
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraction and transport of sulfate using macrocyclic squaramide receptors†
Lei
Qin
 a,
Sacha J. N.
Vervuurt
a,
Robert B. P.
Elmes
ab,
Stuart N.
Berry
a,
Nicholas
Proschogo
a and
Katrina A.
Jolliffe
*a
a,
Sacha J. N.
Vervuurt
a,
Robert B. P.
Elmes
ab,
Stuart N.
Berry
a,
Nicholas
Proschogo
a and
Katrina A.
Jolliffe
*a
aSchool of Chemistry, The University of Sydney, NSW 2006, Australia. E-mail: kate.jolliffe@sydney.edu.au
bDepartment of Chemistry, Maynooth University, National University of Ireland, Maynooth, Co., Kildare, Ireland
First published on 4th November 2019
Abstract
The selective extraction of the hydrophilic sulfate ion from water is highly challenging because the high free energy of hydration of this ion makes it more difficult to extract than less hydrophilic ions such as chloride and nitrate. Lipophilic macrocyclic squaramide receptors 1 and 2 were synthesized. Receptor 2 efficiently extracted sulfate from aqueous sodium sulfate solutions into a chloroform phase, via exchange with nitrate ions, overcoming the Hofmeister bias. The resulting 2·SO42− complex was readily recycled through precipitation of BaSO4. Transport of sulfate across a bulk chloroform membrane by 2 was demonstrated across a wide pH range (pH 3.2–9.4) and in the presence of high concentrations of competing anions (chloride, nitrate and dihydrogenphosphate), opening the door to the use of 2 for the selective removal of sulfate from water across a range of applications.
Introduction
The development of selective receptors capable of extracting sulfate from aqueous solution is of significant interest because of the important roles this anion plays in biological, environmental and industrial processes.1 The removal of sulfate from aqueous solution is of particular importance in oil production and desalination processes where sulfate ions contribute to the formation of scale that clogs pipes and fouls membranes.2–4 It is also of relevance in the nuclear industry where sulfate interferes with the vitrification process required for safe long-term storage of nuclear waste, primarily as a result of the low solubility of sulfate in borosilicate glass.5–7 Precipitation of BaSO4 is frequently used to remove sulfate from solution, but this approach is problematic in removing sulfate from nuclear waste as a result of the co-precipitation of radioactive 228Ra/226Ra and 90Sr ions forming Ba(Ra)SO4 and Ra(Sr)SO4.8–10 Therefore, it has been proposed that the selective extraction of sulfate from nitrate rich solutions by liquid–liquid extraction (LLE) using synthetic receptors could have significant benefits for nuclear waste remediation.11Despite the need to selectively extract sulfate from aqueous media, several key challenges have hindered the development of selective sulfate extraction agents. Sulfate has a very high hydration energy (ΔGhyd = −1080 kJ mol−1),12 which poses a dual challenge for selective extraction of this anion from aqueous solution. Firstly, to extract sulfate from an aqueous phase into an organic phase, a receptor needs to bind sulfate with high affinity to compensate for the large dehydration energy. Secondly, if other anions such as nitrate, are present in high concentrations and are less strongly hydrated (ΔGhyd = −306 kJ mol−1)12 than sulfate, these are easier to extract from aqueous solution than sulfate (commonly referred to as Hofmeister bias) reducing sulfate extraction efficiency. To overcome this bias and allow sulfate extraction in the presence of less hydrophilic anions, receptors must have excellent selectivity for sulfate. A further important challenge lies in the release of sulfate following extraction to allow facile recycling of the receptors and enable commercially viable industrial processes.11
While a number of receptors for selective sulfate recognition have recently been reported,13–25 there are relatively few examples of suitable receptors that overcome the Hofmeister bias to allow LLE of sulfate.26–33 Sessler and co-workers have successfully employed calix[n]pyrroles to extract sulfate into organic media in the presence of methyltrialkylammonium ions.26–28 Wu and co-workers have demonstrated that a tripodal hexaurea receptor is capable of extraction of sulfate ions into chloroform solution in the presence of TBACl and that the sulfate can be back-extracted with aqueous barium chloride to regenerate the receptor as a chloride complex.30 Moyer and coworkers have demonstrated that a simple diiminoguanidinium extractant demonstrates very high sulfate selectivity and compatibility with aliphatic solvents commonly used in LLE processes.31 More recently, Romanski and coworkers have demonstrated that a ditopic receptor extracts potassium sulfate from aqueous solution.33 In related work, the transport of sulfate across a bilayer membrane has been shown to be facilitated by tripodal thioureas.17 However, receptors that can transport sulfate across a bulk liquid membrane to facilitate receptor recycling for real-world applications of sulfate extraction remain unexplored.
We have recently reported the use of macrocyclic squaramides as highly selective sulfate receptors with strong affinity for this anion in aqueous mixtures34 and reasoned that these macrocycles could be readily modified with aliphatic chains to solubilize them in organic solvents without altering their sulfate binding affinity, thereby enabling efficient and selective LLE of sulfate ions and their transport across a bulk liquid membrane. We now demonstrate that suitably functionalized macrocyclic squaramides are able to extract sulfate from aqueous solutions of sodium sulfate across a wide pH range (pH 3.2–9) and are capable of sulfate–nitrate exchange, overcoming the Hofmeister bias. We also show for the first time that dynamic sulfate transport can be achieved across a bulk liquid membrane in the presence of competing anions, demonstrating efficient receptor recyclability.
Results and discussion
The structures of macrocyclic squaramides (MSQs) 1 and 2 are based on our previously reported sulfate selective receptor 3 (Chart 1). In concurrent work,35 we have demonstrated that replacing the benzene spacers in 3 with pyridines provides increased sulfate binding affinity, particularly at low pH where protonation of the pyridine units can occur, without reducing the selectivity that these macrocycles display for sulfate. We therefore chose to use isonicotinamide derived macrocycles in this work. We reasoned that it should be possible to functionalize this macrocyclic core with aliphatic chains to solubilize the macrocycle in organic solvents without impacting the demonstrated high sulfate binding affinity and selectivity of the macrocyclic core.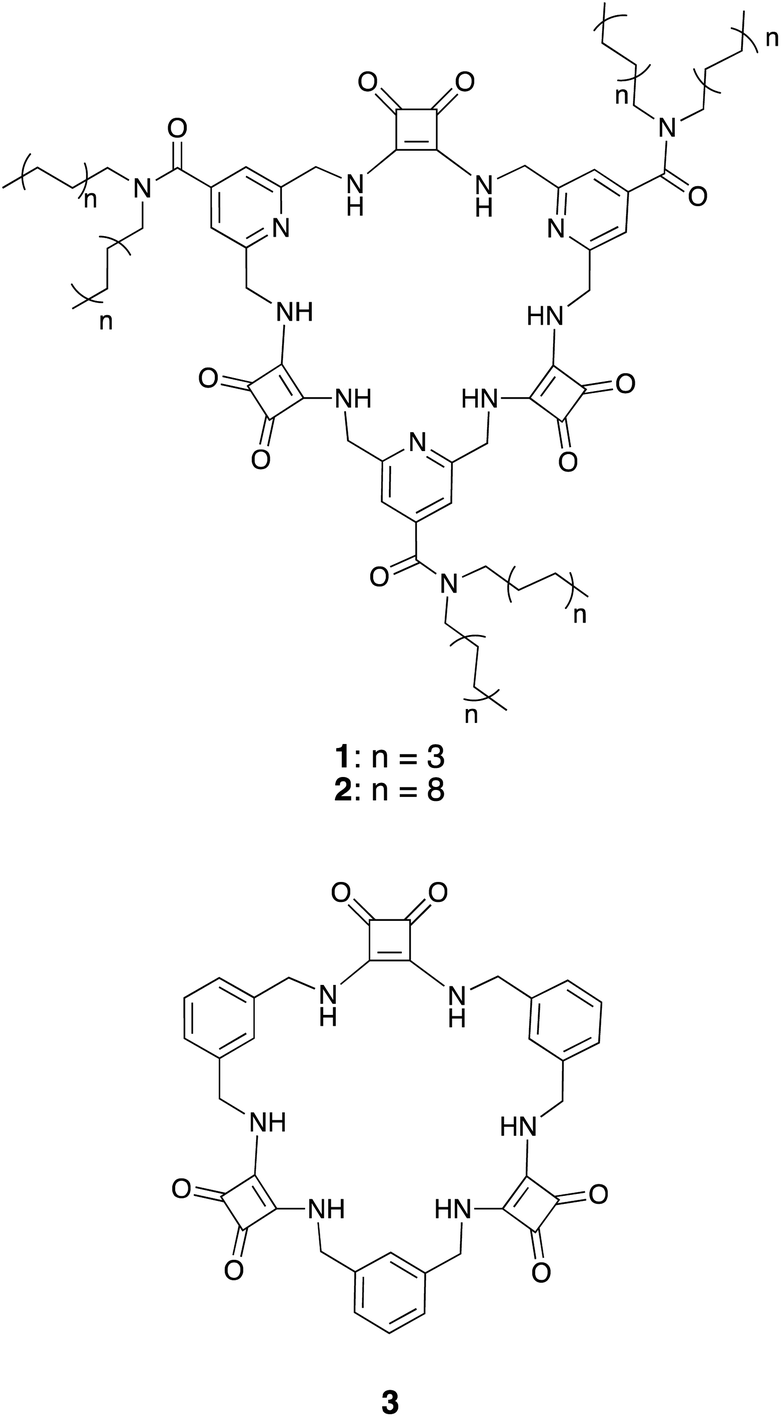 | ||
| Chart 1 Structures of the MSQs 1–3. | ||
Synthesis
The synthesis of macrocycles 1 and 2 followed similar procedures to those described previously for the synthesis of MSQs (Scheme 1).34 Briefly, basic hydrolysis of methyl 2,6-bis(azido)isonicotinate36,37 was followed immediately by reaction of the resulting carboxylic acid with either dioctylamine or dioctadecylamine in the presence of carbodiimide (CDI) to give diazides 4 and 5, respectively. Staudinger reduction of 4 and 5 to form the corresponding diamines 6 and 7 was followed by reaction with two equivalents of diethyl squarate to give disquarates 8 and 9, respectively. Following mono-Boc protection of diamines 6 and 7, the so-formed amines 10 and 11 were immediately reacted with 0.5 equivalents of diethyl squarate in ethanol to give the diisonicotinamide squaramides 12 and 13. Deprotection of compound 12 upon treatment with trifluoroacetic acid and subsequent reaction of diamine 14 with the corresponding disquarate 8 in ethanol provided the desired [3]MSQ 1 in 56% yield over the two steps. In contrast, attempts to condense diamine 15 with disquarate 9 under the same conditions were unsuccessful. However, in a mixed solvent system of EtOH/toluene/hexane (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45:45 v/v/v) to ensure the solubility of all starting materials and reduce the aggregation of the long alkyl chains,38,399 and 15 were successfully condensed in the presence of one equivalent of TBAH2PO4 to form [3]MSQ 2 in 58% yield. We found that dihydrogen phosphate was crucial for the formation of [3]MSQ 2; the addition of a range of other anions (Cl−, ClO4−, I−, BF4−, SO42−) did not lead to isolation of the desired product. In the absence of an anion or in the presence of anions such as ClO4−, I−, BF4− that are known to only weakly coordinate to squaramides,30,40,41 no reaction occurred. In the presence of Cl− and SO42−, which bind to squaramides with relatively high affinities, mixtures of products were observed but all attempts to isolate desired macrocycle 2 (or other discrete species) from these reactions failed. We hypothesize that Cl− and SO42− may bind strongly to the reactants in the non-polar conditions used,40–44 locking them into conformations that do not favour cyclisation, thus promoting the formation of linear oligomers, whereas the weaker binding to H2PO4− allows interconversion of conformers to allow cyclisation to progress.
:45:45 v/v/v) to ensure the solubility of all starting materials and reduce the aggregation of the long alkyl chains,38,399 and 15 were successfully condensed in the presence of one equivalent of TBAH2PO4 to form [3]MSQ 2 in 58% yield. We found that dihydrogen phosphate was crucial for the formation of [3]MSQ 2; the addition of a range of other anions (Cl−, ClO4−, I−, BF4−, SO42−) did not lead to isolation of the desired product. In the absence of an anion or in the presence of anions such as ClO4−, I−, BF4− that are known to only weakly coordinate to squaramides,30,40,41 no reaction occurred. In the presence of Cl− and SO42−, which bind to squaramides with relatively high affinities, mixtures of products were observed but all attempts to isolate desired macrocycle 2 (or other discrete species) from these reactions failed. We hypothesize that Cl− and SO42− may bind strongly to the reactants in the non-polar conditions used,40–44 locking them into conformations that do not favour cyclisation, thus promoting the formation of linear oligomers, whereas the weaker binding to H2PO4− allows interconversion of conformers to allow cyclisation to progress.
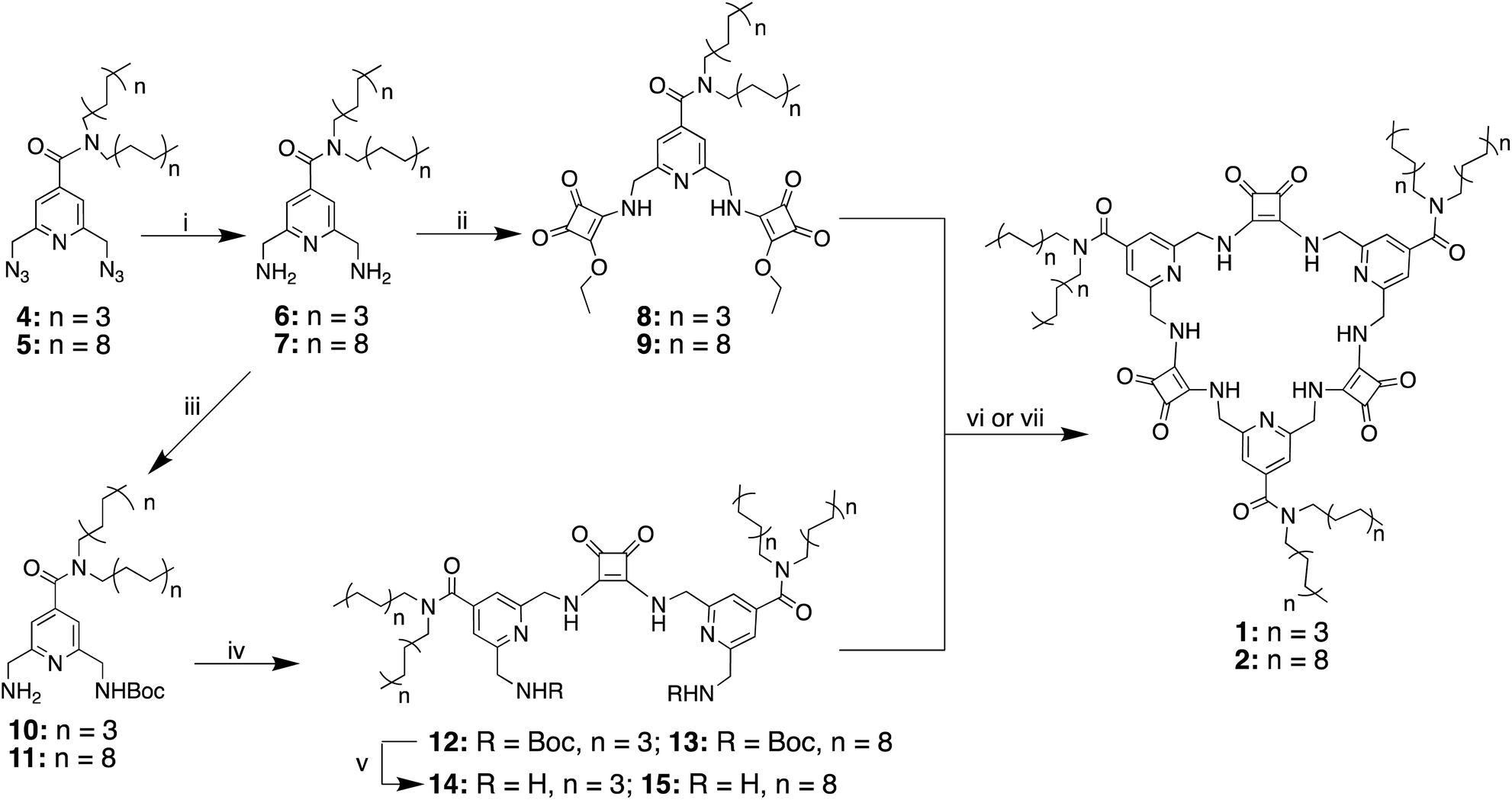 | ||
| Scheme 1 Synthesis of the macrocyclic squaramide based receptors 1 and 2. Conditions: (i) Ph3P, H2O, THF, (6, 79%; 7, 85%); (ii) diethyl squarate, EtOH, RT, 16 h, (8, 82%; 9, 79%); (iii) Boc2O, CH2Cl2, RT, 16 h, (10, 49%; 11, 62%); (iv) diethyl squarate, EtOH, RT, 16 h (12, 46%; 13, 60%); (v) TFA/CH2Cl2, RT, 2 h (14, quant.; 15, quant.); (vi) 8 + 14, EtOH, RT, 48 h, (1, 56%); (vii) 9 + 15, TBAH2PO4, EtOH/toluene/hexane 10/45/45 v/v/v, 60 °C, 48 h (2, 58%). | ||
Sulfate extraction
We first established that appending alkyl chains to the MSQs did not impact their previously observed ability to bind with high affinity to sulfate ions.34 In water-saturated CDCl3, the signal attributable to the squaramide NH protons of MSQ 2 is too broad to observe and the signal for the benzylic protons occurs as a broad multiplet indicating the presence of multiple slowly interconverting conformers of the macrocycle.34 Titration of TBA2SO4 into a solution of 2 in H2O-saturated CDCl3 led to a sharpening and downfield shift of the signal attributable to the squaramide NH with the appearance of a new signal at δ 9.50 ppm after the addition of 1 equiv. of SO42− that further sharpened into a triplet on addition of excess SO42− (Fig. S25†). A sharpening and upfield shift of the signal attributable to the aromatic protons, together with a sharpening and downfield shift of the signal attributable to the benzylic protons were also observed. This indicates the formation of a 2·SO42− complex in CDCl3 with intermediate/slow exchange, suggesting strong binding (Ka > 104 M−1) under these conditions. Titration of TBANO3 into a solution of 2 in H2O-saturated CDCl3 resulted in similar changes to the spectra, however the downfield shift of the signal attributable to the squaramide proton was significantly lower than that observed upon addition of sulfate, with this signal emerging at δ 8.16 ppm after addition of 1 equiv. of nitrate, again suggesting strong 1:1 binding (Ka > 104 M−1) under these conditions.
The ability of 1 and 2 to extract sulfate from aqueous solution using liquid–liquid extraction was next investigated by vigorously shaking an aqueous solution of TBA2SO4 (see ESI† for details) with a CDCl3 solution of either 1 or 2 [45 mM] for 1 minute. The two layers were immediately separated and the organic phase analysed by 1H NMR. For MSQ 1, 1H NMR spectroscopy indicated that none of the MSQ remained in the organic phase. However, a precipitate formed in the aqueous layer and after filtration and redissolution in CDCl3, analysis of the precipitate by 1H NMR (Fig. S28†) indicated the presence of TBA+ and 1·SO4 in a 2:1 ratio, as established through integration of the macrocycle and TBA+ signals, together with the chemical shift of the squaramide NH protons matching that observed in the titration experiments above. This indicates the formation of a TBA2[1·SO4] complex, confirming the 1:1 complexation stoichiometry and suggesting that, while 1 is capable of binding to SO42− at an aqueous–organic interface, the resulting complex is not sufficiently soluble in CDCl3 to extract the SO42− into the organic phase.29 In contrast, with the more lipophilic MSQ 2, analysis of the CDCl3 phase after liquid–liquid extraction indicated that one equiv. of TBA2SO4 was extracted into the organic phase, as determined by comparison of the integrations of the signals attributable to the macrocycle and tetrabutylammonium counterion which gave a ratio of 2 TBA+ ions per macrocycle (Fig. S30 and S31†). Notably, 2 was capable of efficient sulfate extraction, even at sub-stoichiometric sulfate concentrations (Fig. S31†). However, the lipophilic tetrabutylammonium counter ions were required for efficient extraction to take place, as attempts to extract Na2SO4 under the same conditions were unsuccessful.
We next evaluated the ability of MSQ 2 to extract sulfate in the presence of nitrate ions using an anion metathesis approach in which aqueous solutions of Na2SO4 at either pH 3.2 or pH 7.4 were layered onto a solution of [3]MSQ 2 and 2.0 eq. TBANO3 in CDCl3 (pH of the aqueous phase was adjusted using conc. HNO3). The two layers were vigorously shaken for 1 minute, then separated and the organic phases were analyzed using 1H NMR (Fig. 1). The 1H NMR signals corresponding to the 2·NO3− and 2·SO42− complexes are clearly differentiated by the chemical shift of the NH signals and appeared independently in 1:10 and 3:10 ratios of 2·SO42−: 2·NO3− at pH 7.4 and pH 3.2, respectively. This indicates that there is slow exchange between the 2·NO3− and 2·SO42− complexes under these conditions. We speculate that the relative higher proportion of 2·SO42− formed under acidic conditions is due to the partial protonation of the pyridine units in the macrocycle at pH 3.2 as the pKa of isonicotinamide is 3.3,45 which results in increased sulfate binding affinity.34 The 2·SO42− complex in CDCl3 was readily recycled to the nitrate complex upon washing with an aqueous solution of Ba(NO3)2 (Fig. 2d) as a result of the formation of a BaSO4 precipitate (Ksp = 1.1 × 10−10, 25 °C).46 These experiments demonstrate that MSQ 2 is capable of sulfate–nitrate exchange processes at an aqueous–organic interface, indicating that the excellent selectivity demonstrated by MSQ 2 for SO42− overcomes the Hofmeister bias and eliminates the need for lipophilic counter ions in the aqueous phase.
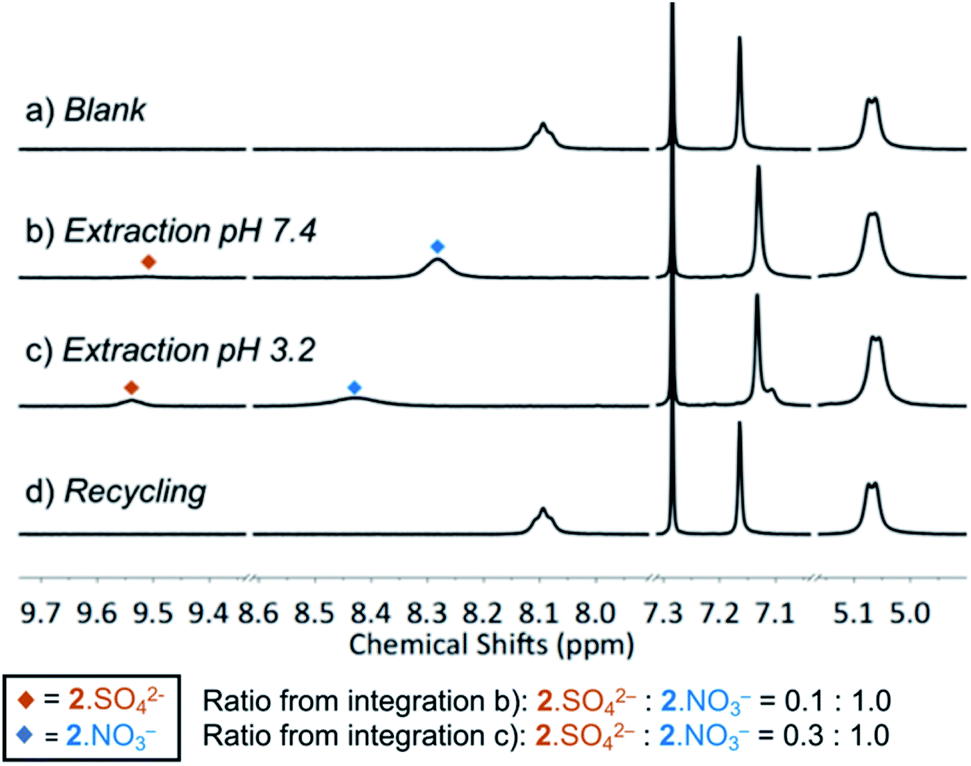 | ||
| Fig. 1 Partial 1H NMR spectra (400 MHz) of a CDCl3 solution of MSQ 2 (5 mM) and TBANO3 (10 mM) after extraction of the following aqueous solutions: (a) blank (20 mM Tris buffer, pH 7.4); (b) 500 mM Na2SO4 in 20 mM Tris buffer, pH 7.4; (c) 500 mM Na2SO4 in 20 mM Tris buffer, pH 3.2, adjusted by addition of conc. HNO3; (d) back extraction of solution (c) through washing with 100 mM aqueous Ba(NO3)2. Back extraction of solution (b) gave an identical spectrum. | ||
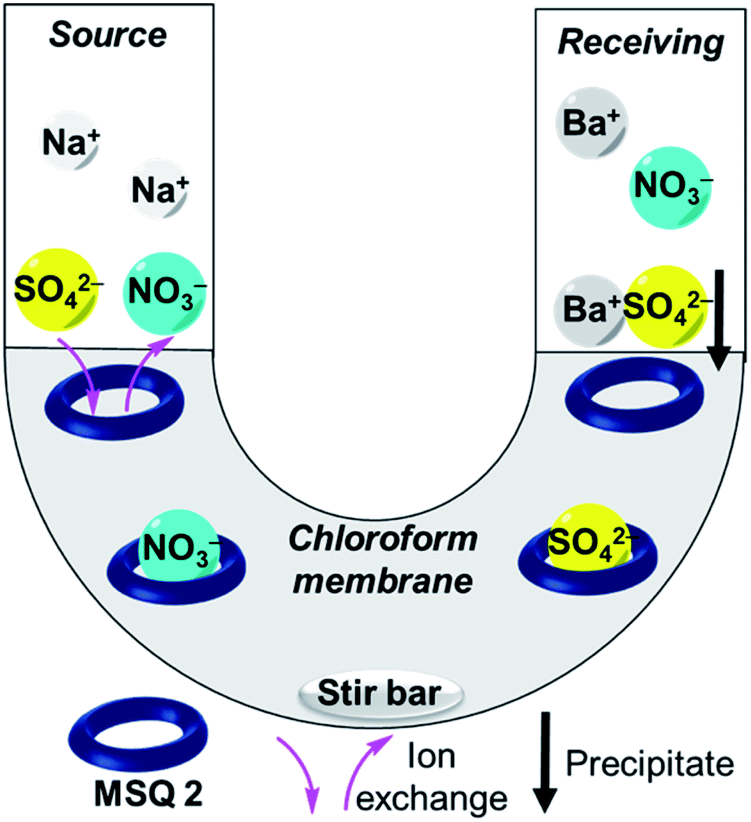 | ||
| Fig. 2 Illustration of U-tube transport experiment (TBA+ cations are omitted for clarity). | ||
Sulfate transport across a bulk liquid membrane
We next investigated the ability of 2 to transport sulfate across a bulk chloroform membrane using classic Cram U-tube experiments (Fig. 2),5,47–49 as proof of principle that the receptor is capable of the dynamic removal of sulfate from aqueous solution through an anion exchange mechanism. In initial experiments the aqueous source and receiving phases were buffered to pH 7.4 (20 mM Tris) with the source phase also containing 500 mM Na2SO4 and the bulk chloroform phase containing 10 mM 2. Sulfate concentrations in both the source and receiving phases were detected using a modified BaSO4 gravimetric analysis method50,51 in which the non-precipitated Ba2+ concentration was measured using inductively coupled plasma mass spectrometry (ICP-MS) after the formation of a BaSO4 precipitate. The final sulfate concentrations in each experiment were also determined by ICP-MS by measuring the concentration of sulfide (Table S2†).52,53 In the absence of any ion source in the organic phase, no transport was observed after 21 days (Fig. 3 and Table S2†), indicating that MSQ 2 is not capable of transporting Na2SO4 across a bulk liquid membrane. However, upon the addition of five equivalents, relative to the receptor, of TBANO3 (tetraalkylammonium ions have previously been shown to facilitate sulfate extraction through formation of ion pair complexes27) to the chloroform phase, a sulfate concentration of 15 mM (all data listed in Table S2†) was detected in the receiving phase after 21 days. No sulfate was detected in the receiving phase in the absence of receptor 2. These results indicate that 2 is capable of efficiently transporting the highly hydrophilic sulfate ion across a bulk liquid membrane with subsequent release into an aqueous phase via an anion exchange mechanism.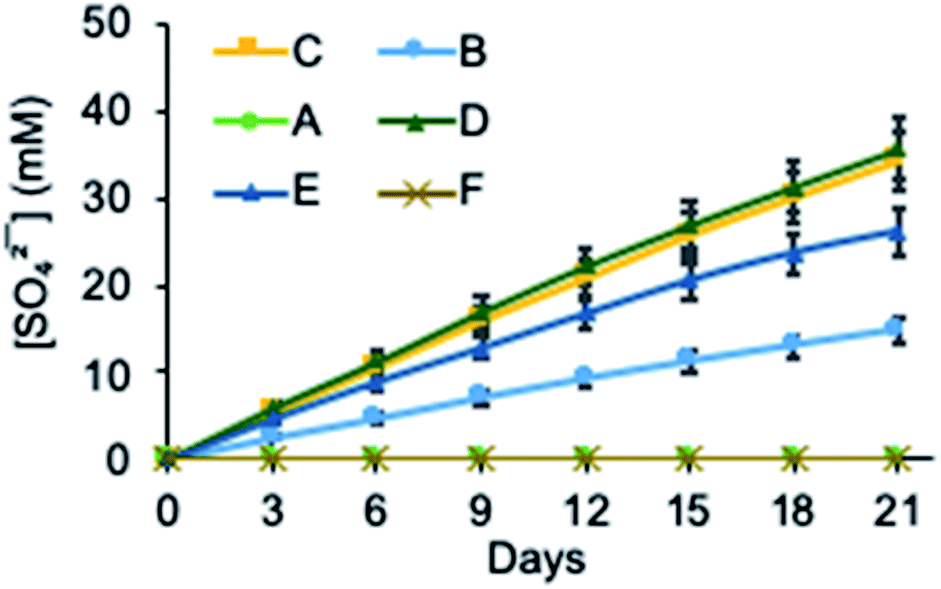 | ||
| Fig. 3 Sulfate transport by 2 across a bulk chloroform membrane determined by the [SO42−] in the receiving phase. Conditions: (A) source phase 500 mM Na2SO4 in 20 mM Tris buffer (pH 7.4); receiving phase 20 mM Tris buffer (pH 7.4); organic phase 10 mM 2 in CHCl3. (B) Source phase 500 mM Na2SO4 in 20 mM Tris buffer (pH 7.4); receiving phase 20 mM Tris buffer (pH 7.4); organic phase 10 mM 2 and 50 mM TBANO3 in CHCl3. (C) Source phase 500 mM Na2SO4 in 20 mM Tris buffer (pH 7.4); receiving phase 300 mM BaCl2 in 20 mM Tris buffer (pH 7.4); organic phase 10 mM 2 and 50 mM TBANO3 in CHCl3. (D) Source phase 500 mM Na2SO4 in H2O (pH 3.2, HNO3); receiving phase 300 mM BaCl2 in H2O (pH 3.2, HNO3); organic phase 10 mM 2 and 50 mM TBANO3 in CHCl3. (E) source phase 500 mM Na2SO4 in H2O (pH 9.4, NaOH); receiving phase 300 mM BaCl2 in H2O (pH 9.4, NaOH); organic phase 10 mM 2 and 50 mM TBANO3 in CHCl3. (F) source phase 500 mM Na2SO4 in 20 mM Tris buffer (pH 7.4); receiving phase 300 mM BaCl2 in 20 mM Tris buffer (pH 7.4); organic phase 50 mM TBANO3 in CHCl3. | ||
In subsequent experiments, BaCl2 was added to the receiving phase. We anticipated this would facilitate sulfate release though precipitation of BaSO4, thereby removing sulfate from the receiving phase and increasing transport rates through Le Chatelier's principle. This resulted in a >2-fold increase in the amount of sulfate transported over the same time period. No change in transport rate was observed upon lowering the pH to 3.2, whereas increasing the pH to 9.4 resulted in a modest reduction in sulfate transport. This may be due to the reduced binding affinity of the isonicotinamide MSQ core at basic pH35 or alternatively might be a result of increased competition from carbonate ions at this higher pH. There was no detectable change in the concentration of sodium ions in the source or receiving phases in any of the transport experiments confirming that, under these conditions, transport occurs via an anion metathesis process. Finally, we evaluated sulfate transport with a mixture of anions in the source phase that mimics that in nuclear waste (100 mM Na2SO4, 100 mM Na2HPO4, 500 mM NaNO3, 500 mM NaCl, pH 7.4). Under these highly competitive conditions, 2 still exhibited sulfate transport, although the rate was diminished, reflecting the ability of 2 to bind strongly to other anions in chloroform (Table S5†). While we have previously established that water-soluble analogues of 2 and related macrocycles bind sulfate with higher affinity than other anions in polar solvents (such as 1:1 v/v DMSO/H2O),34,35 in relatively non-polar solvents such as chloroform, 2 binds to nitrate, chloride and sulfate with Ka > 104 for all three ions. Since both transport and extraction experiments require binding to occur at the interface between the water and chloroform phases, our hypothesis is that the demonstrated higher affinity of the macrocyclic core of 2 for sulfate over other anions in aqueous media results in preferential binding of sulfate by 2 at the aqueous interface, leading to the observed extraction and transport behaviour.
Experimental
Synthesis of macrocycle 1
Compound 12 (76 mg, 0.07 mmol) was dissolved in a solution of TFA/CH2Cl2 (1:1 v/v, 3 mL) before the reaction mixture was stirred at room temperature for 2 hours and then concentrated under reduced pressure. The resulting oil was dissolved in EtOH (3 mL) then a solution of 8 (46 mg, 0.07 mmol) and Et3N (0.5 mL) in EtOH (50 mL) was added and the resulting mixture was stirred at room temperature for 48 h. The solvent was then removed under reduced pressure to give a yellow oil. Subjection of this material to flash silica gel chromatography (5/95 v/v methanol/dichloromethane elution) and concentration of the appropriate fractions (Rf 0.3) gave the macrocycle 1 (57 mg, 56%) as a beige solid. Mp. 262–268 °C (decomp.); 1H NMR (400 MHz, DMSO-d6): 0.79–0.90 (m, 18H), 0.98–1.29 (m, 60H), 1.41 (s, 6H), 1.56 (s, 6H), 3.04 (s, 6H), 3.36 (s, 6H), 4.81 (s, 12H), 7.23 (s, 6H), 8.02 (br s, 6H); 13C NMR (100.6 MHz, DMSO-d6): 14.3, 14.4, 22.4, 22.5, 26.3, 26.8, 27.4, 28.4, 28.9, 29.1, 29.2, 29.5, 31.6, 31.7, 44.5, 48.6, 118.2, 146.8, 158.2, 168.3, 183.3, 2 signals obscured or overlapping; HRMS (ESI, MeOH) calcd for C84H126N12O9Na [M + Na]+ 1447.9845, found 1447.9829; νmax (film) per cm−1: 3237 (broad), 2925, 2851, 1801, 1714, 1609.
Synthesis of macrocycle 2
Compound 13 (53 mg, 0.032 mmol) was dissolved in a solution of TFA/CH2Cl2 (1:1 v/v, 3 mL) and the reaction mixture was stirred at room temperature for 2 hours, then concentrated under reduced pressure. The solid was washed with 5% NaHCO3 solution (5 mL) then dried under stream of N2 (g). The resulting solid was dissolved in 20 mL toluene and then added to a solution of 9 (30 mg, 0.032 mmol) and TBAH2PO4 (10.8 mg, 0.032 mmol) in EtOH/toluene/hexane 10/45/45 v/v/v (500 mL) and stirred at 60 °C for 48 h. The solvent was then removed under reduced pressure to give a yellow solid. Subjection of this material to flash silica gel chromatography (1/99 to 5/95 v/v methanol/dichloromethane elution) and concentration of the appropriate fractions (Rf 0.3) gave compound 2 (42 mg, 58%) as a beige solid. Mp. 252–258 °C (decomp.); 1H NMR (400 MHz, CDCl3): 0.86 (t, J = 6.8 Hz, 18H), 0.98–1.41 (m, 180H), 1.38–1.55 (m, 6H), 1.55–1.69 (m, 6H), 3.08 (t, J = 7.7 Hz, 6H), 3.42 (t, J = 7.7 Hz, 4H), 4.9 (br s, 12H), 7.1 (s, 6H), 7.7 (br s, 6H); 13C NMR (100.6 MHz, CDCl3): δ 189.29, 183.18, 177.99, 172.37, 168.08, 155.74, 147.20, 147.20, 118.25, 69.92, 48.98, 44.97, 31.92, 29.70, 29.65, 29.46, 29.41, 29.35, 29.19, 28.75, 27.46, 27.08, 26.68, 26.62, 22.68, 15.86, 14.10; HRMS (ESI, MeOH) calcd for C144H246N12O9H2 [M + 2H]2+ 1144.9653, found 1144.9645; νmax (film) per cm−1: 3254 (broad), 2920, 2851, 1807, 1598, 1535, 1466.
Conclusions
In summary, we have shown that the neutral MSQ 2 can efficiently extract SO42− from an aqueous Na2SO4 solution into organic solution, via an anion exchange mechanism with nitrate ions, overcoming the Hofmeister bias. This is attributed to the high binding affinity of 2 for sulfate ions. We have further successfully demonstrated that, assisted by a lipophilic cation, MSQ 2 can transport the highly hydrophilic sulfate ion across a bulk chloroform layer via an anion exchange mechanism with nitrate, allowing the extraction of sulfate from sodium sulfate solutions. Notably, receptor 2 is able to transport sulfate across a bulk chloroform membrane even when a complex mixture of anions is present and across a wide pH range (pH 3.2–9.4). Release of the sulfate from the receptor into the receiving phase is facilitated through precipitation of BaSO4 thereby increasing the rate of sulfate transport. These results provide proof-of-principle that neutral receptors for the sulfate ion can be employed in the selective removal of sulfate from aqueous solution in a recyclable manner, overcoming one of the key limitations for the use of such receptors in real-world applications such as the removal of sulfate from nuclear waste.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an Australian Research Council Discovery Project grant (DP170100118).Notes and references
- I. Ravikumar and P. Ghosh, Chem. Soc. Rev., 2012, 41, 3077–3098 RSC.
- M. S. H. Bader, Desalination, 2006, 201, 100–105 CrossRef CAS.
- K. M. Abdullaev, M. M. Agamaliev and D. A. Akhmedova, J. Water Chem. Techno., 2019, 41, 119–124 CrossRef.
- L. F. Greenlee, D. F. Lawler, B. D. Freeman, B. Marrot and P. Moulin, Water Res., 2009, 43, 2317–2348 CrossRef CAS PubMed.
- B. A. Moyer and R. P. Singh, Fundam. Appl. Anion Sep., Springer, 2004 Search PubMed.
- E. A. Katayev, Y. A. Ustynyuk and J. L. Sessler, Coord. Chem. Rev., 2006, 250, 3004–3037 CrossRef CAS.
- R. K. Mishra, K. V. Sudarsan, P. Sengupta, R. K. Vatsa, A. K. Tyagi, C. P. Kaushik, D. Das and K. Raj, J. Am. Ceram. Soc., 2008, 91, 3903–3907 CrossRef CAS.
- T. Y. Zhang, K. Gregory, R. W. Hammack and R. D. Vidic, Environ. Sci. Technol., 2014, 48, 4596–4603 CrossRef CAS.
- P. Medley, P. Martin, A. Bollhöfer and D. Parry, Appl. Radiat. Isot., 2015, 95, 200–207 CrossRef CAS.
- F. Grandia, J. Merino and J. Bruno, Assessment of the radium–barium co-precipitation and its potential influence on the solubility of Ra in the near-field, Technical Report TR-08-07, Swedish Nuclear Fuel and Waste Management Co., Stockholm, 2008 Search PubMed.
- B. A. Moyer, R. Custelcean, B. P. Hay, J. L. Sessler, K. Bowman-James, V. W. Day and S. O. Kang, Inorg. Chem., 2013, 52, 3473–3490 CrossRef CAS PubMed.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 2007, 1321–1340 CrossRef.
- C. Jin, M. Zhang, L. Wu, Y. Guan, Y. Pan, J. Jiang, C. Lin and L. Wang, Chem. Commun., 2013, 49, 2025–2027 RSC.
- R. B. P. Elmes, K. K. Y. Yuen and K. A. Jolliffe, Chem.–Eur. J., 2014, 20, 7373–7380 CrossRef CAS.
- H. Zhou, Y. Zhao, G. Gao, S. Li, J. Lan and J. You, J. Am. Chem. Soc., 2013, 135, 14908–14911 CrossRef CAS.
- V. J. Dungan, H. T. Ngo, P. G. Young and K. A. Jolliffe, Chem. Commun., 2013, 49, 264–266 RSC.
- N. Busschaert, L. E. Karagiannidis, M. Wenzel, C. J. E. Haynes, N. J. Wells, P. G. Young, D. Makuc, J. Plavec, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 1118–1127 RSC.
- A. Schaly, R. Belda, E. Garcia-Espana and S. Kubik, Org. Lett., 2013, 15, 6238–6241 CrossRef CAS.
- Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, A. M. Belenguer, S. Kubik and S. Otto, Chem. Commun., 2011, 47, 9798–9800 RSC.
- J. L. Sessler, E. Katayev, G. D. Pantos and Y. A. Ustynyuk, Chem. Commun., 2004, 1276–1277 RSC.
- P. A. Gale, J. R. Hiscock, C. Z. Jie, M. B. Hursthouse and M. E. Light, Chem. Sci., 2010, 1, 215–220 RSC.
- C. Jia, Q. Q. Wang, R. A. Begum, V. W. Day and K. Bowman-James, Org. Biomol. Chem., 2015, 13, 6953–6957 RSC.
- J. I. Kim, H. Juwarker, X. Liu, M. S. Lah and K. S. Jeong, Chem. Commun., 2010, 46, 764–766 RSC.
- P. Mateus, R. Delgado, V. Andre and M. Teresa Duarte, Org. Biomol. Chem., 2015, 13, 834–842 RSC.
- N. A. Tzioumis, K. K. Y. Yuen and K. A. Jolliffe, Supramol. Chem., 2018, 30, 667–673 CrossRef CAS.
- C. J. Fowler, T. J. Haverlock, B. A. Moyer, J. A. Shriver, D. E. Gross, M. Marquez, J. L. Sessler, M. A. Hossain and K. Bowman-James, J. Am. Chem. Soc., 2008, 130, 14386–14387 CrossRef CAS PubMed.
- C. J. Borman, R. Custelcean, B. P. Hay, N. L. Bill, J. L. Sessler and B. A. Moyer, Chem. Commun., 2011, 47, 7611–7613 RSC.
- S. K. Kim, J. Lee, N. J. Williams, V. M. Lynch, B. P. Hay, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2014, 136, 15079–15085 CrossRef CAS PubMed.
- L. R. Eller, M. Stępień, C. J. Fowler, J. T. Lee, J. L. Sessler and B. A. Moyer, J. Am. Chem. Soc., 2007, 129, 11020–11021 CrossRef CAS.
- C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q. S. Li and X. J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS PubMed.
- N. J. Williams, C. A. Seipp, K. A. Garrabrant, R. Custelcean, E. Holguin, J. K. Keum, R. J. Ellis and B. A. Moyer, Chem. Commun., 2018, 54, 10048–10051 RSC.
- B. Akhuli, I. Ravikumar and P. Ghosh, Chem. Sci., 2012, 3, 1522–1530 RSC.
- D. Jagleniec, L. Dobrzycki, M. Karbarz and J. Romanski, Chem. Sci., 2019, 10, 9542–9547 RSC.
- L. Qin, A. Hartley, P. Turner, R. B. P. Elmes and K. A. Jolliffe, Chem. Sci., 2016, 7, 4563–4572 RSC.
- L. Qin, J. R. Wright, J. D. E. Lane, S. N. Berry, R. B. P. Elmes and K. A. Jolliffe, Chem. Commun., 2019, 55, 12312–12315 RSC.
- S. G. Gouin, M. Roger, N. Leygue, D. Deniaud, K. Julienne, E. Benoist, C. Picard, J. Kovensky and C. Galaup, Bioorg. Med. Chem. Lett., 2012, 22, 2684–2688 CrossRef CAS.
- L. Laurent, B. Hervé and B. Emilie, Novel rare earth element cryptates including a tetraazatriphenylene unit, WO 2010070232, 2008.
- R. O. Dunn and M. O. Bagby, J. Am. Oil Chem. Soc., 1995, 72, 123–130 CrossRef CAS.
- R. O. Dunn and M. O. Bagby, J. Am. Oil Chem. Soc., 1994, 71, 101–108 CrossRef CAS.
- V. E. Zwicker, K. K. Yuen, D. G. Smith, J. Ho, L. Qin, P. Turner and K. A. Jolliffe, Chem.–Eur. J., 2018, 24, 1140–1150 CrossRef CAS.
- V. Amendola, G. Bergamaschi, M. Boiocchi, L. Fabbrizzi and M. Milani, Chem.–Eur. J., 2010, 16, 4368–4380 CrossRef CAS.
- V. Amendola, L. Fabbrizzi, L. Mosca and F. P. Schmidtchen, Chem.–Eur. J., 2011, 17, 5972–5981 CrossRef CAS.
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
- R. B. Elmes, P. Turner and K. A. Jolliffe, Org. Lett., 2013, 15, 5638–5641 CrossRef CAS.
- J. L. Castro, J. F. Arenas, M. R. Lopez-Ramirez, J. Soto and J. C. Otero, J. Colloid Interface Sci., 2013, 396, 95–100 CrossRef CAS.
- C. C. Templeton, J. Chem. Eng. Data, 1960, 5, 514–516 CrossRef CAS.
- D. J. Cram, Angew. Chem., Int. Ed., 1988, 27, 1009–1020 CrossRef.
- G. M. Ritcey, Tsinghua Sci. Technol., 2006, 11, 137–152 CAS.
- G. M. Ritcey and A. W. Ashbrook, Solvent Extraction: Principle and Applications to Process Metallurgy, Part I, Elsevier, Amsterdam, 1984 Search PubMed.
- O. K. Galle and L. R. Hathaway, Appl. Spectrosc., 1975, 29, 518–519 CrossRef CAS.
- F. Torrades and M. Castellvi, Fresenius. J. Anal. Chem., 1994, 349, 734–737 CrossRef CAS.
- M. Colon, M. Iglesias, M. Hidalgo and J. L. Todoli, J. Anal. At. Spectrom., 2008, 23, 416–418 RSC.
- P. R. Craddock, O. J. Rouxel, L. A. Ball and W. Bach, Chem. Geol., 2008, 253, 102–113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data for all new compounds; general methods and data for sulfate extraction and transport experiments. See DOI: 10.1039/c9sc04786g |
| This journal is © The Royal Society of Chemistry 2020 |