, T. Wakihara and T. Okubo, J. Am. Chem. Soc., 2015, 137, 14533–14544 CrossRef CAS.
- P. K. Dutta and M. Puri, J. Phys. Chem., 1987, 91, 4329–4333 CrossRef CAS.
- L. M. Brent, M. A. Celeste, P. L. Robert, G. T. Richard, C. R. Thomas and F. M. Edith, US4440871, 1984.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS.
- L. Grajciar, J. Phys. Chem. C, 2016, 120, 27050–27065 CrossRef CAS.
- M. Yu, J. L. Falconer, T. J. Amundsen, M. Hong and R. D. Noble, Adv. Mater., 2007, 19, 3032–3036 CrossRef CAS.
- Z. Wang, S. Chen, J. W. Y. Lam, Q. Wei, R. T. K. Kwok, X. Ni, Q. Hu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 8238–8245 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00664e |
| ‡ Lei Dong and Dehong Hu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |