
Self-assembled chitosan/rose bengal derivative nanoparticles for targeted sonodynamic therapy: preparation and tumor accumulation†
Yu Gao*a,
Zhihong Lia,
Chaoqun Wanga,
Jiali Youa,
Biyu Jina,
Fan Moa,
Jianzhong Chenb,
Yunquan Zhenga and
Haijun Chen*a
aCollege of Chemistry, Fuzhou University, Fuzhou 350108, China. E-mail: hellogaoyu@126.com; chenhaij@gmail.com
bSchool of Pharmacy, Fujian University of Traditional Chinese Medicine, Fuzhou 350108, China
First published on 5th February 2015
Abstract
Sonodynamic therapy (SDT) is expected to be a promising novel therapeutic strategy for non-invasive cancer treatment. Rose bengal (RB), which serves as a typical photosensitizer, can also be activated by ultrasound and has high potential to be a sonosensitizer for SDT. However, the fast elimination rate and poor tumor accumulation of RB greatly limit its in vivo applications. The aim of this work is to synthesize a rose bengal derivative (rose bengal ω-carboxyheptyl ester, RBD), prepare chitosan (CS)/RBD complex nanoparticles (RBDCNs), and investigate the cellular uptake and tumor accumulation of RBDCNs in cancer cell lines and a transplanted colon cancer mouse model. CS and RBD could form complex nanoparticles with a particle size of 150–200 nm and positive surface charges (>+40 mV) at a mass ratio 1. The addition of CS conferred a remarkable decrease in the fluorescent intensity of RBD, suggesting the inclusion of dye within the complex nanoparticles. RBDCNs showed good stability in the medium containing salt ions with dye leakage of less than 20%, while the high concentration of serum could affect the interaction between CS and RBD, resulting in about 75% dye leakage under a 10% FBS condition. RBDCNs could achieve more efficient cellular uptake in HT29 and HepG2 cells than RB, RBD, or CS/RB complex nanoparticles (RBCNs). The high accumulation of RBDCNs in tumors was also observed when RBDCNs were intravenously injected into CT-26 colon cancer transplanted Balb/c mice. These results suggested that RBDCNs could be efficient and promising tumor targeting delivery systems.
1. Introduction
Non-invasive cancer treatment is a promising alternative to traditional cancer therapies, which targets only cancer cells, leaving healthy cells intact. Photodynamic therapy (PDT), which uses the combination of laser light and a sensitizer, has received considerable attention as a non- or less-invasive and tissue selective tumor treatment.1–3 PDT involves the procedure of an intravenously administered photosensitizer followed by laser light activation at the targeted tissues. Therefore, it has obvious advantages of targeting lesions more precisely than chemotherapy and radiotherapy, and could be repeated multiple times with minimal toxicity to normal tissues. Currently, PDT has been used to treat cancer, and some photosensitizers such as Photofrin®, Levulan®, and Metvixia® have been approved by the FDA.4 However, PDT often meets the problem of the limited penetration of light into deep-seated tissue, and only superficial regions can be treated by PDT.Sonodynamic therapy (SDT) is a new approach derived from PDT for cancer therapy, and employs ultrasound to activate sonosensitizers and to enhance the bioeffects of some chemicals, and shows a superior tissue penetration effect over PDT.2,5 Many photosensitizers such as photolon,6 the titanium dioxide (TiO2) nanoparticle,7 hypocrellin-B derivative SL017,8 and phthalocyanine9,10 have been found not only to be response to laser light, but also to be ultrasonically activated. Rose bengal (RB), 4,5,6,7-tetrachloro-2′,4′,5′,7′-tetraiodofluorescein disodium salt, is one of such photosensitizers with the potential for anticancer therapy. Upon light irradiation, RB could generate cytotoxic singlet oxygen that kill tumor cells, and the ultrasound could further enhance its cell killing ability by certain mechanical stress such as augmentation of physical disruption of cellular membrane.11 However, RB demonstrated fast elimination rate and poor tumor accumulation,12 which greatly limited further in vivo applications.
To overcome its shortcomings, a series of RB derivatives with long aliphatic lipid chains were synthesized by Sugita et al. to facilitate selective tumor accumulation.13 One type of the synthesized RB derivatives with an alkyl chain and a branching carboxyl group demonstrated more than 1 order of magnitude tumor tissue distribution than RB. Although improvement of the amphiphilicity of RB could increase its tumor accumulation to some extent, the introduction of the long carbon chain might significantly decrease its water solubility. In addition, the adjustment of the amphiphilicity could change the biodistribution of RB to increase its tumor accumulation, but it could not enable the tumor site specific delivery. Without suitable formulation, the leakage of sensitizers in the blood circulation and the uptake of sensitizers by the normal tissues could not be avoided.
Nanoparticles as drug delivery systems make it possible to deliver the sonosensitizers to desired tumor site specifically. It has been reported by many research groups that nanoparticles with proper size are expected to accumulate in tumors by passive targeting due to the enhanced permeation and retention (EPR) effect.14,15 Chitosan (CS) is a natural polysaccharides having been widely used in various pharmaceutical and biomedical formulations.16,17 It demonstrates excellent biodegradability and biocompatibility compared with other nanomaterials which have been shown to induce inflammatory response,18 fibrogenesis,19 or genotoxicity.20 Because of its unique polyvalent positive-charged property, chitosan possesses some abilities that other sugar molecules do not have.21–23 It could interact with negatively charged DNA or RNA to serve as non-viral gene delivery vector.24–27 By chemically available functional groups of polyanions, such as phosphoric acid in sodium tripolyphosphate (TPP), small molecular drugs also could be entrapped within CS/TPP nanoparticles.28 We are interested in design and development of a new formulation for RB which has ability of passive targeting of tumor tissue followed by efficiently transport into tumor cells. In this work, CS was chosen for nanoparticle preparation for tumor targeted delivery of RB. The rose bengal derivative (rose bengal ω-carboxyheptyl ester, RBD) with high partition coefficient was synthesized to facilitate transportation of RB into tumor cells after accumulation in tumor tissue. The physicochemical properties of CS/RBD complex nanoparticles (RBDCNs) were characterized, and the in vitro cellular uptake and in vivo distribution in transplanted colon cancer mouse model of RBDCNs were investigated.
2. Experimental section
2.1 Materials
Chitosan (Mw = 60 kDa; degree of deacetylation ≥90%) was purchased from Boao Biotechnologies Co., Ltd. (Shanghai, China). The RPMI 1640 medium, Dulbeccos Modified Eagle Medium (DMEM), McCoy's 5A medium, antibiotics, and fetal bovine serum (FBS) were purchased from Life Technologies GmbH (Darmstadt, Germany). 8-Bromooctanoic acid was obtained from Sigma (St. Louis, USA). Rose bengal sodium salt (RB), sodium hydroxide, N,N-dimethylformamide (DMF), diethyl either, and ethanol were purchased from Sinopharm Group Chemical Reagent Co., Ltd. (China). Rose bengal derivative with an eight-carbon chain (RBD) was synthesized according to the previous report.132.2 Cell culture and animals
The human liver hepatocellular carcinoma cell line (HepG2), the human colon carcinoma cell line (HT29), and the mouse colon carcinoma cell line (CT-26) were purchased from Cell Resource Center of Shanghai Institute for Biological Sciences (Chinese Academy of Sciences, Shanghai, China). HepG2 cells and HT29 cells were grown in RPMI 1640 medium and McCoy's 5A growth medium, respectively, which contained 10% FBS, 100 units per mL penicillin and 100 μg mL−1 streptomycin sulfate. CT-26 cells were grown in RPMI 1640 medium with 20% FBS. All cells were maintained at 37 °C in a humidified and 5% CO2 incubator. Six- to eight-week-old male Balb/c mice obtained from Fuzhou Wushi Animal Center were maintained in standard cages in an air-conditioned room with access to food and water ad libitum, and exposed to a 12 h light/dark cycle. The animal experiment was approved by the local Ethics Committee of Fuzhou University.2.3 Preparation of CS/RBD complex nanoparticles (RBDCNs)
RB was dissolved in water. RBD was dissolved in 0.1 N sodium hydroxide aqueous solution followed by adding hydrochloric acid to adjust the pH of the solution close to neutral. RBDCNs at various weight ratios were formed by slowly dropping RBD of desired concentrations diluted in water into an equal volume of solutions of CS diluted in 5 mM sodium acetate/acetic acid buffer (pH 5.5). The solutions were mixed rigorously to ensure effective formation of RBDCNs. Then the solutions were kept at room temperature for 30 min. The CS/RB complex nanoparticles (RBCNs) were prepared using the same procedures.2.4 Physicochemical characterization of RBDCNs
The morphological examination of RBCNs and RBDCNs was performed using a transmission electron microscope (TEM, H-7650, Hitachi, Japan) after negative staining with sodium phosphotungstate solution (0.2%, w/v). The mean particle size and zeta potential of RBCNs and RBDCNs were determined by dynamic light scattering (DLS) method using Nanotrac® Wave Particle Size and Zeta Potential Analyzer (Microtrac Inc, Montgomeryville, PA). For zeta potential (mV) measurements, 100 μL of each colloidal suspension was diluted into 2 mL of ultrapure water. These measures were carried out at 25 °C.The fluorescence excitation and emission peaks for RB and RBD were determined using the Tecan Infinite™ 200 PRO Microplate Reader (Salzburg, Austria). The physical loading efficiencies of RB and RBD in the complex nanoparticles were quantified using a Hitachi F-4600 fluorescent spectrophotometer. According to the differences of the fluorescent intensity, the drug loading capacity was calculated by the following relationship:
| Drug loading capacity = [1 − (Fsample − Fblank)/(FRBD only − Fblank)] × 100% |
2.5 Salt and serum induced dissociation of RBDCNs
RBDCNs with various weight ratios were prepared as described above and diluted with water to a concentration of 0.5 μg RBD/100 μL solution. A series of water solutions containing various concentrations of NaCl (0.2, 0.4, 0.6, 0.8, 1.0 M) or phosphate buffered saline containing different percentages of FBS (1%, 2%, 5%, 10%, 20%) were prepared. Then, 100 μL of the solution was added to 100 μL RBDCNs solution to achieve the desired gradient of NaCl or FBS. The mixed solution was arrayed into a Corning black flat bottom 96-well plate and the fluorescence of RB (excitation 518 nm, emission 561 nm) or RBD (excitation 535 nm, emission 564 nm) for each sample was measured with a Tecan Infinite™ 200 PRO multimode microplate reader. Fraction drug exclusion was determined by the following relationship:| Drug exclusion = [1 − (Fsample − Fblank)/(FRBD only − Fblank)] × 100% |
The salt and serum induced dissociation of RBCNs were detected using the same procedure.
2.6 Cellular uptake efficiency
HT29 and HepG2 cells were seeded in a 24-well plate with 0.5 mL growth medium and allowed to attach for 24 h. Then, cells were incubated with RBDCNs at weight ratio 1 containing 2.5, 5, 10 μg mL−1 RBD under fresh medium without FBS for 2 h at 37 °C, followed by washing three times with PBS (pH 7.4). The same experiment was performed with cells incubated with RBCNs. Finally, cells were detached, subjected to flow cytometry (Coulter EPICS XL, Beckman Coulter Co. Ltd., FL, USA) and the mean fluorescence intensity of cells were analyzed.The internalization of complex nanoparticles was visualized by an inverted fluorescence microscope (Nikon 50i, Kawasaki, Japan). HepG2 cell monolayers were cultured on 10 mm2 glass coverslips for 24 h. After incubation with RBDCNs at weight ratio 1 containing 10 μg mL−1 RBD or RBCNs at weight ratio 1 containing 10 μg mL−1 RB for 2 h at 37 °C under fresh medium without FBS, cells were washed three times with PBS (pH 7.4). Then, cells were fixed using methanol–acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Subsequently, cells were observed under a fluorescence microscope.
:1, v/v). Subsequently, cells were observed under a fluorescence microscope.
2.7 Tissue distribution
For biodistribution study, Balb/c mice were intravenously transplanted with 100 μL of phosphate buffered saline (pH 7.4) containing 1 × 106 CT-26 cells through the tail vein. After 2 weeks, pulmonary tumor nodules could be clearly observed in the lungs of mice. For quantitative evaluation, RBD or RBDCNs at mass ratio 1 diluted with physiological solution was injected into mice transplanted with CT-26 colon cancer via the tail vein at a dose of 2 mg RBD per kg. Mice were sacrificed at 1 h after injection (n = 3), and the principal organs were isolated (Fig. S1†). The extracted tissues were homogenized with lysis buffer (Promega, Madison, WI, USA), and the samples were centrifuged at 10000 rpm for 5 min. The supernatant was measured using a Tecan Infinite™ 200 PRO multimode microplate reader. The data were normalized to the tissue weight.
2.8 Statistical analysis
Statistical analysis was performed using a Student's t-test. The differences were considered significant for p < 0.05 and p < 0.01 indicative of a very significant difference.3. Results and discussion
3.1 Preparation and characterization of RBDCNs
CS is a linear polysaccharide composed of randomly distributed β-(1-4)-linked D-glucosamine (deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit) with a pKa value of ∼6.5. It is insoluble in water, but soluble in acidic solutions below its pKa, in which it can convert glucosamine units (–NH2) into the soluble protonated form (–NH3+).29 Due to this unique positively charged properties, CS could form polyelectrolyte complexes with other oppositely charged biopolymers by electrostatic interactions with various forms including nanoparticle, gels, microparticles, tablets, beads, as well as films and membranes.29RB is an acid dye in which the charge on the dye ion (chromophore) is negative. Besides the negative chromophore, RBD has an extra carboxyl group which could interact with amino group. We postulated that the positively charged CS could bind to RBD to form nanoparticles through the electronic interaction (Fig. 1). In this work, we chose CS with 90% degree of deacetylation and with moderate molecular weight (60 kDa). The degree of deacetylation (DD) and the molecular weight (MW) of CS are important to their physical and biological properties.30,31 The high DD make CS have more free amino groups and more positive to interact with other negatively charged substances. However, the cytotoxicity of CS was increased by increasing polymer DD especially when CS had high MW.32 Therefore, we chose CS with high DD to guarantee the strong interaction between CS and RBD, and with moderate MW to make the formulation biocompatible.
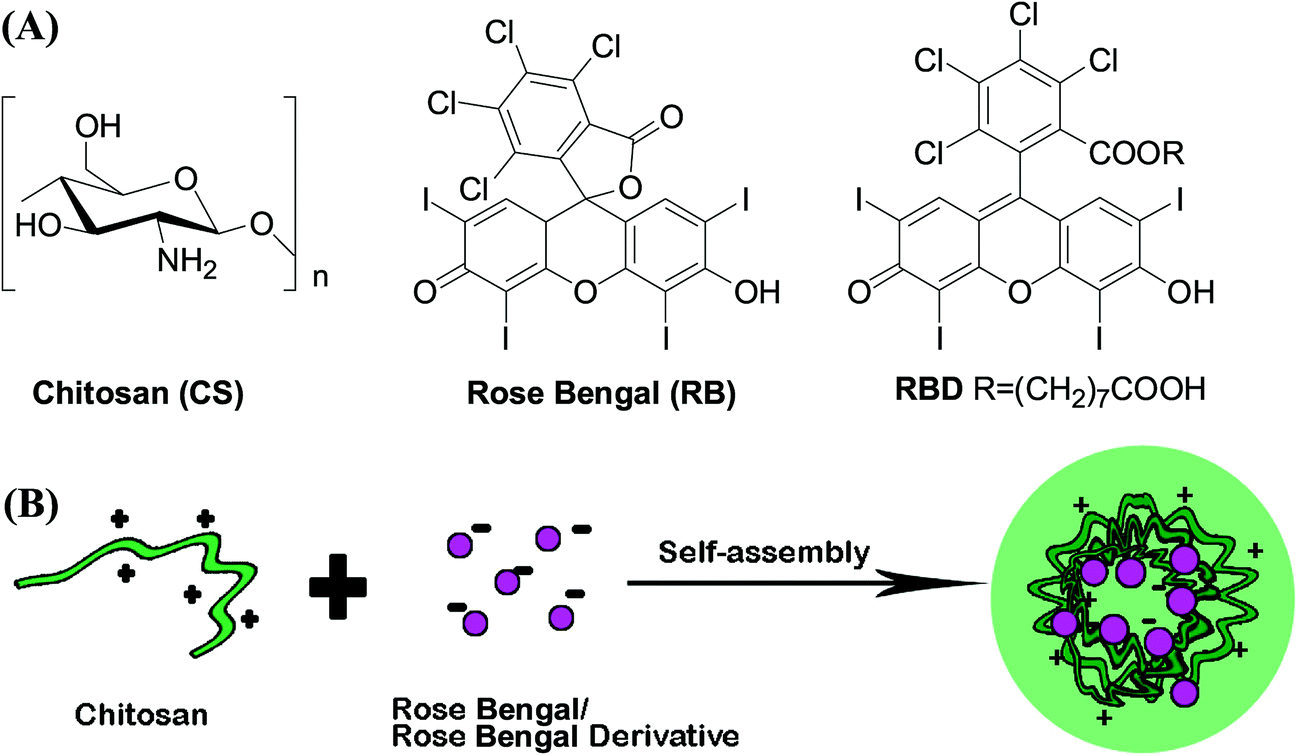 | ||
| Fig. 1 (A) Chemical structures of chitosan (CS), rose bengal (RB) and rose bengal derivative (RBD). (B) Schematic illustration of CS/RB or CS/RBD complex nanoparticles. | ||
By slowly dropping RB or RBD solution to CS solution followed by vigorous votexting, CS/RB or CS/RBD could successfully self-assembled into complex nanoparticles. The mean particle size of RBCNs and RBDCNs at mass ratios from 0.5:1 to 4:1 detected by DLS method were about 75–360 nm, and the zeta potential of RBCNs and RBDCNs at the four mass ratios are all above +10 mV (Fig. 2A and B), indicating the successfully formation of nanoparticles. It is very important to mention that the RB or RBD should be added slowly to the solution of CS. The red precipitate can be formed when quickly mixing the solution of RB or RBD with the solution of CS. The particle sizes of RBDCNs were bigger than RBCNs at all the mass ratios. The diameters of RBCNs slightly decreased with the increase of mass ratio, but the dynamic light scattering analysis of RBDCNs showed a different trend. These results demonstrated that the introduction of an eight-carbon chain to RB could significantly affect its electronic interaction with CS. To find out the reason, the particle size distributions of RBCNs and RBDCNs at different mass ratios were compared. At the mass ratio of 1, RBCNs and RBDCNs showed single peak with normal distribution (Fig. 2C and D), and the dispersion indexes of RBDCNs were smaller than that of RBCNs. The narrower particle size distribution might be due to the extra carboxyl group in RBD which made it have stronger electronic interaction with CS and the extra eight-carbon chain which made it have stronger hydrophobic interaction with CS. When the mass ratio was above 1, the distribution of RBCNs and RBDCNs showed more than one peak (Fig. 2E and F). The peak at around 1 nm could be attributed to the free CS macromolecules. The peak at around 1 μm might due to the slightly flocculation of complex nanoparticles under high CS concentration. It is reported that effective flocculation can be found with highly charged complexes as well as with complexes near the 1:1 ratio of charges.33 The high density of positive charges and the low flexibility of RBD due to the hydrophobic eight-carbon chain might induce the mismatch of opposite charges eventually resulting in flocculation. TEM was performed to characterize the morphologies of RBCNs and RBDCNs. The morphologies of RBCNs and RBDCNs were round and uniform (Fig. 3A and B). The sizes of RBCNs and RBDCNs observed from TEM are smaller than that measured by DLS. This is due to the samples measured by TEM are in dry state which would make the particles shrink.
 | ||
| Fig. 2 The physicochemical characterization of RBCNs and RBDCNs. Particle sizes (A) and zeta potentials (B) of RBCNs and RBDCNs at different mass ratios. Particle size distribution of RBCNs (C) and RBDCNs (D) at the mass ratio of 1. Particle size distribution of RBCNs (E) and RBDCNs (F) at the mass ratio of 2. | ||
 | ||
| Fig. 3 TEM images of complex nanoparticles. (A) TEM image of RBCNs at the mass ratio of 1. (B) TEM image of RBDCNs at the mass ratio of 1. Scale bar = 200 nm. | ||
3.2 Stability of RBDCNs
As we postulated, the positively charged CS could bind to RBD to form nanoparticles, and the RBD could be wrapped inside the nanoparticles. As fluorescent dyes, the entrapment efficiency of RB or RBD could be determined by fluorescence detection method. Through full wavelength scanning using the Tecan Infinite M200 Microplate Reader, RB shows excitation maximum at 518 nm and emission maximum at 561 nm in water (Fig. 4A). The conjugation of eight-carbon chain to RB slightly changed its fluorescence spectrum. RBD displays excitation maximum at 535 nm and emission maximum at 564 nm (Fig. 4B). Using a fluorescent spectrophotometer, we could find that the fluorescence intensity of RB or RBD solution decreased dramatically after adding of the CS (Fig. 4C and D). The decreased fluorescent intensity is due to the entrapment or complexation of RB or RBD inside the nanoparticles. It is almost impossible to measure the fluorescence intensity of RBCNs or RBDCNs at mass ratio 1, suggesting that the physical loading efficiencies of RB and RBD in the complex nanoparticles could be close to 100%. The results demonstrated that there have strong interactions between CS and RB or RBD.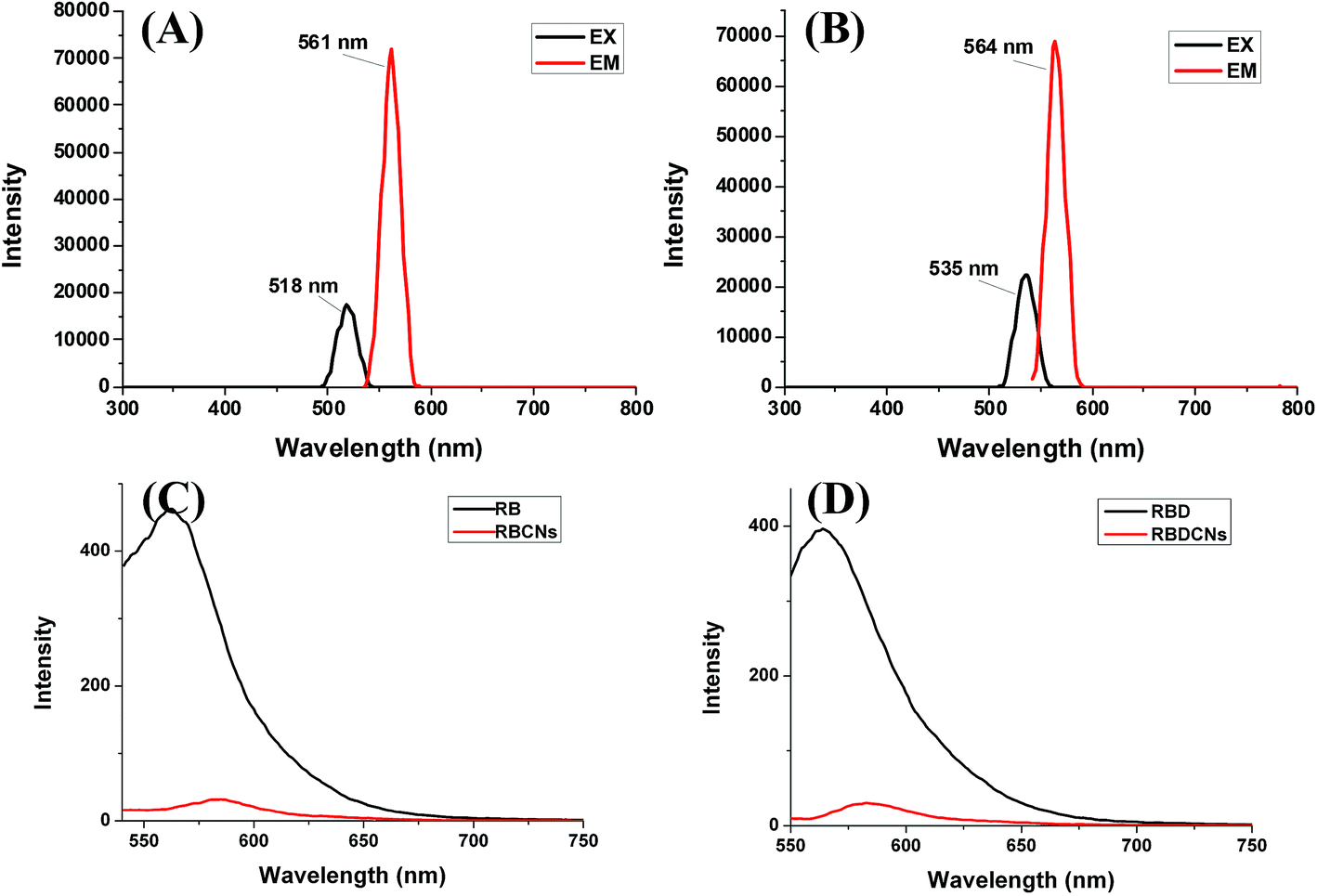 | ||
| Fig. 4 Fluorescence spectra of RB, RBD, RBCNs, and RBDCNs. (A) Determination the fluorescence excitation and emission peaks for RB using the Tecan Infinite M200 Microplate Reader. (B) Determination the fluorescence excitation and emission peaks for RBD using the Tecan Infinite M200 Microplate Reader. (C) Fluorescence spectra of RB and RBCNs at mass ratio 1 (λex = 518 nm). (D) Fluorescence spectra of RBD and RBDCNs at mass ratio 1 (λex = 535 nm). | ||
The stability of nanoparticles is a key parameter influencing their biological behaviors. Zeta potential could be an important tool to predict the long term stability of the nanoparticle. Nanoparticles with high zeta potentials might have high degrees of stability to avoid aggregation due to van der Waal inter-particle attractions. In this work, the zeta potentials of RBCNs and RBDCNs at the mass ratios above 1 are higher than 40 mV, indicating that RBCNs and RBDCNs could be dispersed stably in water due to the strong electric repulsion between particles.34 As a drug delivery system, the nanoparticles not only should be stable in the storage environment, they but also should withstand salt and plasma proteins challenges in the biological environment before they reach the desired action site.35 Many previous studies have shown that serum could reduce the interaction between the polyelectrolyte complexes.25 We then detect the dissociation rate of RBCNs or RBDCNs under different concentrations of salt and serum.
From Fig. 5, it could be easily found that RBCNs and RBDCNs could stand salt challenge effectively. With salt concentration even as high as 0.5 M, the drug exclusion rates of RBCNs and RBDCNs at mass ratio ≥1 were less than 20%. Compared with RBCNs, RBDCNs demonstrated stronger ability to resist the salt induced dissociation. The resistant ability increased gradually with the increase of the mass ratio. However, in the presence of FBS, both RBCNs and RBDCNs demonstrated significant dye leakage from complex nanoparticles. The serum could markedly affect the interaction between CS and dye. The dye exclusion increased gradually with the increase of the mass ratio. At the highest serum concentration we studied, more than 60% of the dye was dissociated from the complex nanoparticles (Fig. 5C and D). The results demonstrated that the effect of serum on the stability of RBCNs or RBDCNs was greater than the effect of salt.
 | ||
| Fig. 5 The salt and serum induced dissociation of RBCNs or RBDCNs. (A) Salt induced dissociation of RBCNs. (B) Salt induced dissociation of RBDCNs. (C) Serum induced dissociation of RBCNs. (D) Serum induced dissociation of RBDCNs. RBCNs or RBDCNs were incubated with different concentrations of NaCl (0.1–0.5 M) or phosphate buffered saline containing different percentages of FBS (0.5–10%). The dissociation of RB or RBD from the complex nanoparticles could be detected by a Tecan InfiniteTM 200 PRO multimode microplate reader. | ||
3.3 Cellular uptake of RBDCNs
From the above study, we found that CS and RB or RBD at mass ratio 1 could form uniform complex nanoparticles with suitable particles sizes (100–200 nm) for in vivo application. Also, at this mass ratio, RB or RBD could be almost completely loaded into the complex nanoparticles. So we chose RBCNs and RBDCNs at mass ratio 1 to investigate their cellular uptake efficiency. Either in HT-29 cells or HepG2 cells, RBD demonstrated significantly higher cellular uptake efficiency than RB (Fig. 6), which indicated that the introduction of the amphiphilic alkyl chain could improve its cellular internalization ability. Amphiphilic molecules might have interactions with cell membrane and are more likely to be uptaken by cells.36,37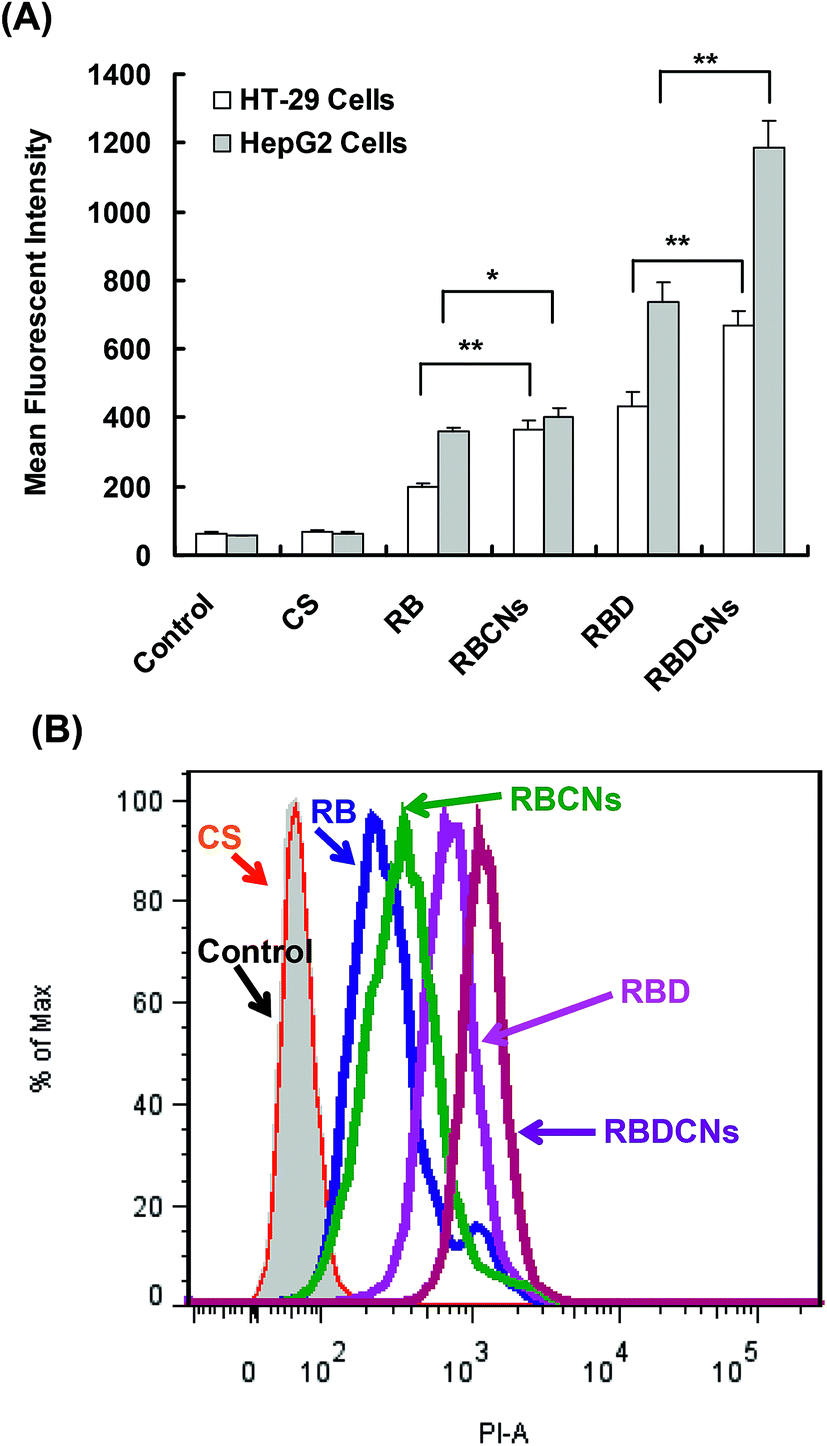 | ||
| Fig. 6 Flow cytometry analysis of cellular uptake of RB or RBD in HT-29 and HepG2 cells. Cells were incubated with 2.5 μg mL−1 RB or RBD or their complex nanoparticles at mass ratio 1 with the same drug concentration for 2 h. (A) Mean fluorescence intensity of cells treated with RB or RBD or their complex nanoparticles. Each data point represents the mean ± SD of three replicates. *p < 0.05, **p < 0.01. (B) Flow histogram of HepG2 cells after incubation with RB, RBCNs, RBD, or RBDCNs for 2 h. | ||
From Fig. 6, we could also find that the mean fluorescent intensity (MFI) of cells treated with RBCNs and RBDCNs were significantly higher than that of cells treated with free RB and RBD, respectively, indicating that the complex nanoparticles could facilitate the cellular uptake of RB and RBD. The MFI of HepG2 cells treated with RBDCNs could be as high as 1186, while the MFI of the cells treated with RBD was only 736. It could be obviously found from the histogram of FACS that the peaks of RB, RBCNs, RBD, and RBDCNs were distributed in high logarithmic scale in turn (Fig. 6B). We also used fluorescence microscope to visualize the cellular uptake and the intracellular distribution of RB, RBCNs, RBD, and RBDCNs in HepG2 cells (Fig. 7). The results were consistent with the results of FACS analysis. RBD demonstrated enhanced fluorescent intensity than RB. Both RB and RBD showed significant weak fluorescent intensity than their complex nanoparticle counterparts. All the above results indicated that modification of RB with an alkyl chain could significantly enhance its cellular uptake efficiency, and fabrication of CS and RBD could further improve cellular uptake of RBD.
 | ||
| Fig. 7 Images of HepG2 cells incubated with RB (A), RBCNs (B), RBD (C) and RBDCNs (D) observed under a fluorescent microscope (20× magnification). RBCNs and RBDCNs were prepared at mass ratio 1. The dosages for all treated groups were 2.5 μg mL−1 RB or RBD. | ||
We also investigated the effect of drug concentration on the cellular uptake efficiency of RBCNs or RBDCNs in HepG2 cells. The cellular uptake of RB, RBCNs, RBD, and RBDCNs in HepG2 cells correlated with the drug concentration (Fig. 8). The MFI of cells treated with RB at 10 μg mL−1 was 661, and the MFI of cells treated with RBDCNs at 10 μg mL−1 could reach as high as 5674.
 | ||
| Fig. 8 The effect of drug concentration on the cellular uptake efficiency of RBCNs or RBDCNs in HepG2 cells. Cells were incubated with different concentration of RB or RBD or their complex nanoparticles at mass ratio 1 for 2 h. (A) Mean fluorescence intensity of cells treated with RB or RBD or their complex nanoparticles. Each data point represents the mean ± SD of three replicates. (B) Flow histogram of HepG2 cells after incubation with RB (a), RBCNs (b), RBD (c), or RBDCNs (d) under different drug concentrations for 2 h. L: low drug concentration, 2.5 μg mL−1; M: middle drug concentration, 5 μg mL−1; H: high drug concentration, 10 μg mL−1. | ||
3.4 Tumor accumulation of RBDCNs
The ideal biodistribution characteristics of anti-cancer drugs are that the drugs are only distributed to tumor tissue while spare normal tissue. Tumor vessels are usually abnormal and lack effective lymphatic drainage, which lead to the tumor vessels permeable to macromolecular drugs and allow greater amounts of the macromolecules to be retained in the tumor.38,39 The EPR effects make nanoparticles substantially accumulate and retain at the tumor site after long systemic circulation times, and ultimately make greater amounts of the drug to be released in tumor tissue compared with free drug.To confirm our assumption that entrapped RBD inside nanoparticles could improve its accumulation in tumor tissue, the biodistribution of RBD and RBDCNs were compared in colon cancer transplanted mouse model. Compared with organ-specific metastasis mouse model, the traditional subcutaneous xenograft tumor model is less than ideal for studying the EPR effects. Fig. 9A and B demonstrated the successfully establishment of the lung-specific colon cancer metastasis model. Because the intravenous administered drugs or nanoparticles could distribute into each tissue only after several minutes, the mice were sacrificed and organs were isolated 1 h after administration before too much of the drugs were eliminated to keep high drug concentrations in each tissue and reduce the measurement error. As shown in Fig. 9, we could find that the distribution pattern of RBDCNs was different with RBD. The biodistribution of RBD did not show tissue specificity. The distribution of RBDCNs in lung was much higher than in heart, spleen, and kidney, which could be attributed to the EPR effect. The distribution of RBDCNs in tumor was much higher than RBD (Fig. 9). The results demonstrated that the self-assembled nanoparticles could be an efficient delivery system for RBD for targeted sonodynamic therapy.
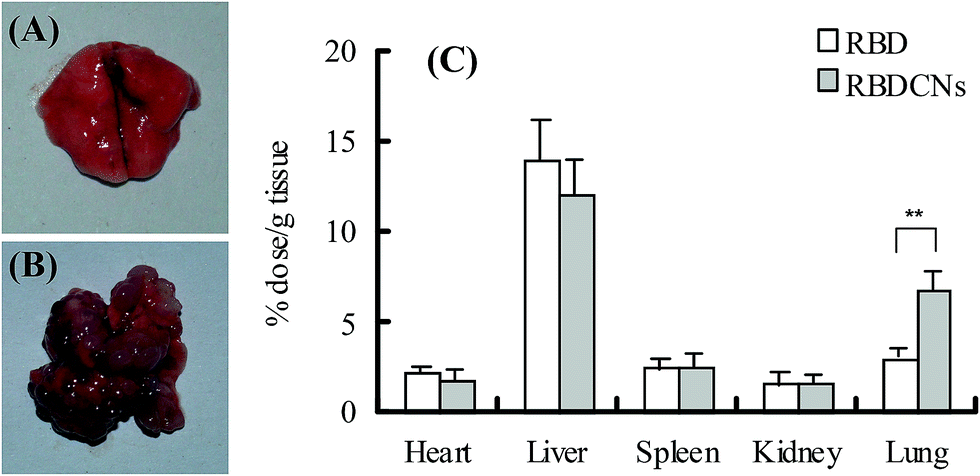 | ||
| Fig. 9 The biodistribution of RBD and RBDCNs in CT-26 colon cancer transplanted Balb/c mouse. (A) Lung of healthy Balb/c mice. (B) Lung of CT-26 colon cancer transplanted mouse model. (C) The distribution of RBD and RBDCNs in the tissues of heart, liver, spleen, kidney and lung in CT-26 colon cancer transplanted Balb/c mouse. Each data point represents the mean ± SD (n = 3). **p < 0.01. | ||
4. Conclusion
In this work, RBD was synthesized and RBDCNs were prepared for targeted delivery of RB. RBDCNs have the ability of passive targeting of tumor tissue followed by efficiently transport into tumor cells. CS and RBD could form complex nanoparticles with particle size of 150–200 nm and positive surface charges (>+40 mV) at mass ratio 1. RBDCNs showed good stability in the medium containing salt ions with dye leakage less than 20%, while the high concentration of serum could affect the interaction between CS and RBD, resulting in about 75% dye leakage under 10% FBS condition. RBDCNs could achieve efficient cellular uptake in HT29 and HepG2 cells than RB, RBD, or CS/RB complex nanoparticles. The high accumulation of RBDCNs in tumor was also observed when RBDCNs were intravenously injected into CT-26 colon cancer transplanted Balb/c mice. These results suggested that RBDCNs could be efficient and promising tumor targeted delivery systems for RB.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. J1103303 and no. 81402781), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, and Technology Development Foundation of Fuzhou University (Project Numbers 2013-XQ-8, 2013-XQ-9, 2014-XY-7, and 2014-XY-8).References
- N. Shirasu, S. O. Nam and M. Kuroki, Anticancer Res., 2013, 33, 2823–2831 CAS.
- H. Shibaguchi, H. Tsuru, M. Kuroki and M. Kuroki, Anticancer Res., 2011, 31, 2425–2429 Search PubMed.
- W. K. Bai, E. Shen and B. Hu, Chin. J. Cancer Res., 2012, 24, 368–373 CrossRef CAS.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- H. Chen, X. Zhou, Y. Gao, B. Zheng, F. Tang and J. Huang, Drug Discovery Today, 2014, 19, 502–509 CrossRef CAS PubMed.
- D. A. Tserkovsky, E. N. Alexandrova, V. N. Chalau and Y. P. Istomin, Exp. Oncol., 2012, 34, 332–335 CAS.
- S. Yamaguchi, H. Kobayashi, T. Narita, K. Kanehira, S. Sonezaki, N. Kudo, Y. Kubota, S. Terasaka and K. Houkin, Ultrason. Sonochem., 2011, 18, 1197–1204 CrossRef CAS PubMed.
- H. E. El-Sikhry, G. G. Miller, M. R. Madiyalakan and J. M. Seubert, Invest. New Drugs, 2011, 29, 1328–1336 CrossRef CAS PubMed.
- H. Kolarova, K. Tomankova, R. Bajgar, P. Kolar and R. Kubinek, Ultrasound Med. Biol., 2009, 35, 1397–1404 CrossRef PubMed.
- H. N. Xu, H. J. Chen, B. Y. Zheng, Y. Q. Zheng, M. R. Ke and J. D. Huang, Ultrason. Sonochem., 2015, 22, 125–131 CrossRef CAS PubMed.
- W. Hiraoka, H. Honda, L. B. Feril Jr, N. Kudo and T. Kondo, Ultrason. Sonochem., 2006, 13, 535–542 CrossRef CAS PubMed.
- E. Fischer and F. Varga, Acta Physiol. Acad. Sci. Hung., 1979, 54, 89–94 CAS.
- N. Sugita, K. Kawabata, K. Sasaki, I. Sakata and S. Umemura, Bioconjugate Chem., 2007, 18, 866–873 CrossRef CAS PubMed.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS PubMed.
- Y. Gao, J. Xie, H. Chen, S. Gu, R. Zhao, J. Shao and L. Jia, Biotechnol. Adv., 2014, 32, 761–777 CrossRef CAS PubMed.
- J. Wu, N. Kamaly, J. Shi, L. Zhao, Z. Xiao, G. Hollett, R. John, S. Ray, X. Xu, X. Zhang, P. W. Kantoff and O. C. Farokhzad, Angew. Chem., Int. Ed., 2014, 53, 8975–8979 CrossRef CAS PubMed.
- J. Wu, X. Wang, J. K. Keum, H. Zhou, M. Gelfer, C. A. Avila-Orta, H. Pan, W. Chen, S. M. Chiao, B. S. Hsiao and B. Chu, J. Biomed. Mater. Res., Part A, 2007, 80, 800–812 CrossRef PubMed.
- J. Wu and C.-C. Chu, J. Mater. Chem. B, 2013, 3, 353–360 RSC.
- A. Manke, S. Luanpitpong, C. Dong, L. Wang, X. He, L. Battelli, R. Derk, T. A. Stueckle, D. W. Porter, T. Sager, H. Gou, C. Z. Dinu, N. Wu, R. R. Mercer and Y. Rojanasakul, Int. J. Mol. Sci., 2014, 15, 7444–7461 CrossRef PubMed.
- K. J. Siegrist, S. H. Reynolds, M. L. Kashon, D. T. Lowry, C. Dong, A. F. Hubbs, S. H. Young, J. L. Salisbury, D. W. Porter, S. A. Benkovic, M. McCawley, M. J. Keane, J. T. Mastovich, K. L. Bunker, L. G. Cena, M. C. Sparrow, J. L. Sturgeon, C. Z. Dinu and L. M. Sargent, Part. Fibre Toxicol., 2014, 11, 6 CrossRef PubMed.
- M. M. Madathil, C. Bhattacharya, Z. Yu, R. Paul, M. J. Rishel and S. M. Hecht, Biochemistry, 2014, 53, 6800–6810 CrossRef CAS PubMed.
- Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- B. R. Schroeder, M. I. Ghare, C. Bhattacharya, R. Paul, Z. Yu, P. A. Zaleski, T. C. Bozeman, M. J. Rishel and S. M. Hecht, J. Am. Chem. Soc., 2014, 136, 13641–13656 CrossRef CAS PubMed.
- S. P. Strand, S. Lelu, N. K. Reitan, C. de Lange Davies, P. Artursson and K. M. Varum, Biomaterials, 2010, 31, 975–987 CrossRef CAS PubMed.
- Y. Gao, Z. Zhang, L. Chen, W. Gu and Y. Li, Biomacromolecules, 2009, 10, 2175–2182 CrossRef CAS PubMed.
- Y. Gao, Z. Zhang, L. Chen, W. Gu and Y. Li, Int. J. Pharm., 2009, 371, 156–162 CrossRef CAS PubMed.
- Y. Gao, Z. Xu, S. Chen, W. Gu, L. Chen and Y. Li, Int. J. Pharm., 2008, 359, 241–246 CrossRef CAS PubMed.
- Y. Cho, R. Shi and R. Ben Borgens, J. Biol. Eng., 2010, 4, 2 CrossRef PubMed.
- Y. Luo and Q. Wang, Int. J. Biol. Macromol., 2014, 64, 353–367 CrossRef CAS PubMed.
- J. K. Park, M. J. Chung, H. N. Choi and Y. I. Park, Int. J. Mol. Sci., 2011, 12, 266–277 CrossRef CAS PubMed.
- M. C. Chen, F. L. Mi, Z. X. Liao, C. W. Hsiao, K. Sonaje, M. F. Chung, L. W. Hsu and H. W. Sung, Adv. Drug Delivery Rev., 2013, 65, 865–879 CrossRef CAS PubMed.
- M. Huang, E. Khor and L. Y. Lim, Pharm. Res., 2004, 21, 344–353 CrossRef CAS.
- E. S. Dragan, M. Mihai and S. Schwarz, ACS Appl. Mater. Interfaces, 2009, 1, 1231–1240 CAS.
- R. H. Muller, C. Jacobs and O. Kayser, Adv. Drug Delivery Rev., 2001, 47, 3–19 CrossRef CAS.
- Y. S. Lin, N. Abadeer and C. L. Haynes, Chem. Commun., 2011, 47, 532–534 RSC.
- A. M. Harmon, M. H. Lash, S. M. Sparks and K. E. Uhrich, J. Controlled Release, 2011, 153, 233–239 CrossRef CAS PubMed.
- D. Casey, K. Charalambous, A. Gee, R. V. Law and O. Ces, J. R. Soc., Interface, 2014, 11, 20131062 CrossRef PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15347b |
| This journal is © The Royal Society of Chemistry 2015 |