
Self-doped 3-hexylthiophene-b-sodium styrene sulfonate block copolymer: synthesis and its organization with CdSe quantum dots†
Jin Wang*a,
Chenchen Guoa,
Yongqiang Yub,
Huabing Yinc,
Xueting Liua and
Yang Jiangd
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei 23009, P. R. China. E-mail: jinwang@hfut.edu.cn
bSchool of Electronic Science and Applied Physics, Hefei University of Technology, Hefei 23009, P. R. China
cDepartment of Electronics and Electrical Engineering, University of Glasgow, G12 8QQ, UK
dSchool of Materials Science and Engineering, Hefei University of Technology, Hefei 23009, P. R. China
First published on 22nd January 2015
Abstract
A new self-doped 3-hexylthiophene-b-sodium styrene sulfonate block copolymer (P3HT-b-PSSNa) was obtained via solution copolymerization of sodium styrene sulfonate (SSNa) with vinyl end-group poly(3-hexylthiophene) (v-P3HT), which was synthesized under Grignard metathesis (GRIM) reaction conditions. The self-doping structure can be controlled by altering the feed amounts of SSNa monomer, and it exhibited tunable properties. In comparison with v-P3HT, P3HT-b-PSSNa shows good solubility, broad absorption, higher electrical conductivity and good donor/acceptor (D/A) interface binding with CdSe quantum dots (QDs). Composites of P3HT-b-PSSNa and CdSe exhibit uniform dispersion, good compatibility and highly efficient fluorescence quenching, and surface chemical state analysis confirmed the chemical bonding between copolymer and QDs. This is helpful for rapid charge transfer of different semiconductors in the two-phase D/A interface. Here, we developed a strategy for producing a conjugated polymer with both doped stability during repeated electric cycles and compatibility with inorganic semiconductor materials.
Introduction
Polythiophene derivatives have been widely investigated due to their good electrical properties and stability, and regioregular poly(3-alkylthiophene)s (rrP3ATs) are some of the most widely studied conducting polymers. Grignard metathesis1 (GRIM), developed by McCullough, provides a simple and economical method for rrP3ATs, and much recent research and many commercial products in organic polymer electronics or photoelectronics have been based on rrP3ATs. Potential and practical applications include chemical and optical sensors, electrochromic devices, field effect transistors, and solar cells.2–5Conjugated polymers with an extended π-electron system exhibit a pseudo-band gap in the range 1.5–2.5 eV, and have low conductivity and mobility.6 Generally, the conductivity of undoped conjugated polymers is 10−7–10−11 S cm−1. However, for the application of conjugated polymers instead of inorganic or traditional semiconductors, higher conductivity is required. Doping is an effective method to produce conducting polymers. In contrast to inorganic semiconductor systems, doping in the case of conjugated polymers refers to oxidation or reduction of the π-electron system, p-doping and n-doping, respectively. Polymers having conjugated double bonds can be oxidized or reduced using charge transfer agents (dopants) more easily for better electrochemical activity.
The issue with doped conductive polymers at the present time is their relative instability under atmospheric conditions. The use of external dopants has always been a problem in many practical applications due to their poor stability, especially under thermal and electric cycling. Self-doping in conducting polymers has been proved to improve their stability upon cycling in neutral and alkaline conditions.7 Self-doped polyanilines (PAns) have been studied extensively for this reason. This involves the use of a small-molecule acidic group for internal doping such as fuming sulfuric acid treatment of PAn,8 and copolymers of an acid and aniline, such as poly(aniline-co-N-benzoylsulfonic aniline), which is obtained by post-synthesis treatment of PAn in the presence of o-sulfobenzoic anhydride.9 Self-doping of polythiophene and its derivatives has also been explored. Ratna et al.10 synthesized poly(thiophene-3-oligoethylene glycol sulfonate), which was obtained by incorporating long sulfonate-terminated tethers into the conducting polymer polythiophene, and had a reversible doping and dedoping effect. The sulfonated polythiophene derivative poly[2-(3-sulfinopropyl)-2,3-dihydro-thieno[3,4-b][1,4]-dioxin] was synthesized from (3,4-ethylenedioxythiophene)sulfonate monomer in the presence of FeCl3.11 Due to the immobilization of a covalently linked sulfonate group, the thin polymer films were shown to be highly stable in air in their doped state, in contrast to the thiophene derivatives. In brief, people have recognized that the nature of the interaction between a dopant and a conjugated polymer has a strong influence on the stability in the doped state. Chemical bonding, such as hydrogen bonding, covalent bonding, and coordination bonding, is beneficial for stability, although it may not further extend the electroactivity.
Another issue with conducting polymers of wide concern is their solubilities from the real applications point of view. Grafting of pendant side groups onto a polymer backbone, such as a flexible alkyl chain and alkoxy groups, has also been investigated in order to enhance its solubility.12–14 However, researchers have observed that whilst the presence of alkyl constituent groups on the conducting polymer unit increases its solubility but decreases its conductivity. Polythiophene derivatives, such as P3HT, only dissolve in a limited number of organic solvents, such as chloroform. This is not good for blending with inorganic semiconductors, for example when used for hybrid heterojunctions. It has been reported that when sulfonic acid groups (–SO3−) were added to a conjugated polymeric backbone to form a self-doped copolymer it had better solubility and its photoelectric performance was improved after film-forming.15,16 Polythiophene derivatives also incorporate inorganic semiconductors to form active materials for photoelectronics applications.17–19 In inorganic–organic hybrid photoelectron conversion devices, one limitation to their efficiency is electron transfer from an excited state of a donor polymer to an inorganic electron-acceptor material. Possible nanoparticle aggregation often limits the energy transfer pathway and leads to uncontrollable phase separation of both components on a micrometer scale. It is obvious that ideal nanostructures will facilitate efficient charge photogeneration and device performance for organic semiconductors with short diffusion lengths. The relationship between the nanomorphology and photophysical properties of in situ grown CdS:P3HT blends has been researched,20 showing that charge generation from P3HT excitons is heavily nanomorphology-dependent. In our previous research,21 the interrelationship between the D/A type 3-hexylthiophene/pyridine copolymer (P3HT/PY) and CdSe QDs was studied, and it was shown that good compatibility between P3HT/PY copolymer and CdSe QDs is advantageous for electron transfer.
In this paper, we synthesized self-doped poly(3-hexylthiophene)-b-poly(sodium styrene sulfonate) (P3HT-b-PSSNa), in which sodium styrene sulfonate (SSNa) doping segments were bonded chemically with the P3HT molecular chain. On the one hand, the dopant sulfonic acid groups can improve the conductive properties of conjugated polymers greatly and form a stable and reversible redox system; on the other hand, the hydrophilic styrene sulfonate segments can improve the compatibility between thiophene polymers and inorganic semiconductor materials effectively. The good dispersion of inorganic QDs into the conjugated polymer promotes charge transfer at their D/A interface, accordingly increasing the photoelectric conversion efficiency of the heterojunction structure.
Experimental section
Materials
Butylmagnesium chloride solution (C4H9ClMg, 2 M in THF), vinylmagnesium bromide (C2H3BrMg, 1 M in THF), sodium styrene sulfonate (SSNa), [1,3-bis(diphenylphosphino)propane]dichloronickel(II) (Ni(dppp)Cl2) (99%), cadmium oxide (CdO), selenium dioxide (SeO2), palmitic acid and octadecylene were purchased from Aladdin Reagent Database Inc. Tetrahydrofuran (THF), methyl alcohol, trichloromethane, benzoyl peroxide (BPO) and N-methylpyrrolidone (NMP) were purchased from Sinopharm Chemical Reagent Co. Ltd. 2,5-dibromo-3-hexylthiophene was purchased from Puyang Huicheng Electronic Material Co. Ltd. THF was purified to obtain anhydrous THF before use. Other reagents and medicines were used directly. All reactions took place under high-purity nitrogen protection; all glassware was dried without water.Synthesis of v-P3HT
A dry three-necked flask was vacuumed, filled with nitrogen and charged with 2,5-dibromo-3-hexylthiophene (2.14 mL, 10 mmol) and anhydrous THF (10 mL). 5 mL C4H9ClMg (2 M in THF) was injected by a syringe and the mixture stirred at room temperature for 2 h. 0.1 g Ni(dppp)Cl2 was added quickly, the mixture stirred for 10 minutes at room temperature, then C2H3BrMg (1 M in THF) (2–3 mmol) and THF (10 mL) were added to the reaction mixture, which was stirred continually for 5 minutes then poured into methanol to precipitate the polymer at low temperature. The polymer was filtered into an extraction thimble and then washed by Soxhlet extraction with methanol, hexanes, and chloroform, respectively, then purified by extraction with chloroform. 1H NMR (500 MHz, CDCl3): δH 6.96 (s, 1H), 6.83 (m, 1H), 5.49 (d, 1H), 5.11 (d, 1H), 2.807–0.915 (m, 13H). GPC: Mn: 6575, Mw: 9051, Mw/Mn: 1.5.Synthesis of P3HT-b-PSSNa
v-P3HT powder was added to a dry three-necked flask and dissolved in a suitable amount of a chlorobenzene and trichloromethane mixture under nitrogen protection, then BPO (10 to 15 mol% of the monomer) was put into the flask as the initiator, stirred and heated to 70 °C slowly. The correct amount of SSNa dissolved in NMP was added to the flask dropwise. The reaction mixture was stirred for 6 to 8 h at 70 °C. The final product was purified by precipitation with methanol and distilled water successively, and then filtration. The residue was then washed by distilled water, filtered and then vacuum-dried for 12 h.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 815, Mw/Mn: 1.6.
815, Mw/Mn: 1.6.Preparation of composite films of P3HT-b-PSSNa/CdSe
CdSe QDs were synthesized according to the literature.22 Composite films were prepared by tuning the mass ratio of P3HT-b-PSSNa-0.2 to CdSe to 1:0.2, 1:0.5, and 1:1, respectively. The mixtures were ultrasonicated for 5 min at 25 °C to obtain a uniform liquid and were then spin-coated onto an ITO substrate.
Characterization and measurement
1H NMR spectra were recorded using a VNMRS 600 superconducting NMR spectrometer from Agilent Technologies Group. The molecular weights of polymers were measured by gel permeation chromatography (GPC), using a Waters 410 differential refractometer at a flow rate of 1 mL min−1, in which THF was used as the eluent and polystyrene as the standard. The fluorescence properties of the copolymer were tested via a F4500 fluorescence spectrometer from the Hitachi company and UV-vis was tested by the UV-2550 from the Shimadzu Group. X-ray photoelectron spectroscopy (XPS) analysis was conducted by a ThermoFisher ESCALAB 250, in which the electronic binding energies of the samples were measured. Cyclic voltammetry (CV) was conducted on a CHI660B electrochemical workstation with two Pt plates and Ag/Ag+ as working electrode, counter electrode and reference electrode, respectively. Electrolyses were performed using a tetrabutylammonium hexafluorophosphate (Bu4NPF6) acetonitrile solution (0.1 M) at a scan rate of 50 mV s−1.Results and discussion
Synthesis and structure
The synthetic routes to P3HT-b-PSSNa are depicted in Scheme 1. v-P3HT was synthesized through the GRIM method. Subsequently, v-P3HT with its reactive vinyl end group was copolymerized with SSNa monomer via a simple solution polymerization, producing P3HT-b-PSSNa. The chemical structures of v-P3HT and P3HT-b-PSSNa were confirmed by 1H NMR spectroscopy. The 1H NMR spectra expansions in the region 8.5–5 ppm for v-P3HT and P3HT-b-PSSNa are shown in Fig. 1(a) and (b), respectively. As shown in Fig. 1(a), the peak at 6.96 ppm (g) is assigned to resonance of protons on the thiophene ring with head-to-tail (HT) coupling,23 which confirms the highly regioregular structure of v-P3HT. The appearance of new signals at 5.49 ppm (i) and 5.11 ppm (i) is due to the terminal vinyl protons, and the signal at 6.83 ppm (h) is due to the vinyl proton which is adjacent to the thiophene ring, respectively. In contrast to the 1H NMR spectrum of the v-P3HT polymer, the signals of vinyl protons at 5.49 ppm and 5.11 ppm have disappeared in the 1H NMR spectrum of P3HT-b-PSSNa; the appearance of new signals at 8.19 ppm (a) and 8.07 ppm (b) in Fig. 1(b) is due to the hydrogens on the benzene ring on either side of the sulfonic acid group, respectively. The signals at 7.43 ppm (d) and 7.63 ppm (c) belong to the hydrogens on the benzene ring on one side of the ethylidene group. From the 1H NMR spectra, it can be confirmed that SSNa units have bonded with P3HT by means of reacting with the vinyl end group, i.e., the P3HT-b-PSSNa block copolymer was synthesized successfully. The GPC test also shows that the P3HT-b-PSSNa block copolymer has a narrow molecular weight distribution.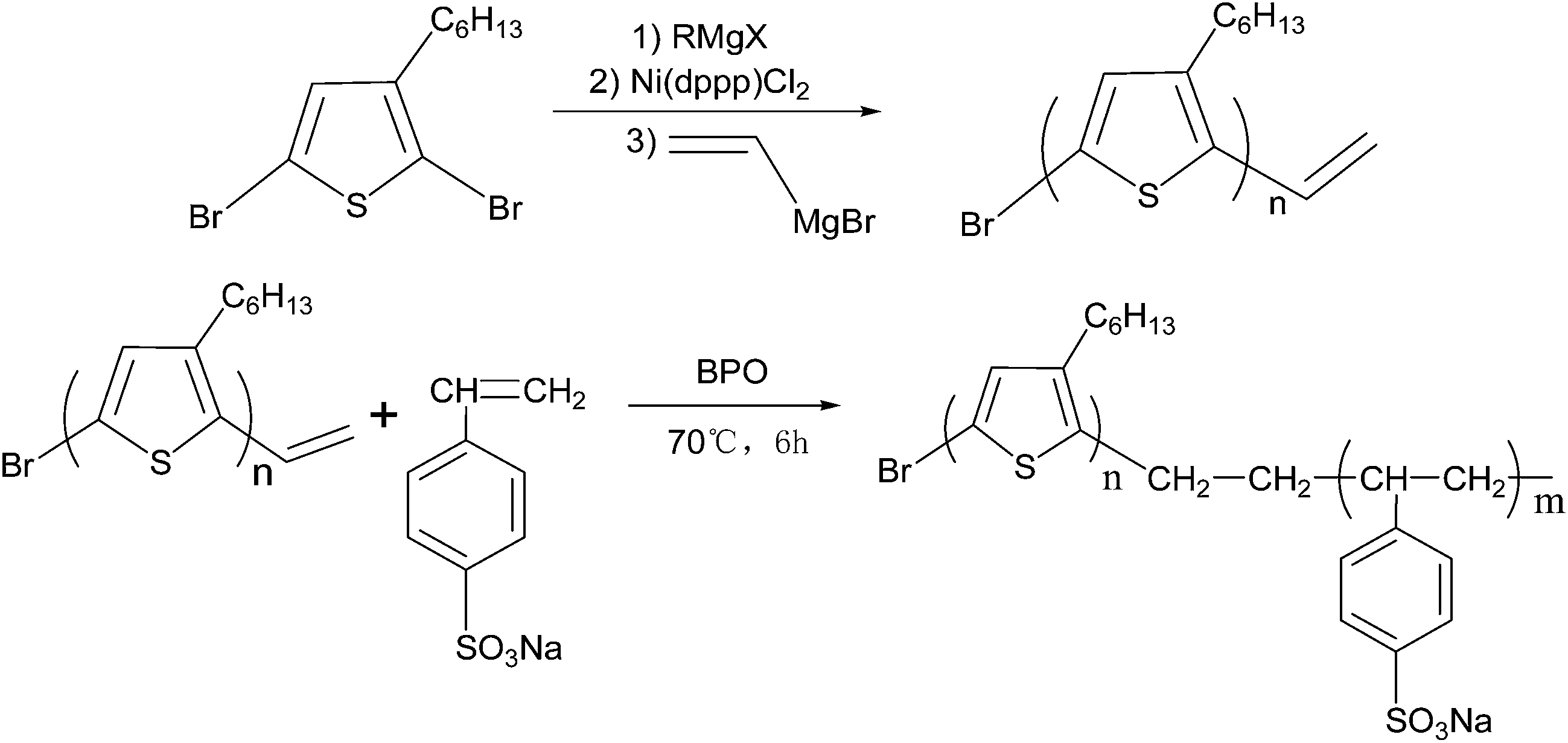 | ||
| Scheme 1 Synthesis methods of v-P3HT and P3HT-b-PSSNa. | ||
 | ||
| Fig. 1 1H NMR spectra of (a) v-P3HT and (b) P3HT-b-PSSNa block copolymer. | ||
The composition of the block copolymer can be determined from the integral ratio of the signals of P3HT and PSSNa in the 1H NMR spectrum. According to the 1H NMR spectrum of P3HT-b-PSSNa, the signals at 8.5–7.5 ppm correspond to the protons of PSSNa. Thus, the ratio of P3HT to PSSNa segments in P3HT-b-PSSNa was determined to be 1:0.2 (named as P3HT-b-PSSNa-0.2). P3HT-b-PSSNa block copolymers with different ratios can be copolymerized by adding different amounts of SSNa monomer; 1H NMR spectra can be seen in ESI Fig. S1 and S2.† The ratios of P3HT to PSSNa segments were 1:0.4 and 1:1.7 (named as P3HT-b-PSSNa-0.4 and P3HT-b-PSSNa-1.7), respectively. The composition ratio of P3HT to PSSNa in the block copolymers can also be calculated from the GPC test and this is consistent with the test result of the 1H NMR spectrum.
UV-vis spectra
The as-synthesized P3HT-b-PSSNa block copolymers were dispersible in a wide range of solvents because of the amphiphilic nature of the polymers. The solvent effects on the UV-vis absorption of the P3HT-b-PSSNa block copolymers are shown in Fig. 2. Fig. 2(a) and (b) are the UV-vis absorption spectra of v-P3HT and P3HT-b-PSSNa in different solutions with the same mixing concentration, respectively. The saturating concentration of the polymer is lowered when in a poor solvent. By contrasting Fig. 2(a) with Fig. 2(b), the block polymers show typical amphiphilic properties. In contrast to the water-insoluble v-P3HT, P3HT-b-PSSNa exhibits aqueous solubility. The aqueous solubility depends on the proportion of the PSSNa segment in P3HT-b-PSSNa. Fig. 3 shows photos of v-P3HT and P3HT-b-PSSNa in different solutions, respectively. The solubilities in different solvents can be observed directly. P3HT-b-PSSNa-0.4 exhibits good solubility in DMF and water in comparison with that of v-P3HT. P3HT-b-PSSNa-0.4 dissolves in DMF solvent completely and the solution takes on a dark red color, and dissolves in water in large part and takes on a pink color. For P3HT-b-PSSNa-0.2, the solubility still shows no marked improvement in DMF and water (as shown in ESI Fig. S3 and S4†).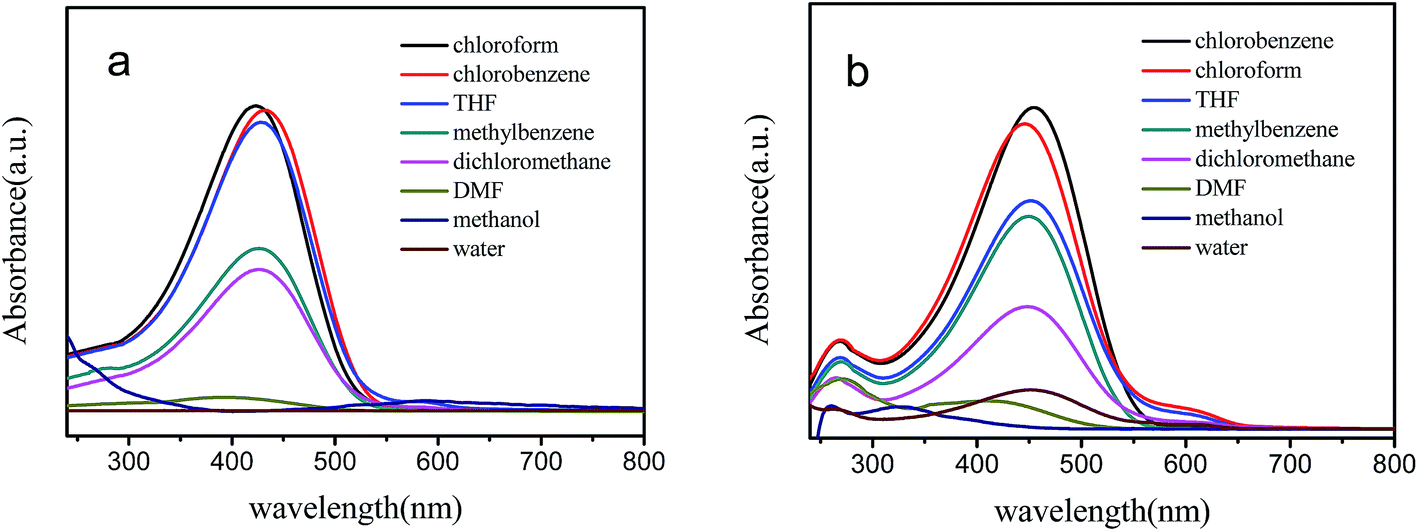 | ||
| Fig. 2 UV-vis spectra of v-P3HT (a) and P3HT-b-PSSNa-0.4 (b) in different solutions. | ||
 | ||
| Fig. 3 Digital images of v-P3HT (a) and P3HT-b-PSSNa-0.4 (b) in different solvents. | ||
A UV-vis absorption shift and a variation in intensity are induced by electronic energy migration along the backbone between adjacent segments (i.e., intra-chain) or energy hopping between segments (i.e., inter-chain) in close proximity.24 As shown in Fig. 2(a) and (b), v-P3HT in non-selective good solvents, such as chloroform, exhibits broad absorption from 300 to 550 nm. The peak of the absorption spectrum of v-P3HT is at ca. 430 nm, which is due to intra-chain π–π* transition in P3HT. The characteristic absorption peaks of P3HT-b-PSSNa in chloroform are at ca. 265 nm and ca. 450 nm, respectively, which correspond to π–π* transitions of benzene and thiophene rings, respectively. In comparison with v-P3HT, the absorption spectra of P3HT-b-PSSNa block copolymers show a red shift by ca. 20 nm, which can be ascribed to the incorporation of the doping sulfonic acid group. The molecular chain of the polymer tends to be stretched in a good solvent and tends to be curled in a poor solvent. P3HT, with sulfonic acid groups, has typical amphiphilic properties which increase its solubility in different solutions. This favors macromolecular chain stretching in solution and a better coplanar molecular structure. Especially, P3HT-b-PSSNa-0.4 exhibits a broad extended absorption edge from 550 nm to 650 nm in chloroform and THF; this indicates that P3HT-b-PSSNa-0.4 has stronger electronic coupling and π–π* interactions in chloroform and THF solutions.
The UV-vis spectra of v-P3HT and P3HT-b-PSSNa solutions and films are shown in Fig. 4. The UV-vis spectra of v-P3HT and P3HT-b-PSSNa in the film phase have both become broader and undergone a red shift compared with their solutions. This could be ascribed to conformational changes increasing the degree of conjugation in the crystalline polymer backbone in the condensed state. P3HT-b-PSSNa film exhibits a broader absorption than that of v-P3HT, and the maximum-absorption wavelength is red-shifted by about 42 nm. This can be ascribed to an increase in the effective conjugation length of the polymer chain. Moreover, alignment of P3HT-b-PSSNa block copolymer segments occurs more easily and leads to a more regular structure.
 | ||
| Fig. 4 UV-vis spectra of v-P3HT and P3HT-b-PSSNa-0.2 block copolymer. | ||
Electrochemical analysis
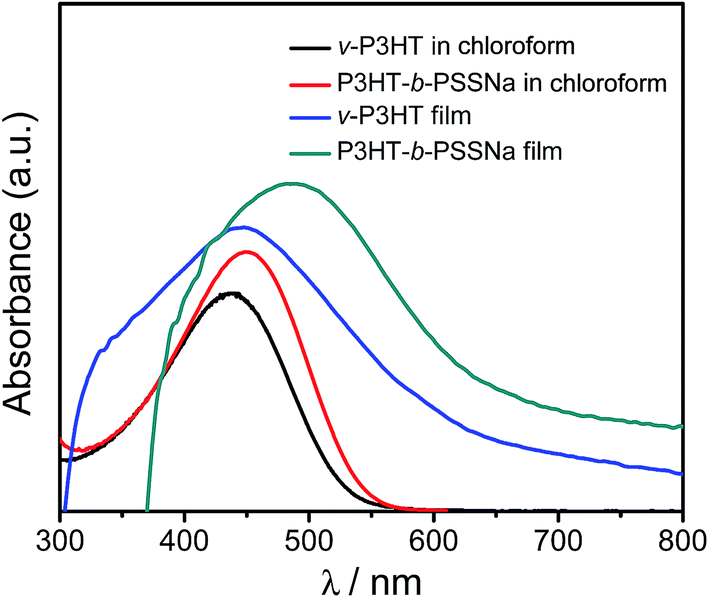
The electrochemical properties of v-P3HT and P3HT-b-PSSNa were investigated using cyclic voltammetry. Fig. 5 depicts the CV curves of both p-doping and n-doping processes. During the cathodic scan, the potential value of P3HT-b-PSSNa shifted with different amounts of SSNa units. The reduction onset potential of P3HT-b-PSSNa is in the range −0.83 to −0.95 eV, which is lower than that of v-P3HT. It is noted that reduction of these block copolymers is more facile than that of electron-donating v-P3HT, due to the presence of doping sulfonate units, i.e. the copolymer exhibits better electron-transporting properties. The electron affinity (Ea) of these copolymers is higher than that of v-P3HT (Ea = 3.71 eV). This may be attributed to the presence of electron-withdrawing doping moieties in the polymer backbone which lowers the lowest unoccupied molecular orbital (LUMO). Consequently, these copolymers may provide more facile electron injection when used as active materials in photoelectric devices. The n-doping of copolymers was accompanied by an obvious color change (electrochromism) from dark blue to orange in the n-doped polymer films. In addition, all polymers exhibited a similar p-doping process. The oxidation peak values are in the range 0.75–1.76 eV for P3HT-b-PSSNa. The oxidation onset potentials of P3HT-b-PSSNa decrease in turn, which may be attributed to the introduction of PSSNa segments; the asymmetry causes the oxidation onset potential to undergo a negative shift. P3HT-b-PSSNa-0.4 has the lowest potential and is shown to be easily oxidized. With an increase in the proportion of PSSNa segments, conjugation and electron-withdrawing effects are dominant, and these lead to the oxidation potentials shifting positively. During the oxidation process, the color of P3HT-b-PSSNa films changed from orange in the original films to dark blue. | ||
| Fig. 5 Cyclic voltammograms of v-P3HT (a) and the block copolymers P3HT-b-PSSNa-0.2 (b), P3HT-b-PSSNa-0.4 (c), and P3HT-b-PSSNa-1.7 (d). | ||
The band gap of the copolymer could be estimated from the difference in onset potential between the reduction and oxidation processes. The highest occupied molecular orbital (HOMO) and LUMO are calculated according to the equations reported by de Leeuw et al.:25 EHOMO = −(Eonset,ox + 4.68 eV) and ELUMO = −(Eonset,red + 4.68 eV), where Eonset,ox and Eonset,red are the onset potentials for oxidation and reduction versus Ag/AgCl. The cyclic voltammograms of v-P3HT and P3HT-b-PSSNa are summarized in Table 1. In comparison to v-P3HT, P3HT-b-PSSNa self-doping copolymers have lower band gaps and lower HOMO energies. The band gap of v-P3HT is 1.92 eV, and the band gaps of the P3HT-b-PSSNa block copolymers are in the range 1.61–1.75 eV. This is due to the introduction of the self-doping styrene sulfonate segments, which improve the efficiency of charge transfer at the D/A interface, according to the above results. The beneficial point associated with the lower HOMO energies of the self-doping P3HT-b-PSSNa copolymers is the advantage of a higher open-circuit voltage (Voc) in photovoltaic devices.26
| Polymer | Eoxonset | EHOMO (eV) | Ip (eV) | Eredonset | ELUMO (eV) | Ea (eV) | Eecg (eV) |
|---|---|---|---|---|---|---|---|
| v-P3HT | 0.95 | −5.63 | 5.63 | −0.97 | −3.71 | 3.71 | 1.92 |
| P3HT-b-PSSNa-0.2 | 0.76 | −5.44 | 5.44 | −0.95 | −3.73 | 3.73 | 1.71 |
| P3HT-b-PSSNa-0.4 | 0.75 | −5.43 | 5.43 | −0.86 | −3.82 | 3.82 | 1.61 |
| P3HT-b-PSSNa-1.7 | 0.92 | −5.60 | 5.60 | −0.83 | −3.85 | 3.85 | 1.75 |
Conducting polymers undergo major physical and chemical changes when oxidized (doped) or reduced (dedoped). These changes are accompanied by charge transference and neutralization processes in electrolytes, involving ion movements to and from the polymer main chains. The proposed mechanism of charge balancing is shown in Scheme 2. The “self-doping” in P3HT conducting polymers, in which the anionic group sulfonate is attached to the polymer main chain, provides rapid charge neutralization and transference when the polymer is electrochemically switched between its oxidation states.27,28 In the oxidation reaction, thienyl groups with positive charges attract negatively charged sulfonic acid groups to form intermolecular ion pairs, whereas in the reduction reaction, due to the thienyl groups not being charged, this makes the negatively charged sulfonic acid groups leave, with a reversible doping and dedoping effect.
 | ||
| Scheme 2 Doping mechanism of P3HT-b-PSSNa block copolymers. | ||
For further understanding, the electrochemical impedance of v-P3HT and P3HT-b-PSSNa block copolymers was measured in the frequency range 100 kHz–1 Hz. The impedance spectra are shown as Nyquist plots in Fig. 6. A single semicircle in the high-frequency region and a straight line in the low-frequency region for all spectra can be observed. Based on the Nyquist plots, the equivalent series resistance (ESR) of v-P3HT and P3HT-b-PSSNa block copolymers can be obtained from the intersection point of the curves with the real impedance axis. A difference in the ESR of electrodes can be attributed to the different conductance of electrode materials. The relatively low conductivity of v-P3HT resulted in its higher charge transfer resistance. In comparison, P3HT-b-PSSNa block copolymers have a lower ESR because of their better conductivity. The semicircle in the figure represents the solution resistance and the resistance of charge transfer (RΩ + Rα); since the RΩ of the system can be regarded as a fixed value, therefore the diameter of the semicircular figure could be directly compared with the value of the semicircle-based Rα. It can be seen from the Nyquist plots that P3HT-b-PSSNa block copolymers have a smaller diameter in comparison to that of v-P3HT and P3HT-b-PSSNa-0.4 has the smallest diameter among all the materials, which indicates that the P3HT-b-PSSNa-0.4 block copolymer has higher conductivity and faster charge transfer rates than the other samples. It is known that doping–dedoping of polythiophene involves protons, the diffusion process of which often limits the reaction rate. PSSNa is a proton-rich compound with higher proton conductivity. Its abundant protons available on the surface should remove the diffusion limit imposed by the solution phase to allow a fast reaction rate.29
 | ||
| Fig. 6 Nyquist plots of v-P3HT and P3HT-b-PSSNa block copolymer. | ||
P3HT-b-PSSNa/CdSe blending
The morphologies and structures of CdSe QDs and a P3HT-b-PSSNa/CdSe blend are shown in Fig. 7(a) and (b). The TEM image in Fig. 7(a) shows that the CdSe QDs are uniform in size and shape. The magnified HRTEM image of CdSe QDs shows a diameter of 4–5 nm and apparent lattice planes can be distinguished. The lattice fringe spacings between two adjacent crystal planes of the particles were respectively determined to be 0.214 and 0.213 nm, corresponding to the (202) and (220) planes of CdSe. The CdSe QDs structures exhibited typical single-crystal features, as revealed by the selected area electron diffraction (SAED) pattern in the inset of Fig. 7(b). This has clear diffraction rings that correspond to the cubic phase of CdSe. As shown in Fig. 7(b), CdSe QDs can disperse in a P3HT-b-PSSNa polymer matrix uniformly, suggesting good compatibility between the self-doped conjugated polymer and inorganic CdSe QDs. Moreover, the P3HT-b-PSSNa/CdSe blend exhibits good film-forming ability, in contrast to CdSe QDs. The presence of Se, Cd, S, C and O is evidenced by the energy-dispersive X-ray (EDX) pattern, which was produced on the surface of the P3HT-b-PSSNa/CdSe blend film, in Fig. 7(c). | ||
| Fig. 7 TEM image of CdSe QDs (a), inset: magnified HRTEM image of an individual dot; TEM image and EDX pattern of P3HT-b-PSSNa/CdSe blend (b and c), inset in (b): SAED pattern. | ||
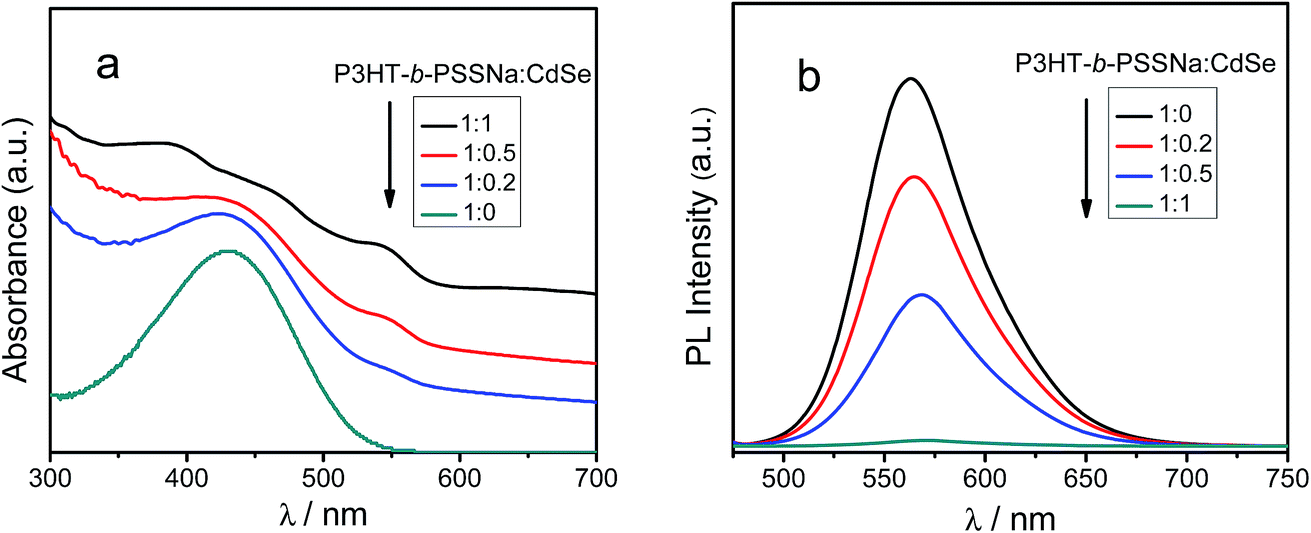
The charge transfer of different semiconductors at the two-phase D/A interface was studied by means of blending a self-doped P3HT-b-PSSNa block copolymer with CdSe QDs. The UV-vis and PL spectra of P3HT-b-PSSNa/CdSe blends are shown in Fig. 8. As shown in Fig. 8(a), the UV-vis spectrum of P3HT-b-PSSNa in chloroform solution presents a strong absorption peak in the range 430–450 nm, while the absorption band became broader and was red-shifted after blending incorporation of CdSe QDs, and the absorption intensity increased by increasing the amount of CdSe. This also indicates that P3HT-b-PSSNa/CdSe blends have highly efficient light absorption capability in the visible spectrum, which is conducive to better matching the solar spectrum. The efficiency of charge generation was further investigated by photoluminescence measurement. As shown in Fig. 8(b), 35% quenching of photoluminescence occurred at a 1:0.2 mass ratio of P3HT-b-PSSNa:CdSe. The quenching of photoluminescence reached 65% at a 1:0.5 mass ratio, and the P3HT-b-PSSNa/CdSe blend film exhibits nearly complete quenching of more than 95% at a 1:1 mass ratio. Photoluminescence quenching is expected, due to attachment of the sulfonic acid dopant to the surface of CdSe, because the aromatic π-electrons can act as efficient acceptors for photogenerated holes, thus hindering the radiative recombination process.30 The high efficiency of quenching can be attributed to the presence of self-doped sulfonic acid groups in P3HT-b-PSSNa improving the dispersibility of CdSe QDs in the polymer matrix, which is helpful for effective charge separation and transfer at the two-phase D/A interface. The highly efficient photoluminescence quenching of P3HT-b-PSSNa/CdSe blends suggests ultrafast photoinduced charge transfer from P3HT-b-PSSNa to CdSe QDs.
 | ||
| Fig. 8 UV-vis (a) and PL spectra (b) of P3HT-b-PSSNa/CdSe blend films. | ||
Combination between CdSe QDs and P3HT-b-PSSNa could be further detected by the XPS technique. XPS patterns of P3HT, P3HT-b-PSSNa block copolymer and P3HT-b-PSSNa/CdSe blends are presented in Fig. 9(a–f), respectively. The energy scales were calibrated by the C 1s resonance peak, which was fixed at 284.8 eV. Fig. 9(a) shows the binding energy peaks of S 2p in the thiophene ring at 164.2 eV and 165.2 eV in bulk P3HT. In Fig. 9(b), the peaks at 168.9 eV and 164.4 eV correspond to the binding energy of S 2p in the –SO3− group and the thiophene ring,31 respectively. They also confirmed the existence of a doping –SO3− group, and that a self-doped P3HT-b-PSSNa block copolymer was obtained. The appearance at 532.4 eV in Fig. 9(c) is ascribed to the binding energy of O 1s in the –SO3− group. In Fig. 9(d), the peaks at 405.2 eV and 412.0 eV are ascribed to the binding energy of Cd 3d5/2 and Cd 3d3/2, respectively.32 The difference between these two peak positions is 6.8 eV, which is in agreement with the ref. 33. The surface chemical state of Cd is not easy to change in different chemical environments. The chemical bonding between CdSe QDs and P3HT-b-PSSNa can be detected from the change in the binding energy of S 2p. As can be seen in Fig. 9(e), in comparison with that of S 2p in the –SO3− group, the absence of the peak centered at 168.9 eV in P3HT-b-PSSNa illustrates that the S atom in the –SO3− group has been chemically shifted and may have undergone combination with CdSe QDs. Further information can be obtained in Fig. 9(f); the binding energy value of O 1s in P3HT-b-PSSNa/CdSe blends shows a peak at 531.9 eV. There is a chemical shift of about 0.5 eV in comparison with that in P3HT-b-PSSNa, which means that the surface electronic states of –SO3− groups in the P3HT-b-PSSNa/CdSe blends have been redistributed. That is, –SO3− groups can be doped in the P3HT conjugated polymer on one hand, on the other hand the doped –SO3− groups favor compatibility with CdSe QDs. Measurement of the binding energy provides evidence for the chemical bonding between CdSe QDs and P3HT-b-PSSNa. The XPS results coincide with the morphology observations in TEM images and the photoluminescence quenching between P3HT-b-PSSNa and CdSe QDs. The chemical bonding and good dispersion of CdSe QDs in the polymer matrix would help photogenerated excitons reach the D/A interface within their lifetime.
 | ||
| Fig. 9 XPS spectra of S 2p peak in v-P3HT (a), S 2p and O 1s peaks in P3HT-b-PSSNa block copolymer (b and c), Cd 3d, S 2p and O 1s peaks in P3HT-b-PSSNa/CdSe blends (d–f). | ||
Conclusions
A new self-doped P3HT-b-PSSNa block copolymer was obtained successfully. The segment of P3HT was synthesized via Grignard metathesis (GRIM), which ensures a regular structure and narrow molecular weight distribution. The segment of SSNa, which was synthesized via solution copolymerization, can be controlled by altering the feed amounts of SSNa monomer, and it exhibited tunable self-doping and amphiphilic properties. On the one hand, the dopant sulfonic acid groups can improve the conductive properties of the conjugated polymer; however, hydrophilic sulfonic acid groups with longer chains do not help to further increase the conductivity. v-P3HT and P3HT-b-PSSNa block copolymers both display sharp reversible peaks corresponding to reversible n-doping and p-doping in their cyclic voltammograms, and show electrochemical activity. In comparison to v-P3HT, P3HT-b-PSSNa self-doping copolymers have lower band gaps and lower HOMO energies. Self-doping by chemical bonding can also provide permanent stability during alternating thermal and electric cycles. On the other hand, the introduction of sulfonic acid groups improves the solubility of the conjugated polymer in polar solutions and provides good compatibility with inorganic QDs. P3HT-b-PSSNa exhibits much better solubility in DMF and water in comparison with that of v-P3HT. The UV-vis absorption of P3HT-b-PSSNa film shows a broader absorption than that of v-P3HT, and the maximum-absorption wavelength is red-shifted by about 42 nm. The characteristic of good compatibility can greatly increase the utility of the conjugated polymer such as for organic–inorganic hybrid photoelectronic devices. In a P3HT-b-PSSNa/CdSe QDs blend system, CdSe QDs uniformly disperse in the P3HT-b-PSSNa polymer matrix. The fluorescence quenching of the copolymer reached more than 95% when the mass ratio of CdSe QDs to P3HT-b-PSSNa copolymer was 1:1. The highly efficient photoluminescence quenching of P3HT-b-PSSNa/CdSe blends suggests ultrafast photoinduced charge transfer from P3HT-b-PSSNa to CdSe QDs.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no. 61076040), Anhui Provincial Natural Science Foundation (Grant no. 1208085MB23).References
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed
.
- M. Kimura, R. Sakai, S. Sato, T. Fukawa, T. Ikehara, R. Maeda and T. Mihara, Adv. Funct. Mater., 2012, 22, 469–476 CrossRef CAS
- R. C. Evans, M. Knaapila, N. Willis-Fox, M. Kraft, A. Terry, H. D. Burrows and U. Scherf, Langmuir, 2012, 28, 12348–12356 CrossRef CAS PubMed
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2010, 23, 397–415 CrossRef
- C. B. Nielsen and I. McCulloch, Prog. Polym. Sci., 2013, 38, 2053–2069 CrossRef CAS PubMed
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed
- L. V. Lukachova, E. A. Shkerin, E. A. Puganova, E. E. Karyakina, S. G. Kiseleva, A. V. Orlov, G. P. Karpacheva and A. A. Karyakin, J. Electroanal. Chem., 2003, 544, 59–63 CrossRef CAS
- W. J. Ke, G. H. Lin, C. P. Hsu, C. M. Chen, Y. S. Cheng, T. H. Jen and S. A. Chen, J. Mater. Chem., 2011, 21, 13483–13489 RSC
- J. W. Jung, J. U. Lee and W. H. Jo, J. Phys. Chem. C, 2010, 114, 633–637 CAS
- B. D. Martin, J. Naciri, M. H. Moore, A. L. Daniel, A. D. Michael, C. P. Erica and B. Ratna, Electrochem. Commun., 2009, 11, 169–173 CrossRef CAS PubMed
- F. Tran-Van, M. Carrier and C. Chevrot, Synth. Met., 2004, 142, 251–258 CrossRef CAS PubMed
- T. Hirai, S. Osumi, H. Ogawa, T. Hayakawa, A. Takahar and K. Tanaka, Macromolecules, 2014, 47, 4901–4907 CrossRef CAS
- L. Gao, S. D. Tang, L. Zhu and G. Sauve, Macromolecules, 2012, 45, 7404–7412 CrossRef CAS
- J. Yuan, L. Xiao, B. Liu, Y. F. Li, Y. H. He, C. Y. Pan and Y. P. Zou, J. Mater. Chem. A, 2013, 1, 10639–10645 CAS
- Y. A. Udum, K. Pekmez and A. Yıldız, Eur. Polym. J., 2005, 41, 1136–1142 CrossRef CAS PubMed
- Y. A. Udum, K. Pekmez and A. Yildiz, Eur. Polym. J., 2004, 40, 1057–1062 CrossRef CAS PubMed
- A. Bruno, C. Borriello, S. A. Haque, C. Minarini and T. D. Luccio, Phys. Chem. Chem. Phys., 2014, 16, 17998–18003 RSC
- J. Y. Lek, G. C. Xing, T. C. Sum and Y. M. Lam, ACS Appl. Mater. Interfaces, 2014, 6, 894–902 CAS
- T. T. Xu, M. Yang, J. D. Hoefelmeyer and Q. Q. Qiao, RSC Adv., 2012, 2, 854–862 RSC
- S. A. Dowland, L. X. Reynolds, A. MacLachlan, U. B. Cappel and S. A. Haque, J. Mater. Chem. A, 2013, 1, 13896–13901 CAS
- J. Wang, X. X. Luo, D. L. Kang, Z. F. Zhou, W. B. Xu and Y. Jian, J. Nanosci. Nanotechnol., 2013, 13, 523–528 CrossRef CAS PubMed
- C. Wang, Y. Jiang, L. Chen, S. Li, G. Li and Z. Zhang, Macromol. Chem. Phys., 2009, 116, 388–391 Search PubMed
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS
- E. Collini and G. D. Dcholes, Science, 2009, 323, 369–373 CrossRef CAS PubMed
- D. M. de Leeuw, M. M. J. Simenon, A. B. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS
- J. F. Lee, S. L. C. Hsu, P. I. Lee, H. Y. Chuang, M. L. Yang, J. S. Chen and W. Y. Chou, Sol. Energy Mater. Sol. Cells, 2011, 95, 2795–2804 CrossRef CAS PubMed
- S. Suematsu, Y. Oura, H. Tsujimoto, H. Kanno and K. Naoi, Electrochim. Acta, 2000, 45, 3813–3821 CrossRef CAS
- F. Tran-Van, M. Carrier and C. Chevrot, Synth. Met., 2004, 142, 251–258 CrossRef CAS PubMed
- Z. M. Cui, C. X. Guo, W. Y. Yuan and C. M. Li, Phys. Chem. Chem. Phys., 2012, 14, 12823–12828 RSC
- K. A. Mazzio, K. Okamoto, Z. Li, S. Gutmann, E. Strein, D. S. Ginger, R. Schlaf and C. K. Luscombe, Chem. Commun., 2013, 49, 1321–1323 RSC
- J. Y. Kim, J. H. Jung, D. E. Lee and J. Joo, Synth. Met., 2002, 126, 311–316 CrossRef CAS
- J. E. B. Katari, V. L. Colvin and A. P. Alivisatos, J. Phys. Chem., 1994, 98, 4109–4117 CrossRef CAS
- J. F. Yan, Q. Ye and F. Zhou, RSC Adv., 2012, 2, 3978–3985 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15195j |
| This journal is © The Royal Society of Chemistry 2015 |