
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dynamic nature of Cu sites in Cu-SSZ-13 and the origin of the seagull NOx conversion profile during NH3-SCR†
A. R.
Fahami
ad,
T.
Günter
a,
D. E.
Doronkin
ab,
M.
Casapu
 a,
D.
Zengel
a,
T. H.
Vuong
e,
M.
Simon
c,
F.
Breher
c,
A. V.
Kucherov
f,
A.
Brückner
e and
J.-D.
Grunwaldt
*ab
a,
D.
Zengel
a,
T. H.
Vuong
e,
M.
Simon
c,
F.
Breher
c,
A. V.
Kucherov
f,
A.
Brückner
e and
J.-D.
Grunwaldt
*ab
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu
bInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany
dDipartimento di Energia, Laboratorio di Catalisi e Processi Catalitici, Politecnico di Milano, Via La Masa 34, 20133 Milano, Italy
eLeibniz-Institut für Katalyse e. V. an der Universität Rostock (LIKAT), Albert-Einstein-Str. 29a, 18059 Rostock, Germany
fN.D. Zelinsky Institute of Organic Chemistry, Leninsky pr. 47, 119991 Moscow, Russia
First published on 3rd January 2019
Abstract
Cu-Zeolites with chabazite structure show a peculiar dual-maxima NO conversion profile, also known as a seagull profile, during the selective catalytic reduction by ammonia. In order to understand the origin of this behavior, systematic catalytic tests and operando spectroscopy were applied to derive structure–performance relationships for Cu-SSZ-13 catalysts with low and high Cu loading. Operando X-ray absorption, X-ray emission and in situ electron paramagnetic resonance spectroscopy measurements, including novel photon-in/photon-out techniques, demonstrated the interconversion of isolated Cu sites and dimeric bis(μ-oxo) Cu species, the former occurring via formation of ammonia Cu2+/Cu+ complexes and the latter in an oxidizing gas mixture. The formation of dimeric Cu+–O2–Cu+ species by involving Cu sites in close vicinity was linked to the high activity at low temperatures of the highly loaded Cu-SSZ-13 sample. In contrast, the isolated Cu sites present at very low Cu loadings are strongly poisoned by adsorbed NH3. The activity decrease around 350 °C that gives rise to the seagull shaped NO conversion profile could be attributed to a more localized structure of mono(μ-oxo)dicopper complexes. Above this temperature, which corresponds to partial NH3 desorption from Cu sites, the isolated Cu sites migrate to form additional dimeric entities thus recovering the SCR activity.
1. Introduction
Selective catalytic reduction (SCR) of nitrogen oxides (NOx) with NH3 is currently a leading technology to reduce NOx emissions from diesel engines to nitrogen and water.1,2 The main part of NOx emissions from a diesel engine is nitric oxide (NO), which is mostly removed via the so-called standard SCR reaction that involves equimolar conversion of NO and NH3.3 If NO2 is present in the gas mixture, e.g. from an upstream positioned diesel oxidation catalyst, it has been shown that the catalytic reduction occurs via a different reaction, Fast SCR, where the reduction of NOx by NH3 takes place at a relatively lower temperature and with a higher reaction rate.4Zeolites and zeotypes with chabazite structure (CHA) like SSZ-13 or SAPO-34 ion-exchanged with Cu have shown high activity and resistance to harsh hydrothermal ageing, which attracted tremendous attention of industry and academia.5–7 Despite commercial applications of Cu-SSZ-13, the nature and location of the active Cu sites, the interaction of the reactants in different reaction regimes, and the mechanism of the SCR reactions have not yet been fully understood.8 There is a general agreement on the redox cycle of Cu in Standard SCR9,10 and several mechanisms for the SCR reactions were proposed.10–16 The basis of this controversial point of view is the nature of Cu species and the difficulties of monitoring the active sites under reaction conditions. Many studies have been carried out to observe the active species, suggesting Cu dimers (e.g., [Cu–O–Cu]2+),16,17 monomers (e.g., Cu2+),18–20 or multinuclear clusters (e.g., CuxOy).11,13,21 Furthermore, their preferential location at the six or eight member ring (MR) as well as the coordination to a single or two adjacent Al sites is not yet clarified.11,22,23 This scope becomes more complex since depending on the Cu/Al and Si/Al ratios, and even the utilized method to synthesise the catalyst, different forms of Cu can emerge, which seem to be mobile in the cage of the zeolite under certain conditions.9,16
An atypical phenomenon recognized for Cu-SSZ-13 during Standard SCR reaction is the decrease followed by the increase of the NO conversion, generally between 250–350 °C,7 which is referred to as the “seagull” profile. The origin of this effect that appears for intermediate Cu loadings (0.5–3 wt%)24 is a strongly debated topic.13,25 It was observed that the seagull profile is more pronounced when the space velocity is high, O2 content is low, for lower Cu loadings as well as when hydrocarbons are present in the feed.7,25–27 Joshi et al.25 claimed that the competition between SCR and NH3 oxidation is the reason of the activity drop, and the contribution of the NO oxidation enables Fast SCR at higher temperatures. A different nature of the Cu sites at low and high temperature has been also discussed by several authors. Based on kinetic tests for a catalyst series and DFT calculations, Gao and co-workers13,24 proposed transient Cu-dimers as relevant species at low temperature, which are then split to monomeric sites with lower deNOx activity at higher temperature. Such dimeric Cu intermediates seem to facilitate fast reoxidation of Cu+ sites, which are formed during reaction of NO and NH3,12,14 thus, allowing a closed catalytic cycle. The idea of forming dimeric Cu species was supported by the molecular dynamics (MD) calculations of Paolucci et al.16 The presence of different sites at low and high temperature was also tackled by Lomachenko et al.,28 who proposed mobile NH3-solvated Cu+/Cu2+ species as active sites up to 200 °C and the zeolite framework-coordinated Cu2+ (Z-Cu2+) to be the dominant active species above 250 °C. In the low-temperature range, the standard SCR rate shows a linear dependency for intermediate Cu concentrations but a quadratic variation for very low Cu loadings. For the last case, recent studies reported that such isolated sites are not able to activate O2 for NO oxidation to NO2,29 a step still considered necessary for the standard SCR reaction. The activation of O2 by two [Cu(NH3)2]+ combined with a facilitated formation of such multinuclear sites were also proposed as mandatory in describing the SCR mechanism.16
In the present work, we aimed at understanding the seagull profile of the SCR conversion for Cu-SSZ-13 by analysing the dynamic structural and electronic changes of the Cu sites during interaction with the SCR and related gas mixtures, at different temperatures below and above the seagull point. Systematic catalytic tests under various reaction conditions were applied for two Cu-SSZ-13 catalysts with different Cu loadings and strong variations in SCR activity around the seagull region. By using operando XAS and XES spectroscopy in a spatially resolved manner along the catalyst bed located in a plug-flow reactor, accurate structure–activity correlations could be derived. This was supplemented by in situ EPR spectroscopy measurements, to uncover variations in nuclearity of Cu sites under relevant reaction conditions.
2. Experimental part
2.1 Catalyst synthesis
Na-SSZ-13 was prepared by a method similar to Deka19 and Zones30 that has been described earlier.12 First, a mixture of 0.67 g sodium hydroxide, 41.1 g deionised water, 14.8 g N,N,N-trimethyladamantylammonium hydroxide (TMAdOH, 25 wt%, Sachem) and 0.43 g aluminium hydroxide was stirred for 30 min. 13.0 g colloidal silica (Ludox® AS-40) was added and mixed for another 10 min. The as prepared gel was transferred into a 200 ml Teflon-lined autoclave and aged at room temperature for 2 h before heating it statically for 4 days at 160 °C. The resulting slurry was filtered, washed with 1 L deionised water and dried at 80 °C before calcination at 550 °C for 2 h. To remove the sodium ions, the as prepared Na-SSZ-13 was ion exchanged with aqueous 1 M NH4NO3 solution (20 mL g−1 zeolite) for 2 h at 75 °C, washed with deionised water, dried at 80 °C and calcined at 550 °C for 2 h. These steps were repeated for further two times without calcination after the final step to receive NH4-SSZ-13. A Cu ion exchange was conducted with 2 g NH4-SSZ-13 in 200 ml 0.001 M or 0.005 M Cu(OAc)2 solution at room temperature for 24 h. The suspension was filtered, washed with 1 L deionised water, dried at 80 °C and calcined at 550 °C for 8 h. The elemental analysis gave a Si/Al-ratio of 16 and a Cu-loading of 0.5 wt% for the low loaded sample and 1.2 wt% for the higher loaded sample (about 45% ion exchange degree). The BET-surface area of the resulting 1.2% Cu-SSZ-13 sample is 590 m2 g−1. For the sake of simplicity, 0.5% Cu-SSZ-13 and 1.2% Cu-SSZ-13 are referred to as Cu-0.5 and Cu-1.2 respectively in the rest of the text.2.2 Catalytic tests
The catalytic measurements were performed in a laboratory setup with a fixed-bed plug-flow quartz tube reactor (inner diameter: 8 mm). 250 mg of the sample (sieve fraction: 125–250 μm) was mixed with 250 mg quartz sand (same sieve fraction) and loaded into the reactor to obtain a bed length of about 1 cm, which was hold in position by quartz wool plugs. The temperature was measured with thermocouples at the beginning and end of the catalyst bed. Gases were dosed with individual mass flow controllers via heated lines to get a mixture of 0–1000 ppm NO, 0–1000 ppm NH3, 10 vol% O2, 5 vol% H2O and N2 balance. The gas hourly space velocity (GHSV) was kept at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 during all measurements. The outlet gas was analysed by MKS MultiGas 2030 FTIR analyser.
000 h−1 during all measurements. The outlet gas was analysed by MKS MultiGas 2030 FTIR analyser.
To evaluate the extent of the potential ammonia inhibition the following experiments have been carried out: standard SCR was evaluated with the NH3 to NO ratio ranging from 0.1 to 1.25 (feeding 1000 ppm NO, 100–1250 ppm NH3, 2 vol% H2O and 8 vol% O2 with a GHSV of 200000 h−1 and N2 as balance) at several temperatures of 190, 235, 285 and 330 °C. The effect of water addition was also investigated by varying the water concentration in the standard SCR feed between 0–5 vol%.
2.3 Catalyst characterization
000 h−1. Gases were dosed via mass flow controllers, whereas water was fed via a saturator. The temperature was varied from room temperature to 500 °C. The gas composition was analysed by a MKS MultiGas 2030 FTIR analyser after diluting the resulting gas mixture (50 ml min−1) to the minimum recommended gas flow for the FTIR instrument (∼350 ml min−1). XAS spectra were recorded at six and five positions along the catalyst bed for Cu-0.5 and Cu-1.2 respectively with a beam size of about 200 × 200 μm2, while applying different SCR-related gas mixtures. For temperature programmed reduction by ammonia combined with XANES (TPR-XANES) the catalysts were first pre-treated in flow of 10% O2 in He at 550 °C for 10 min, then cooled down to 30 °C and exposed to a flow of 1000 ppm NH3, 0–1.5% H2O in He (GHSV of 200000 h−1) while heating with 10 K min−1 rate up to 550 °C.
Linear combination analysis (LCA) of the XANES region from −20 eV to +30 eV around the absorption edge was used to determine the state of Cu in spatially-resolved XAS spectra measured during SCR. ATHENA software from the IFFEFIT package was used for data analysis.34 Principal Component Analysis (PCA, as available in the Demeter package) identified the presence of three components in the spectra, one of these components proved to be Cu2+. As references, the spectra of Cu-0.5 and Cu-1.2 measured in 1000 ppm NO, 10% O2, 1.5% H2O in He at the temperature of the corresponding experiment were used for representing Cu2+. Therefore, any nitrate/nitrite species formed under the given conditions were considered in the LCA. Two other references spectra for Cu+ with and without directly adsorbed ammonia could not be measured separately and instead were extracted from the set of spectra obtained during temperature-programmed reduction of Cu-0.5 by NH3 using multivariate curve resolution-alternating least squares method (MCR-ALS).35,36 This mathematical procedure used in chemometrics allows extraction of a priori unknown spectra of reference compounds from a set of spectra of a mixture with changing concentrations. The technique is similar to PCA, but it allows setting appropriate physical constraints to the components which permits obtaining meaningful spectra. In our case constraint of non-negativity was applied to the matrix of reference spectra and constraints of unimodality (i.e., having only one maximum) were applied to concentration profiles. The obtained reference spectra indeed demonstrated all the features of Cu+ with and without directly adsorbed ammonia as previously described in ref. 9 and 12 and were assigned correspondently. MCR-ALS was required because the direct use of spectra available in literature as references for the LCA is not possible due to differences in spectral resolution, and Cu-0.5 dataset was chosen since the individual features of the spectra were better resolved in this case. In the case of Cu-1.2, even at high temperature the last spectrum shows the contribution of Cu+ with directly adsorbed ammonia.
EXAFS spectra were background subtracted, normalized, k2-weighted and Fourier transformed in the k range 2–11.5 Å−1 using ATHENA and fitting was performed using ARTEMIS.34E0 was selected at 8989 eV corresponding to the inflection point of the rising edge not taking into account the shoulder at 8983 eV related to Cu+ species. The data fitting was performed in R-space between 1 and 3.5 Å (uncorrected for the phase shift) on the k1-, k2-, and k3-weighted data (corresponding to the first Cu–O and Cu–Cu shells). From the fits of reference spectrum of CuO the amplitude reduction factor S02 = 0.7 was obtained and used to analyse the catalyst spectra.
For the HERFD-XANES measurements the monochromator energy was scanned while the spectrometer was kept fixed at the maximum of the Kβ1,3 emission line (8903.6 eV), the spectra were then normalized based on the edge step. For the vtc-XES measurements the incident energy was kept constant above the Cu K absorption edge at 9100.0 eV and the spectrometer scanned the X-rays emitted by the sample. The valence-to-core spectra were first normalized based on the area of the Kβ1,3 emission lines. Then, the valence-to-core data was subtracted from the tail of the Kβ1,3 emission line with the background approximated by four pseudo Voigt functions.38 Gas flow dosing unit and the capillary microreactor were identical to the ones used for conventional XAS (section 2.3.3). The X-ray beam size was 0.2 × 1 mm and the measurements were performed at a distance of approx. 0.5 mm from the beginning of the catalyst bed unless stated otherwise.
3. Results
3.1 Catalytic tests of two differently loaded Cu-SSZ-13 catalysts
Cu-Chabazites with Cu loadings below and above approx. 1 wt% typically show markedly different catalytic activity in NH3-SCR, with higher NOx conversion at lower temperatures over the higher loaded zeolites and also showing the “seagull” shape (dual-maxima) conversion profile.7,24,25 In this study two samples were selected as representative for these two categories containing 0.5 and 1.2 wt% Cu, respectively. Although characteristic for the commercial Cu-chabazite catalysts, an even higher concentration of Cu was not considered to avoid the formation of excessive “spectator” Cu species,39 which could attenuate relevant spectroscopic variations. The activity profiles recorded for the Cu-1.2 and Cu-0.5 samples (Fig. 1a) illustrate the positive effect of a higher Cu loading, especially for the low temperature region. At 300 °C about 90% of NO is reduced over the Cu-1.2 catalyst whereas only 23% NOx conversion was obtained with the Cu-0.5 sample. In addition, the highly loaded sample shows a better defined seagull profile, with the first conversion maximum at approx. 300 °C and the second one above 400 °C. On the contrary, considerable NOx conversion over the low loaded Cu-0.5 is obtained only at higher temperatures (>400 °C), corresponding to the second maximum in the activity profile of Cu-1.2 catalyst. Even by decreasing the GHSV, to have a similar flow vs. amount of Cu during the activity tests (Fig. S1†), the low loaded catalyst showed a significantly lower NOx conversion. The consumption of NH3 during the standard SCR reaction is for both catalysts close to the stoichiometric ratio up to 350 °C (Fig. 1b), which minimizes any correlation between the decrease of the activity around 350 °C and overconsumption of NH3 due to parasitic oxidation.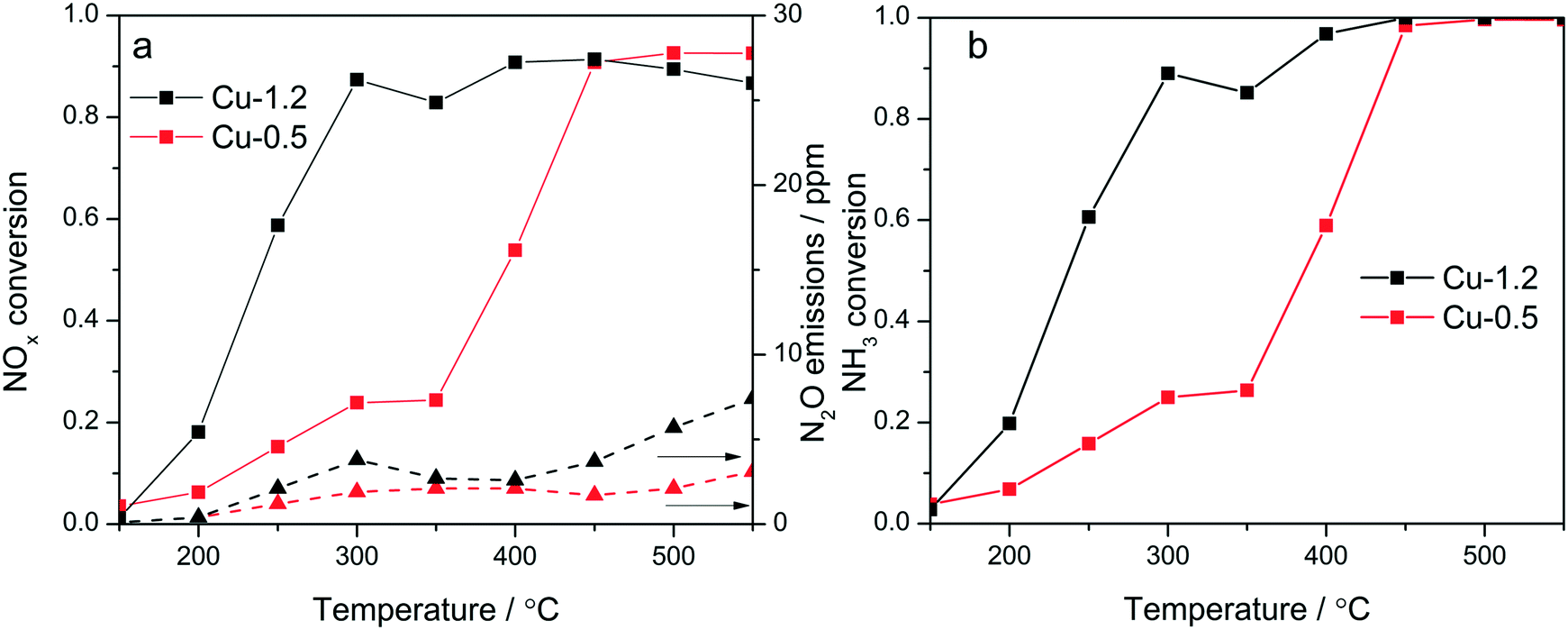 | ||
| Fig. 1 Conversion of NOx and production of N2O (a), and conversion of NH3 (b) measured during NH3–SCR over the tested Cu-zeolites measured in the laboratory plug-flow reactor. Conditions: 1000 ppm NO, 1000 ppm NH3, 10% O2, 5% H2O, balance N2, GHSV 200000 h−1. | ||
Similar screening of Cu loading was previously performed and comparable results were obtained for Cu SSZ-13 catalysts with different Si:Al ratios.18,20,23 This difference in activity was so far related to the presence of different Cu sites: close to the 6-member-rings of CHA framework (6MR) for low loadings and close to the 8MR for zeolites with an increased Cu content,11 to formation and splitting of Cu sites of different nuclearity and reactivity,24 or attributed to a change in the rate limiting step.40 In contrast to the SCR performance, only minor differences were observed during oxidation of NH3 and NO (Fig. S2†). Both catalysts convert less than 10% NO even at 500 °C. NH3 conversion is significant only above 400 °C, and reaches 55% over Cu-1.2 and 30% over Cu-0.5 at 450 °C.
In order to elucidate whether the NH3-inhibition effect, previously reported for Fe-exchanged zeolite and V-based SCR catalysts,41,42 and recently also for Cu-SSZ-13,43 could be the reason for the low activity of the low loaded Cu-0.5 sample, the NH3 concentration was varied between 0.1–1.25 NH3:NO molar ratios during activity tests at 190–330 °C. Fig. 2 shows the corresponding conversions of NO for Cu-0.5 and Cu-1.2 catalysts. According to the measured activity variations, an ammonia inhibition effect was identified at low temperatures for both catalysts. At 245 °C the activity of Cu-1.2 increased from 45% (stoichiometric NH3:NO ratio) to 53% (0.5 NH3:NO ratio, additional 3% of NO conversion is attributed to oxidation to NO2 which does not require NH3) but decreased when more NH3 was dosed. For Cu-0.5 the NO conversion varied from about 9% to 16% for the same sub-stoichiometric NH3:NO ratios and decreased at higher NH3:NO ratios. At 285 °C only the Cu-0.5 catalyst suffered slightly from NH3 inhibition. The Cu-1.2 catalyst, which above this temperature converts more than 70% of NO, does not seem to be affected by the excessive NH3 dosage. Additional experiments performed with higher Cu loadings (∼3 wt% Cu, not shown) indicate further diminishment of the inhibition effect at low temperatures.
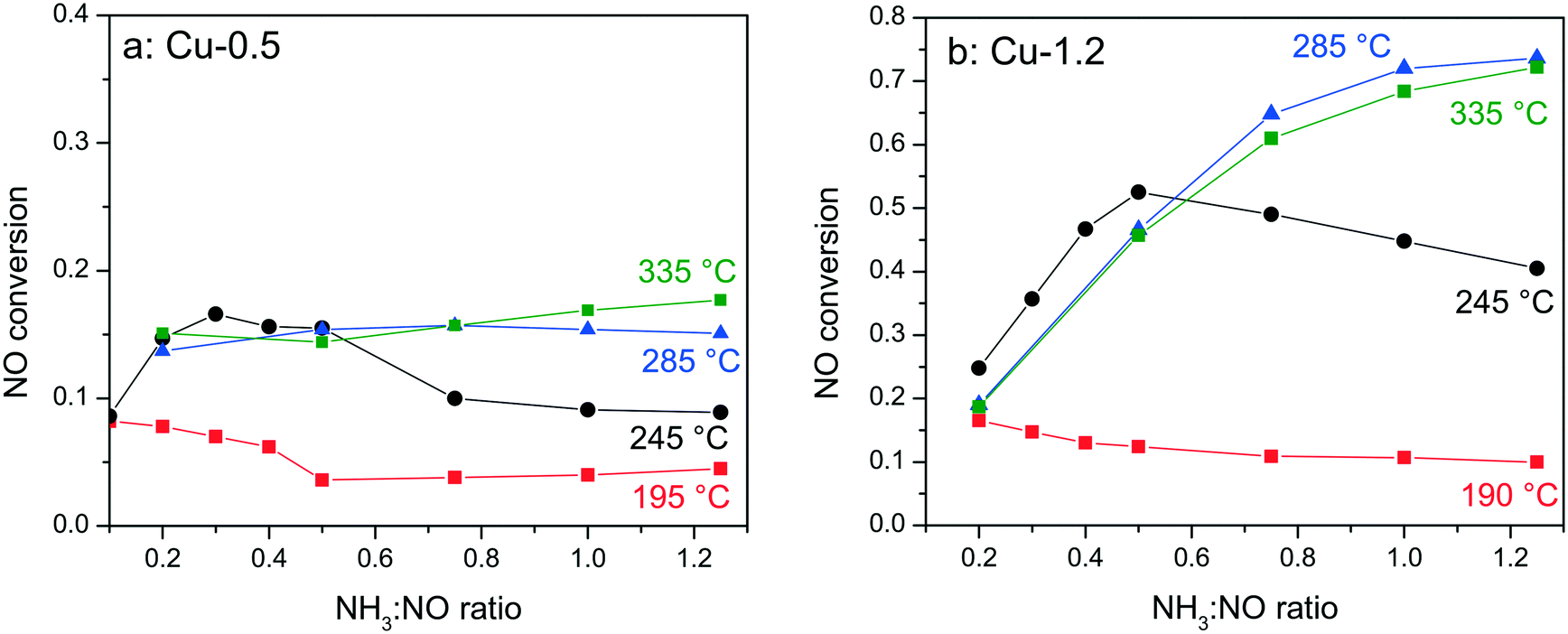 | ||
| Fig. 2 Conversion of NO during NH3–SCR of NOx at different NH3:NO ratios over (a) Cu-0.5 and (b) Cu-1.2 catalysts. Conditions: 1000 ppm NO, 100–1250 ppm NH3, 10% O2, 2% H2O, balance N2, GHSV 200000 h−1. | ||
The presence of water in the SCR feed also affects the catalytic activity but in a reversed manner as compared to the NH3 effect, i.e. NO conversion increases with increasing water content (Fig. S3a†). This increase is more pronounced at temperatures below 350 °C (about 20% increase at 250 °C for Cu-1.2 if 1.5 vol% H2O is present), i.e. in the low temperature part of the seagull shaped conversion profile. Such a behavior could be explained by an attenuation of the NH3 inhibition effect, as the amount of adsorbed NH3 decreases in the presence of H2O. This argument is supported by the NH3-TPD experiments performed in the presence or absence of H2O (Fig. S4†), and also often reported in previous studies.44 It is interesting to notice that for the same experiment the variation of water concentration has almost no impact on the low temperature N2O emissions (Fig. S3b†). Such a behavior could be explained based on recent studies on N2O formation over Cu-SSZ-13 catalysts,45,46 which claim two different paths: via NH4NO3 at low temperatures and via NH3 oxidation at high temperatures. Also the participation of different active sites for the SCR reaction and N2O formation cannot be ruled out. Both possibilities are, however, difficult to prove considering the rather low N2O emissions over Cu-SSZ-13 catalysts.
Hence, the activity profiles obtained for different NO:NH3 ratios as well as in the presence or absence of water suggest that NH3 inhibition contributes to the lower SCR activity of Cu-0.5 sample but, since this effect is present also for the highly loaded catalyst, it cannot solely explain the large difference in performance at low temperature and the “seagull” effect. Therefore, complementary ex situ and in situ/operando spectroscopic methods were applied to elucidate the specific structure of the Cu sites and the variations appearing at low and high temperature during interaction with the SCR-reactants, as their potential has been previously demonstrated.18,19
3.2 Ex situ and in situ characterization: EXAFS, UV-vis, vtc-XES and EPR
At first, the Cu sites in Cu-0.5 and Cu-1.2 zeolites were probed ex situ by EXAFS and diffuse reflectance (DR) UV-vis (Fig. 3). EXAFS spectra of the two catalysts, when measured as pellets without any pretreatment (Fig. 3a), are almost identical. The fit of the ex situ FT-EXAFS spectra of Cu-1.2 performed in ref. 12 resulted in identification of the first shell (O) with Cu–O bond distance of 1.96 Å and a coordination number of 4. The same result was obtained in this study for the Cu-0.5 sample. This outcome is in agreement with ref. 9 and 26 where the resemblance of the untreated hydrated Cu sites in SSZ-13 to hydrated Cu2+ ions in aqueous solution was noted. UV-vis spectra (Fig. 3b) of both catalysts show two groups of absorption bands.18 The band at 800 nm is a d–d transition band which usually appears in hydrated Cu2+ zeolites and was related to d–d transition occurring in distorted octahedral aqua complexes of Cu2+,47,48 while ligand-to-metal charge transfer (LMCT, O2−Cu2+ → O−Cu+) bands are to be seen at 220 and 300 nm. The former band has been attributed to isolated Cu(II),49 the latter one was previously seen in low-loaded Cu-SSZ-13 but was not clearly ascribed.18 In the case of other zeolite types such as Cu-BEA and Cu-ZSM-5, also the bands at 320 nm and 440 nm were found but only for dehydrated samples, and were related to mono(μ-oxo) dicopper [Cu–O–Cu]2+ sites.48–50 Most probably due to the formation of hydrated species, the presence of such sites was not observed in this study. Whereas the intensity of the absorption bands was different, confirming different concentration of Cu2+, the band positions remained the same, i.e. ex situ UV-vis also could not shed light on the differences in nature of Cu sites.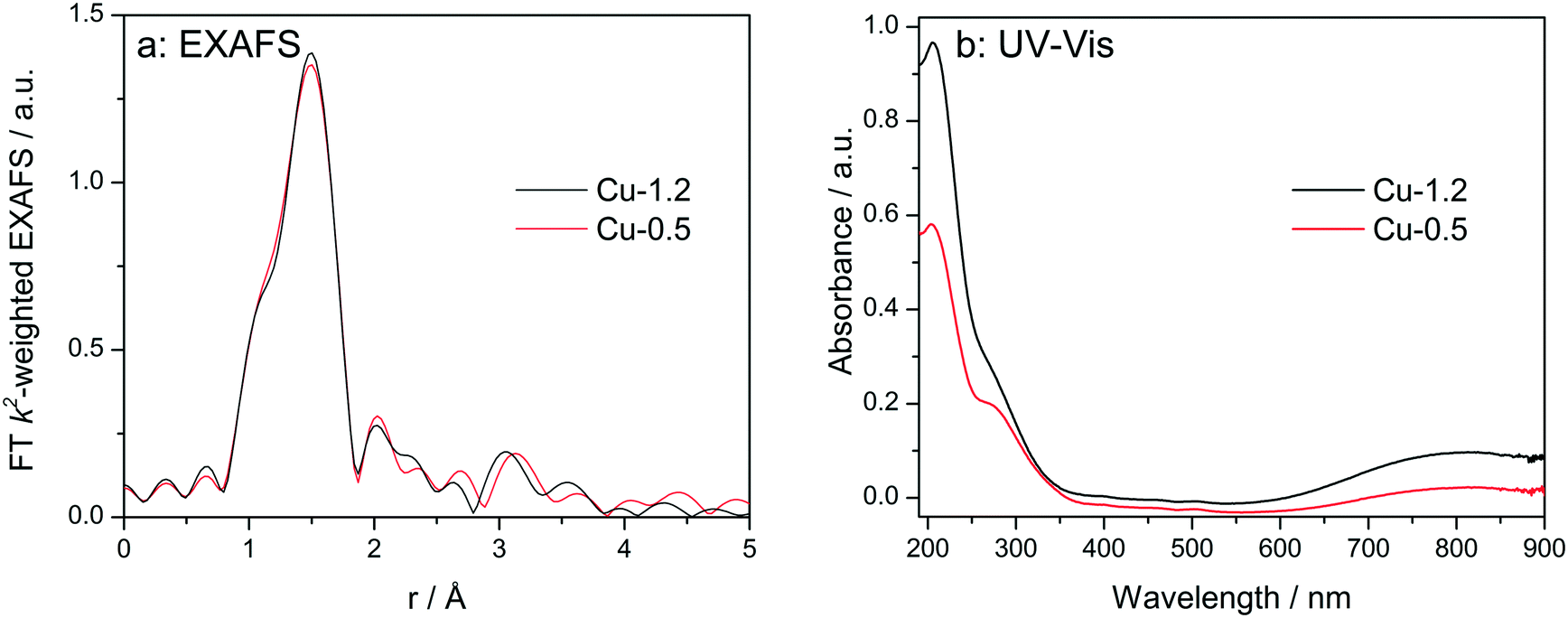 | ||
| Fig. 3 Ex situ FT k2-weighted EXAFS spectra (a, uncorrected for the phase shift) and DR UV-vis (b) spectra of Cu-0.5 and Cu-1.2 catalysts. | ||
Next, EPR measurements were conducted for the two catalysts in fresh state and after dehydration of Cu species at 300 °C for 1 h in 20% O2/Ar, and the results are presented in Fig. 4 and Table 1. The EPR spectra of the hydrated catalysts are dominated by an isotropic signal at g = 2.17 characteristic for isolated [Cu(H2O)n]2+ ions,51–56 the signals in Cu-1.2 being more intense. After dehydration in 20% O2/Ar at 300 °C, the characteristic axial signal of isolated Cu2+ with well-resolved hyperfine structure (hfs) connected directly to the CHA framework was recorded, in agreement with Godiksen et al.53 The sample Cu-1.2 contains two different types of isolated Cu2+ species (I and II) with the following spin Hamiltonian parameters, derived by spectra simulation (Table 1, A⊥ not resolved): type I at g1∥ = 2.368, A1∥ = 427 MHz (152.4 G), g1⊥ = 2.071 and type II at g2∥ = 2.321, A2∥ = 445 MHz (158.8G), g2⊥ = 2.071. Besides, a broad isotropic background signal III had to be superimposed to obtain proper fits, the contribution of which is however very weak. Both isolated species can be assigned to Cu2+ with coordination to 4 oxygen donors in distorted tetragonal planar geometry, possibly located in 6MR or in 6MR and 8MR that contain Al sites in different vicinity.57–59 Considering the Cu loading of the two catalysts and the Si:Al ratio in the SSZ-13 that is 16, the two different sites could be a mixture of Cu2+ exchanged either at paired Al sites or at different isolated Al sites, as e.g. Z2Cu and ZCuOH.23 For the Cu-0.5 catalyst mainly the type I of the isolated Cu species could be identified that probably corresponds to Cu coordinated to two Al sites. At the same time, a pronounced decrease of the Cu2+ signals after dehydration to almost the same intensity was observed for both the Cu-0.5 and Cu-1.2 samples. Despite the higher Cu loading in the Cu-1.2 catalyst, the same amount of Cu single sites seems to be EPR active in both samples. Cu sites may become EPR-silent in two major ways, either via reduction of Cu2+ or via interaction of several neighboring paramagnetic Cu2+ species. Partial autoreduction of Cu2+ sites in Cu-SSZ-13 is known to occur during heating in inert atmosphere or in vacuum.53 Considering that in this study the two samples were exposed to Ar after dehydration in 20% O2/Ar at 300 °C, the auto-reduction of the Cu sites was checked under similar conditions by in situ XAS. The results obtained indicate a virtually identical extent of Cu2+ to Cu+ reduction in both catalysts at 300 °C (not shown). Therefore, such a process cannot explain the almost perfect overlap of the EPR spectra for the dehydrated low and highly loaded Cu catalysts. Hence, the broad isotropic line III (Table 1) might reflect magnetically interacting Cu2+ species, e.g. dimeric species or Cu sites located in close vicinity. Such species bridged by hydroxyl groups or oxygen have been previously claimed in literature for Cu-SSZ-13 and other Cu-exchanged zeolites.49,50
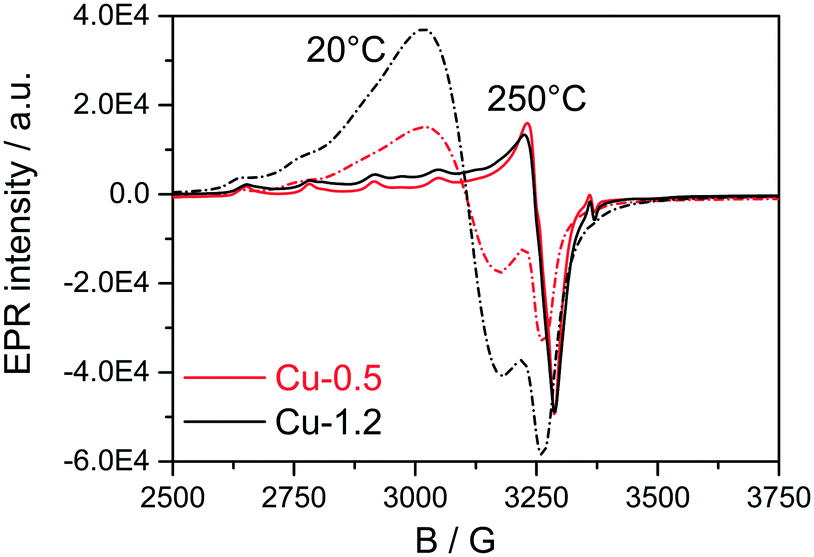 | ||
| Fig. 4 EPR spectra of Cu-0.5 (black lines) and Cu-1.2 (red lines) samples measured in hydrated state at 20 °C (dashed lines) and at 250 °C after 1 h pretreatment in 20% O2/Ar at 300 °C (solid lines). | ||
| Sample | Site | I rel/% | g ⊥ | g || | A ||/MHz (G) | Treatment |
|---|---|---|---|---|---|---|
| a Spectra could only be simulated properly by assuming superhyperfine coupling to 3 or 4 14N nuclei. | ||||||
| Cu-0.5 | I | 97 | 2.071 | 2.368 | 427 (152) | 1 h in 20% O2/Ar at 300 °C |
| III | 3 | 2.17 | ||||
| Cu-1.2 | I | 63.9 | 2.071 | 3.368 | 427 (152) | |
| II | 33.8 | 2.071 | 2.321 | 445 (159) | ||
| III | 2.3 | 2.17 | ||||
| Cu-0.5 and Cu-1.2 | IVa [Cu(NH3)3]2+ or [Cu(NH3)4]2+ | 2.057 | 2.263 | 550 (196) shfs to 14N A⊥ = 35 MHz A|| = 65 MHz | 2000 ppm NH3/Ar | |
Vtc-XES spectra of the dehydrated samples at 350 °C in dry air (Fig. 5a) show nearly identical Kβ′′ (8958 eV) and Kβ2,5 (8970–8980 eV) spectral features, which are typical for oxidized Cu species in Cu-SSZ-13 directly linked to oxygen.33,60,61 However, in the high-energy side of the spectra clear differences were observed. All features above 8980 eV are much more pronounced for the low-loaded Cu-0.5 sample in comparison to the highly-loaded Cu-SSZ-13 (note that both spectra are normalized by the maximum intensity of Kβ1,3 emission line). This once again suggests alterations in the structure of Cu due to the presence of another Cu site in close vicinity. Nonetheless, DFT calculations of the XES spectra would be necessary in the future to precisely elucidate their nature.
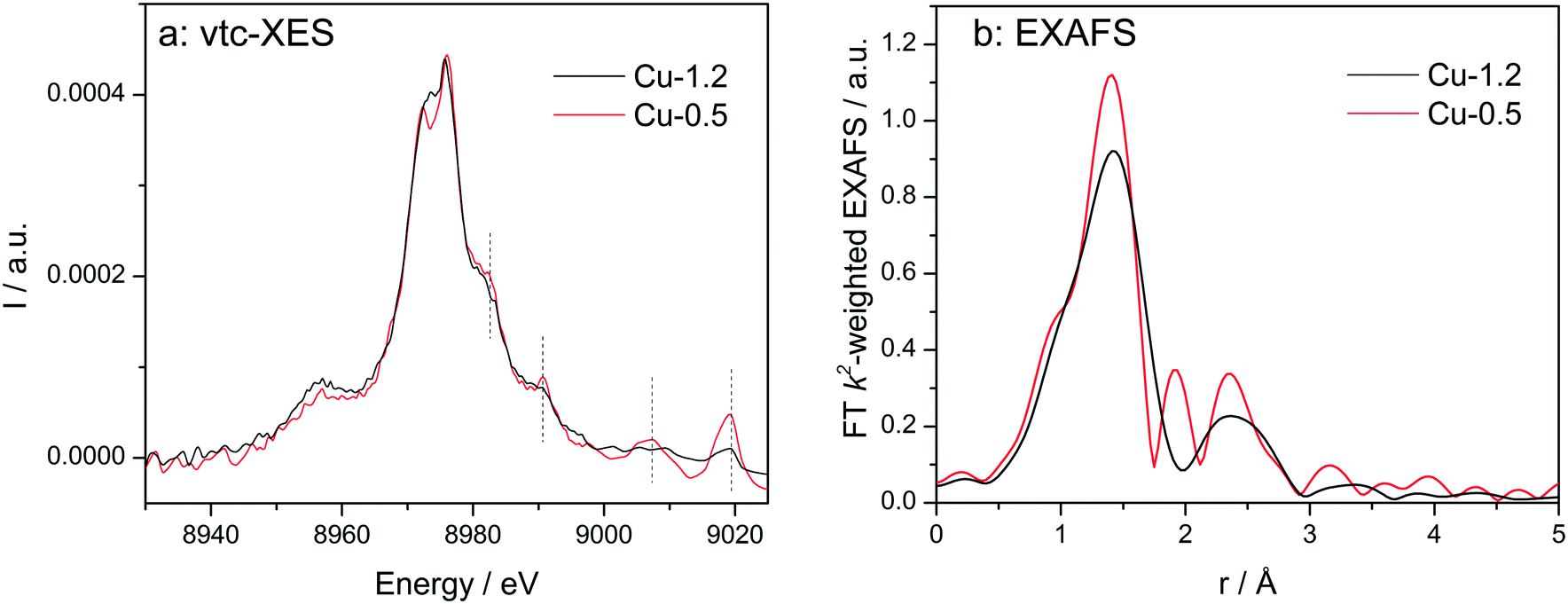 | ||
| Fig. 5 Vtc-XES (a) and FT k2-weighted EXAFS (b) spectra of Cu-0.5 and Cu-1.2 catalysts measured under dry air at 350 °C. | ||
EXAFS of dehydrated catalysts is presented in Fig. 5b and fitting results can be found in the ESI† (Table S1). For both samples a coordination number of about three was obtained for the first coordination shell corresponding to Cu–O bonding. This decrease of the coordination number has been previously assigned to dehydration of Cu2+ coordinated to isolated Al sites.23 In the spectrum of Cu-1.2 the second shell of lower intensity but clearly visible could be attributed only to a neighboring Cu atom during EXAFS analysis. However, we cannot exclude that a less defined interaction with the zeolite framework could be the reason that prevents proving the presence of Al or Si in the second coordination sphere. The fit quality of the Cu-0.5 spectrum was worse and only a minor contribution from Cu in the second shell was found according to the EXAFS fit. Thus, EXAFS data, although of medium quality, allows identifying the Cu–O–Cu interaction, particularly in the dehydrated Cu-1.2 sample.
The formation of dimeric or clustered sites is also supported by a statistical analysis on a similar metal-exchanged system by Brandenberger et al.62 They have demonstrated that the increase of ion exchange degree enhances the probability to form dimeric or clustered Fe sites. Analogously, at very low concentrations such an interaction between two Cu sites is less favored as they are too far away from each other. In contrast, for higher Cu loadings the formation of dimeric Cu–O–Cu entities in addition to the isolated monomeric species for higher Cu-concentrations is plausible. This evolution of the nuclearity directly correlates with the improved SCR activity, as observed in this study and also by the similar case of BEA and ZSM-5 zeolites, which start to be more active at higher Cu loadings.63 Furthermore, Guo et al. also observed by Raman spectroscopy the formation of Cu–O–Cu dimers upon dehydration of Cu-chabazite.64 However, since Cu sites seem to be very mobile,50,61,65 one has to follow their structural dynamics under realistic reaction conditions, i.e. operando.
3.3 Comparison of Cu-0.5 and Cu-1.2 by operando XANES
By using a quartz capillary-based catalytic reactor with plug flow geometry that allowed obtaining meaningful activity data and direct correlations between catalyst structure and catalytic activity, operando XAS and vtc-XES measurements were conducted for both catalysts under various SCR-related conditions around the seagull lower conversion point. In both cases several positions along the catalyst bed were monitored with a small X-ray beam (max. 1 × 0.2 mm size) at selected temperatures around the seagull point, while collecting the corresponding NOx conversion (Fig. 6 and 7). In contrast to the standard SCR activity reported in Fig. 1 for the laboratory tests, the seagull profile during the operando experiment (Fig. 7) is slightly less visible due to the dilution of the gas mixture before the FTIR instrument, as described in the experimental section. However, the two characteristic low- and high-temperature regions can be clearly distinguished also in this case. Consequently, distinct variations of the local structure were noticed under SCR reaction conditions at increasing temperatures: (i) inhibition of the SCR reaction due to the high NH3 concentration at the inlet of the catalyst bed,66 (ii) reduction of the Cu2+ sites during the standard SCR reaction and (iii) reoxidation of the Cu+ sites at the end of the catalyst bed at higher temperatures, when the NOx conversion occurs mainly at the inlet and mid positions.12,61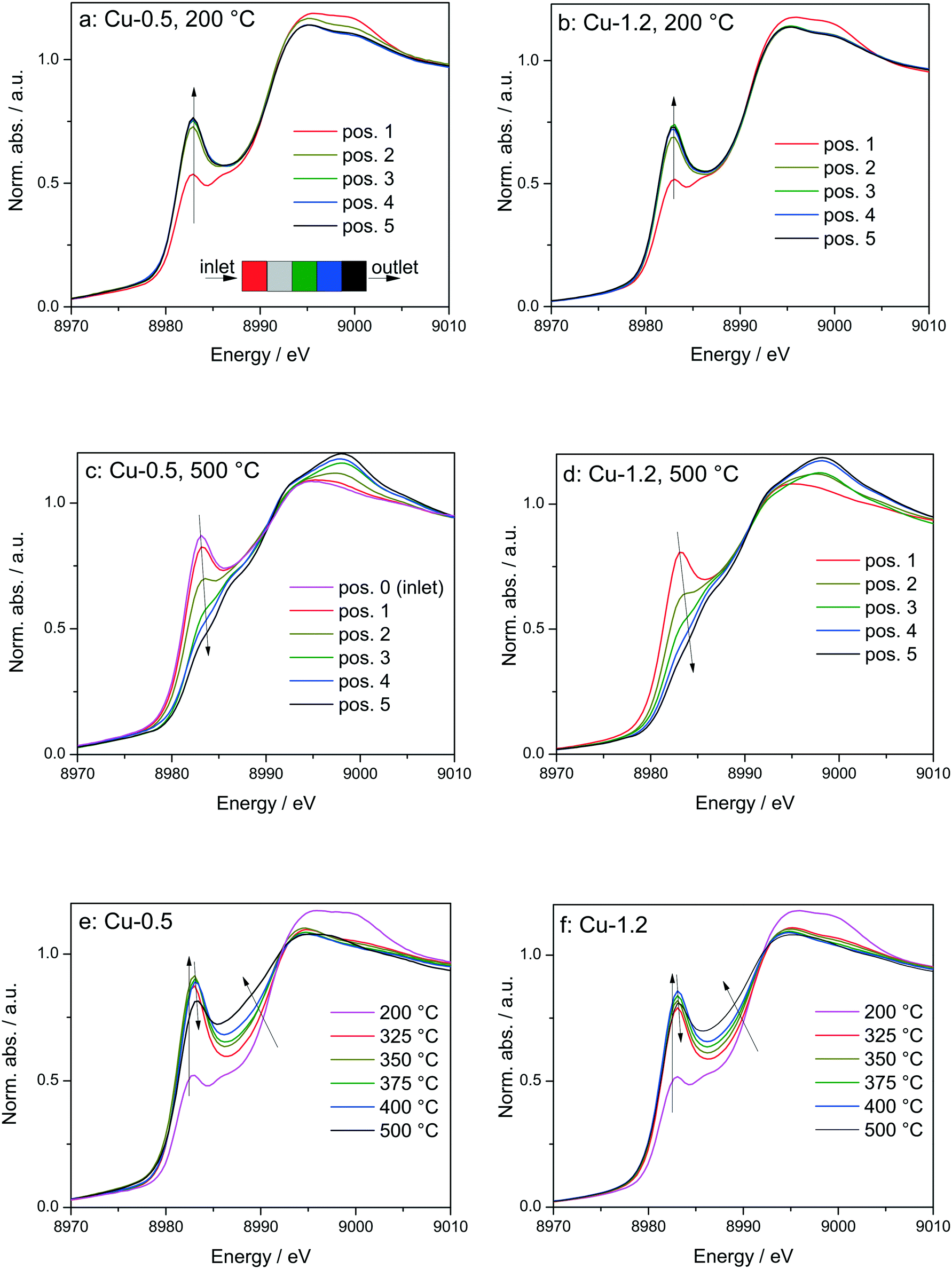 | ||
| Fig. 6
Operando XANES spectra of (a, c and e) Cu-0.5 and (b, d and f) Cu-1.2 measured at different temperatures under SCR feed. Parts (a–d) show spatially resolved spectra measured at equidistant points from inlet to the outlet of a catalyst bed at 200 and 500 °C (7 mm long catalyst bed, positions illustrated by the different color code in 6a), while parts (e and f) present spectra measured at position 1 (0.5 mm from the inlet) of a catalyst bed. Conditions: 1000 ppm NO, 1000 ppm NH3, 10% O2, 1.5% H2O, balance He, GHSV 200000 h−1. Corresponding catalytic data are reported in Fig. 7. | ||
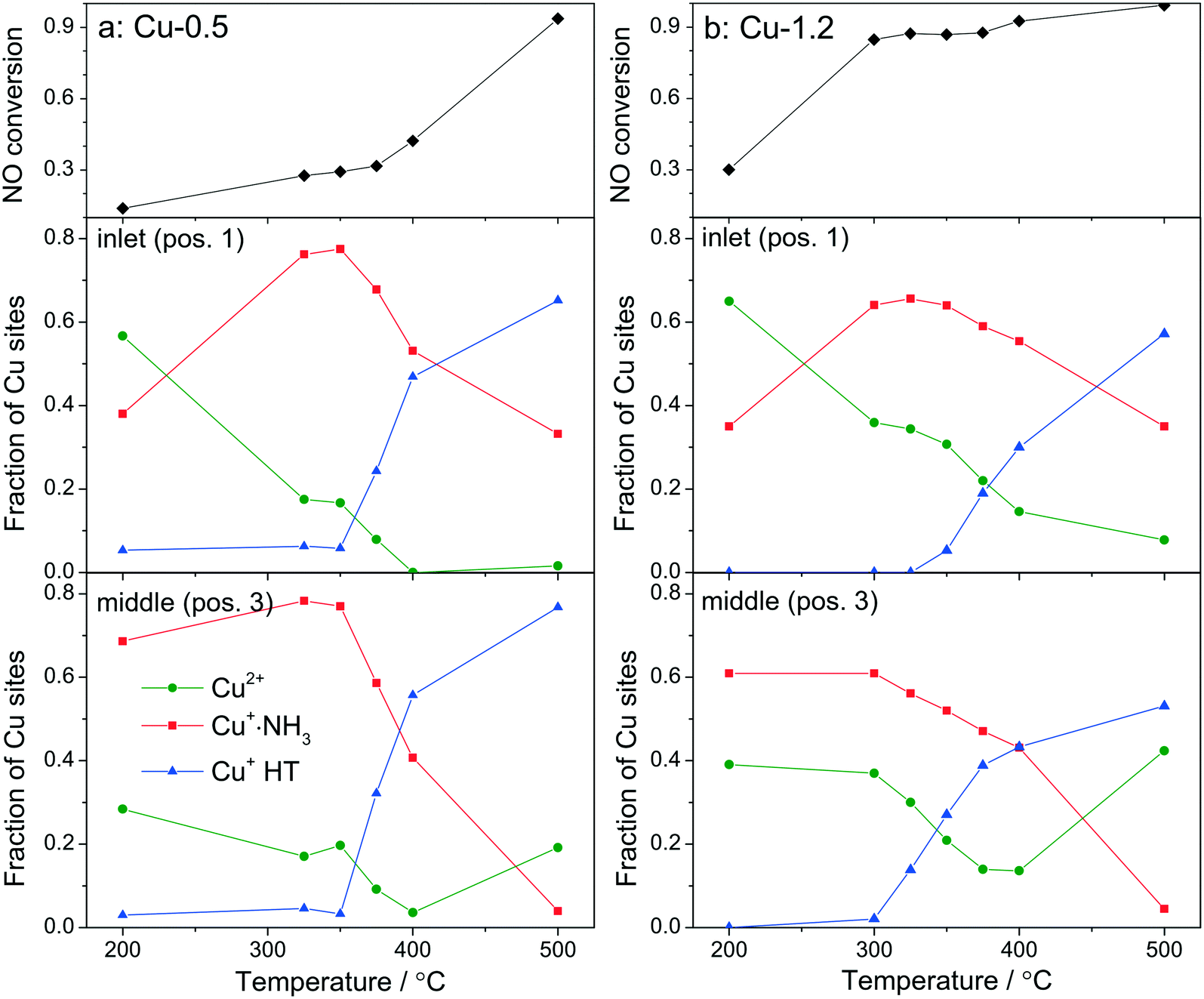 | ||
| Fig. 7 Conversion of NO and fraction of Cu+ coordinated with NH3 (with linear geometry) and the HT (high temperature) Cu+, both normalized to all Cu species, obtained by LCA of XANES spectra measured during operando XAS studies at SLS. Values reported for (a) Cu-0.5 and (b) Cu-1.2 catalysts. XANES data are taken at positions 1 (near the inlet) and 3 (middle) of the catalyst bed. The fractions of Cu+ are reported for the inlet of the catalyst bed. The error bars of the LCA are within 10%. Conditions: 1000 ppm NO, 1000 ppm NH3, 10% O2, 1.5% H2O, balance He, GHSV 200000 h−1. | ||
In spite of markedly different catalytic activity (Fig. 1 and 7) similar spectral dynamics were observed for both Cu-0.5 and Cu-1.2 catalysts, especially illustrated by variation of the feature at 8982.7 eV (Fig. 6), previously attributed by conventional and HERFD-XANES studies to linearly coordinated Cu+ species.12,61,66 Only between 325–400 °C the intensity of this feature is significantly higher for the Cu-0.5 sample, which clearly indicates the presence under standard SCR conditions of a higher number of linearly coordinated Cu+ sites in the low-loaded sample.
As illustrated for several temperatures in Fig. 6 and S5,† the XANES spectra collected at several positions along the catalyst bed (the measurement positions are chosen in the same way as in ref. 66) indicate the absence of gradient in oxidation state/local structure for the low loaded Cu-SSZ-13 up to 400 °C but pronounced differences were observed at 500 °C. In contrast, changes in the XANES spectral features at several positions along the catalyst bed were noticed for the highly-loaded Cu-SSZ-13 starting at 325 °C. This evolution is in line with the SCR activity of the two samples.
In order to see how the Cu+/Cu2+ species evolve during the SCR at different positions in the catalyst bed, we have conducted a linear combination fit analysis of the XANES region. Unlike the previous work where the edge shape of Cu-SAPO-34 XAS spectra could be modelled with a linear combination of just two species, namely Cu2+ and Cu+ sites from CuO and Cu2O reference compounds,66 here we noticed not just changing intensity of the shoulder at 8982.7 eV but also an increase in the overall absorbance between 8985 and 8990 eV above 350 °C. Principal component analysis suggested that at least three different components need to be used to fit the obtained operando XANES spectral sets. This is in agreement with the recent study of Lomachenko et al.,28 who also observed the evolution of three different Cu species during the SCR process between 150–400 °C. Based on the ex situ and in situ characterization performed in this study we propose the interchange of the following monomeric or dimeric components, depending on the gas mixture and temperature: (ii) Cu2+ species, (ii) Cu+ linearly coordinated to NH3 and (iii) NH3-free Cu+ sites and 3-fold coordinated.67 The presence of Cu+ linearly coordinated alongside with Cu2+ was previously demonstrated12 at low temperatures whereas the third type of sites appear during the SCR process at higher temperatures.
For fitting the operando XANES spectra linear combination analysis with the reference spectra was employed. The dehydrated Cu2+ reference spectrum was acquired under oxidizing conditions at about 200 °C.66 This selection was done based on a dehydration experiment up to 550 °C and also on previous published results.12 The Cu+ references were obtained by recording several series of in situ XANES during temperature programmed reduction (TPR) of pre-oxidized catalysts with ammonia as a reductant, and applying multivariate curve resolution-alternating least squares method (MCR-ALS, see section 2.3.3 and Fig. S6†).35,36 The obtained spectra used for linear combination analysis of operando XANES are shown in Fig. S7† together with the HERFD-XANES spectra recorded previously12 under model conditions in order to help the attribution of spectral features. As previously mentioned, the obtained reference spectra were assigned to Cu+·NH3 complexes with linear geometry (with either two NH3 ligands or NH3 and H2O)9 and to Cu+ without direct coordination to ammonia and 3-fold coordinated,12 referred below as Cu+ HT (high temperature). The results of the linear combination analysis of the XANES spectra (Fig. 7a and b) collected at 0.5 mm from the inlet (pos. 1) and in the middle (pos. 3) of the catalyst bed revealed that for both samples the presence of a high portion of Cu+ sites among all Cu species even at low temperatures under standard SCR conditions (as can be seen by the appearance of the shoulder at 8982.7 eV in Fig. 6). Although it is expected that the reduction of Cu2+ and the formation of Cu+(NH3)x species occur mainly due to the SCR reaction, its extent does not seem to correlate with the measured performance, e.g. NOx conversion was not observed over Cu-0.5 at temperatures below 350 °C. On the other hand, given the possibility to distinguish between two different Cu+ sites, i.e. coordinated or uncoordinated to NH3, we could also compare their fraction depending on temperature. Alongside, conversion of NO measured during XAS acquisition is shown. The data obtained for Cu-0.5 (Fig. 7a and S8†) suggests a direct correlation of the SCR activity with the amount of NH3-free Cu+ sites or not linearly coordinated, which appear above 350 °C irrespective of the position in the catalyst bed, and an inverse correlation with the concentration of Cu+·NH3 sites. Since the NH3 inhibition at low temperature (<300 °C) was detected for both catalysts, the decrease of NH3 amount at the Cu sites seems to lead to an increase of the SCR activity at higher temperatures. This appears to be particularly important for the low loaded Cu-SSZ-13 sample, predominantly containing isolated Cu sites. Another indication for SCR activity is the formation of Cu2+ especially observed towards the outlet of the catalyst bed.
The situation is different for the highly-loaded Cu-1.2 catalyst. Its higher SCR activity at low temperatures (Fig. 1 and 7b) does not correlate to the amount of NH3-free Cu+ sites in this catalyst, which appear only in the high temperature region of the seagull-shaped conversion profile. However, the amount of Cu2+ is significantly larger for this sample at all positions of the catalyst bed and for all investigated temperatures, which is also an indicator for less NH3 inhibition at the Cu sites and, especially, for the occurrence of the reoxidation step of the SCR mechanism. On the one side this points out that not the reduction of the Cu sites but rather the reoxidation is a rate limiting step for NH3–SCR on Cu-0.5. On the other side, different Cu species seem to be responsible for the low-temperature activity of Cu-SSZ-13 with high Cu loading.
3.4 Structure of Cu species analyzed by operando EXAFS
In addition to the differences in the oxidation and reduction of Cu species along the catalyst bed, especially at low temperatures significant differences were observed during the analysis of the corresponding spatially resolved Fourier Transformed (FT) EXAFS spectra (Table 2). The appearance of a second coordination sphere at the uncorrected distance of about 2.4 Å around Cu central atom reported by Lomachenko et al.,28 was also observed during this study under SCR conditions for Cu-1.2 catalyst but only at the end of the catalyst bed, and starting with 325 °C (Fig. S9†). In line with the EPR and XES measurements of the dehydrated Cu-SSZ-13 samples, this second shell in the FT EXAFS could indicate the bond with the zeolite framework and/or with another Cu atom as a dimeric entity, e.g. mono or bis(μ-oxo)dicopper species.49 In contrast to the results in ref. 28 we did not observe any variation below this temperature, e.g. 200 °C, most probably due to the lower Cu concentration in our sample and consequently to the extension of the reaction zone over the whole catalyst bed. It results that the changes in the structure occur at the positions in the catalyst bed where the NOx conversion is almost complete, which correspond to low NH3 concentrations. This hypothesis is supported by the FT EXAFS spectra obtained for the low loaded catalyst but only at 400 °C, as a second coordination shell emerged at positions towards the end of the catalyst bed.| CN(O)/d(Cu–O) | CN(Cu)/d(Cu–O–Cu) | CN(Al)/d(Cu–O–Al) | σ 2 (10−3 Å2) | δE0 (eV) | ρ (%) | |
|---|---|---|---|---|---|---|
| Cu-0.5 NH3 + O2 200 °C | 3.2 ± 0.3 1.93 ± 0.01 Å | n.a. | n.a. | 5.8 ± 1.5 | −1.2 ± 0.9 | 0.9 |
| Cu-0.5 NO + O2 200 °C | 4.5 ± 0.3 1.95 ± 0.01 Å | n.a. | n.a. | 6.2 ± 1.4 | −1.2 ± 0.8 | 0.6 |
| Cu-0.5 NH3 + O2 375 °C | 2.4 ± 0.2 1.92 ± 0.01 Å | n.a. | n.a. | 5.6 ± 1.3 | −1.2 ± 0.8 | 0.8 |
| Cu-0.5 NO + O2 375 °C | 3.9 ± 0.4 1.94 ± 0.01 Å | 1.3 ± 0.5 3.03 ± 0.02 Å | 1.0 ± 0.4 2.74 ± 0.04 Å | 6.5 ± 1.5 | −0.6 ± 0.9 | 0.5 |
| Cu-1.2 NH3 + O2 200 °C | 3.4 ± 0.4 1.94 ± 0.01 Å | n.a. | n.a. | 7.1 ± 1.7 | 0.2 ± 1.0 | 1.1 |
| Cu-1.2 NO + O2 200 °C | 4.6 ± 0.4 1.94 ± 0.01 Å | 0.5 ± 0.4 2.92 ± 0.04 Å | n.a. | 7.4 ± 1.2 | −2.0 ± 0.7 | 0.4 |
| Cu-1.2 NH3 + O2 400 °C | 2.5 ± 0.1 1.92 ± 0.01 Å | n.a. | n.a. | 6.3 ± 1.2 | −1.4 ± 0.6 | 0.4 |
| Cu-1.2 NO + O2 400 °C | 4.2 ± 0.4 1.93 ± 0.1 Å | 0.85 ± 0.4 2.97 ± 0.03 Å | 0.7 ± 0.3 2.72 ± 0.04 Å | 9.3 ± 1.6 | −1.2 ± 0.8 | 0.4 |
In order to elucidate whether the appearance of the second coordination shell is a step of the SCR mechanism and not only a fingerprint for the dehydrated Cu sites, additional XAS and XES investigations were performed at different temperatures by exposing the catalysts to the NH3-only and NO-only gas mixtures. This allows an easier interpretation by separately identifying the spectral features characteristic for the interaction of Cu with an oxidizing feed in comparison to the reducing NH3-containing feed, without mixing the corresponding spectral features as we have seen earlier that it will lead to an averaged information.12 Catalytic activity of Cu-0.5 and Cu-1.2 in oxidation of NO or NH3 is reported in Fig. S2.† As the differences in the XANES spectra under these conditions are very similar to the results reported in a previous study,12 here only the EXAFS region is discussed.
Fig. 8 displays the FT EXAFS spectra recorded for Cu-1.2 catalyst at low and high temperatures of 200 and 400 °C, i.e. below and above the seagull shaped NOx conversion minimum. Similarly as observed at the end of the catalyst bed during SCR, a second coordination shell appears in the FT EXAFS spectra of Cu-1.2 under the NO oxidation feed (Fig. 8a). Although the feature is small, it is reproducible and has been seen not only at the inlet of the reactor but for several points along the catalyst bed. In contrast, if NH3 and O2 are dosed at 200 °C only the first coordination shell could be detected.
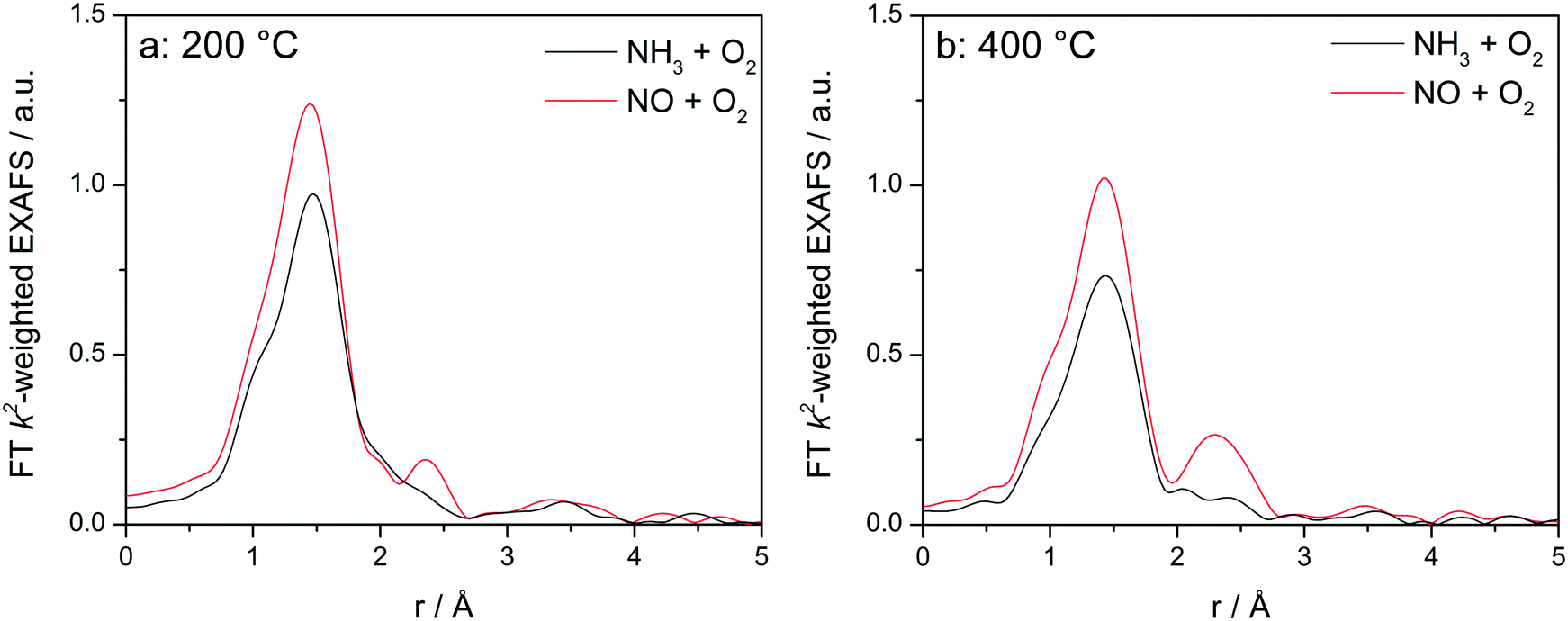 | ||
| Fig. 8
Operando FT k2-weighted EXAFS spectra measured on Cu-1.2 at (a) 200 °C and (b) 400 °C. Conditions: 1000 ppm NO or 1000 ppm NH3, 10% O2, 1.5% H2O/He, GHSV 200000 h−1. | ||
While differences in the spectra of Cu-1.2 in the absence/presence of NH3 are small at low temperatures, they become much more pronounced at temperatures above the seagull point (Fig. 8b). The second coordination shell in the FT EXAFS becomes very pronounced at 400 °C for the inlet position under the NO oxidation gas mixture but disappears in NH3/O2. Therefore, any bonding to another Cu site or to the zeolite framework seems to be lost in the presence of NH3, and the dimeric Cu species are converted to single Cu sites coordinated to NH3, as for example linearly coordinated Cu+(NH3)2 complexes. However, the data obtained in NO/O2 indicate that the Cu+–O–Cu+ dimers linked to the zeolite framework constantly reappear if NH3 is consumed during the SCR process.
In contrast, the operando FT-EXAFS spectra of Cu-0.5 (Fig. 9) revealed the presence of a single coordination sphere at low temperatures and the appearance of the second coordination sphere (2.5 Å, not corrected for the phase shift) only above 375 °C. Since this structural change occurs just within the SCR activity window of the low loaded Cu-SSZ-13 catalyst, which is analogous to Cu-1.2 sample, it suggests the migration of Cu sites at high temperature13 and formation also in this case of Cu dimeric species during the reoxidation step. As 375 °C corresponds to the NOx SCR onset over the low loaded Cu-0.5 but also to the temperature of the conversion minimum at the seagull SCR profile over high-loaded Cu-1.2, one could correlate the NH3–SCR activity of Cu-0.5 and Cu-1.2 with the amount of specific Cu sites present in the catalyst at different temperatures.
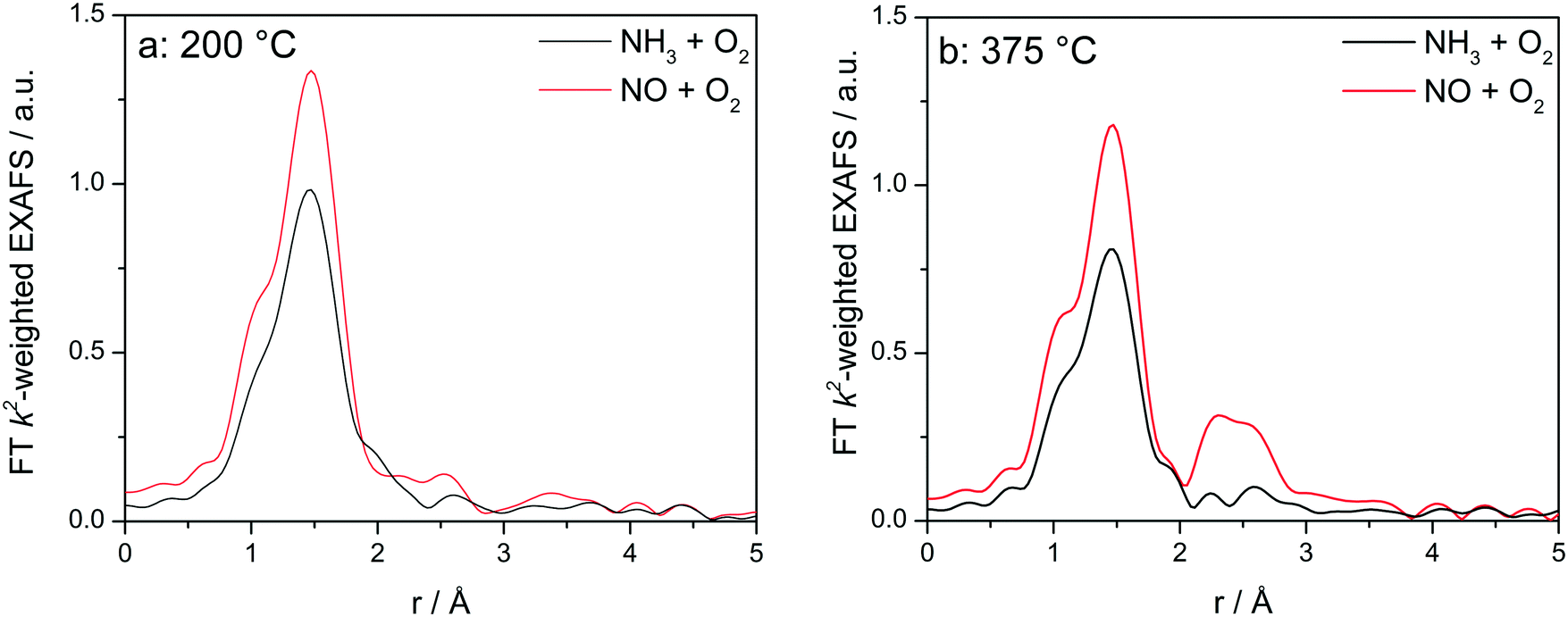 | ||
| Fig. 9
Operando FT k2-weighted EXAFS measured on Cu-0.5 in the temperature regions with (a) low NOx conversion (200 °C) and (b) NOx conversion onset (375 °C). Conditions: 1000 ppm NO or 1000 ppm NH3, 10% O2, 1.5% H2O/He, GHSV 200000 h−1. | ||
The analysis results the presented FT EXAFS spectra are summarized in Table 2. All spectra measured under NH3 oxidation conditions for both catalysts can be fitted assuming just one shell composed of O or N atoms. These elements cannot be distinguished in EXAFS because of similar scattering factors, but recent vtc-XES measurements60 allowed us to identify both, the O and N neighbors, in the first coordination shell under these reaction conditions. For the highly loaded Cu-SSZ-13 catalyst the number of ligands varies between 3.4 (below 350 °C) and 2.5 (above 350 °C). For Cu-0.5 values between 3.2 (low temperature) and 2.4 (high temperature) were calculated. These results correspond well to observations of predominantly linear single Cu sites coordinated with ammonia made by Giordanino et al.61 and also observed during our EPR measurements (reported in the following section and in Fig. 12).
Under NO oxidation feed the spectrum of Cu-0.5 at 200 °C may also be fitted with just one coordination shell but in this case the coordination number is higher than 4, which could correspond to hydrated Cu sites. The situation slightly changes for Cu-1.2 catalyst and the second coordination sphere could be fitted with a low number of Cu atoms. In this case the low intensity could be due to the presence of hydrated μ-oxo dicopper complexes within this temperature range, without strong interaction with the zeolite framework (solvated sites). As proposed by Gao et al.,13 such transient Cu+–O–Cu+ sites, which are present only in highly loaded SSZ-13, could be reoxidized by O2 to Cu2+–O–Cu2+ completing the NH3–SCR cycle.
At high temperatures, the fitting of the second shell requires the presence of both, Cu and Al atoms (or Si because of similar scattering factors), at slightly different distances, since the fitting with two atoms of the same element does not result in meaningful parameters of a good fit. Therefore, the results at temperatures above 350 °C suggest for both Cu-1.2 and Cu-0.5 the formation of dehydrated μ-oxo dicopper complexes with each Cu atom probably bound to a zeolite T-atom via oxygen, as was proposed by Woertnik et al.68 for Cu-ZSM-5 zeolite. The obtained Cu–O–Cu distances are only slightly longer, but comparable with 2.87–2.95 Å found in Cu-ZSM-5 zeolites by Groothaert et al.49 and Grünert et al.69 Regarding the contribution at 2.72–2.74 Å, Cu neighbors were suggested by Palomino et al.70 and Kuroda et al.71 for Cu-ZSM-5, but Paolucci et al. attributed it to a zeolite T atom (Al or Si) in Cu-SSZ-13,10 which also agrees with our fitting results. Overall, the outcome clearly indicates for both Cu loadings at high temperatures (above the seagull point) a more localized Cu-dimeric structure in the NO oxidizing atmosphere.
3.5 Structure and evolution of Cu species analyzed by operando vtc-XES
The switch between two structures as a function of the NH3 or NO presence at low and high temperatures around the seagull point has been confirmed for the highly loaded Cu-SSZ-13 catalyst also by operando vtc-XES measurements (Fig. 10). All spectral features appearing above 8980 eV were significantly more pronounced in NH3 oxidation mixture and under SCR conditions (Fig. S10†), which could suggest that monomeric linearly coordinated Cu+(NH3)x complexes are formed (Fig. 10 and 11).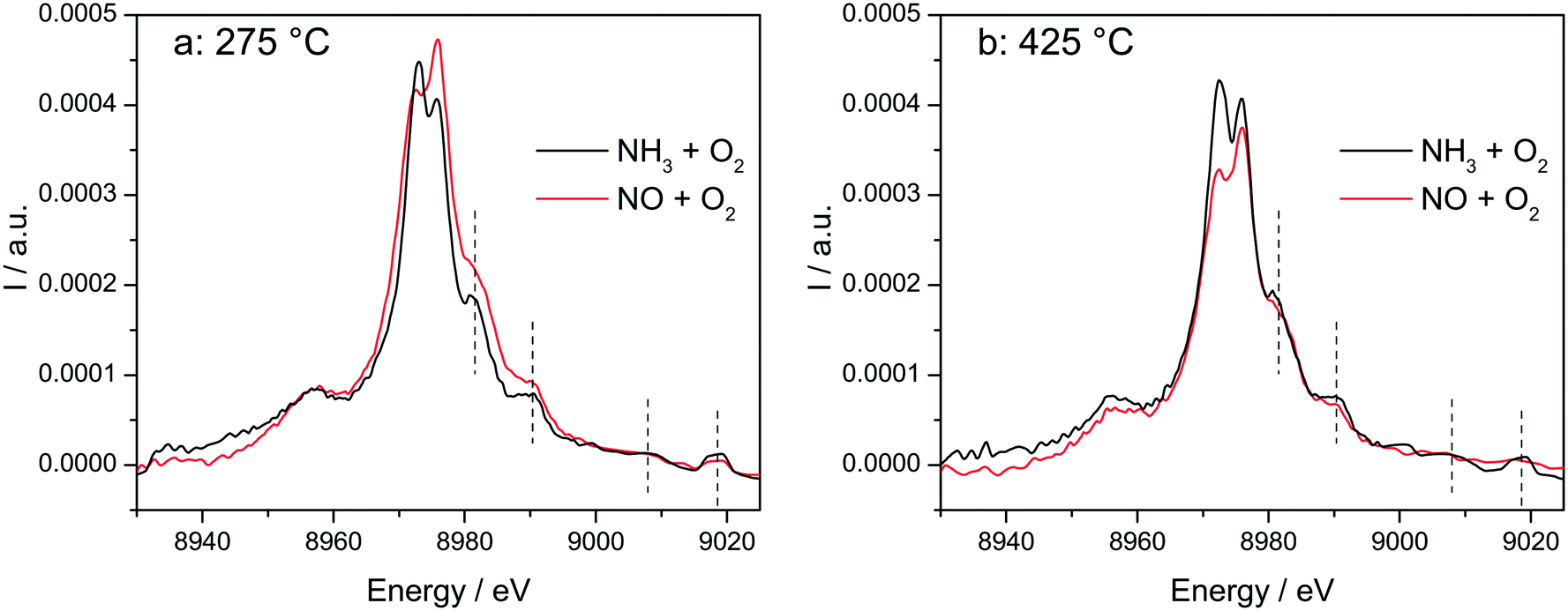 | ||
| Fig. 10
In situ vtc-XES measured on Cu-1.2 at (a) 275 °C and (b) 425 °C. Conditions: 1000 ppm NO or 1000 ppm NH3, 10% O2, 1.5% H2O, balance He, GHSV 200000 h−1. | ||
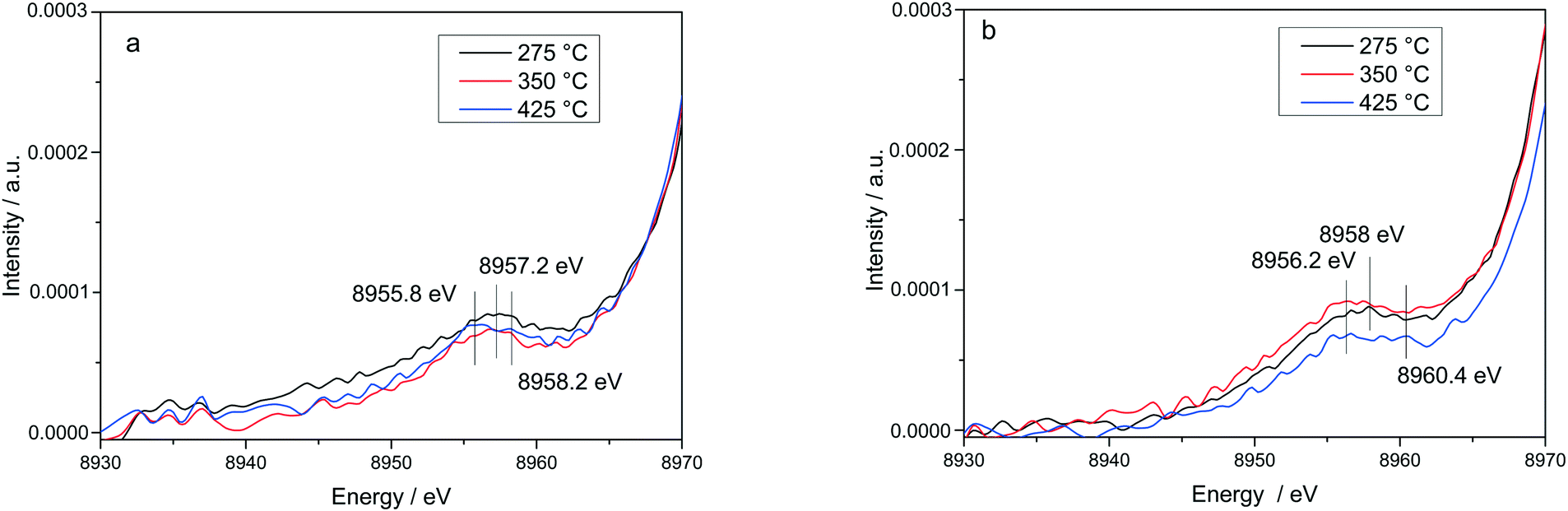 | ||
| Fig. 11 Evolution of vtc-XES spectra Kβ′′ region at different temperatures for Cu-1.2 catalyst. Conditions: (a) 1000 ppm NH3, 10% O2, 1.5% H2O, He; (b) 1000 ppm NO, 10% O2, 1.5% H2O, He, GHSV 200000 h−1. | ||
The stability of such complexes even in the gas phase has been theoretically and also experimentally demonstrated.72,73 This might lead to their formation and also to the weakening of the bond with the zeolite framework. Moreover, their mobility within the chabazite framework has been often claimed and it seems to be enhanced by the SCR reaction mixture and temperature increase.65,74 Furthermore, whereas the variations in the Kβ′′ and Kβ2,5 regions under different feeds were discussed earlier for very low temperatures (200 °C) and correlated for the SCR process with a direct coordination of NH3 to the Cu sites and of NO via the neighboring O atom,12,61 significant structural differences could be observed at higher temperatures for both NH3 and NO oxidation feeds. Thus, at 275 °C, below the seagull lower conversion point, the direct coordination of NH3 to the Cu sites is clearly visible in the shift of Kβ′′ line towards higher energy (8957.2 eV vs. 8956.9 eV in O2 + H2O mixture). At 425 °C due to NH3 oxidation and also desorption, the intensity of the Kβ′′ region also indicates a decrease of NH3 concentration at the Cu sites (Fig. 11a), similar as reported in ref. 28, and two features can be distinguished at 8955.8 eV and at 8958.2 eV.
The fingerprint of the direct NO coordination at the Cu sites in the form of nitrites/nitrates, which is not visible during the low temperature onset of NOx conversion as it is strongly inhibited by H2O,12,60 appears clearly at 275 °C. A shift of the Kβ′′ from about 8956.9 eV in 10% O2, 1.5% H2O/N2 (ref. 75) to 8958 eV was observed if 1000 ppm NO were added (Fig. 11b). Although slightly broader towards lower energies, the shift was maintained at 350 °C. At 425 °C, above the seagull point, two features become visible in the Kβ′′ region with maxima around 8956.2 eV and 8959.6 eV. Whereas the first one is probably due to a neighboring O atom, the second feature indicates still some interaction with NOx. In addition, a strong decrease of the Kβ2,5 region was observed. Hence, in line with the FT EXAFS data, not only the adsorption/desorption of NH3 and NOx but a different local structure is also indicated by the vtc-XES measurements at higher temperatures. However, further DFT calculations are necessary to elucidate all the variations in the XES spectra and to demonstrate this hypothesis.
3.6 Structure of Cu species analyzed by in situ EPR
Fig. 12 depicts the EPR spectra collected for the two catalysts at 250 °C in the presence of NH3-only, NO + O2 and SCR gas mixture. In comparison to the EPR spectra recorded after dehydration in 20% O2/Ar (Fig. 4), exposure to NH3/Ar flow at 250 °C decreased the intensity of the isolated Cu2+ species in both samples, indicating that Cu2+ is almost completely reduced to EPR silent Cu+ (Fig. 12, red lines). However, the reduction extent of sample Cu-1.2 is slightly lower compared to sample Cu-0.5. After 30 min flushing with NH3, the EPR spectra of both samples contain only one type of Cu2+ species, namely type IV (Table 1), with the same spin Hamiltonian parameters. They are probably coordinated by NH3 molecules. This confirms the XAS and vtc-XES results presented above, indicating that NH3 is adsorbed at Cu2+ species and reduces them partly to Cu+.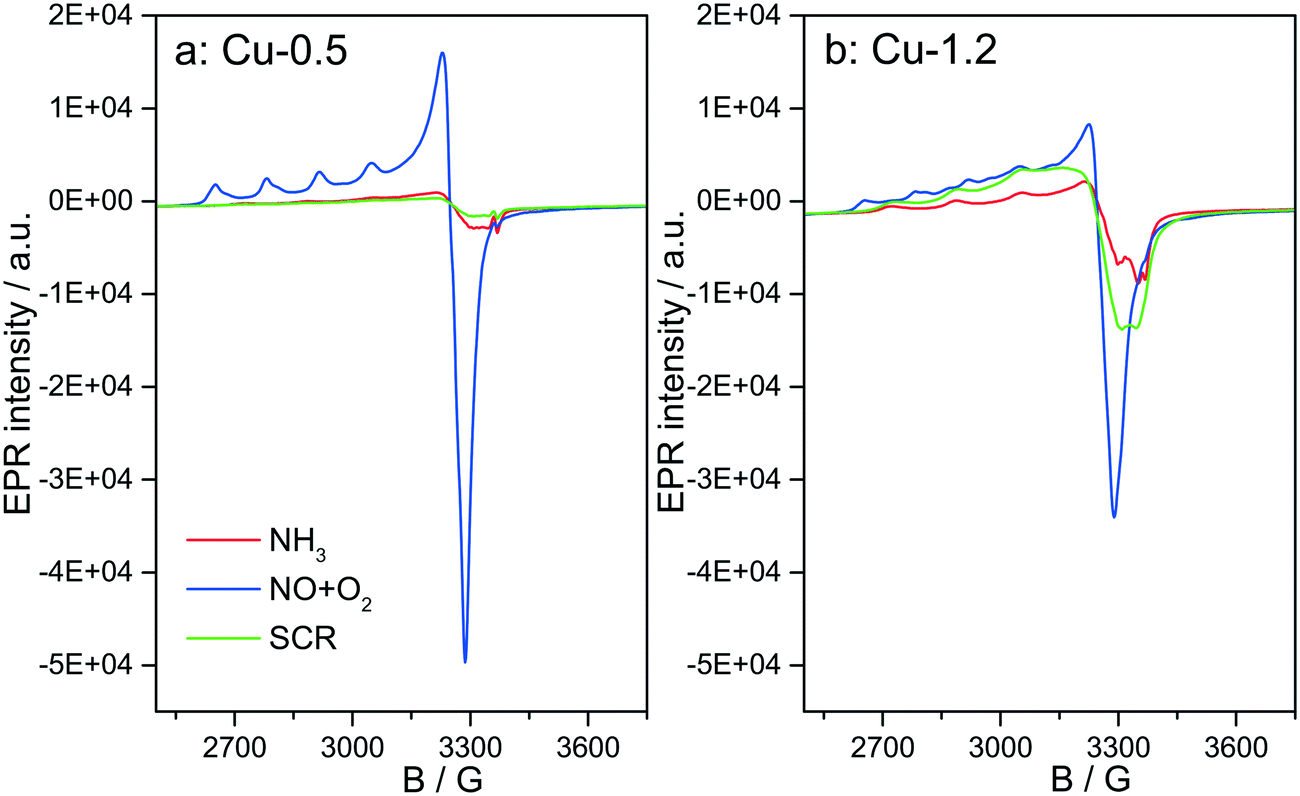 | ||
| Fig. 12 In situ EPR spectra of (a) Cu-0.5 and (b) Cu-1.2 samples measured at 250 °C in: 2000 ppm NH3/Ar (red line); 2000 ppm NO, 10% O2/Ar (blue line) and 2000 ppm NH3, 2000 ppm NO, 10% O2/Ar (green line). | ||
The presence of Cu sites in close vicinity, e.g. dimers or cluster-like CuOx species, at low temperatures in the absence of NH3 is evident from the EPR spectra acquired at 250 °C (below the seagull point) for the two catalysts in a gas mixture containing only NO and O2. As shown in Fig. 12, both Cu2+ species identified for the dehydrated samples (I and II in Table 1) also appear in the NO + O2 dry gas mixture. However, whereas the hfs signal of species I in sample Cu-0.5 is identical to that recorded in inert atmosphere, the hfs signals of both species I and II in the highly loaded Cu-1.2 sample loose intensity in comparison to their initial state in Ar (Fig. 4), in favor of the broad background signal of magnetically interacting Cu2+. Moreover the total intensity of the EPR signal decreases for sample Cu-1.2 but not for sample Cu-0.5. This points to an antiferromagnetic interaction of neighboring Cu2+ sites in Cu-1.2, which may be dimers. Although this result supports the findings of EXAFS, it must be mentioned that by EPR Cu2+ dimers cannot be discerned from other Cu2+Ox species of small nuclearity just based on a broad isotropic line.
Switching from NO/O2 to the SCR feed at 250 °C leads to a strong decrease of the signal intensity in sample Cu-0.5, which is even more pronounced than in the presence of NH3 only. In sample Cu-1.2, this intensity loss is much smaller and the signal under SCR feed is significantly higher than under NH3 only, which is opposite to sample Cu-0.5. This is clearly linked to the faster reoxidation of Cu+ species for the catalyst with a higher Cu loading, as also indicated by the operando XANES results (Fig. 6).
4. Discussion
Summing up the activity and in situ/operando characterization data obtained for the 0.5 wt% and 1.2 wt% Cu loaded SSZ-13 catalysts, the following insights were found:• While Cu sites in both low- and highly-loaded hydrated samples are at room temperature virtually identical [Cu(H2O)x]2+ solvated species,76 the in situ XES, XAS and EPR data as well as the ex situ characterization upon dehydration show different species linked to the zeolite framework: isolated/monomeric Cu sites, present especially for low Cu-loadings, and species with higher nuclearity, e.g. Cu mono or bis(μ-oxo) dimers for higher Cu concentrations.
• A stronger NH3-inhibition effect was identified at low temperatures especially for the low loaded Cu-SSZ-13 catalyst, predominantly containing isolated Cu sites. This effect is diminished above 300 °C, which corresponds to the high-temperature region of the seagull profile.
• By performing the LCA based on the references obtained by MCR-ALS, the participation to the SCR mechanism of Cu2+, [Cu(NH3)x]+ and NH3-free Cu+ sites could be demonstrated. A higher concentration of Cu2+ and a lower amount of [Cu(NH3)x]+ species was found for Cu-1.2 catalysts over the seagull region, which indicates a lower NH3 inhibition and an easier reoxidation of the reduced sites during the SCR mechanism.
• The dimeric Cu species are converted to linearly coordinated monomeric [Cu(NH3)x]+ sites by ammonia at low as well as at high temperatures during the SCR mechanism. This could be clearly demonstrated by the spatially resolved operando XAS measurements. H2O at low temperatures (<200 °C) may also stabilize monomeric Cu sites, e.g., as solvated Cu2+ species, which is in line with previous studies.22,23
• The dimeric Cu species are stable at high temperatures (>200 °C) in the presence of the NO oxidation feed. The formation of nitrites/nitrates as intermediate species was captured in the vtc-XES spectra at temperatures above 200 °C. The variation in the local structure of the Cu sites is also suggested by the different interaction of NOx with Cu at temperatures below and above the seagull point.
• At higher temperatures, in the second region of the seagull profile, both the monomeric and the dimeric species are more linked to the zeolite framework, as also previously shown.58 However, our results indicate that a certain mobility is maintained in the presence of NH3, which leads to the migration of the isolated species and formation of the dimeric sites, identified in this study by operando XAS and in situ EPR measurements. This reaction path is in line with that proposed by Paolucci et al.,16 describing a dynamic multinuclear site formation during the reoxidation step of Cu+ to Cu2+.
• The prerequisite of the SCR activity, especially at low temperatures, seems to be the presence of Cu sites in close vicinity, e.g. dimeric Cu species, as they are most probably required during the reoxidation of the Cu+ sites by O2. The overall higher concentration of Cu2+ species during the SCR reaction, uncovered for the catalyst containing a higher Cu loading by the spatially resolved operando XAS and in situ EPR measurements, further supports this assumption.
• The partial loss of mobility at higher temperature due to the desorption of H2O leads to the formation of a more rigid and localized dimeric structure, linked to the zeolite framework, which could explain the decrease of activity for these species above 300 °C in the seagull profile.
• The mobility gained by the monomeric sites at high temperature (>350 °C) results in the formation of additional dimeric species, as indicated by the operando FT EXAFS and XES data. This behavior could explain the high temperature SCR activity of the low loaded Cu-SSZ-13 catalyst but also to the activity increase for the Cu-1.2 sample in the second region of the SCR seagull profile.
Based on these results, the following the transformations of the active Cu species during NH3–SCR is proposed (Scheme 1). According to our current observations and those of other groups,61 even at low temperatures Cu species are readily reduced by ammonia to form mobile linear [Cu(NH3)x]+ complexes. However, this transformation does not make Cu sites SCR-active (cf.Fig. 7). Instead, at low temperatures we noticed rather strong ammonia poisoning by the formation of stable but inactive [Cu(NH3)x]+ complexes. As reported in our recent study,76 especially isolated Cu sites located at a 6MR tend to stronger retain NH3 in comparison to those sites located at the 8MR. In the present study, we could demonstrate for the Cu-1.2 sample the formation of Cu dimers in the absence of NH3 by operando EXAFS, vtc-XES and in situ EPR measurements. This fits well to the study by the group of Sachtler observing Cu dimers formation under oxidizing conditions and splitting when reductant is present in the feed,50 in that case CO or hydrogen was the reductant. Reductive splitting of Cu dimers by hydrocarbons may also account for the more visible seagull NOx conversion profile in the presence of hydrocarbons.7 Dimeric Cu sites have been proposed several times to be SCR active,15 especially during Cu+ to Cu2+ reoxidation, this nicely follows from correlation of low temperature (below 250 °C) activity with Cu loading.24 Necessity of Cu dimer formation at lower temperatures might be attributed to the fact that two neighboring Cu sites are required to be oxidized from Cu+ to Cu2+ by O2.15 Poor activity of Cu-0.5 under these conditions could be due to the rather large diffusion distance to the next neighboring site16 and insufficient mobility to form active dimeric species.
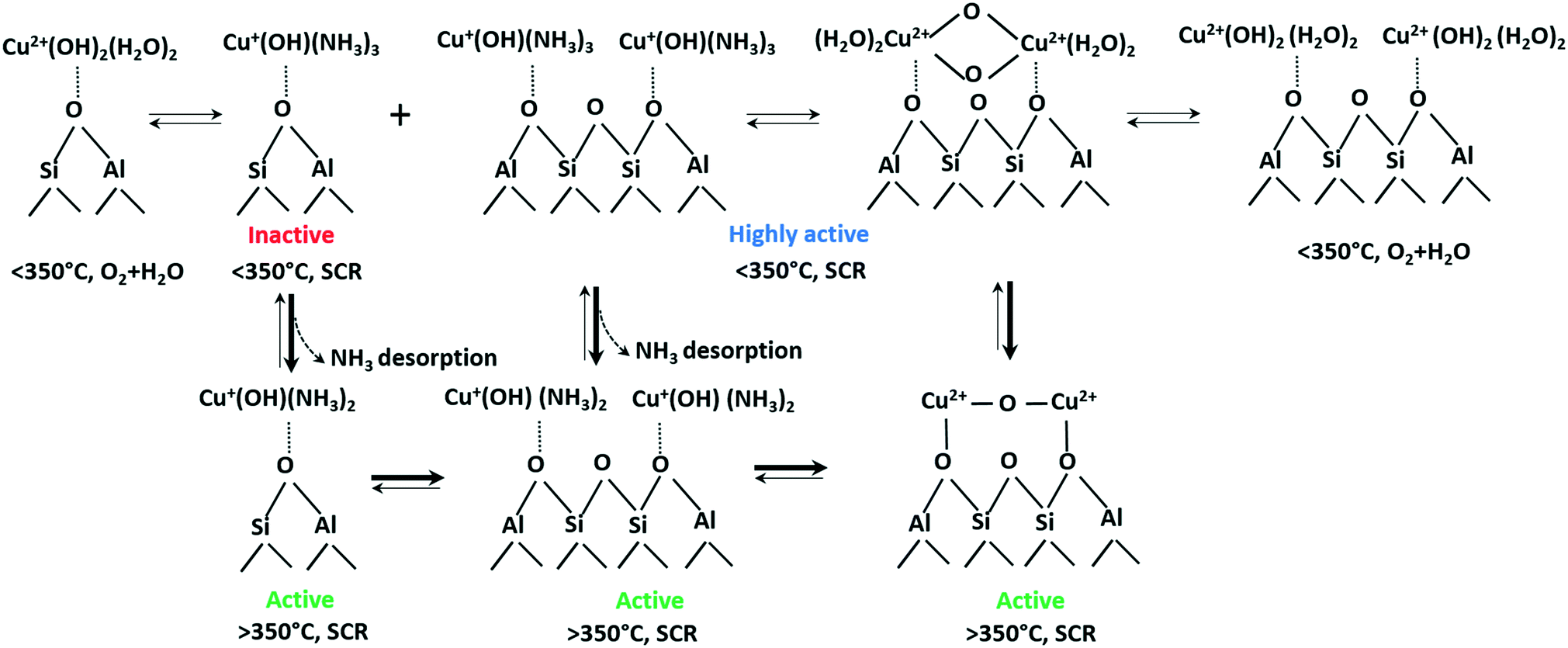 | ||
| Scheme 1 Transformations of Cu species in Cu-SSZ-13 under different SCR-related conditions. An approximate number of adsorbed NH3 or H2O molecules at low and high temperature is suggested based on the calculated coordination numbers in Table 2 and on previously published results.76 | ||
The structure and activity of Cu sites changes at higher temperatures. MD simulations predicted the formation of cationic OH-bridged Cu dimers.17 In fact, we observed Cu dimer formation under oxidizing conditions even in the low-loaded Cu-0.5 catalyst (Fig. 9 and Table 2). We also observed that the SCR activity correlates with the amount of NH3-free Cu+ (Fig. 7), which corresponds to sites that are not poisoned by NH3 and are probably involved in dimeric Cu+–O2–Cu+ reoxidation intermediates. Furthermore, the operando EXAFS, and vtc-XES results demonstrated a higher probability of Cu dimers formation under the oxidizing NO + O2 feed, which is clearly visible even in low-loaded Cu-0.5 catalyst at 375 °C (Fig. 9 and Table 2). Hence, we propose that the mixture of Cu sites in close proximity (able to quickly form dimers) as well as isolated Cu species coexist at high temperatures and are both SCR active, which boosts the NOx conversion above 350 °C. In contrast to Gao et al.,24 who suggest the splitting of Cu dimers at high temperature under oxidizing conditions, we regard this process as an intermediate step of the SCR mechanism taking place especially in the presence of NH3. As a result, while it was previously suggested that the SCR activity decrease at 300–350 °C stems from temperature-induced splitting of active Cu dimers,24 we propose that it is rather caused by the formation of more localized Cu species. This limited mobility due to the interaction with the zeolite framework was also suggested in the recent study of Paolucci et al.16 On the other side, Joshi et al. also proposed that the increased NOx conversion at high temperature can be attributed to the onset of NO oxidation to NO2 that enhances NOx reduction via fast SCR route.40 According to our XES investigations, NO interacts with Cu sites above 200 °C even in the presence of water (Fig. 11), this interaction being hindered by water at lower temperatures.12,77 Hence, NO oxidation over bis(μ-oxo)dicopper species with the formation of nitrates/nitrite species that contribute to the SCR mechanism is highly probable, especially at higher temperature where NH3 partially desorbs from Cu sites.
According to the structural changes of the Cu species identified in this study and presented in Scheme 1, the standard SCR reaction involves Cu dimeric species that are formed either between Cu sites in close proximity or by migration of [Cu(NH3)x]+ isolated species. This is represented in Scheme 2 for temperatures below and above the seagull point together with other illustratively proposed reaction steps. The presence of neighboring Cu sites is mandatory for the low-temperature activity of Cu-SSZ-13 catalysts (<300 °C), since it is required during the slow reoxidation of Cu+ to Cu2+ through the formation of Cu+–O2–Cu+ dimers, as shown here and by other studies.16 Such a path explains the poor activity at low temperatures of the Cu-0.5 catalyst. In this sample, the isolated Cu sites adsorb NH3 and form [Cu(NH3)x]+ complexes with limited mobility, which are not reacting further also due to NH3 inhibition. In contrast, the formation of dimeric species is facilitated at higher loadings. Although interacting in a similar manner with NH3, the resulting Cu+(OH)(NH3)x neighboring species react with NO, probably by formation of an unstable nitrosamine intermediate that is quickly converted to N2 and H2O. In a next step, the resulting Cu+ sites (not represented in Scheme 2) are reoxidized via a dimeric species, closing the SCR cycle.
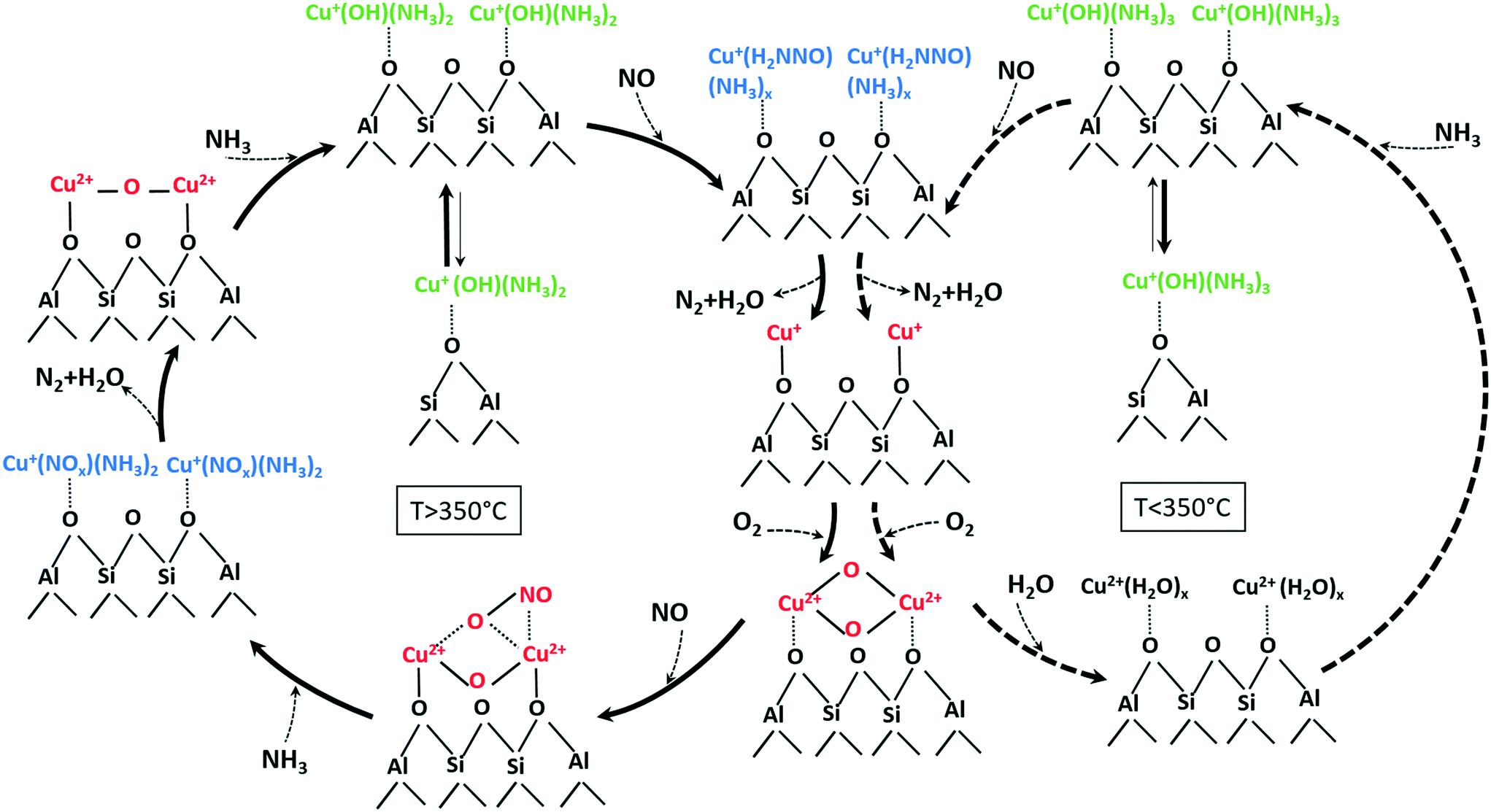 | ||
| Scheme 2 Involved species and mechanistic aspects of NH3–SCR over low and highly loaded Cu-SSZ-13 samples at temperatures below (right side) and above (left side) the seagull profile minimum (unbalanced reactions for better overview on evolved species). An approximate number of adsorbed NH3 or H2O molecules at low and high temperature is suggested based on the calculated coordination numbers in Table 2 and on previously published results.76 | ||
The reaction steps/species identified at high temperatures by the different complementary characterization methods used in this study are reported in a similar illustrative way in Scheme 2 (left side) but with some distinct differences. First of all, the desorption of NH3 and the increased mobility of [Cu(NH3)x]+ brings the isolated Cu sites next to each other and results in the formation of additional Cu dimers during the SCR process. At the same time, the lower NH3 solvation degree leads to more localized and less active structures. Possibly at high temperatures, NH3 is mainly stored on the Brønsted acid sites of the zeolite. Spillover of NH3 from zeolite to the reaction site may additionally slow down SCR, which we observe as seagull-shape NOx conversion decrease at 350 °C. On the contrary, NO adsorbs directly on Cu sites and is oxidized to NO2 by the activated O2 molecule in the bis(μ-oxo)dicopper complex, probably via the unstable reaction intermediate shown in Scheme 2. In a next step, the resulting nitrate/nitrites quickly react with NH3 producing localized Cu2+–O–Cu2+ dimers species. Such a transition from a delocalized bis(μ-oxo)dicopper structure, e.g. solvated by NH3 or H2O, to mono(μ-oxo)dicopper species linked to the zeolite framework is supported also by the study of Alayon et al.,78 which indicates by DFT calculations that rather a mono(μ-oxo)dicopper structure is favored at the Cu dimeric site in the zeolite.
5. Conclusions
By combining operando XAS, operando vtc-XES and in situ EPR we could identify structural changes at Cu sites in Cu-SSZ-13 zeolites under various conditions related to NH3–SCR of NO, which could be correlated to the seagull profile of the NOx conversion. The formation of oxygen bridged (μ-oxo)dicopper complexes was observed for the catalyst containing 1.2 wt% Cu under oxidizing conditions at low and high temperatures, while these complexes were readily split to monomeric Cu sites when NH3 was introduced in the feed. The SCR cycle implies monomeric [Cu(NH3)x]+ and dimeric Cu2+–O2–Cu2+ species. For the low loaded Cu-0.5 sample, which contains mainly monomeric Cu species, further adsorption of NH3 at low temperatures makes the single Cu sites SCR inactive (NH3-inhibition). Moreover, since the reoxidation step seems to require two Cu+ sites in close vicinity, this step is constrained at low Cu loadings. Upon desorption of NH3 at higher temperatures and gained increased mobility the monomeric Cu sites become SCR-active, which corresponds to the second maximum of the seagull profile. However, the desorption of NH3 combined with dehydration leads to a more localized structure of the dimeric species, which could be related to the decrease of the NOx conversion at about 350 °C. Finally, the outcome ratifies X-ray absorption and emission spectroscopy techniques as advantageous for the in situ and operando studies due to the penetration depth of X-rays which allows building in situ cells with desired geometry, allowing fine tuning of process parameters and facilitating easier kinetic measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the DFG (GR 3987/5-1) and the Federal Ministry of Education and Research (BMBF, projects 05K10VKB and 05K13VK2). A. Fahami is grateful to the Erasmus program. T. Günter and D. Zengel thank DBU for provided scholarships. We thank ESRF for providing the beamtime at the ID26 beamline and financial support at ESRF and Dr. L. Amidani, Dr. P. Glatzel, and C. Lapras for the support during measurements. We also thank SLS for providing the beamtime at the SuperXAS beamline and Dr. O. Safonova and Dr. M. Nachtegaal for the support. Finally, Prof. I. Nova and Prof. E. Tronconi (Politecnico di Milano) are greatly acknowledged for their cooperation, support and fruitful discussions in this work.References
- M. Koebel, M. Elsener and M. Kleemann, Catal. Today, 2000, 59, 335–345 CrossRef CAS.
- L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef.
- T. V. Johnson, Int. J. Engine Res., 2009, 10, 275–285 CrossRef CAS.
- M. P. Ruggeri, A. Grossale, I. Nova, E. Tronconi, H. Jirglova and Z. Sobalik, Catal. Today, 2012, 184, 107–114 CrossRef CAS.
- P. G. Blakeman, E. M. Burkholder, H.-Y. Chen, J. E. Collier, J. M. Fedeyko, H. Jobson and R. R. Rajaram, Catal. Today, 2014, 231, 56–63 CrossRef CAS.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. Peden and D. H. Kim, Catal. Today, 2012, 184, 252–261 CrossRef CAS.
- Q. Ye, L. Wang and R. T. Yang, Appl. Catal., A, 2012, 427, 24–34 CrossRef.
- J. H. Kwak, H. Zhu, J. H. Lee, C. H. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- E. Borfecchia, K. Lomachenko, F. Giordanino, H. Falsig, P. Beato, A. Soldatov, S. Bordiga and C. Lamberti, Chem. Sci., 2015, 6, 548–563 RSC.
- C. Paolucci, A. A. Verma, S. A. Bates, V. F. Kispersky, J. T. Miller, R. Gounder, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11828–11833 CrossRef CAS PubMed.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. Peden, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS.
- T. Günter, H. W. Carvalho, D. E. Doronkin, T. Sheppard, P. Glatzel, A. J. Atkins, J. Rudolph, C. R. Jacob, M. Casapu and J.-D. Grunwaldt, Chem. Commun., 2015, 51, 9227–9230 RSC.
- F. Gao, D. Mei, Y. Wang, J. n. Szanyi and C. H. Peden, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- T. V. Janssens, H. Falsig, L. F. Lundegaard, P. N. Vennestrøm, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko and C. Lamberti, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS.
- M. P. Ruggeri, I. Nova, E. Tronconi, J. A. Pihl, T. J. Toops and W. P. Partridge, Appl. Catal., B, 2015, 166, 181–192 CrossRef.
- C. Paolucci, I. Khurana, A. A. Parekh, S. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets and J. T. Miller, Science, 2017, 357, 898–903 CrossRef CAS PubMed.
- G. M. Psofogiannakis, J. F. McCleerey, E. Jaramillo and A. C. Van Duin, J. Phys. Chem. C, 2015, 119, 6678–6686 CrossRef CAS.
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 CrossRef CAS.
- U. Deka, I. Lezcano-Gonzalez, S. J. Warrender, A. L. Picone, P. A. Wright, B. M. Weckhuysen and A. M. Beale, Microporous Mesoporous Mater., 2013, 166, 144–152 CrossRef CAS.
- D. Wang, L. Zhang, J. Li, K. Kamasamudram and W. S. Epling, Catal. Today, 2014, 231, 64–74 CrossRef CAS.
- J. Connerton and R. W. Joyner, in Stud. Surf. Sci. Catal., ed. N. Kruse, A. Frennet and J. M. Bastin, Elsevier, 1998, vol. 116, pp. 327–334 Search PubMed.
- F. Gao and C. Peden, Catalysts, 2018, 8, 140 CrossRef.
- C. Paolucci, J. Di Iorio, F. Ribeiro, R. Gounder and W. Schneider, in Adv. Catal., Elsevier, 2016, vol. 59, pp. 1–107 Search PubMed.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. Peden, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- L. Olsson, K. Wijayanti, K. Leistner, A. Kumar, S. Y. Joshi, K. Kamasamudram, N. W. Currier and A. Yezerets, Appl. Catal., B, 2015, 174, 212–224 CrossRef.
- J. H. Kwak, T. Varga, C. H. Peden, F. Gao, J. C. Hanson and J. Szanyi, J. Catal., 2014, 314, 83–93 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed.
- A. A. Verma, S. A. Bates, T. Anggara, C. Paolucci, A. A. Parekh, K. Kamasamudram, A. Yezerets, J. T. Miller, W. N. Delgass, W. F. Schneider and F. H. Ribeiro, J. Catal., 2014, 312, 179–190 CrossRef CAS.
- S. I. Zones, US Pat., 06519954, 1983 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- R. Frahm, M. Nachtegaal, J. Stötzel, M. Harfouche, J. A. van Bokhoven and J.-D. Grunwaldt, AIP Conf. Proc., 2010, 1234, 251–255 CrossRef CAS.
- J.-D. Grunwaldt, M. Caravati, S. Hannemann and A. Baiker, Phys. Chem. Chem. Phys., 2004, 6, 3037–3047 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. de Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964–4976 RSC.
- J. Jaumot, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- P. Glatzel, M. Sikora, G. Smolentsev and M. Fernández-García, Catal. Today, 2009, 145, 294–299 CrossRef CAS.
- E. Gallo and P. Glatzel, Adv. Mater., 2014, 26, 7730–7746 CrossRef CAS PubMed.
- T. Günter, PhD Thesis, Karlsruhe Institute of Technology, 2016 Search PubMed.
- S. Y. Joshi, A. Kumar, J. Luo, K. Kamasamudram, N. W. Currier and A. Yezerets, Appl. Catal., B, 2015, 165, 27–35 CrossRef CAS.
- I. Nova, C. Ciardelli, E. Tronconi, D. Chatterjee and B. Bandl-Konrad, AIChE J., 2006, 52, 3222–3233 CrossRef CAS.
- I. Nova, M. Colombo, E. Tronconi, V. Schmeisser and M. Weibel, SAE Int. J. Engines, 2011, 4, 1822–1838 CrossRef.
- A. Marberger, A. W. Petrov, P. Steiger, M. Elsener, O. Kröcher, M. Nachtegaal and D. Ferri, Nat. Catal., 2018, 1, 221–227 CrossRef.
- M. Niwa and N. Katada, Catal. Surv. Asia, 1997, 1, 215–226 CrossRef CAS.
- H.-Y. Chen, Z. Wei, M. Kollar, F. Gao, Y. Wang, J. Szanyi and C. H. F. Peden, J. Catal., 2015, 329, 490–498 CrossRef CAS.
- D. Zhang and R. T. Yang, Energy Fuels, 2018, 32, 2170–2182 CrossRef CAS.
- J. Dědeček and B. Wichterlová, J. Phys. Chem. B, 1997, 101, 10233–10240 CrossRef.
- F. Giordanino, P. N. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741–12761 RSC.
- M. H. Groothaert, K. Lievens, J. A. van Bokhoven, A. A. Battiston, B. M. Weckhuysen, K. Pierloot and R. A. Schoonheydt, ChemPhysChem, 2003, 4, 626–630 CrossRef CAS PubMed.
- G. Lei, B. Adelman, J. Sarkany and W. Sachtler, Appl. Catal., B, 1995, 5, 245–256 CrossRef CAS.
- J. Dedecek, Z. Sobalik, Z. Tvaruazkova, D. Kaucky and B. Wichterlova, J. Phys. Chem., 1995, 99, 16327–16337 CrossRef CAS.
- F. Gao, E. D. Walter, E. M. Karp, J. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. Peden, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- A. Godiksen, F. N. Stappen, P. N. Vennestrøm, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- A. Kucherov, G. Gerlock, H.-W. Jen and M. Shelef, Catal. Today, 1996, 27, 79–84 CrossRef CAS.
- A. Kucherov, H. G. Karge and R. Schlögl, Microporous Mesoporous Mater., 1998, 25, 7–14 CrossRef CAS.
- A. Kucherov, A. Shigapov, A. Ivanov and M. Shelef, J. Catal., 1999, 186, 334–344 CrossRef CAS.
- A. Godiksen, P. Vennestrom, S. Rasmussen and S. Mossin, Top. Catal., 2017, 60, 13–29 CrossRef CAS.
- A. Godiksen, F. N. Stappen, P. N. R. Vennestrom, F. Giordanino, S. B. Rasmussen, L. F. Lundegaard and S. Mossin, J. Phys. Chem. C, 2014, 118, 23126–23138 CrossRef CAS.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- A. Boubnov, H. W. Carvalho, D. E. Doronkin, T. Günter, E. Gallo, A. J. Atkins, C. R. Jacob and J.-D. Grunwaldt, J. Am. Chem. Soc., 2014, 136, 13006–13015 CrossRef CAS PubMed.
- F. Giordanino, E. Borfecchia, K. A. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. V. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 CrossRef CAS PubMed.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Appl. Catal., A, 2010, 373, 168–175 CrossRef CAS.
- C. Henriques, M. Ribeiro, C. Abreu, D. Murphy, F. Poignant, J. Saussey and J. Lavalley, Appl. Catal., B, 1998, 16, 79–95 CrossRef CAS.
- Q. Guo, F. Fan, D. M. Ligthart, G. Li, Z. Feng, E. J. Hensen and C. Li, ChemCatChem, 2014, 6, 634–639 CrossRef CAS.
- S. Shwan, M. Skoglundh, L. F. Lundegaard, R. R. Tiruvalam, T. V. Janssens, A. Carlsson and P. N. Vennestrøm, ACS Catal., 2014, 5, 16–19 CrossRef.
- D. E. Doronkin, M. Casapu, T. Günter, O. Müller, R. Frahm and J.-D. Grunwaldt, J. Phys. Chem. C, 2014, 118, 10204–10212 CrossRef CAS.
- R. Sarangi, Coord. Chem. Rev., 2013, 257, 459–472 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- W. Grünert, N. W. Hayes, R. W. Joyner, E. S. Shpiro, M. Siddiqui and G. Baeva, J. Phys. Chem., 1994, 98, 10832–10846 CrossRef.
- G. Turnes Palomino, P. Fisicaro, S. Bordiga, A. Zecchina, E. Giamello and C. Lamberti, J. Phys. Chem. B, 2000, 104, 4064–4073 CrossRef PubMed.
- Y. Kuroda, R. Kumashiro, T. Yoshimoto and M. Nagao, Phys. Chem. Chem. Phys., 1999, 1, 649–656 RSC.
- C. W. Bauschlicher Jr, S. R. Langhoff and H. Partridge, J. Chem. Phys., 1991, 94, 2068–2072 CrossRef.
- K. Inoue, K. Ohashi, T. Iino, K. Judai, N. Nishi and H. Sekiya, Phys. Chem. Chem. Phys., 2007, 9, 4793–4802 RSC.
- A. Clemens, A. Shishkin, P.-A. Carlsson, M. Skoglundh, F. Martínez-Casado, Z. Matĕj, O. Balmes and H. Harelind, ACS Catal., 2015, 5, 6209–6218 CrossRef CAS.
- T. Günter, D. E. Doronkin, A. Boubnov, H. W. P. Carvalho, M. Casapu and J.-D. Grunwaldt, Top. Catal., 2016, 59, 866–874 CrossRef.
- B. Kerkeni, D. Berthout, D. Berthomieu, D. E. Doronkin, M. Casapu, J.-D. Grunwaldt and C. Chizallet, J. Phys. Chem. C, 2018, 122, 16741–16755 CrossRef CAS.
- A. R. Fahami, I. Nova and E. Tronconi, Catal. Today, 2017, 297, 10–16 CrossRef CAS.
- E. M. C. Alayon, M. Nachtegaal, A. Bodi, M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 7681–7693 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00290h |
| This journal is © The Royal Society of Chemistry 2019 |