
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper arylnitrene intermediates: formation, structure and reactivity†
Noël R. M.
de Kler
and
Jana
Roithová
 *
*
Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: jana.roithova@ru.nl
First published on 15th September 2020
Abstract
The mechanism of oxidation of arylamines by copper enzymes is not clarified yet. Here, we explored a reaction between a possible high-valent copper(II)-oxyl intermediate and arylamine. We have employed a TPA ligand (TPA = tris(2-pyridylmethyl)amine) with the NH2 group in position 2 of one of the pyridine rings (TPANH2). This model system allows generation of [(TPANH2)Cu(O)]+ in the gas phase, which immediately undergoes a reaction between the arylamino group and the copper oxyl moiety. The reaction leads to elimination of H2O and formation of a copper–nitrene complex. The structure of the resulting copper–nitrene complex was confirmed by infrared spectroscopy in the gas phase. We show that the copper–nitrene complex reacts by hydrogen atom transfer with 1,4-cyclohexadiene and by an order of magnitude faster by a double hydrogen atom transfer with ethanethiol and methanol. DFT calculations explain the formation of the copper nitrene as well as its reactivity in agreement with the experimental findings.
Aniline containing chemicals are a class of industrially produced chemicals that are toxic and harmful for the environment and need to be processed in an effective and economic approach. Oxidation of aniline substrates with enzymes is one of the green strategies to process aniline-based waste streams.1,2 For example, multi-copper laccases are known for the oxidization of phenol- and aniline-type pollutants.3 The large biodiversity of this enzyme family can be linked to different selectivities in the oxidations of various aniline and phenol derivatives.4
Laccases contain a mononuclear copper active site (T1) where the substrate oxidation takes place and a trinuclear copper cluster (T2/T3) that mediates four-electron reduction of oxygen to form water.5–7 Both active sites are interconnected and thereby capable of oxidising various substrates. The electron transfer pathway has been well studied, but the overall oxidation mechanisms of aniline-type molecules is not clarified yet.8–11 One of the possible scenarios is that the reaction with oxygen leads to the formation of reactive [copper(II)-oxyl]+ (CuO+) intermediates that subsequently oxidize aniline-type molecules. CuO+ has been thought to be an important reactive species in enzymes such as particulate methane monooxygenases and polysaccharide monooxygenases.12–14 However, copper(II)-oxyl complexes were not directly detected in solution because of their high reactivity; so far the only structural and reactivity assessment comes from gas phase studies.15–19 The copper(II)-oxyl functionality is extremely reactive and oxidizes a broad spectrum of substrates even in the gas phase.18 Here we will show how the nascent copper(II)-oxyl with the TPA ligand (TPA = tris(2-pyridylmethyl)amine) reacts with aniline-type molecules and investigate the mechanism of this oxidation.
The gaseous copper(II)-oxyl complexes can be best generated by electrospray ionization from their copper(II)chlorate precursors.18 Electrospray ionization of methanol solution of [(TPA)Cu(ClO3)2] yields initially the [(TPA)Cu(ClO3)]+ ion (1a, m/z 436) that can lose a ClO2 radical in collision-induced dissociation (CID) to generate the desired [(TPA)Cu(O)]+ complex (2a, Fig. 1a). The extreme reactivity of the copper(II)-oxyl species leads to oxidation of any C–H bond in proximity, which usually results in oxidation of the ligand.15,16,20 Accordingly, we have spectroscopically proved that the nascent [(TPA)Cu(O)]+ complex immediately activates a C–H bond of one of the pyridine ligands leading to an isobaric copper(I) complex with hydroxylated TPA ligand [(TPAOH)Cu]+ (see Fig. S13, S14 and the related discussion in the ESI†).
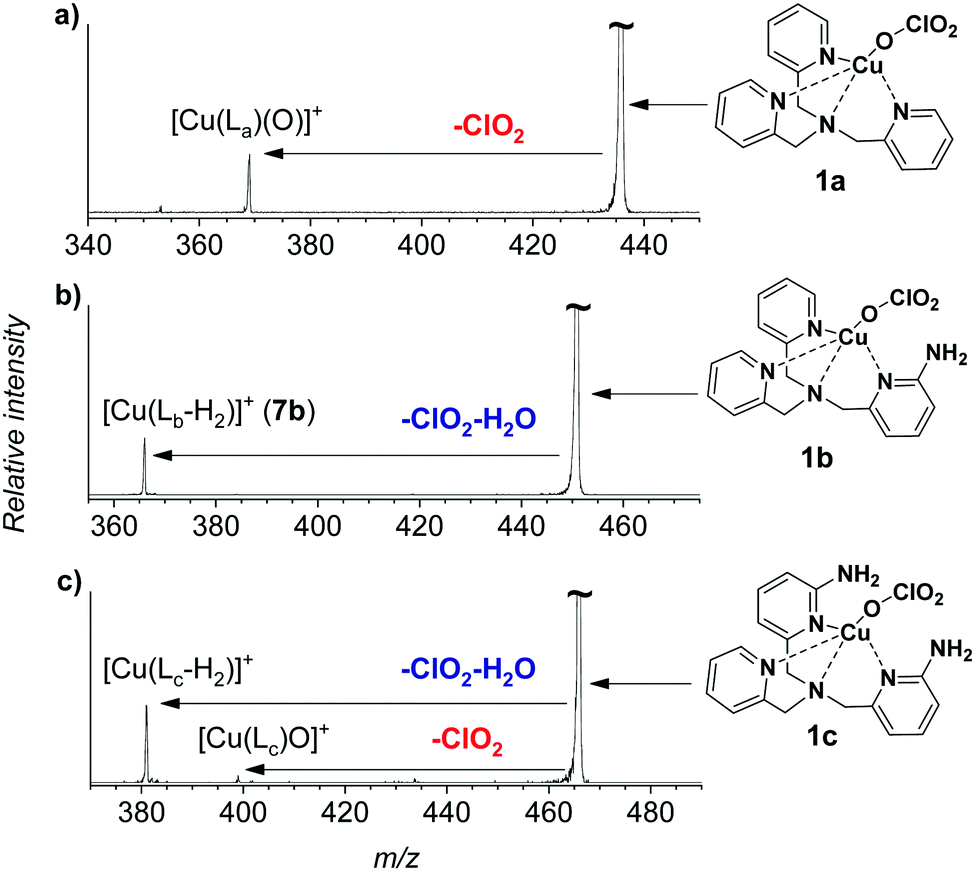 | ||
| Fig. 1 Collision induced dissociation spectra of [(Ligand)Cu(ClO3)]+ complexes with xenon as the collision gas (PXe = 0.1 mTorr) at a collision energy of ∼−4.2 eV in the centre of mass frame. | ||
In order to be able to study the arylamine oxidation, we had to link the amine functionality directly to the ligand. Hence, we functionalized the TPA ligand with one (TPANH2, b) and two (TPA(NH2)2, c) amino group(s) according to a slightly modified literature procedure.21–24 The precursor complexes [Cu(TPANH2)(ClO3)]+ (1b, m/z 451) and [Cu(TPA(NH2)2)(ClO3)]+ (1c, m/z 466) were generated by electrospray ionization of a methanol solution of the corresponding ligand and Cu(ClO3)2 (Fig. S1, ESI†). Dissociation of complexes 1b and 1c (Fig. 1b and c) led to the expected ClO2 radical elimination which, however, was associated with H2O elimination. Presumably, the initial formation of [(TPANH2)CuO]+ (2b) or [(TPA(NH2)2)CuO]+ (2c) is followed by hydrogen atom transfer (HAT) from the amino group(s) leading to the formation of [Cu(TPA-nitrene)]+ derivatives.
We have attempted to trap the [(TPANH2)CuO]+ (2b) reactive intermediate by slowing down the subsequent water elimination by deuteration of the amino group. However, we have observed selective elimination of ClO2 and D2O yielding again the nitrene product ions (Fig. S2, ESI†). For the complexes having the ligand with two amino groups, [Cu(TPA(NH2)2)(ClO3)]+ (1c), we were able to trap a small amount of the ions prior to water elimination (2c, Fig. 1c), but the abundance was too small to perform further experiments with these ions. Most likely, both NH2 groups participate in the HAT reactions leading to stabilization of the intermediate. In the following, we will focus on the system with ligand b.
We have further investigated the structure of the nitrene products. Exploratory DFT calculations suggest that H2O elimination proceeds via a step-wise transfer of two hydrogen atoms from nitrogen to oxygen (Fig. 2). The reaction proceeds in the triplet state and it initially leads to copper(II)hydroxo complex 33b with an aminyl radical at the ligand. The second hydrogen atom transfer leads to copper(I) complex with the ligand bearing nitrene and coordinated to H2O (34b). The alternative coupling of the OH and NH groups in 33b must be associated with a spin flip and leads to the hydroxyl aryl amine product 18b (see Fig. S15, ESI† for further details). Probably, due to the large internal energy, this complex cannot be isolated in the gas phase and instead the reaction proceeds further towards H2O elimination. Either 18b or 34b can lose H2O to form the detected nitrene ions.
 | ||
| Fig. 2 Potential energy surface for the fragmentation and rearrangements of [Cu(TPANH2)(ClO3)]+ (1b). The structures were calculated at the B3LYP/6-31+G* level and the relative energies refer to 0 K and are in kcal mol−1. Some structures are displayed with a truncated ligand, but all calculations were performed with the full ligand. | ||
We have further considered rearrangement of the initially formed nitrene 35b to more stable isomers.25,2635b can undergo a ring expansion associated with a spin flip to form 16b, which is however 9.6 kcal mol−1 higher in energy than 35b (see also Fig. S15, ESI†). Alternatively, the nitrene 35b can change coordination mode and form isomer 37b, which is energetically preferred by 5.9 kcal mol−1. For more possibilities see Fig. S15 and S16 in the ESI.†
In order to assign the structure of the formed nitrene product, we have measured the IR spectrum of the mass selected ion (m/z 366) using the helium tagging photodissociation technique (Fig. 3).27,28 The spectra belong to the ions that are cooled to the ground vibrational state and are isolated under vacuum. Hence, they can be straightforwardly assigned based on comparison with theoretically predicted IR spectra of possible isomers (5b–7b). The experimental spectrum clearly agrees with the theoretical spectrum predicted for 37b. In particular, the bands at ∼1505 cm−1 and ∼1220 cm−1 correspond to the stretching vibrations of the pyridine ring bearing the nitrene functionality and to the stretching vibration of the nitrogen–pyridyl bond, respectively (highlighted in red and blue in Fig. 3). This vibration is not present in any other theoretical spectrum. The Cu–nitrene vibration is predicted at 600 cm−1; this range is unfortunately not accessible in our instrument (more details in Fig. S3, ESI†). The isotopic shifts of this band are predicted by the theory as −3 cm−1 for 15N labelling and −0.1 cm−1 for 65Cu labelling, which makes the experimental confirmation of this bond next to impossible. This spectral range in photodissociation spectroscopy can be studied only with free electron lasers.29
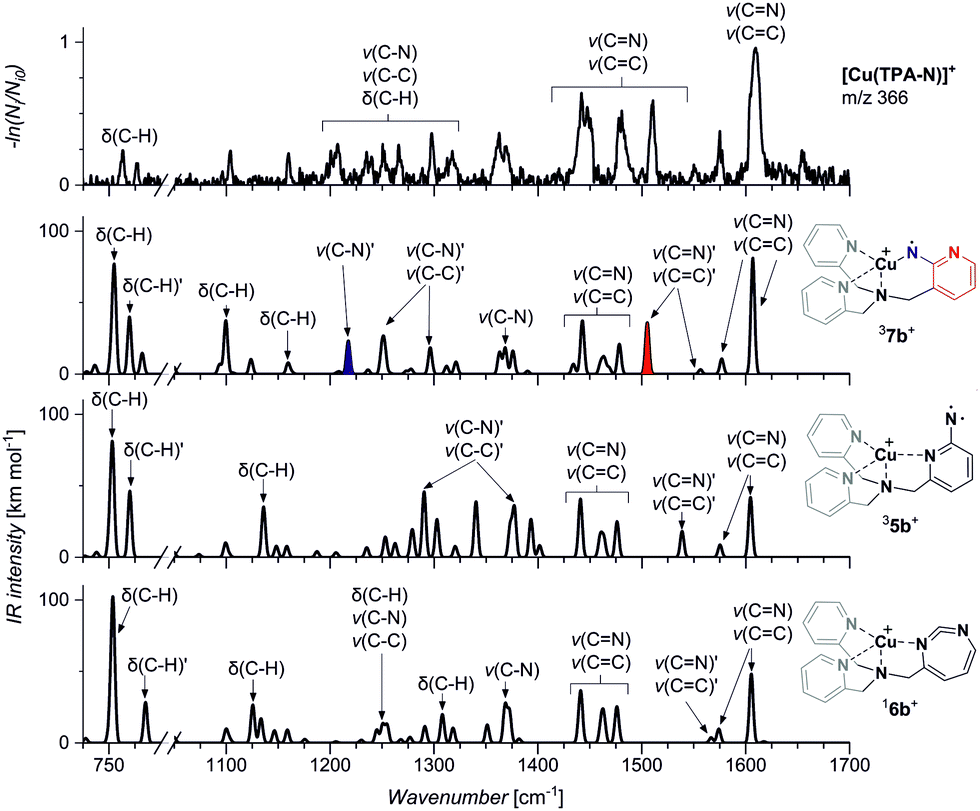 | ||
Fig. 3 Helium tagging infrared photodissociation spectrum of [Cu(TPA-N)]+ (m/z 366) (top panel). Theoretical IR spectra of 37b, 35b and 16b were calculated for the optimized structures at the B3LYP/6-31+G* level and scaled by 0.97. δ(C–H), v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and v(CN) correspond to the absorption bands in the pyridine rings whereas δ(C–H)′, v(CC)′ and v(CN)′ correspond to the absorption bands in the nitrene-pyridine ring. C) and v(CN) correspond to the absorption bands in the pyridine rings whereas δ(C–H)′, v(CC)′ and v(CN)′ correspond to the absorption bands in the nitrene-pyridine ring. | ||
The results show that the nascent copper(II)-oxyl intermediate readily reacts with aromatic amines to form copper-arylnitrenes. The reaction between an amine and copper(II)-oxyl is a new pathway of how to form copper nitrenes. The previous reports show the formation of copper nitrene complexes from copper azide precursors and from the reaction between a copper complex and PhINTs.30–34 These reactive complexes are used for direct C–H amination reactions.35,36 That is why we have also tested whether 7b is reactive in hydrogen atom transfer reactions (Fig. S11, ESI†).
First, we tested the reactivity of 7b toward 1,4-cyclohexadiene with a weak C–H bond. The reaction in the gas phase proceeds by single hydrogen atom transfer demonstrating that 7b could indeed participate in direct C–H amination (Fig. 4). A KIE of ∼2.5 was determined for HAT via the reaction of 7b with partially deuterated 1,4-cyclohexadiene-1,2,3,4,5,6-d6 in the gas phase (see Fig. S9, ESI† for more details). We also observe an adduct formation, which can result from the aziridination reaction, but we cannot confirm it based on the available data.
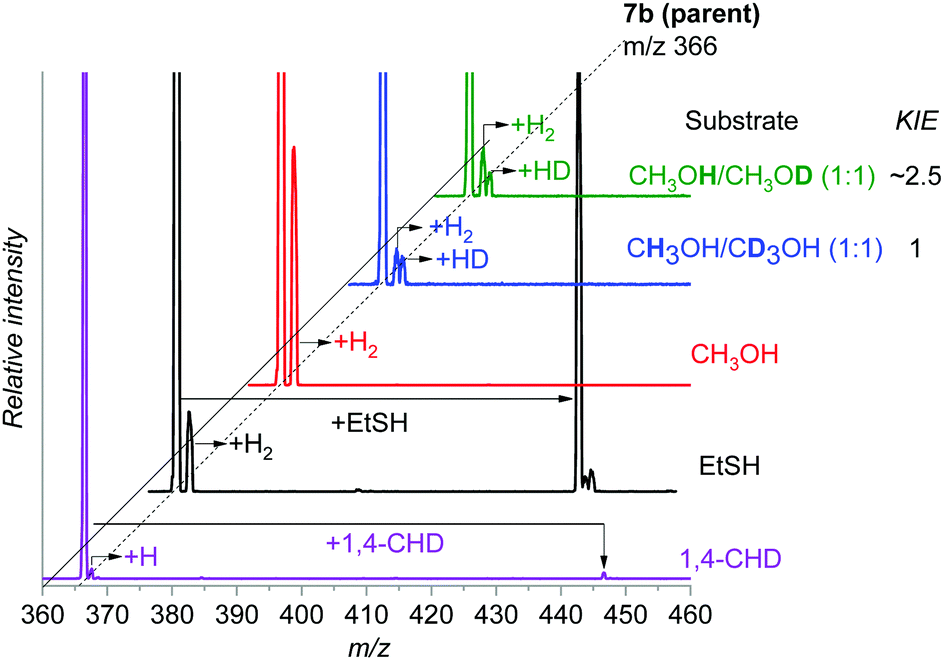 | ||
Fig. 4 Gas phase reactivity of 7b with 1,4-cyclohexadiene (purple), ethanethiol (black), methanol (red), CH3OH/CD3OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (blue), and CH3OH/CH3OD (1:1) (green). The experiments were performed at zero collision energy with a pressure of the reactant gas of 0.2 mTorr, except for 1,4-cyclohexadiene for which it was raised to 0.3 mTorr (see also Fig. S11, ESI†). The relative concentrations of the isotopically labelled methanols were determined by an association reaction with the [Au(PMe3)]+ cation (see the ESI† for details). :1) (blue), and CH3OH/CH3OD (1:1) (green). The experiments were performed at zero collision energy with a pressure of the reactant gas of 0.2 mTorr, except for 1,4-cyclohexadiene for which it was raised to 0.3 mTorr (see also Fig. S11, ESI†). The relative concentrations of the isotopically labelled methanols were determined by an association reaction with the [Au(PMe3)]+ cation (see the ESI† for details). | ||
We reported previously that the reactive complexes preferring the proton-coupled electron transfer (PCET) mechanism for hydrogen atom transfer react more readily with thiols than with activated C–H bonds.37 Hence, we tested this type of reactivity with ethanethiol (EtSH). The reaction of 7b and EtSH leads to the formation of an adduct and to double hydrogen atom abstraction from EtSH (black spectrum in Fig. 4). The rate of the reaction is an order of magnitude larger than the rate of the reaction with 1,4-cyclohexadiene (see Fig. S11, ESI†). The selective occurrence of double hydrogen atom transfer suggests that the aminyl radical formed from the nitrene in the first step can readily abstract a hydrogen atom from the carbon atom of the thiolate. The analogous dehydrogenation proceeds with methanol also (Fig. 4), which we investigated further in more detail.
The reaction of 7b with methanol leads selectively to dehydrogenation of methanol without formation of an adduct (Fig. 4). The labelling experiments showed that the kinetic isotope effect (KIE) for the hydrogen atom abstraction from the oxygen atom is ∼2.5, whereas KIE for the HAT from the methyl group is about 1 (compare the red, blue and green spectra in Fig. 4, detailed evaluation of KIEs using different approaches is in the ESI:† Fig. S5–S7). Exploratory DFT calculations confirm that the initial O–H bond activation is the rate determining step followed by HAT from the methyl group leading via a smaller energy barrier (Fig. 5 and Fig. S8, ESI†), while the first HAT proceeds in the triplet state, the second HAT from the methyl group is associated with a spin flip to the singlet state of the products.
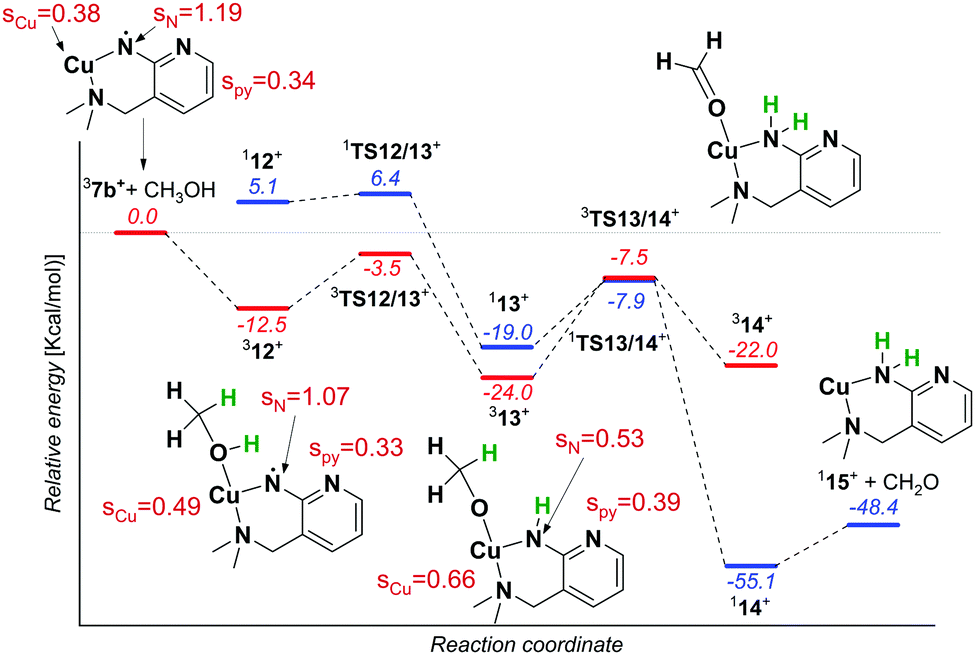 | ||
| Fig. 5 Potential energy surface for the reaction of 37b+ with methanol calculated at the B3LYP level with the 6-311+G(2d,p) basis set for copper and 6-31+G* basis set for all other atoms. The relative energies refer to 0 K and are in kcal mol−1. The spin densities were obtained from Mulliken analysis at the B3LYP/6-311G(2d,p) level. The structures are displayed with a truncated ligand, but all the calculations were performed with the full ligand. | ||
The distribution of the spin density in 7b shows that one of the unpaired electrons stays localized at the nitrene nitrogen atom. The electronic structure can be probably best represented as a mixture of two configurations: copper(I)/nitrene diradical and copper(II)/aminyl radical (note that an analogous structure has been found for copper(II)-oxyl complexes).19 Hence, the first HAT with methanol leads to a complex between copper(II) methanolate and the aminyl radical. The final step leads to a copper(I) complex with an amine ligand.
In conclusion, we have shown that a copper(II)-oxyl complex reacts readily with arylamines by hydrogen atom transfer reactions. In the gas phase, this reaction leads to the formation of copper nitrene complexes. We have characterized the formed copper nitrene complex 7b by its IR spectra and tested its reactivity. 7b reacts with 1,4-cyclohexadiene by hydrogen atom transfer with a KIE of ∼2.5. In addition, 7b reacts readily with methanol or ethanethiol by two subsequent hydrogen atom transfers. With methanol, the hydrogen atom transfer from the oxygen atom is the rate determining step with the kinetic isotope effect of ∼2.5.
The project was funded by the European Research Council (ERC CoG No. 682275).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 Search PubMed.
- B. Yang, Y. Wang, Z. Liu, J. Liu and J. Cai, J. Environ. Health Sci. Eng., 2019, 17, 135–140 Search PubMed.
- F. Mirazizi, A. Bahrami, S. Soleimani Asl, A. Zaribafan, K. Haghbeen and S. Aminzadeh, Int. J. Environ. Sci. Technol., 2018, 15, 1679–1686 Search PubMed.
- R. Reiss, J. Ihssen, M. Richter, E. Eichhorn, B. Schilling and L. Thöny-Meyer, PLoS One, 2013, 8, e65633 Search PubMed.
- E. I. Solomon, A. J. Augustine and J. Yoon, Dalton Trans., 2008, 3921–3932 Search PubMed.
- E. I. Solomon, Inorg. Chem., 2016, 55, 6364–6375 Search PubMed.
- K. Ray, Y. M. Lee and W. Nam, Coord. Chem. Rev., 2017, 334, 25–42 Search PubMed.
- S. Shleev, A. Jarosz-Wilkolazka, A. Khalunina, O. Morozova, A. Yaropolov, T. Ruzgas and L. Gorton, Bioelectrochemistry, 2005, 67, 115–124 Search PubMed.
- I. Pardo and S. Camarero, Cell. Mol. Life Sci., 2015, 72, 897–910 Search PubMed.
- S. M. Jones and E. I. Solomon, Cell. Mol. Life Sci., 2015, 72, 869–883 Search PubMed.
- A. Sekretaryova, S. M. Jones and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 11304–11314 Search PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 Search PubMed.
- V. V. Vu and S. T. Ngo, Coord. Chem. Rev., 2018, 368, 134–157 Search PubMed.
- M. O. Ross, F. MacMillan, J. Wang, A. Nisthal, T. J. Lawton, B. D. Olafson, S. L. Mayo, A. C. Rosenzweig and B. M. Hoffman, Science, 2019, 364, 566–570 Search PubMed.
- D. Schröder, M. C. Holthausen and H. Schwarz, J. Phys. Chem. B, 2004, 108, 14407–14416 Search PubMed.
- L. Jašóková, E. Hanikýřová, D. Schröder and J. Roithová, J. Mass Spectrom., 2012, 47, 460–465 Search PubMed.
- N. J. Rijs, P. González-Navarrete, M. Schlangen and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 3125–3135 Search PubMed.
- G. Yassaghi, E. Andris and J. Roithov, ChemPhysChem, 2017, 18, 2217–2224 Search PubMed.
- M. Srnec, R. Navrátil, E. Andris, J. Jašík and J. Roithová, Angew. Chem., Int. Ed., 2018, 57, 17053–17057 Search PubMed.
- N. Dietl, C. Van Der Linde, M. Schlangen, M. K. Beyer and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 4966–4969 Search PubMed.
- M. Bhadra, J. Y. C. Lee, R. E. Cowley, S. Kim, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2018, 140, 9042–9045 Search PubMed.
- J. C. Mareque-Rivas, R. Prabaharan and R. T. Martín de Rosales, Chem. Commun., 2004, 76–77 Search PubMed.
- D. E. Diaz, D. A. Quist, A. E. Herzog, A. W. Schaefer, I. Kipouros, M. Bhadra, E. I. Solomon and K. D. Karlin, Angew. Chem., Int. Ed., 2019, 58, 17572 Search PubMed.
- D. A. Quist, D. E. Diaz, J. J. Liu and K. D. Karlin, J. Biol. Inorg. Chem., 2017, 22, 253–288 Search PubMed.
- A. Maltsev, T. Bally, M. L. Tsao, M. S. Platz, A. Kuhn, M. Vosswinkel and C. Wentrup, J. Am. Chem. Soc., 2004, 126, 237–249 Search PubMed.
- C. Wentrup, Acc. Chem. Res., 2011, 44, 393–404 Search PubMed.
- J. Roithová and D. Gerlich, Int. J. Mass Spectrom., 2013, 355, 204–210 Search PubMed.
- J. Roithová, A. Gray, E. Andris, J. Jašík and D. Gerlich, Acc. Chem. Res., 2016, 49, 223–230 Search PubMed.
- L. Jašóková and J. Roithová, Chem. – Eur. J., 2018, 24, 3374–3390 Search PubMed.
- S. Kundu, E. Miceli, E. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710–14713 Search PubMed.
- T. Corona, L. Ribas, M. Rovira, E. R. Farquhar, X. Ribas, K. Ray and A. Company, Angew. Chem., Int. Ed., 2016, 55, 14005–14008 Search PubMed.
- A. G. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 Search PubMed.
- J. Moegling, A. Hoffmann, F. Thomas, N. Orth, P. Liebhäuser, U. Herber, R. Rampmaier, J. Stanek, G. Fink, I. Ivanović-Burmazović and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2018, 57, 9154–9159 Search PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S. L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138–1143 Search PubMed.
- M. R. Fructos, S. Trofimenko, M. Mar Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2006, 128, 11784–11791 Search PubMed.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., 2008, 120, 10109–10112 Search PubMed.
- E. Andris, R. Navrátil, J. Jašík, M. Puri, M. Costas, L. Que and J. Roithová, J. Am. Chem. Soc., 2019, 140, 14391–14400 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting results obtained by NMR, MS and DFT calculations. See DOI: 10.1039/d0cc05198e |
| This journal is © The Royal Society of Chemistry 2020 |