
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Battery waste-derived functional materials for the capture and removal of harmful gases
Nishesh Kumar
Gupta
 *
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal-462066, India. E-mail: nishesh0602@gmail.com
First published on 18th June 2024
Abstract
The persistent use of primary alkaline batteries in electronic gadgets and lithium-ion batteries in electric vehicles is creating a large volume of battery waste. Proper management and processing are necessary to prevent the dumping of used batteries in landfills. Valuable metals such as lithium, cobalt, nickel, and zinc can be extracted and purified from spent batteries. Alternatively, they can be used in synthesising functional materials. This review explores a promising solution for battery waste management by repurposing it to create materials capable of removing harmful gases. Reusing battery components such as electrodes, electrolytes, and polymer separators leads to the development of innovative strategies for creating adsorbents and catalysts. These materials are capable of efficiently capturing or catalysing harmful gases into harmless gases or ions. The review outlines various methods for converting battery waste into valuable materials, structural modifications, performance evaluations, and underlying mechanisms responsible for the removal of harmful gases. This review highlights the potential of battery waste as a sustainable resource for addressing rising air pollution and promoting a circular economy.
 Nishesh Kumar Gupta | Dr Nishesh Kumar Gupta is currently a SERB-funded National Postdoctoral Fellow at the Indian Institute of Science Education and Research Bhopal, India. He earned his PhD from the University of Science and Technology, Korea and Integrated MSc from the National Institute of Technology Rourkela, India. His current research focuses on the synthesis of metalated COFs for CO2 fixation. He has authored more than 70 peer-reviewed articles, secured 5 patents, and contributed to 4 book chapters, accumulating over 1900 citations. His achievements have led to recognition on the Stanford List of Top 2% World Scientists for both 2022 and 2023. |
Environmental significanceRepurposing battery waste for toxic gas removal minimizes environmental harm from electronic waste and mitigates air pollution. Transforming discarded battery components into functional materials reduces the reliance on raw materials and enhances air quality by efficiently neutralizing toxic gases. This innovative approach aligns well with the circular economy principles, promoting sustainable battery waste management while addressing severe environmental challenges. |
Introduction
Given the increasing global demand for clean energy, managing waste from energy storage systems, particularly batteries, has become a major environmental concern. Primary alkaline batteries like Zn–MnO2 batteries are extensively used to power portable electronic devices. However, post-exhaustion, billions of these single-use batteries end up in landfills, posing severe threats to the environment. Moreover, dumping these batteries in landfills leads to a significant loss of metal resources such as Zn and Mn.1 Rechargeable batteries like lithium-ion batteries (LIBs) are considered the backbone of the electric vehicle (EV) industry. The need to transition from fossil fuel-powered vehicles to EVs is driving LIBs production at an unprecedented scale. It is projected that annual production will surpass the ‘one million tons’ mark by 2025.2 The International Energy Agency estimated that EVs manufactured in 2019 alone generated 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of LIBs waste. By 2040, this figure could escalate to a staggering 8 million tons of waste.3 With improper disposal mechanisms, most spent LIBs are either temporarily stored or end up in landfills, causing severe contamination of the land and local ecosystem. These spent LIBs are valuable commodities, as the elements required for making the cathode of LIBs, including Li, Co, Ni, and Cu, hold significant market value.4
000 tons of LIBs waste. By 2040, this figure could escalate to a staggering 8 million tons of waste.3 With improper disposal mechanisms, most spent LIBs are either temporarily stored or end up in landfills, causing severe contamination of the land and local ecosystem. These spent LIBs are valuable commodities, as the elements required for making the cathode of LIBs, including Li, Co, Ni, and Cu, hold significant market value.4
Recycling is considered the most sought-after method for extracting valuable metals from spent batteries.5–8 However, battery recycling faces severe restrictions due to limited metal recovery, non-uniformity in cathode composition, overuse of acids, bases, and organic solvents, high energy costs, and the emission of toxic gases during processing.9,10 Another approach to reducing battery waste is repurposing it into valuable functional materials, offering a promising avenue for upcycling. This could provide significant commercial value to battery waste before recycling, thereby reducing overall recycling costs. In the literature, numerous reports have demonstrated promising applications of battery waste-derived functional materials in heterogeneous catalysis,11,12 energy storage devices,13,14 and heavy metal sequestration.15,16 Liu briefly reviewed these applications in 2018.17 However, the review may be outdated, as battery-derived functional materials have since found new applications in gas capture and removal. Thus, there is a need for a review to highlight the diversified use of battery waste-derived functional materials for air decontamination applications.
This review focuses on the emerging field of battery waste-derived functional materials, specifically highlighting their applications in capturing and removing harmful gases. Emissions from industrial facilities, thermal power plants, incineration sites, and vehicles contain a mixture of harmful gases, including sulphur oxides (SOx), nitrogen oxides (NOx), carbon oxides (COx), volatile organic compounds (VOCs), and hydrogen sulphide (H2S). The complex interaction of these gases in the atmosphere, influenced by solar radiation and water vapour, leads to the formation of photochemical smog and acid rain, contributing to climate change and posing threats to both human health and the environment. Prolonged exposure to these airborne pollutants is associated with various adverse health outcomes, such as asthma, lung cancer, psychological disorders, autism, and low birth weight.18,19 Addressing this urgent issue requires the development of effective and cost-efficient technologies for capturing and eliminating gaseous contaminants. By utilizing the inherent properties of battery waste constituents, including cathodes, polymers, anodes, and electrolytes, researchers are exploring novel opportunities to develop sustainable materials suitable for treating harmful neutral and acidic gases. This review analyses recent progress in the synthesis, characterization, and performance assessment of battery waste-derived functional materials for treating VOCs, toxic neutral gases (CO and NO), and acidic gases (CO2, NO2, SO2, and H2S). This review aims to highlight the concept of ‘waste-to-wealth’ in the context of battery waste, which could set a precedent for processing future battery wastes, including those from newly developed LIBs and sodium-ion batteries (NIBs).
Battery waste-derived functional materials
Batteries are typically divided into two categories, i.e., primary and secondary batteries. The key difference lies in the nature of their chemical reactions. Primary batteries, such as Zn–MnO2 and Zn–carbon batteries, are discarded once they can no longer provide sufficient electrical energy. In contrast, secondary batteries, like LIBs and NIBs, can convert chemical energy into electrical energy through reversible chemical reactions. This means the original chemical state can be restored by reversing the current flow, i.e., by charging from an external source. However, with continuous use, even secondary batteries experience performance fading and are discarded when their performance reaches a minimum threshold. These LIBs are poorly collected and recycled, resulting in most of them being disposed of in landfills.Alkaline Zn–MnO2 batteries and LIBs are frequently used for synthesising functional materials. While multi-step synthetic approaches are commonly employed in developing these materials, some studies have reported the direct use of battery waste without significant pre-treatment. Here, a brief discussion has been included to inform readers about conventional approaches that have been adopted for developing functional materials derived from battery waste with some details about synthesis processes.
While discharging a Zn–MnO2 alkaline battery, Zn oxidizes to produce ZnO, while MnO2 undergoes reduction to form MnOOH and Mn(OH)2. The MnOOH reacts with Mn(OH)2 and Zn(OH)42− to yield Mn3O4 and ZnMn2O4, respectively.20 Thus, the black mass derived from spent alkaline Zn–MnO2 batteries is rich in ZnO and MnO2 phases with minor presence of other metal and metal oxides, such as Zn, MnO, Mn2O3, Mn3O4, and ZnMn2O4, along with KOH electrolyte and graphitic carbon.21 This black mass could be modified based on the requirements of the target gas molecule and the treatment process. For capturing highly acidic gases like H2S, SO2, and NO2, the KOH electrolyte along with the ZnO–MnO2 composite in the black mass is sufficient for gas oxidation, and thus, no pre-treatment is required.21,22 However, for the thermal oxidation of gases like VOCs, researchers have treated the black mass with strong mineral/organic acids either to neutralize the KOH electrolyte23–25 or to completely/selectively dissolve metals.26,27 In some cases, a hydrometallurgical process like bioleaching is used to eliminate the use of strong mineral acids.28,29 The leached metal solution was further processed to obtain the desired functional materials like ZnxMn3−xO4,30 MnO2,27 and Mn2O3.26 Further modifications have been made to the black mass-derived materials to improve the catalytic properties, such as by adding photoactive TiO2,31 and catalytically active metals like Pd,32–34 Cu,27 and Ag.35 Doping functional materials with active metals can improve physicochemical properties, such as surface area,27 and lower the activation energy barrier associated with the catalytic elimination of gaseous pollutants.
The metal leachate obtained from the treated black mass of Zn–MnO2 batteries could be used as a metal solution for developing new oxide materials, providing better control over the phase composition, morphology, and other desirable properties like surface area and porosity. In two such reports, Cu-loaded27 and Ag-loaded MnO2 catalysts35 were prepared from the selective leaching of black mass obtained from discarded Zn–MnO2 batteries. The black mass was treated with HCl (2 mol L−1) at 80 °C for 4 h. The precipitate was filtered out and the filtrate (Mn-rich solution) was oxidised by NaClO solution at 45 °C for 6 h. The formed MnO2 was phase-separated and washed several times with dilute HCl. While Cu loading over MnO2 was performed by the wet impregnation method using copper nitrate salt, Ag was loaded by in situ reduction of Ag+ ions (from the silver nitrate salt) using NaBH4 as the reducing agent (Fig. 1). The formed catalysts possessed a higher surface area than the black mass, which is beneficial for the catalytic removal of gaseous pollutants.
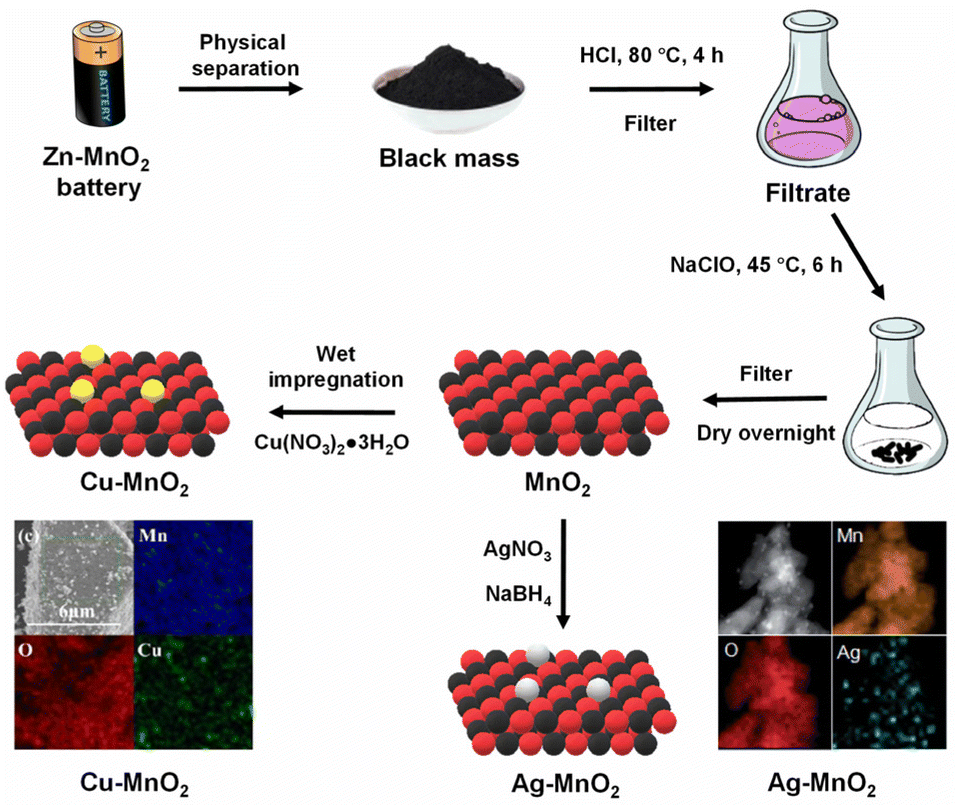 | ||
| Fig. 1 Synthesis methodology for preparing Cu–MnO2 (ref. 27) and Ag–MnO2 catalysts.35 EDX mapping of Cu–MnO2 reproduced from ref. 27 with permission from Elsevier, copyright 2024. EDX mapping of Ag–MnO2 reproduced with minor modification from ref. 35 with permission from The Royal Society of Chemistry, copyright 2024. | ||
LIBs consist of a cathode, an anode, an electrolyte, a separator, and current collectors, along with packaging components. There are five major types of cathodes used in LIBs, namely lithium cobalt oxide (LiCoO2), lithium iron phosphate (LiFePO4), lithium manganese oxide (LiMn2O4), lithium aluminium cobalt nickel oxide (LiAlxCoyNi1−x−yO2), and lithium cobalt manganese nickel oxide (LiCoxMnyNi1−x−yO2).3 Since the cathodes of LIBs are composed of valuable metals, much research is focused on developing processes to leach out metals, extract Li as a soluble salt (with low Li purity), and synthesise transition metal-based functional materials.36–38 In one such report, spent ternary LIBs were discharged in a 10% NaCl (aq.) solution for 24 h and manually disassembled to obtain the cathodes. The cathodes were calcined at 450 °C for 3 h to peel off active materials from the Al foils. The active cathode powder was treated with oxalic acid at 180 °C for 16 h in a Teflon-lined autoclave to extract Ni, Co, and Mn as insoluble oxalates. After phase separation and drying, the oxalate salt mixture was calcined at 550 °C for 3 h to yield NiCoMnOx catalysts (Fig. 2).37 In another study, the active cathode powder was treated with an H2SO4–H2O2 solution to leach Co, Ni, Mn and Li. While the obtained leachate was treated with KMnO4 solution or ozone to form Co/Ni-doped MnO2, the majority of Li, Co, and Ni remained in the solution phase.36
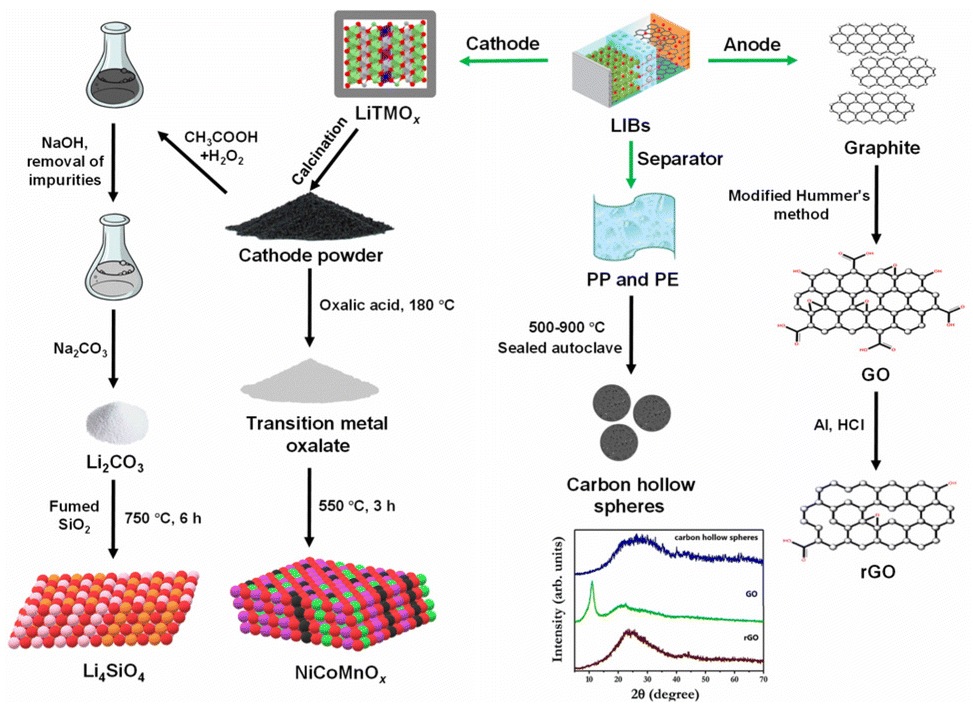 | ||
| Fig. 2 A schematic illustration of synthesis methodologies adopted for processing LIB's components into functional materials.37,39,40 The PXRD pattern of synthesised carbon materials reproduced with minor modification from ref. 40 with permission from The Royal Society of Chemistry, copyright 2024. | ||
While some researchers have developed novel processes to extract highly pure Li (as Li2CO3) along with transition metal oxides,41,42 others have used the extracted Li2CO3 as a precursor for synthesising functional materials.39,43,44 Qin and co-workers have extensively worked on extracting Li as pure Li2CO3 from LIBs' cathodes and subsequently using it as a precursor for synthesising Li4SiO4 sorbent for CO2 capture. One such process involved discharging LIBs in a NaCl solution and manually disassembling them to obtain the cathodes. After treating the cathodes with NaOH solution to remove Al, the cathode material was placed in a leaching solution (H2SO4–H2O2, Na2S2O8, or CH3COOH–H2O2). The pH was adjusted with NaOH or NH4OH to remove impurities and obtain a Li-rich solution. Na2CO3 was added to the solution phase at 95 °C to extract Li as a carbonate salt. The recovered Li2CO3, along with commercially available fumed silica, was mixed and calcined at 750 °C for 6 h to yield Li4SiO4 (Fig. 2).39,44
Apart from the cathode of LIBs, the anode (graphitic carbon) after proper chemical treatment has been upcycled to yield functionalized carbon adsorbents.40,45 Aravindan and co-workers developed reduced graphene oxide (rGO) from the graphitic anode by first oxidising graphite using a modified Hummer's method and then reducing the GO with hydrochloric acid and the outer metallic Al casing of the battery. The group also reported the template-free synthesis of porous carbon hollow spheres by one-step carbonization of the recovered polymer separators (polypropylene (PP) and polyethene (PE)) at elevated temperatures (Fig. 2).40 Many methods have been reported to modulate the physiochemical properties of LIBs waste-derived materials by metal doping,46 integration with porous substrates,47,48 and acid etching.49,50 Although these methods have not been discussed in detail in this section, their role in the catalytic properties of materials will be discussed in subsequent sections.
Gas capture and decontamination
In the literature, battery waste-derived functional materials have been primarily used as catalysts for the thermal oxidation of gaseous pollutants such as VOCs, NOx, and COx. However, some recent studies have demonstrated that these materials could also be used as room-temperature adsorbents/catalysts for treating harmful acidic gases. In the subsequent subsections, these functional materials will be discussed in the context of capturing and removing various neutral and acidic gaseous pollutants.Removal of VOCs
Volatile organic compounds (VOCs) are a diverse group of carbon-based chemicals that evaporate easily at ambient temperatures. They are found in common household products, including paints, solvents, cleaning agents, building materials, and furnishings. While many VOCs are only mildly irritating at low levels, some can have toxic effects on human health and the environment. Some VOCs are known carcinogens, while many are considered precursors of ozone, photochemical smog, and secondary aerosols. Low-temperature catalytic oxidation is considered a suitable method for eliminating VOCs from the environment. Supported noble metals and transition metal oxides are promising catalysts for the total oxidation of VOCs at low temperatures.51 Battery waste is rich in oxides of 3d-series transition metals and could be utilized for the purpose after suitable modification of the cathode material.The black mass extracted from discarded Zn–MnO2 alkaline batteries is rich in ZnO and MnOx, which, after neutralization with mineral acids, was used for the thermal oxidation of VOCs at low-to-mid temperatures.23,24,32 Black mass, after neutralization with H2SO4, catalysed the total oxidation of VOCs, including benzene, toluene, and o-xylene (collectively BTX) below 400 °C.23 The type of acid used for neutralization played an important role in the catalytic activity. The catalytic activity followed this order: sulphuric acid > nitric acid > oxalic acid > hydrochloric acid > phosphoric acid > no acid, when these acids were used for neutralization. Neutralization with different acids yielded black mass with varying proportions of Mn and Fe (catalytically active metals) and surface area, where H2SO4-treated black mass possessed superior physicochemical properties.24 The catalytic activity of acid-treated black mass was further improved by impregnating noble metals like Pd.32 After Pd loading, the black mass effectively catalysed benzene (T90 ∼ 298 °C), toluene (T90 ∼ 253 °C), and o-xylene (T90 ∼ 239 °C) (note: T90 is the temperature at which 90% conversion of VOC was observed). Without Pd, the T90 value was 385, 331, and 377 °C for benzene, toluene, and o-xylene, respectively. Increased Pd loading enhanced the lattice oxygen mobility available for effective oxidation of aromatic VOCs. ZnO, from the acid-treated zinc rod (from spent alkaline batteries), was used as a support over which catalytically active Mn52 and Pd33 sites were dispersed to oxidise VOCs. Increased MnOx loading on ZnO improved the lattice oxygen mobility and strong acid-site density. BTX molecules adsorbed on the acidic sites reacted with the lattice oxygen to form CO2 and H2O. The formed O-vacancies were replenished by oxygen atoms formed by the dissociation of molecular oxygen (Fig. 3a).52 MnOx/ZnO with 30 wt% Mn loading had a T90 value of 411, 347, and 381 °C for benzene, toluene, and o-xylene, respectively. Contrarily, just 1.0 wt% of Pd loading over ZnO was sufficient for the total oxidation of BTX molecules at the same feed concentration, where the T90 for benzene, toluene, and o-xylene was 389, 325, and 318 °C, respectively.33 Increased metallic Pd loading favoured the catalytic process by improving the lattice oxygen mobility. On the electron-rich Pd sites, BTX molecules were adsorbed and subsequently reacted with lattice oxygen near the Pd–ZnO interface to form CO2 and H2O (Fig. 3b). Though only a minuscule amount of Pd is sufficient for catalytic process, the high cost of Pd compared to MnOx could offset the overall catalytic gain.
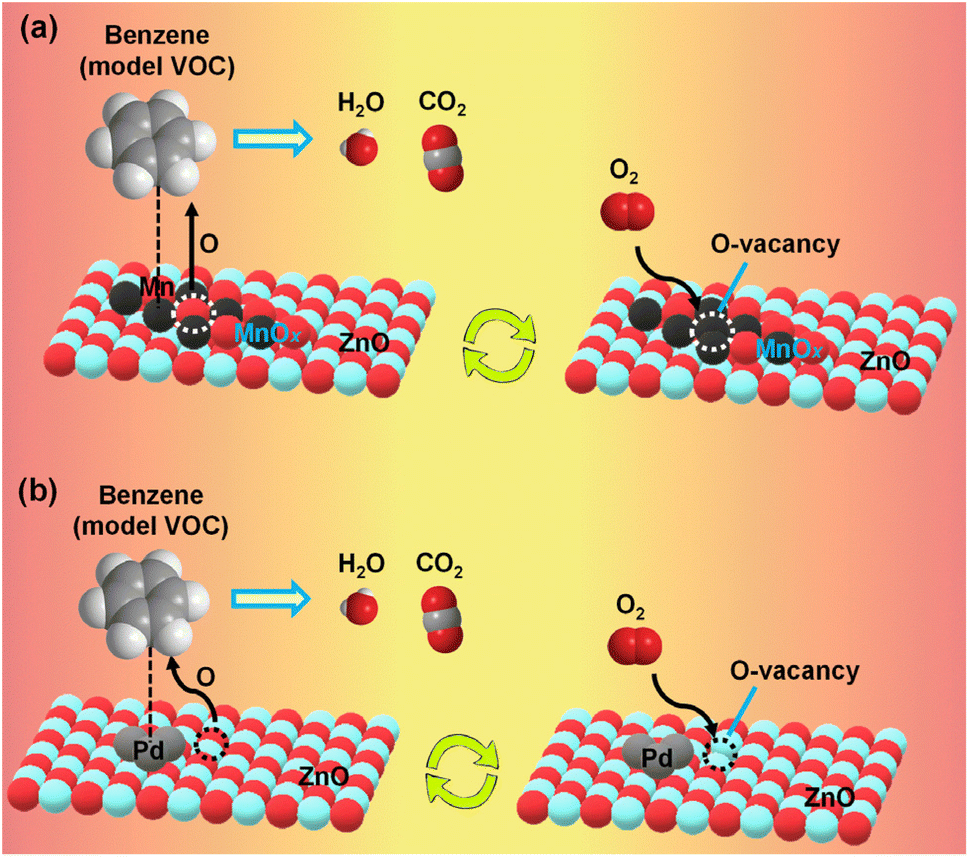 | ||
| Fig. 3 Proposed reaction mechanism for complete oxidation of BTX molecules over (a) MnOx–ZnO52 (b) Pd–ZnO.33 | ||
After comparing the T90 values, it is evident that the thermal oxidation of benzene is more difficult than that of toluene or o-xylene as the lower ionization potential of methylated benzene derivatives makes their oxidation much easier than benzene.32 Another observation is that the oxidation of oxygenated VOCs like ethanol (T50 ∼ 170 °C) over MnOx catalyst is more favourable than that of aromatic VOCs like toluene (T50 ∼ 280 °C). Ethanol is strongly chemisorbed on the MnOx surface through the coordination of non-bonding oxygen electrons compared to the weak bonding observed for toluene through π-type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C orbitals. Since strong chemisorption of ethanol lowers its activation energy, oxidation of ethanol occurs at a lower temperature than toluene.29
C orbitals. Since strong chemisorption of ethanol lowers its activation energy, oxidation of ethanol occurs at a lower temperature than toluene.29
Cathodes of all different types of LIBs have been upcycled to transition metal oxides for the thermal oxidation of VOCs. Artificially created Li, Co, and O vacancies in LiCoO2 through partial Co3+ leaching and de-intercalation of Li+ ions by HNO3-etching yielded catalysts possessing excellent oxidation efficiency for benzene. While pristine LiCoO2 catalysed only 10% of benzene at 300 °C, 100% oxidation was observed for the acid-treated LiCoO2 from synthetic air. The Li and O vacancies in LiCoO2 facilitated benzene adsorption and activation, while the Co and O vacancies induced the formation of active surface oxygen species. The number of vacancies and subsequently the catalytic activity was further improved by increasing the acid-etching time. Also, the acidic solution rich in Li and Co could be further processed to recover valuable metals.50 Mixed metal oxides, such as Co3−xMxO4 (M = Li, Ni, Cu, Al, Mn), synthesised from LiCoO2, catalysed toluene (T90 ∼ 274 °C) better than the Co3O4 synthesised from nitrate salt (T90 ∼ 282 °C). The superior activity of Co3−xMxO4 was related to a higher surface area, better low-temperature reactivity, a higher density of weak acid sites, and a higher proportion of high-valent cations and lattice oxygen. While impurities like Cu and Mn in the catalyst improved the activity, Li negatively affected it.38 Several other mixed metal oxides like CoMnNiOx for the oxidation of propane,53 MnOx (CoMn2O4–NiCo2O4–Cu1.2Mn1.8O4–Mn2O3) for the oxidation of 1-methoxy-2-propanol,47 CuNi0.5Mn1.5O4 for the oxidation of toluene, ethylbenzene, and formaldehyde,54 and Mn5O8/CeO2, CuNi0.5Mn1.5O4/CeO2, and Co3O4/CeO2 for the oxidation of 2-ethoxy ethyl acetate,55 have shown the potential of LIBs-derived catalysts in VOC elimination. In all these catalytic systems, a higher surface area, abundant high-valent transition metal sites, and a higher proportion of adsorbed oxygen species in catalysts determined their VOC oxidation activity.
On numerous occasions, LIBs-derived manganese oxides (MnOx), especially MnO2, have been reported for VOC oxidation. It is noteworthy that high-valent Mn-sites in MnOx catalysts (in MnO2) catalyse VOCs better than the low-valent Mn-sites (in Mn2O3 and Mn3O4).28,56 Even the presence of different phases of MnO2 and dopants could alter the catalytic activity of MnOx. In one such study, Co/Ni-doped α-MnO2 and β-MnO2 catalysts were synthesised by the oxidative precipitation of Mn-rich leachate using KMnO4 and ozone, respectively. While doped catalysts performed better than the pure MnO2 catalysts, doped α-MnO2 catalysed the oxidation of toluene and formaldehyde better than doped β-MnO2.36 α-MnO2 with a [2 × 2] tunnel structure is more active than β-MnO2 with a [1 × 1] tunnel structure, as the [2 × 2] tunnel structure could facilitate the absorption and diffusion of VOC molecules towards active sites.57 An appropriate proportion of transition metal dopants like Co and Ni in MnO2 could improve the catalytic activity by increasing the concentration and mobility of lattice oxygen. Similar observations have been made with Cu-doped α-MnO2 during the oxidation of toluene and chlorobenzene. Cu-sites in α-MnO2 favoured the catalytic process by increasing the surface oxygen species and active oxygen species through the Cu2+/Cu+ redox cycle.58 Even the morphologies of catalysts could influence their catalytic activity, as was the case with morphologically different MnO2 catalysts, MnO2-CM (microspheres), MnO2-HM (stacked nanorods), and MnO2/SBA-15 (microspheres), synthesised by hydrothermal, co-precipitation, and impregnation methods, respectively, for the oxidation of toluene. MnO2-HM showed 100% conversion of toluene even with the lowest surface area due to its better redox ability and the large presence of adsorbed oxygen species at a low synthesis temperature of 140 °C.48
On rare occasions, spent ternary LIBs have been processed to synthesise perovskite catalysts, LaMn1−xBxO3 (B = Co, Ni, Cu, Al) for the thermal oxidation of toluene59 and SmMnO3 and SmCoO3 for propylene glycol methyl ether.60 The LIBs-derived perovskites performed better than those synthesised from commercial salts, as the former has a higher surface area, Mn4+/Mn3+ ratio (or Co3+/Co2+ ratio for Co-perovskite), and lattice oxygen species. The partial substitution of Mn/Co sites with Ni, Co, Al, and Cu ions improved catalytic performance. However, the presence of Li as an impurity in the catalyst severely inhibited the activity.
Plasma-assisted catalytic oxidation of VOCs is another method that has been studied using battery waste-derived catalysts like Mn2O3 (ref. 26) and Mn2O3/Al2O3 (ref. 61) with BTX pollutants. Though the method effectively catalysed a quantitative amount of benzene and toluene, the oxidation of o-xylene was below 75% even under the best experimental conditions. The catalytic activity was significantly affected by synthesis conditions, including the type of acid, acid concentration, black mass-to-acid volume ratio, calcination temperature, and Mn content over the alumina substrate.26,61 Other than thermal oxidation, photocatalytic oxidation of toluene (∼50 ppm) has been performed over TiO2-modified black mass (from Zn–MnO2 alkaline battery).31,62 The composite photo-oxidized 100% of toluene within 3 h of irradiation, better than TiO2 alone, black mass alone, or even the TiO2–ZnMn oxide composite synthesised from black mass leachate. The TiO2–black mass composite, with higher photon absorption efficiency, better charge transfer properties, and low charge-recombination rate, effectively absorbed photons and generated superoxide and hydroxide radicals for rapid photo-oxidation of toluene.62 Even the calcination temperature during the synthesis of TiO2–black mass composite played a major role in the photocatalytic efficiency. The photo-catalyst synthesised at a lower calcination temperature (200 °C) showed the highest catalytic activity due to the highest surface area, highest photon absorption property, narrowest band gap, and lowest charge recombination efficiency.31 Thus, low-concentration VOC sources are effectively treated by photo-oxidation using TiO2–black mass composites.
Removal of CO and NO
Carbon monoxide (CO) is a highly poisonous gas produced from the incomplete combustion of organic matter. CO must be removed from tail gases, and for that, thermal oxidation of CO to CO2 is a viable method. Battery waste-derived catalysts could efficiently oxidise CO to CO2 at low-to-mid temperatures. Zn–MnO2 battery-derived Cu–MnO2 (ref. 63) and Ag–MnO2 (ref. 35) completely oxidised CO at 120 and 150 °C, respectively, performing better than MnO2 (240 °C). The inclusion of Ag and Cu in MnO2 played a synergistic role and improved the catalytic activity. Importantly, MnO2 with higher Cu loading performed better by providing a large surface area, more surface-active oxygen species and oxygen vacancies, and a higher proportion of Mn4+ and Cu2+ ions for catalysis.63 CoFe2O4–LIB (CoFe2O4–Co3O4), derived from LIBs' cathode, has been used for CO oxidation. The composite oxidised 98% of CO at 300 °C, better than pure CoFe2O4 (60% CO oxidation at the same temperature). Detailed spectroscopic investigations suggested that both CoFe2O4 and Co3O4 were essential for CO oxidation, where CO was oxidised to CO2 by interacting with the adsorbed oxygen species and lattice oxygen over CoFe2O4 and Co3O4, respectively (Fig. 4).41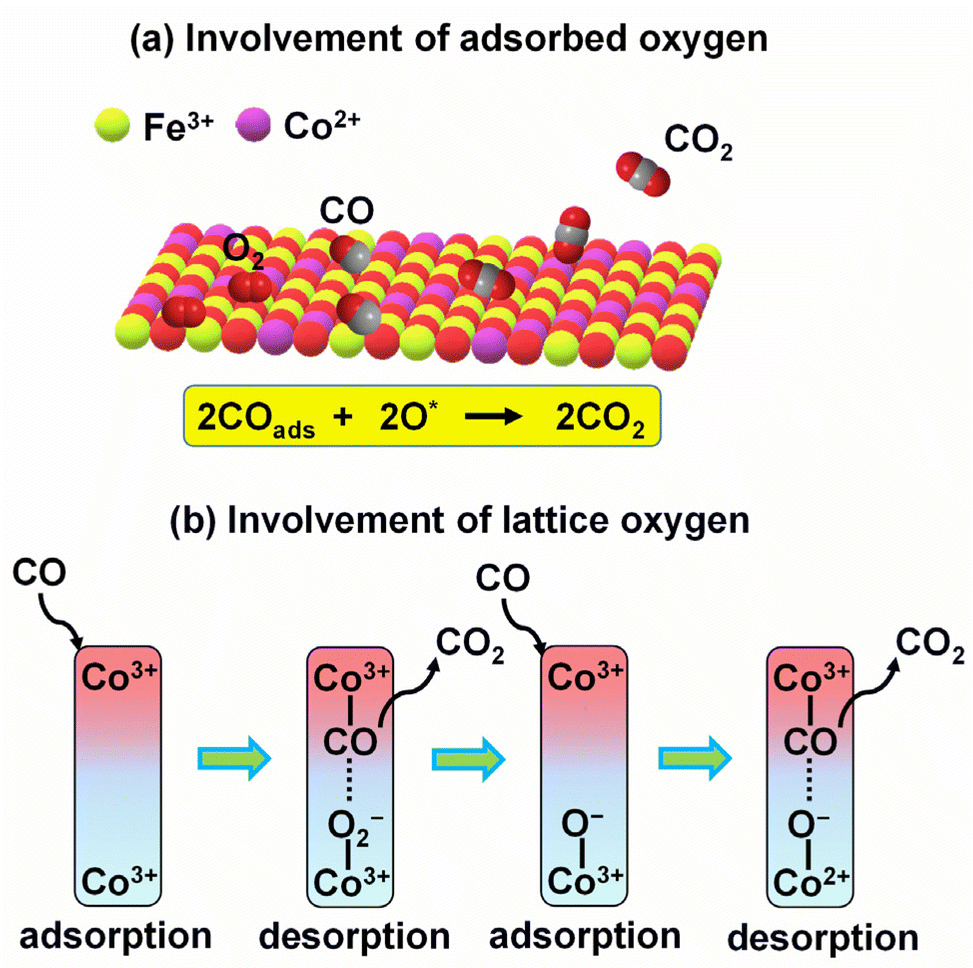 | ||
| Fig. 4 The role of CoFe2O4–LIBs catalyst in CO oxidation involves the interaction of CO with (a) adsorbed oxygen species over CoFe2O4 and (b) lattice oxygen over Co3O4.41 | ||
Nitrogen oxides (NOx), emitted from thermal power plants and incinerators, are some of the toxic gaseous pollutants responsible for acid rain and photochemical smog. Low-temperature NH3-selective catalytic reduction (NH3-SCR) technology is promising in reducing the NOx concentration from stationary sources due to its high de-nitration efficiency. The process involves NH3-driven reduction of NO to N2 at low temperatures over a suitable catalyst. Though V2O5–WO3/TiO2 (an industrial catalyst) is used for the process, the high cost of V and W, V toxicity, low SO2 resistance, and poor low-temperature activity make it essential to explore other economical and effective catalysts. Mn-based catalysts are inexpensive and perform well even at low temperatures.64 Spent alkaline Zn–MnO2 batteries are a good source for extracting necessary Mn for synthesising catalysts. Chang and co-workers estimated that CO2 emission during carbothermal extraction of Mn from spent batteries was 1.0 kg per kg of black mass, which was ∼15% of the CO2 emitted during traditional Mn manufacturing (6.7 kg per kg Mn). Moreover, the recovered Mn cost is much lower than the commercial Mn salts. Thus, processing primary batteries for Mn seems to be an economical and environmentally benign approach. The battery-derived Mn/TiO2 catalyst was found superior to commercial salt-based Mn/TiO2. The superior catalytic activity and high N2/N2O selectivity of Mn/TiO2 at 160–200 °C were ascribed to a higher Mn4+ proportion and favourable presence of Fe3+ impurity, which improved the oxidation ability of battery-derived Mn/TiO2. However, certain impurities like carbon and Zn in treated black mass could reduce the activity of Mn/TiO2 by decreasing the Mn4+ concentration and acidity of the catalytic surface. Thus, caution must be taken during the processing of black mass for Mn extraction using the carbothermal method.65
After Li recovery from LIBs cathode using oxalic acid, the residual precipitate was upcycled to yield NiCoMnOx catalyst for NOx reduction. NiCoMnOx (Co3O4–NiCo2O4–MnCo2O4.5–NiO) catalysed 90% of NOx in a broad temperature range of 110–230 °C, better than the catalyst synthesised with metal salts. The LIBs-derived catalyst possessed a higher surface area and abundant oxygen defects, which favoured the adsorption of reactant gases, i.e., NO and NH3 (Fig. 5). Though the LIBs-derived catalyst performed well in the presence of moisture, SO2 in the feed gas severely hampered its catalytic activity by reacting with the adsorbed NH3 to form ammonium sulphate and bisulphate species over the catalyst surface.37 Polyethylene glycol (PEG), a water-soluble polymer, was used as a templating agent during the LIBs processing to synthesise NiCoMnOx catalysts with modulated physicochemical properties. The NiCoMnOx–PEG catalyst possessed a higher surface area and a higher proportion of Co3+, Mn4+, and adsorbed oxygen species, which favoured the oxidation of NO to NO2 (fast SCR reaction). Though NiCoMnOx catalysts synthesised with or without PEG catalysed 90% of NO in a similar temperature window of 80–230 °C, NiCoMnOx–PEG performed better in the presence of H2O and SO2 in the feed gas. A higher SO2 tolerance of NiCoMnOx catalyst was due to the presence of more acidic sites over the surface, which delayed SO2 adsorption and subsequent conversion to sulphate species.66 Zhang and co-workers modulated the physiochemical properties of LiMn2O4 (simulated cathode of LIBs) by exchanging some of the Li+ ions with protons (acetic acid treatment) and/or by surface loading with a small amount of V2O5. While the inclusion of protons increased the surface acidity, V2O5 regulated the redox behaviour and increased the number of acidic sites. The LixH1−xMn2O4–V2O5 (dual-modulated) catalyst showed the best combination of NO reduction activity and N2 selectivity than singly modulated catalysts due to improved NH3 adsorption, superior nitrite decomposition, and reduced surface nitrate species formation.49 These studies have highlighted that after suitable modifications, cathodes of waste LIBs could be upcycled as efficient NH3-SCR catalysts.
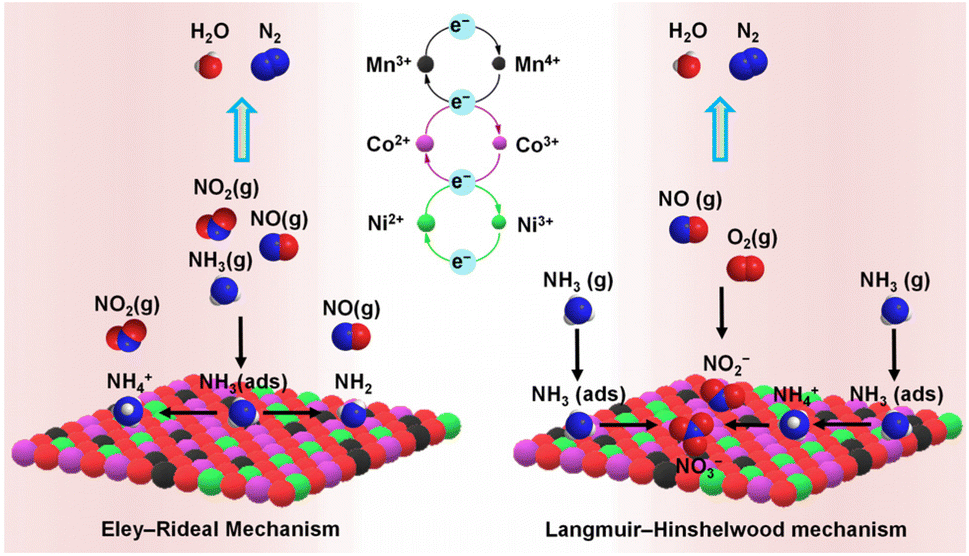 | ||
| Fig. 5 Proposed NH3-SCR mechanism for NO conversion to N2 over waste LIBs-derived NiCoMnOx catalyst.37 | ||
Removal of acidic gases
Although carbon dioxide (CO2) is a weakly acidic gas, it is considered an important greenhouse gas responsible for rising global temperatures and climate change. Carbon capture is a leading technology that relies on porous materials for preferential sequestration of CO2 from flue gas or even air.67 Carbon-based porous materials like reduced graphene oxide (rGO, surface area ∼ 374 m2 g−1) and porous carbon hollow spheres (surface area ∼ 402 m2 g−1) have been developed from LIBs' anodes (graphitic carbon) and polymer separators, respectively. These materials have shown potential in capturing CO2 gas at 25 °C and 40 bar, with adsorption capacities reaching 61.2 and 170.3 cm3 g−1 for rGO and carbon spheres, respectively.40 Although these adsorbents store a large volume of CO2 at 40 bar, for practical purposes, the material must be functional at low CO2 pressures. A K2CO3-impregnated carbon residue has been demonstrated as a reusable adsorbent for capturing CO2 from indoor air (1000–2000 ppm). In moist conditions, hydrated K2CO3 reacted with CO2 to form bicarbonates. Through this reaction, the material adsorbed ∼9.7 wt% of CO2 at 25 °C. Regeneration of the adsorbent requires heating to 150 °C and was regenerable for four cycles.45High-temperature CO2 chemisorption over alkali ceramics is a well-established approach where CO2 is chemisorbed as a carbonate salt of an alkali metal. Lithium orthosilicate (Li4SiO4) is a lithium ceramic that has been explored as a CO2 captor and possesses high capacity and regenerability.68 More often, different types of cathodes from spent LIBs have been used to recover Li as Li2CO3, which was further utilized as a Li precursor for synthesising Li4SiO4. In one such strategy, CO2 sequestration was done by using CO2 as a leaching agent to extract Li out of the spent LiFePO4 cathode as highly pure Li2CO3. Moreover, the FePO4 residue was converted to Na4Fe3(PO4)2P2O7 (a cathode for NIBs). This approach effectively fixed ∼120 kg of CO2 for every ton of recycled LiFePO4.42 A more direct approach involved using extracted Li2CO3 and a Si source (commercial silica or rice husk ash) to synthesise Li4SiO4 by a solid-state route. Through this, the cost of producing Li4SiO4 was reduced by 25–95% compared to the conventional route involving commercial precursors.39,43,44,69 Different types of cathodes, carbon sources for pyrolysis of cathodes,39 and leaching medium43 affected the composition of Li2CO3, further influencing the physicochemical properties of Li4SiO4. The best CO2 uptake was recorded for LiFePO4 battery-derived Li4SiO4, where the material possessed a capacity of 270–280 mg g−1 for 80 cycles in a 15 vol% CO2 feed.43 Also, waste (battery and rice husk)-derived Li4SiO4 was found to be a better CO2 captor than those synthesised with commercial precursors,69 further highlighting the importance of battery waste-derived materials.
Spent alkaline Zn–MnO2 batteries have been upcycled as adsorbents/catalysts for treating highly acidic gases like H2S, SO2, and NO2. ZnxMn3−xO4 and ZnO synthesised from the leachate of Zn–MnO2 batteries were used for treating H2S gas at 400 °C. These transition metal oxides successfully removed H2S from simplified gas mixtures and real syngas by reacting with H2S to form α-MnS, γ-MnS, ZnS, elemental sulphur, polysulfide, and MnSO4. The binary metal oxide with a high Mn/Zn ratio possessed superior catalytic activity and favoured the oxidation of sulphide to elemental sulphur and sulphate.30 Since black mass derived from Zn–MnO2 batteries is highly alkaline and has active ZnO, MnO2, and ZnMn2O4 phases, it was possible to use the raw black mass directly for H2S removal at 20 °C and high humidity. The black mass possessed an adsorption capacity of 60 mg g−1 for five cycles. Over the black mass, H2S molecules dissociated to sulphide and further oxidised by H2O and molecular O2 to sulphur and sulphate. Theoretical calculations confirmed that ZnO was more reactive than MnO2, and black mass derived from a fully discharged battery is expected to show even higher adsorption capacity.22 In a separate study, untreated black mass from Zn–MnO2 batteries of different brands was used as catalysts for treating 100 ppm of SO2 and NO2. These black mass samples have different phase composition, surface area, and alkalinity, which influenced their SO2 uptake capacity. For three cycles, the best material exhibited an average SO2 and NO2 adsorption capacity of 30.2 and 13.8 mg g−1, respectively, at 20 °C and high humidity. Further performance improvement was achieved by integrating black mass with calcium alginate to form hydrogel beads. The black mass catalysed SO2 to sulphate and NO2 to nitrite and nitrate ions.21 These studies highlighted the potential of black mass in treating a wide range of acidic gases at room temperature. Unlike CO2 captors like Li4SiO4, which require high energy input for both material synthesis and CO2 chemisorption, direct use of black mass (ZnO–MnO2–KOH) in treating acidic gases at low temperatures should be promoted for sustainable processing and upcycling of spent batteries (Fig. 6).
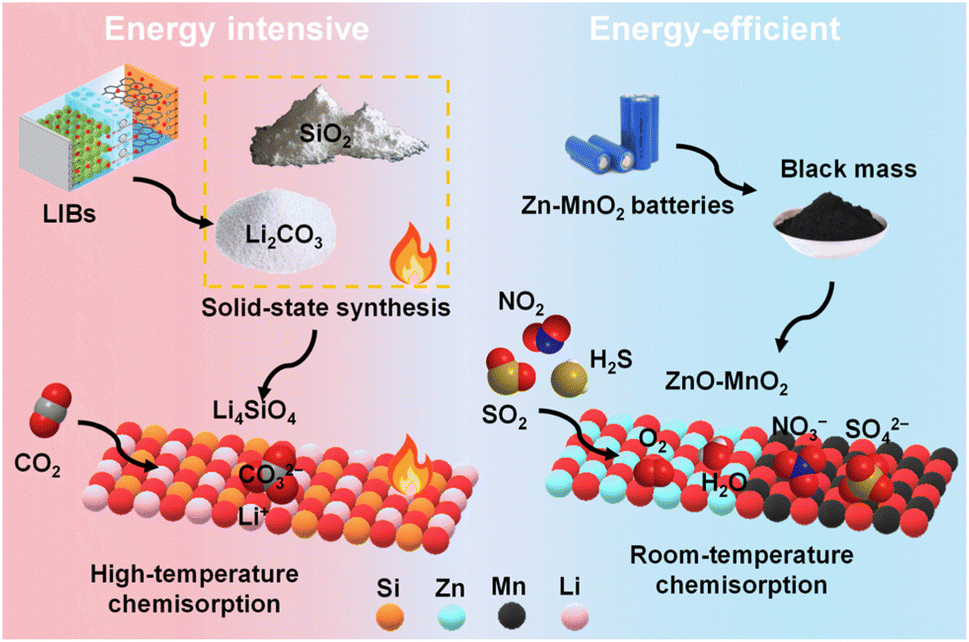 | ||
| Fig. 6 An illustration showing energy-intensive and energy-efficient upcycling of battery waste for acidic gas removal. | ||
Challenges and prospects
Waste LIBs and alkaline batteries are processed to develop functional materials for air decontamination applications. The synthesised materials serve as catalysts for the thermal oxidation of VOCs to CO2 and H2O, NH3-SCR catalysts for reducing NOx to N2, and room-temperature adsorbents/catalysts for eliminating acidic gases like CO2, SO2, NO2, and H2S. Many of the battery-derived catalysts have shown higher catalytic performance than those synthesised with commercial salts. Methods have been developed to sequestrate CO2 either by adsorbing or by converting it into carbonates. Additionally, CO2 has been employed as a leaching agent to extract pure and high-valued Li2CO3 from waste LiFePO4 batteries. To a greater extent, the main objectives of processing battery waste and developing functional materials for gas capture and removal applications have been fulfilled by the current literature. However, challenges persist for its translation from lab to industry. The biggest challenge in waste battery processing is the variation in the types of batteries available in the market. There is no one method which could be applied to all kinds of battery waste. This underscores the importance of consumer awareness in segregating battery waste at its source before collection by relevant authorities.The second challenge associated with battery waste processing is the excessive consumption of concentrated mineral acids like sulphuric or nitric acid for the leaching of desired or undesired metals from the battery's cathodes. This practice must be discouraged as mineral acids are toxic, require large energy input during production, and upon unchecked disposal are catastrophic to the environment.70 In such a scenario, the use of hydrometallurgical methods like bioleaching of metals using sulphuric acid-producing bacteria like Acidithiobacillus thiooxidans must be encouraged, which could reduce the volume of secondary waste generated from the cathode processing.28,29 Another issue faced during the upcycling of battery waste to functional materials is excessive energy consumption during the calcination process. Some studies have highlighted that materials synthesised at lower temperatures are more efficient than those synthesised at higher temperatures. An energy optimization during the synthesis process could greatly reduce the overall cost of upcycling and CO2 emissions associated with thermal energy consumption. Another approach is to upcycle battery waste with minimum steps to target a desired gas pollutant. It was observed that the black mass from alkaline batteries could be directly used for capturing H2S at room temperature. Thus, the need to upcycle the black mass to metal oxide catalysts for mid-temperature removal of H2S could be avoided. Moreover, many energy-intensive processes involved in gas treatment, like high-temperature CO2 chemisorption over alkali ceramics, must be discouraged. Room-temperature sequestration of CO2 over porous carbonaceous materials derived from polymer separators is a viable option for carbon capture, which should be explored more and extended to a larger scale after a systematic cost-to-performance evaluation. Though catalytic oxidation of aromatic VOCs is possible only at mid-temperatures, other VOCs like formaldehyde can be degraded even at room temperature.71 Even photocatalytic oxidation using solar radiation is a feasible option for eliminating aromatic hydrocarbons like toluene over battery-derived catalysts. Implementing these energy-efficient strategies could reduce the generation of secondary waste, overconsumption of energy, and associated CO2 emissions during the upcycling of battery waste. All these strategies are scarcely reported in the literature and need detailed investigation before their implementation in real conditions.
The urgent need to transition from fossil fuel to a renewable energy-driven economy is fuelling the growth of energy storage systems. Much of the research is concentrated on developing LIBs that could hold sufficiently large amounts of energy and are safe during operation. While research and development in battery manufacturing is a must to achieve the green economy targets, attention should also be given to the processing of battery waste. So far, a large volume of used batteries are either stored or disposed of in landfills. The situation for disposal of primary batteries is even worse as these batteries are consumed in billions with poor mechanisms for waste battery collection and processing in most of the countries. Though it is doubtful that the extraction of valuable metals like Li, Co, Ni, and Cu in LIBs could offset the entire cost of recycling, upcycling battery waste as functional materials and using them for gas pollutant removal could be an economical measure towards a cleaner environment. In the future, millions of tons of LIBs and Zn–MnO2 batteries are expected to be available for processing. Public and private entities must act responsibly in tackling this environmental issue by educating the public about battery segregation and recycling. The government must incentivise the entities focused on recycling and repurposing of battery waste. “A better segregation strategy is the golden step towards processing any waste”. Soon, more efficient and safer batteries like NIBs are expected to be rolled out in the market, which will further increase the volume of battery waste. Unlike LIBs, NIB's recycling cost will be even harder to offset as these are made with inexpensive metals like Na and Mn. Though some of the recent works have highlighted the potential of NIBs' cathode (Na–Mn oxides) in cleaning acidic gases in ambient conditions,72–74 more efforts are needed in identifying and developing functional materials capable of degrading harmful organic/inorganic gaseous pollutants in ambient conditions. The future of upcycling battery waste is bright as the research is moving in the right direction and targeting the most important issues of rising battery waste and air pollution.
Conclusions
The upcycling of discarded primary alkaline batteries and rechargeable LIBs to functional materials for harmful gas removal represents a promising approach to addressing the rising concentration of gaseous pollutants in the atmosphere. Through innovative approaches, electrodes, electrolytes, and polymer separators of battery waste could be processed into various functional materials, including metal oxides, composites, and carbonaceous materials. The synthesised materials have been effectively utilized as catalysts for thermal oxidation of VOCs and CO, NH3-SCR catalysts for NOx reduction, and room-temperature adsorbents/catalysts for capturing and mineralizing acidic gases. In many cases, the battery waste-derived materials were found more efficient than those synthesised by metal salts for a desired application. Numerous strategies, like metal-doping, integration with porous substrates, and acid/base-etching have been implemented to further improve the performance of these materials. These research findings have shown immense potential in repurposing battery waste streams for creating high-performing materials capable of effectively removing harmful gases from various sources. As research in this field continues to advance, it holds the promise of not only improving air quality but also promoting the circular economy by transforming waste into valuable resources.Author contributions
NKG is responsible for the literature survey and manuscript writing and reviewing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NKG would like to acknowledge Prof. Srungarpu N. Achary, Scientific Officer at the Solid State Chemistry Division, Bhabha Atomic Research Centre (BARC), Mumbai, India, for valuable scientific discussions.References
- S. Xará, J. Delgado, M. F. Almeida and C. Costa, J. Mater. Cycles Waste Manage., 2013, 15, 61–72 CrossRef.
- K. Hantanasirisakul and M. Sawangphruk, Glob Chall., 2023, 7, 2200212 CrossRef PubMed.
- Z. J. Baum, R. E. Bird, X. Yu and J. Ma, ACS Energy Lett., 2022, 7, 712–719 CrossRef CAS.
- S. Orangi, N. Manjong, D. P. Clos, L. Usai, O. S. Burheim and A. H. Strømman, J. Energy Storage, 2024, 76, 109800 CrossRef.
- A. Sobianowska-Turek, W. Szczepaniak, P. Maciejewski and M. Gawlik-Kobylińska, J. Power Sources, 2016, 325, 220–228 CrossRef CAS.
- K. K. Jena, A. AlFantazi and A. T. Mayyas, Energy Fuels, 2021, 35, 18257–18284 CrossRef CAS.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- M. Bhar, S. Ghosh, S. Krishnamurthy, Y. Kaliprasad and S. K. Martha, RSC Sustainability, 2023, 1, 1150–1167 RSC.
- X. Yu, W. Li, V. Gupta, H. Gao, D. Tran, S. Sarwar and Z. Chen, Glob Chall., 2022, 6, 2200099 CrossRef PubMed.
- H. Bae and Y. Kim, Mater. Adv., 2021, 2, 3234–3250 RSC.
- E. Paone, M. Miceli, A. Malara, G. Ye, E. Mousa, E. Bontempi, P. Frontera and F. Mauriello, ACS Sustainable Chem. Eng., 2022, 10, 2275–2281 CrossRef CAS.
- X. Wang, H. Qiu, H. Liu, P. Shi, J. Fan, Y. Min and Q. Xu, Green Chem., 2018, 20, 4901–4910 RSC.
- J. Zhou, S. Wang, X. Wang, C. Zhang, Z. Gu, T. Zhou, Z. Yuan, T. Long, J. Yin, Y. Yang and L. Yang, CrystEngComm, 2022, 24, 7540–7544 RSC.
- J. Meng, F. Liu, Z. Yan, F. Cheng, F. Li and J. Chen, Inorg. Chem. Front., 2018, 5, 2167–2173 RSC.
- W. Zou, X. Feng, W. Wei, Y. Zhou, R. Wang, R. Zheng, J. Li, S. Luo, H. Mi and H. Chen, Inorg. Chem., 2021, 60, 9496–9503 CrossRef CAS PubMed.
- T. Zhao, Y. Yao, M. Wang, R. Chen, Y. Yu, F. Wu and C. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 25369–25376 CrossRef CAS PubMed.
- P. Liu, ACS Sustainable Chem. Eng., 2018, 6, 11176–11185 CrossRef CAS.
- I. Manisalidis, E. Stavropoulou, A. Stavropoulos and E. Bezirtzoglou, Front. Public Health, 2020, 8, 14 CrossRef PubMed.
- V. V. Tran, D. Park and Y.-C. Lee, Int. J. Environ. Res. Public Health, 2020, 17, 2927 CrossRef PubMed.
- R. Patrice, B. Gérand, J. B. Leriche, L. Seguin, E. Wang, R. Moses, K. Brandt and J. M. Tarascon, J. Electrochem. Soc., 2001, 148, A448 CrossRef CAS.
- N. K. Gupta, K. Rajput, S. N. Achary, E. J. Kim, B. R. Mehta, D. R. Roy and K. S. Kim, Energy Fuels, 2024, 38, 545–554 CrossRef CAS.
- N. K. Gupta, K. Rajput, S. N. Achary, E. J. Kim, B. R. Mehta, D. R. Roy and K. S. Kim, Energy Fuels, 2024, 38, 7431–7435 CrossRef CAS.
- B.-S. Kim, S.-C. Jung, H.-Y. Jung, M. A. Khan, B.-H. Jeon and S. C. Kim, J. Ind. Eng. Chem., 2022, 114, 323–330 CrossRef CAS.
- Y.-K. Park, H. Song, M. K. Kim, S.-C. Jung, H. Y. Jung and S. C. Kim, J. Hazard. Mater., 2021, 403, 123929 CrossRef CAS PubMed.
- Y.-K. Park, W. G. Shim, S.-C. Jung, H.-Y. Jung and S. C. Kim, Korean J. Chem. Eng., 2022, 39, 161–166 CrossRef CAS.
- S. Hoseini, N. Rahemi, S. Allahyari, M. Tasbihi and E. Ghareshabani, Adv. Powder Technol., 2020, 31, 4187–4196 CrossRef CAS.
- X. Zhang, H. Li, Y. Yang, T. Zhang, X. Wen, N. Liu and D. Wang, J. Environ. Chem. Eng., 2017, 5, 5179–5186 CrossRef CAS.
- M. V. Gallegos, L. R. Falco, M. A. Peluso, J. E. Sambeth and H. J. Thomas, Waste Manage., 2013, 33, 1483–1490 CrossRef CAS PubMed.
- M. V. Gallegos, M. A. Peluso, E. Finocchio, H. J. Thomas, G. Busca and J. E. Sambeth, Chem. Eng. J., 2017, 313, 1099–1111 CrossRef CAS.
- M. Maroño, I. Ortiz, J. M. Sánchez, L. Alcaraz, F. J. Alguacil and F. A. López, Chem. Eng. J., 2021, 419, 129669 CrossRef.
- Z. Zhao, H. Du, B. Shen, P. Gao, C. Huang and S.-Q. Guo, Environ. Res., 2022, 212, 113300 CrossRef CAS PubMed.
- S. C. Kim, M. K. Kim, S.-C. Jung, H.-Y. Jung, H. Kim and Y.-K. Park, Chemosphere, 2021, 276, 130209 CrossRef CAS PubMed.
- S. C. Kim and B.-S. Kim, Environ. Pollut., 2023, 338, 122678 CrossRef CAS PubMed.
- Y.-K. Park, S.-C. Jung, H.-Y. Jung, S. Y. Foong, S. S. Lam and S. C. Kim, Environ. Sci. Pollut. Res., 2021, 28, 24552–24557 CrossRef CAS PubMed.
- J. Mao, G. Zhao, D. Wang and Y. Li, RSC Adv., 2014, 4, 25384–25388 RSC.
- X. Min, M. Guo, L. Liu, L. Li, J. Gu, J. Liang, C. Chen, K. Li, J. Jia and T. Sun, J. Hazard. Mater., 2021, 406, 124743 CrossRef CAS PubMed.
- N. Wu, M. Li, Q. Zhang, G. Xue, Y. Wang and C. Gong, Chem. Eng. J., 2024, 481, 148564 CrossRef CAS.
- M. Guo, K. Li, L. Liu, H. Zhang, W. Guo, X. Hu, X. Min, J. Jia and T. Sun, Ind. Eng. Chem. Res., 2020, 59, 194–204 CrossRef CAS.
- Y. Tong, C. Qin, L. Zhu, S. Chen, Z. Lv and J. Ran, Environ. Sci. Technol., 2022, 56, 5734–5742 CrossRef CAS PubMed.
- S. Natarajan, H. C. Bajaj and V. Aravindan, J. Mater. Chem. A, 2019, 7, 3244–3252 RSC.
- M. He, W. Liu, M. Gao, P. Zhang, X. Jin, H. Wu and Q. Liu, Green Chem., 2023, 25, 9969–9980 RSC.
- C. Xu, X. Hu, Y. Yang, Z. Jian, W. Chen, L. Yang, C. Yang, H. Liu, J. Zhao, H. Cao and Y.-S. Hu, Energy Storage Mater., 2023, 60, 102819 CrossRef.
- Y. Tong, C. Qin, X. Zhu, Z. Lv, X. Huang and J. Chen, ACS Sustainable Chem. Eng., 2023, 11, 6722–6730 CrossRef CAS.
- J. Ruan, Y. Tong, J. Ran and C. Qin, ACS Sustainable Chem. Eng., 2023, 11, 14158–14166 CrossRef CAS.
- Y.-R. Lee, A. Ra Cho, S. Kim, R. Kim, S. Wang, Y. Han, H. Nam and D. Lee, Chem. Eng. J., 2023, 470, 144232 CrossRef CAS.
- M. Guo, K. Li, H. Zhang, X. Min, J. Liang, X. Hu, W. Guo, J. Jia and T. Sun, Sci. Total Environ., 2020, 740, 139951 CrossRef CAS PubMed.
- M. Guo, X. Wang, L. Liu, X. Min, X. Hu, W. Guo, N. Zhu, J. Jia, T. Sun and K. Li, Environ. Res., 2021, 193, 110563 CrossRef CAS PubMed.
- X. Min, M. Guo, K. Li, J. Gu, X. Hu, J. Jia and T. Sun, Environ. Res., 2022, 215, 114299 CrossRef CAS PubMed.
- X. Yang, K. Liu, X. Han, J. Xu, M. Bian, D. Zheng, H. Xie, Y. Zhang and X. Yang, J. Hazard. Mater., 2023, 459, 132209 CrossRef CAS PubMed.
- T. Dai, H. Zhou, Y. Liu, R. Cao, J. Zhan, L. Liu and B. W.-L. Jang, ACS Sustainable Chem. Eng., 2019, 7, 5072–5081 CrossRef CAS.
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC.
- B.-S. Kim, M. Ki Kim and S. Chai Kim, Chem. Eng. J., 2023, 475, 146003 CrossRef CAS.
- G. Li, K. He, F. Zhang, G. Jiang, Z. Zhao, Z. Zhang, J. Cheng and Z. Hao, Appl. Catal., B, 2022, 309, 121231 CrossRef CAS.
- M. Guo, K. Li, L. Liu, H. Zhang, W. Guo, X. Hu, X. Meng, J. Jia and T. Sun, J. Hazard. Mater., 2019, 380, 120905 CrossRef CAS PubMed.
- J. Sun, X. Min, J. Gu, J. Liang and M. Guo, J. Environ. Chem. Eng., 2021, 9, 104964 CrossRef CAS.
- H. Xu, N. Yan, Z. Qu, W. Liu, J. Mei, W. Huang and S. Zhao, Environ. Sci. Technol., 2017, 51, 8879–8892 CrossRef CAS PubMed.
- J. Zhang, Y. Li, L. Wang, C. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC.
- X. Min, M. Guo, K. Li, J. Gu, X. Hu, J. Jia and T. Sun, Sep. Purif. Technol., 2022, 295, 121316 CrossRef CAS.
- M. Guo, K. Li, L. Liu, H. Zhang, X. Hu, X. Min, J. Jia and T. Sun, J. Taiwan Inst. Chem. Eng., 2019, 102, 268–275 CrossRef CAS.
- J. Sun, L. Liu, Y. Zhang, M. Guo and B. Zhou, Environ. Sci. Pollut. Res., 2021, 28, 38829–38838 CrossRef CAS PubMed.
- S. Hoseini, N. Rahemi, S. Allahyari and M. Tasbihi, J. Cleaner Prod., 2019, 232, 1134–1147 CrossRef CAS.
- Z. Zhao, B. Shen, Z. Hu, J. Zhang, C. He, Y. Yao, S.-Q. Guo and F. Dong, J. Hazard. Mater., 2020, 400, 123236 CrossRef CAS PubMed.
- X. Zhang, H. Li, Y. Yang, T. Zhang, X. Wen, N. Liu and D. Wang, J. Environ. Chem. Eng., 2017, 5, 5179–5186 CrossRef CAS.
- G. Xu, X. Guo, X. Cheng, J. Yu and B. Fang, Nanoscale, 2021, 13, 7052–7080 RSC.
- J. Jan, C.-L. Chang and S. Chang, J. Hazard. Mater., 2024, 472, 134497 CrossRef CAS PubMed.
- N. Wu, Q. Zhang, W. Wang, G. Xue, Y. Wang and C. Gong, J. Environ. Chem. Eng., 2024, 12, 112598 CrossRef CAS.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. AlBahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- M. J. Venegas, E. Fregoso-Israel, R. Escamilla and H. Pfeiffer, Ind. Eng. Chem. Res., 2007, 46, 2407–2412 CrossRef CAS.
- C. Zhang, C. Li, W. Li and X. Guo, Sep. Purif. Technol., 2023, 326, 124730 CrossRef CAS.
- C. Agarwal and A. K. Pandey, Environ. Sci.: Adv., 2023, 2, 1306–1339 CAS.
- W. Liu, T. Yu, Z. Dai, M. Zhang, H. Jin, H. Ge, X. Wang, D. Jin and H. Lou, J. Inorg. Organomet. Polym., 2023, 33, 451–461 CrossRef CAS.
- N. K. Gupta, K. Rajput, S. N. Achary, R. P. Dhavale, B. R. Mehta, D. R. Roy and K. S. Kim, New J. Chem., 2024, 48, 4670–4674 RSC.
- N. K. Gupta, S. N. Achary, H. Viltres, J. Bae and K. S. Kim, ACS Omega, 2022, 7, 37774–37781 CrossRef CAS PubMed.
- I. Yanase and T. Takano, Inorg. Chem. Commun., 2019, 104, 212–218 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |