
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in catalytic and non-catalytic epoxidation of terpenes: a pathway to bio-based polymers from waste biomass
Mohamad Faiz Mukhtar Gunam Resulab,
Abdul Rehman *ac,
Faisal Saleemac,
Muhammd Usmanc,
Ana María López Fernándeza,
Valentine C. Ezea and
Adam P. Harveya
*ac,
Faisal Saleemac,
Muhammd Usmanc,
Ana María López Fernándeza,
Valentine C. Ezea and
Adam P. Harveya
aSchool of Engineering, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
bDepartment of Chemical and Environmental Engineering, Faculty of Engineering, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
cDepartment of Chemical and Polymer Engineering, University of Engineering and Technology Lahore, Faisalabad Campus, Pakistan. E-mail: a.rehman2@uet.edu.pk
First published on 8th November 2023
Abstract
Epoxides derived from waste biomass are a promising avenue for the production of bio-based polymers, including polyamides, polyesters, polyurethanes, and polycarbonates. This review article explores recent efforts to develop both catalytic and non-catalytic processes for the epoxidation of terpene, employing a variety of oxidizing agents and techniques for process intensification. Experimental investigations into the epoxidation of limonene have shown that these methods can be extended to other terpenes. To optimize the epoxidation of bio-based terpene, there is a need to develop continuous processes that address limitations in mass and heat transfer. This review discusses flow chemistry and innovative reactor designs as part of a multi-scale approach aimed at industrial transformation. These methods facilitate continuous processing, improve mixing, and either eliminate or reduce the need for solvents by enhancing heat transfer capabilities. Overall, the objective of this review is to contribute to the development of commercially viable processes for producing bio-based epoxides from waste biomass.
1. Introduction
Terpenes are multifaceted compounds amenable to selective functionalization via epoxidation, thereby yielding a broad spectrum of molecules with diverse properties. Epoxides, characterized by their oxirane rings and high reactivity, hold a pivotal role in both the polymer industry and broader chemical synthesis. Traditionally, petrochemical-derived epoxides such as cyclohexene oxide, ethylene oxide, styrene oxide, and propylene oxide have been extensively employed in a range of industrial applications, including textiles, electronics, and packaging. However, there is an escalating demand for the development of alternative strategies that leverage waste biomass as a feedstock, aiming to mitigate environmental impact.1Waste biomass serves as a renewable feedstock and presents a promising avenue for the production of bio-based polymers, including polyamides, polyesters, polyurethanes, and polycarbonates (Fig. 1). Lignocellulosic biomass, a major constituent of waste biomass, has garnered significant attention in this domain. Moreover, terpenes, a prevalent class of natural compounds, can be sourced from waste byproducts of forestry and agricultural activities to produce unsaturated monoterpenes. For instance, the pulp and paper industry generates α-/β-pinene, while limonene is a byproduct of the citrus juice manufacturing sector.2
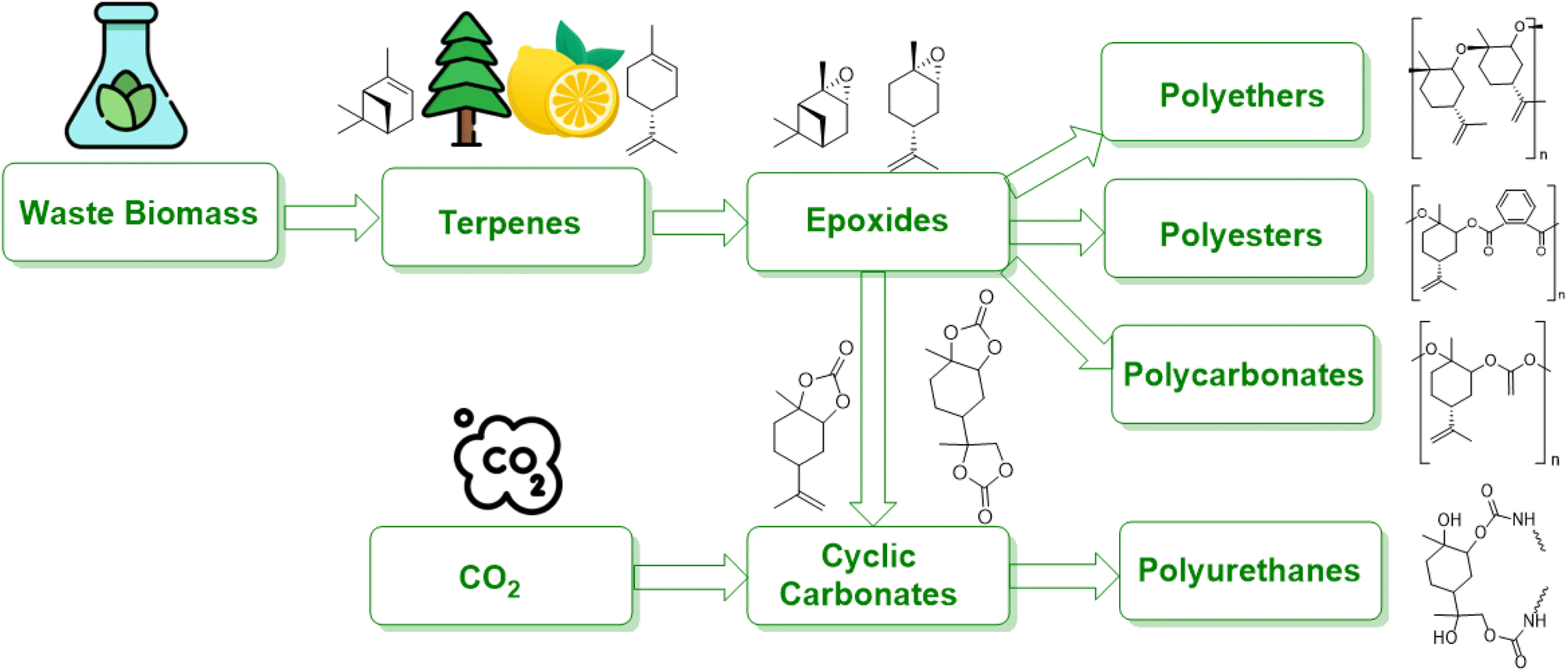 | ||
| Fig. 1 Multiple routes to polymerize epoxides derived from biomass. | ||
Among renewable monoterpenes, turpentine is particularly noteworthy as it is the most abundant byproduct of the Kraft paper pulping process, with the annual production of crude sulfate turpentine (CST) estimated at 230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes. Additionally, the distillation of resin from forest plants yields approximately 100000 tonnes of gum turpentine annually. These significant monoterpene feedstocks present an excellent opportunity for the development of efficient catalytic methods to convert them into versatile synthetic intermediates.3,4 Enhancing the range of bio-renewable products generated by terpene biorefineries can improve the economic feasibility of these processes, while simultaneously reducing waste and overall carbon dioxide (CO2) emissions.3
000 tonnes. Additionally, the distillation of resin from forest plants yields approximately 100000 tonnes of gum turpentine annually. These significant monoterpene feedstocks present an excellent opportunity for the development of efficient catalytic methods to convert them into versatile synthetic intermediates.3,4 Enhancing the range of bio-renewable products generated by terpene biorefineries can improve the economic feasibility of these processes, while simultaneously reducing waste and overall carbon dioxide (CO2) emissions.3
Terpenes sourced from biomass, such as (R)-(+)-limonene and α-pinene (as illustrated in Scheme 1), serve as valuable resources with wide applications in flavourings, fragrances, and pharmaceuticals.2,5–7 Beyond these traditional uses, these terpenes can also be transformed through chemical synthesis into other beneficial compounds. The epoxidation of waste terpenes like limonene-1,2-epoxide and α-pinene oxide offers an economically viable and environmentally sustainable approach for generating valuable intermediates for diverse industrial applications.8,9 For example, limonene-1,2-epoxide has demonstrated its potential as a monomer in the synthesis of limonene polycarbonate, while α-pinene oxide can be isomerized to produce campholenic aldehyde, a key ingredient in the fragrance industry.4,10
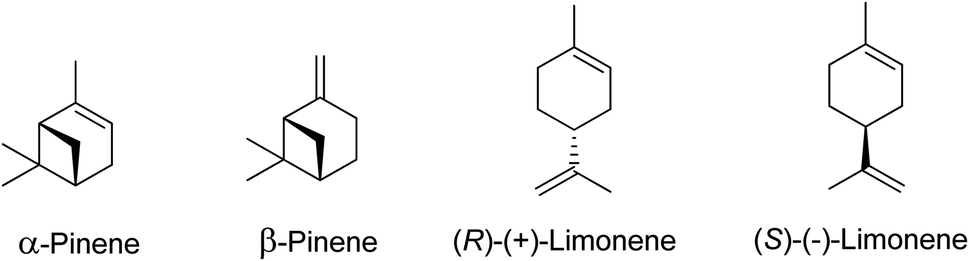 | ||
| Scheme 1 Common monoterpenes found in turpentine oil. | ||
Despite growing environmental concerns, current industrial methods for producing epoxides often rely on outdated techniques that use harmful chemicals, leading to issues of toxicity and stability. As a result, there has been an increasing focus on research into the epoxidation of (R)-(+)-limonene and α-pinene using environmentally benign oxidants like hydrogen peroxide (H2O2).11–21 Recognized as the ‘greenest’ terminal oxidant, hydrogen peroxide generates only water (H2O) as a by-product, as illustrated in Scheme 2. Consequently, the development of cleaner epoxidation methods, particularly those based on hydrogen peroxide, has become a significant area of research in recent years.
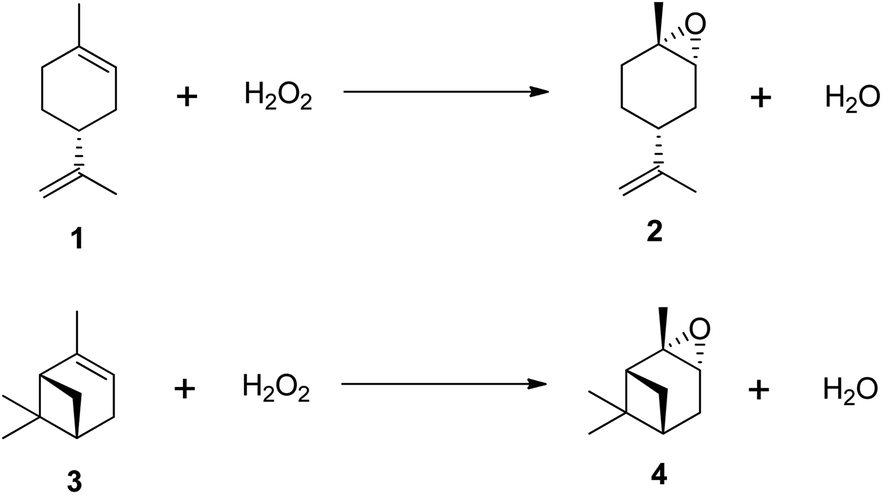 | ||
| Scheme 2 Epoxidation of terpenes with H2O2. (1) (R)-(+)-limonene, (2) trans-limonene-1,2-epoxide, (3) α-pinene and (4) trans-α-pinene oxide. | ||
However, the use of H2O2 for terpene epoxidation presents several challenges, including low selectivity due to the formation of multiple oxidative products, limited reaction rates arising from the biphasic nature of the reaction, and the immiscibility of hydrophobic terpenes with aqueous H2O2. Additionally, the highly exothermic nature of the reaction complicates heat removal.22–24 To overcome these challenges, there is a pressing need to develop more efficient, environmentally friendly, and intensified processes that improve both mass and heat transfer. One promising avenue is the development of continuous processes, which can mitigate mass and heat transfer limitations. Flow chemistry and innovative reactor designs, such as the Oscillatory Baffled Reactor (OBR), offer a multi-scale approach for industrial applications. The OBR, a tubular reactor equipped with baffles, facilitates efficient multi-phase fluid mixing and heat removal by generating and dissipating vortices through the interaction between oscillating fluids and baffles. Employing catalysts like polyoxometalates can lead to the highly selective epoxidation of (R)-(+)-limonene and α-pinene with H2O2.5
This review offers a comprehensive overview of recent developments in both catalytic and non-catalytic methods for terpene epoxidation, employing a variety of oxidizing agents and process intensification techniques. It also examines the generalizability of findings from limonene epoxidation studies to other terpenes. A key focus of this review is the need for continuous processes to overcome mass and heat transfer limitations inherent in bio-based terpene epoxidation. To this end, the review explores the utility of flow chemistry and innovative reactor designs, such as the Oscillatory Baffled Reactor, for achieving continuous processing, enhanced mixing, and reduced solvent use. Ultimately, the review aims to contribute to the development of commercially viable processes for the production of bio-based epoxides from waste biomass. While there are existing reviews on the epoxidation of terpenes, such as the work published by Mahamat et al. (2021),6 this review offers a unique perspective by focusing on the catalytic oxidative upgrades of renewable terpene olefins. This review provides a comprehensive analysis of the challenges in achieving high selectivity and yields, particularly in the context of complex reaction mixtures, the addition of organic solvents, and high concentrations of oxidants.
2. Waste biomass as a resource for bio-based polymers
Synthetic polymers are widely used in industries such as packaging, textiles, toys, and electronics, and in 2014, approximately 300 Mt of polymers produced globally were fossil fuel-derived.7 However, the stocks of these feedstocks are declining despite an increase in demand, and environmental concerns have raised awareness of the raw materials used and the end-of-life options of these polymers.8 While the literature does not provide a direct answer to the decrease in the production of fossil fuel-derived polymers, there are indications of a shift towards bio-based alternatives. For instance, Sheldon and Norton (2020) in “Green Chemistry and the Plastic Pollution Challenge: Towards a Circular Economy” discuss how substituting plastics derived from fossil resources with bio-based alternatives can reduce emissions of greenhouse gases.9 Similarly, Vogli et al. (2020) in “Life Cycle Assessment and Energy Balance of a Novel Polyhydroxyalkanoates Production Process” compare the environmental impacts of bio-based polymers with fossil-based ones, suggesting a move towards more sustainable options.25 To address these concerns and the diminishing supply of fossil fuels, alternative measures focusing on the development of renewable polymers are needed. The increasing interest in bio-based polymers has been fueled by recent developments in this field, and policy and legislation, such as the 2015 climate agreement by the United Nations, are supporting the growth of sustainable bio-based polymers.26 Waste biomass is being used to develop sustainable bio-based polymers that can reduce waste and CO2 emissions. Many chemicals can be produced from waste biomass, such as agricultural waste, forestry waste, waste CO2, and paper waste, which can be used as feedstocks for bio-based polymers. It should be noted that bio-based polymers can vary in their biodegradability; they can be categorized as either biodegradable (e.g., starch, PHA) or non-biodegradable (e.g., bio-based PP).11Fig. 2 illustrates bio-based alternatives to petrochemicals for the manufacturing of polymers, such as poly (limonene carbonate) and vegetable oil-derived triglycerides.12 Recent research has focused on using carbohydrates in various forms, such as starch, cellulose, glucose, and fructose, to synthesize molecules like lactide, furan dicarboxylic acid (FDCA), ethylene glycol, and ethylene, which can be converted to bio-based polymers like polylactide (PLA), poly(ethylene furanoate) (PEF), bio-derived poly(ethylene terephthalate) (bio-PET), and bio-derived polyethene (bio-PE). Terpenes, diacids, and levoglucosenone are some common platform chemicals used in this process, with terpenes being used as feedstock due to their abundance.13
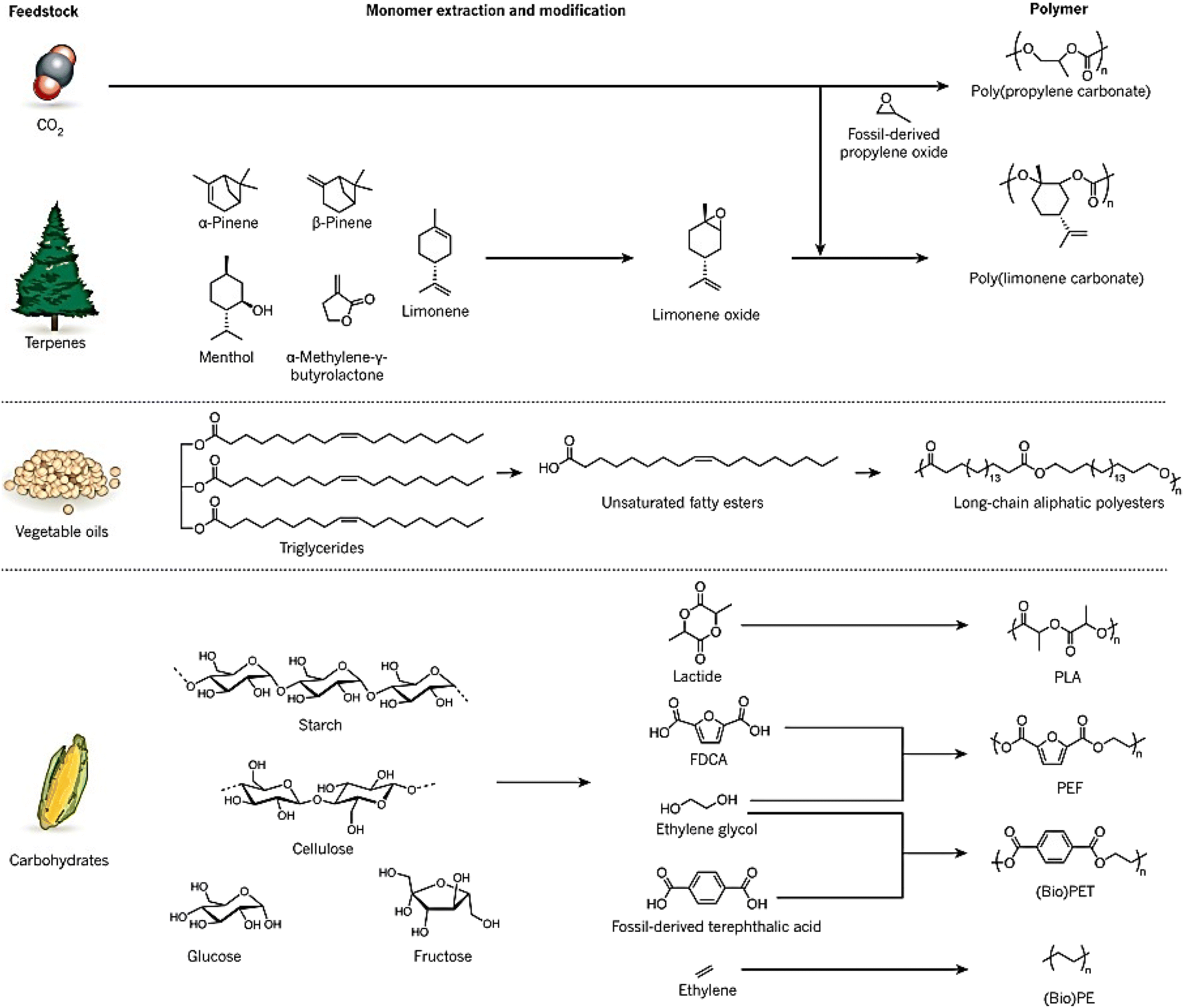 | ||
| Fig. 2 Bio-based alternatives to petrochemicals for the manufacturing of polymers.1 | ||
2.1 Terpenes derived from waste biomass
Terpenes are hydrocarbons that occur naturally in plants, animals, and insects.14,15 They can be used as bio-based feedstock to produce several valuable products. To reduce dependence on fossil-based feedstocks and minimize greenhouse gas emissions, scientists have sought viable plant-based routes. Terpenes derived from waste biomass have been gaining attention for their potential in the synthesis of fine chemicals and various applications.16 The utilization of waste biomass for terpene extraction not only provides a sustainable method for chemical production but also adds value to underutilized plant-based wastes.17 Modified terpenes that contain functional groups are known as terpenoids. Terpenes can be classified based on the number of isoprene units they contain, with the general formula (C5H8)n. Table 1 lists the classification of terpenes and provides examples for each class. The global pulping industry produces an estimated 3.5 × 105 tonnes of turpentine oil per year,18 and this number is expected to rise with the current surge in the industry. Turpentine oil is produced per tonne of pulp in quantities ranging from 0.3–1.0 kg,19 and the most commonly produced monoterpene is α-pinene (∼70%).20 Limonene (∼8.6%, 0.3 × 105 tonnes per annum) is obtained worldwide primarily from turpentine oil production.18 The crude turpentine oil obtained from steam-distilling pine typically contains 75–85% α-pinene, with the remaining percentages being 0–3% β-pinene and 5–15% limonene.21 Lemon peels are one of the main sources of limonene, containing 90 wt% limonene.19 Citrus fruit peels yield limonene in the steam extraction process, and orange peels and waste products from orange harvesting are the primary sources of limonene.22 Bio-based polymer production can benefit from the abundance of α-pinene and limonene available globally at low cost.23 The agricultural and timber industries produce an estimated 330000 tonnes of terpenes each year.
| Classification | General formula | Examples | Molecular structure |
|---|---|---|---|
| Hemiterpenes | C5H8 | Isoprene | 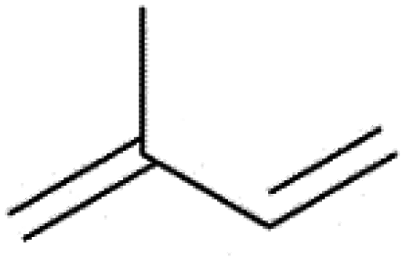 |
| Monoterpenes | C10H16 | (R)-(+)-limonene | 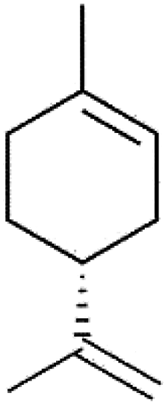 |
| Sesquiterpenes | C15H24 | Humulene | 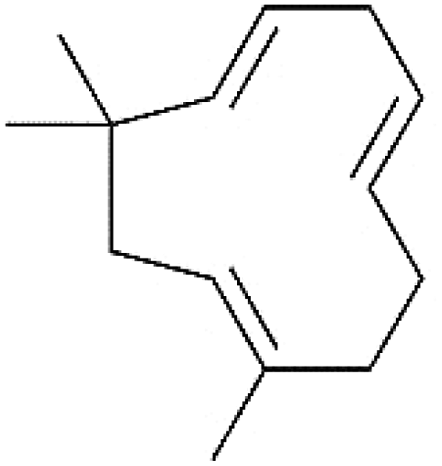 |
| Diterpenes | C20H32 | Cembrene |  |
| Triterpenes | C30H48 | Squalene |  |
| Tetraterpenes | C40H64 | Carotenoids |  |
| Polyterpenes | >8 isoprene units | Rubber |  |
Low molecular weight terpenes, such as monoterpenes, are commonly used as fragrances due to their high volatility, while some of the heavier terpenes like rubbers find use in the latex industry. Natural rubbers, which are polyisoprenes, are the most abundant bio-based terpenes.14 Despite having a common structural skeleton, plants produce structurally diverse monoterpenes24 Scheme 3 illustrates the biosynthesis routes and skeletons of monoterpenes in plants.
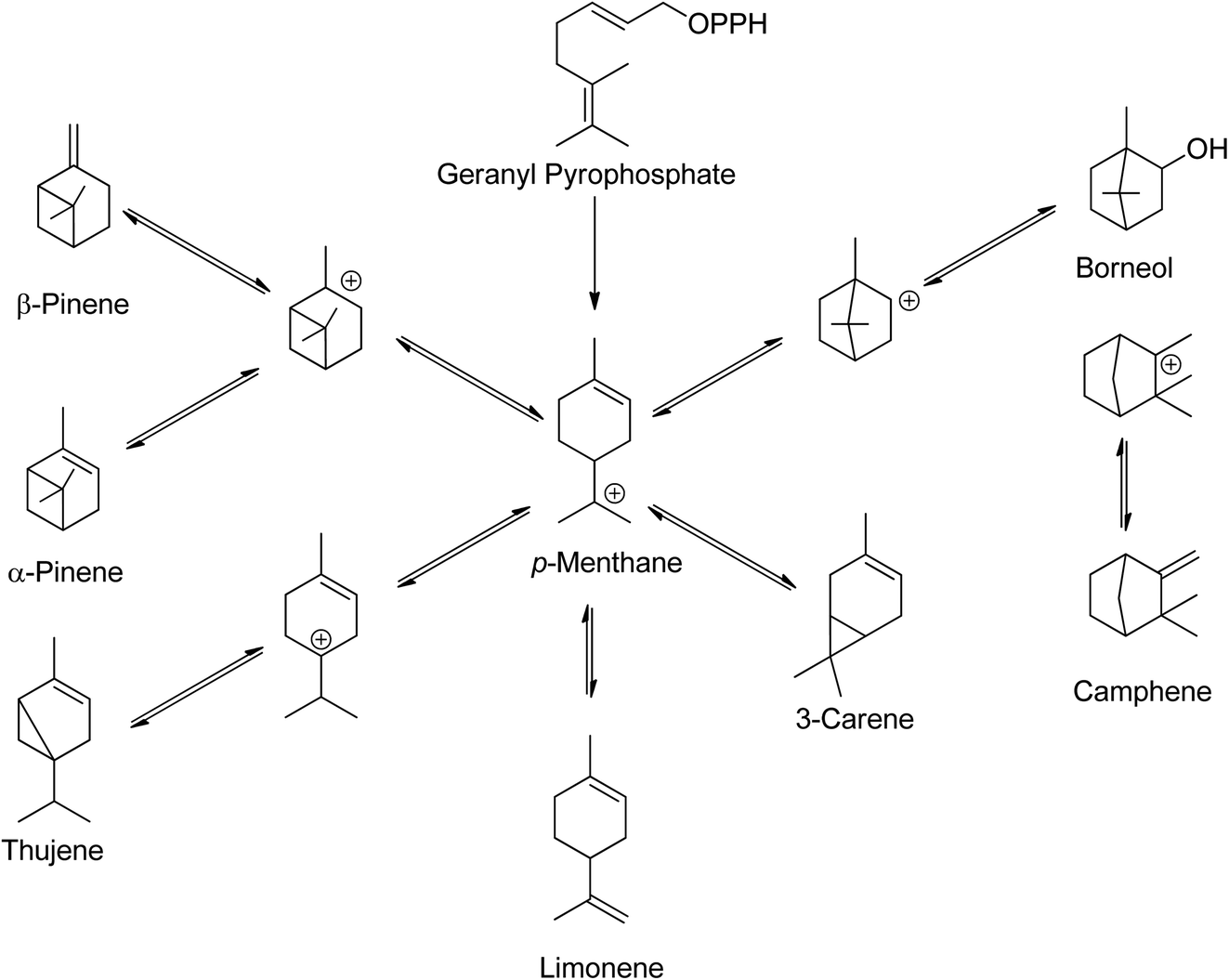 | ||
| Scheme 3 In plants, monoterpenes have a common skeleton and biosynthetic route.24 | ||
Among the many uses of monoterpenes, α-pinene and (R)-(+)-limonene find application in the pharmaceutical industry, as well as in flavours and fragrances.14,27 Polyterpenes derived from these terpenes can also be used as polymers.2 Turpentine oil, which mainly consists of α-pinene (45–97%), β-pinene (0.5–28%), and other monoterpenes including (R)-(+)-limonene, is a major source of these monoterpenes.28 Recently, α-pinene has been extracted from waste biomass, specifically from wood pulp processing, while (R)-(+)-limonene has been extracted from citrus peel waste.29 Orange peel waste comprises 60% of the orange mass in the orange juice industry30 and orange oil extracted from orange peel is up to 96 wt% (R)-(+)-limonene.31 As a result, there is significant potential to convert this waste into valuable products such as bio-based polymers.32 Furthermore, (R)-(+)-limonene can be converted into useful chemicals that find application as food, cosmetic, or pharmaceutical additives.
α-Pinene, another waste biomass-derived terpene, finds application in several flavours, fragrances, and pharmaceutical precursors.2,24,33–37 Turpentine oil, a by-product of the wood and paper industries, is composed mainly of α-pinene. Epoxidation of α-pinene yields valuable products such as verbenol, verbenone, and α-pinene oxide.38,39 α-Pinene oxide is an invaluable intermediate due to its many uses in various processes. It can be isomerized into products like campholenic aldehyde, which is useful in the production of sandalwood fragrances.5
3. Epoxidation of terpenes
3.1 Introduction to epoxidation of terpenes
Epoxide is a highly reactive and versatile cyclic ether consisting of a three-membered ring. Due to their versatility, epoxides are suitable as intermediates in many chemical reactions,40 and the polymer industry heavily relies on petrochemical-derived epoxides such as ethylene oxide, styrene oxide, and propylene oxide (Scheme 4), which as of 2014 had a combined estimated world production of 33 million tonnes/year.41 However, concerns for environmental and sustainability issues have led to the emergence of possible alternatives to conventional epoxides using waste biomass and other non-petrochemical-based sources.1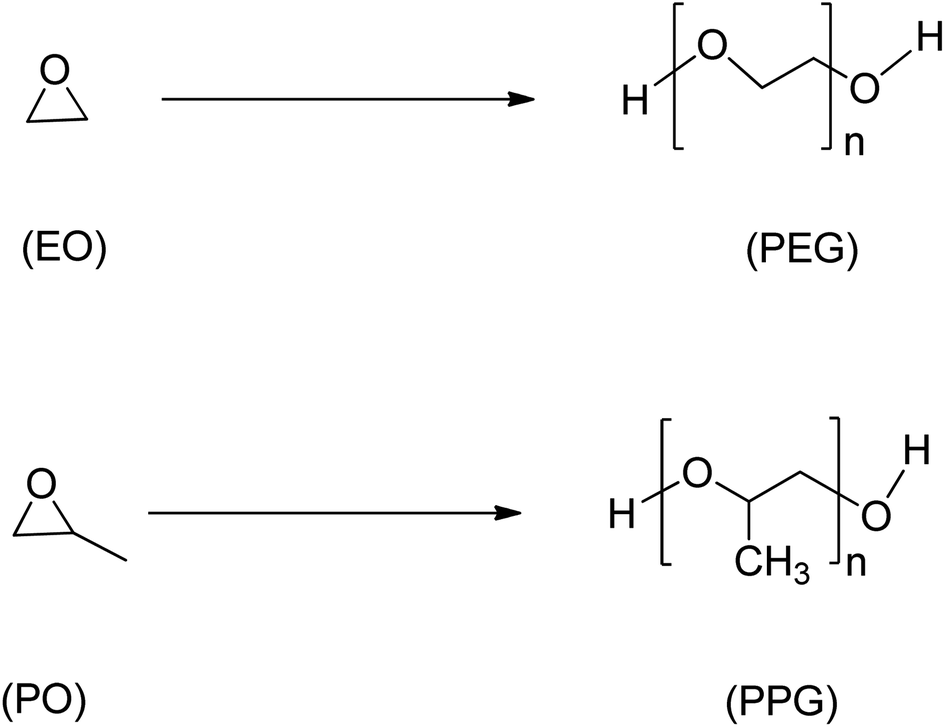 | ||
| Scheme 4 Ethylene oxide (EO), propylene oxide (PO) and their respective polyethers, polyethene glycol (PEG) and polypropylene glycol (PPG). | ||
Epoxides are used in various chemical processes such as the production of polyester, polyurethane, epoxy resins, glycols, alcohols, and carbonyl compounds. Long-chain alkenes like soybean oil are commercially epoxidised to produce plasticisers and stabilisers for PVC manufacturing.12 There are several processes to produce epoxides, including the Prilezhaev route (using peracids as oxidants), sharpless epoxidation (using organic hydroperoxides), Jacobsen-Katsuki epoxidation (using hypochlorites), Shi epoxidation (using dioxiranes), and the use of aqueous hydrogen peroxides (H2O2),12 with various starting materials commonly used such as alkenes, chlorocarbons, carbonyl compounds, epichlorohydrin, and substituted hydroxyls.42,43 One of the conventional epoxidation processes involves the oxidation of ethylene to ethylene oxide, which occurs in the gaseous phase using a silver-based catalyst at temperatures between 220 and 330 °C,1 as shown in Scheme 5.
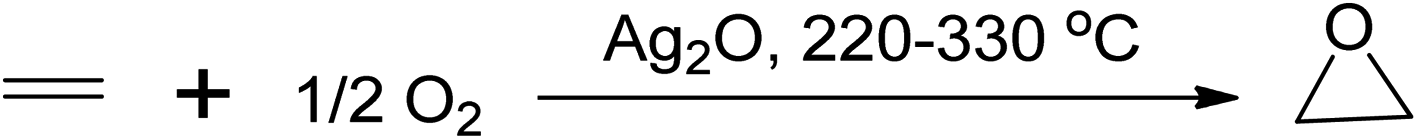 | ||
| Scheme 5 Epoxidation of ethylene-to-ethylene oxide with molecular oxygen using a silver catalyst. | ||
Similarly, propylene oxide can be produced via several routes, with the peroxide-based process being used commercially at a temperature between 100 and 130 °C using either a molybdenum (Mo) or titanium (Ti) based catalyst,40,44 as shown in Scheme 6. However, these hydroperoxide processes have limitations in terms of the formation of side products, and stoichiometric processes using peracids and catalytic processes using oxidants such as O2, organic peroxides, and H2O2 are more favourable.45
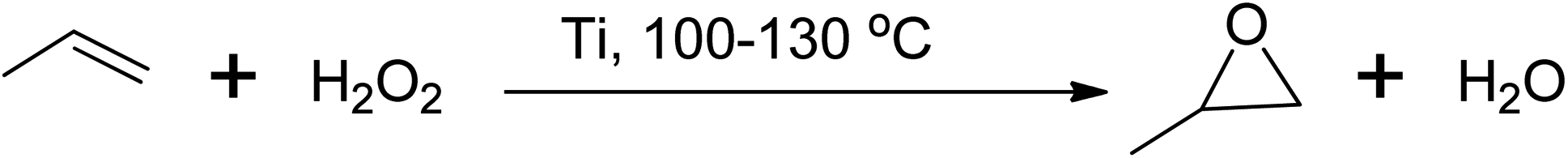 | ||
| Scheme 6 Peroxide-based epoxidation of propylene to corresponding propylene oxide using titanium catalyst. | ||
Mahamat et al. (2021)6 emphasize the critical role of terpene epoxides as primary intermediates in the synthesis of various green polymers, including epoxy resins and non-isocyanate polyurethanes. They outline the efforts in developing both catalytic and non-catalytic processes for terpene epoxidation, primarily focusing on limonene. They highlight that epoxidation is a key process that needs to be adapted for natural products like terpenes, as it has been traditionally used to valorize fossil-origin compounds. It can be deduced that it is important to consider technical feasibility and scalability alongside scientific understanding, highlighting the necessity of epoxidation processes that are both efficient and adaptable to a changing industrial landscape.
3.2 Oxidants used in terpene epoxidation
Epoxidation of alkenes can be achieved through three main routes: Prilezhaev, hydroperoxide, and catalytic routes. The Prilezhaev route typically uses peracids as oxidants, the hydroperoxide route uses organic peroxides such as tert-butyl hydrogen peroxide (TBHP), while the catalytic route usually employs molecular oxygen or H2O2 as oxidants. The active oxygen from these oxidants is transferred to the double bonds, resulting in the formation of an epoxide.Molecular oxygen is a green oxidant that offers many advantages, but it also has some limitations. One of the limitations is that molecular oxygen is less reactive toward organic molecules at low temperatures and has less selectivity toward the main products due to the formation of radicals.47 This can lead to the substrate being oxidized into many side products.47–49 To address these limitations, Pena et al. (2012) used nickel-aluminium hydrotalcite as a catalyst to oxidize (R)-(+)-limonene in the liquid phase using molecular oxygen at temperatures between 70 and 90 °C under atmospheric pressure and in solvent-free conditions (Scheme 7).47 They achieved a (R)-(+)-limonene conversion of up to 50%, producing many oxidative products including limonene-1,2-epoxide, limonene-8,9-epoxide, carveol, and carvone. However, they also found that the selectivity to epoxides was lower when using molecular oxygen due to the thermal decomposition of limonene hydroperoxides that initiate autooxidation.47
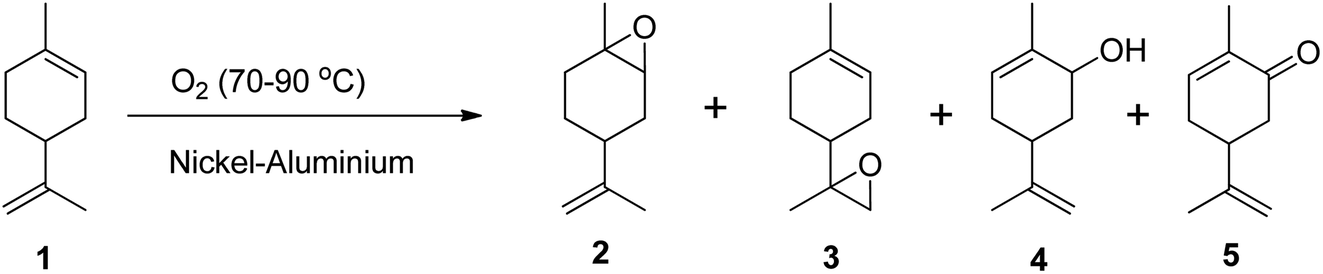 | ||
| Scheme 7 Epoxidation of (1) (R)-(+)-limonene with molecular oxygen-producing (2) limonene-1,2-epoxide, (3) limonene-8,9-epoxide, (4) carveol and (5) carvone. | ||
In a study on epoxidizing limonene with oxygen, Co/SBA-16 was created via post-grafting impregnation using a cobalt acetylacetonate solution. Isobutyraldehyde and ethyl acetate were used as green solvents, and the catalyst showed great activity under mild conditions.50 The metal composition of the catalyst was found to affect epoxide selectivity, with Co2+ being more active than Fe2+, Ni2+, and Mn2+. Co2+ was found to catalyze chain initiation and branching of alkene epoxidation by O2 in the presence of aldehydes, which activate dioxygen through coordination with the metal core. Scheme 8 shows a radical mechanism for epoxidizing limonene aerobically by an aldehyde catalyzed by SBA-16-supported Co2+. The reaction between isobutyraldehyde (RCHO) and molecular oxygen generates the acylperoxy radical, which acts as the primary epoxidizing agent. This species directly converts limonene to epoxide through pathway A or converts isolated Co2+ to Co-peroxy species through pathway B. Oxygen is transferred through the reaction between Co-peroxy and limonene in a concerted mechanism.
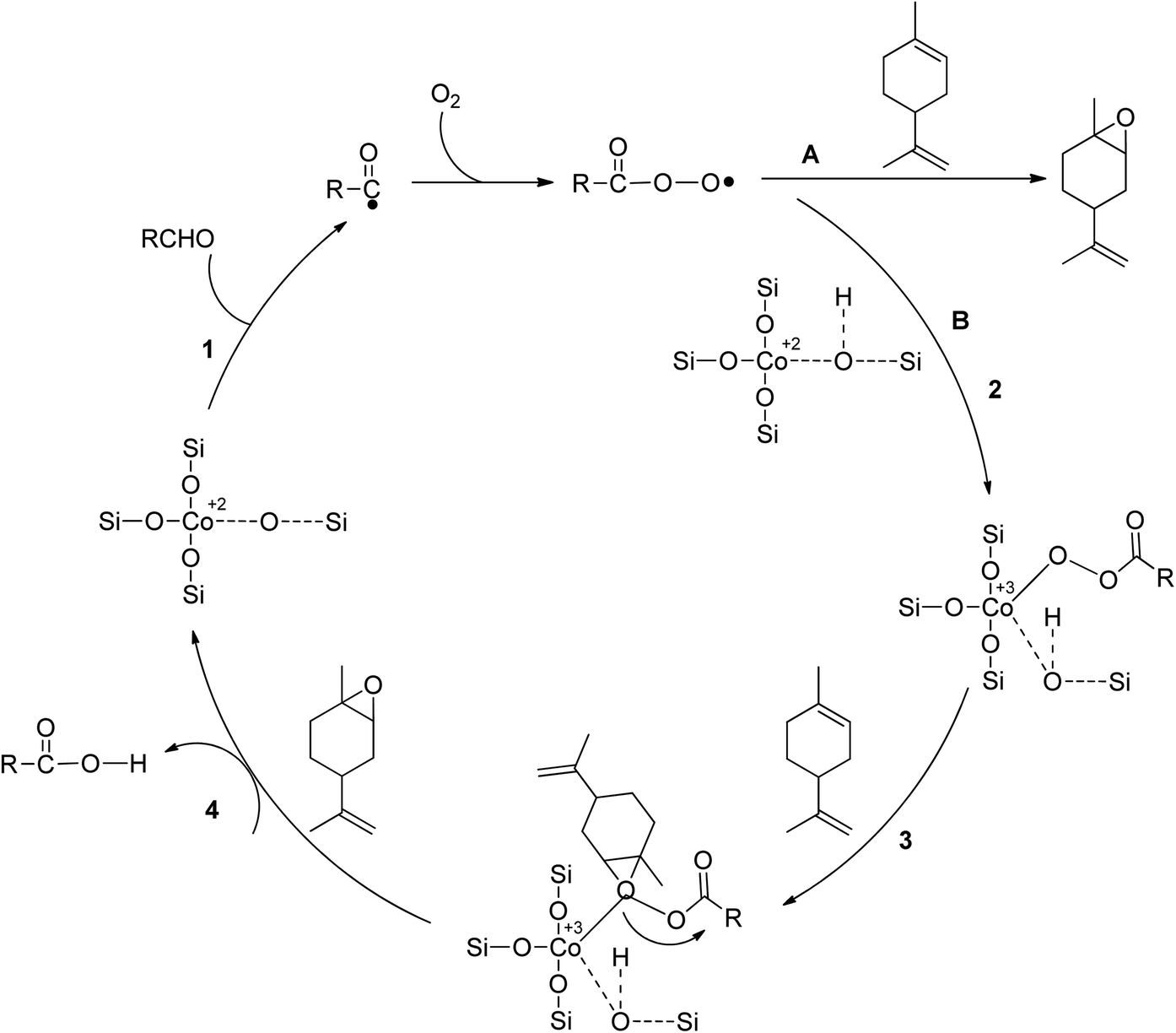 | ||
| Scheme 8 An aerobic limonene epoxidation method is proposed in the literature, reproduced from ref. 51. | ||
In a study using mesoporous silicon silica (SBA-16) grafted with cobalt, limonene was epoxidized aerobically using a new “pH-adjusting” technique.51 The study examined various cobalt loadings to optimize Co/SBA-16's catalytic performance. The results showed that Co/SBA-16 (1.1%) had the best-optimized epoxide yield (85%) and a lower cobalt loading than ideal Co/SBA-16 (4.5%) produced by post-grafting Co(acac)2.50 This was because the cobalt precursor had not been introduced before calcining the SBA-16 support, making Co2+ ions more accessible to silanol groups on the catalyst surface and increasing the density of tetrahedral Co2+ species. However, as cobalt loading increased, a decreasing trend in epoxide yield was observed, possibly due to equivalent oxidation facilitated by greater coordination of Co2+ ions at high cobalt loading.50 Increasing the oxygen pressure from 15–44 psi resulted in the highest yield of limonene epoxides, followed by a decrease at a higher oxygen pressure of 58 psi, likely due to subsequent epoxide oxidation or ring-opening. The reaction mechanism was proposed based on a kinetic study under optimum reaction conditions, with the proposed pathway C employing the peroxy intermediate Co3+OO− in Scheme 9 being significant only at extremely low limonene concentrations due to the peculiar impact of a sharp decrease in the initial rate of limonene epoxidation rate at increasing limonene concentrations. Therefore, the method is not suitable for large-scale use.
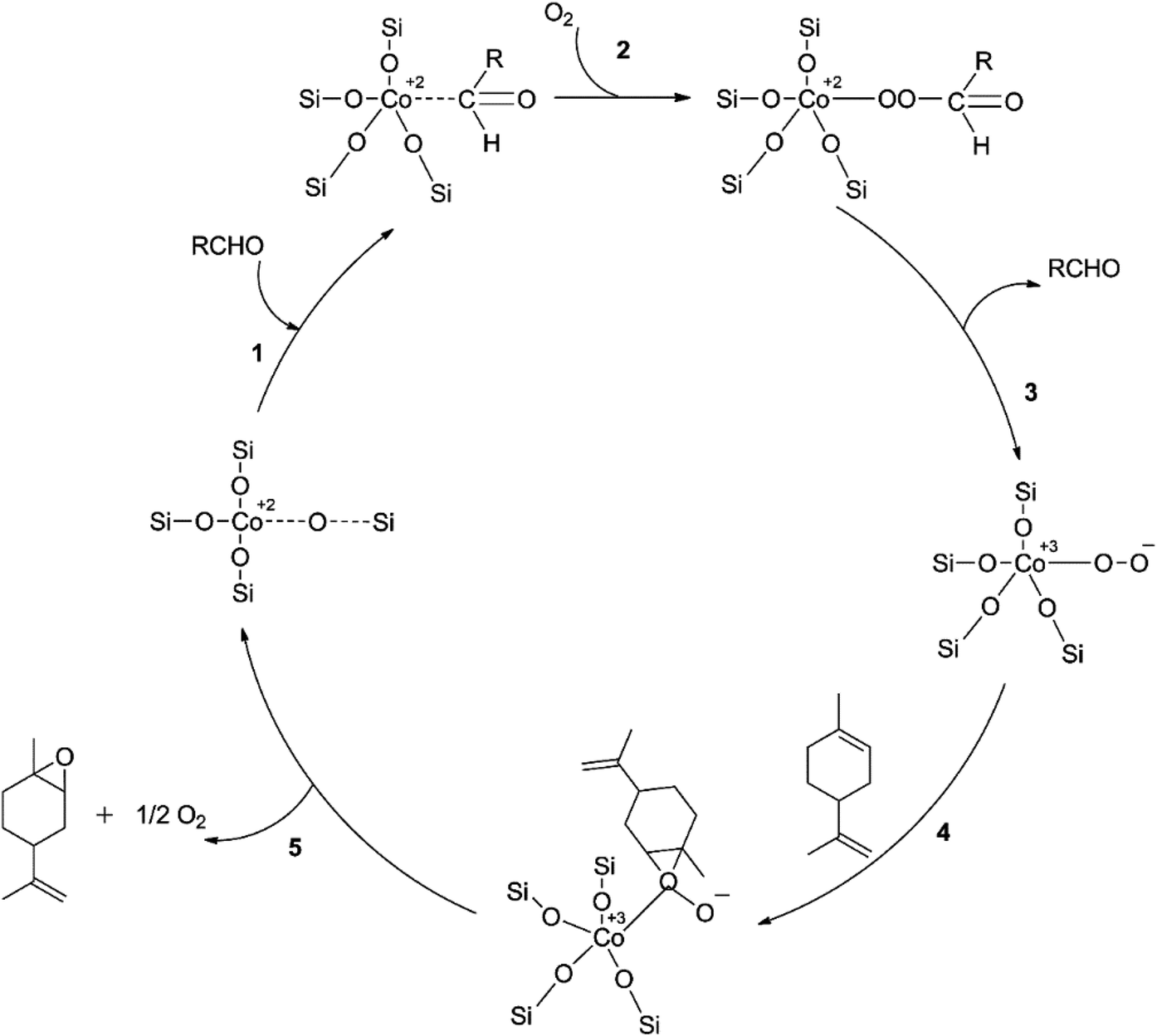 | ||
| Scheme 9 A proposed method for limonene aerobic epoxidation reproduced from ref. 50. | ||
A study investigated the direct epoxidation of limonene using highly dispersed ruthenium (Ru) supported on activated carbon (AC) prepared by several immobilisation techniques, without the use of solvents.52 The study found that in mild reaction conditions, Ru/AC showed excellent efficacy in the solvent-free epoxidation of limonene. Furthermore, the selectivity of the catalyst towards epoxidation was significantly impacted by the method of catalyst preparation. The cation exchange catalyst showed the greatest selectivity and yield of epoxidation products, despite having a lower conversion of limonene (40.4%) compared to the catalyst made using the sol-immobilization (SI) method. The oxidation of olefins with dioxygen can result in both epoxidation and allylic oxidation products. The ratio of allylic oxidation to epoxide products depends on the competition between alkyl peroxy radical incorporation and hydrogen removal. Limonene was found to be electrophilically attacked by ions leading to the formation of 1,2- and 8,9-epoxy limonene, the major epoxidation products, and 1,2,8,9-diepoxy limonene, a secondary epoxidation product. Allylic H-abstraction caused the formation of carveol and carvone. Scheme 10 depicts the two chemical routes that could be used to produce the main oxygenated byproducts.
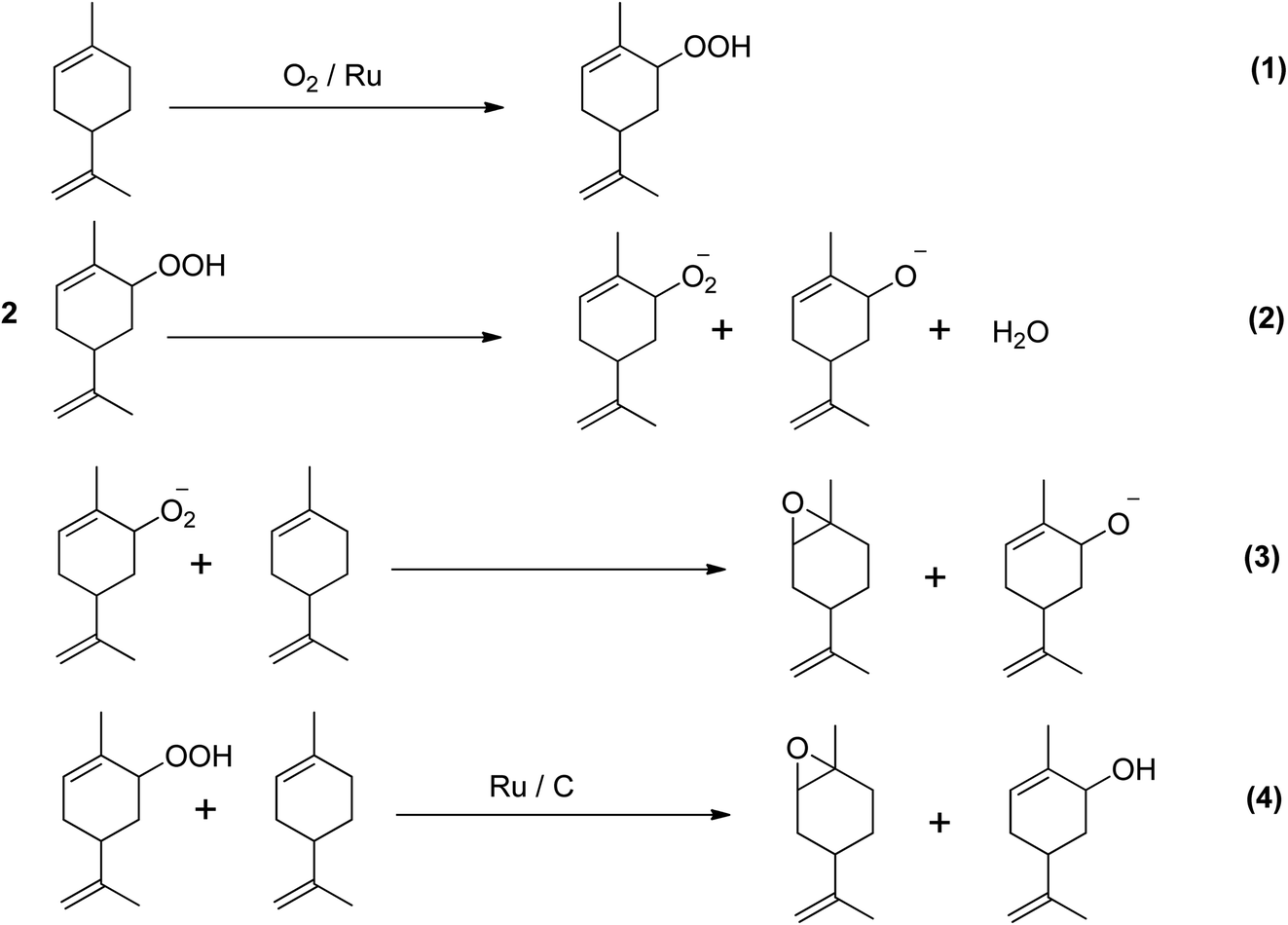 | ||
| Scheme 10 Proposed chemical route for solvent- and initiator-free aerobic limonene epoxidation.31 | ||
The study investigated two possible pathways for the epoxidation of limonene using ruthenium catalysts. The first pathway involves the activation of limonene and oxygen by ruthenium, leading to the formation of metal-alkene adducts on the catalyst surface. The second pathway is the autoxidation of limonene via a free radical chain mechanism, where the allylic C–H bonds of limonene are activated to produce limonene hydroperoxide, which can then break down into epoxides and allylic products when reacting with oxygen. The study found that the ratio of epoxides to allylic oxidation products was higher, indicating a preference for the first pathway. Observations have been made on the epoxidation of α-pinene using molecular oxygen. Patil et al. (2007) utilized a cobalt(II)-based heterogeneous catalyst in dimethylformamide (DMF) solvent, which resulted in a 47% conversion rate and 61% selectivity to α-pinene oxide. However, the selectivity to epoxides was reduced due to a free radical mechanism.53
In contrast, Almeida et al. (2022) investigated green solvents such as organic carbonates for catalytic aerobic epoxidation of biodegradable alkenes.31 Various catalysts were created using a conventional impregnation technique, including manganese, iron, and cobalt supported on alumina and silica. The cobalt catalyst supported by silica calcined at 550 °C produced significant results, producing a wide range of epoxides from biomass-based substrates with moderate to outstanding yields in mild reaction conditions. This method relies on the in situ production of active peroxy species from aldehydes and molecular oxygen using a non-noble metal catalyst, allowing for the basic limitations of conventional industrial techniques to be resolved. Moreover, dimethyl carbonate is highly recommended as a means of replacing undesirable solvents in industrial processes, as stated in current solvent sustainability guides. The reaction proceeds without any toxicity or reagent stability issues, resulting in the desired epoxide compounds.
Studies have recently focused on epoxidizing terpenes using the dimethyldioxirane (DMDO) epoxidation method. Ahmat et al. (2023) conducted a study on the DMDO-induced epoxidation of limonene and pinenes in microemulsions.54 The study produced DMDO in situ in a microemulsion system to assess the efficacy of using the surfactant cetyltrimethylammonium hydrogen sulphate (CTAHS) to homogenize limonene and pinene in aqueous solutions. Using CTAHS offered significant benefits for the epoxidation process. Molar ratios of 1.6 and 0.9 of oxone/limonene and oxone/α-pinene resulted in 100% yields of limonene bis-epoxide and α-pinene oxide after 30 minutes. This method could be highly useful on an industrial scale. The main drawback of this method is the need to buffer the reaction mixture at a pH of 7–8 with sodium bicarbonate. Nonetheless, this approach has several advantages, including excellent selectivity for epoxides, no by-product formation, no solvent requirement in the aqueous medium, and no previous metal catalyst production. Acetone was employed in small amounts as a catalyst since it is easy to recycle and does not require deactivation like metal catalysts. Furthermore, this epoxidation method was carried out under ambient conditions as part of a green process.
Michel et al. (2012) used methyltrioxorhenium (MTO) as a suitable catalyst for epoxidizing (R)-(+)-limonene with H2O2 in dichloromethane (DCM) at 25 °C, showing a selectivity of 96% toward epoxide with a yield of 77% after 1 hour.63 According to their findings, the catalyst requires a pyridine-based compound to lower its acid centre and prevent diol formation. The catalyst exhibited enhanced selectivity toward limonene-1,2-epoxide and limonene bis-epoxides at lower and higher temperatures (>25 °C), respectively.
Cagnoli et al. (2005) demonstrated the epoxidation of (R)-(+)-limonene with H2O2 using a titanium-based heterogeneous catalyst (Ti-MCM-41) in acetonitrile at 70 °C, reporting up to 60% conversion of (R)-(+)-limonene to several oxidative products, including limonene-1,2-epoxide, limonene bis-epoxide, limonene-1,2-diol, carveol, and carvone (Scheme 11).69 The catalyst showed only 60% selectivity to the epoxide, but it had better activity than other titanium-based catalysts with minimal leaching.
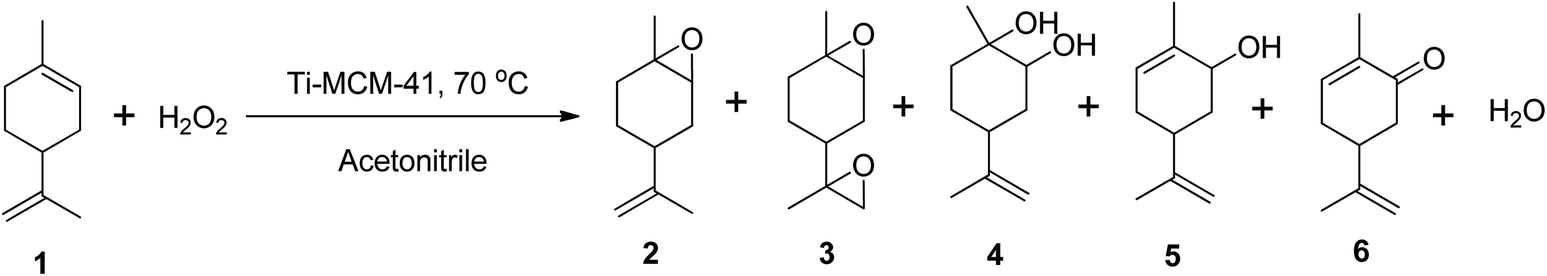 | ||
| Scheme 11 Epoxidation of (1) (R)-(+)-limonene with Ti-MCM-41 producing (2) limonene-1,2-epoxide, (3) limonene bis-epoxide, (4) limonene-1,2-diol, (5) carveol and (6) carvone. | ||
Epoxidizing α-pinene to α-pinene oxide selectively using a metal catalyst is rarely reported in the literature. Cánepa et al. (2011) investigated the use of a titanium-based catalyst (Ti-MCM-41) in the epoxidation of α-pinene with H2O2 at 70 °C in acetonitrile. They found that the reaction showed reduced selectivity to α-pinene oxide and was more selective to verbenone (41%), verbenol (16%), and campholenic aldehyde (27%), as shown in Scheme 12.60 In a more recent study, Cánepa et al. (2015) used a heterogeneous vanadium-based catalyst to epoxidize α-pinene with H2O2 at 70 °C in acetonitrile. They observed a 91% conversion of H2O2 (with α-pinene in a higher molar ratio of 4:1) after 7 hours to yield several oxidative products, mainly verbenones (46%) but achieved less than 3% selectivity to α-pinene oxide.70
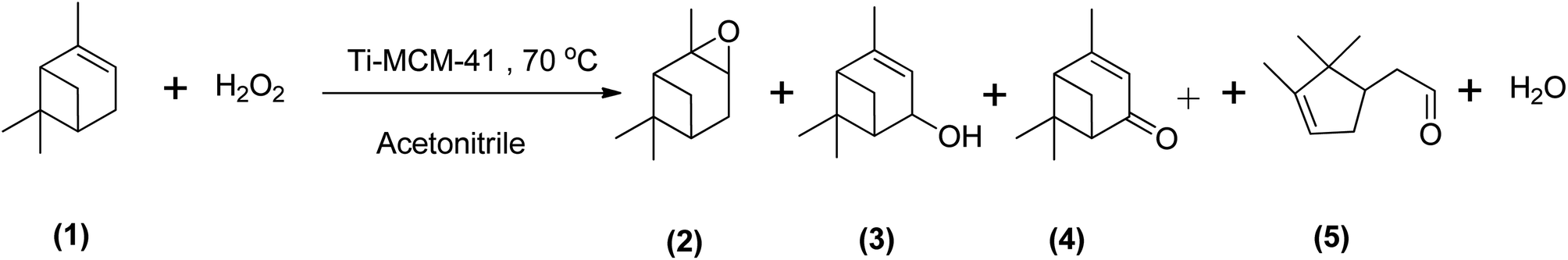 | ||
| Scheme 12 Epoxidation of (1) α-pinene with Ti-MCM-41 catalyst producing (2) α-pinene oxide, (3) verbenol, (4) verbenone and (5) campholenic aldehyde. | ||
Tungsten-based polyoxometalates are known as effective catalysts for epoxidation reactions using H2O2 as the oxidant.71 Jimtaisong and Luck (2006) compared the efficacy of molybdenum-based and tungsten-based catalysts for the epoxidation of cyclooctene using both TBHP and H2O2. They found that the tungsten-based catalyst was more active than the molybdenum-based catalyst when H2O2 was used as an oxidant, but less active when TBHP was used.72
Ishii et al. (1988) and Venturello and D'Aloisio (1988) demonstrated efficient epoxidation of alkenes using tungsten-based polyoxometalates and H2O2 in a biphasic environment containing quaternary ammonium salts as a phase transfer catalyst.73 Venturello and D'Aloisio achieved up to 98% conversion of 1-dodecene with 96% selectivity to epoxides at 70 °C in 1,2-dichloroethane (Scheme 13),74 while Ishii et al. achieved 96% conversion of cyclooctene at 98% selectivity to epoxides in chloroform.73 However, both methods require chlorinated solvents, making them less environmentally friendly, so alternative approaches have been developed.
 | ||
| Scheme 13 Epoxidation of 1-dodecene to 1,2-epoxydodecane using quaternary ammonium salt (Q+X−) as phase transfer catalyst. | ||
An improved method for the epoxidation of alkenes was developed using tungsten-based polyoxometalates in an organic solvent-free system, which employed aminomethylphosphonic acid (NH2CH2PO3H2) and a halide-free quaternary ammonium hydrogensulfate (Q+HSO4−) as phase transfer catalyst, as depicted in Scheme 14.75 This method significantly impacted further research on epoxidation reactions utilizing tungsten-based catalysts and H2O2 as an oxidant, as the process was conducted in an environmentally friendly manner. However, the hydrolytic decomposition of acid-labile epoxides in acidic conditions was a drawback, and the acid used in this method was costly.
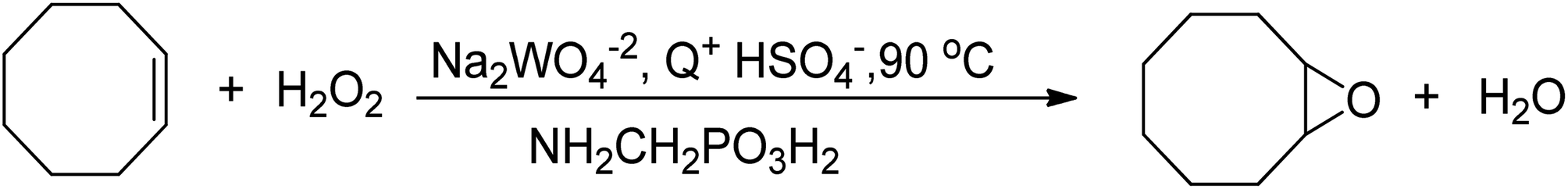 | ||
| Scheme 14 The epoxidation of cyclooctene with H2O2 in a halide- and solvent-free system. | ||
Grigoropoulou et al.76 improved the ‘Noyori’ method for the epoxidation of (R)-(+)-limonene without using expensive aminomethylphosphonic acid. They achieved 94% conversion of (R)-(+)-limonene with 81% selectivity to limonene-1,2-epoxide by studying the epoxidation of (R)-(+)-limonene with H2O2 using a sodium tungstate (Na2WO4) catalyst at a temperature of 70 °C in organic solvent-free conditions. To increase the selectivity to epoxide and to prevent the ring-opening reaction in acidic conditions, they used an inorganic salt (Na2SO4). Their findings had a substantial impact on the further development of using H2O2 as an oxidant for selective epoxidation. Hachiya et al. (2012) and Takumi et al. (2014)77 also added inorganic salt to the solvent-free epoxidation of (R)-(+)-limonene with H2O2 using a tungsten-based catalyst and produced a stable epoxide in acidic environments.78 However, the effect of Na2SO4 on the stability of epoxides requires further understanding and will be discussed in the relevant section (Section 4.6).
Maheswari et al. (2005) developed an improved version of the ‘Noyori’ method for the epoxidation of cyclooctene with H2O2. They used a tungsten-based (Na2WO4/H2WO4) catalyst at 60 °C in a biphasic system without solvents. They added chloroacetic acids to mimic the effect of peracids, which resulted in a 90% conversion of cyclooctene at 99% selectivity to cyclooctene oxide. The residual acidity of the aqueous medium was crucial for the catalytic performance of the tungsten-based catalyst. The study found that acidity positively affects the epoxidation rate, but this approach may not be suitable for acid-labile epoxides such as terpenes oxide, as increasing acidity can lead to hydrolytic decomposition.79 To address this, Kaur et al. (2010)80 proposed a safer approach for the epoxidation of terpenes using solid urea hydrogen peroxide (UHP) with a tungsten-based catalytic system, which produced a yield comparable to conditioned aqueous H2O2-based epoxidations.
Although the authors cited above have reported methods for producing limonene-1,2-epoxide with high selectivity and minimal hydrolytic decomposition in recent years, the process has not been fully optimized, and the reaction kinetics remain unclear. Additionally, little attention has been given to conditions that promote the ring-opening of the epoxides. To address these issues, we utilized aqueous H2O2 as an oxidant for the epoxidation of (R)-(+)-limonene and α-pinene. This choice was made due to H2O2's higher selectivity for epoxides and its environmentally friendly properties. We employed a tungsten-based polyoxometalates catalyst, which showed superior activity with H2O2 compared to other catalysts. Section 3.8 provides a further discussion of the chemistry, structure, and mechanism of the polyoxometalate catalyst. Many oxidative products can be generated by epoxidation of (R)-(+)-limonene with H2O2 (oxidant). Scheme 15 shows products of (R)-(+)-limonene epoxidation, including their respective stereoisomers.
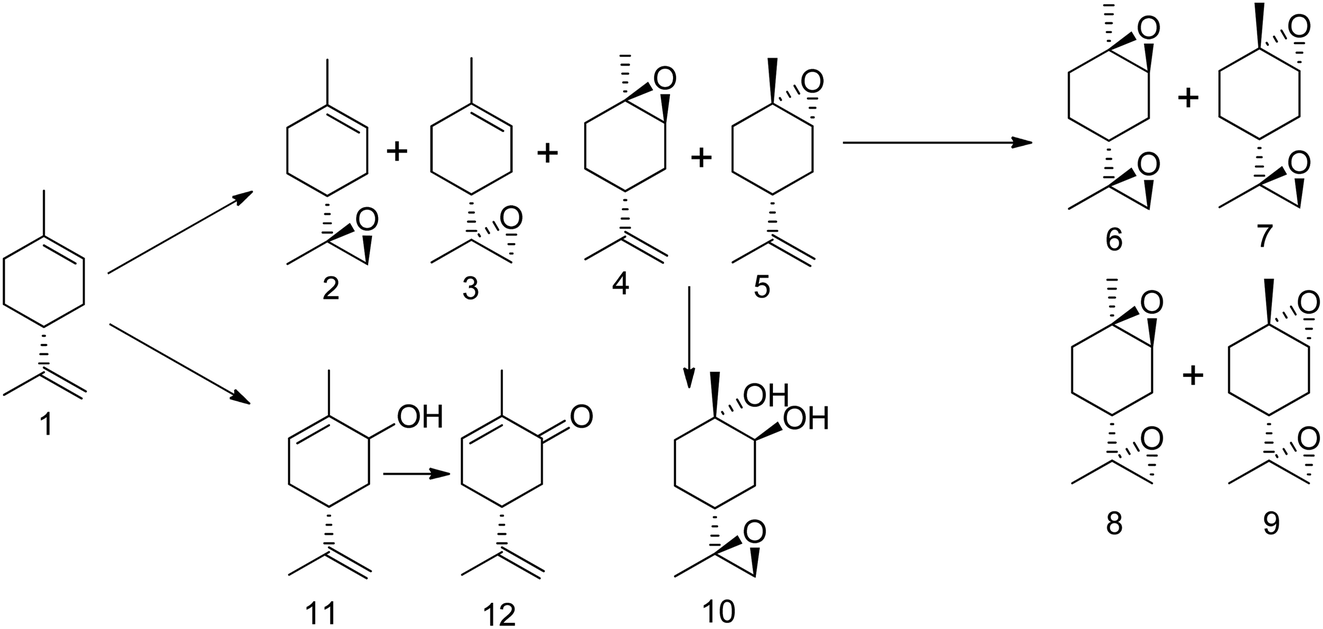 | ||
| Scheme 15 Products of (R)-(+)-limonene epoxidation with hydrogen peroxide (H2O2) as oxidant in the presence of tungsten-based catalyst; (1) (R)-(+)-limonene, (2) cis-limonene-8,9-epoxide, (3) trans-limonene-8,9-epoxide, (4) cis-limonene-1,2-epoxide, (5) trans-limonene-1,2-epoxide, (6) RSS limonene bis-epoxide, (7) SRS limonene bis-epoxide, (8) RSR limonene bis-epoxide, (9) SRR limonene bis-epoxide, (10) trans-limonene-1,2-diol, (11) carveol and (12) carvone. | ||
H2O2-based epoxidation of (R)-(+)-limonene (1) with tungsten-based polyoxometalates yields both limonene-8,9-epoxide (2 and 3) and limonene-1,2-epoxide (4 and 5), which are a mixture of cis- and trans-isomers. Limonene-1,2-epoxide is the primary product due to the electrophilic nature of the oxidative species, which likely causes epoxidation at highly substituted double bonds.81 To produce Limonene-bis-epoxide (6, 7, 8 and 9; a mixture of four stereoisomers), external double bonds of limonene-1,2-epoxide must be epoxidized. The hydrolytic decomposition of limonene-1,2-epoxide can also result in the formation of limonene-1,2-diol (10) facilitated by acid (H+) and H2O. Additionally, allylic oxidation of (R)-(+)-limonene primarily produces carveol (11), which can be further oxidatively dehydrogenated to produce carvone (12).82 The well-known H2O2-based epoxidation reaction of α-pinene with a metal catalyst produces several oxidative products. Scheme 16 presents a simplified version of the reaction scheme.
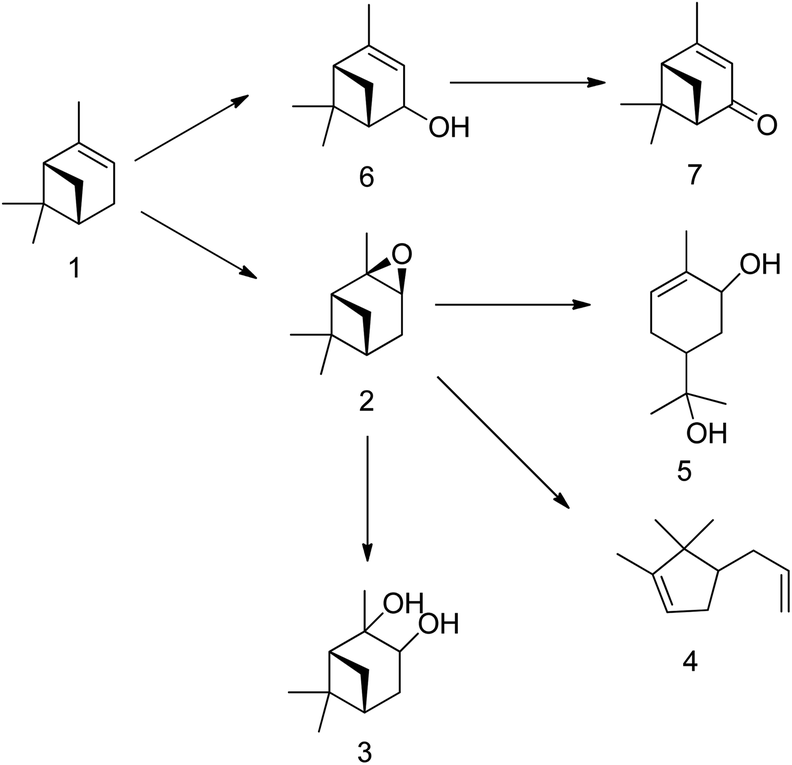 | ||
| Scheme 16 Product of α-pinene epoxidation with hydrogen peroxide (H2O2) as an oxidant in the presence of a tungsten-based catalyst; (1) α-pinene, (2) α-pinene oxide, (3) pinanediol, (4) campholenic aldehyde, (5) sobrerol, (6) verbenol and (7) verbenone. | ||
The tungsten-based catalyst used in the epoxidation of α-pinene (1) with H2O2 primarily produces α-pinene oxide (2) due to the electrophilic nature of the peroxo species, attracting substitution of π bonds.75 The hydrolysis of α-pinene oxide yields pinanediol (3) and sobrerol (5), assisted by H+ and H2O. α-Pinene oxide can also undergo rearrangement to form campholenic aldehyde (4) and allylic oxidation to produce verbenol (6), which can then be oxidatively dehydrogenated to form verbenone (7). The nature of the catalyst and radical formation determine whether epoxidation or allylic oxidation predominates.29 Various hydroperoxides, including verbenyl hydroperoxides, can also be formed as intermediates using an active catalyst.83,84 Research has shown that α-pinene can isomerize during the oxidation process to produce several products, including β-pinene, (R)-(+)-limonene, terpineol, camphene, and 3-carene.85,86 The rearrangement of α-pinene oxide has produced many products, such as pinocarveol, isopinocamphone, and trans-carveol,10,87 Additionally, oxidation can also lead to the formation of polymeric compounds, as shown in Scheme 17.
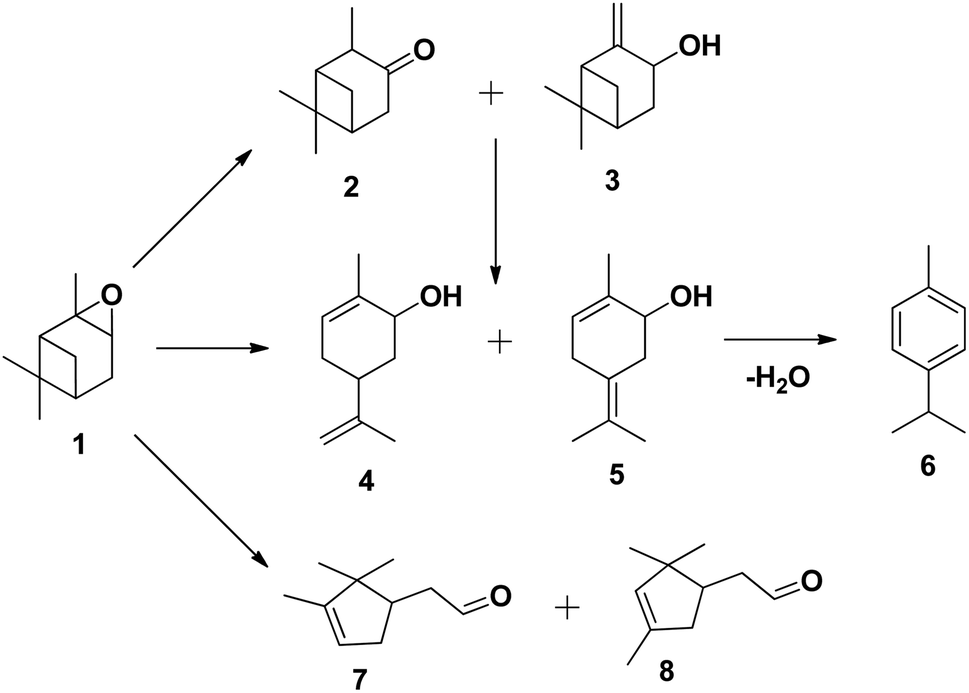 | ||
| Scheme 17 Products of isomerisation of α-pinene oxide; (1) α-pinene oxide, (2) isopinocamphone, (3) pinocarveol, (4) trans-carveol, (5) 2-methyl-5-(propan-2-ylidene) cyclohex-2-enol, (6) p-cymene, (7) campholenic aldehyde and (8) fencholenic aldehydes.10 | ||
In recent work, a novel approach to rapidly and selectively epoxidize α-pinene using H2O2 in a single step without any solvent was reported.23 Tungsten-based polyoxometalates were used as a catalyst to epoxidize α-pinene with H2O2, and variables in the screening parameters included temperatures (30–70 °C), oxidant concentrations (100–200 mol%), acid concentrations (0.02–0.09 M), and solvent types (e.g., p-cymene, toluene, acetonitrile, and 1,2-dichloroethane). Using process conditions of a 50 °C temperature, a solvent-free environment, and α-pinene/H2O2/catalyst molar ratio of 5:1:0.01, almost 100% selective epoxidation of α-pinene to α-pinene oxide was achieved with negligible side product formation within a short processing time (20 min). 93% Conversion of H2O2 with 100% selectivity to α-pinene oxide was achieved using a single-step addition of the oxidant without a solvent. The study also demonstrated that allylic oxidation rates increased as oxidant concentrations increased. A kinetic study suggested that this reaction was first-order in terms of both the concentrations of the catalyst and α-pinene, but fractional order (0.5) in terms of the concentration of H2O2. An activation energy of 35 kJ mol−1 was required for the epoxidation reaction of α-pinene. The key advantages of the described epoxidation procedure include the ability to perform the reaction at a constant temperature under a solvent-free environment to speed up the reaction rate and achieve selectivity to α-pinene oxide of over 100%.
We recently reported a study on the synthesis of α-pinane carbonate from α-pinanediol.88 This work represents the first example in the literature of producing α-pinane carbonate from α-pinanediol, which is a by-product of the most common monoterpene found in nature, α-pinene. Prior attempts to make pinane carbonate by reacting α-pinene epoxide with CO2 failed because the epoxide lacked reactivity in ring-opening polymerization due to steric hindrance. In this case, we combined α-pinanediol and DMC to create α-pinane carbonate under the influence of a TBD guanidine catalyst. The resulting white, crystalline solid known as α-pinane carbonate had a melting point of 86 °C. After recrystallization, the highest yield of α-pinane carbonate obtained was 81.1 ± 2.8%, with 98.7% purity. We used several techniques, including FTIR, NMR, GC, GCMS, and QTOF mass spectrometers, to characterise the produced α-pinane carbonate.
To establish a feasible process for potential resource recovery of the α-pinanediol starting material, we examined the reactivity of the α-pinane carbonate for base hydrolysis using NaOH in ethanol/water. The saponification of α-pinane into α-pinanediol and sodium carbonate was completed in less than 30 minutes. At an industrial scale, the α-pinane carbonate can potentially be polymerized to produce poly(α-pinane carbonate), which is a bio-based thermoplastic, like poly(limonene carbonates). To recycle the initial feedstock for α-pinanediol, the used poly(α-pinane carbonate) might be hydrolyzed in the same manner as the α-pinane carbonate monomer.
The hydrolysis of alkyl esters has been reported previously. The reaction mechanism for the hydrolysis of α-pinane carbonate is illustrated in Scheme 18.88 A rate-determining step involves the bimolecular collision of the α-pinane carbonate and hydroxide ion to create a tetrahedral intermediate, which decomposes to give the products.89 For intermediate stabilization, a molecule of water is required, but the same job can be performed using a protic solvent like simple alcohols, which are the product of alkyl esters hydrolyzed with a base.89,90 The hydroxyl group acts as a nucleophile during the reaction as illustrated in Scheme 18, reacting with α-pinane carbonate (a) to produce intermediate (b), which disintegrates and thus the carbonyl group reforms to form 2-carboxylate-3-pinanol (d) using intermediate (c) in the basic ethanol/water solution, which further rearranges to provide final product (e). The hydroxyl group most likely attacks on the opposite side in the most electropositive position since one side of α-pinane carbonate is more crowded, but further investigation is still required to establish the precise mechanism of the reaction.
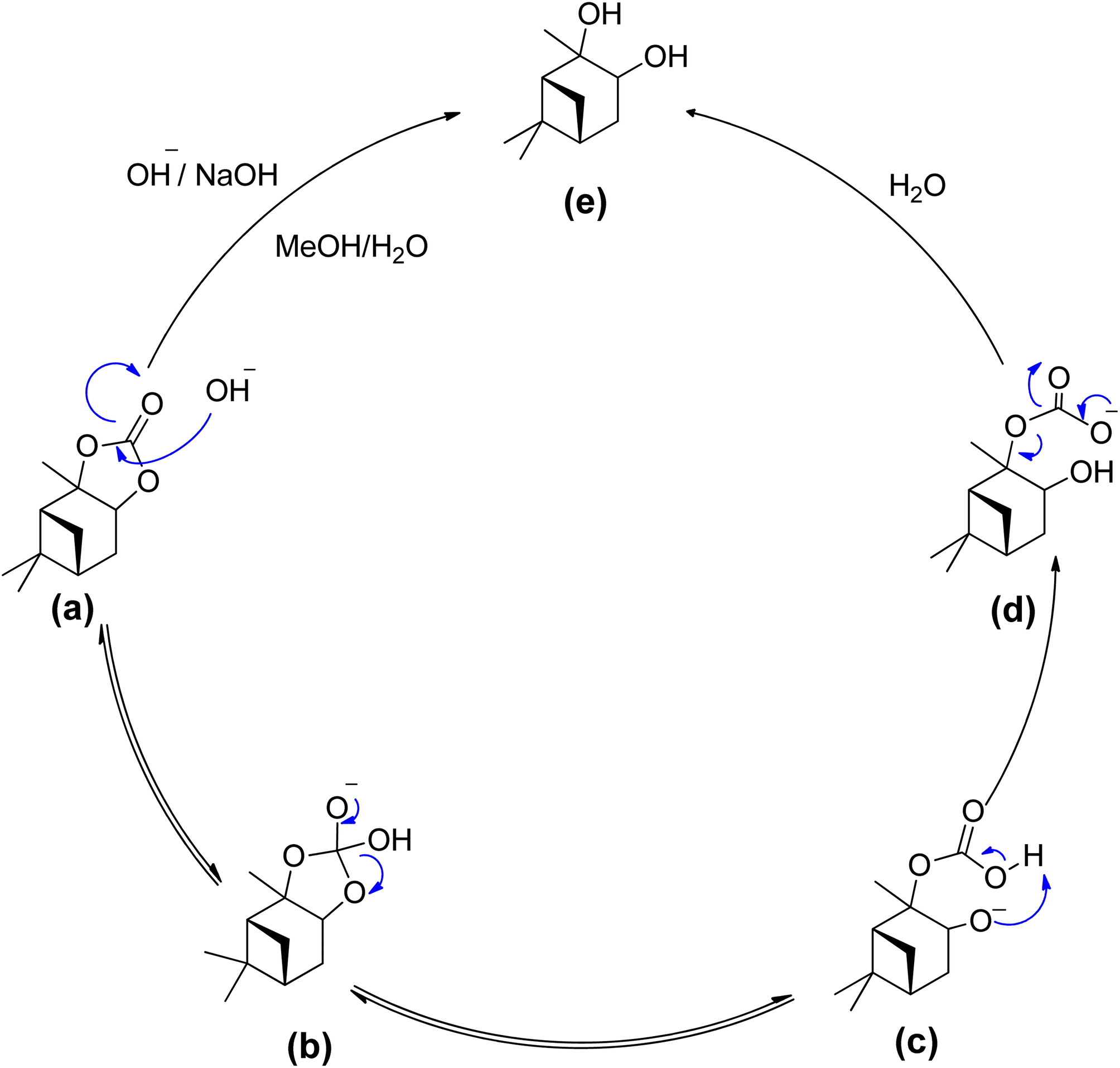 | ||
| Scheme 18 Proposed reaction mechanism - a bimolecular collision of solvated hydroxide ion with the alky esters carbonyl carbon.88 | ||
3.2.3.1 Prilezhaev epoxidation. The prilezhaev epoxidation of alkenes is typically achieved using peracids as oxidants. Prileschajew (1909)
91 first demonstrated this method, which has since become widely accepted as the most common technique for synthesizing epoxides. Commonly used peracids include peroxyacetic acid, hydroxybenzoic acid, peroxyformic acid, m-chloroperoxybenzoic, and p-nitroperoxybenzoic (Scheme 19), due to their relative availability, lower cost, the stoichiometric yield of epoxides, and stability at mild temperatures.12
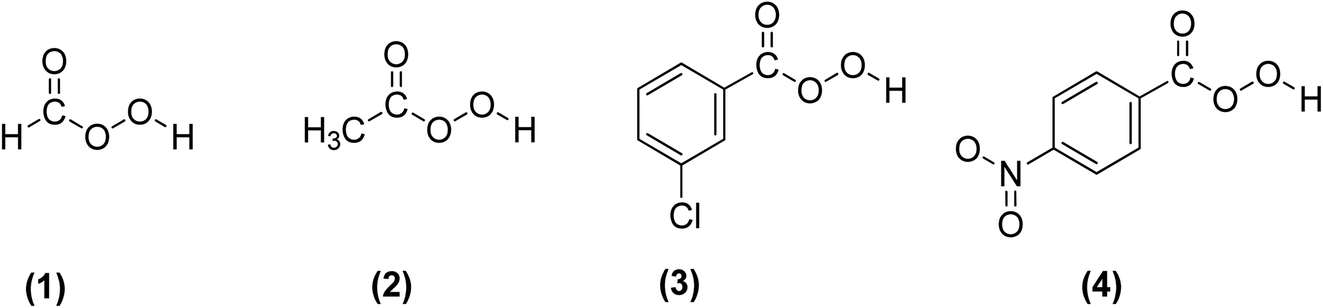 | ||
| Scheme 19 Commonly used peracids for epoxidation; (1) peroxyformic acid, (2) peroxyacetic acid, (3) m-chloroperoxybenzoic and (4) p-nitroperoxybenzoic. | ||
Peracids offer the flexibility of reaction medium, as they can be used in aqueous, organic, heterogeneous, and homogeneous media. However, they are hazardous due to the generation of acid waste. The reaction scheme and mechanism of the epoxidation of alkene using peracid are shown in Scheme 20.
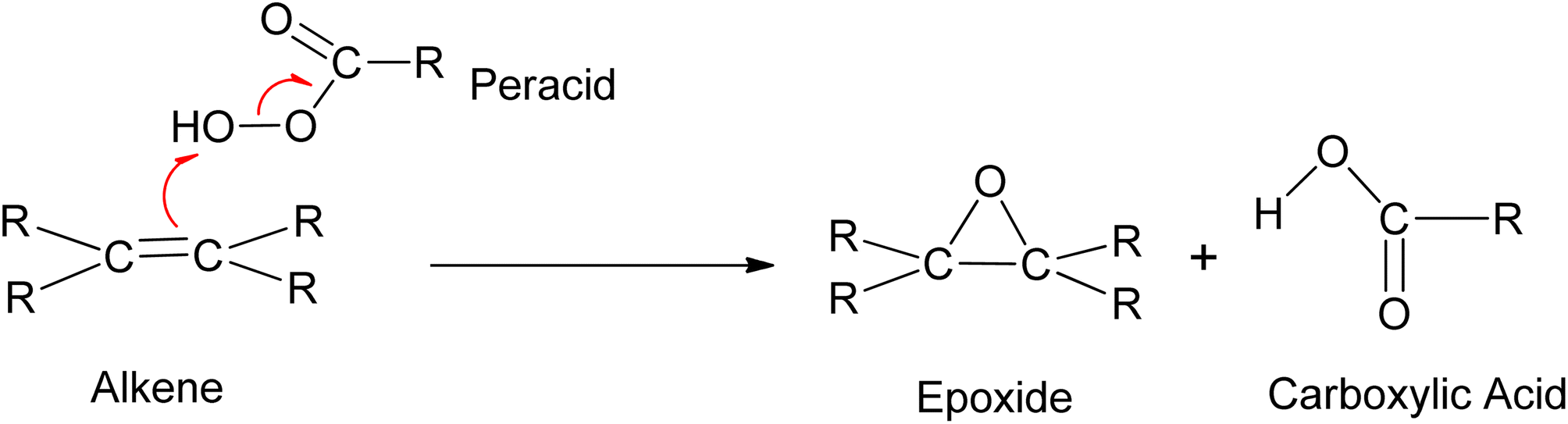 | ||
| Scheme 20 The reaction scheme and mechanism of the epoxidation of alkene using peracid. | ||
There are two methods for performing epoxidation using peracids: preformed peracid epoxidation and in situ peracid epoxidation. In both methods, aqueous H2O2 is reacted with carboxylic acid over an acidic catalyst (e.g., H2SO4) to form a peracid.92 Preformed peracids require rigorous storage procedures to minimize the risk of explosions due to exothermic decomposition. In situ peracids are favoured because they require minimal amounts of reactants to generate the oxidative species in the reaction, making the process much safer. The oxidative species are normally generated using a mineral acid as a catalyst, such as H2SO4. Alternatively, a heterogeneous catalyst such as an ion-exchange resin (IER) could be used to overcome separation and purification issues usually associated with homogeneous catalysts. The recyclability and selectivity of heterogeneous IERs for in situ peracids are better than for preformed peracids for the epoxidation of undecyclinic acid.12
Researchers have been seeking an alternative to the peracid epoxidation method that is selective, safe, and “clean” as peracids produce significant amounts of acid waste, which poses safety and environmental issues. Goud et al. (2017) compared the performance of two types of peracids formed in situ, peroxyacetic acid and peroxyformic acid, for the epoxidation of jatropha oil using IER (Amberlite IR-120) as a catalyst at a temperature of up to 70 °C. They investigated the performance of the peracids with and without the presence of solvent (toluene) and found that the efficiency of peroxyformic acid in the in situ epoxidation of peracid is higher than peroxyacetic acid at temperatures below 80 °C. However, at temperatures above 80 °C, peroxyacetic acid was as effective as peroxyformic acid. Peracids have also been shown to be unsuitable for acid-sensitive epoxides, such as terpene oxide, as they cause severe hydrolysis of the epoxides.93
The peracetic acid route, which uses peracetic acids, such as m-chloroperoxybenzoic acid, as an oxidant, is one of the most common industrial epoxidation methods.42 These chemicals produce significant amounts of acid waste, which poses safety and environmental issues.92,94 Peracids have also been shown to be unsuitable for acid-sensitive epoxides, such as terpene oxide, as they cause severe hydrolysis of the epoxides.76 As a result, researchers have been seeking an alternative to the peracid epoxidation method that is selective, safe, and “clean”.
3.2.3.2 Hydroperoxide routes. The epoxidation of alkenes using hydroperoxides, such as cumene hydroperoxides (CHP), ethylbenzene hydroperoxides (EBHP) and tert-butyl hydroperoxides (TBHP), is a better alternative to peracids as they are easier to handle and less hazardous for industrial epoxidation.95 Hydroperoxides are significantly superior to H2O2, as they are soluble in hydrocarbons, while the dissolution of hydrogen peroxide is only possible in polar solvents. Organic peroxides have also been successfully used for the epoxidation of acid-labile epoxides, resulting in a better yield than peracids.12,42 Balula and Bruno studied the epoxidation of (R)-(+)-limonene with TBHP and reported a 99% conversion with 100% selectivity, highlighting the advantage of using organic hydroperoxides.75 Chiker et al. showed that when supported titanium is employed as the catalyst, TBHP is significantly more selective towards epoxides than H2O2.55
Studies have also focused on developing a heterogeneous catalyst for epoxidation using organic peroxides as oxidants. Charbonneau and Kaliaguine reported the epoxidation of (R)-(+)-limonene with TBHP using a titanium-based heterogeneous catalyst at a temperature of 75 °C in acetonitrile.81 They reported an (R)-(+)-limonene conversion of 80% at 79% selectivity to limonene-1,2-epoxide. The reaction parameters, such as temperature, TBHP concentration, and solvent polarity, have been optimized. Solvents with higher polarity facilitate a higher activity for the epoxidation reaction with TBHP compared to solvents with lower polarity. Very little progress was made while utilizing TBHP to increase transformation to limonene bis-epoxide above Ti/SBA-16 catalytical system.96 Recent research by Cunningham et al. produced a comparable outcome using H2O2 as the oxidizing agent in an attempt to epoxidize limonene.97
Using TBHP as an oxidant produces radicals during the epoxidation reaction that are the same as observed for the epoxidation of α-pinene using other similar kinds of oxidants. Becerra et al. demonstrated the epoxidation of α-pinene with TBHP by applying an iron-based heterogeneous catalyst at a temperature of 50 °C in acetone. They obtained an α-pinene conversion of 83% after 23 hours to many oxidative products, mainly verbenone. Their study concluded that under both catalytic and non-catalytic conditions, TBHP decomposes to form radicals that promote allylic oxidation of α-pinene (Scheme 21).29
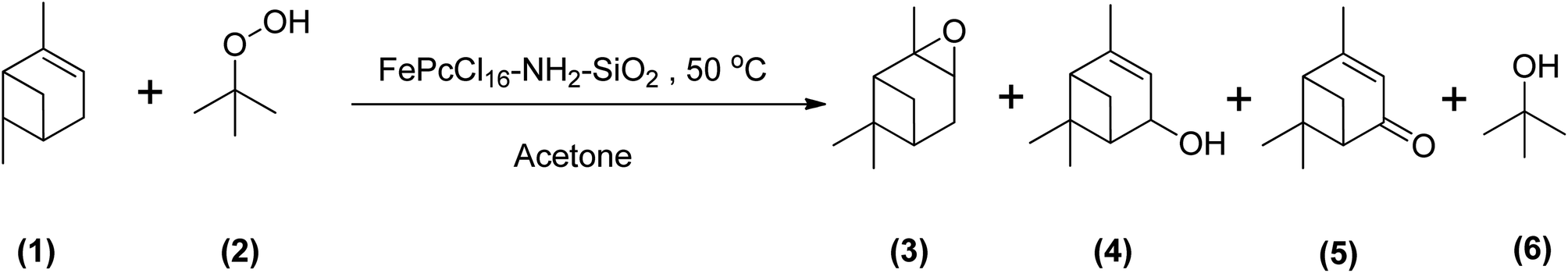 | ||
| Scheme 21 Epoxidation of (1) α-pinene with (2) TBHP producing (3) α-pinene oxide, (4) verbenol, (5) verbenone and (6) tert-butanol. | ||
The use of organic peroxides, such as TBHP, generates organic by-products that require additional separation processes. For instance, in the presence of TBHP, (R)-(+)-limonene and α-pinene epoxide produce tert-butanol, which necessitates the further separation of the alcohol from the epoxide products.98 In this regard, a greener and more selective oxidant is required to develop a selective epoxidation process for (R)-(+)-limonene and α-pinene.
3.3 Strategies to enhance reaction efficiency
Fig. 3 illustrates that the formation of a Keggin structure occurs at a pH of approximately 1. This structure is easily synthesized and highly stable with the general formula [Xn+M12O40](8−n)−. Where P or silica (Si) is commonly used as the central atom (X) and molybdenum (Mo) and W are the addenda atoms (M). Increasing the pH to about 2.2 leads to the hydrolysis of the Keggin structure, resulting in the formation of a lacunary Keggin with the general formula [XM11O39]n−. Other metal ions, such as iron (Fe2+), nickel (Ni2+), or cobalt (Co2+), can be incorporated into the lacunary Keggin structure. Balula et al. (2013) reported the use of lacunary Keggin phosphotungstates as a catalyst for the epoxidation of (R)-(+)-limonene with hydrogen peroxide at 80 °C, using acetonitrile as a solvent. They found that the catalyst exhibited high activity, achieving complete conversion of (R)-(+)-limonene to limonene-1,2-epoxide, bis-epoxide, and limonene-1,2-diol. In the pH range between 3.5–5, a Wells-Dawson structure with the general formula [X2n+M18O62](2n−16)− and a planar Anderson structure with the general formula [Xn+M6O24](12−n)− are typically obtained.
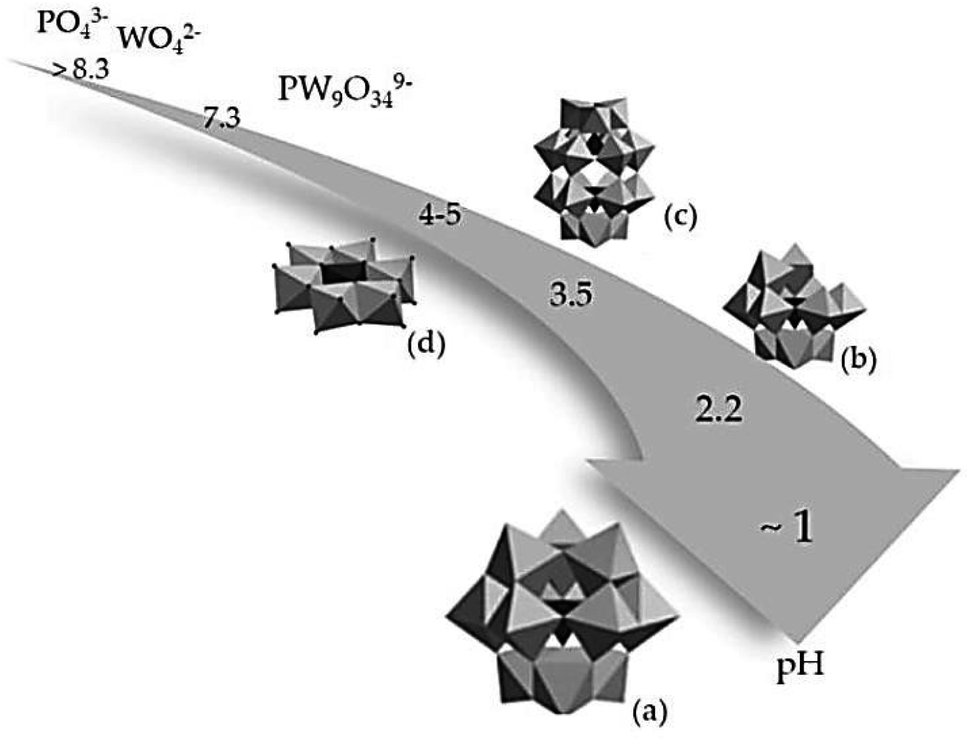 | ||
| Fig. 3 Influence of acidity on the structure of the polyoxometalates complex, (a) Keggin, (b) lacunary Keggin, (c) Wells–Dawson and (d) Anderson structure.102 | ||
Polyoxometalates (POMs) with transition metal substitutions have gained considerable interest as oxidatively stable catalysts. However, achieving high reaction rates and epoxide yields typically requires the use of chlorinated solvents. Heteropolyanions, formed by fusing an anion at the complex centre under acidic conditions, have become a focus of research. Various anions, including beryllium (Be), boron (B), aluminium (Al), silicon (Si), germanium (Ge), tin (Sn), and phosphorus (P), can be fused. When counter-ions such as protons (H+), Na+, NH4+, K+, and Cs+ associate with these structures, a secondary structure (salts) can form. Heteropolyacids (HPAs) are solid POMs composed of heteropolyanions, hydration water, and protons as counter-ions. HPAs have strong acidity, reportedly stronger than mineral acids. H3PW12O40, a commonly used POM/HPA species, is a superacid employed in many catalytic processes, including alkylation, acylation, and oxidation.103 POM catalysts are gaining popularity in oxidation due to their high Brønsted acidity, which provides efficient redox capability enabling rapid reactions and regeneration.104 The redox mechanism of POMs in oxidation reactions can use either di-oxygen (O2) or hydrogen peroxide (H2O2) as oxidants, where following the oxidation of the substrate, the POM is re-oxidized by the oxidant.
Numerous studies have investigated the suitability of different compounds, such as Fe,105 Mn,106 Ru,107 Re,108 W,109 alumina,110 in addition to hydrotalcite,111 with hydrogen peroxide for the epoxidation of alkenes. Among these, W-based POM was found to be the most effective H2O2.112 whereas commonly used Mo and Ti-based catalysts were less effective with H2O2.113 The use of POM in epoxidation was first discussed by Venturello et al. (1983),71 who reported the epoxidation of 1-dodecane using H2O2 oxidant with Na2WO4 and H3PO4 in biphasic conditions. They identified the active oxidative species [PO4[W(O)(O2)2]4]3–, which is commonly referred to as the Venturello anion (PW4).74 The mechanism of alkene epoxidation with the Venturello anion is presented in Scheme 22.
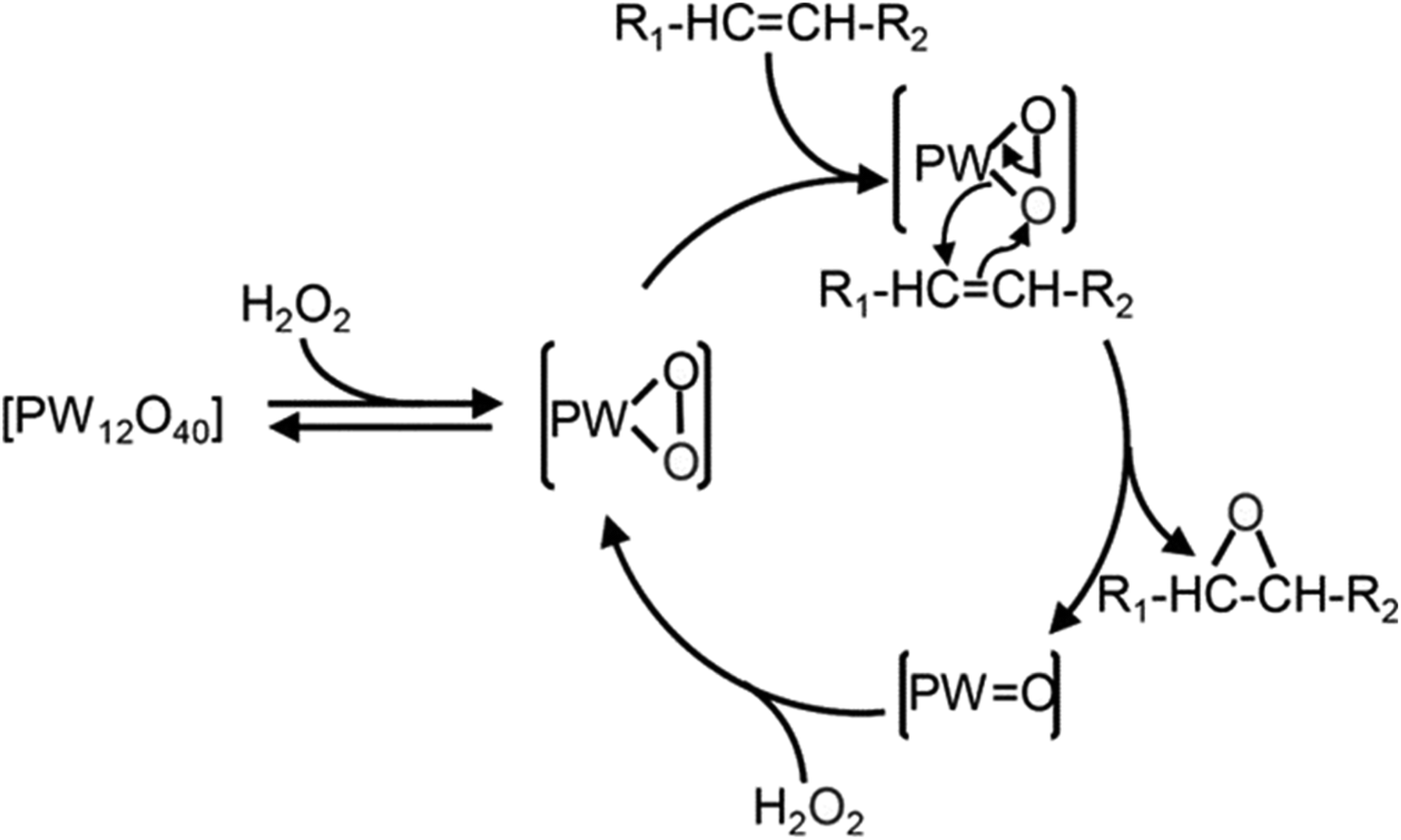 | ||
| Scheme 22 Reaction mechanism of alkene epoxidation using Venturello anion as the catalyst.114 | ||
Ishii et al. (1988) also investigated the suitability of POM for alkene epoxidation.73 They tested the activity of three POM species, namely phosphotungstate (H3PW12O40), H4SiW12O40, and H3PMO12O12, in biphasic conditions using H2O2 as an oxidant in chloroform solvent. Among all POM tested, phosphotungstate was found to be the most active. Like Venturello's work, the use of a PTC (quaternary ammonium salts) as a counter-ion was necessary. Fig. 4 depicts the structure of the Venturello anion.
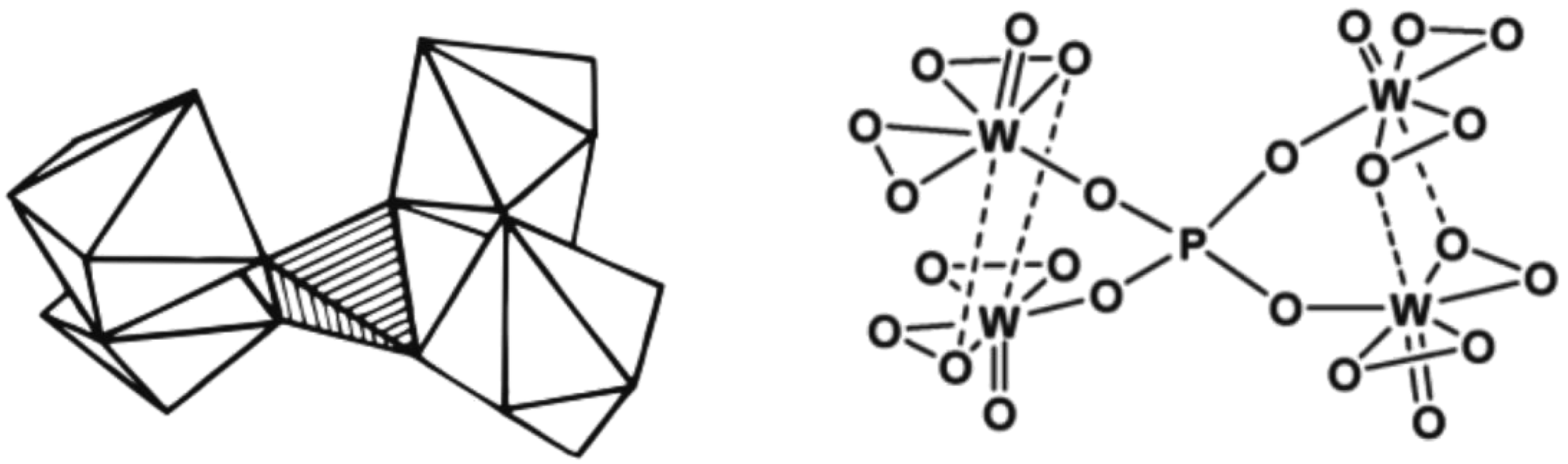 | ||
| Fig. 4 The visualisation of the structure of Venturello anion (PW4).116 | ||
Ishii-Venturello chemistry has gained interest in alkene epoxidation. A study by Duncan et al. (1995)43 investigated the active species, the Venturello anion, in the epoxidation of 1-octene with H2O2. They confirmed the presence of PW4 as the active species and compared the activity of 11 POMs. The Venturello anion required at least a 50-fold excess of H2O2 to form from the P–W precursors. Interestingly, they observed the formation of subsequent peroxo species (SPS) following the interaction of PW4 species and the substrate. The SPS was rapidly regenerated to form PW4 species by H2O2, as shown in Scheme 23. The PW4 species was found to be the most active POM for epoxidation, exhibiting more than double the activity of the peroxotungstate species ([[WO(O2)2(H2O)]2O]2–). However, the use of chlorinated solvents is required to facilitate the catalyst's acid character, which is a disadvantage. Also, the irreversible adsorption of epoxide and diol can deactivate the catalyst.114,115
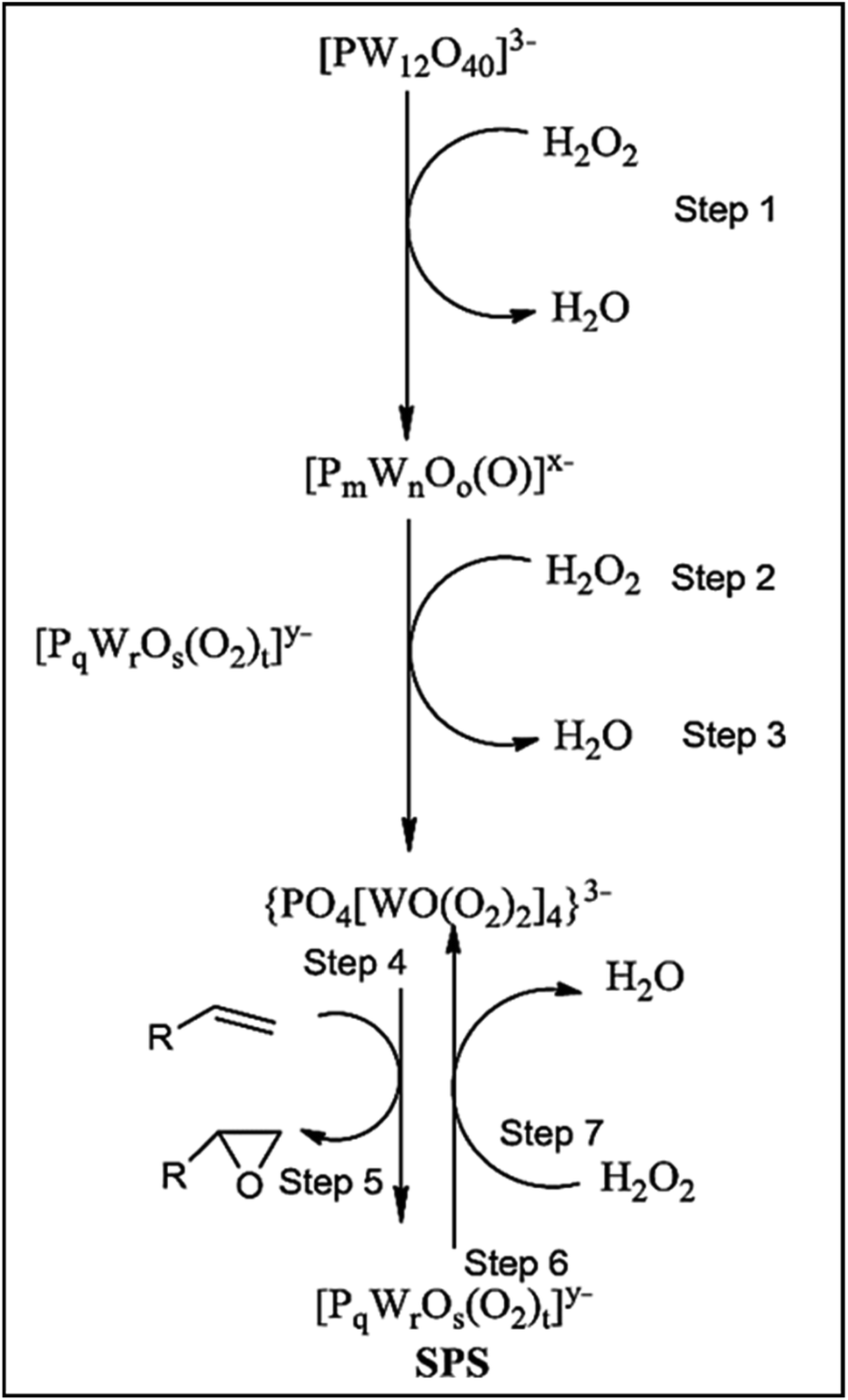 | ||
| Scheme 23 Mechanism of active species formation.12 | ||
Polytungstophosphate and PTC were used to adapt the mechanism for epoxidizing (R)-(+)-limonene and α-pinene using H2O2, as shown in Fig. 5. The biphasic nature of the epoxidation of (R)-(+)-limonene and its oxidative products requires the use of a hydrophobic substrate that is not miscible with aqueous H2O2. Fig. 5 summarizes all the reactions involved in the epoxidation of ((R)-(+)-limonene. The formation of the oxidative species follows the Ishii-Venturello chemistry, which has been extensively studied in the literature.12,43 The formation of the polytungstophosphate catalyst from H2O2, tungstate, and phosphate is facilitated by shuttles from the organic phase through PTC (Q+). During the epoxidation reaction, both (R)-(+)-limonene and limonene-1,2-epoxide receive active oxygen. The H+ ions participate in the formation of the oxidative species as well as the protonic attack on the epoxides at the interphase. When a protonic attack occurs between the epoxides and H2O (nucleophile), a diol is formed. POM has been used as a catalyst in significant research on alkene epoxidation, as shown in Table 4.
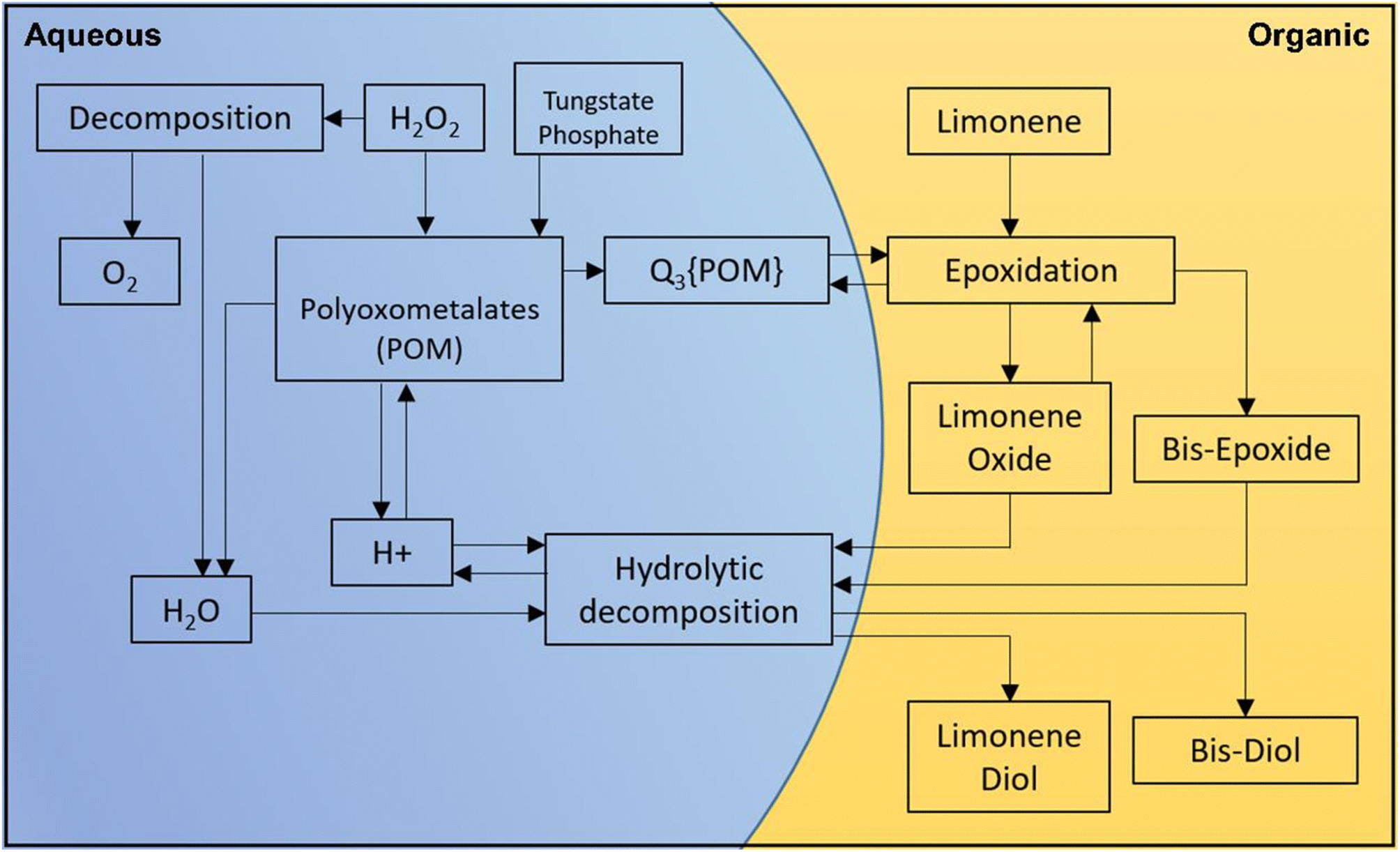 | ||
| Fig. 5 Overall reaction scheme for the epoxidation of (R)-(+)-limonene, the formation of oxidative species, the decomposition of H2O2 and the hydrolytic decomposition of the epoxides. Each reaction is visualized in the schematic diagram (an aqueous phase is shown in blue, while an organic phase is shown in gold). | ||
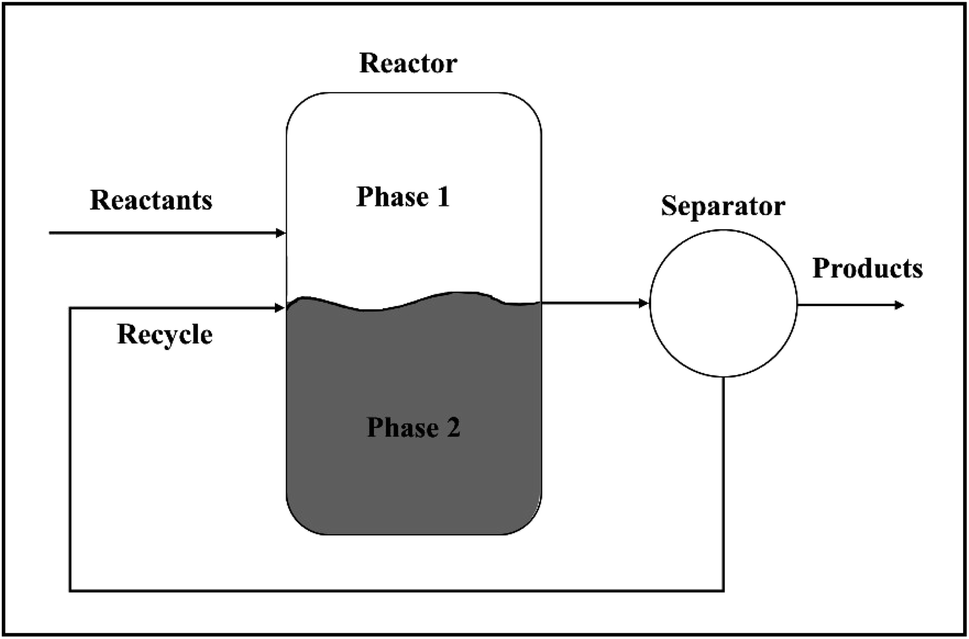 | ||
| Fig. 6 A simplified version of a biphasic reaction system showing product separation and recycling. | ||
Typically, one of the phases in a biphasic mixture is a water-based aqueous phase because of the high immiscibility of the organic phase.117 However, biphasic systems consisting of two fluorous organic phases or involving ionic liquids have also been used. Examples of biphasic reactions involving the aqueous-organic system include alkylation,118 carbonylation,119 Diels–Alder reaction,120 epoxidation,71 and hydrogenation.121 Most organic reactions in a biphasic system are performed using a phase transfer catalyst or in emulsification. Both approaches have many benefits, especially their effect on reaction rates.122–127 Although one method may be dominant over another, there are instances where the use of one method significantly alters the reaction rates. For example, Menger and Elrington (1991) found that the oxidation of mustard was 80 times faster in emulsification than in a non-emulsified system using a phase transfer catalyst.128 Some studies have investigated the prospect of having both methods in one system.
Emulsification in a biphasic reaction can also be achieved in a microfluidic and membrane reactor. De Bellefon et al. (2000) demonstrated that the isomerisation of allylic alcohol could be emulsified using a microfluidic reactor.129 They concluded that the application of a microfluidic reactor enhances emulsification, allowing for rapid screening of process variables.
An example of a biphasic epoxidation reaction involving (R)-(+)-limonene was reported by Villa et al. (2002). They studied the biphasic epoxidation of (R)-(+)-limonene using oxidant (H2O2) with heterogeneous peroxotungstophosphate (Venturello anion) on Amberlite IRA-900 in acetonitrile. According to their results, acetonitrile increased the (R)-(+)-limonene concentration in the aqueous phase, highlighting the relationship between the concentration of (R)-(+)-limonene and the mass transfer rate in the biphasic system.130
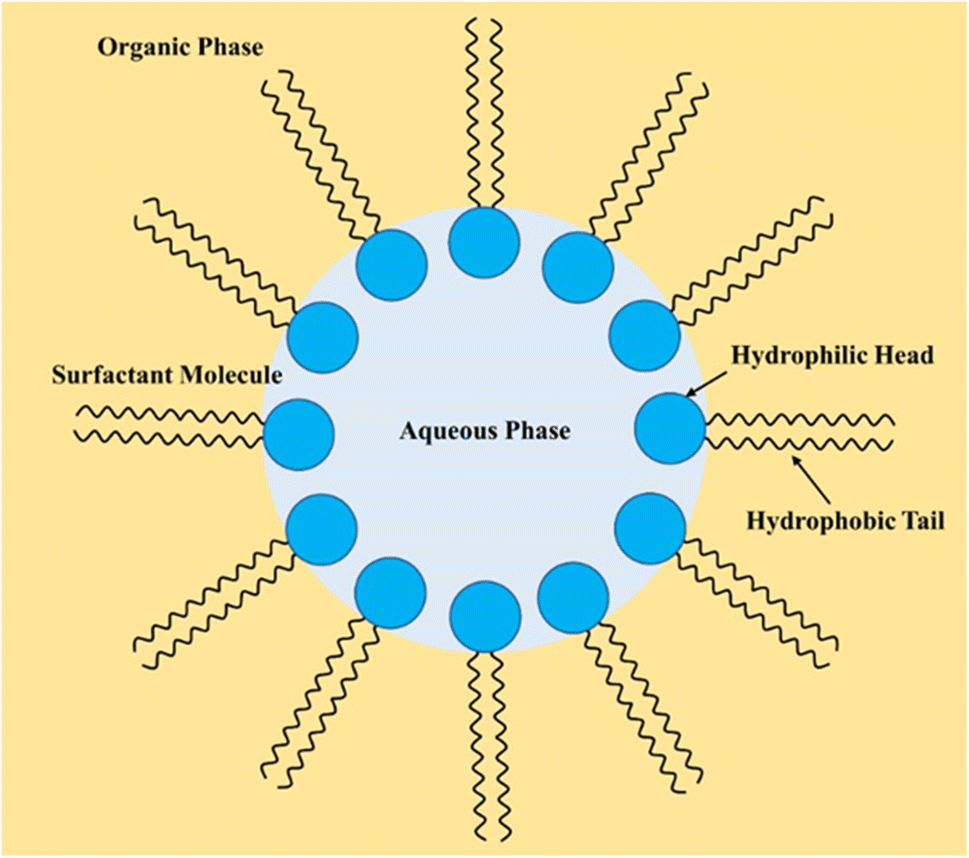 | ||
| Fig. 7 Schematic of a ‘water-in-oil’ emulsion where the hydrophobic tail of the surfactant stretches towards the organic phase (gold background). In the meantime, the hydrophilic head remains in the aqueous phase (blue background). | ||
| Class | Surfactant head net charge | Examples |
|---|---|---|
| Nonionic | No charge | Alkylphenol ethoxylates (Triton™ X-100) (Doong et al., 1998),140 sorbitan monolaurate (SPAN® 20), polyethene glycol sorbitan monolaurate (TWEEN® 20) (Bishopp et al., 2014)141 |
| Anionic | Negative | Sulfonate: perfluorooctanesulfonate (PFOS) (Vecitis et al., 2008),142 carboxylate: sodium stearate (Wang et al., 2010)143 |
| Cationic | Positive | Quaternary ammonium salts: cetyl methyl ammonium bromide (CTAB) (Doong et al., 1998),140 methyltrialkyl (C8–C10) ammonium chloride (Adogen® 464) (Lee and chang, 1978)144 |
| Zwitterionic | Negative and positive (two heads) | Cocamidopropyl betaine (CAPB) (Hunter and Fowler Jr, 1998),131 phosphatidylethanolamine (Attwood et al., 1992)145 |
| Type | Example reaction | References |
|---|---|---|
| Benzyltriethylammonium chloride | Alkylation | (Kimura et al., 1983)146 |
| Methyltricaprylammonium chloride/methyltrioctylammonium chloride | Oxidation | (Venturello and D'Aloisio, 1988; Neumann and Gara, 1994)74,147 |
| Methyl tributyl ammonium chloride | Esterification | (Desikan and Doraiswamy, 2000)148 |
| POM catalyst | Substrate | Oxidant | Temperature | Solvent | Conversion (%) | Selectivity (%) | Reference |
|---|---|---|---|---|---|---|---|
| [M4(H2O)2-(PW9O34)2]n−, M = Co(II), Mn(II) and Fe(III) | (R)-(+)-Limonene | 30 wt% H2O2 | 80 °C | Acetonitrile | Co(II), Mn(II), Fe(III) = 86%, 68%, 89% | Co(II), Mn(II), Fe(III) = 51%, 82%, 77% | Santos et al., 2017 (ref. 167) |
| K10[M4(H2O)2-(PW9O34)2]·20H2O M = Cu(II) | (R)-(+)-Limonene | 30 wt% H2O2 | 2 °C | 1,2-Dichloroethane | 96% | 95% to limonene-1,2-epoxide | Egusquiza et al., 2012 (ref. 62) |
| [SeO4[WO(O2)2]s2] | Cyclohexene | 30 wt% H2O2 | Ambient | Acetonitrile | 99% | 97% to cyclohexene oxide | Kamata et al., 2010 (ref. 168) |
| [Ti2(OH)2As2–W19O67(H2O)]8− | (R)-(+)-Limonene | 30 wt% H2O2 | 50 °C | Acetonitrile | 76% | 75% to limonene-1,2-epoxide | Donoeva et al., 2010 (ref. 169) |
| [PW4O32] | Cyclooctene | 30 wt% H2O2 | 65 °C | Ethyl acetate | 99% | 99% to cyclooctene oxide | Ding et al., 2008 (ref. 170) |
| Na2WO4/H2WO4 | Cyclooctene | 30 wt% H2O2 | 60 °C | Solvent-free | 99% | 99% to cyclooctene oxide | Maheswari et al., 2006 (ref. 171) |
| Na8H[ΔPW9O34]·19 H2O | (R)-(+)-Limonene | 35 wt% H2O2 | 2 °C | 1,2-Dichloroethane | 100% | 96% to limonene-1,2-epoxide | Casuscelli et al., 2005 (ref. 57) |
The general mechanism of a PTC in a biphasic mixture is shown in Fig. 8, where the Q+ ion pairs with the anionic reactants or oxidants (Y−) in the aqueous phase before shuttling to the organic phase. Additionally, alkenes (R) absorb active oxygen in the organic phase, allowing the Q+ to return to the aqueous phase to complete the cycle. The high conversions and yields associated with PTC epoxidations, which typically occur in two-phase systems, have attracted attention. PTCs can be immobilized as a solid support, making them reusable and avoiding waste problems completely. Furthermore, the process can be made more commercially viable as PTCs are stable at high temperatures, enabling higher conversion rates in less time.
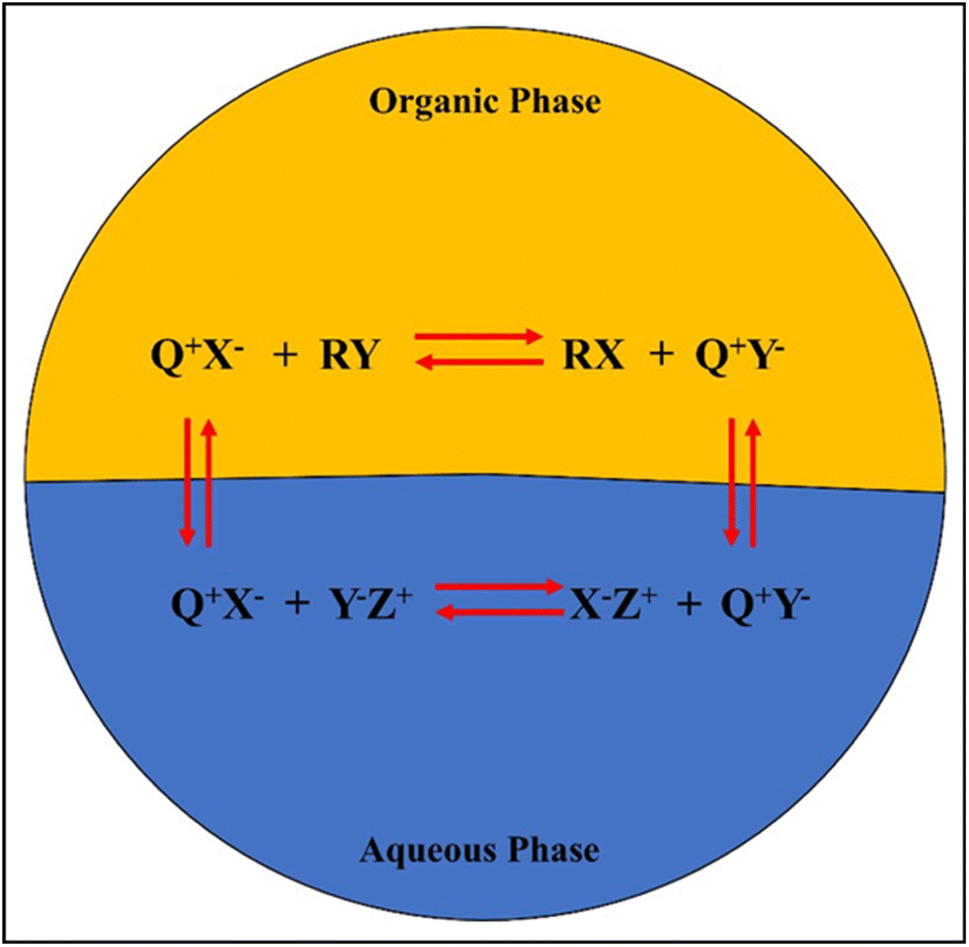 | ||
| Fig. 8 Mechanism of PTC in a biphasic mixture. | ||
Ahmat and Kaliaguine (2023) present a significant advancement in the field of terpene epoxidation, specifically focusing on limonene and pinene.54 The main finding of the study is the successful epoxidation of these hydrophobic terpenes using dimethyldioxirane (DMDO) generated in situ in a microemulsion system. The study highlights the efficacy of cetyltrimethylammonium hydrogen sulfate (CTAHS) as a phase transfer catalyst to homogenize the terpenes in the aqueous reaction medium. The study by Murugan and Siva (2005) presents a novel soluble multi-site phase transfer catalyst (MPTC) named 1,3,5-tris(4-(2,2′-bis(N-triethylammoniummethylene chloride)eth-1-ene)phenoxymethyl)benzene (TBTEAPB).139 The main finding is that this newly synthesized MPTC significantly enhances the C-alkylation of α-pinene with epichlorohydrin, especially when compared to single-site and di-site phase transfer catalysts (PTCs).
The TBTEAPB catalyst showed rate constants approximately 6 times higher than single-site PTCs and 3 times higher than di-site PTCs. This indicates a substantial improvement in catalytic efficiency, which is particularly significant for the field of terpene modification. The study also found that the reaction could be facilitated even under mild conditions, such as a low concentration of base (20% NaOH) and lower MPTC concentrations, making the process potentially more sustainable and cost-effective.
Apart from terpenes, PTC is widely used for various alkenes. For instance, Duncan et al. (1995) demonstrated the epoxidation of alkenes, such as 1-octene, using heteropolyacids in the presence of a PTC.43 Using a PTC system, tungsten(VI) and molybdenum(IV) based catalysts were investigated for the epoxidation of alkenes. The amount and nature of PTC determine alkene oxidation, in addition to the assembling anion of the catalyst.149 Similarly, Sato et al. (1996) described the catalytic epoxidation of alkenes using H2O2 in a system comprised of sodium tungstate catalyst, aminomethyl-phosphonic acid, and methyl tri-n-octyl ammonium hydrogen sulphate as the PTC, where both solvent and solvent-free epoxidation showed high product yield corresponding to the PTC amount. One study found that using quaternary ammonium heteropolymer-tungstate degraded quickly to oxo-peroxophosphate ions, which increased the yield of the epoxidation reaction system.150
Chen et al. (2004) reported the epoxidation of alkenes in a PTC system using acidic media and an organic solvent.125 In their study, they found that H2O2 is the best oxidant and sodium tungstate, and phosphoric acid are the best catalysts for this process. Phosphate and sulphate anions were tested in a recent study to assess how they affected the epoxidation of alkene in Arquad 2HT® systems.132 In the reaction, both anions were found to be effective. The phase transfer catalyzed system also showed that tungsten(IV) had a stronger activity than sodium tungstate. Bishopp et al. (2014) used tungsten catalysts to epoxide sunflower seed oil with H2O2, without the presence of common PTC. To overcome the mass transfer limitation associated with the biphasic system, a non-ionic surfactant was used to create emulsions. The emulsification reduced the diameter of droplets and facilitated the reaction. The researchers discovered that H2O2 decomposition increased exponentially above a certain catalyst concentration, which reduced the epoxidation rate by starving the system of oxidants. However, when the catalyst concentration was lower, the H2O2 decomposition was exceptionally low. By adding acetic acid to the tungsten catalyst, the H2O2 decomposition was almost negligible. They found that H2O2 was first order at lower catalyst concentrations.142
The separation of PTC from the reaction mixture has become a significant concern, leading to research on polymer-supported PTC. Desikan and Doraiswamy (2000) used aqueous sodium acetate to esterify benzyl chloride. They compared the activity of methyl tributyl ammonium chloride dissolved in a polymer with that of methyl tributyl ammonium chloride supported on a polymer. The supported PTC showed about 50% higher activity than the soluble form and was easy to separate post-reaction.148
The PTC agent typically consists of a cation part (e.g., quaternary ammonium, Q+) and an anion part (e.g., chloride, X−). Fig. 8 shows the general mechanism of a PTC in a biphasic mixture.
3.4 Stereoselective terpene epoxidation
Orange peel essential oils are rich in the (R)-enantiomer of limonene, which is also known as D-limonene (+)-limonene), a chiral molecule. Limonene bis-epoxide produces two isomers with distinct arrangements of the epoxide bridges for each limonene enantiomer, resulting in diverse reactivities relevant to subsequent polymerizations. Cunningham et al. (2020) used an aqueous H2O2 oxidant to epoxidize a variety of trisubstituted alkene bond-containing terpenes using tungsten-based polyoxometalate catalysts, which always produced two diastereomers, racemic mixtures of the cis- and trans-epoxides of each terpene.97 Similarly, mesoporous SBA-16 substituted with cobalt was used to produce cis- and trans-diastereomers of limonene by aerobic epoxidation, and the products had a precise distribution of 60:40 cis:trans limonene bis-epoxide (Scheme 24).50
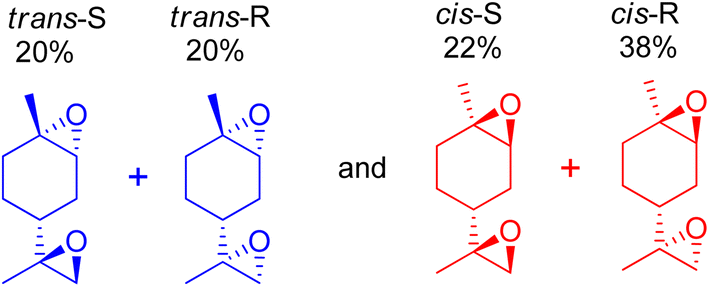 | ||
| Scheme 24 Various isomers of limonene bis-epoxide are produced from (R)-(+)−limonene. | ||
Zinc β-diminate complex was used as a catalyst to synthesize poly(limonene carbonate) from a cis/trans combination of (R)- and (S)-limonene oxide, which showed that trans-diastereomers of the epoxide are easily polymerized while the cis-diastereomers are left unreacted, resulting in high regio, diastereoisomer, and enantiomerically pure polymer.154–156 Recent studies have shown that the limonene-1,2-epoxide mixture (available commercially i.e. 40:60, cis:trans) can be used to synthesize polycarbonates and cyclic carbonates with a higher conversion of the trans-isomer than the cis-isomer in the presence of tetrabutylammonium halides (TBAX) as catalysts.155,157 The diastereomeric cis-has lower reactivity than the trans-due to the “boat-like” form of its transition state, which is correlated with its higher activation energy. A stereoselective epoxidation method needs to be developed to continue producing terpenes in their trans-form. Hauenstein et al. (2016) proposed a large-scale stereoselective synthesis method for the trans-isomer of limonene-1,2-epoxide (>1 kg per batch) by brominating (R)-(+)-limonene with N-bromosuccinimide (NBS) in an aqueous acetone solution.155 Endo-cyclic trans-bromohydrin is formed, which is readily oxidized into subsequent trans-limonene-1,2-epoxide by using a 60 °C aqueous sodium hydroxide solution for 2 hours (Scheme 25).
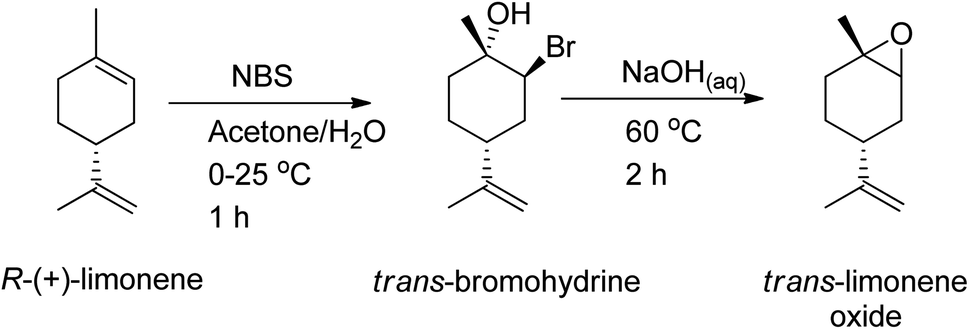 | ||
| Scheme 25 As a bromination source, NBS is used to undergo stereoselective epoxidation of (R)-(+)-limonene. | ||
The amount of trans-limonene-1,2-epoxide in the resulting products was found to be more than 85%. Recent studies have shown that both the exocyclic and endocyclic double bonds of (R)-(+)-limonene can be stereoselectively di-bromohydrated to form trans-limonene bis-epoxide (Scheme 26).
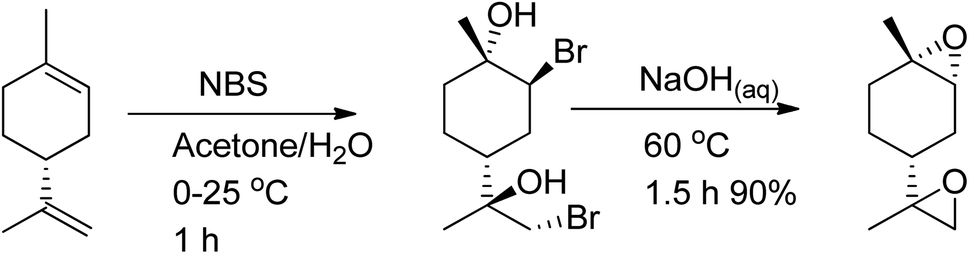 | ||
| Scheme 26 Using both (R)-(+)-limonene's exocyclic and endocyclic double bonds as starting materials, trans-LBE is synthesized via stereoselective di-bromohydration. | ||
Various parameters, such as reaction temperature, time, and the molar ratio of (R)-(+)-limonene to N-bromosuccinimide (NBS), were examined to produce trans-limonene bis-epoxide. The optimal conditions were found to be a 1:2 (R)-(+)-limonene:NBS molar ratio at 60 °C for 5 minutes, which resulted in a 97% yield. Furthermore, a stereoselective epoxidation mechanism for (R)-(+)-limonene was proposed, leading to the formation of trans-limonene bis-epoxide. The mechanism involves the production of a bromonium ion intermediate from the reaction between (R)-(+)-limonene and NBS in acetone and water (Step 1 in Scheme 27), which is then attacked by the hydroxyl group of water to form bromohydrin (Step 2) and eventually trans-limonene bis-epoxide. The excess NBS causes both double bonds to be bromohydrated (Step 3). Faster epoxidation reactions were observed, and it was suggested that the production of trans-limonene bis-epoxide occurred through an intramolecular SN2 reaction in which NaOH deprotonated OH to form an alkoxide intermediate, which then attacked the C–Br to replace the bromide ion as a leaving group (Step 4).
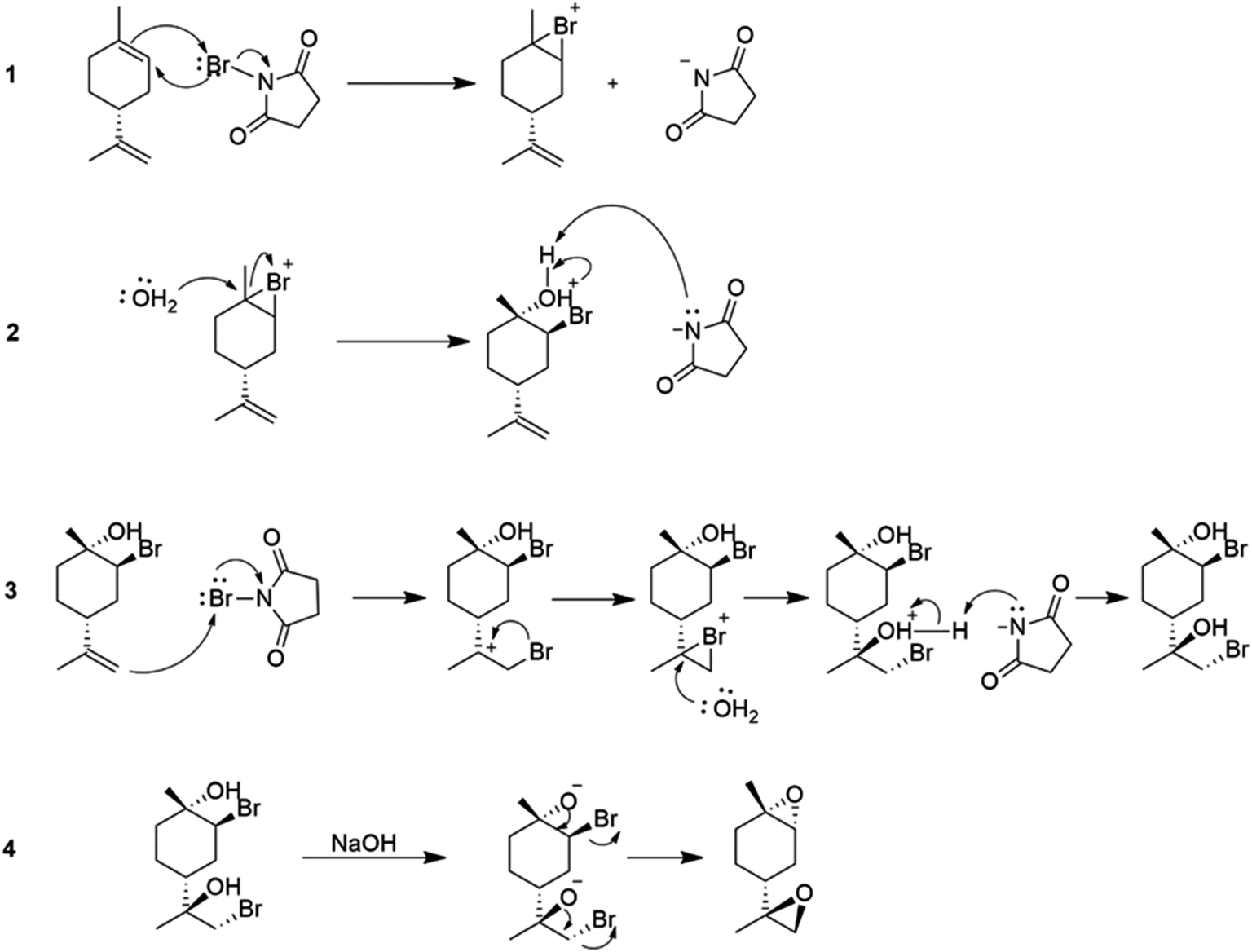 | ||
| Scheme 27 NBS-mediated stereoselective epoxidation of (R)-(+)-limonene, reaction mechanism. | ||
3.5 Reactor optimisation
Ranganathan and Sieber (2017) present an innovative approach to epoxidizing olefins like monoterpenes (3-carene, limonene, and α-pinene) by coupling the anthraquinone autooxidation process for hydrogen peroxide (H2O2) production with lipase-mediated epoxidation.163 Utilizing Candida antarctica lipase B (CALB) and a unique hybrid reactor setup, the study overcomes the limitation of continuous H2O2 addition, a common drawback in traditional methods. The process achieved high yields of epoxides (up to 88.8% for 3-carene and 83.8% for α-pinene) and allowed for the reuse of the catalyst for up to five cycles with minimal loss of activity. The study concludes that this semi-continuous approach is scalable to industrial standards and offers a broad range of applications due to its efficiency and sustainability.
The flow chemistry method also allows efficient photochemical reactions to be started by passing reaction streams through UV lamps or other light sources, and solid-supported reagents can be used as reactants to facilitate the purification process. Moreover, the ability to handle hazardous or toxic reagents safely is another benefit of continuous processes.
Microreactors offer an effective solution to reduce waste production and provide excellent reaction and thermal control due to their narrow channel diameters and static mixers, which allow for extremely good mixing in the millisecond range. Concentrated solvents or no solvents at all can be used, which reduces the amount of waste produced.162 Real-time process analytical technology (PAT) provides in-depth data for mechanistic research and ensures high-quality chemical outputs. The benefits of flow chemistry and continual processing methods can be increased by synthesizing chemicals in miniature micro- or milli-reactors, which typically have volume capacities between nanoliters and millilitres.164 Scaling up a continuous flow reactor can be achieved by operating it for a longer time or by using a “numbering up” method to execute reactions in several reactors simultaneously. Alternatively, large continuous flow reactors can be built to produce large quantities of product but may require more process development.165 In conclusion, flow chemistry and continuous processing provide sustainable and environmentally friendly alternatives to traditional batch synthesis methods, offering benefits such as efficient mixing, better temperature control, and reduced waste production. These methods can be further optimized using microreactors and real-time process analytical technology.
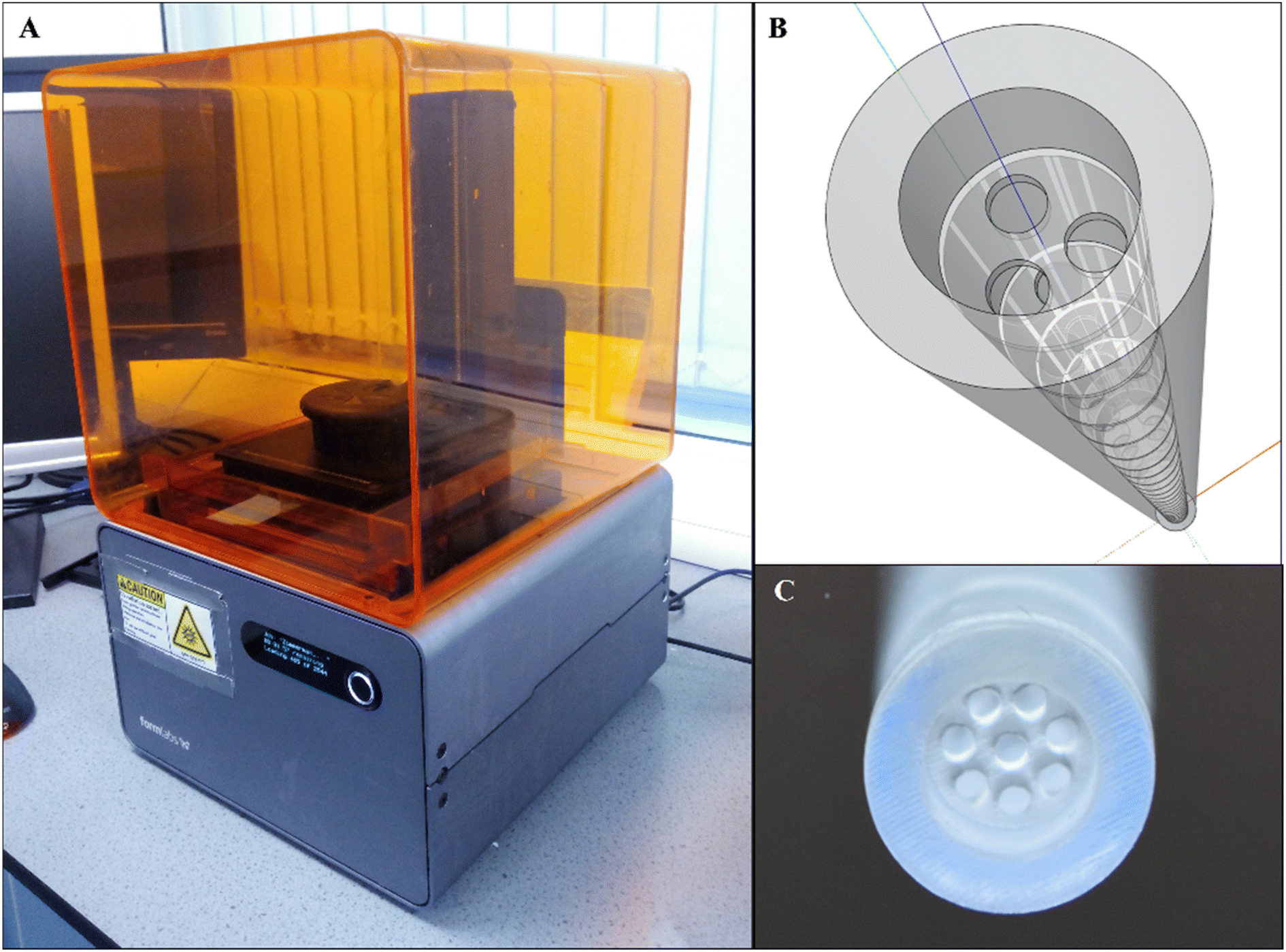 | ||
| Fig. 9 An application of additive manufacturing to fabricate millimetre-scale orifice baffles. (A) Form2 SLA 3D printer (B) a SketchUp drawing model showing a tri-orifice baffle (C) a printed, cured and cleaned multi-orifice baffled meso-OBR reactor.5 | ||
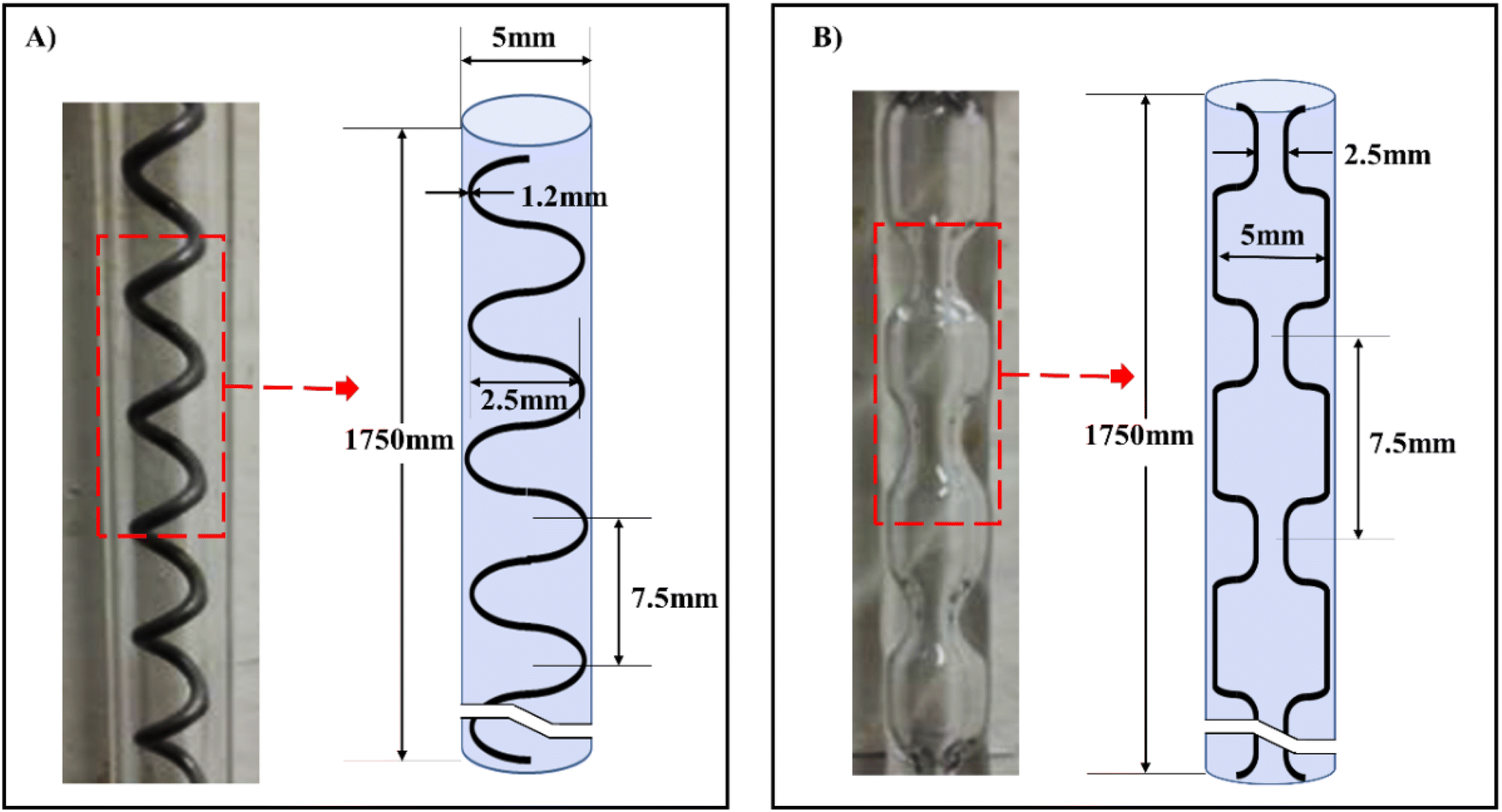 | ||
| Fig. 10 Meso-OBRs designs configuration and schematics of the conventional, (A) helical baffles (B) integral baffles.5 | ||
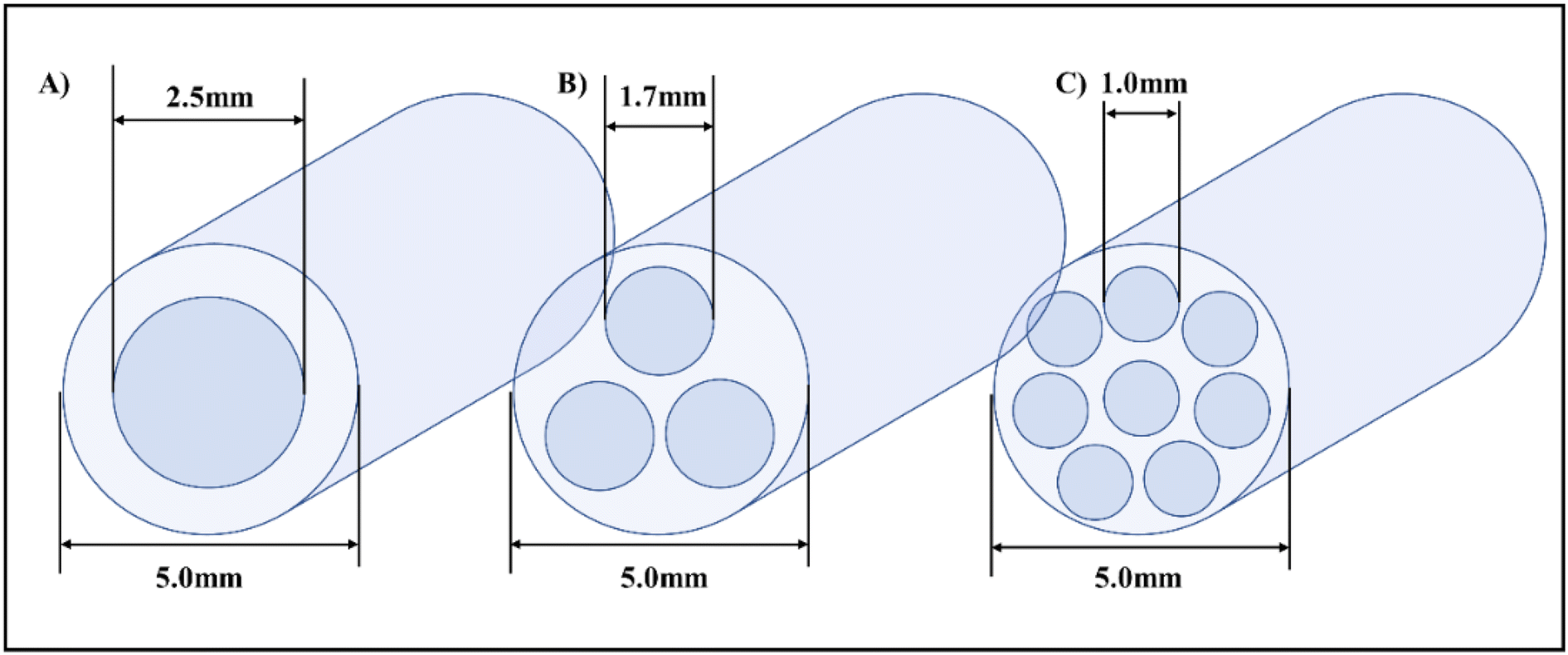 | ||
| Fig. 11 Orifice meso-OBR design schematics and configurations for 3D printing, (A) single orifice (B) tri-orifice (C) Multi-orifice.5 | ||
The smaller orifice diameters of tri- and multi-orifice baffles induce smaller drop sizes while increasing the surface at the same time, resulting in enhanced reaction rates. Multi-orifice baffles showed fewer variances in limonene epoxide yields than other baffle designs, indicating a high extent of plug flow and reproducibility. In a multi-steady-state screening of residence time, multi-orifice baffles could screen the parameter in half the time as helical baffles and just a fraction of the time as batch baffles. Tight control of process parameters enabled clear steady states to be attained at all residence times. The meso-OBR designs demonstrated efficient heat transfer capability under solvent-free conditions, enabling the reaction to become isothermal with a temperature change of just ±1 °C. By doing so, the need for solvent is eliminated, resulting in a 5-fold decrement in required reaction volume. Using multi-orifice meso-OBRs, terpenes can be epoxidized with H2O2, by up to an 80-fold intensification factor. We used helically baffled meso-OBRs to continuously epoxidize terpenes with H2O2 (Fig. 12). With multi-orifice meso-OBRs, the epoxidation of terpenes with H2O2 can be completed in just 30 minutes instead of the 8 hours required by conventional processes.
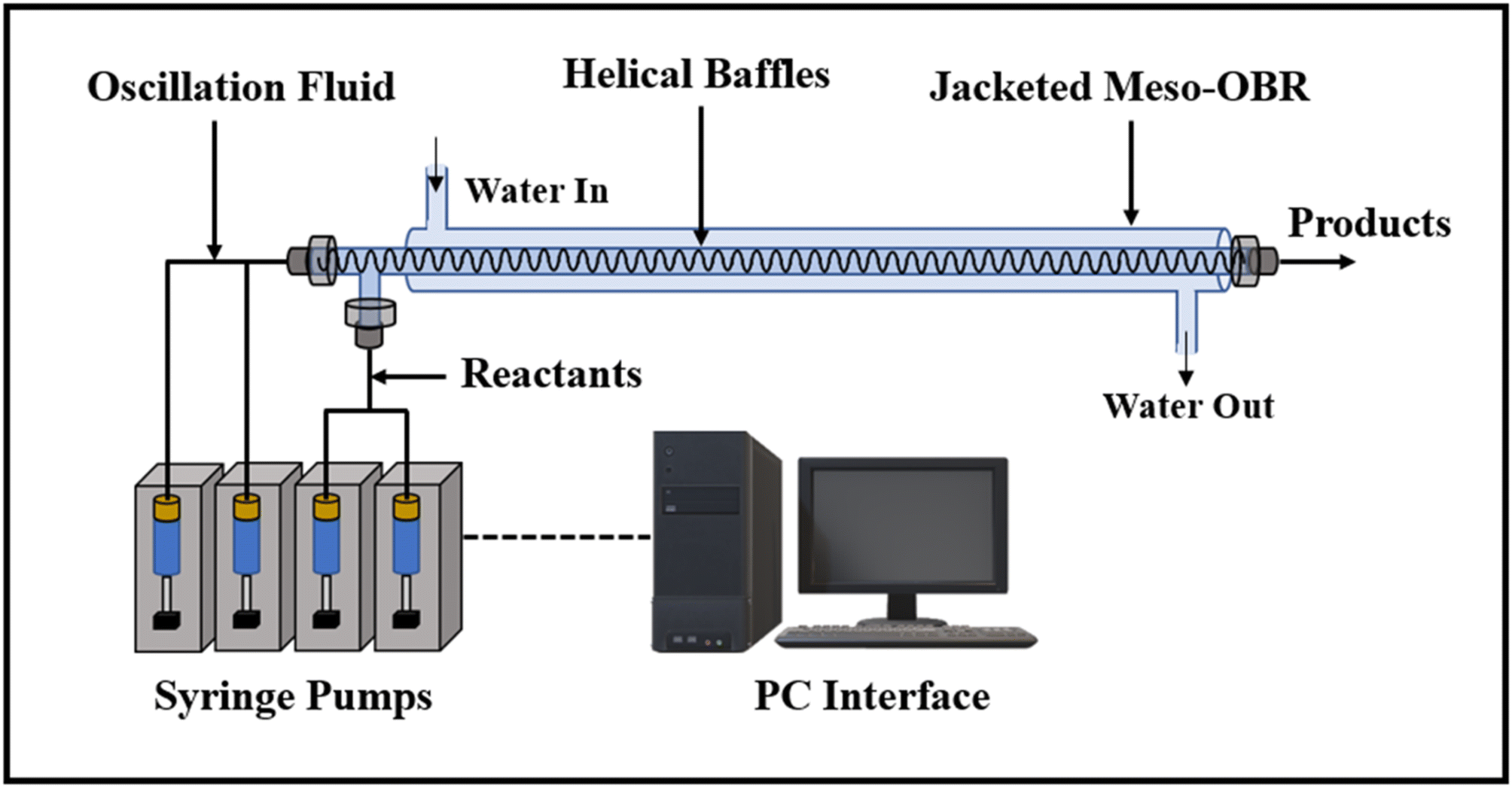 | ||
| Fig. 12 The helically baffled meso-OBR is used for the flow epoxidation of terpenes with H2O2.5 | ||
Tibbetts et al. (2021) demonstrated a solvent-free continuous flow epoxidation process of bio-renewable terpene feedstocks using a recyclable polyoxometalate (POM) phase transfer catalyst (PTC) made of tungsten and aqueous H2O2.166 The flow epoxidation was performed on a variety of bio-renewable terpene substrates, resulting in the production of large amounts of limonene 1,2-epoxide (Fig. 13). The use of static mixing channels in the flow microreactor enhanced heat and mass transfer rates between organic and aqueous streams, resulting in rapid epoxidation reactions with temperature control. The recovered catalyst can be recycled to produce additional epoxides, making it possible to obtain milligrams of a variety of epoxides by epoxidizing di- and tri-substituted alkenes. In contrast to biphasic batch epoxidation conditions, which can have potential hazards of thermal runaway reaction due to poor temperature control and ineffective mixing, flow epoxidation has a multitude of advantages suitable for large-scale epoxide production. H2O2 is an effective oxidant for catalytic epoxidation reactions in flow reactors for petroleum-based substrates. The use of static mixing channels in the flow microreactor enhances heat and mass transfer rates, making it possible to achieve 89% conversion of limonene to limonene 1,2-oxide and 90% selectivity with a residence time of 16.7 minutes. To achieve this, a 7.5 mL volume microreactor containing 2 mol% VPTC and 1.6 equivalents of H2O2 (pH 7.0) flowing at 27 mL h−1 was used under optimal conditions.168
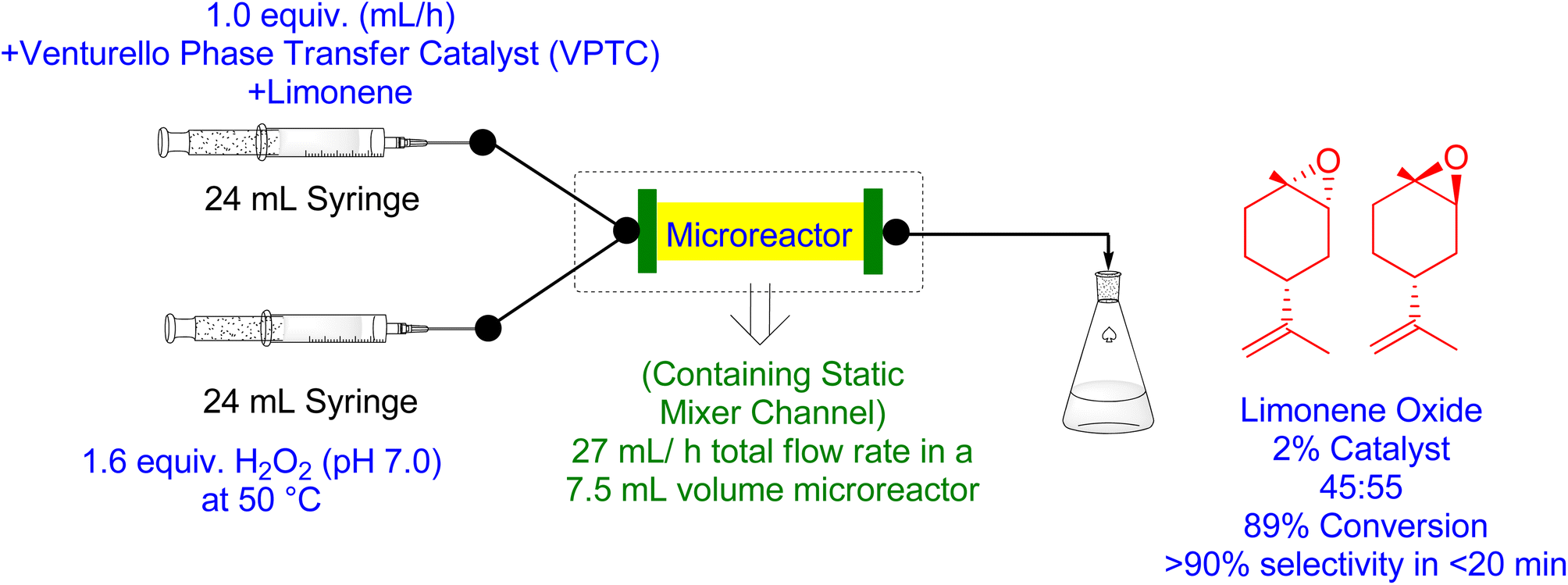 | ||
| Fig. 13 Catalytic flow epoxidation of limonene using Venturello Phase Transfer Catalyst (VPTC) (reproduced from ref. 166). | ||
In previous studies, α-pinene oxide produced with H2O2/VPTC in batch epoxidation was found to be hydrolyzed to its anti-diol. However, under flow conditions, the addition of 30 mol% Na2SO4 to aqueous H2O2 (pH 7.0) with a high catalytic loading of 3 mol% resulted in a 95% conversion and selectivity for the corresponding oxide (Fig. 14). Similarly, using a high temperature and catalytic loading (75 °C and 5 mol%) with a shorter static mixer residency time of 1.65 min, a total conversion of β-pinene into its respective β-pinene oxide with 55% conversion and 99% selectivity was achieved. We applied the same flow epoxidation protocol to untreated industrial CST, the world's largest renewable monoterpene feedstock, with high catalytic loading and the equivalent of H2O2 (5 mol% and 2.2) and the addition of 30 mol% Na2SO4. After a total residence time of 15 min at 60 °C, the three main components (α-pinene, 3-carene, and β-pinene) were consumed up to 95%, resulting in the production of α-pinene oxide (63%) and 3-carene oxide with excellent selectivity.166
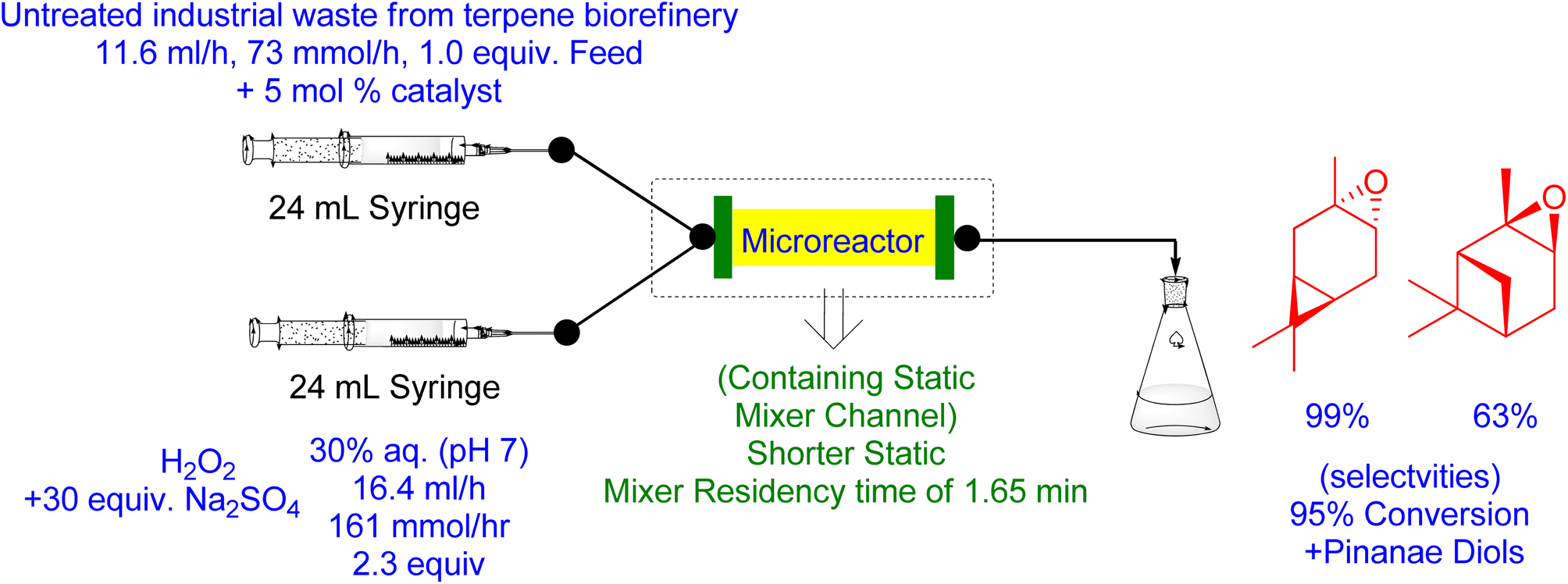 | ||
| Fig. 14 Catalytic flow epoxidation of Untreated Industrial waste from biorefinery using Venturello Phase Transfer Catalyst (VPTC) (Reproduced from ref. 166) | ||
4. Conclusions and prospects
The olefinic terpene family is a promising renewable resource that could lead to significant economic gains in various chemical industries. Terpenes can be found in the resin of coniferous trees and the essential oils of citrus fruits, and epoxidation reactions are a promising approach for transforming them into value-added compounds for the pharmaceutical, fragrance, and flavouring industries. This critical review analyzes catalytic oxidative upgrades of renewable terpene olefins and discusses the challenges in achieving high selectivity and yields with complex reaction mixtures, hazardous organic solvents, and high concentrations of oxidants. While the review by Mahamat et al. (2021)6 offers valuable insights into limonene epoxidation using various oxidants, our work uniquely addresses the complexities of achieving high selectivity and yields in the presence of multiple challenges such as hazardous solvents and oxidants. We believe that our focus on these aspects, as well as on the potential synergy between heterogeneous and homogeneous catalysis, adds a distinctive layer of understanding to the field. Further advancements are needed to achieve industrial demands, and thorough knowledge of reaction mechanisms is necessary to design epoxidation catalysts that are highly active, selective, environmentally friendly, and cost-effective. The synergy between heterogeneous and homogeneous catalytic properties could be highly beneficial, but satisfactory catalyst systems that offer rates and selectivity equivalent to those of homogeneous counterparts are still lacking. Research on surfactants, phase transfer catalysts, inorganic salts, and reaction kinetics is necessary to guide the future design of innovative catalysts for green epoxidation that are simple to separate and regenerate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The lead author expresses his gratitude for the financial support provided by an Engineering and Physical Sciences Research Council (EPSRC) grant (Sustainable Polymers, grant number EP/L017393/1) and the Ministry of Higher Education, Malaysia.References
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540(7633), 354–362 CrossRef CAS PubMed.
- A. J. Silvestre and A. Gandini, Terpenes: major sources, properties and applications, in Monomers, polymers and composites from renewable resources, Elsevier, 2008, p. 17–38 Search PubMed.
- M. N. Belgacem and A. Gandini, Monomers, polymers and composites from renewable resources, Elsevier, 2011 Search PubMed.
- G. Fráter, J. A. Bajgrowicz and P. Kraft, Fragrance chemistry, Tetrahedron, 1998, 54(27), 7633–7703 CrossRef.
- M. F. M. G. Resul, et al., Continuous process for the epoxidation of terpenes using mesoscale oscillatory baffled reactors, Chem. Eng. Process, 2022, 177, 108998 CrossRef CAS.
- Y. Mahamat Ahmat, et al., Epoxidation of terpenes, Catalysts, 2021, 11(7), 847 CrossRef CAS.
- L. Shen, E. Worrell and M. Patel, Present and future development in plastics from biomass, Biofuel. Bioprod. Biorefin., 2010, 4(1), 25–40 CrossRef CAS.
- P. T. Anastas and M. M. Kirchhoff, Origins, current status, and future challenges of green chemistry, Accounts Chem. Res., 2002, 35(9), 686–694 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green chemistry and the plastic pollution challenge: towards a circular economy, Green Chem., 2020, 22(19), 6310–6322 RSC.
- M. Stekrova, et al., Isomerization of α-pinene oxide using Fe-supported catalysts: selective synthesis of campholenic aldehyde, Appl. Catal., A, 2014, 470, 162–176 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- G. Yadav and D. Satoskar, Kinetics of epoxidation of alkyl esters of undecylenic acid: comparison of traditional routes vs. Ishii-Venturello chemistry, J. Am. Oil Chem. Soc., 1997, 74(4), 397–407 CrossRef CAS.
- P. Gallezot, Process options for converting renewable feedstocks to bioproducts, Green Chem., 2007, 9(4), 295–302 RSC.
- E. Breitmaier, Terpenes: flavors, fragrances, pharmaca, pheromones, John Wiley & Sons, 2006 Search PubMed.
- A. Gandini, The irruption of polymers from renewable resources on the scene of macromolecular science and technology, Green Chem., 2011, 13(5), 1061–1083 RSC.
- A. Llevot, et al., Renewability is not enough: recent advances in the sustainable synthesis of biomass-derived monomers and polymers, Chem.–Eur. J., 2016, 22(33), 11510–11521 CrossRef CAS PubMed.
- M. R. Thomsett, et al., Progress in the synthesis of sustainable polymers from terpenes and terpenoids, Green Mater., 2016, 4(3), 115–134 CrossRef.
- M. Colonna, et al., Synthesis and radiocarbon evidence of terephthalate polyesters completely prepared from renewable resources, Green Chem., 2011, 13(9), 2543–2548 RSC.
- M. Firdaus, L. Montero de Espinosa and M. A. Meier, Terpene-based renewable monomers and polymers via thiol–ene additions, Macromolecules, 2011, 44(18), 7253–7262 CrossRef CAS.
- P. Knuuttila, Wood sulphate turpentine as a gasoline bio-component, Fuel, 2013, 104, 101–108 CrossRef CAS.
- M. a. F. Gscheidmeier and H. Turpentines, in Ullmann's Encyclopedia of Industrial Chemistry, ed. M. a. F. Gscheidmeier, 2000 Search PubMed.
- R. Ciriminna, et al., Limonene: a versatile chemical of the bioeconomy, Chem. Commun., 2014, 50(97), 15288–15296 RSC.
- M. F. M. G. Resul, et al., Development of rapid and selective epoxidation of α-pinene using single-step addition of H2O2 in an organic solvent-free process, RSC Adv., 2021, 11(52), 33027–33035 RSC.
- W. Erman, Chemistry of monoterpenes. Part A. Edited by PG Gassmann, Marcel Dekker, New York, 1985 Search PubMed.
- L. Vogli, et al., Life cycle assessment and energy balance of a novel polyhydroxyalkanoates production process with mixed microbial cultures fed on pyrolytic products of wastewater treatment sludge, Energies, 2020, 13(11), 2706 CrossRef CAS.
- J. C. Philp, et al., Bioplastics science from a policy vantage point, New Biotechnol., 2013, 30(6), 635–646 CrossRef CAS PubMed.
- R. Albert, S. Traynor, and R. Webb, Fragrance and flavor chemicals. Naval Store-Production, Chemistry, Utilization, ed. D. F. Zinkel and J. Russell, PULP Chemical Association, New York, 1989, p. 479 Search PubMed.
- J. Derfer and S. Traynor, Chemistry of Turpentine in Naval Stores: Production, Chemistry and Utilization, New York, 1989 Search PubMed.
- J.-A. Becerra, L.-M. González and A.-L. Villa, A bio-inspired heterogeneous catalyst for the transformation of limonene from orange peel waste biomass into value-added products, Catal. Today, 2018, 302, 250–260 CrossRef CAS.
- E. M. Garcia-Castello, et al., Reverse osmosis concentration of press liquid from orange juice solid wastes: flux decline mechanisms, J. Food Eng., 2011, 106(3), 199–205 CrossRef CAS.
- L. D. Almeida, et al., Catalytic aerobic epoxidation of bio-renewable alkenes using organic carbonates as green solvents, Mol. Catal., 2022, 527, 112400 CrossRef CAS.
- R. Wikandari, et al., Improvement of biogas production from orange peel waste by leaching of limonene, BioMed Res. Int., 2015, 2015, 494182 Search PubMed.
- N. Fdil, et al., Terpenic olefin epoxidation using metals acetylacetonates as catalysts, J. Mol. Catal. A: Chem., 1996, 108(1), 15–21 CrossRef CAS.
- H. Mimoun, Catalytic opportunities in the flavor and fragrance industry, Chimia, 1996, 50(12), 620 CrossRef CAS.
- A. Calogirou, B. Larsen and D. Kotzias, Gas-phase terpene oxidation products: a review, Atmos. Environ., 1999, 33(9), 1423–1439 CrossRef CAS.
- C. Chapuis and D. Jacoby, Catalysis in the preparation of fragrances and flavours, Appl. Catal., A, 2001, 221(1–2), 93–117 CrossRef CAS.
- K. Bauer, D. Garbe and H. Surburg, Common fragrance and flavor materials: preparation, properties and uses, John Wiley & Sons, 2008 Search PubMed.
- P. A. Wender and T. P. Mucciaro, A new and practical approach to the synthesis of taxol and taxol analogs: the pinene path, J. Am. Chem. Soc., 1992, 114(14), 5878–5879 CrossRef CAS.
- E. Juaristi and L. Paquette, Handbook of Reagents for Organic Synthesis. Chiral Reagents for Asymmetric Synthesis, Wiley, Chichester, 2003 Search PubMed.
- G. Sienel, R. Rieth and K. Rowbottom, Ulmann's Encyclopedia of Industrial Chemistry, Verlag Chemie, Weinheim, 2003 Search PubMed.
- J. Herzberger, et al., Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: synthesis, novel polymer architectures, and bioconjugation, Chem. Rev., 2016, 116(4), 2170–2243 CrossRef CAS PubMed.
- D. Swern, Organic Peroxides in Organic Peroxides, ed. D. Swern, Wiley-Interscience, New York, 1971 Search PubMed.
- D. C. Duncan, et al., Mechanism and dynamics in the H3 [PW12O40]-catalyzed selective epoxidation of terminal olefins by H2O2. Formation, reactivity, and stability of {PO4[WO(O2)2]4}3, J. Am. Chem. Soc., 1995, 117(2), 681–691 CrossRef CAS.
- J. Kobe, et al., Epoxidation–Industrial. Encyclopedia of catalysis, 2002 Search PubMed.
- K. Kamata, et al., Efficient epoxidation of olefins with≥ 99% selectivity and use of hydrogen peroxide, Science, 2003, 300(5621), 964–966 CrossRef CAS PubMed.
- W. Zhong, et al., Synergistic hollow CoMo oxide dual catalysis for tandem oxygen transfer: preferred aerobic epoxidation of cyclohexene to 1, 2-epoxycyclohexane, Appl. Catal. B Environ., 2018, 225, 180–196 CrossRef CAS.
- A. Pena, et al., Limonene oxidation by molecular oxygen under solvent-free conditions: the influence of peroxides and catalysts on the reaction rate, React. Kinet. Mech. Catal., 2012, 107(2), 263–275 CrossRef CAS.
- Y. Ishii, S. Sakaguchi and T. Iwahama, Innovation of hydrocarbon oxidation with molecular oxygen and related reactions, Adv. Synth. Catal., 2001, 343(5), 393–427 CrossRef CAS.
- L. Vanoye, et al., Epoxidation using molecular oxygen in flow: facts and questions on the mechanism of the Mukaiyama epoxidation, Catal. Sci. Technol., 2016, 6(13), 4724–4732 RSC.
- S. Madadi, et al., Aerobic epoxidation of limonene using cobalt substituted mesoporous SBA-16 Part 1: Optimization via Response Surface Methodology (RSM), Appl. Catal. B Environ., 2020, 260, 118049 CrossRef CAS.
- S. Madadi, J.-Y. Bergeron and S. Kaliaguine, Kinetic investigation of aerobic epoxidation of limonene over cobalt substituted mesoporous SBA-16, Catal. Sci. Technol., 2021, 11(2), 594–611 RSC.
- S. Madadi and S. Kaliaguine, Activated carbon-supported ruthenium as a catalyst for the solvent-and initiator-free aerobic epoxidation of limonene, ACS Sustain. Chem. Eng., 2021, 9(31), 10557–10568 CrossRef CAS.
- M. V. Patil, M. K. Yadav and R. V. Jasra, Catalytic epoxidation of α-pinene with molecular oxygen using cobalt (II)-exchanged zeolite Y-based heterogeneous catalysts, J. Mol. Catal. A: Chem., 2007, 277(1–2), 72–80 CrossRef CAS.
- Y. M. Ahmat and S. Kaliaguine, Epoxidation of limonene and pinenes by dimethyldioxirane in microemulsions, Catal. Today, 2023, 407, 146–155 CrossRef.
- F. Chiker, et al., Green and selective epoxidation of alkenes catalysed by new TiO 2–SiO 2 SBA mesoporous solids, Green Chem., 2003, 5(3), 318–322 RSC.
- S. Casuscelli, et al., Effect of reaction conditions on limonene epoxidation with H2O2 catalyzed by supported Keggin heteropolycompounds, Appl. Catal., A, 2004, 274(1–2), 115–122 CrossRef CAS.
- S. Casuscelli, et al., Application of complex heteropolytungstates in limonene epoxidation by H2O2 in biphasic medium, Catal. Today, 2005, 107, 230–234 CrossRef.
- J. M. d. S. e Silva, et al., The chemical reactivity of some terpenes investigated by alumina catalyzed epoxidation with hydrogen peroxide and by DFT calculations, J. Mol. Catal. A: Chem., 2006, 252(1–2), 186–193 CrossRef.
- R. Barrera Zapata, A. L. Villa and C. Montes de Correa, Kinetic modeling of limonene epoxidation over PW-Amberlite, Ind. Eng. Chem. Res., 2009, 48(2), 647–653 CrossRef CAS.
- A. L. Cánepa, et al., H2O2 based α-pinene oxidation over Ti-MCM-41. A kinetic study, J. Mol. Catal. A: Chem., 2011, 347(1–2), 1–7 CrossRef.
- Y. Kon, et al., An effective synthesis of acid-sensitive epoxides via oxidation of terpenes and styrenes using hydrogen peroxide under organic solvent-free conditions, Synthesis, 2011, 2011(7), 1092–1098 CrossRef.
- M. G. Egusquiza, et al., Advance in the study of limonene epoxidation with H2O2 catalyzed by Cu (II) complex heteropolytungstates, Catal. Commun., 2012, 26, 117–121 CrossRef CAS.
- T. Michel, et al., Selective epoxidation of (+)-limonene employing methyltrioxorhenium as catalyst, J. Mol. Catal. A: Chem., 2012, 358, 159–165 CrossRef CAS.
- M. Zheng, et al., Oxidation of α-Pinene with Hydrogen Peroxide Catalyzed by VO(OAc)2, in Advanced Materials Research, Trans Tech Publ, 2012 Search PubMed.
- A. J. Bonon, et al., Limonene epoxidation with H2O2 promoted by Al2O3: Kinetic study, experimental design, J. Catal., 2014, 319, 71–86 CrossRef CAS.
- J.-P. Schirmann and S. Y. Delavarenne, Hydrogen peroxide in organic chemistry, 1981, Edition et documentation industrielle Search PubMed.
- G. Strukul, Catalytic oxidations with hydrogen peroxide as oxidant, Springer Science & Business Media, 2013, vol. 9 Search PubMed.
- C. W. Jones, Applications of hydrogen peroxide and derivatives, Royal Society of Chemistry, 1999, vol. 2 Search PubMed.
- M. Cagnoli, et al., “Clean” limonene epoxidation using Ti-MCM-41 catalyst, Appl. Catal., A, 2005, 287(2), 227–235 CrossRef CAS.
- A. L. Cánepa, et al., Biomass toward fine chemical products: oxidation of α-pinene over sieves nanostructured modified with vanadium, J. Mol. Catal. A: Chem., 2015, 404, 65–73 CrossRef.
- C. Venturello, E. Alneri and M. Ricci, A new, effective catalytic system for epoxidation of olefins by hydrogen peroxide under phase-transfer conditions, J. Org. Chem., 1983, 48(21), 3831–3833 CrossRef CAS.
- A. Jimtaisong and R. L. Luck, Synthesis and Catalytic Epoxidation Activity with TBHP and H2O2 of Dioxo-, Oxoperoxo-, and Oxodiperoxo Molybdenum (VI) and Tungsten (VI) Compounds Containing Monodentate or Bidentate Phosphine Oxide Ligands: Crystal Structures of WCl2(O)2 (OPMePh2)2, WCl2(O)(O2)(OPMePh2)2, MoCl2(O)2dppmO2⊙C4H10O, WCl2(O)2dppmO2, Mo(O)(O2)2dppmO2, and W(O)(O2)2dppmO2, Inorg. Chem., 2006, 45(25), 10391–10402 CrossRef CAS PubMed.
- Y. Ishii, et al., Hydrogen peroxide oxidation catalyzed by heteropoly acids combined with cetylpyridinium chloride. Epoxidation of olefins and allylic alcohols, ketonization of alcohols and diols, and oxidative cleavage of 1, 2-diols and olefins, J. Org. Chem., 1988, 53(15), 3587–3593 CrossRef CAS.
- C. Venturello and R. D'Aloisio, Quaternary ammonium tetrakis (diperoxotungsto) phosphates (3-) as a new class of catalysts for efficient alkene epoxidation with hydrogen peroxide, J. Org. Chem., 1988, 53(7), 1553–1557 CrossRef CAS.
- R. Noyori, M. Aoki and K. Sato, Green oxidation with aqueous hydrogen peroxide, Chem. Commun., 2003,(16), 1977–1986 RSC.
- G. Grigoropoulou, J. Clark and J. Elings, Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant, Green Chem., 2003, 5(1), 1–7 RSC.
- K. Takumi, N. Sasagawa, Y. Ezaki, K. O. N. Yoshihiro, Y. Ono and K. Sato, US Pat., 8,742,145, US Patent and Trademark Office, Washington, DC, 2014 Search PubMed.
- H. Hachiya, et al., Unique Salt Effect on Highly Selective Synthesis of Acid-Labile Terpene and Styrene Oxides with a Tungsten/H2O2 Catalytic System under Acidic Aqueous Conditions, Synthesis, 2012, 44(11), 1672–1678 CrossRef CAS.
- P. U. Maheswari, et al., A Na2WO4/H2WO4-Based Highly Efficient Biphasic Catalyst towards Alkene Epoxidation, using Dihydrogen Peroxide as Oxidant, Adv. Synth. Catal., 2005, 347(14), 1759–1764 CrossRef CAS.
- D. Kaur, et al., Epoxidation studies of terpenes with urea hydrogen peroxide and phosphotungstic acid, Indian J. Chem., 2010, 49(5), 598–603 Search PubMed.
- L. Charbonneau and S. Kaliaguine, Epoxidation of limonene over low coordination Ti in Ti-SBA-16, Appl. Catal., A, 2017, 533, 1–8 CrossRef CAS.
- A. Wróblewska, The epoxidation of limonene over the TS-1 and Ti-SBA-15 catalysts, Molecules, 2014, 19(12), 19907–19922 CrossRef PubMed.
- U. Neuenschwander, F. Guignard and I. Hermans, Mechanism of the Aerobic Oxidation of α-Pinene, ChemSusChem, 2010, 3(1), 75–84 CrossRef CAS PubMed.
- U. Neuenschwander, E. Meier and I. Hermans, Peculiarities of β-Pinene Autoxidation, ChemSusChem, 2011, 4(11), 1613–1621 CrossRef CAS PubMed.
- K. A. Swift, Catalytic transformations of the major terpene feedstocks, Top. Catal., 2004, 27(1), 143–155 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chemical routes for the transformation of biomass into chemicals, Chem. Rev., 2007, 107(6), 2411–2502 CrossRef CAS PubMed.
- G. Carr, et al., Ring opening of α-pinene epoxide, J. Chem. Soc., Perkin Trans. 2, 1994,(7), 1419–1422 RSC.
- V. C. Eze, et al., Synthesis of cyclic α-pinane carbonate–a potential monomer for bio-based polymers, RSC Adv., 2022, 12(27), 17454–17465 RSC.
- J. F. Mata-Segreda, Hydroxide as general base in the saponification of ethyl acetate, J. Am. Chem. Soc., 2002, 124(10), 2259–2262 CrossRef CAS PubMed.
- V. Theodorou, et al., A simple method for the alkaline hydrolysis of esters, Tetrahedron Lett., 2007, 48(46), 8230–8233 CrossRef CAS.
- N. Prileschajew, Oxydation ungesättigter Verbindungen mittels organischer Superoxyde, Berichte der deutschen chemischen Gesellschaft, 42(4), 4811–4815 CrossRef CAS.
- M.-Y. Yao, et al., Highly efficient silica-supported peroxycarboxylic acid for the epoxidation of unsaturated fatty acid methyl esters and vegetable oils, ACS Sustain. Chem. Eng., 2016, 4(7), 3840–3849 CrossRef CAS.
- V. V. Goud, et al., Kinetics of epoxidation of jatropha oil with peroxyacetic and peroxyformic acid catalysed by acidic ion exchange resin, Chem. Eng. Sci., 2007, 62(15), 4065–4076 CrossRef CAS.
- G. Grigoropoulou and J. H. Clark, A catalytic, environmentally benign method for the epoxidation of unsaturated terpenes with hydrogen peroxide, Tetrahedron Lett., 2006, 47(26), 4461–4463 CrossRef CAS.
- G. Yadav and A. Pujari, Epoxidation of Styrene to Styrene Oxide: Synergism of Heteropoly Acid and Phase-Transfer Catalyst under Ishii–Venturello Mechanism, Org. Process Res. Dev., 2000, 4(2), 88–93 CrossRef CAS.
- L. Charbonneau, et al., Catalyst-free epoxidation of limonene to limonene dioxide, ACS Sustain. Chem. Eng., 2018, 6(4), 5115–5121 CrossRef CAS.
- W. B. Cunningham, et al., Sustainable catalytic protocols for the solvent free epoxidation and anti-dihydroxylation of the alkene bonds of biorenewable terpene feedstocks using H2O2 as oxidant, Green Chem., 2020, 22(2), 513–524 RSC.
- M. Guidotti, et al., Heterogeneous Catalytic Epoxidation: High Limonene Oxide Yields by Surface Silylation of Ti-MCM-41, Chem. Eng. Technol., 2011, 34(11), 1924–1927 CrossRef CAS.
- D. E. Katsoulis, A survey of applications of polyoxometalates, Chem. Rev., 1998, 98(1), 359–388 CrossRef CAS PubMed.
- C. L. Hill, Progress and challenges in polyoxometalate-based catalysis and catalytic materials chemistry, J. Mol. Catal. A: Chem., 2007, 262(1–2), 2–6 CrossRef CAS.
- F. Cavani, Heteropolycompound-based catalysts: a blend of acid and oxidizing properties, Catal. Today, 1998, 41(1–3), 73–86 CrossRef CAS.
- Z. Zhu, R. Tain and C. Rhodes, A study of the decomposition behaviour of 12-tungstophosphate heteropolyacid in solution, Can. J. Chem., 2003, 81(10), 1044–1050 CrossRef CAS.
- N. Mizuno and M. Misono, Heterogeneous catalysis, Chem. Rev., 1998, 98(1), 199–218 CrossRef CAS PubMed.
- C. L. Hill, Introduction: polyoxometalates multicomponent molecular vehicles to probe fundamental issues and practical problems, Chem. Rev., 1998, 98(1), 1–2 CrossRef CAS PubMed.
- B. Dutta, et al., γ-Fe2O3 nanoparticle in NaY-zeolite matrix: preparation, characterization, and heterogeneous catalytic epoxidation of olefins, Inorg. Chim. Acta, 2010, 363(4), 696–704 CrossRef CAS.
- K. C. Gupta, A. K. Sutar and C.-C. Lin, Polymer-supported Schiff base complexes in oxidation reactions, Coord. Chem. Rev., 2009, 253(13–14), 1926–1946 CrossRef CAS.
- D. E. De Vos, B. F. Sels and P. A. Jacobs, Practical heterogeneous catalysts for epoxide production, Adv. Synth. Catal., 2003, 345(4), 457–473 CrossRef CAS.
- A. Di Giuseppe, et al., Chitin-and chitosan-anchored methyltrioxorhenium: an innovative approach for selective heterogeneous catalytic epoxidations of olefins, J. Catal., 2010, 276(2), 412–422 CrossRef CAS.
- M. Amini, M. M. Haghdoost and M. Bagherzadeh, Monomeric and dimeric oxido–peroxido tungsten (VI) complexes in catalytic and stoichiometric epoxidation, Coord. Chem. Rev., 2014, 268, 83–100 CrossRef CAS.
- B. S. Lane and K. Burgess, Metal-catalyzed epoxidations of alkenes with hydrogen peroxide, Chem. Rev., 2003, 103(7), 2457–2474 CrossRef CAS PubMed.
- M. Uguina, et al., Alumina as heterogeneous catalyst for the regioselective epoxidation of terpenic diolefins with hydrogen peroxide, J. Mol. Catal. A: Chem., 2006, 256(1–2), 208–215 CrossRef CAS.
- S. T. Oyama, Rates, kinetics, and mechanisms of epoxidation: homogeneous, heterogeneous, and biological routes, in Mechanisms in homogeneous and heterogeneous epoxidation catalysis, Elsevier, 2008, pp. 3–99 Search PubMed.
- I. Arends and R. Sheldon, Recent developments in selective catalytic epoxidations with H2O2, Top. Catal., 2002, 19(1), 133–141 CrossRef CAS.
- A. Mouret, et al., Eco-friendly solvents and amphiphilic catalytic polyoxometalate nanoparticles: a winning combination for olefin epoxidation, Green Chem., 2014, 16(1), 269–278 RSC.
- C. Palumbo, et al., An efficient epoxidation of terminal aliphatic alkenes over heterogeneous catalysts: when solvent matters, Catal. Sci. Technol., 2016, 6(11), 3832–3839 RSC.
- N. Mizuno, K. Yamaguchi and K. Kamata, Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates, Coord. Chem. Rev., 2005, 249(17–18), 1944–1956 CrossRef CAS.
- E. Wiebus and B. Cornils, Biphasic Systems: Water—Organic, in Catalyst Separation, Recovery and Recycling, Springer, 2006, pp. 105–143 Search PubMed.
- C. Rabeyrin and D. Sinou, Catalytic asymmetric alkylation in water in the presence of surfactants: influence of the nature of the nucleophile and the allylic acetate on the activity and enantioselectivity, Tetrahedron: Asymmetry, 2003, 14(24), 3891–3897 CrossRef CAS.
- R. G. da Rosa, J. D. R. de Campos and R. Buffon, The effect of water on the rhodium-catalyzed carbonylation of isopropylallylamine, J. Mol. Catal. A: Chem., 2000, 153(1–2), 19–24 CrossRef CAS.
- C. Loncaric, K. Manabe and S. Kobayashi, AgOTf-Catalyzed Aza-Diels–Alder Reactions of Danishefsky's Diene with Imines in Water, Adv. Synth. Catal., 2003, 345(4), 475–477 CrossRef CAS.
- Z. Yang, M. Ebihara and T. Kawamura, Homogeneous hydrogenation of olefins catalyzed by a novel tetrarhodium (II) complex as precursor in aqueous solution, J. Mol. Catal. A: Chem., 2000, 158(2), 509–514 CrossRef CAS.
- E. Duynstee and E. Grunwald, Organic reactions occurring in or on micelles. I. Reaction rate studies of the alkaline fading of triphenylmethane dyes and sulfonphthalein indicators in the presence of detergent salts1, J. Am. Chem. Soc., 1959, 81(17), 4540–4542 CrossRef CAS.
- K. Letts and R. Mackay, Reactions in microemulsions. I. Metal ion incorporation by tetraphenylporphine, Inorg. Chem., 1975, 14(12), 2990–2993 CrossRef CAS.
- W. P. Weber and G. W. Gokel, Reactions of Dichlorocarbene With Non-Olefinic Substrates, in Phase Transfer Catalysis in Organic Synthesis, Springer, 1977, pp. 44–57 Search PubMed.
- Y. Chen, et al., On the nature of reaction-controlled phase transfer catalysts for epoxidation of olefin: a 31P NMR investigation, Catal. Lett., 2004, 93(1), 41–46 CrossRef CAS.
- X. Yang, S. Gao and Z. Xi, Reaction-controlled phase transfer catalysis for styrene epoxidation to styrene oxide with aqueous hydrogen peroxide, Org. Process Res. Dev., 2005, 9(3), 294–296 CrossRef CAS.
- K. Holmberg, Organic reactions in microemulsions, Eur. J. Org Chem., 2007, 2007(5), 731–742 CrossRef.
- F. Menger and A. Elrington, Organic reactivity in microemulsion systems, J. Am. Chem. Soc., 1991, 113(25), 9621–9624 CrossRef CAS.
- C. de Bellefon, et al., Microreactors for dynamic, high throughput screening of fluid/liquid molecular catalysis, Angew. Chem., 2000, 112(19), 3584–3587 CrossRef.
- d. P. Villa, A. Taborda and d. C. C. Montes, Kinetics of limonene epoxidation by hydrogen peroxide on PW-Amberlite, J. Mol. Catal. A: Chem., 2002, 185(1–2), 269–277 CrossRef.
- J. E. Hunter and J. F. Fowler Jr, Safety to human skin of cocamidopropyl betaine: a mild surfactant for personal-care products, J. Surfactants Deterg., 1998, 1(2), 235–239 CrossRef CAS.
- Y. Mahha, et al., Environmentally friendly epoxidation of olefins under phase-transfer catalysis conditions with hydrogen peroxide, J. Catal., 2007, 249(2), 338–348 CrossRef CAS.
- M.-L. Wang and V. Rajendran, Ultrasound assisted phase-transfer catalytic epoxidation of 1, 7-octadiene–A kinetic study, Ultrason. Sonochem., 2007, 14(1), 46–54 CrossRef CAS PubMed.
- T. Seki and A. Baiker, Catalytic oxidations in dense carbon dioxide, Chem. Rev., 2009, 109(6), 2409–2454 CrossRef CAS PubMed.
- D. C. Albanese, F. Foschi and M. Penso, Sustainable oxidations under phase-transfer catalysis conditions, Org. Process Res. Dev., 2016, 20(2), 129–139 CrossRef CAS.
- C. M. Starks, Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts, J. Am. Chem. Soc., 1971, 93(1), 195–199 CrossRef CAS.
- W. P. Weber and G. W. Gokel, Altered Reactivity, in Phase Transfer Catalysis in Organic Synthesis, Springer, 1977, pp. 242–251 Search PubMed.
- M. Makosza, Phase-transfer catalysis. A general green methodology in organic synthesis, Pure Appl. Chem., 2000, 72(7), 1399–1403 CrossRef CAS.
- E. Murugan and A. Siva, Preparation of a novel soluble multi-site phase transfer catalyst and the kinetic study for the C-alkylation of α-pinene, J. Mol. Catal. A: Chem., 2005, 235(1–2), 220–229 CrossRef CAS.
- R.-a. Doong, Y.-W. Wu and W.-g. Lei, Surfactant enhanced remediation of cadmium contaminated soils, Water Sci. Technol., 1998, 37(8), 65–71 CrossRef CAS.
- S. D. Bishopp, J. L. Scott and L. Torrente-Murciano, Insights into biphasic oxidations with hydrogen peroxide; towards scaling up, Green Chem., 2014, 16(6), 3281–3285 RSC.
- C. D. Vecitis, et al., Kinetics and mechanism of the sonolytic conversion of the aqueous perfluorinated surfactants, perfluorooctanoate (PFOA), and perfluorooctane sulfonate (PFOS) into inorganic products, J. Phys. Chem. A, 2008, 112(18), 4261–4270 CrossRef CAS PubMed.
- L.-M. Wang, et al., Sodium stearate-catalyzed multicomponent reactions for efficient synthesis of spirooxindoles in aqueous micellar media, Tetrahedron, 2010, 66(1), 339–343 CrossRef CAS.
- D. G. Lee and V. S. Chang, Oxidation of hydrocarbons. 8. Use of dimethyl polyethylene glycol as a phase transfer agent for the oxidation of alkenes by potassium permanganate, J. Org. Chem., 1978, 43(8), 1532–1536 CrossRef CAS.
- D. Attwood, C. Mallon and C. Taylor, Phase studies on oil-in-water phospholipid microemulsions, Int. J. Pharm., 1992, 84(2), R5–R8 CrossRef CAS.
- Y. Kimura, P. Kirszensztejn and S. L. Regen, Poly(ethylene glycol)-graft copolymers as synthetic equivalents of benzyltriethylammonium chloride for triphase catalytic alkylation, J. Org. Chem., 1983, 48(3), 385–386 CrossRef CAS.
- R. Neumann and M. Gara, Highly active manganese-containing polyoxometalate as catalyst for epoxidation of alkenes with hydrogen peroxide, J. Am. Chem. Soc., 1994, 116(12), 5509–5510 CrossRef CAS.
- S. Desikan and L. Doraiswamy, Enhanced activity of polymer-supported phase transfer catalysts, Chem. Eng. Sci., 2000, 55(24), 6119–6127 CrossRef CAS.
- J.-Y. Piquemal, C. Bois and J.-M. Brégeault, Synthesis, reactivity, X-ray crystal and molecular structures of a novel dimeric tungsten (vi) polyoxoperoxo complex: [PPh4]2[Ph2SiO2{W2O2(μ-O2)2(O2)2}] and of a molybdenum (vi) analogue, Chem. Commun., 1997,(5), 473–474 RSC.
- J. Gao, et al., A spectroscopic study on the reaction-controlled phase transfer catalyst in the epoxidation of cyclohexene, J. Mol. Catal. A: Chem., 2004, 210(1–2), 197–204 CrossRef CAS.
- A. M. Hyde, et al., General principles and strategies for salting-out informed by the Hofmeister series, Org. Process Res. Dev., 2017, 21(9), 1355–1370 CrossRef CAS.
- A. Campanella and M. A. Baltanás, Degradation of the oxirane ring of epoxidized vegetable oils in liquid–liquid heterogeneous reaction systems, Chem. Eng. J., 2006, 118(3), 141–152 CrossRef CAS.
- M. Randall and C. F. Failey, The Activity Coefficient of Non-Electrolytes in Aqueous Salt Solutions from Solubility Measurements. The Salting-out Order of the Ions, Chem. Rev., 1927, 4(3), 285–290 CrossRef CAS.
- F. Auriemma, et al., Stereocomplexed poly(limonene carbonate): a unique example of the cocrystallization of amorphous enantiomeric polymers, Angew. Chem., 2015, 127(4), 1231–1234 CrossRef.
- O. Hauenstein, et al., Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency, Green Chem., 2016, 18(3), 760–770 RSC.
- F. Parrino, et al., Polymers of limonene oxide and carbon dioxide: polycarbonates of the solar economy, ACS Omega, 2018, 3(5), 4884–4890 CrossRef CAS PubMed.
- A. Rehman, et al., Highly selective, sustainable synthesis of limonene cyclic carbonate from bio-based limonene oxide and CO2: a kinetic study, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- N. G. Anderson, Practical use of continuous processing in developing and scaling up laboratory processes, Org. Process Res. Dev., 2001, 5(6), 613–621 CrossRef CAS.
- T. Illg, P. Löb and V. Hessel, Flow chemistry using milli-and microstructured reactors—From conventional to novel process windows, Bioorg. Med. Chem., 2010, 18(11), 3707–3719 CrossRef CAS PubMed.
- S. G. Newman and K. F. Jensen, The role of flow in green chemistry and engineering, Green Chem., 2013, 15(6), 1456–1472 RSC.
- M. Colombo and I. Peretto, Chemistry strategies in early drug discovery: an overview of recent trends, Drug discovery today, 2008, 13(15–16), 677–684 CrossRef CAS PubMed.
- N. G. Anderson, et al., Org. Process Res. Dev., 2012, 16, 852–869 CrossRef CAS.
- S. Ranganathan and V. Sieber, Development of semi-continuous chemo-enzymatic terpene epoxidation: combination of anthraquinone autooxidation and the lipase-mediated epoxidation process, React. Chem. Eng., 2017, 2(6), 885–895 RSC.
- X. Zhang, S. Stefanick and F. J. Villani, Application of microreactor technology in process development, Org. Process Res. Dev., 2004, 8(3), 455–460 CrossRef CAS.
- B. P. Mason, et al., Greener approaches to organic synthesis using microreactor technology, Chem. Rev., 2007, 107(6), 2300–2318 CrossRef CAS PubMed.
- J. D. Tibbetts, et al., Sustainable catalytic epoxidation of biorenewable terpene feedstocks using H2O2 as an oxidant in flow microreactors, Green Chem., 2021, 23(15), 5449–5455 RSC.
- G. d. S. Botas, et al., Baccharis reticularia DC and limonene nanoemulsions: promising larvicidal agents for Aedes aegypti (Diptera: Culicidae) control, Molecules, 2017, 22(11), 1990 CrossRef PubMed.
- K. Kamata, et al., Efficient stereo-and regioselective hydroxylation of alkanes catalysed by a bulky polyoxometalate, Nat. Chem., 2010, 2(6), 478–483 CrossRef CAS PubMed.
- B. G. Donoeva, et al., Epoxidation of Alkenes with H2O2 Catalyzed by Dititanium-Containing 19-Tungstodiarsenate (III): Experimental and Theoretical Studies, Wiley Online Library, 2010 Search PubMed.
- Y. Ding, et al., [π-C5H5N(CH2)15CH3]3[PW4O32]/H2O2/ethyl acetate/alkenes: a recyclable and environmentally benign alkenes epoxidation catalytic system, Green Chem., 2008, 10(9), 910–913 RSC.
- P. U. Maheswari, et al., The role of carboxylic acids on a Na2WO4/H2WO4-based biphasic homogeneous alkene epoxidation, using H2O2 as oxidant, J. Mol. Catal. A: Chem., 2006, 258(1–2), 295–301 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |