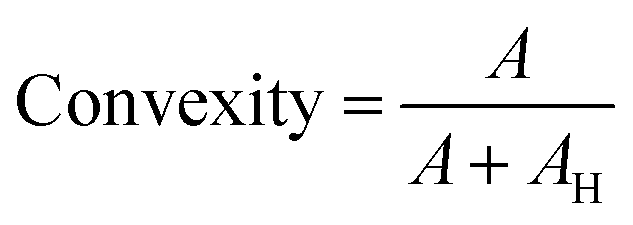DOI:
10.1039/D4TB00670D
(Paper)
J. Mater. Chem. B, 2024,
12, 10221-10240
Modelling methacrylated chitosan hydrogel properties through an experimental design approach: from composition to material properties†
Received
29th March 2024
, Accepted 8th August 2024
First published on 26th August 2024
Abstract
Hydrogels of biopolymers are gradually substituting synthetic hydrogels in tissue engineering applications due to their properties. However, biopolymeric hydrogels are difficult to standardize because of the intrinsic variability of the material and the reversibility of physical crosslinking processes. In this work, we synthesized a photocrosslinkable derivative of chitosan (Cs), namely methacrylated chitosan (CsMA), in which the added methacrylic groups allow the formation of hydrogels through radical polymerization triggered by UV exposure. We then performed a systematic study to link the physical properties of the materials to its preparation parameters to standardize its preparation according to specific applications. We studied the properties of CsMA solutions and the derived hydrogels using a statistical method, namely, response surface method, which allowed us to build empirical models describing material properties in terms of several selected processing factors. In particular, we studied the viscosity of CsMA solutions as a function of CsMA concentration, temperature, and shear rate, while hydrogel compression modulus, morphology, degradation and solubilization were investigated as a function of CsMA concentration, photoinitiator concentration and UV exposure. CsMA solutions resulted in shear thinning and were thus suitable for extrusion-based 3D printing. The CsMA hydrogel was found to be highly tunable, with a stiffness in the 12–64 kPa range, and was stable over a long timeframe (up to 60 days). Finally, the possibility to engineer hydrogel stiffness through an empirical model allowed us to hypothesize a number of possible applications based on the mechanical properties of several biological tissues reported in the literature.
1. Introduction
Hydrogels are three-dimensional (3D) polymeric networks embedding large amount of water.1 The water weight can reach thousands of times the weight of the dry polymeric network, making them the ideal candidate for various applications in tissue engineering.2,3 Hydrogels have a structure that has been used to mimic the natural extracellular matrix (ECM), favorably affecting cell adhesion, growth, and migration, especially in the case of gels made of biological or natural precursors.4 While synthetically derived hydrogels have stable and controllable chemical physical properties, including but not limited to stiffness, degradation, and swelling, naturally derived hydrogels suffer from the lack of standardization and the difficulty in controlling their properties. In fact, natural polymers to be formed into hydrogels usually rely on physical crosslinking, which is difficult to control due to the natural variability of the material itself.5 Natural hydrogels also exhibit mechanical properties with lower performance when compared to their synthetic counterparts.6 However, due to their unique set of properties that include biocompatibility, low cytotoxicity, and their similarity to the physiological environment, natural hydrogels are substituting synthetic hydrogels, especially in the applications in which the interaction with biological tissues is required.7
Among the different sources of biomaterials, chitosan-based hydrogels have a wide range of applications, from drug delivery to tissue engineering.8 Chitosan (Cs) is a natural polymer derived from chitin, which is a structural component found in the exoskeletons of crustaceans such as shrimp, crabs, and lobsters.9–11 Cs is a highly versatile material, with a range of properties that can be tuned depending on factors such as the degree of deacetylation, molecular weight, and crosslinking.9–11 For example, chitosan can form hydrogels with a high water content, which makes them ideal for drug delivery and tissue engineering applications.12–15 It also possesses antimicrobial properties, which make it useful for wound healing applications.16–19 Additionally, chitosan has been shown to be biodegradable, since it can be easily broken down and absorbed by the body over time. This makes it an attractive material for non-permanent implantable medical devices or drug delivery systems that require a temporary presence in the body.9–11
The Cs hydrogels are commonly obtained by gelation through physical crosslinking of the Cs chains.20 This includes a neutralization process by the increase of the pH value (using sodium hydroxide, NaOH, or ammonia, NH3); the increase in temperature, which allows the loss of the hydration water of the Cs chains and thus their interaction to form strong hydrophobic bonding (with the addition of organic phosphates (β-glycerophosphate, β-GP) or weak bases (sodium bicarbonate, NaHCO3) to avoid the Cs precipitation); the ionotropic gelation, which induces the formation of bridges between the anionic groups of the crosslinker (i.e., sodium tripolyphosphate, TPP) and the cationic groups of the Cs chains.20 Physical crosslinking is usually reversible, less stable, and less controllable than chemical crosslinking.21 Chemical crosslinking relies on chemical reactions that provide stable irreversible covalent bonds between the polymer chains.5,21,22 A common method to obtain a chemical crosslinking is to chemically modify the Cs chains to add methacrylic groups that, when triggered by UV exposure, allow for a radical polymerization. This reaction is performed using methacrylic acid (MA), a reactive monomer commonly used to functionalize biopolymers. The resultant material is known as methacrylated chitosan (CsMA).23–25 Using CsMA, 2D and 3D structures can be easily developed by photolithography and 3D printing.26,27 In addition, CsMA, in contrast to Cs, is water-soluble,23–25 which makes this material suitable for applications that require the embedding of cells.25,28
CsMA hydrogels have been investigated, alone or as a matrix to include other materials, for a range of tissue engineering applications, including bone, cartilage, and nerve regeneration. CsMA composite hydrogels promoted the differentiation of stem cells into osteogenic and chondrogenic lineages, making them promising candidates for bone and cartilage regeneration.27 Additionally, CsMA hydrogels, used in neuronal tissue engineering, induced the differentiation of Neural stem cells (NSCs) into tubulin-positive neurons and astrocytes.29 CsMA hydrogels have also been studied for drug delivery applications, where they offer the controlled release of drugs over time.30,31 These hydrogels can be used to deliver a variety of drugs, including small molecules, proteins, and nucleic acids. The hydrophilic nature of the methacrylated chitosan hydrogels allows for high drug loading and efficient drug release.30,31
Besides showing interesting biological properties in terms of biocompatibility and cell differentiation, hydrogels made of CsMA offer several advantages from a physico-chemical point of view, including tunable mechanical properties, high water content, and controlled degradation profiles.32,33 However, previous works reported in the literature did not explore the range in which the CsMA hydrogel properties can be tuned. There was also no available method for predicting the hydrogel properties based on its preparation process parameters. The tunability of the hydrogel rheology may allow for adapting the hydrogel to an extrusion-based printing process, while the control of the stiffness and degradation rate can be used to fulfil different needs in terms of cell culture, and to more faithfully mimic the biological tissue of interest. The main concept driving this study is that by experimentally manipulating the selected parameters of hydrogel preparation, specifically focusing on a single material like CsMA, and by in silico modelling of the full parameter space, it is possible to greatly tune the properties of the resulting hydrogels. This versatility may allow them to be used in a wide range of tissue engineering applications, especially in the case in which, e.g., a specific mechanical strength is needed to simulate a biological tissue.
In this work, we first prepared CsMA, and then performed FTIR analysis to confirm that the Cs chains were correctly chemically modified by addition of the MA groups. We subsequently in silico simulated a fraction of the CsMA chemical structure in order to evaluate its water solubility. Then, applying a statistical method, namely response surface method (RSM), we interpolated empirical equations describing the CsMA solution and hydrogel properties with the scope to prove the possibility of engineering them accordingly to specific tissue engineering applications, and to match the stiffness of natural biological tissue. RSM is a method used to model and analyze the relationships between multiple process variables and the outcoming properties of interest.34,35 In RSM, the studied system is treated as a black box, in which the variables of interest (called factors) are the inputs, and the studied properties (called responses) are the outputs. The advantage of this methodology is to avoid any consideration about the underlining chemical and physical mechanisms that are usually taken into account in mechanistic approaches.36 In our specific case, we modelled through RSM the viscosity of the CsMA solution in terms of the CsMA concentration, test temperature, and shear rate. This allowed us to evaluate the rheological properties of CsMA, which are crucial in the case of its use as a bioink in extrusion-based bioprinting technologies, largely employed for tissue engineering applications. Furthermore, we investigated the CsMA hydrogel properties as a function of the (i) CsMA concentration, (ii) amount of photoinitiator, and (iii) UV exposure time, i.e., all parameters related to the crosslinking degree. Among the hydrogel physico-chemical properties, the compression modulus, morphology, swelling ratio and degradation profile were analyzed. Finally, comparing the range of the tunable compression modulus with the values reported in the literature for biological tissues, we hypothesized a set of biological applications from neuronal cell culture to pre-calcified bone.
2. Materials and methods
2.1. Materials
Low molecular weight chitosan (Cs, 50–190 kDa) in powder form with a deacetylation degree of 75–85%, methacrylic anhydride (MA), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), and sodium bicarbonate were bought from Sigma Aldrich.
2.2. Methacrylated chitosan (CsMA) synthesis
Chitosan was reacted with excess methacrylic anhydride (Fig. 1A) to achieve methacrylation of both amine and methane hydroxyl units. The reaction was performed following a protocol of a previously published work with slight modifications.25Fig. 1B illustrates the schematic production of the lyophilized batch of CsMA, and the subsequent production of a crosslinked CsMA hydrogel. Briefly, MA was added dropwise to 2% (w/v) Cs acetic hydrochloric solution (0.1 M). MA (4 mL) was added for each gram of Cs used. The reaction included two phases. First, the prepared CsMA stock was kept under stirring for 6 h at 60 °C. Then, the temperature was lowered to room temperature for 24 h. The solution was then neutralized by adding a saturated solution of sodium bicarbonate (NaHCO3), and subsequently dialyzed against ultrapure water to eliminate the unreacted reagents for 4 days, changing the dialysis water once a day. The products were lyophilized at −50 °C (LIO 5 Pa, 5 pascal) for 3 days and maintained at 4 °C before its use. To prepare the crosslinked hydrogel samples, a defined amount of CsMA (in accordance with the chosen concentration) was solubilized in water. The solubilization, as in the case of Cs, required up to several hours (24 h in the case of highly concentrated solutions). Afterwards, LAP (lithium phenyl-2,4,6-trimethylbenzoylphosphinate) was added as the photoinitiator and stirred until dissolution. The solution was then poured in PDMS cylindrical molds and crosslinked under UV-light, controlling the exposure time.
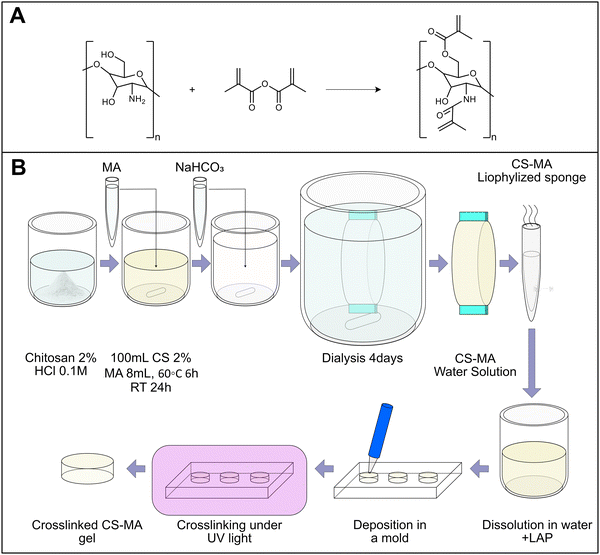 |
| | Fig. 1 (A) The proposed chemical reaction is extrapolated from previous works in the literature.25 The added methacrylic groups allow the photocrosslinking of the solution to form a hydrogel. (B) Preparation of CsMA stock and successive preparation of CsMA crosslinked hydrogels. | |
2.3. Chemical structure analysis
To qualitatively estimate the functionalization of Cs with MA groups, FTIR spectra of both Cs and CsMA polymers were collected. FTIR was performed using a FT/IR-6300 type A spectrophotometer (JASCO, Easton, MD, USA) on the lyophilized material in attenuated total reflectance mode (ATR-FTIR) in the 4000–600 cm−1 range with a resolution of 1 cm−1. The spectra were corrected by subtracting their baseline, and by normalizing them in the [0,100] range. Peaks were then assigned following the literature.
CsMA was characterized by 1H and 13C nuclear magnetic resonance (NMR) analysis run on a Bruker AVIII-400 MHz spectrometer using 99.9% deuterium oxide (D2O) as the solvent.
Moreover, to evaluate CsMA conformation and the presence of intramolecular hydrogen bonding, the theoretical structures of both Cs and CsMA polymers were simulated using the free version of SAMSON Connect 2022 R1 (https://www.samson-connect.net/, v.3.0.0). Fragments of Cs and CsMA composed of 14 units were drawn, both with 3 acetyl groups. The acetyl groups were added considering a deacetylation of 80%, as reported by the producer. The potential energy of the molecular system was interactively minimized using the universal force field (UFF) algorithm. Then, the presence of hydrogen bonding was visualized through the free extension (hydrogen bond finder, https://www.samson-connect.net/element/e0caae67-7422-ef1a-bf97-bcb88f1d2a11.html, v.0.9.3). Their strength was then roughly evaluated and color-coded based on the interatomic distances (strong H-bond, coded in gray, d ≤ 2.5 Å; moderate H-bond, coded in light blue, 2.5 Å < d ≤ 3.2 Å; weak H-bond, coded in blue 3.2 Å < d ≤ 4 Å).
2.4. Solution preparation
CsMA solutions for both the determination of the UV calibration curve and the rheology analysis were prepared by dissolving the lyophilized CsMA in ultrapure water, increasing the temperature up to 60 °C, and sonicating for 30 min. It should be noted that the time required for the complete solubilization strongly depends on the concentration. The higher concentrations used in this study required almost 24 h to be completely dissolved in water. The concentrations chosen for this test were: 0.1%, 0.25%, 0.5%, 0.75%, 1%, 1.5%, 2%, 2.5%, 3.5%.
2.5. Mechanical characterization
To verify the dependence of the CsMA solution viscosity to both polymer concentration and temperature, a rheological analysis was performed on all the previously reported CsMA concentrations at the following three temperatures: 4 °C, 25 °C and 37 °C (Physica MCR 301, Anton Paar). The viscosity against the shear rate was evaluated by a 25 mm parallel plate geometry, the gap was set to 0.3 mm, 200 μL of solution was used in each test, and the shear rate was linearly increased in the [1 s−1, 100 s−1] range. The temperature was set by using a Peltier plate stage mounted on the rheometer, and the samples deposited on the stage were left to equilibrate at the set temperature for 10 min. The test was performed in duplicate for each hydrogel solution.
The crosslinked hydrogels were tested in compression to determine their Youngs’ modulus using a universal testing machine (ZwickiLine 1 kN, Zwick Roell). For each composition, described in Table 1, n = 5 crosslinked cylinders were tested. To prepare the cylinders, the CsMA solutions were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 m to remove the bubbles present in the densest compositions. A 5 mm thick PDMS membrane with circular holes of 5 mm of diameter was used as the mold. This membrane adhered on a glass coverslip, and each hole was filled with a 300 μL of CsMA solution mixed with the photoinitiator (LAP, lithium phenyl-2,4,6-trimethylbenzoylphosphinate). After UV-exposure, the membrane was detached from the coverslip to reveal the cylindrical crosslinked hydrogels. The crosslinking follows the radical reaction described in Fig. 1C, and reported in previous works.37 The different samples were compared using an ANOVA test with Tukey's post hoc comparison.
000 rpm for 5 m to remove the bubbles present in the densest compositions. A 5 mm thick PDMS membrane with circular holes of 5 mm of diameter was used as the mold. This membrane adhered on a glass coverslip, and each hole was filled with a 300 μL of CsMA solution mixed with the photoinitiator (LAP, lithium phenyl-2,4,6-trimethylbenzoylphosphinate). After UV-exposure, the membrane was detached from the coverslip to reveal the cylindrical crosslinked hydrogels. The crosslinking follows the radical reaction described in Fig. 1C, and reported in previous works.37 The different samples were compared using an ANOVA test with Tukey's post hoc comparison.
Table 1 Compositions used to perform the degradation test. It should be noted that the amount of LAP in percentage is expressed in terms of weight. To get the weight over volume, the number should be divided by a factor of ten. The volume for each hydrogel should be adjusted based on the dimension of the vial
| Sample |
A
|
B
|
C
|
Compression |
Degradation |
| CsMA (%) |
CsMA (mg mL−1) |
LAP (% w/w) |
LAP (mg mL−1) |
t exp. (s) |
V (μL) |
Rep. |
V (μL) |
Rep. |
| 1 |
1 |
10 |
1 |
0.1 |
180 |
300 |
5 |
500 |
3 |
| 2 |
3 |
30 |
1 |
0.3 |
180 |
300 |
5 |
500 |
3 |
| 3 |
1 |
10 |
10 |
1 |
180 |
300 |
5 |
500 |
3 |
| 4 |
3 |
30 |
10 |
3 |
180 |
300 |
5 |
500 |
3 |
| 5 |
1 |
10 |
1 |
0.1 |
300 |
300 |
5 |
500 |
3 |
| 6 |
3 |
30 |
1 |
0.3 |
300 |
300 |
5 |
500 |
3 |
| 7 |
1 |
10 |
10 |
1 |
300 |
300 |
5 |
500 |
3 |
| 8 |
3 |
30 |
10 |
3 |
300 |
300 |
5 |
500 |
3 |
2.6. Morphological characterization
The n = 8 hydrogel formulations at different CsMA concentrations were observed by secondary electron microscopy (FESEM, Zeiss Gemini 300, Germany). One cylinder of crosslinked hydrogel was analyzed for each of the 8 compositions. The hydrogels were frozen by immersion in liquid nitrogen, and then lyophilized (Lio 5 Pa, 5 pascal) until the water was completely removed. To reveal the cross-section, the dried hydrogels were immersed in liquid nitrogen and cut with a scalpel. This allowed a fragile fracture, avoiding any structural deformation. Prior to SEM imaging, the samples were coated with a 10 nm layer of gold by sputtering (CCU-010 LV, Safematic GmbH). Five images, at 1000× of magnification, were collected for each sample (a total of 40 images). The dimensions and shape factors were calculated by an automated procedure built on the Knime platform (v. 4.7.2, Knime AG, Zurich, Switzerland). A scheme of the performed analysis is reported in Fig. 2A, while the implemented workflow in Knime within the used nodes is shown in Fig. 2B. The area was calculated as the number of pixels occupied by each pore and multiplied by the conversion factor (3.775 px μm−1), then the equivalent diameter was calculated using eqn (1) under the assumption of perfect circularity. Two shape factors were also calculated, the circularity and the convexity, using eqn (2) and (3), respectively. In these equations, A is the calculated area and P is the calculated perimeter, while AH is the total area of the convex hulls. In this case, the samples were compared using a Kruskal–Wallis one-way analysis of variance, which does not require the normality of the residuals with a Dunn's post hoc test.| | 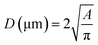 | (1) |
| | 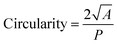 | (2) |
| | 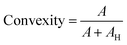 | (3) |
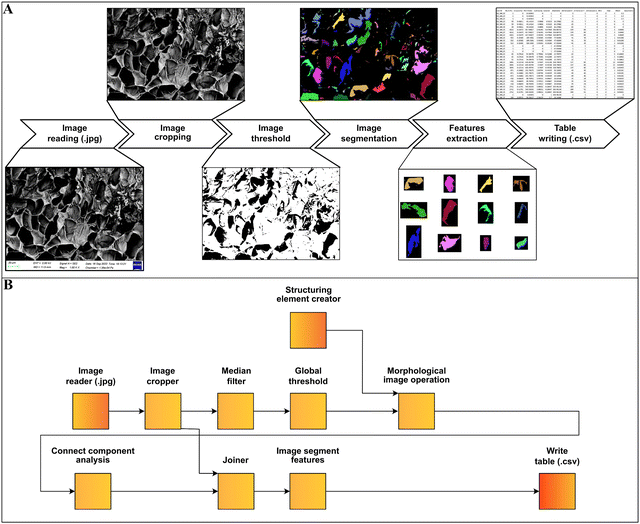 |
| | Fig. 2 (A) Pore analysis of SEM images and an automated procedure. Forty images were collected at 1000× magnification, cropped, thresholded and segmented. Once segmented, the features were extracted, and the dimension and shape parameters calculated. (B) Implemented workflow using Knime showing the used nodes. | |
2.7. Optical characterization
The optical transmittance in the UV-visible region of the solution with increasing concentration of CsMA was determined by UV-vis spectroscopy (microplate reader Clario Star Plus, BMG Labtec), performing the measurement in the 200–1000 nm range with 1 nm resolution. A constant volume of 200 μL of each solution was inserted in a 96-well plate, and the spectra were collected (see Fig. S1, ESI†).
2.8. Degradation and solubilization
The NanoDrop One microvolume UV-vis spectrophotometer (Thermo Scientific, USA) was used to collect the spectra. A calibration curve was determined by solubilizing different percentages of Cs (from 0.1% to 3.5%) in water, and collecting 3 UV-vis spectra for each sample in the 190–800 nm range. The spectra were baseline corrected, and then the intensity at 215 nm was used for the calibration curve (Fig. 3A). It should be noted that we did not use the intensity at the peak (about 200 nm) due to the saturation of the instrument response at the higher concentrations. The result of the calibration is reported in Section 1.1 of the ESI.†
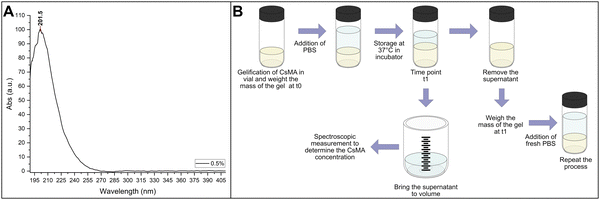 |
| | Fig. 3 (A) UV-vis spectra of a 0.5% water solution of CsMA; the maximum absorbance was detected at 201.5 nm. To avoid signal saturation, the intensity was measured at 215 nm. (B) Methodology to determine the amount of CsMA released into the PBS solution (solubilization), and the amount of CsMA hydrogel broken in smaller pieces and removed at each time point (degradation). | |
The degradation/swelling and solubilization were conducted by a single experiment, with a procedure that allowed us to separate these two contributions. The method proposed here is a modified version of a procedure previously reported in the literature and applied on methacrylated silk fibroin scaffolds,37 where it was proved that the presence of LAP was insignificant in terms of the UV intensity due to its negligible quantity. The steps of the experimental procedure are schematized in Fig. 3B. Briefly, 2 mL glass vials were weighed with their caps to determine their mass, indicated as mvial. 500 μL of water solution of CsMA with LAP as photoinitiator were inserted in each vial and crosslinked. Three vials for each of the compositions reported in Table 1 were prepared. The mass of the vial within the gel was weighed, and then the mass of the gel at time t0 was calculated using eqn (4). 1 mL of PBS was added to each vial, which was then sealed hermetically with parafilm and stored in an incubator at 37 °C. Once the timepoint was reached, the supernatant was accurately removed and taken to 1 mL volume in a volumetric flask by the addition of PBS. Form this solution, 100 μL was collected and used to record the UV-vis spectra (subtracting as background the signal intensity from pure PBS). The concentration of CsMA in solution was then calculated by eqn (S1) (ESI†). With the supernatant volume equal to 1 mL, the amount of CsMA was calculated using eqn (5). Then, the vial containing the hydrogel was weighed again and the mass of the hydrogel was calculated at time t1 using eqn (6). Finally, 1 mL of fresh PBS was inserted into the vial that was hermetically closed and stored at 37 °C until the following time point. This procedure was repeated at each time point. The mass of the hydrogel at t1 is the combination of the degradation of the gel and the absorption of PBS, as defined in eqn (7). It should be noted that our gel, being mechanically strong over a period of time, was broken into smaller pieces that were lost during the experimental procedures. We took this into account with the term (mCSMA(detached)(t1)), which is unknown, as it is also unknown how much PBS was absorbed (mPBS(absorbed)(t1)). Overall, the unknown term of eqn (7) is the composed term (mCSMA(detached)(t1) + mPBS(absorbed)(t1)) that was calculated by eqn (8). Then, at each time point, the solubilization was calculated by eqn (9), and the combination of the degradation and swelling by eqn (10). Negative results for eqn (10) indicate that the absorption of PBS overcomes the loss of detached hydrogel pieces, while positive results indicate that the loss of the detached hydrogel pieces overcomes the absorption of PBS. In the first case, we refer to the phenomena as net swelling (SWnet), and the second case is referred to as net degradation (Degnet).
| | | mgel(t0) = mvial+gel − mvial | (4) |
| |  | (5) |
| | | mgel(t1) = mvial+gel(t1) − mvial | (6) |
| | | mgel(t1) = mgel(t0) − (mCSMA(detached)(t1) + mPBS(absorbed)(t1)) − mCsMA(solubilized)(t1) | (7) |
| | | mCSMA(detached)(t1) + mPBS(absorbed)(t1) = mgel(t0) − mgel(t1) − mCsMA(solubilized)(t1) | (8) |
| |  | (9) |
| |  | (10) |
2.9. Statistical methods
The properties of the CsMA solution and hydrogel were studied by design of experiment (DoE). Specifically, a response surface method was used in the case of the CsMA solution viscosity, where seven solutions with increasing CsMA concentration were tested (factor C, 0.5%, 1%, 1.5%, 2%, 2.5%, 3%, 3.5%) in a range of shear rate (factor B) between 1 (1 s−1) and 100 (1 s−1) at three different temperatures (factor A, 4 °C, 25 °C, and 37 °C). The complete model is reported in eqn (11). The properties of the hydrogel were instead investigated as a function of the factors influencing the crosslinking degree, i.e., two factors related to the hydrogel composition (A – the CsMA concentration (%), B – the LAP concentration (% w/w)) and one process factor (C – the exposure time (s)) using a 23 full factorial DoE. The complete model is reported in eqn (12). In Table 1, the 8 different compositions are reported within the factors’ levels.
The entire statistical analysis has been done using the programming language R (v. 4.3.1) and the RStudio IDE (2023.06.1, Build 524), following the strategy described in previous works.38 An initial analysis was done by calculating a correlation matrix, in which the value of r2 was reported for a couple of variables. This was used to verify the presence of linear correlation among them (correlation of first order). Then, a model was built by selecting the significantly relevant factors, tested by analysis of variance (ANOVA). The significant level was assigned as follows: p ≤ 0.1 (.), p ≤ 0.05 (*), p ≤ 0.01 (**), p ≤ 0.001 (***). Only factors with p ≤ 0.1 were included in the model, while the model was considered significant if p ≤ 0.05. This choice has been made to take into account the natural variability of the biopolymer. The hierarchy has been preserved in all models, which means that insignificant lower-order terms were included if higher-order terms are found to be significant. The model function (indicated in the equation as F) was chosen to achieve two scopes; the first was to normalize the model residues, and the second was to make the model residues pattern less. After evaluating different models, the grade of the polynomial equation for the RSM was determined. The polynomial grade that was selected aims to maximize R2, the adjusted R2 (RA2), and the predicted R2 (RP2), while simultaneously ensuring the minimization of the predicted residual error sum of squares (PRESS). RA2 refers to the adjusted R2 index, which takes into account the number of terms included in the model, allowing for a fair comparison between models with different numbers of terms. On the other hand, RP2 measures the accuracy of the regression model in predicting responses for new observations. RP2 was determined by removing a single data point from the database, using the modified dataset to interpolate the model, and then assessing the accuracy of the modified model in predicting the excluded data point. The process was iteratively repeated for all datapoints. The PRESS was calculated in the same way, where a single observation was removed from the dataset and the remaining observations were reanalyzed. In each scenario, the value predicted for the omitted observation was calculated, and the PRESS statistic was determined as the sum of squares of all the resulting prediction errors.
| | | F(η) = c0 + c1A + c2B + c3C + c4AB + c5AC + c7BC + c8ABC + A2 + B2 + C2 + A2B + A2C + AB2 + AC2 + B2C + BC2 + A3 + B3 + C3 | (11) |
| | | F(Y) = c0 + c1A + c2B + c3C + c4AB + c5AC + c7BC + c8ABC | (12) |
3. Results
3.1. Chemical structure analysis
FTIR spectra, reported in Fig. 4A, were collected to check the Cs chain functionalization with MA groups. In the Cs spectrum, a strong band in the region 300–3700 cm−1 corresponds to the N–H and O–H stretching modes, as well as the intramolecular hydrogen bonds.39 The absorption bands, characteristics of polysaccharide,40–42 were individuated at around 292 cm−1 and 2881 cm−1 and attributed to C–H symmetric and asymmetric stretching, respectively. The presence of the residual N-acetyl groups was confirmed by the peaks at around 1656 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of amide I) and 1321 cm−1 (C–N stretching of amide III), and the shoulder peak at 1562 cm−1 (N–H bending of amide II). This last one was probably overlapped by the other bands. The N–H bending of the primary amine was assigned to the peak at 1593 cm−1. The CH2 bending and CH3 symmetrical deformations were assigned to the peaks at around 1423 cm−1 and 1377 cm−1, respectively. The absorption band at 1153 cm−1 was attributed to asymmetric stretching of the C–O–C bridge. The bands at 1066 and 1028 cm−1 were assigned to the C–O stretching. All these assignments were found in the Cs spectra of earlier works.17,39,43,44 After the reaction, we observed several diagnostic peaks, indicating the successful functionalization. In the CsMA spectra, the shoulder peak centered at 1705 cm−1 confirmed the ester CO group.45 The modification of the spectra in the region between 1585 cm−1 and 1680 cm−1 can be attributed to the superposition in the same band of the amide absorption and the stretching of the alkenes CC bond. Both peaks centered at 1649 cm−1 and at 1615 cm−1 can be related to this superposition.46 A peak at 1454 cm−1 was detected, which could be attributed to CH2 bending, and it was recognized as a shoulder in the Cs spectrum. We also observed the modification of the spectra in the low frequency region between 1000 cm−1 and 600 cm−1, where the C–H bend is usually observed. We assigned this bend to the small peak centered at 835 cm−1.47 The 1H NMR spectrum (Fig. 5A) shows the peaks at 3.16–4.89 ppm, which are ascribed to the glucosamine unit. Four methylene protons of MMA are observed between 5.5 and 5.32 ppm for the functionalization of both amino and hydroxymethyl groups, respectively. It was also confirmed by 13C NMR spectrum (Fig. 5B), which shows two peaks centered at 181.43 and 178.29 ppm assigned to the carbonyl ester (–CH2OCO–) and carbonyl amide (–NHCO–), respectively.
O stretching of amide I) and 1321 cm−1 (C–N stretching of amide III), and the shoulder peak at 1562 cm−1 (N–H bending of amide II). This last one was probably overlapped by the other bands. The N–H bending of the primary amine was assigned to the peak at 1593 cm−1. The CH2 bending and CH3 symmetrical deformations were assigned to the peaks at around 1423 cm−1 and 1377 cm−1, respectively. The absorption band at 1153 cm−1 was attributed to asymmetric stretching of the C–O–C bridge. The bands at 1066 and 1028 cm−1 were assigned to the C–O stretching. All these assignments were found in the Cs spectra of earlier works.17,39,43,44 After the reaction, we observed several diagnostic peaks, indicating the successful functionalization. In the CsMA spectra, the shoulder peak centered at 1705 cm−1 confirmed the ester CO group.45 The modification of the spectra in the region between 1585 cm−1 and 1680 cm−1 can be attributed to the superposition in the same band of the amide absorption and the stretching of the alkenes CC bond. Both peaks centered at 1649 cm−1 and at 1615 cm−1 can be related to this superposition.46 A peak at 1454 cm−1 was detected, which could be attributed to CH2 bending, and it was recognized as a shoulder in the Cs spectrum. We also observed the modification of the spectra in the low frequency region between 1000 cm−1 and 600 cm−1, where the C–H bend is usually observed. We assigned this bend to the small peak centered at 835 cm−1.47 The 1H NMR spectrum (Fig. 5A) shows the peaks at 3.16–4.89 ppm, which are ascribed to the glucosamine unit. Four methylene protons of MMA are observed between 5.5 and 5.32 ppm for the functionalization of both amino and hydroxymethyl groups, respectively. It was also confirmed by 13C NMR spectrum (Fig. 5B), which shows two peaks centered at 181.43 and 178.29 ppm assigned to the carbonyl ester (–CH2OCO–) and carbonyl amide (–NHCO–), respectively.
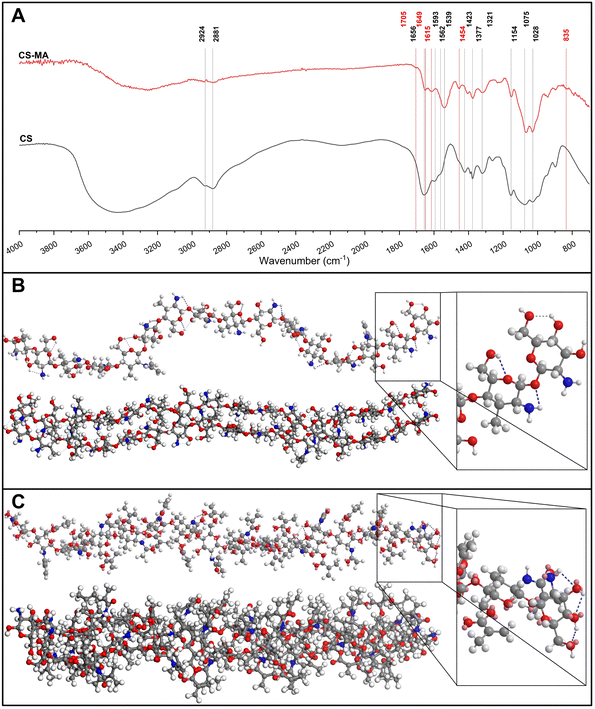 |
| | Fig. 4 Comparison between the equilibrium structures of a fragment of Cs (A) and CsMA (B) composed of 14 repetitive units. The intramolecular H-bonding interactions were reported with different colors based on their strength: weak interactions in gray, medium interactions in light blue, and strong interactions in blue. It can be observed that in Cs, the intramolecular hydrogen bonding bends the molecule into an helicoidal structure, while in CsMA, it is almost linear. Interestingly, upon optimizing the geometry of two molecules, we can observe the presence of several intermolecular hydrogen bonds in the case of Cs, which contributes toward retaining a helicoidal structure, and a few hydrogen bonds in the case of CsMA. (C) FTIR spectra collected on Cs and CsMA. The red peaks can be considered as a diagnostic for the functionalization. | |
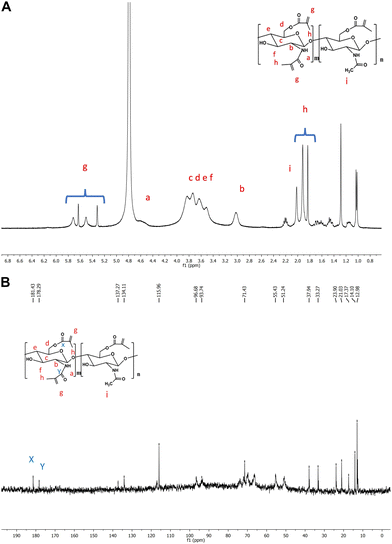 |
| | Fig. 5 (A) 1H-NMR NMR of chitosan–methacrylate. (B) 13C-NMR NMR of chitosan–methacrylate. | |
The simulation of Cs and CsMA molecules by geometrical optimization allowed us to hypothesize the reason behind the different water solubilities of these two materials. In fact, the empirical results show that Cs does not dissolve in water, while CsMA does. This difference is a consequence of the different molecular structures. The strong and medium hydrogen interactions of Cs (indicated as dotted lines) allow it to assume an α-helix structure and to interact with other Cs molecules. Consequently, this structure needs a mild acid condition to be solubilized, as observable in Fig. 4B. In contrast, the structure of CsMA, reported in Fig. 4C, is almost linear and the interaction with other CsMA molecules is less pronounced. For this reason, CsMA was subsequently water-soluble.
3.2. Mechanical characterization
The set of CsMA solutions with increasing concentration was characterized by rheology to determine the relationship between their viscosity and the shear rate and the testing temperature. We tested 7 uncrosslinked solutions of CsMA with concentrations varying from 0.5% to 3.5% through shear-sweep tests at different temperatures. Regardless of the concentration, all solutions were extrudable from a Gauge 22 nozzle (∼430 μm internal diameter), as reported in the ESI.† The results of the rheological tests are shown in Fig. 6A in terms of shear stress vs. shear strain. As a general observation, the solution acts as a non-Newtonian shear thinning fluid (as shown in the viscosity plot reported in Fig. S2, ESI†), which implies its printability by extrusion-based technologies.48
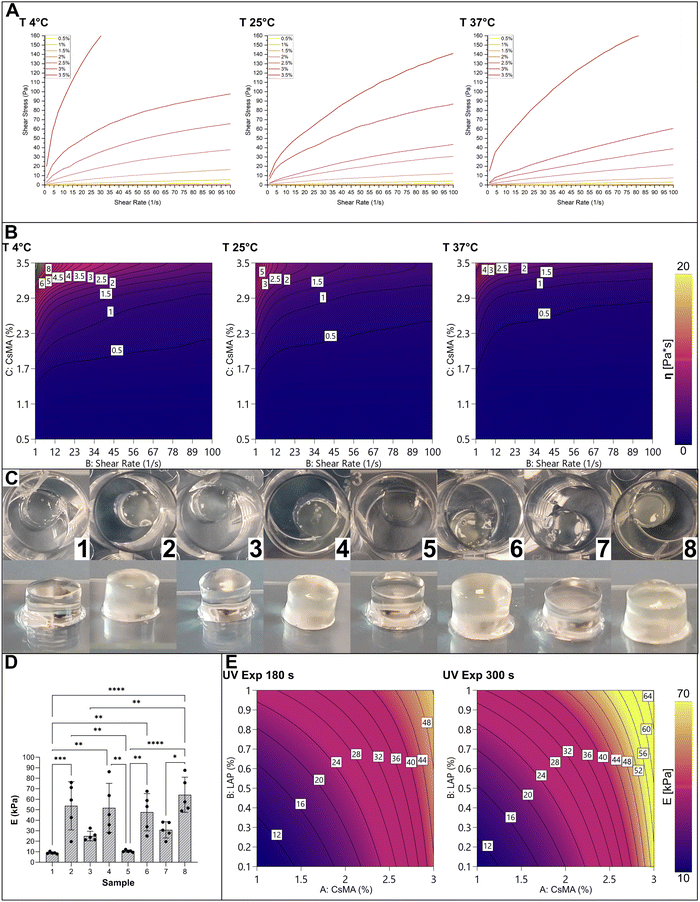 |
| | Fig. 6 (A) Rheological curves (shear stress vs. shear rate) of CsMA solutions at three different temperatures (4, 25 and 37 °C), showing a shear-thinning behaviour in all cases. (B) Contour plot of the empirical RSM model of the viscosity built based on A: the CsMA concentration (%), B: shear rate (1 s−1) and, C: temperature (°C). The model was sliced along the temperature axis to allow its visualization. (C) CsMA crosslinked hydrogels. (D) The compressive Young's modulus of the prepared samples: almost all differences were statistically significant, indicating the strong influence of the considered factors on the mechanical strength. (E) Contour plot of the empirical DoE model of the mean compressive Young's modulus build based on A: CsMA concentration (%), B: LAP concentration (% w/w) and C: UV exposure time (s). In this case, the model was sliced along one axis (UV exposure time) to allow its visualization. | |
The adopted response surface method (ANOVA Table S1, ESI†) allowed us to avoid physical considerations in the nature of the hydrogel, and to provide an empirical equation describing the rheological behaviour. We modeled the viscosity (η) in terms of the CsMA concentration, shear rate, and temperature, using a model chosen based on some preliminary statistical analysis (Table S2, ESI†). The results are shown in Fig. 6B as a contour plot at the testing temperatures. The modelled eqn (S2) (ESI†) is reported in the ESI,† within the ANOVA (Table S1, ESI†), the coefficient of the high and low 95% confidence intervals (CI), reported in Table S3 (ESI†), and the actual vs. predicted plot, reported in Fig. S3 (ESI†). It should be noted that even if the model is continuous in relation to the temperature, a slicing along the temperature axes was performed to visualize it as a four-dimensional model. Moving from the lower temperature contour plot (from the left to the right), the general trend was a decrease in the solution viscosity that instead increased when the polymer concentration increased. The trend with the increase of the shear rate is typical of a shear thinning fluid. At low concentration, the viscosity remains almost the same by increasing the shear rate. Instead, at higher concentration, the viscosity decreases with increasing shear rate.
Cylinders of crosslinked CsMA hydrogels were tested in mechanical compression. A single cylinder per typology is shown in Fig. 6C. Interestingly, the low concentration resulted in clear, transparent hydrogels, while the higher concentration resulted in semi opaque, yellowish hydrogels. The compression Young's modulus (E) is shown in the boxplot of Fig. 6D (the mechanical stress–stain curves shown in Fig. S4 of the ESI†), while the descriptive statistic is reported in Table 2. Interestingly, almost all the means were statistically different, and a trend could be recognized. Samples of the trials 2, 4, 6 and 8 had a higher compression modulus due to the higher CsMA concentration. Among these samples, the one produced with the 8th trial had the highest compression modulus due to a combination of a high CsMA (30%) and LAP (10% w/v) concentration, and the long exposure time (300 s). This indicates that all the considered terms were significant in determining the compression modulus, as highlighted by the ANOVA (Table S4, in the ESI†). The DoE-modelled empirical equation of E is reported in eqn (S3) (ESI†), while the coefficients of the 95% high and low CI are reported in Table S5 (ESI†) and the actual versus predicted plot is shown in Fig. S5 (ESI†). The model is shown as a contour plot in Fig. 6E. Also in this case, being as the model is four-dimensional, a slicing was done on the exposure time axes to allow the graphical representation. E increases diagonally with the increase of both CsMA and LAP concentrations. The impact of the UV curing time depends on the other two factors (CsMA and LAP concentrations). In fact, at low CsMA and LAP concentrations, the impact is negligible (at the lowest level of concentrations, the model predicts a E value of 9.4 kPa in the case of 180 s of exposure and 10.4 kPa in the case of 300 s of exposure). Meanwhile, upon increasing the concentration, it becomes relevant (at the highest level, the prediction is 52.3 kPa for 180 s of exposure, and 67.2 kPa for 300 s of exposure). Overall, the compression modulus ranged from a minimum mean value of about 9 kPa to a maximum mean value of about 65 kPa. In this case, the mean and median were close enough to consider the distributions as normal (Table 2). It should be noted that, as expected, higher compression moduli were accompanied with a higher standard deviation (StD, Table 2) and interquartile range (IQR, Table 2).
Table 2 Descriptive statistics of Young's compressive modulus. The mean value, standard deviation (StD), minimum (Min.) and maximum (Max.) values, first (Q1) and third (Q3) quartiles, median value, and interquartile range (IQR) were calculated
| Sample |
Mean (kPa) |
StD (kPa) |
Min. (kPa) |
Q1 (kPa) |
Median (kPa) |
Q3 (kPa) |
Max. (kPa) |
IQR (kPa) |
| 1 |
8.96 |
0.92 |
7.96 |
8.58 |
8.79 |
9.04 |
10.45 |
0.46 |
| 2 |
53.79 |
22.99 |
19.71 |
42.63 |
59.39 |
71.24 |
75.97 |
28.61 |
| 3 |
25.01 |
4.58 |
21.36 |
21.46 |
23.77 |
25.99 |
32.47 |
4.53 |
| 4 |
51.81 |
23.44 |
28.02 |
35.74 |
45.37 |
63.58 |
86.35 |
27.84 |
| 5 |
10.67 |
1.03 |
9.55 |
10.23 |
10.39 |
10.92 |
12.29 |
0.69 |
| 6 |
47.71 |
17.75 |
24.97 |
33.94 |
53.10 |
58.87 |
67.69 |
24.93 |
| 7 |
30.87 |
7.70 |
19.73 |
27.90 |
30.55 |
37.91 |
38.27 |
10.01 |
| 8 |
64.28 |
16.73 |
48.88 |
51.62 |
57.51 |
75.75 |
87.63 |
24.13 |
3.3. Morphological characterization
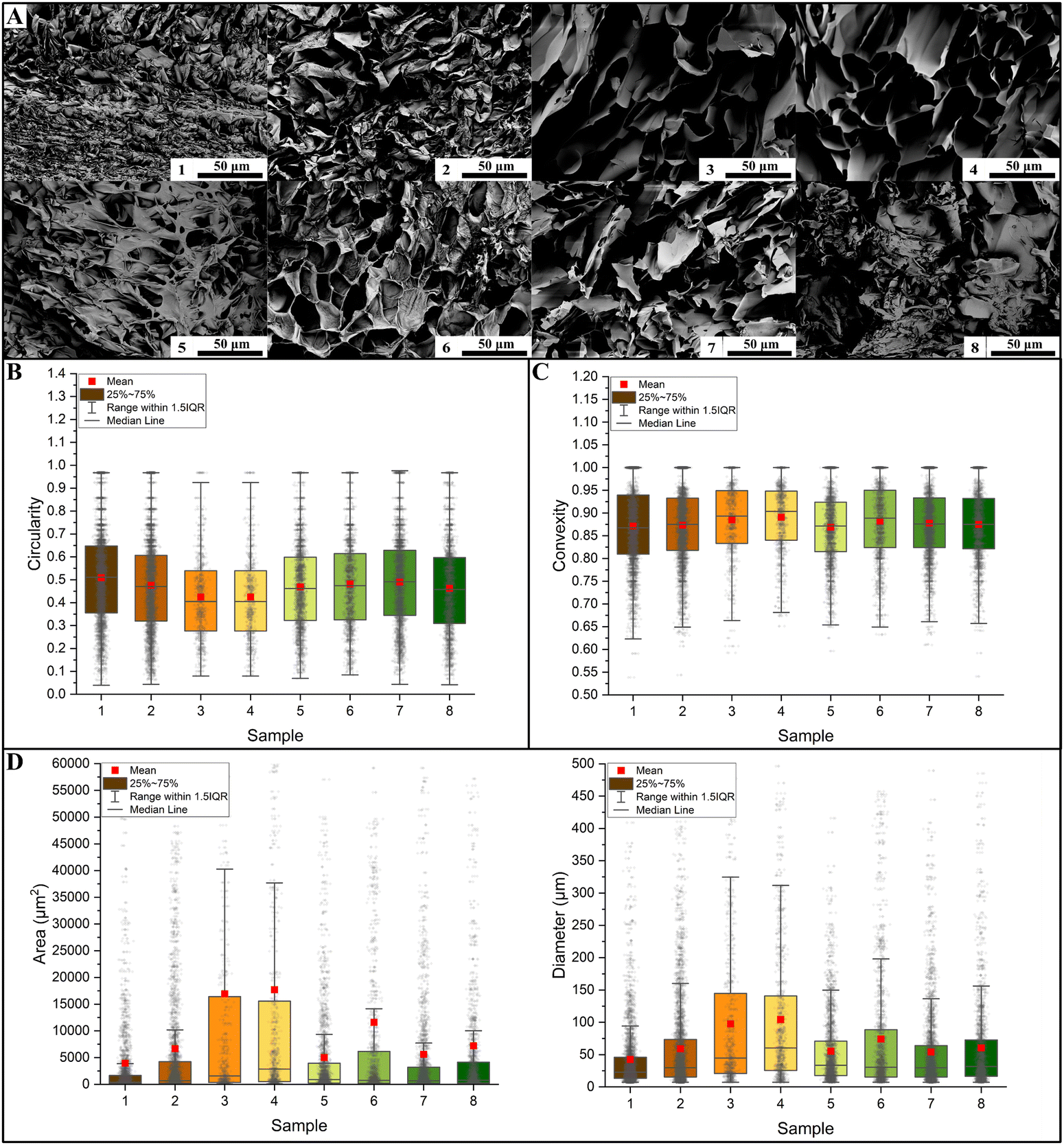
SEM micrographs were used to evaluate the effect of the factors on the samples’ morphology. It is clear from Fig. 7A that sample 3 and sample 4 resulted in larger pore dimensions. In terms of shape factors, both circularity (Fig. 7B) and convexity (Fig. 7C) did not reveal statistical differences among the different samples. For this reason, we were not able to build a statistical model on these shape factors.
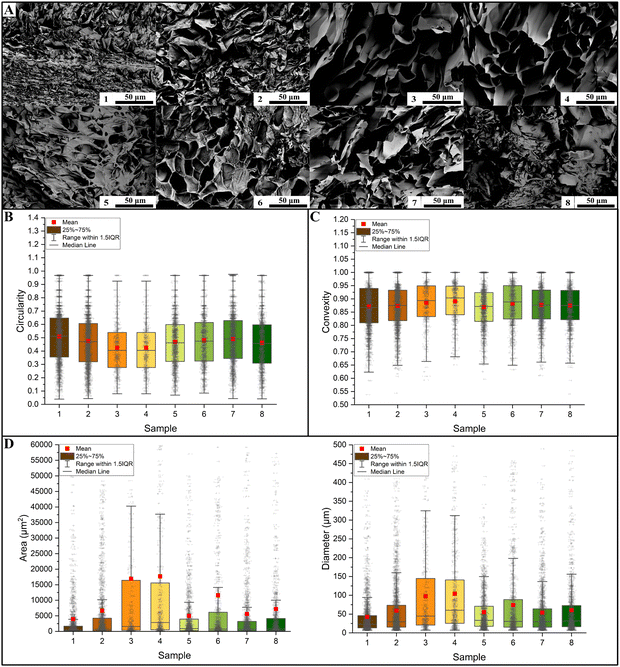 |
| | Fig. 7 (A) SEM images of the hydrogels’ microstructure. A single image for each sample is reported here; the dimensional analysis were conducted on 5 images per sample. The microstructure results depend on the chosen study factors. (B) Quantitative analysis of pore circularity shows that there is a large variability in all considered samples. (C) This is also true in terms of convexity. Both shape factors resulted in wide distributions. (D) In terms of the pore area, the samples were significantly different in most cases (in this case, the asterisk notation was not reported because of a high number of significant comparison), as reported by Table S6 in the ESI.† As expected, being a derived response, an identical result was obtained by analyzing equivalent diameters (Table S7, ESI†). | |
The morphological characterizations revealed statistically significant differences in terms of dimensions (both area and diameter, Fig. 7D). Almost all the differences turned out to be statistically significant, as proved by the Kruskal–Wallis tests reported in Tables S6 and S7 (ESI†) for the area and the diameter, respectively. From the performed quantitative analysis reported in Table 3 (area) and Table 4 (diameter), sample 3 (mean area of 16947 μm2, mean diameter of 97.4 μm) and sample 4 (17714 μm2, 104.2 μm) resulted in samples with larger pores. Meanwhile, sample 1 (mean area of 3955 μm2, mean diameter of 42.6 μm) resulted in smaller pores. It should be noted that sample 3 and sample 4 also had larger distributions, both in terms of standard deviations (StD) and interquartile range (IQR). The StD on the area was 33498.6 μm2 for sample 3 and 33022.3 μm2 for sample 4, while the IQR was 16074.7 μm2 and 15062.9 μm2, respectively. The StD of the diameter was 110.0 μm for sample 3 and 108.2 μm for sample 4, and the IQR was 123.7 μm and 115.3 μm respectively. Sample 1 had smaller distribution with an area StD of 3955.2 μm2 (IQR 1510.6 μm2).
Table 3 Descriptive statistics of the area distributions for different samples. The mean value, standard deviation (StD), minimum (Min.) and maximum (Max.) values, first (Q1) and third (Q3) quartiles, median value, and interquartile range (IQR) were calculated. Samples 3 and 4 resulted in higher areas
| Sample |
Mean (μm2) |
StD (μm2) |
Skew |
Kurt |
Min. μm2 |
Q1 (μm2) |
Median (μm2) |
Q3 (μm2) |
Max. (μm2) |
IQR (μm2) |
| 1 |
3955.2 |
14819.3 |
7.6 |
68.7 |
42.8 |
142.5 |
413.3 |
1653.1 |
181838.0 |
1510.6 |
| 2 |
6664.0 |
16918.4 |
4.7 |
26.9 |
42.8 |
185.3 |
691.2 |
4232.4 |
165207.5 |
4047.2 |
| 3 |
16946.9 |
33498.6 |
2.9 |
8.8 |
42.8 |
342.0 |
1560.4 |
16416.7 |
195404.6 |
16074.7 |
| 4 |
17714.5 |
33022.3 |
2.7 |
7.6 |
42.8 |
513.0 |
2850.1 |
15575.9 |
193438.0 |
15062.9 |
| 5 |
5017.6 |
11714.9 |
5.2 |
36.8 |
42.8 |
242.3 |
883.5 |
3947.4 |
135780.0 |
3705.2 |
| 6 |
11587.9 |
28326.5 |
3.5 |
12.4 |
42.8 |
185.3 |
733.9 |
6156.3 |
164110.2 |
5971.0 |
| 7 |
5608.8 |
15839.4 |
5.6 |
38.9 |
42.8 |
185.3 |
684.0 |
3206.4 |
188108.3 |
3021.1 |
| 8 |
7199.9 |
20483.3 |
5.1 |
30.3 |
42.8 |
213.8 |
783.8 |
4146.9 |
184702.4 |
3933.2 |
Table 4 Descriptive statistics of the diameter distributions for the different samples. The mean value, standard deviation (StD), minimum (Min.) and maximum (Max.) values, first (Q1) and third (Q3) quartiles, median value, and interquartile range (IQR) were calculated. Samples 3 and 4 resulted in higher diameters
| Sample |
Mean (μm) |
StD (μm) |
Skew (μm) |
Kurt (μm) |
Min. (μm) |
Q1 (μm) |
Median (μm) |
Q3 (μm) |
Max. (μm) |
IQR (μm) |
| 1 |
42.6 |
56.8 |
3.8 |
18.6 |
7.4 |
13.5 |
22.9 |
45.9 |
481.2 |
32.4 |
| 2 |
59.2 |
70.6 |
2.3 |
6.1 |
7.4 |
15.4 |
29.7 |
73.4 |
458.6 |
58.1 |
| 3 |
97.4 |
110.0 |
1.6 |
1.8 |
7.4 |
20.9 |
44.6 |
144.6 |
498.8 |
123.7 |
| 4 |
104.2 |
108.2 |
1.5 |
1.5 |
7.4 |
25.6 |
60.2 |
140.8 |
496.3 |
115.3 |
| 5 |
55.4 |
57.6 |
2.2 |
6.0 |
7.4 |
17.6 |
33.5 |
70.9 |
415.8 |
53.3 |
| 6 |
74.1 |
96.2 |
2.1 |
4.2 |
7.4 |
15.4 |
30.6 |
88.5 |
457.1 |
73.2 |
| 7 |
54.1 |
64.9 |
2.7 |
9.0 |
7.4 |
15.4 |
29.5 |
63.9 |
489.4 |
48.5 |
| 8 |
60.4 |
74.3 |
2.7 |
8.8 |
7.4 |
16.5 |
31.6 |
72.7 |
484.9 |
56.2 |
The mean and median diameter were modelled through a DoE approach, and the contour plots of the empirical models are shown in Fig. 8A (eqn (S4), ESI†) and Fig. 8B (eqn (S5), ESI†), respectively. The ANOVA tables (Tables S8 and S10, ESI†) within the coefficient tables (Tables S9 and S11, ESI†), and the actual versus predicted plots (Fig. S6 and S7, ESI†) are reported in the ESI.† The modelled trends of the mean and median values were almost the same. It should be noted that the model shows the presence of a saddle point at around 3% of LAP and 285 s of UV exposure. Tracing the two diagonals passing through the saddle point along one diagonal moving from the saddle point, it can be seen that the diameter increases. Meanwhile, moving on the other, the diameter decreases. In particular, the maximum diameter was obtained at a lower UV exposure and higher LAP and CsMA concentrations. A small increase in the diameter was detected, moving the CsMA concentration from 1% to 3%. It should be noted that models produced from the StD and the IQR (as a measure of the distribution dispersion) were not significant, and, were thus not reported for this reason.
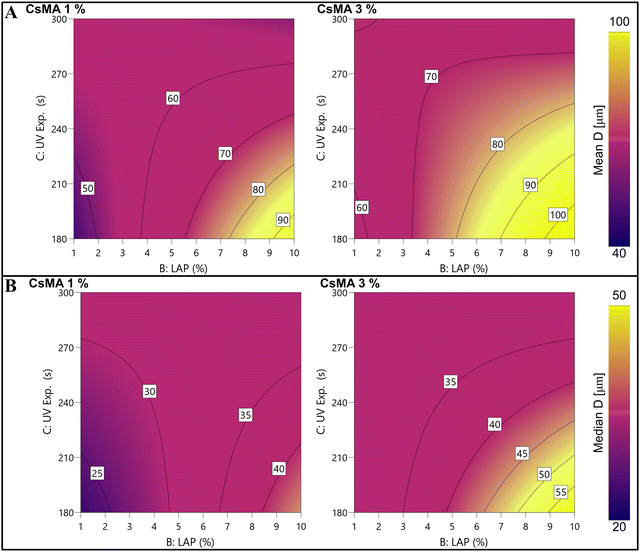 |
| | Fig. 8 (A) Contour plot of the empirical model of the mean diameter. A low UV exposure (180 s) and a high amount of LAP (10% w/w) gave the higher mean pore diameter. (B) Contour plot of the empirical model of the median diameter. As expected, the trend is almost the same as the modelled mean diameter. | |
3.4. Optical characterization
Optical characterization was conducted to verify the possibility of using the UV-vis spectrum to determine the protein concentration in the incubation solutions. Several solutions were tested, as shown in Fig. 9A, starting from a CsMA concentration of 0.1% up to 3.5%. The increasing concentration resulted in a denser solution with a more intense yellowish colour. Samples with higher concentrations were able to entrap the air bubbles formed during the CsMA dissolution. The UV-vis spectra of the solutions, shown in Fig. 9B, indicate that an increase in CsMA slightly decreased both the curve flatness and the percentage transmittance in the visible region (400–800 nm). However, all the samples had a transmittance higher than 80% in the Vis range, resulting in an acceptable transparency.
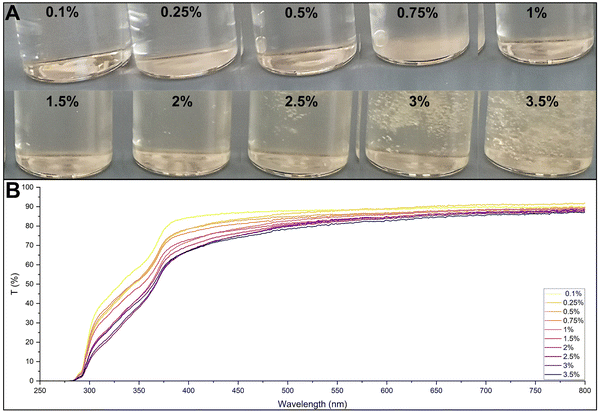 |
| | Fig. 9 (A) CsMA water solutions tested with a UV-vis spectrometer. The CsMA solutions resulted in a yellowish colour with an intensity that increased with an increase in CsMA concentration. In addition, the increase in concentration increased the solution viscosity, resulting in an entrapment of air bubbles. (B) UV-vis transmittance spectra: the increasing in concentration slightly reduced the transmittance in the visible region. Overall, in the visible region (400–800 nm), all samples showed a flat response with a transmittance higher than 80%. | |
3.5. Degradation and solubilization
The degradation/swelling and solubilization were determined by a single continuous experiment on the samples. The curves for the 8 samples are shown in Fig. 10. The tables of the mean values and the standard deviations of the bare weight loss, the degradation/swelling and the solubilization are reported in Tables S12–S14 (ESI†), respectively. We distinguished two types of mechanism. We called degradation/swelling (curve reported in Fig. 10A) the decomposition of the gel due to its breaking into pieces. These pieces were removed at each change of the supernatant (at each time point), and had a major impact on the gel decomposition. It should be noted that in terms of analysis, we could not distinguish this phenomenon from the gain in weight due to the absorption of PBS (swelling). So, we defined the negative percentages as swelling and the positive percentages as degradation. We called solubilization (curve reported in Fig. 10B) the amount of gel that was solubilized into the supernatant and could be detected through spectroscopy. Both degradation and decomposition were calculated as a relative percentage on the initial gel weight.
 |
| | Fig. 10 (A) Degradation of the CsMA hydrogels for up to 2 months; the magnified image shows the results of the first 5 hours. Negative results indicate an increase in the weight, which in the initial phases, can be attributed to the absorption of water. It should be noted that the hydrogel was broken into smaller pieces (missing piece highlighted by the black arrow). (B) Solubilization of the CsMA hydrogels evaluated by spectroscopy up to 2 months. (C) Contour plot of the empirical model of the solubilization. The increase in CsMA and LAP concentration slightly increased the solubilization, which remained below 2% overall. | |
The degradation was the driving force of the gel decomposition. At the end of 2 months (1440 h), the percentage of weight loss was between 38% (sample 8) and 57% (sample 3). It should be noted that in the first hours, some samples gained weight (magnification in Fig. 10A, a negative percentage corresponds to an increase in weight). This is because they were able to incorporate PBS in the first hours of incubation when the hydrogel degradation was not started yet. In two samples (sample 2 and sample 6) the incorporation of PBS was greater than the loss of weight, resulting in a net swelling (SWnet). In all the other cases, the weight loss exceeded the incorporation of PBS, resulting in a net degradation (Degnet). All the curves reached a plateau after 22 days (538 h). This plateau indicates an equilibrium between the lost weight and the incorporation of water. The large standard deviations indicate that this mechanism of degradation was barely controllable.
The solubilization (shown in Fig. 10B) increased with increasing time, spanning (1440 hours) from 0.5% (sample 1) to 2.1% (sample 4) after 2 months. The overall impact on the CsMA hydrogel decomposition was minor, considering the breaking of the gels (degradation). The standard deviations were smaller due to the intrinsic precision of the method that was not based on weight. We modelled the solubilization at 30 days (720 h). However, the result could be considered as representative of the overall behaviour, being the differences in term of solubilization among the different samples constant during time. The contour plot of the DoE model is shown in Fig. 10C. The corresponding eqn (S6) (ESI†) is reported in the ESI,† within the ANOVA (Table S15, ESI†), the coefficient (Table S16, ESI†), and the actual versus predicted plot is shown in Fig. S8 (ESI†). A higher concentration of CsMA and LAP resulted in a higher percentage of solubilized CsMA, while a higher curing time slightly decreased it.
4. Discussion
The control of the properties of a biomaterial, such as hydrogels for biomedical applications, is of vital importance to determine their possible usages. The use of statistical methods such as DoE and RSM allows for the development of empirical model equations that, in turn, allow for the design of the material properties, starting from the fabrication process by tuning some key parameters, i.e., compositions and physical factors. In this work, we applied these methodologies to study a known biomaterial, the methacrylated chitosan (CsMA), with the purpose of providing guidance on the material design. We firstly demonstrated that the reaction performed to modify the bare chitosan by introducing methacrylic groups was successful through IR spectroscopy, that revealed vibrations peaks related to MA groups added into the structure, as previously reported in other studies.45–47 We also hypothesized a possible mechanism behind the CsMA solubility in water by performing a molecular simulation, which revealed stronger intramolecular bonding in the case of Cs compared to CsMA. It should be noticed that the water solubility was also reported in the case of other reactions (as glycidyl methacrylate), providing the same functional groups.49 The solubility of CsMA can be related to the loss of the semicrystalline network of Cs due to the intra- and intermolecular hydrogen bonding,16,50 that were inhibited by the presence of the methacrylic groups. This effect is known not only for chitosan, but also for other biopolymers. For example, silk fibroin is not water-soluble due to its crystalline β secondary structure. However, once methacrylated, it increases its water solubility because of the partial impossibility to recover its crystallinity after the reaction.37
A series of solutions with increasing CsMA concentration were tested by rheometry, revealing a shear thinning behaviour at all tested temperatures. This feature is extremely important in applications that require an extrusion. In fact, shear thinning fluids become less viscous when a deforming force is applied, causing the fluid to flow more easily through the nozzle.51,52 This rheological behaviour is particularly desired in the case of bioprinting, allowing the reduction of the extrusion pressure, which is essential to assure a suitable cell viability.53 The produced RSM model of the viscosity allows us to predict the viscosity of the CsMA solution in advance by knowing the concentration, temperature, and shear rate. This will be extremely useful in adapting the material for a specific biological application.
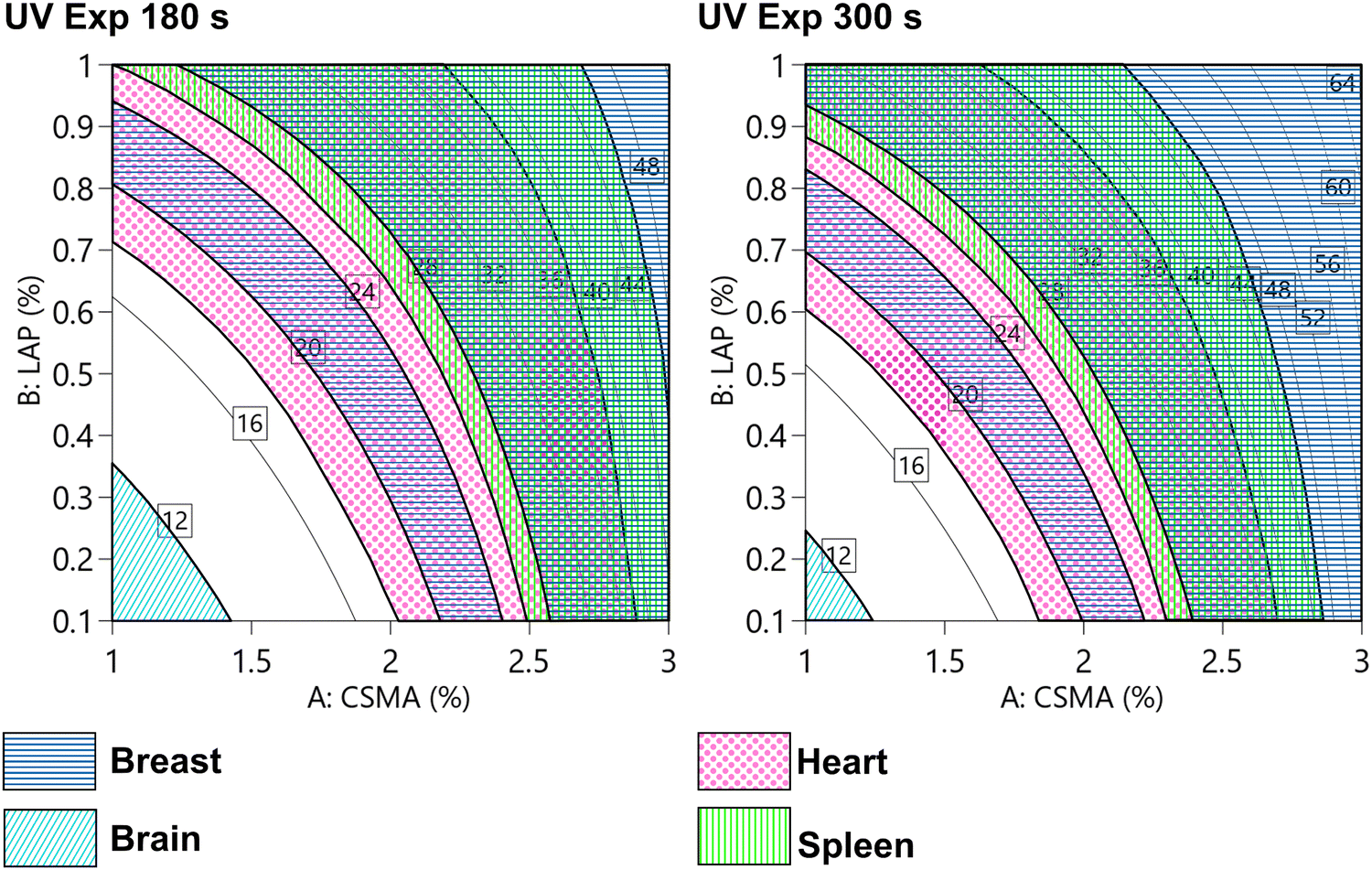
A full factorial experimental design was used to study the Young's modulus in compression based on the composition and the processing parameters. Interestingly, we were able to tune E on a wide range, spanning from 9 kPa to 65 kPa, just by tuning the composition and the UV exposure time. Chitosan radical crosslinked hydrogels were previously reported to allow a tuneable Young's modulus and to provide, under certain crosslinking conditions, a high mechanical strength (up to 30 kPa).54 Hybrid thermal-photo crosslinking,55 dual networks56,57 and the addition of charges58,59 were reported to enhance the compression modulus of chitosan hydrogels. The highest modulus reported in the literature is almost 300 kPa, obtained with a fine control over the dissolution mechanism and avoiding the use of acid solutions.60 The discrepancy with our highest value is justified by the solubilization of chitosan that we performed in an alkali solution. However, we were able to cover a wide range of elastic modulus with a single material, and to model it on the base of the fabrication parameters. In Table 5, we reported a comparison of several human tissues within their elastic modulus ranges (obtained by different methods).61–65 Some of these ranges were also visualized in Fig. 11, superimposed with the contour plot reported in Fig. 6E. It should be noted that in the literature, there is a plethora of different measurement protocols and systems that often disagree when applied on the same tissue. The reason behind this is the lack of standardization and the variability occurring in natural biological tissues. However, in the investigated factorial space, our hydrogel covers a wide range of possible applications. The lower stiffness was obtained with a low CsMA and LAP concentration, and the shorter exposure time is compatible with neural cell cultures and model brain-like tissue. Instead, the higher stiffness, which can be obtained by using a CsMA concentration higher than 2.5%, is appropriate for other tissues, such as breast cancer. Moving outside the considered ranges, increasing both concentrations (CsMA and LAP) and the exposure time, we may reach the requirements for muscular and bone tissues.66 The requirement to simulate the microenvironment for a precalcified bone tissue can be reached in the higher stiffness region of our hydrogel (>30 kPa). However, for the sake of clarity, it was not reported in the figure.66
Table 5 Elastic modulus of several human tissues measured using different methods. It should be noted that a standardized method to assess the mechanical properties of biological tissues does not exist. Several of the ranges reported herein resulted in being compatible with our hydrogel
| Tissue |
Modulus |
Test |
Ref. |
| Brain |
8.83 ± 1.94 kPa (30 mm s−1) |
Compression |
61
|
| 12.8 ± 3.10 kPa (60 mm s−1) |
| 16.0 ± 1.41 kPa (90 mm s−1) |
| Breast |
23.91 ± 4.57 kPa (healthy) |
Elastography |
62
|
| 39.4 ± 12.00 kPa (benign) |
| 55.4 ± 7.02 kPa (malign) |
| Heart |
1.27 ± 0.63 kPa (media) |
Tensile |
63
|
| 27.90 ± 10.59 kPa (intima) |
| Spleen |
14 ± 1.8 kPa (low strain) |
Compression |
64
|
| 35 ± 11 kPa (high stain) |
| Skeletal muscle |
447 ± 97.7 kPa (longitudinal) |
Tensile |
65
|
| 22.4 ± 14.7 kPa (transverse) |
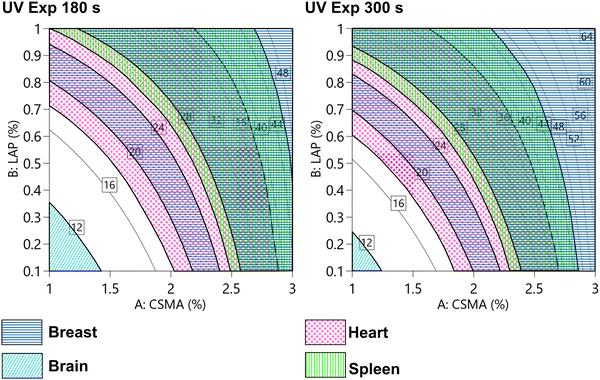 |
| | Fig. 11 Modelled compressive modulus (as reported in Fig. 5E) and compatible areas (shown as patterned areas) with different tissues, such as the breast, brain, heart, and spleen tissues. Other tissues, such as pre-calcified bone (>30 kDa), are not shown. The wide distribution of elastic modulus allows to design the hydrogel that can be adapted for different purposes. Scale reported in kPa. | |
The different hydrogels showed the same pore morphology in terms of shape factors (circularity and convexity), regardless of how the pores were affected dimensionally during the fabrication process. The dependence of the hydrogel microstructure on the biopolymer concentration was previously observed in several studies67,68 focused on the unmodified Cs, where the increase in concentration led to a decrease in the pore size. What we observed instead was an increase in the pore size due to the increase in concentration. This could be explained considering that our material is methacrylated and is stabilized by UV crosslinking. A higher concentration of CsMA ensured the presence of a higher amount of reactive sites, while a higher amount of LAP ensured a rapid reaction, improving the stability of larger pores. It should be noted that, to our knowledge, this is one of the first modelling studies of hydrogel porosity. The mean porosity was between 40 μm and 100 μm with a large distribution and a maximum pore diameter above 400 μm in all the tested conditions. This type of porosity is reported to be suitable for cell culturing. In fact, different pore sizes can influence the cell processes: the nanoporous membranes (100 nm) have been shown to promote the formation of collagen fibers and ECM, whereas macropores (>100 μm) play an important role in cell seeding, distribution, migration and further neo-vascularization in vivo.69,70
We were not able to detect a tangible swelling in the first hours, if not for a few samples, likely because of an equilibrium between the adsorbed and released water, impeding additional water to be retained. In literature, the decrease of the swelling ratio with the increase of the crosslinking of the hydrogels is well reported,71–73 and, in some cases, even a lack of swelling has been observed.74 This can be explained by considering the chemical crosslinking bonds created during the UV exposure that stiffen the structure, allowing for the shape preservation.71–73 The deterioration of the samples was mainly due to breakage into smaller pieces (we referred to this phenomenon as degradation), instead of the solubilization of the material into the testing media. Interestingly, almost all the tested hydrogels were not completely degraded after 2 months, which suggests the possibility of using them for long-term experiments.
5. Conclusion
CsMA is a chemically modified Cs, in which methacrylic groups are added to make Cs crosslinkable upon exposure to UV light. This may be particularly useful in applications in which the mechanical behaviour should be accurately controlled, as in the case of mechanobiology, and in adapting the material to address the requirements of a specific cell typology. In this study, we synthesize CsMA, verified the successful functionalization and empirically modelled its properties. We demonstrated the shear thinning behaviour of the CsMA solutions, and then their viscosity was modelled as a function of CsMA concentration, temperature and shear rate, providing a useful tool to design CsMA solutions according to the needs of extrusion based bioprinting. Starting from different compositions (in terms of CsMA concentration and photoinitiator amount) and UV exposure times, we studied and statistically modelled the CsMA hydrogel properties in terms of mechanical response, morphological characteristics, degradation and solubilization. The CsMA hydrogel properties were proven to be highly tuneable, and we were able to hypothesize several possible applications by matching the hydrogel compressive modulus to the modulus reported in the literature for different biological tissues. In particular, the compression modulus was evaluated in the range of 10–65 kPa, which is suitable for mimicking the mechanical properties of a range of biological tissues, such as the breast, brain, heart, spleen and precalcified bone. The mean pore diameter varied between 40 μm and 100 μm with highly skewed distributions with a portion of the diameters in the range of 200–400 μm, which is suitable for cell culturing. All tested hydrogels were not completely degraded after 60 days in PBS at 37 °C. Also in this case, we were able to model the mean, the median pore diameter, and the solubilization as a function of the fabrication process parameters. For the other considered properties (the standard deviation and interquartile range of the diameter distributions, and the degradation), we could not interpolate a model because there was no statistical difference among the 8 different samples. This indicates that we still have poor control over these yields. The main limitation of this study is the lack of a biological validation that will be assessed in our next work, to verify the possibility of using CsMA as a platform for different cell cultures and tissue engineering applications.
Author contributions
A. B., F. G, conceptualization. A. B., methodology. A. B., validation. A. B., N. S., C. C., A. C. L., F. G., formal analysis. A. B., N. S., C. C., A. R., A. C. L, F. G., investigation. A. B., N. S., C. C., data curation. A. B., writing – original draft. A. B., N. S., C. C., A. P., A. R., A. C. L., F. G., writing – review & editing. A. B., visualization. A. P., A. R., L. M., F. G., Supervision. A. P., L. M., F. G., project administration. A. P., A. R., L. M., G. G. Funding acquisition.
Data availability
Data for this article, including raw data and videos, are available at https://osf.io at DOI: https://doi.org/10.17605/OSF.IO/F2ZNB.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are grateful to the “Tecnopolo per la medicina di precisione” (TecnoMed Puglia) – Regione Puglia: DGR n.2117 del 21/11/2018, CUP: B84I18000540002 and “Tecnopolo di Nanotecnologia e Fotonica per la medicina di precisione” (TECNOMED) – FISR/MIUR-CNR: delibera CIPE n.3449 del 7-08-2017, CUP: B83B17000010001, and to the Italian Ministry of Research (MUR) under the Project PON-SHINE (POTENZIAMENTO DEI NODI ITALIANI IN E-RIHS, CIG: Z902D25DCE – CUP: B27E19000030007) and the complementary actions to the NRRP “FIT4MedRob” Grant (contract number CUP B53C22006960001) and “D34 Health” (contract number, CUP B53C22006100001). A. P. and F. G. gratefully acknowledge the support from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 953121 (FLAMIN-GO). A. B. was supported by IRCCS Istituto Ortopedico Rizzoli “Ricerca Corrente”.
References
- E. M. Ahmed, Hydrogel: preparation, characterization, and applications: a review, J. Adv. Res., 2015, 6, 105–121, DOI:10.1016/j.jare.2013.07.006.
- A. S. Hoffman, Hydrogels for biomedical applications, Adv. Drug Delivery Rev., 2012, 64, 18–23, DOI:10.1016/j.addr.2012.09.010.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Smart Hydrogels in Tissue Engineering and Regenerative Medicine, Materials, 2019, 12, 3323, DOI:10.3390/ma12203323.
- E. González-Díaz and S. Varghese, Hydrogels as Extracellular Matrix Analogs, Gels, 2016, 2, 20, DOI:10.3390/gels2030020.
- X. Xue, Y. Hu, S. Wang, X. Chen, Y. Jiang and J. Su, Fabrication of physical and chemical crosslinked hydrogels for bone tissue engineering, Bioact. Mater., 2022, 12, 327–339, DOI:10.1016/j.bioactmat.2021.10.029.
- Z. Ahmad, S. Salman, S. A. Khan, A. Amin, Z. U. Rahman, Y. O. Al-Ghamdi, K. Akhtar, E. M. Bakhsh and S. B. Khan, Versatility of Hydrogels: From Synthetic Strategies, Classification, and Properties to Biomedical Applications, Gels, 2022, 8, 167, DOI:10.3390/gels8030167.
- A. Mahmood, D. Patel, B. Hickson, J. DesRochers and X. Hu, Recent Progress in Biopolymer-Based Hydrogel Materials for Biomedical Applications, Int. J. Mol. Sci., 2022, 23, 1415, DOI:10.3390/ijms23031415.
- R. Eivazzadeh-Keihan, E. B. Noruzi, S. F. Mehrban, H. A. M. Aliabadi, M. Karimi, A. Mohammadi, A. Maleki, M. Mahdavi, B. Larijani and A. E. Shalan, Review: the latest advances in biomedical applications of chitosan hydrogel as a powerful natural structure with eye-catching biological properties, J. Mater. Sci., 2022, 57, 3855–3891, DOI:10.1007/s10853-021-06757-6.
- I. Aranaz, A. R. Alcántara, M. C. Civera, C. Arias, B. Elorza, A. Heras Caballero and N. Acosta, Chitosan: An Overview of Its Properties and Applications, Polymers, 2021, 13, 3256, DOI:10.3390/polym13193256.
- S. (Gabriel) Kou, L. Peters and M. Mucalo, Chitosan: a review of molecular structure, bioactivities and interactions with the human body and micro-organisms, Carbohydr. Polym., 2022, 282, 119132, DOI:10.1016/j.carbpol.2022.119132.
- V. Zargar, M. Asghari and A. Dashti, A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications, ChemBioEng Rev., 2015, 2, 204–226, DOI:10.1002/cben.201400025.
- A. D. Gholap, S. Rojekar, H. S. Kapare, N. Vishwakarma, S. Raikwar, A. Garkal, T. A. Mehta, H. Jadhav, M. K. Prajapati and U. Annapure, Chitosan scaffolds: expanding horizons in biomedical applications, Carbohydr. Polym., 2024, 323, 121394, DOI:10.1016/j.carbpol.2023.121394.
- G. Morello, G. De Iaco, G. Gigli, A. Polini and F. Gervaso, Chitosan and Pectin Hydrogels for Tissue Engineering and In Vitro Modeling, Gels, 2023, 9, 132, DOI:10.3390/gels9020132.
- B. Canciani, F. Semeraro, V. R. Herrera Millar, F. Gervaso, A. Polini, A. Stanzione, G. M. Peretti, A. Di Giancamillo and L. Mangiavini, In Vitro and In Vivo Biocompatibility Assessment of a Thermosensitive Injectable Chitosan-Based Hydrogel for Musculoskeletal Tissue Engineering, Int. J. Mol. Sci., 2023, 24, 10446, DOI:10.3390/ijms241310446.
- F. Bisconti, B. Vilardo, G. Corallo, F. Scalera, G. Gigli, A. Chiocchetti, A. Polini and F. Gervaso, An Assist for Arthritis Studies: A
3D Cell Culture of Human Fibroblast-Like Synoviocytes by Encapsulation in a Chitosan-Based Hydrogel, Adv. Ther., 2024, 2400166, DOI:10.1002/adtp.202400166.
- C. Qin, H. Li, Q. Xiao, Y. Liu, J. Zhu and Y. Du, Water-solubility of chitosan and its antimicrobial activity, Carbohydr. Polym., 2006, 63, 367–374, DOI:10.1016/j.carbpol.2005.09.023.
- S.-H. Lim and S. M. Hudson, Synthesis and antimicrobial activity of a water-soluble chitosan derivative with a fiber-reactive group, Carbohydr. Res., 2004, 339, 313–319, DOI:10.1016/j.carres.2003.10.024.
- C. Bergonzi, A. Bianchera, G. Remaggi, M. C. Ossiprandi, R. Bettini and L. Elviri, 3D Printed Chitosan/Alginate Hydrogels for the Controlled Release of Silver Sulfadiazine in Wound Healing Applications: Design, Characterization and Antimicrobial Activity, Micromachines, 2023, 14, 137, DOI:10.3390/mi14010137.
- F. Scalera, S. I. A. Pereira, A. Bucciarelli, D. M. Tobaldi, A. Quarta, F. Gervaso, P. M. L. Castro, A. Polini and C. Piccirillo, Chitosan-hydroxyapatite composites made from sustainable sources: a morphology and antibacterial study, Mater. Today Sustainability, 2023, 21, 100334, DOI:10.1016/j.mtsust.2023.100334.
- M. L. Pita-López, G. Fletes-Vargas, H. Espinosa-Andrews and R. Rodríguez-Rodríguez, Physically cross-linked chitosan-based hydrogels for tissue engineering applications: a state-of-the-art review, Eur. Polym. J., 2021, 145, 110176, DOI:10.1016/j.eurpolymj.2020.110176.
- J. Maitra and V. K. Shukla, Cross-linking in Hydrogels – A Review, Am. J. Polym. Sci., 2014, 4(2), 25–31, DOI:10.5923/j.ajps.20140402.01.
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Advances in crosslinking strategies of biomedical hydrogels, Biomater. Sci., 2019, 7, 843–855, 10.1039/C8BM01246F.
- B. G. Amsden, A. Sukarto, D. K. Knight and S. N. Shapka, Methacrylated Glycol Chitosan as a Photopolymerizable Biomaterial, Biomacromolecules, 2007, 8, 3758–3766, DOI:10.1021/bm700691e.
- Y. Zhou, K. Liang, C. Zhang, J. Li, H. Yang, X. Liu, X. Yin, D. Chen, W. Xu and P. Xiao, Photocrosslinked methacrylated chitosan-based nanofibrous scaffolds as potential skin substitute, Cellulose, 2017, 24, 4253–4262, DOI:10.1007/s10570-017-1433-4.
- B. Li, L. Wang, F. Xu, X. Gang, U. Demirci, D. Wei, Y. Li, Y. Feng, D. Jia and Y. Zhou, Hydrosoluble, UV-crosslinkable and injectable chitosan for patterned cell-laden microgel and rapid transdermal curing hydrogel in vivo, Acta Biomater., 2015, 22, 59–69, DOI:10.1016/j.actbio.2015.04.026.
- Y. Shen, H. Tang, X. Huang, R. Hang, X. Zhang, Y. Wang and X. Yao, DLP printing photocurable chitosan to build bio-constructs for tissue engineering, Carbohydr. Polym., 2020, 235, 115970, DOI:10.1016/j.carbpol.2020.115970.
- T. Cebe, N. Ahuja, F. Monte, K. Awad, K. Vyavhare, P. Aswath, J. Huang, M. Brotto and V. Varanasi, Novel 3D-printed methacrylated chitosan-laponite nanosilicate composite scaffolds enhance cell growth and biomineral formation in MC3T3 pre-osteoblasts, J. Mater. Res., 2020, 35, 58–75, DOI:10.1557/jmr.2018.260.
- C. Tonda-Turo, I. Carmagnola, A. Chiappone, Z. Feng, G. Ciardelli, M. Hakkarainen and M. Sangermano, Photocurable chitosan as bioink for cellularized therapies towards personalized scaffold architecture, Bioprinting, 2020, 18, e00082, DOI:10.1016/j.bprint.2020.e00082.
- C. M. Valmikinathan, V. J. Mukhatyar, A. Jain, L. Karumbaiah, M. Dasari and R. V. Bellamkonda, Photocrosslinkable chitosan based hydrogels for neural tissue engineering, Soft Matter, 2012, 8, 1964–1976, 10.1039/C1SM06629C.
- E. Barbu, L. Verestiuc, M. Iancu, A. Jatariu, A. Lungu and J. Tsibouklis, Hybrid polymeric hydrogels for ocular drug delivery: nanoparticulate systems from copolymers of acrylic acid-functionalized chitosan and N-isopropylacrylamide or 2-hydroxyethyl methacrylate, Nanotechnology, 2009, 20, 225108, DOI:10.1088/0957-4484/20/22/225108.
- J. Han, K. Wang, D. Yang and J. Nie, Photopolymerization of methacrylated chitosan/PNIPAAm hybrid dual-sensitive hydrogels as carrier for drug delivery, Int. J. Biol. Macromol., 2009, 44, 229–235, DOI:10.1016/j.ijbiomac.2008.12.009.
- Z.-K. Cui, S. Kim, J. J. Baljon, B. M. Wu, T. Aghaloo and M. Lee, Microporous methacrylated glycol chitosan-montmorillonite nanocomposite hydrogel for bone tissue engineering, Nat. Commun., 2019, 10, 3523, DOI:10.1038/s41467-019-11511-3.
- S. Maiz-Fernández, L. Pérez-Álvarez, U. Silván, J. L. Vilas-Vilela and S. Lanceros-Mendez, Photocrosslinkable and self-healable hydrogels of chitosan and hyaluronic acid, Int. J. Biol. Macromol., 2022, 216, 291–302, DOI:10.1016/j.ijbiomac.2022.07.004.
-
D. C. Montgomery, Design and Analysis of Experiments, 8th edn, 2012 DOI:10.1198/tech.2006.s372.
- A. Bucciarelli, E. E. E. Olivetti, A. Adami and L. Lorenzelli, Design of Experiment Rational Optimization of an Inkjet Deposition of Silver on Kapton, IEEE Sens. J., 2021, 21, 26304–26310, DOI:10.1109/JSEN.2021.3058543.
- A. Bucciarelli, G. Greco, I. Corridori, A. Motta and N. M. Pugno, Tidy dataset of the experimental design of the optimization of the alkali degumming process of Bombyx mori silk, Data Brief, 2021, 38, 107294, DOI:10.1016/J.DIB.2021.107294.
- A. Bucciarelli, T. Muthukumar, J. S. Kim, W. K. Kim, A. Quaranta, D. Maniglio, G. Khang and A. Motta, Preparation and Statistical Characterization of Tunable Porous Sponge Scaffolds using UV Cross-linking of Methacrylate-Modified Silk Fibroin, ACS Biomater. Sci. Eng., 2019, 5, 6374–6388, DOI:10.1021/acsbiomaterials.9b00814.
- F. Perin, E. Spessot, A. Famà, A. Bucciarelli, E. Callone, C. Mota, A. Motta and D. Maniglio, Modeling a Dynamic Printability Window on Polysaccharide Blend Inks for Extrusion Bioprinting, ACS Biomater. Sci. Eng., 2023, 9(3), 1320–1331, DOI:10.1021/acsbiomaterials.2c01143.
- M. Fernandes Queiroz, K. Melo, D. Sabry, G. Sassaki and H. Rocha, Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation?, Mar. Drugs, 2014, 13, 141–158, DOI:10.3390/md13010141.
- R. F. Melo-Silveira, G. P. Fidelis, M. S. S. P. Costa, C. B. S. Telles, N. Dantas-Santos, S. de, O. Elias, V. B. Ribeiro, A. L. Barth, A. J. Macedo, E. L. Leite and H. A. O. Rocha, In Vitro Antioxidant, Anticoagulant and Antimicrobial Activity and in Inhibition of Cancer Cell Proliferation by Xylan Extracted from Corn Cobs, Int. J. Mol. Sci., 2011, 13, 409–426, DOI:10.3390/ijms13010409.
- W. F. Wolkers, A. E. Oliver, F. Tablin and J. H. Crowe, A Fourier-transform infrared spectroscopy study of sugar glasses, Carbohydr. Res., 2004, 339, 1077–1085, DOI:10.1016/j.carres.2004.01.016.
- F. R. F. Silva, C. M. P. G. Dore, C. T. Marques, M. S. Nascimento, N. M. B. Benevides, H. A. O. Rocha, S. F. Chavante and E. L. Leite, Anticoagulant activity, paw edema and pleurisy induced carrageenan: action of major types of commercial carrageenans, Carbohydr. Polym., 2010, 79, 26–33, DOI:10.1016/j.carbpol.2009.07.010.
- A. B. Vino, P. Ramasamy, V. Shanmugam and A. Shanmugam, Extraction, characterization and in vitro antioxidative potential of chitosan and sulfated chitosan from Cuttlebone of Sepia aculeata Orbigny, 1848, Asian Pac. J. Trop. Biomed., 2012, 2, S334–S341, DOI:10.1016/S2221-1691(12)60184-1.
- C. Song, H. Yu, M. Zhang, Y. Yang and G. Zhang, Physicochemical properties and antioxidant activity of chitosan from the blowfly Chrysomya megacephala larvae, Int. J. Biol. Macromol., 2013, 60, 347–354, DOI:10.1016/j.ijbiomac.2013.05.039.
- D. Sugumaran, K. Juhanni and A. Karim, Removal of copper(II) ion using chitosan-graft-poly(methyl methacrylate) as adsorbent, Eproc. Chem., 2017, 2, 1–11 Search PubMed (accessed February 2, 2022).
- S. Derkaoui, D. E. Kherroub and M. Belbachir, Green synthesis, anionic polymerization of 1,4-bis(methacryloyl)piperazine using Algerian clay as catalyst, Green Process Synth., 2019, 8, 611–621, DOI:10.1515/gps-2019-0031.
- T. Qu, X. Duan, X. Pang, J. Huang, L. Gao, J. Wang, Y. Zhu, X. Liang, L. Chen, K. Zhang, H. Wang and J. Huang, Commonalities and characteristics of aqueous extracts from three Uighur medicines were analyzed by using three-stage infrared spectroscopy combined with ultra-performance liquid chromatography-time of flight-mass spectra, J. Tradit. Chin. Med. Sci., 2019, 39, 118–126 Search PubMed.
- S. C. Lee, G. Gillispie, P. Prim and S. J. Lee, Physical and Chemical Factors Influencing the Printability of Hydrogel-based Extrusion Bioinks, Chem. Rev., 2020, 120, 10834–10886, DOI:10.1021/acs.chemrev.0c00015.
- B. G. Amsden, A. Sukarto, D. K. Knight and S. N. Shapka, Methacrylated Glycol Chitosan as a Photopolymerizable Biomaterial, Biomacromolecules, 2007, 8, 3758–3766, DOI:10.1021/bm700691e.
- I. A. Sogias, V. V. Khutoryanskiy and A. C. Williams, Exploring the Factors Affecting the Solubility of Chitosan in Water, Macromol. Chem. Phys., 2010, 211, 426–433, DOI:10.1002/macp.200900385.
- F. Perin, E. Spessot, A. Famà, A. Bucciarelli, E. Callone, C. Mota, A. Motta and D. Maniglio, Modeling a Dynamic Printability Window on Polysaccharide Blend Inks for Extrusion Bioprinting, ACS Biomater. Sci. Eng., 2023, 9, 1320–1331, DOI:10.1021/acsbiomaterials.2c01143.
- R. Sánchez-Sánchez, J. M. Rodríguez-Rego, A. Macías-García, L. Mendoza-Cerezo and A. Díaz-Parralejo, Relationship between shear-thinning rheological properties of bioinks and bioprinting parameters, Int. J. Bioprint., 2023, 9(2), 687, DOI:10.18063/ijb.687.
- S. Boularaoui, G. Al Hussein, K. A. Khan, N. Christoforou and C. Stefanini, An overview of extrusion-based bioprinting with a focus on induced shear stress and its effect on cell viability, Bioprinting, 2020, 20, e00093, DOI:10.1016/j.bprint.2020.e00093.
- A. A. Nada, E. A. Ali and A. A. F. Soliman, Biocompatible chitosan-based hydrogel with tunable mechanical and physical properties formed at body temperature, Int. J. Biol. Macromol., 2019, 131, 624–632, DOI:10.1016/j.ijbiomac.2019.03.093.
- J. W. Seo, S. R. Shin, M.-Y. Lee, J. M. Cha, K. H. Min, S. C. Lee, S. Y. Shin and H. Bae, Injectable hydrogel derived from chitosan with tunable mechanical properties via hybrid-crosslinking system, Carbohydr. Polym., 2021, 251, 117036, DOI:10.1016/j.carbpol.2020.117036.
- Y. Wen, F. Li, C. Li, Y. Yin and J. Li, High mechanical strength chitosan-based hydrogels cross-linked with poly(ethylene glycol)/polycaprolactone micelles for the controlled release of drugs/growth factors, J. Mater. Chem. B, 2017, 5, 961–971, 10.1039/C6TB02201D.
- G. Morello, A. Polini, F. Scalera, R. Rizzo, G. Gigli and F. Gervaso, Preparation and Characterization of Salt-Mediated Injectable Thermosensitive Chitosan/Pectin Hydrogels for Cell Embedding and Culturing, Polymers, 2021, 13, 2674, DOI:10.3390/polym13162674.
- P. Panyamao, W. Ruksiriwanich, P. Sirisa-ard and S. Charumanee, Injectable Thermosensitive Chitosan/Pullulan-Based Hydrogels with Improved Mechanical Properties and Swelling Capacity, Polymers, 2020, 12, 2514, DOI:10.3390/polym12112514.
- K. Kosowska, P. Domalik-Pyzik, M. Sekuła-Stryjewska, S. Noga, J. Jagiełło, M. Baran, L. Lipińska, E. Zuba-Surma and J. Chłopek, Gradient Chitosan Hydrogels Modified with Graphene Derivatives and Hydroxyapatite: Physiochemical Properties and Initial Cytocompatibility Evaluation, Int. J. Mol. Sci., 2020, 21, 4888, DOI:10.3390/ijms21144888.
- L. Zhou, H. Ramezani, M. Sun, M. Xie, J. Nie, S. Lv, J. Cai, J. Fu and Y. He, 3D printing of high-strength chitosan hydrogel scaffolds without any organic solvents, Biomater. Sci., 2020, 8, 5020–5028, 10.1039/D0BM00896F.
- B. Rashid, M. Destrade and M. D. Gilchrist, Mechanical characterization of brain tissue in compression at dynamic strain rates, J. Mech. Behav. Biomed. Mater., 2012, 10, 23–38, DOI:10.1016/j.jmbbm.2012.01.022.
-
A. C. Saavedra, J. Guerrero, P. Montenegro, J. Aguilar, J. A. Pinto, J. Lobo, T. Salcudean, R. Lavarello, B. Castañeda and J. Arroyo, Breast elastography: identification of benign and malignant cancer based on absolute elastic modulus measurement using vibro-elastography, in Medical Imaging 2018: Image Perception, Observer Performance, and Technology Assessment, ed. R. M. Nishikawa and F. W. Samuelson, SPIE, 2018, p. 48. DOI:10.1117/12.2293664.
- G. A. Holzapfel, G. Sommer, C. T. Gasser and P. Regitnig, Determination of layer-specific mechanical properties of human coronary arteries with nonatherosclerotic intimal thickening and related constitutive modeling, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289, H2048–H2058, DOI:10.1152/ajpheart.00934.2004.
- S. Umale, C. Deck, N. Bourdet, P. Dhumane, L. Soler, J. Marescaux and R. Willinger, Experimental mechanical characterization of abdominal organs: liver, kidney & spleen, J. Mech. Behav. Biomed. Mater., 2013, 17, 22–33, DOI:10.1016/j.jmbbm.2012.07.010.
- D. A. Morrow, T. L. Haut Donahue, G. M. Odegard and K. R. Kaufman, Transversely isotropic tensile material properties of skeletal muscle tissue, J. Mech. Behav. Biomed. Mater., 2010, 3, 124–129, DOI:10.1016/j.jmbbm.2009.03.004.
- L. R. Smith, S. Cho and D. E. Discher, Stem Cell Differentiation is Regulated by Extracellular Matrix Mechanics, Physiology, 2018, 33, 16–25, DOI:10.1152/physiol.00026.2017.
- P. Sánchez-Cid, M. Jiménez-Rosado, M. Alonso-González, A. Romero and V. Perez-Puyana, Applied Rheology as Tool for the Assessment of Chitosan Hydrogels for Regenerative Medicine, Polymers, 2021, 13, 2189, DOI:10.3390/polym13132189.
- C. Li, Q. Dang, Q. Yang, D. Chen, H. Zhu, J. Chen, R. Liu and X. Wang, Study of the microstructure of chitosan aerogel beads prepared by supercritical CO 2 drying and the effect of long-term storage, RSC Adv., 2022, 12, 21041–21049, 10.1039/D2RA01875F.
- I. Bružauskaitė, D. Bironaitė, E. Bagdonas and E. Bernotienė, Scaffolds and cells for tissue regeneration: different scaffold pore sizes—different cell effects, Cytotechnology, 2016, 68, 355–369, DOI:10.1007/s10616-015-9895-4.
- V. Karageorgiou and D. Kaplan, Porosity of 3D biomaterial scaffolds and osteogenesis, Biomaterials, 2005, 26(27), 5474–5491, DOI:10.1016/j.biomaterials.2005.02.002.
- K. Zhang, S. Lin, Q. Feng, C. Dong, Y. Yang, G. Li and L. Bian, Nanocomposite hydrogels stabilized by self-assembled multivalent bisphosphonate-magnesium nanoparticles mediate sustained release of magnesium ion and promote in situ bone regeneration, Acta Biomater., 2017, 64, 389–400, DOI:10.1016/j.actbio.2017.09.039.
- S. Xiang, W. Qian, T. Li, Y. Wang, M. Chen, P. Ma and W. Dong, Hierarchical structural double network hydrogel with high strength, toughness, and good recoverability, New J. Chem., 2017, 41, 14397–14402, 10.1039/C7NJ03263C.
- T. Begam, A. K. Nagpal and R. Singhal, A study on copolymeric hydrogels based on acrylamide-methacrylate and its modified vinyl-amine-containing derivative, Des. Monomers Polym., 2004, 7, 311–330, DOI:10.1163/1568555041475329.
- Q. Xing, K. Yates, C. Vogt, Z. Qian, M. C. Frost and F. Zhao, Increasing Mechanical Strength of Gelatin Hydrogels by Divalent Metal Ion Removal, Sci. Rep., 2014, 4, 4706, DOI:10.1038/srep04706.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Nora
Selicato
b,
Chiara
Coricciati
bc,
Alberto
Rainer
a,
Nora
Selicato
b,
Chiara
Coricciati
bc,
Alberto
Rainer