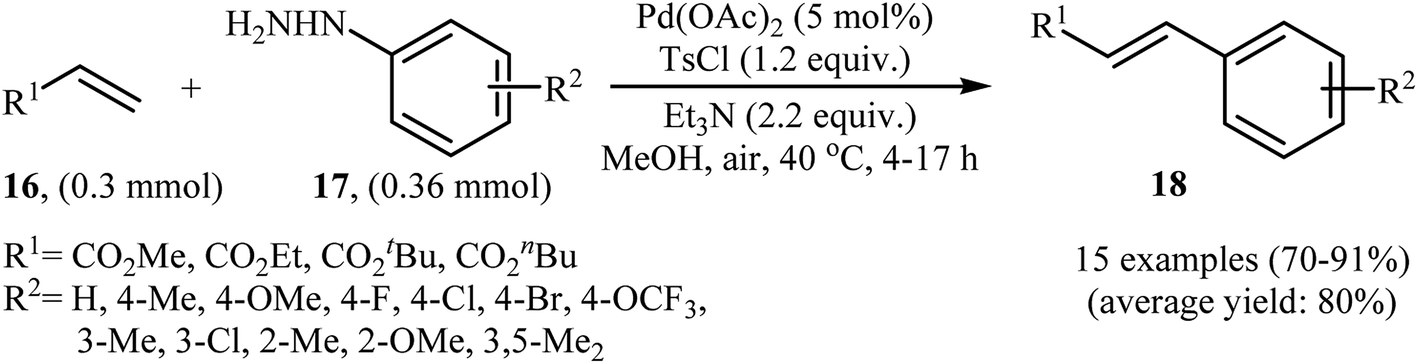DOI:
10.1039/C8RA06423G
(Review Article)
RSC Adv., 2018,
8, 33828-33844
Arylhydrazines: novel and versatile electrophilic partners in cross-coupling reactions
Received
30th July 2018
, Accepted 6th September 2018
First published on 1st October 2018
Abstract
Arylhydrazines are extremely valuable compounds in organic chemistry that are widely used for the synthesis of a variety of biologically active molecules such as indoles, indazoles, pyrazoles, aryltriazoles, β-lactams and quinazolines. These compounds have also been widely utilized as arylation agents in oxidative cross-coupling reactions. In this review, we will highlight the most important explorations and developments in the carbon–carbon and carbon–heteroatom (nitrogen, phosphorus, sulfur, and selenium) cross-coupling of arylhydrazines. The literature has been surveyed from 2001 to June 2018.
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed her PhD degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Associate Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Robab Mohammadi | Robab Mohammadi was born in Tabriz, Iran, in 1979. She received her B.S. degree in Pure Chemistry from University of Tabriz, Iran, and her M.S. degree in applied chemistry from Islamic Azad University, Tabriz Branch, Tabriz, Iran, in 2008 under the supervision of Dr L. Edjlali. She received her PhD degree in 2012 under the supervision of Prof. M. Rabani and Prof. B. Massoumi from Islamic Azad University, Tehran Shomal Branch, Tehran, Iran. Now she is working at Payame Noor University of Tabriz as Associate Professor. Her research interests include nano materials synthesis and new methodologies in material synthesis. |
 Sheida Ahmadi | Sheida Ahmadi was born in Miyaneh, Iran. He has received her B.S. degree in Applied Chemistry from Islamic azad University, Tehran, Iran, in 1997, and her M.S. degree in inorganic chemistry from Shahid Bahonar University, kerman, Iran, in 2007 under the supervision of Prof. S. J. Fatemi. She received her PhD degree in 2017 under the supervision of Prof. M. Hakimi in Payam Noor university, Tehran, Iran. Now she is working at Payame Noor University of Tehran as assistant professor. Her research interests include inorganic polymer, synthesis of the inorganic complexes and bioinorganic chemistry. |
 Aazam Monfared | Aazam Monfared was born in Tehran, Iran, in 1965. She received her B.S. degree in Pure Chemistry from University of Shahid Beheshti, Tehran, Iran, and her M.S. degree in Organic chemistry from Shahid Beheshti University, Tehran, Iran, in 1991 under the supervision of Prof. A. Rustaiyan. She received her PhD degree in 1999 under the supervision of Prof. A. Rustaiyan in Shahid Beheshti University, Tehran, Iran. Now, she is working at Payame Noor University of Tehran as Associate Professor. Her research interests include organic synthesis, phytochemistry, drug synthesis, nano chemistry, methodologies and theoretical chemistry. |
 Zahra Rahmani | Zahra Rahmani was born in Takab, Iran, in 1985. She received her B.S. degree in Applied chemistry from University of Urmia, Iran, and her M.S. degree in organic chemistry from Payam Noor university, Zanjan, Iran, in 2012 under the supervision of prof. E. Vessally. She currently is a PhD student in organic chemistry under the supervision of prof. E. Vessalli in Islamic Azad University, Tabriz, Iran. Her doctoral thesis examines the interaction of boron nitride and aluminum nitride nano structures with some sulfonamide drugs. Now, she is working at Zarshuran Gold Company of Takab as Laboratory supervisor. |
1 Introduction
Cross-coupling reactions have emerged as one of the most important and versatile synthetic tools in modern organic synthesis which facilitate the formation of either carbon–carbon1 or carbon–heteroatom bonds2 via the connection of electrophilic and nucleophilic fragments. Although various carbon and heteroatom-centered nucleophilic partners have been explored in this arena during the past few decades, electrophilic counterparts have been substantially limited to organic halides and triflates.3 Unfortunately, the cross-coupling reactions with the use of organic halides as electrophilic partners are associated with some drawbacks such as: (i) the high cost of organic bromides and iodides, (ii) very low reactivity of organic chlorides, and (iii) environmental toxicity of some organic halides. These disadvantages can make the organic halides undesirable for industrial applications.4 Triflates are excellent coupling partners in most cross-coupling reactions. However, they are unstable, expensive, and liberate potentially genotoxic triflic acid as a by-product.5 The use of diazonium salts as alternative or complementary electrophiles for cross-coupling reactions have been the subject of a number of studies in recent years.6 However, the high reactivity of these salts towards a broad range of nucleophiles7 and instability of some of them at room temperature8 limited their practical synthetic applications. Therefore, there is still further need for broadening the scope of electrophilic coupling partners to achieve unique organic transformations that are not obtainable through known approaches.
Arylhydrazine as the alternative or complementary electrophilic partners for the cross-coupling reactions have attracted considerable attention in recent years.7 These compounds have been successfully utilized as coupling partners in various carbon–carbon and carbon–heteroatom cross-coupling reactions. Interestingly, nitrogen gas and water are the only by-products of C–N bond cleavage of arylhydrazines thus these compounds can be considered as environmentally friendly arylating agents for oxidative cross-coupling reactions. It should be mentioned that a variety of methods have been developed for the synthesis of these compounds. Some of the most popular methods for their preparation include: (i) the oxidation of anilines to the corresponding diazonium salts, followed by reduction with tin(II) compounds;9 (ii) aromatic nucleophilic substitution reaction of aryl halides with hydrazine;10 and (iii) transition metal catalyzed cross-coupling reaction between aryl halides and hydrazine.11
Since a large number of developments in the denitrogenative coupling of aryl hydrazines with various carbon and heteroatom-centered nucleophilic partners have occurred from 2001 to present, a comprehensive review on this explosively growing field seems to be timely. In continuation of our recent published reviews,12 herein, we will highlight the most important developments in the arena of C–C and C–X cross-coupling of arylhydrazines by hoping that it will stimulate researchers to further research in this interesting field.
2 Carbon–carbon cross-couplings
2.1 Suzuki coupling
Palladium-catalyzed Suzuki cross-coupling reaction between arylboronic acids and aryl (pseudo)halides is one of the most powerful methods for the formation of bi(het)aryls.13 High tolerance to functional groups, low toxicity of the arylboronic acids and commercial availability of many organoboron compounds were the advantages, mentioned for this reaction. Arylhydrazines as efficient electrophilic partners for Suzuki coupling reactions have attracted considerable interest in recent years.
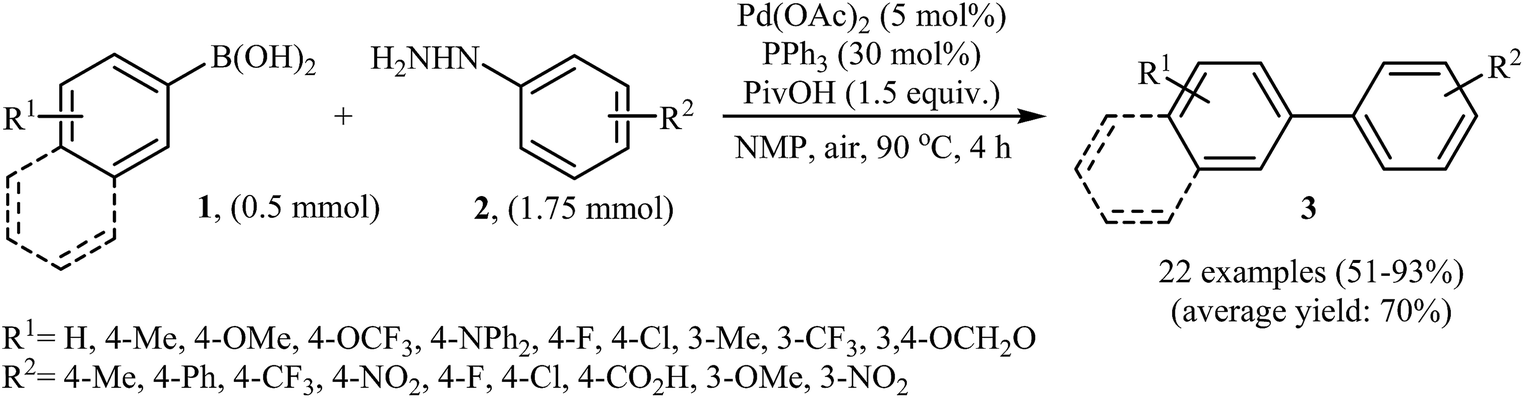
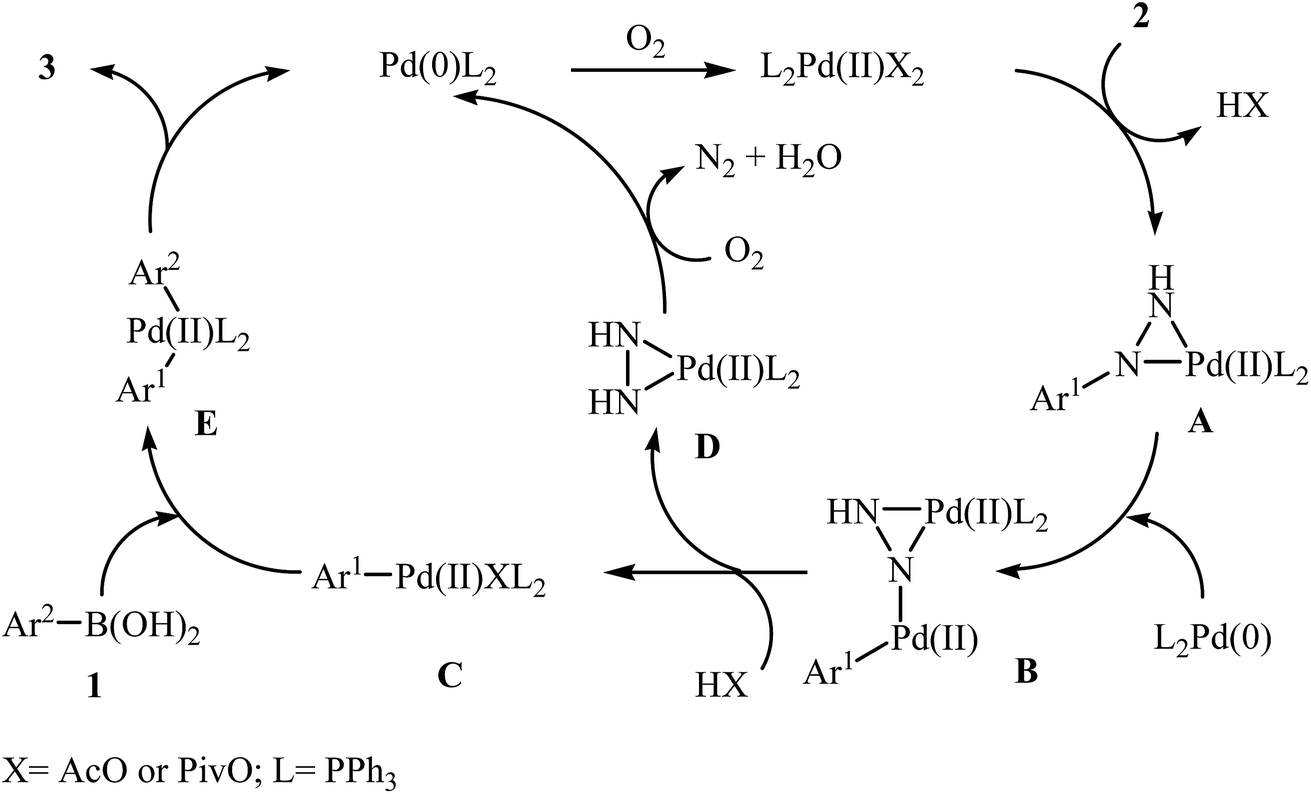
In 2014, Xu and Gao along with their co-workers discovered the first Pd-catalyzed cross-coupling reaction of arylboronic acids 1 with arylhydrazines 2 via C–N bond cleavage to provide the biaryls 3 (Scheme 1).14 Various transition-metal catalysts, ligands, acids, and solvents were carefully tested in order to optimize the reaction conditions and the combination of Pd(OAc)2/PPh3/PivOH as a catalytic system in NMP proved most effective. The optimized protocol resulted in functionalized biaryls 3 in moderate to excellent yields and tolerated a broad range of common functional groups such as chloro, fluoro, nitro, amino, acid, and ether functionalities. This made possible the further elaboration of the products. It should be mentioned that boronic esters and potassium aryltrifluoroborates failed to participate in this coupling reaction. In addition the reaction did not give good yields with heteroarylhydrazines. The authors have proposed a mechanistic cycle (Scheme 2) which starts with the formation of palladiaziridine complex A by the reaction between arylhydrazine 2 and Pd(II)2L2, which undergoes oxidative addition with Pd(0)L2 to furnish the palladium(II)-centered complex B via C–N bond cleavage. Next, protonolysis of complex B leads to the aryl palladium complex C and the palladiaziridine complex D, which was decomposed into Pd(0)L2, N2, and H2O by air. Subsequently, the reaction of arylpalladium complex C with arylboronic acid 1 affords palladium(II) biaryl intermediate E which undergoes C–C reductive elimination to yield the expected product 3 and Pd(0)L2. Finally, reoxidation of Pd(0) by air with the assistance of a ligand and an acid regenerates the Pd(II) catalyst.
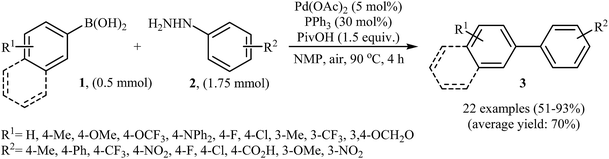 |
| | Scheme 1 Direct denitrogenative cross-coupling of arylboronic acids 1 with arylhydrazines 2. | |
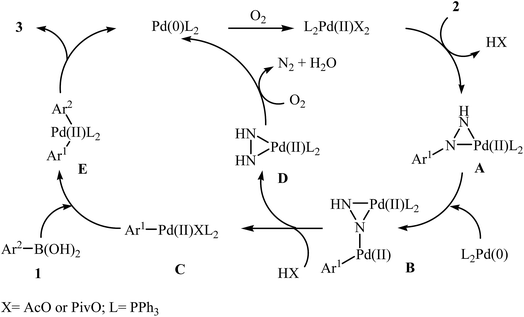 |
| | Scheme 2 Mechanism that accounts for the formation of biaryls 3. | |
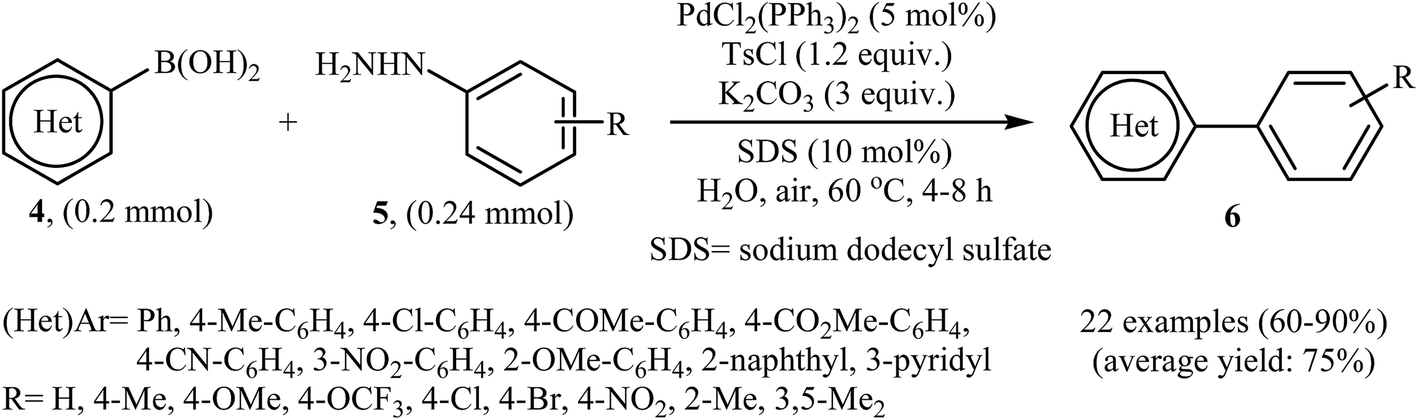
In the same year, Liu, Zhou, and Peng independently reported a similar Suzuki coupling of arylhydrazines using PdCl2(PPh3)2/K2CO3 as the catalyst system and p-toluenesulfonyl chloride (TsCl) as the activation reagent in the most significant green solvent, water (Scheme 3).15 5 mol% of Pd catalyst was sufficient to catalyze the Suzuki cross-coupling of several (het)aryl boronic acids 4 with various ortho, meta, and para substituted arylhydrazines 5 to give corresponding biaryls 6 in good to high yields. It is noted that other palladium catalysts such as Pd(OAc)2, PdCl2, Pd(PPh3)4, and PdCl2(PhCN)2 were also found to promote this coupling reaction; however, in lower yields. No reaction occurred in the absence of the catalyst.
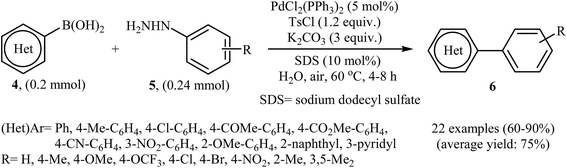 |
| | Scheme 3 Pd-catalyzed Suzuki cross-coupling of (het)aryl boronic acids 4 with arylhydrazines 5 in water. | |
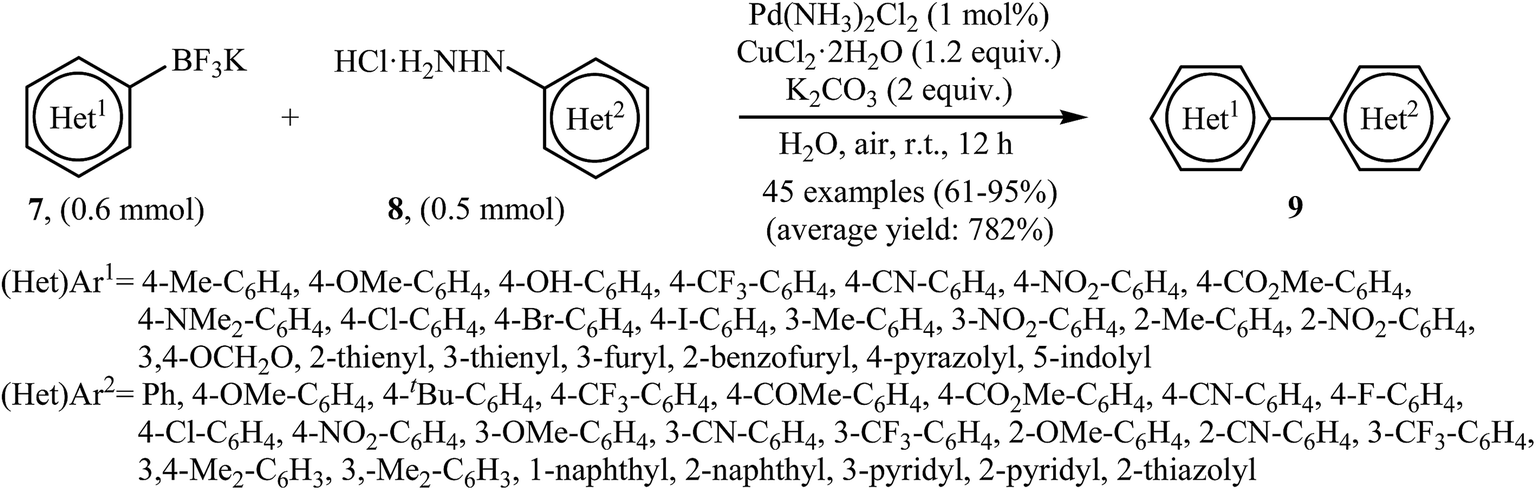
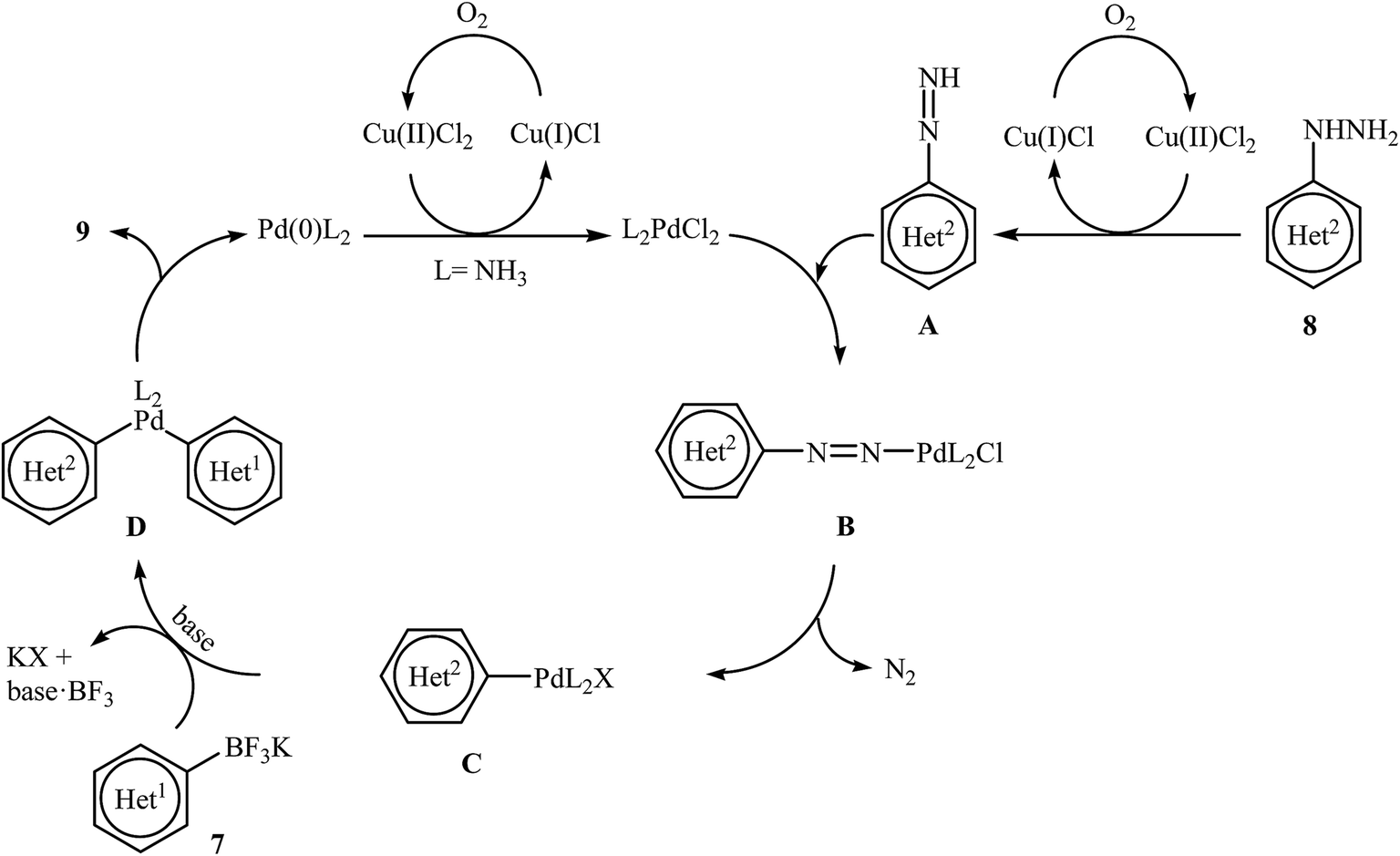
Very recently, Wang and co-workers reported an extension of Pd-catalyzed Suzuki coupling of arylhydrazines to the potassium aryltrifluoroborate coupling partners.16 Thus, in the presence of only 1 mol% of water-soluble Pd(NH3)2Cl2 as a catalyst, 2 mol% of CuCl2·2H2O as a co-catalyst and 1.0 equiv. of K2CO3 as a base in water at room temperature, C–C coupling of (het)aryltrifluoroborates 7 with (het)arylhydrazide hydrochlorides 8 furnished functionalized biaryls 9 in good to excellent yields (Scheme 4). The author proposed mechanism for this denitrogenative cross-coupling reaction is given in Scheme 5.
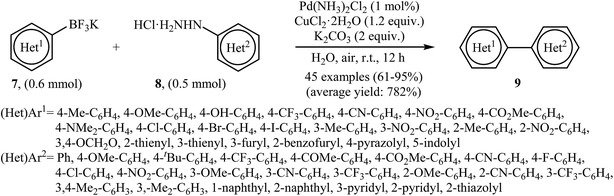 |
| | Scheme 4 Wang's synthesis of biaryls 9. | |
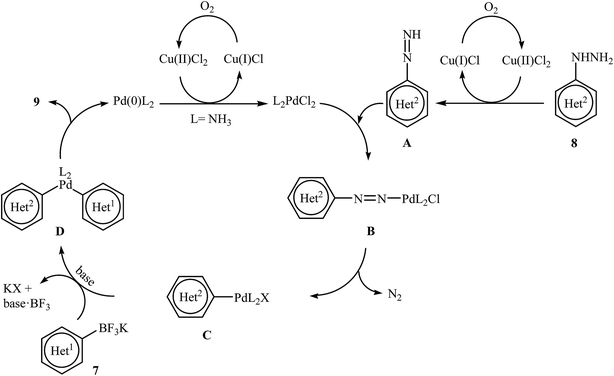 |
| | Scheme 5 Plausible mechanism for the formation of biaryls 9. | |
2.2 Hiyama coupling
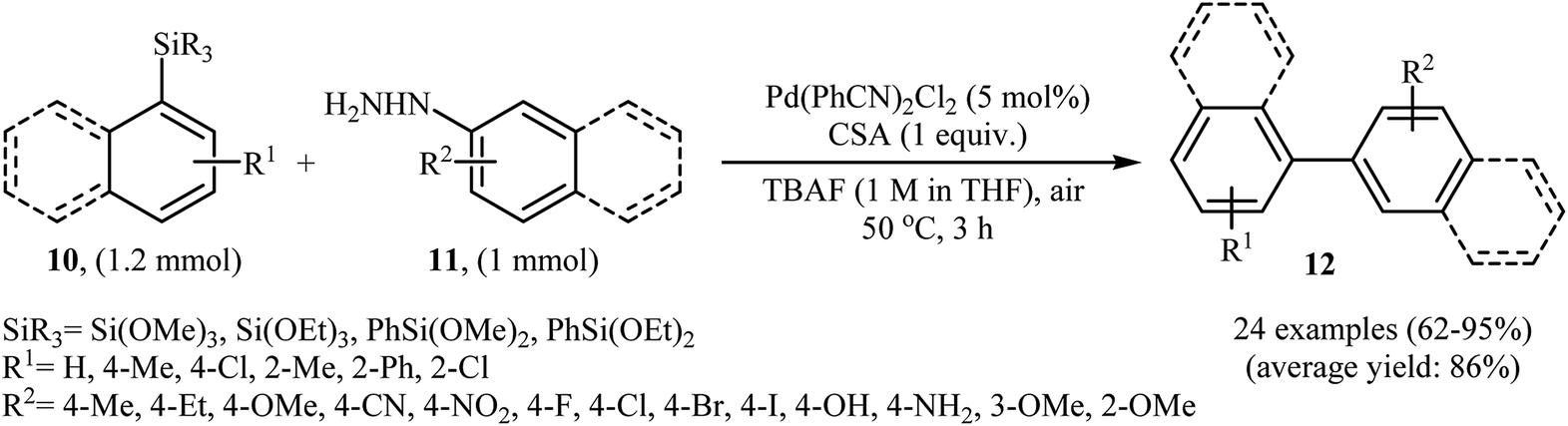
Denitrogenative Hiyama cross-coupling of aryl silanes with arylhydrazines has been scarcely studied; in fact, only one example of such a reaction was reported in the literature. In this report, Zhang and Wang along with their co-workers synthesized a range of biaryls 12 through the reaction between aryl silanes 10 and arylhydrazines 11 employing Pd(PhCN)2Cl2 as a catalyst, tetra-n-butylammonium fluoride (TBAF) as a fluoride source and camphorsulfonic acid (CSA) as proton source (Scheme 6).17 The reactions were carried out in THF at 50 °C, tolerated a variety of sensitive functional groups (e.g., F, Br, Cl, I, OMe, OH, NH2, NO2, CN) and generally provided the desired biaryls 12 in good to excellent yields. Heteroaromatic substrates, such as 2-thienylhydrazine, 2-pyridylhydrazine, 5-hydrazinyl-1H-tetrazole, and 3-(triethoxysilyl)pyridine were also well tolerated by the reaction condition employed. It is noted that the author proposed mechanism for this transformation is most similar to the one described in Scheme 2.
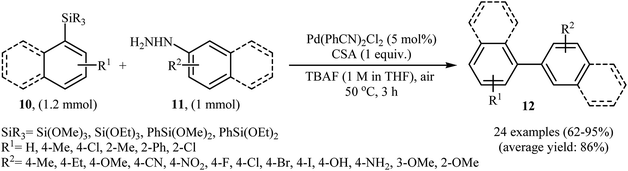 |
| | Scheme 6 Denitrogenative Hiyama cross-coupling of aryl silanes 10 with arylhydrazines 11 developed by Zhang. | |
2.3 Arylation of alkenes
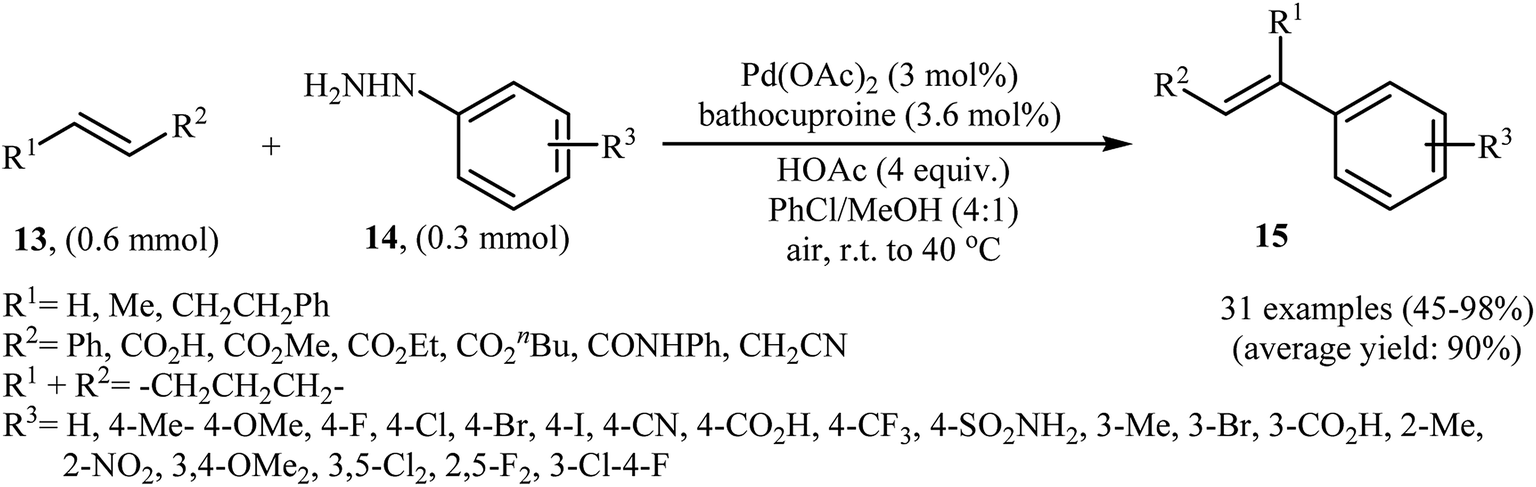
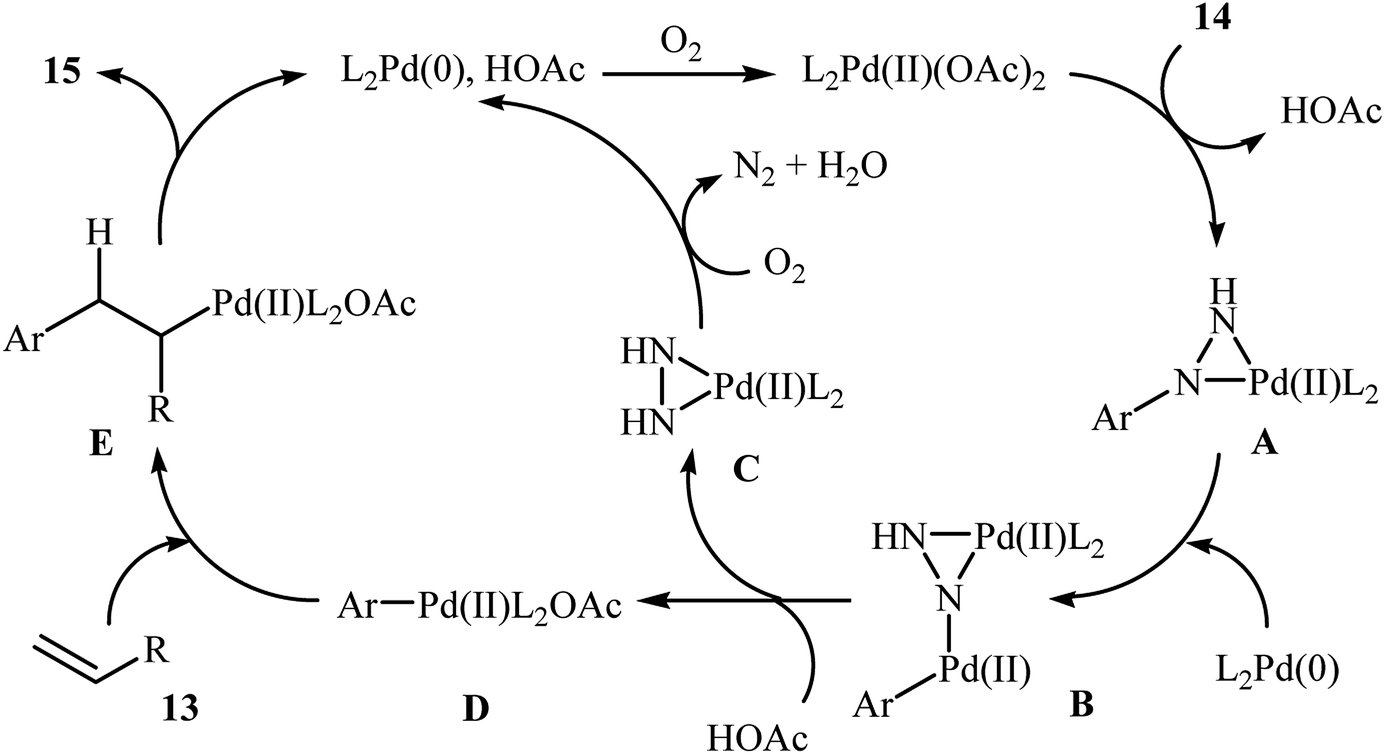
One of the most important C–C coupling reactions is the palladium catalyzed reaction between activated alkenes and unsaturated halides, known as the Heck coupling reaction.18 In 2011, the group of Loh reported the first example of palladium-catalyzed Heck-type cross-coupling of olefins with arylhydrazines, in which the C–N bond was selectively cleaved via oxidative addition.19 The combination of Pd(OAc)2, bathocuproine and HOAc showed a good reactivity towards this reaction. Both electron-donating and electron-withdrawing alkynes 13 and a variety of ortho-, para-, and meta-substituted arylhydrazines 14 were well tolerated and transformed to the corresponding arylalkenes 15 in moderate to almost quantitative yields (Scheme 7). It is worth noting that excellent chemoselectivities were observed when halophenylhydrazines were employed in this coupling reaction, which indicated that the C–N bond was much more facile to be cleaved under these conditions. The proposed mechanism for this Heck-type cross-coupling reaction is a little bit complex involving an initial interaction between the arylhydrazine 14 with palladium catalyst to give palladiaziridine complex A, which inserts another atom of Pd generating the two palladium(II) centered complex B. Protonolysis of this intermediate forms the palladiaziridine complex C and the aryl palladium complex D. Next, intermediate D reacts with olefin 13 in a Heck reaction manner to generate desired arylalkene 15 and the Pd(0) species. Simultaneously, in the presence of oxygen, palladiaziridine complex C collapses to give palladium(0), nitrogen gas, and water (Scheme 8). Shortly afterwards, X. Liu and his team applied this methodology in the synthesis of biologically important aryl-C-glycosides by direct coupling of inactivated glycals with aryl hydrazines.20
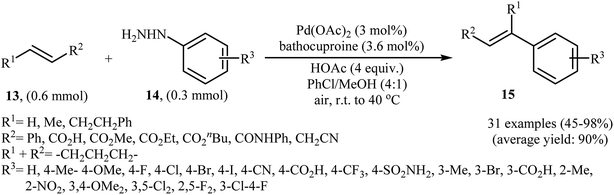 |
| | Scheme 7 Pd(II)-catalyzed C–C bond formation of alkynes 13 with arylhydrazines 14 reported by Loh. | |
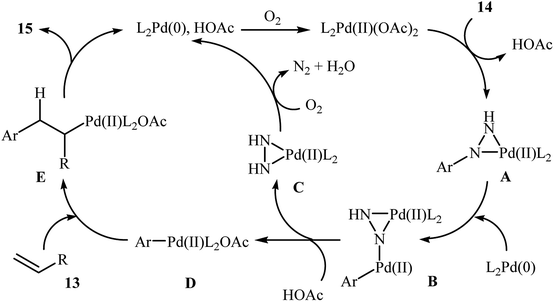 |
| | Scheme 8 Mechanistic proposal for the reaction in Scheme 7. | |
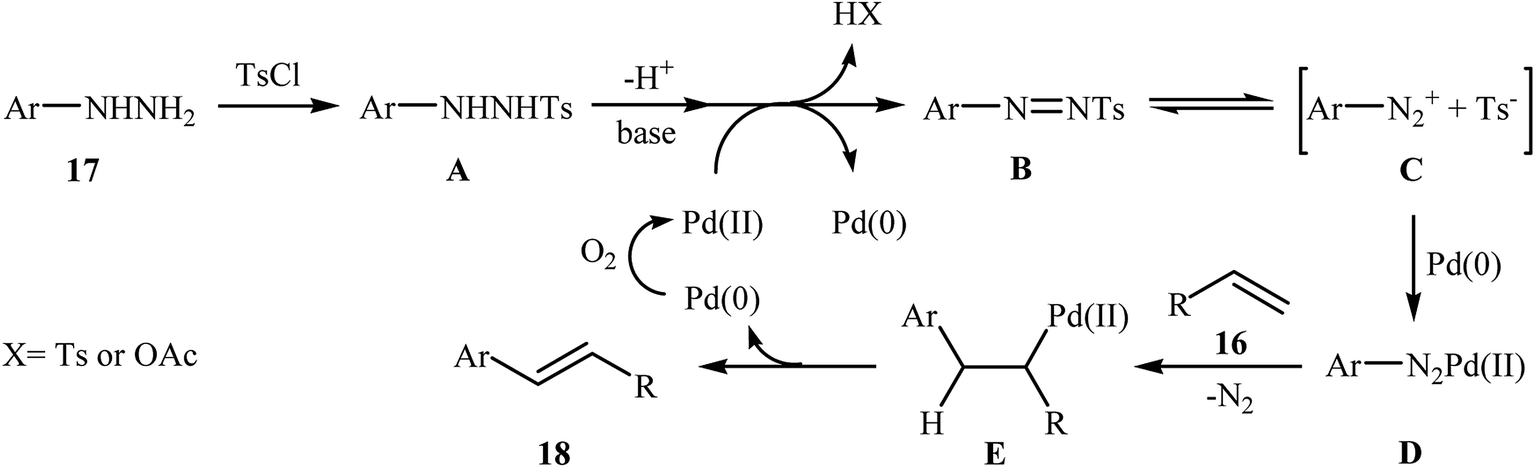
In a related investigation, recently, J. Liu and co-workers also showed that arylalkenes 18 were successfully formed from corresponding terminal alkenes 16 and arylhydrazines 17 in a simple process employing Pd(OAc)2 as a catalyst, Et3N as a base, and TsCl as an activation reagent (Scheme 9).21 It is worth noting that the presence of TsCl is crucial to the success of the reaction, no product was observed in the absence of TsCl. According to the authors proposed mechanism, this transformation proceeds through the formation of a N′-tosyl arylhydrazine intermediate A from the reaction of arylhydrazine 17 with TsCl, followed by dehydrogenation of this intermediate A in the presence of base and Pd(II)-catalyst to give the diazene intermediate B that is in equilibrium with the corresponding diazonium ion C. Next, oxidative addition of intermediate C to the in situ generated Pd(0) yields the organopalladium intermediate D, which reacts with olefin 16 to form intermediate E. Finally, degeneration of E affords the observed coupling product 18. This process allows the regeneration of Pd(0), which is further oxidized by air to provide the Pd(II)-catalyst (Scheme 10).
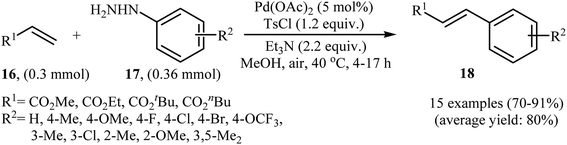 |
| | Scheme 9 Synthesis of arylalkenes 18 through Pd(II)-catalyzed coupling of terminal alkynes 16 with arylhydrazines 17. | |
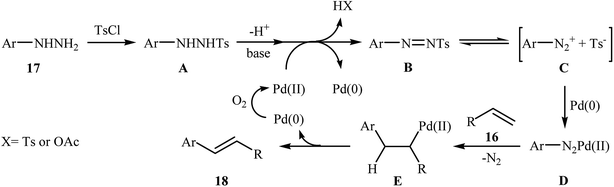 |
| | Scheme 10 Mechanistic proposal for the formation of arylalkenes 18. | |
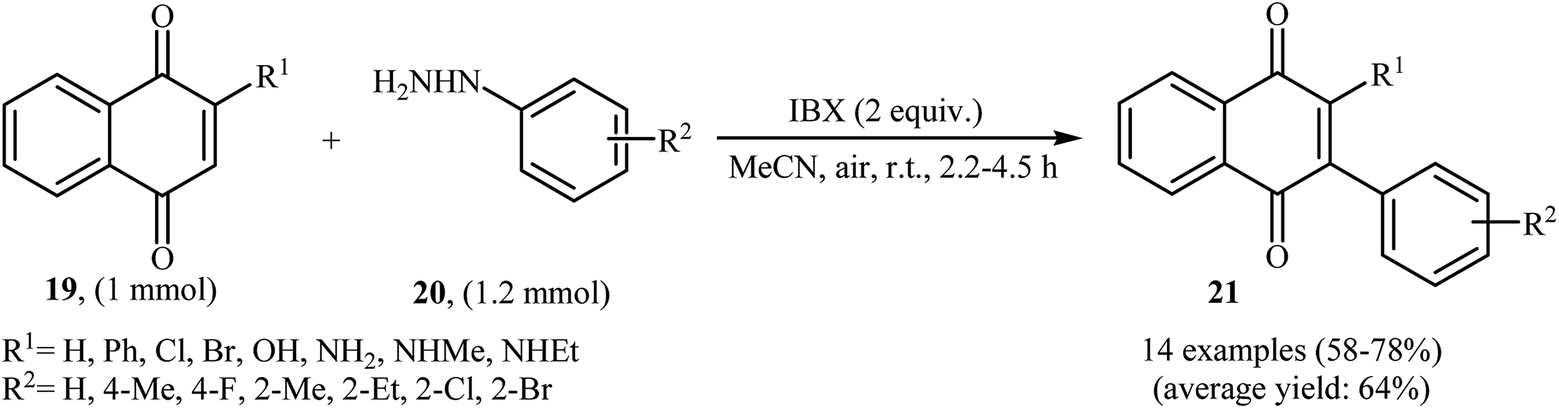
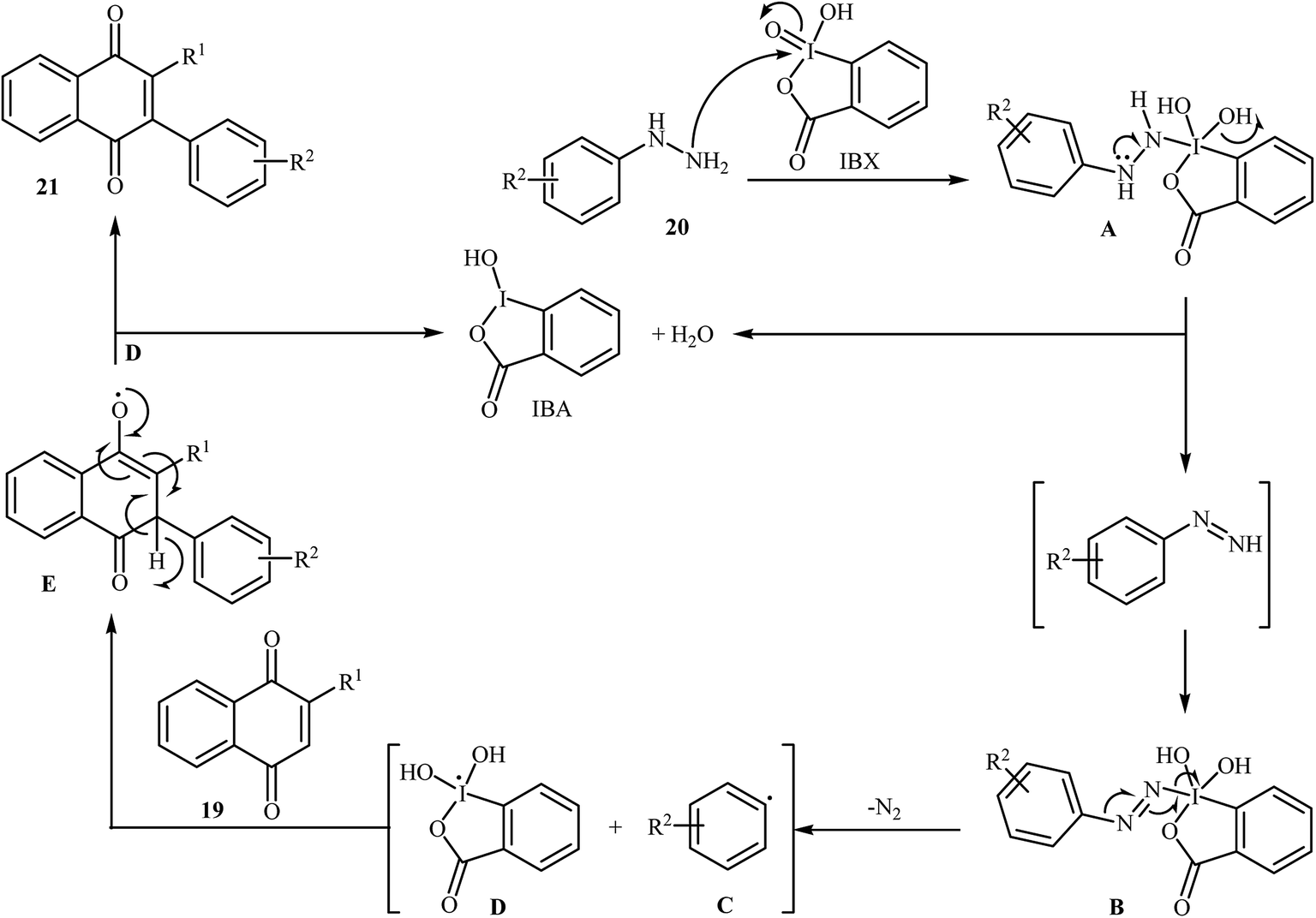
An interesting study about radical mediated arylation of 2-substituted naphthoquinones 19 in the preparation of 3-arylated naphthoquinones 21, using arylhydrazines 20 as the aryl source, and o-iodoxybenzoic acid (IBX) as oxidant, was published by Akamanchi and co-workers (Scheme 11).22 Solvent has a dramatic role in the reaction and among the common solvents [e.g., MeCN, DMSO, THF, CHCl3, and toluene]; acetonitrile was the most effective solvent since it provided a better solubility of IBX. The reaction in DMSO was found to be vigorous and resulted in multiple side products with exothermicity. Under optimized conditions, the reaction tolerated various common functional groups such as fluoro, chloro, bromo, amino, and hydroxy functionalities and gave the expected products in good yields. It is noted that unsubstituted 1,4-naphthoquinone provided a mixture of the mono and di-arylated products. The author proposed mechanism for the formation of arylated naphthoquinones 21 is shown in Scheme 12.
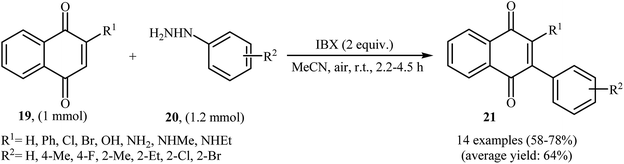 |
| | Scheme 11 Akamanchi's synthesis of 3-arylated naphthoquinones 21. | |
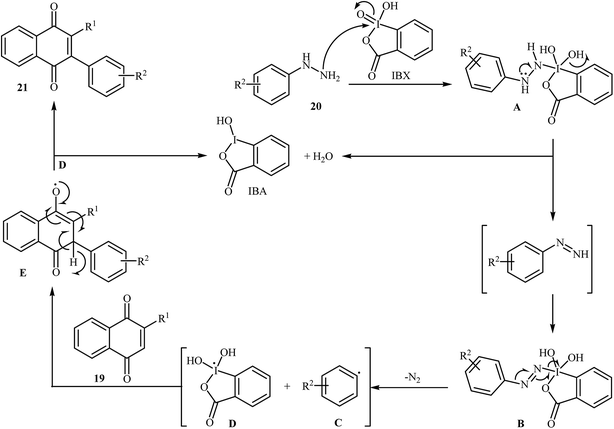 |
| | Scheme 12 Mechanistic explanation of the synthesis of 3-arylated naphthoquinones 21. | |
2.4 Sonogashira coupling
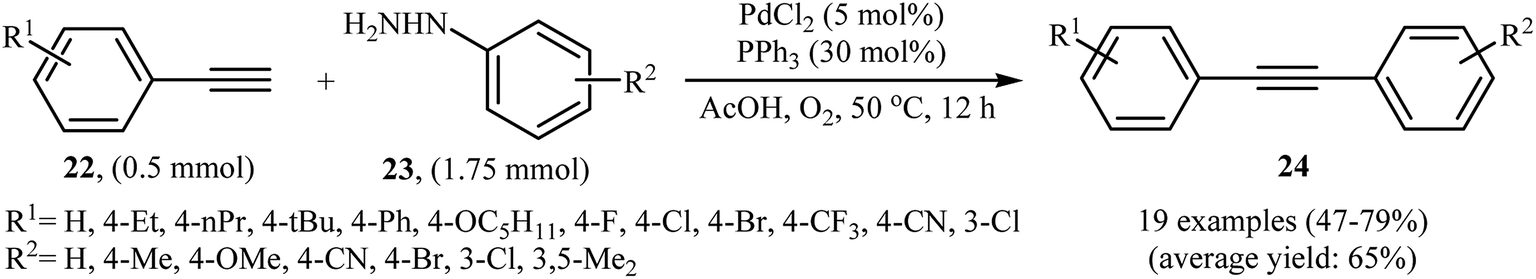
The palladium-catalyzed cross-coupling between sp-hybridized carbon atoms of terminal acetylenes and sp2-hybridized carbon atoms of aryl (pseudo)halides is an important method for C(sp)–C(sp2) bonds formation and has been widely employed in an array of synthetic reactions.23 Recently, Zhao and Song reported a protocol for direct arylation of terminal alkynes with arylhydrazines.24 With the PdCl2/PPh3 catalytic system, acetic acid and DMF solvent, a range of aromatic terminal alkynes 22 were directly arylated with various electron-rich and electron-poor arylhydrazines 23 to give corresponding internal aromatic alkynes 24 in moderate to good yields (Scheme 13). However, aliphatic terminal alkynes did not work well in this reaction. High yielding synthesis of bromine-substituted internal alkynes, which could not be readily obtained in traditional Sonogashira couplings, mild reaction conditions, and scalability were the main advantages mentioned for this synthetic strategy. It should be mentioned that the direct use of commercially available arylhydrazine hydrochlorides as the substrates did not lead to the desired products. Thus, pretreatment of these hydrochloride salts with NaOH is required. According to the author proposed mechanism this reaction proceeds along the similar mechanistic pathway that described in Scheme 8. To the best of our awareness, this is the only example of Sonogashira-type aerobic oxidative coupling of arylhydrazines with terminal alkynes reported so far.
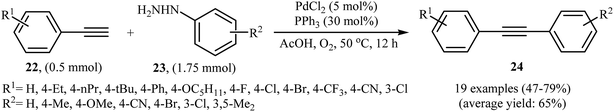 |
| | Scheme 13 Palladium-catalyzed Sonogashira reaction of aromatic terminal alkynes 22 with functionalized arylhydrazines 23. | |
2.5 Direct C–H arylation of arenes
One of the first general reports on the direct C–H arylation of simple arenes with arylhydrazines was published in 2001, by Demir et al.25 They showed that heating of functionalized arylhydrazines 25 in refluxing benzene in the presence of 3 equiv. of Mn(OAc)3·2H2O gave the desired biaryls 26 in good to high yields (Scheme 14). It is worthy to note that the electronic character of the substituents in the arylhydrazines had little effect on the facility of the reaction. Generally, both electron-rich and electron-poor arylhydrazines were well tolerated. Moreover, the substrate scope was successfully extended to use heteroaryl hydrazines coupling components. Seven years later, the same authors have further improved the efficiency of this protocol by performing the process in the presence of 3 equiv. of KMnO4 in benzene–AcOH (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).26
:1).26
 |
| | Scheme 14 Mn(OAc)3-mediated oxidative coupling of benzene with arylhydrazines 25. | |
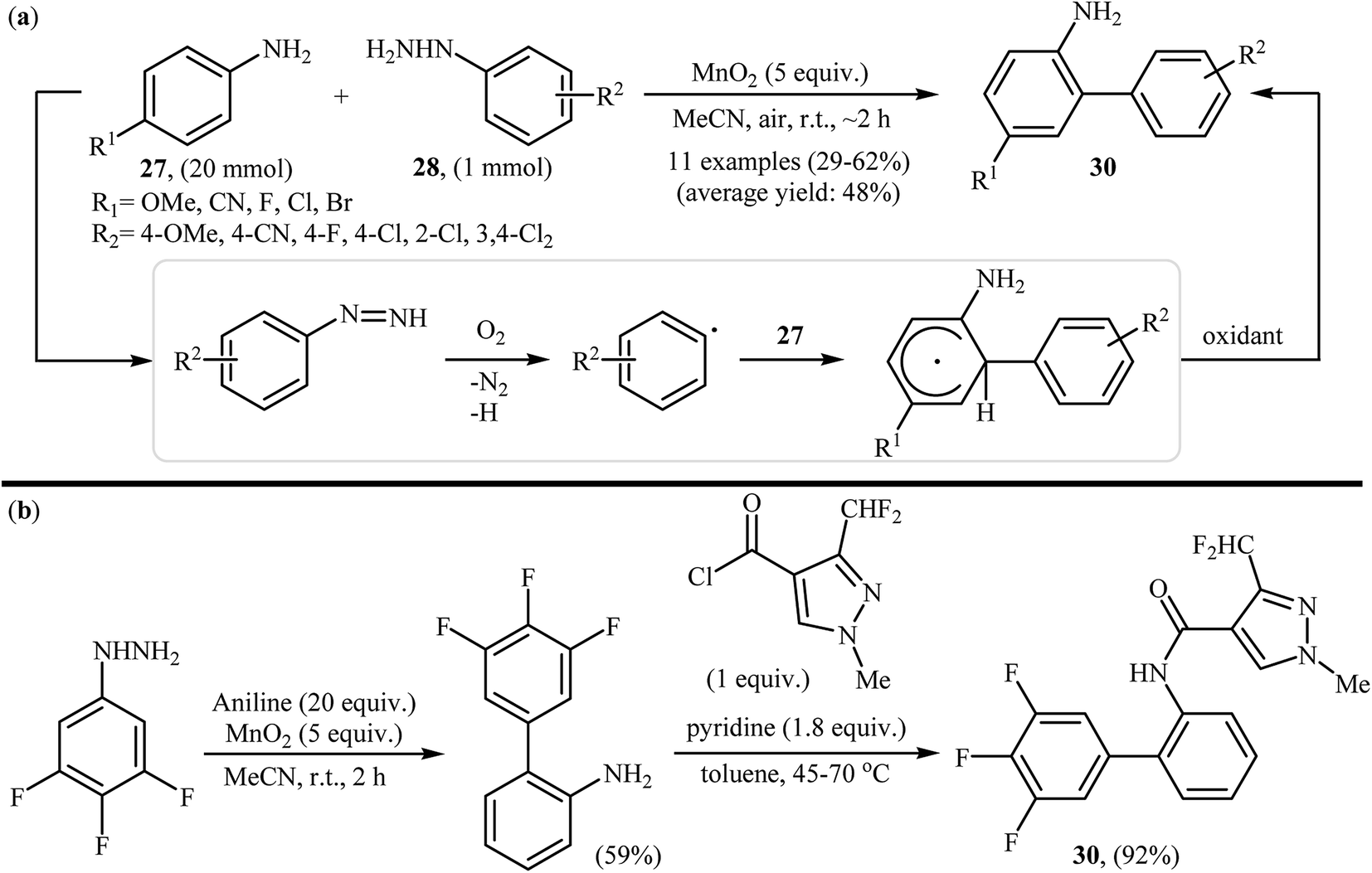
Shortly afterwards, the group of Heinrich extended the substrate scope of this chemistry to a variety of anilines and obtained a range of 2-aminobiphenyls 29 in modestly to good yields via the treatment of para-substituted anilines 27 with arylhydrazines 28 in the presence of over stoichiometric amounts of MnO2 as the oxidant in MeCN at room temperature (Scheme 15a).27 Notably, in the cases of unsubstituted aniline a mixture of 2-aminobiphenyls and 4-aminobiphenyls were formed. Other oxidants were also found to promote the reaction [e.g., Mn(OAc)3, KMnO4, KO2, NaIO4, H2O2]; however, in lower yields. It is interesting to note that comparative studies showed that anilines were significantly better aryl radical acceptors than nitrobenzenes or phenyl ethers. The authors also successfully showed the application of this procedure for the high yielding syntheses of Xemium 30; a novel commercially available fungicide (Scheme 15b). Subsequent study by the same group showed that the same set of 2-aminobiphenyls could be prepared in moderate yields by performing the reaction in aqueous sodium hydroxide (1 M) under air atmosphere.28 In a closely related investigation, Zeng and Zou along with their co-workers also described that air-promoted direct radical arylation of 4-substituted anilines with arylhydrazines in the presence of NaOH as a base in binary solvent MeCN/H2O with ratio 3:2 produced corresponding 2-aminobiphenyls in moderate to good yields.29 Shortly afterwards, the same research team slightly improved the efficiency of this reaction in the terms of yield employing CoPc as a catalyst and Et3N as the base in MeCN.30
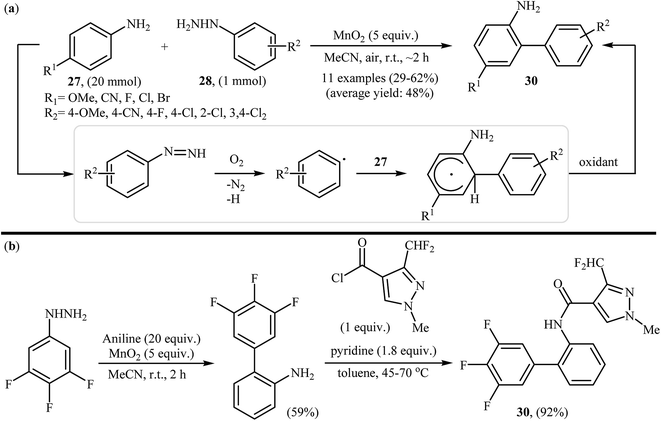 |
| | Scheme 15 (a) MnO2-mediated regioselective C2-arylation of anilines 27 with arylhydrazines 28; (b) Heinrich's synthesis of Xemium 30. | |
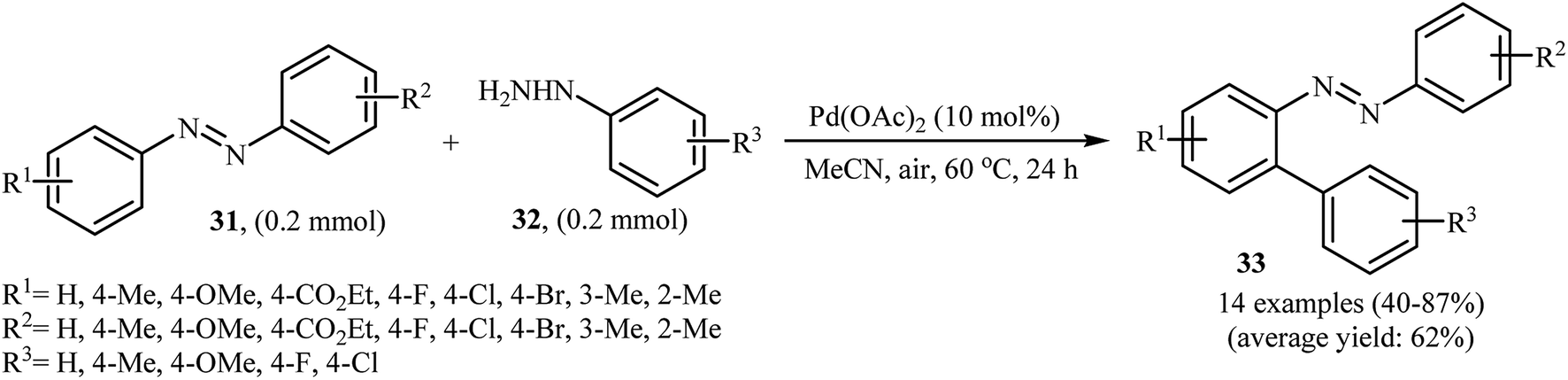
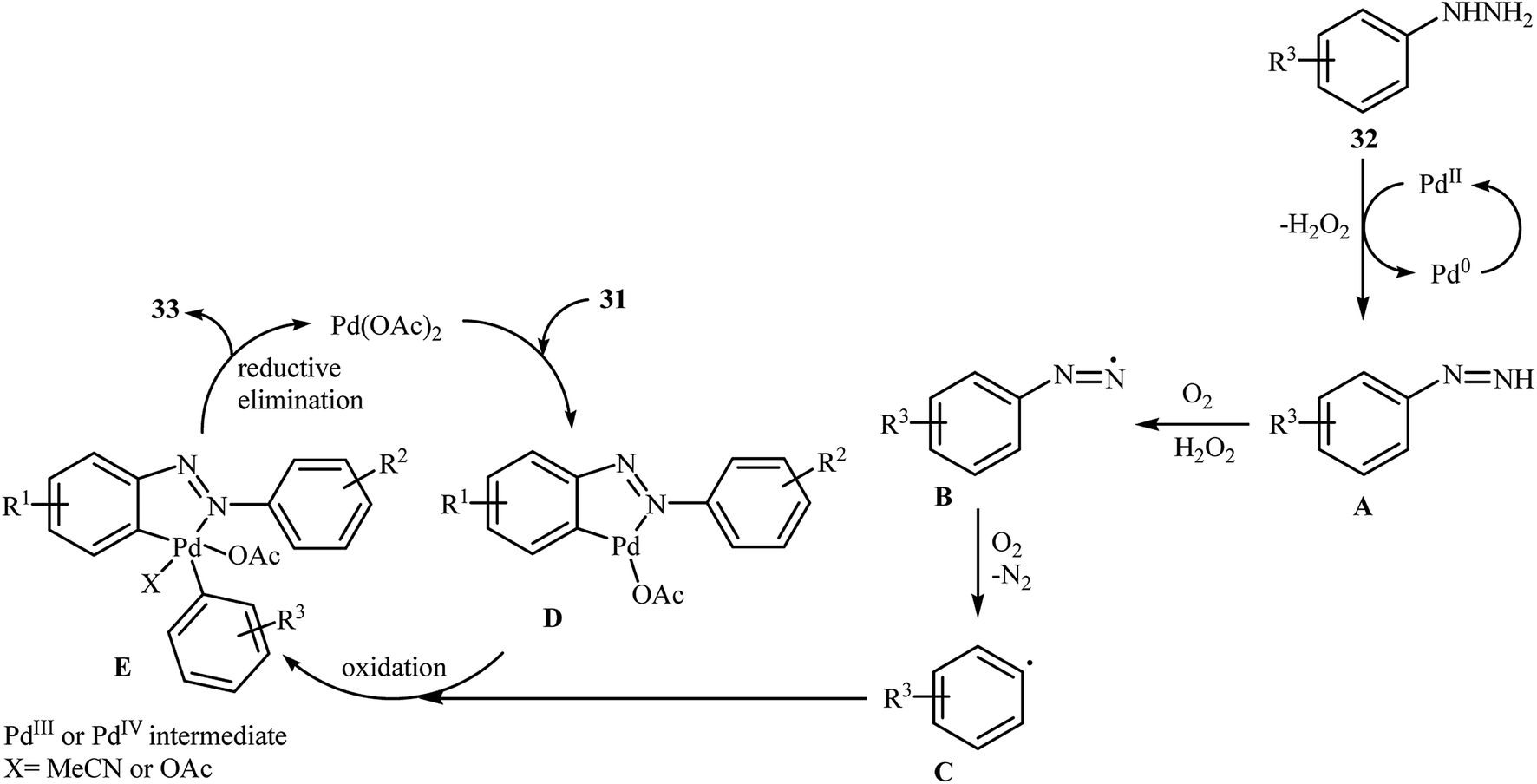
Inspired by these works, Li and Ye presented an elegant Pd-catalyzed regioselective ortho-sp2 C–H arylation of azoarenes 31 with arylhydrazines 32 using 10 mol% of commercially available Pd(OAc)2 as the catalyst under ligand-free conditions.31 The reaction were carried out in MeCN under air atmosphere, tolerated a variety of functional groups, and generally provided the expected coupling products 33 in moderate to good yields (Scheme 16). However, sterically hindered ortho-substituted arylhydrazines failed to enter into the reaction. The results proved that the electronic character of the substituents in azoarenes had a strong effect on the rate of reaction. While electron-donating groups gave the desired products in good yields, the electron-withdrawing groups resulted in a relatively poor to moderate yields. Notably, no para-substituted compound was found by the reaction conditions employed. The author proposed reaction mechanism is outlined in Scheme 17. A possible process for the formation of compounds 33 should involve the formation of diazene A via oxidation of arylhydrazine 32 with PdII, followed by oxidation of this intermediate with air oxygen to give the radical intermediate B, which after release of a molecular nitrogen delivers the aryl radical C. Simultaneously, the reaction of PdII catalyst with azoarene 31 affords a palladacyclic intermediate D. Then, reaction of radical C with intermediate D under heating and oxygen produces a PdIV or PdIII intermediate E. Finally, reductive elimination of this intermediate affords the desired product 33. Recently, the methodology was extended to a series of aromatic diamines, using K2CO3 as the base and DMSO as the solvent, and the bicyclic diamines were obtained in reasonable yields.32
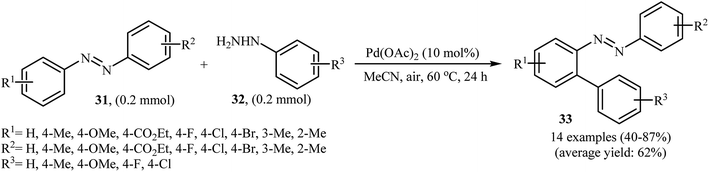 |
| | Scheme 16 Pd-catalyzed regioselective 2-arylation of azoarenes 31 with arylhydrazines 32. | |
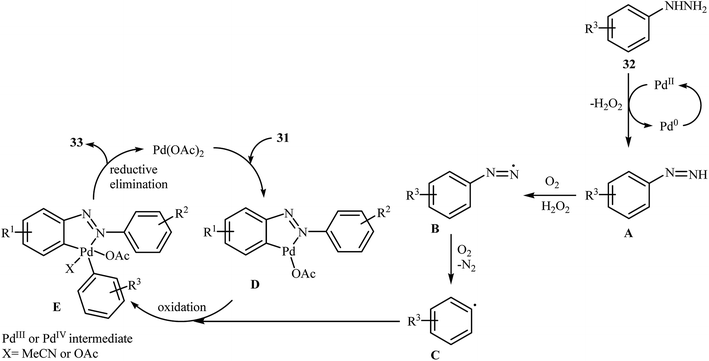 |
| | Scheme 17 Mechanistic proposal for the reaction in Scheme 16. | |
2.6 Direct C–H arylation of heteroarenes
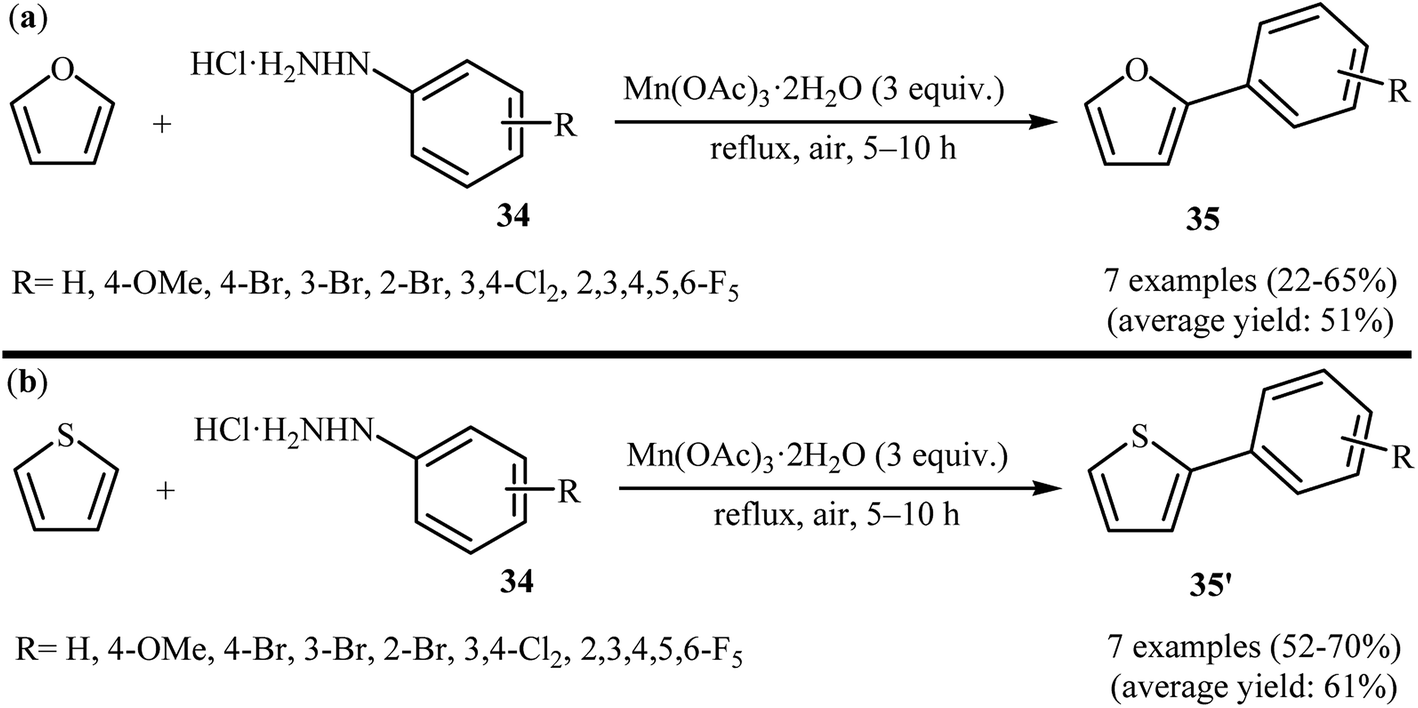
2.6.1 O-Heteroaromatics. One of the earliest reports on the direct C–H arylation of heteroarenes with arylhydrazines, has been reported by Demir and co-workers in 2002, when furan and thiophene underwent radical arylation with arylhydrazines 34 in the presence of 3 equiv. of Mn(OAc)3 as an oxidant to form corresponding 2-arylfurans 35 and 2-arylthiophenes 35′, respectively (Scheme 18).33 The expected heterobiaryls were obtained in poor to almost moderate yields. It is noted that the reaction required high temperature (reflux) and a large excess of heteroarene (∼200 equiv.), used as both reactant and solvent.
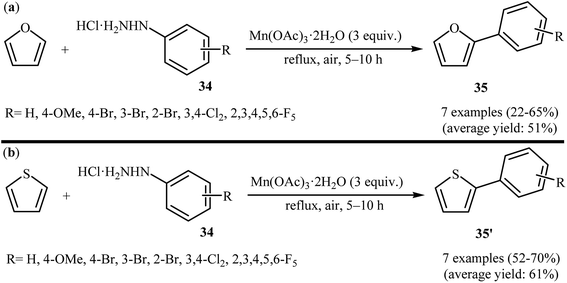 |
| | Scheme 18 Demir's synthesis of (a) 2-arylfurans 35; (b) 2-arylthiophenes 35′. | |
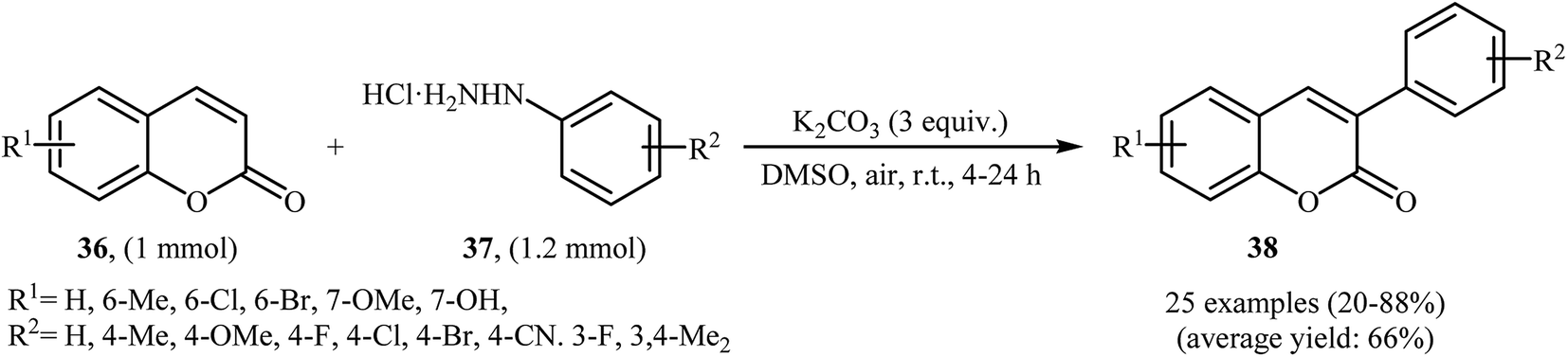
Thirteen years later, Yadav and co-workers developed a simple and efficient base-mediated coupling reaction between functionalized coumarins 36 and arylhydrazines 37 for the regioselective synthesis of α-arylated coumarins 38 (Scheme 19).34 K2CO3 was more suitable compared to the other base (e.g. Na2CO3, Ag2CO3, Cs2CO3, NaOEt, PIDA, TBHP, K2S2O8) for this transformation. Solvent has a dramatic role in the reaction and among the common solvents (e.g. MeOH, DMF, DMA, DMSO, NMP); DMSO was the most effective solvent. This base-mediated reaction was applicable for both the electron-rich as well as electron-poor arylhydrazines and for various substituted coumarins. However, 6-nitro substituted coumarin failed to enter into this reaction. The authors also found that this reaction condition was very efficient for the preparation of α-arylated 2-pyridones via the reaction of 2-pyridones and arylhydrazines. Subsequently, the group of Yuan–Xiao improved the efficiency of this reaction in the terms of yield and reaction time employing KMnO4 as oxidant in MeCN.35
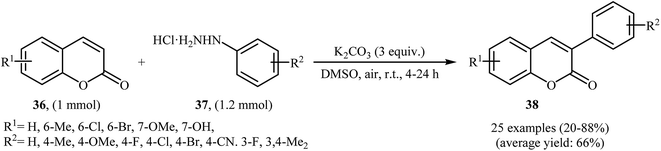 |
| | Scheme 19 K2CO3-mediated regioselective α-arylation of coumarins 36 with arylhydrazines 37. | |
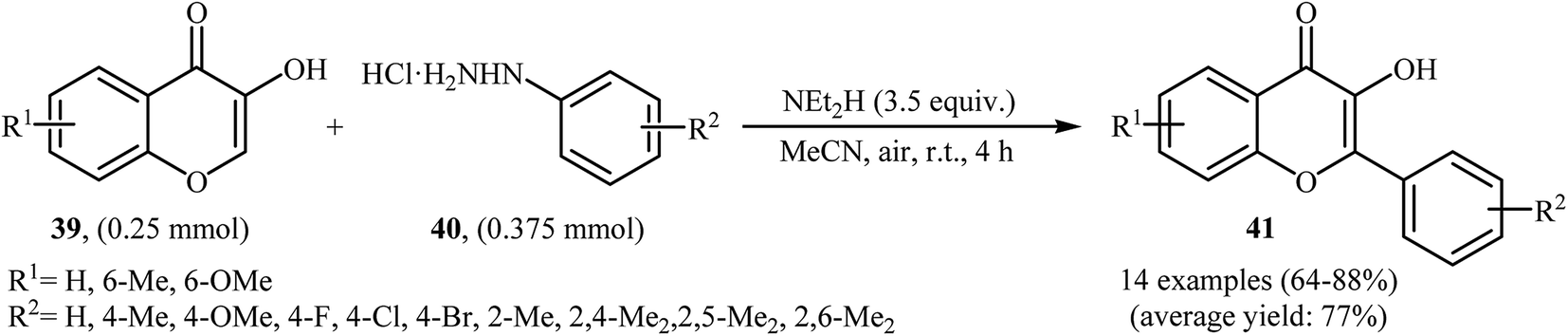
Very recently, Paul and Bhattacharya described an efficient transition-metal free direct C-arylation of 3-hydroxychromones with arylhydrazines employing NEt2H as a base and air as oxidant.36 This oxidative C–H arylation reaction afforded the optimum yield in MeCN at room temperature. Various 3-hydroxychromones 39 and arylhydrazines 40 were used to establish the general applicability of the method (Scheme 20). It is noted that C–C cross-coupling of 5-hydroxy pyran-4-ones with arylhydrazines was also examined using this system.
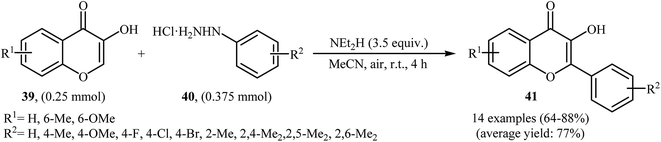 |
| | Scheme 20 Base-mediated direct C-arylation of 3-hydroxychromones 39 with arylhydrazines 40. | |
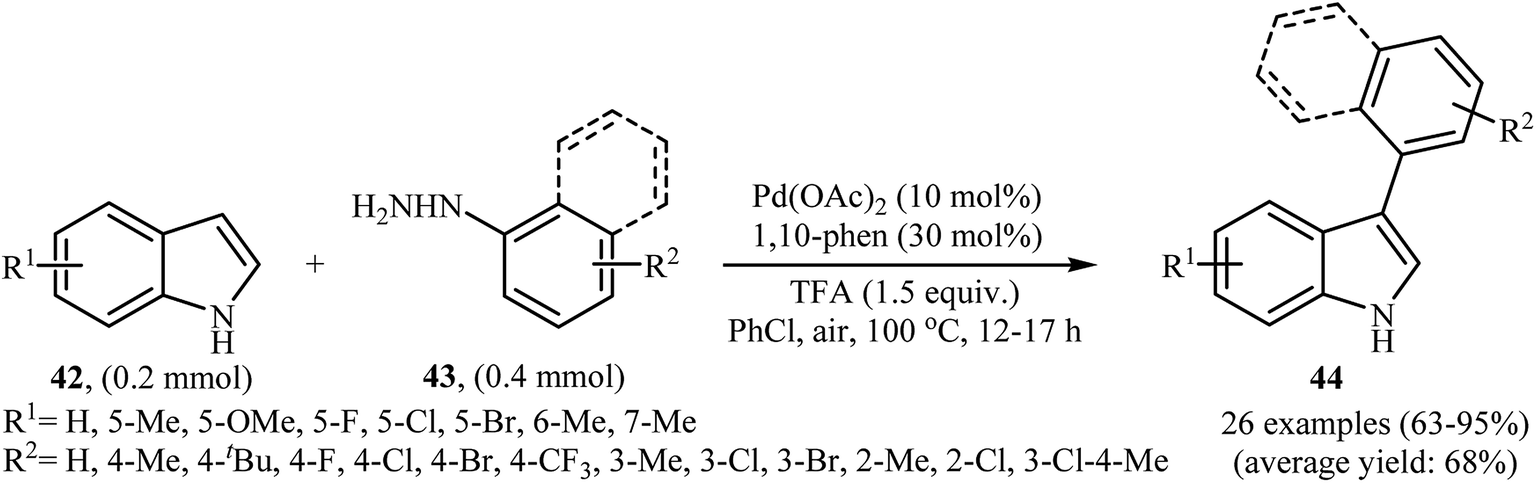
2.6.2 N-Heteroaromatics. In 2013, the group of Chen reported a palladium-catalyzed direct C-3-arylation of 1H-indoles 42 with arylhydrazines 43 that afforded functionalized 3-aryl-1H-indoles 44 in good to excellent yields (Scheme 21).37 The use of Pd(OAc)2 as catalyst and 1,10-phen as ligand was key to obtaining good results, while the use of other palladium complexes [e.g., PdCl2, PdCl2(PPh3)2, Pd(dba)2] or ligands [e.g., 2,2′-bipy] resulted in a dramatic decrease of the reaction yield. In this methodology, a series of oxidants and solvents were used by the authors for a comparative study. In these experiments, the best result was obtained when PhCl solvent and air oxidant was used. This direct denitrogenative C-3-arylation methodology tolerated a wide range of substituents on the 1H-indole partner and was efficient for the use of different arylhydrazines with diverse steric and electronic properties. However, the strongly electron-withdrawing nitro- and cyano-substituted indoles and nitro-substituted arylhydrazines failed to afford the expected products. Moreover, methoxy substituted arylhydrazines did not work well under this reaction conditions.
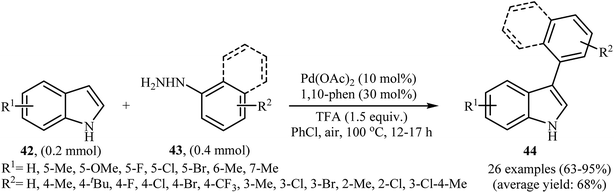 |
| | Scheme 21 Pd-catalyzed regioselective C3-arylation of 1H-indoles 42 with arylhydrazines 43. | |
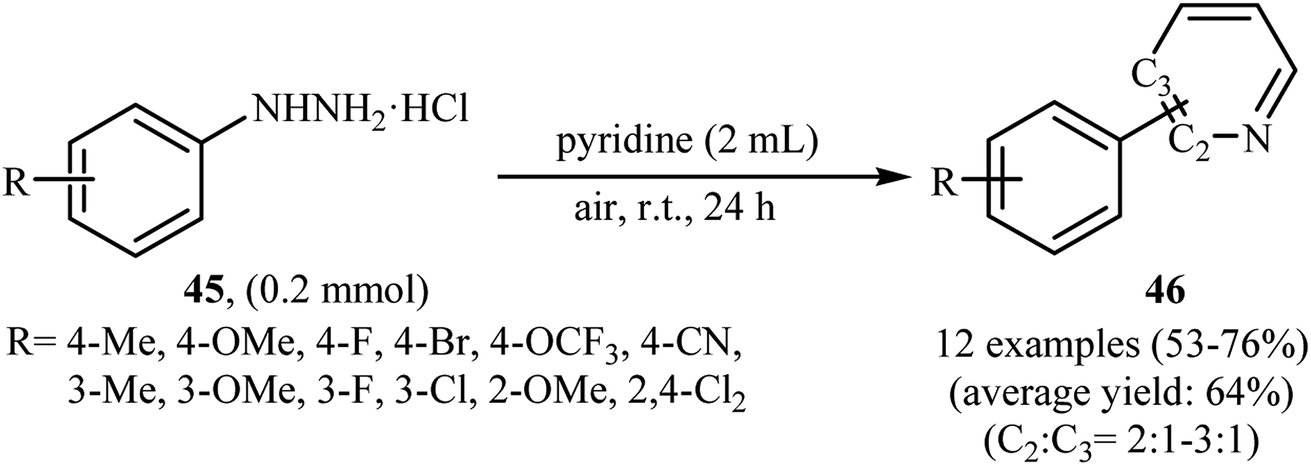
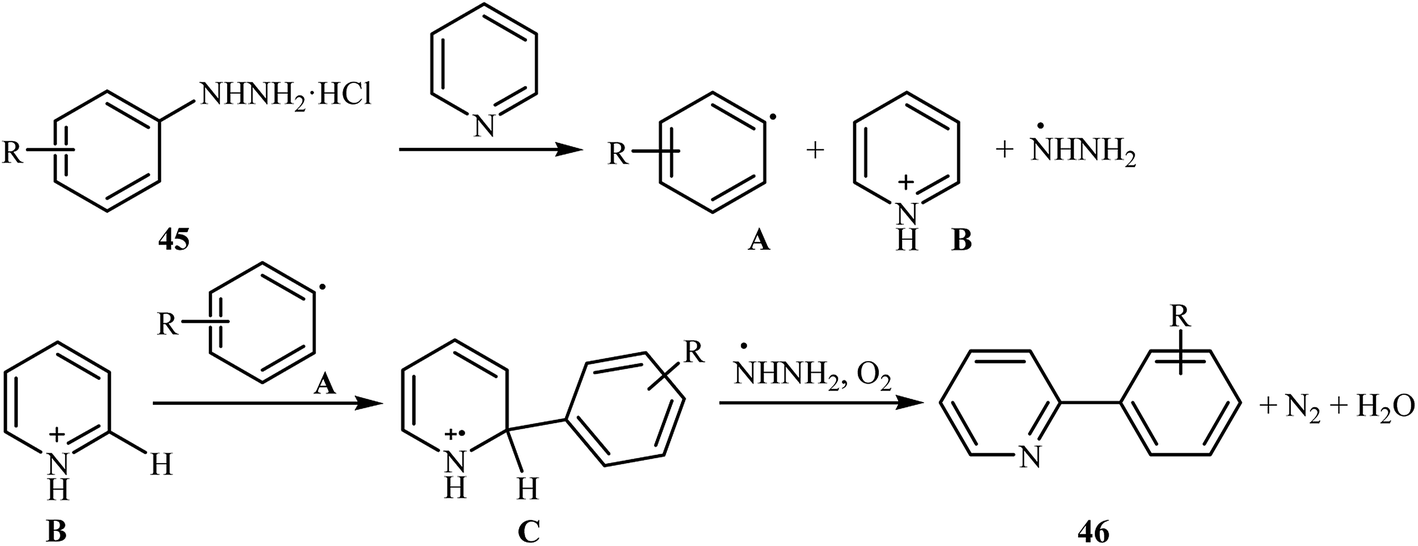
Shortly thereafter, Li, Liu and Kuang developed a methodology for the selective arylation of pyridine with arylhydrazine hydrochlorides 45 without any catalyst, ligand and base.38 The reaction proceeded under mild conditions (room temperature) in air and afforded a mixture of C2- and C3-arylated pyridines 46 in moderate to good yields (Scheme 22). It is noted that in all cases, the C2-arylated products were obtained in higher amount than the C3-arylated products. The authors claimed that pyridine playing a dual role in this reaction; the solvent and the reactant. To explain the formation of the products, the authors proposed a mechanism in which, after formation of aryl radical A via the decomposition of arylhydrazine 45 in the presence of pyridine, the reaction of this radical with protonated pyridine B provides the radical cation C that, after oxidation by air oxygen affords the desired product 2 (Scheme 23). In a related study, Taniguchi and Mizuno along with their co-workers showed that the treatment of amino-substituted pyridine derivatives with arylhydrazines in the presence of K2CO3 (3 equiv.) in DMSO at room temperature produced corresponding arylated pyridines in moderately to high yields.39 They found that the reaction is also compatible with amino-substituted pyrazines, quinolines, and pyrimidines. Previously, the group of Yadav applied this reaction condition in the regioselective C-3 arylation of quinoline-4-one derivatives with arylhydrazines.40
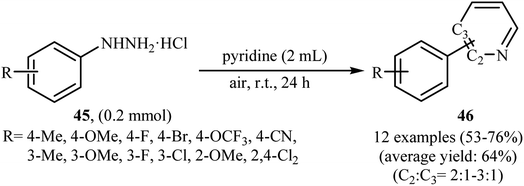 |
| | Scheme 22 Transition-metal free arylation of pyridine with arylhydrazine hydrochlorides 45. | |
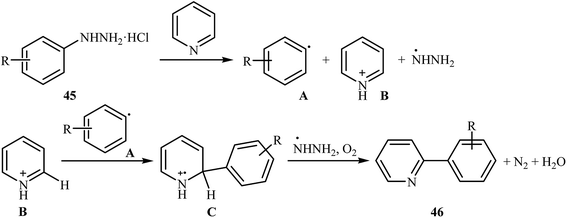 |
| | Scheme 23 Mechanistic proposal for the reaction in Scheme 22. | |
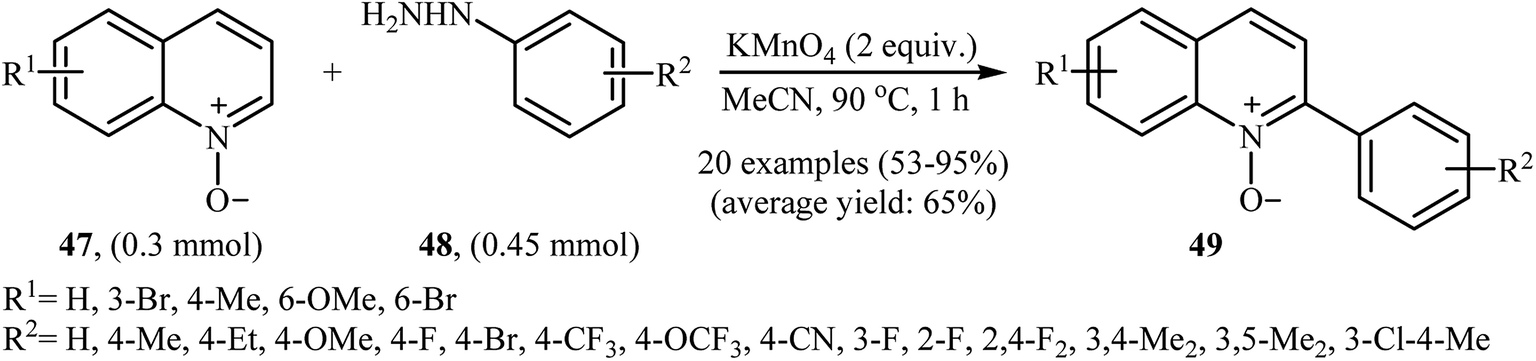
In 2016, Yuan, Li, and Xiao reported an analogous methodology for the direct C2-selective C–H arylation of quinoline N-oxides 47 with arylhydrazines 48 (Scheme 24).41 In their optimization study, the authors found that the use of 2.0 equiv. of KMnO4 as an oxidant in MeCN gave the best results. Examination of the scope of the reaction revealed that a range of arylhydrazines bearing both electron-withdrawing and electron-donating groups and a number of functionalized quinoline N-oxides afforded the 2-arylquinoline N-oxides 49 in moderate to excellent yields. However, quinoline N-oxides possessing a strong electron-withdrawing –NO2 group at the C6 position failed to participate in this reaction. Interestingly, aliphatic hydrazines were also well tolerated in the transformation. In 2017, similar chemistry as that described for quinoline N-oxides was used by Lee's research team to prepare 3-arylquinoxalin-2(H)-one derivatives via direct C3-selective C–H arylation of quinoxalin-2(H)-ones with arylhydrazines employing iodosobenzene (PhIO) as oxidant in MeCN.42
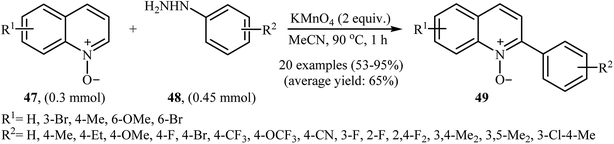 |
| | Scheme 24 Direct C2-arylation of quinoline N-oxides 47 with arylhydrazines 48. | |
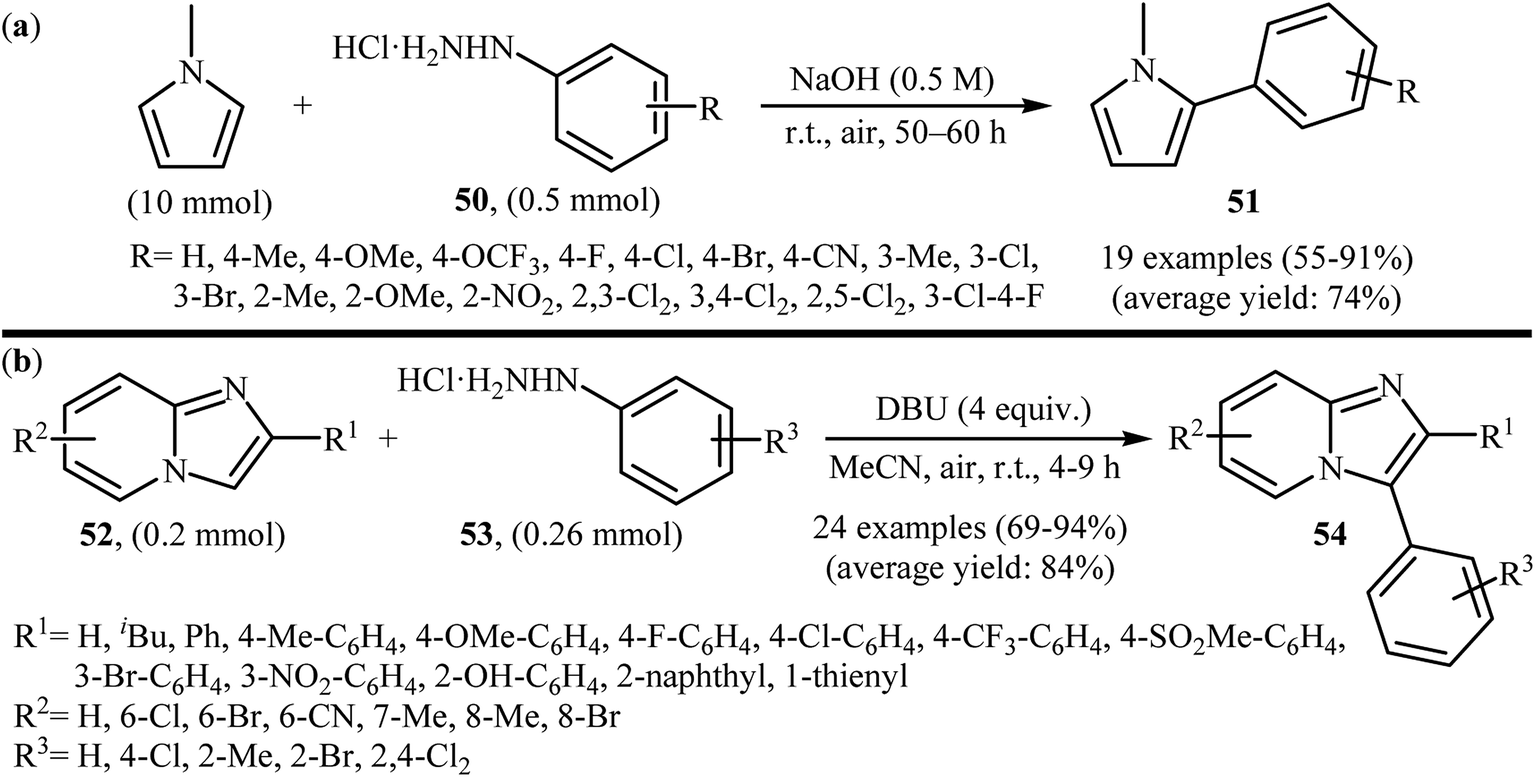
Recently, using the same oxidative C–N bond activation strategy, an efficient base-mediated oxidative radical arylation of N-methylpyrrole was reported by Talaz and co-workers.43 In the presence of NaOH as a base and air oxygen as an environmentally benign oxidant, various arylhydrazines 50 could react with N-methylpyrrole to give the desired C-2 arylated pyrroles 51 in moderate to excellent yields (Scheme 25a). Very recently, using an analogous methodology, Hajra and co-workers reported the synthesis of 3-arylated imidazopyridine derivatives 54 from the reaction between 2-substituted imidazopyridines 52 and arylhydrazines 53 using DBU as a base in MeCN (Scheme 25b).44
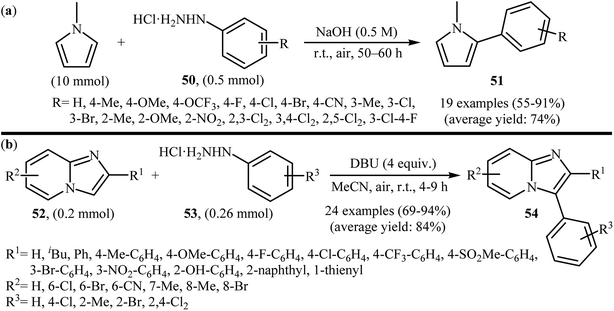 |
| | Scheme 25 (a) Talaz's synthesis of 2-arylated pyrroles 51; (b) Hajra's synthesis of 3-arylated imidazopyridines 54. | |
3 Carbon–heteroatom cross-couplings
3.1 C–N bond formation
In 2013, the group of Akamanchi reported the first example of denitrogenative C–N cross-coupling of amines with arylhydrazines.45 They showed that primary aromatic amines 55 underwent rapid C–N cross-coupling with various arylhydrazines 56 in the presence of IBX/Cu(OAc)2 combination as a catalytic system in MeCN to provide the corresponding diarylamines 57 in good yields. It is interesting to note that the electronic and steric characters of the substituents in the phenyl ring periphery of both amines and hydrazines had remarkably little effect on the facility of reaction. Various substrates were examined involving electron-donating and withdrawing groups in the para, ortho, and meta positions according to Scheme 26. The authors proposed that the reaction proceeds via a similar mechanistic pathway that described in Scheme 12.
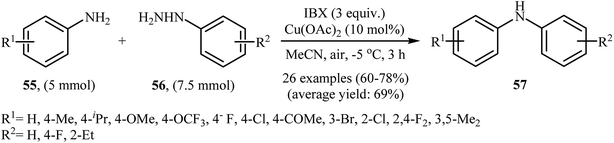 |
| | Scheme 26 Synthesis of diarylamines 57 through N-arylation of aromatic amines 55 with arylhydrazines 56. | |
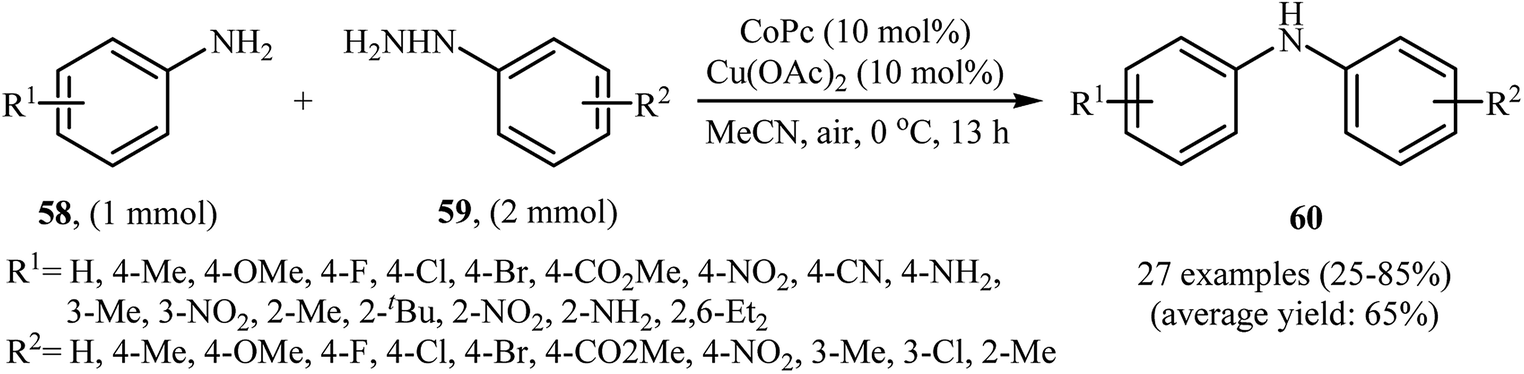
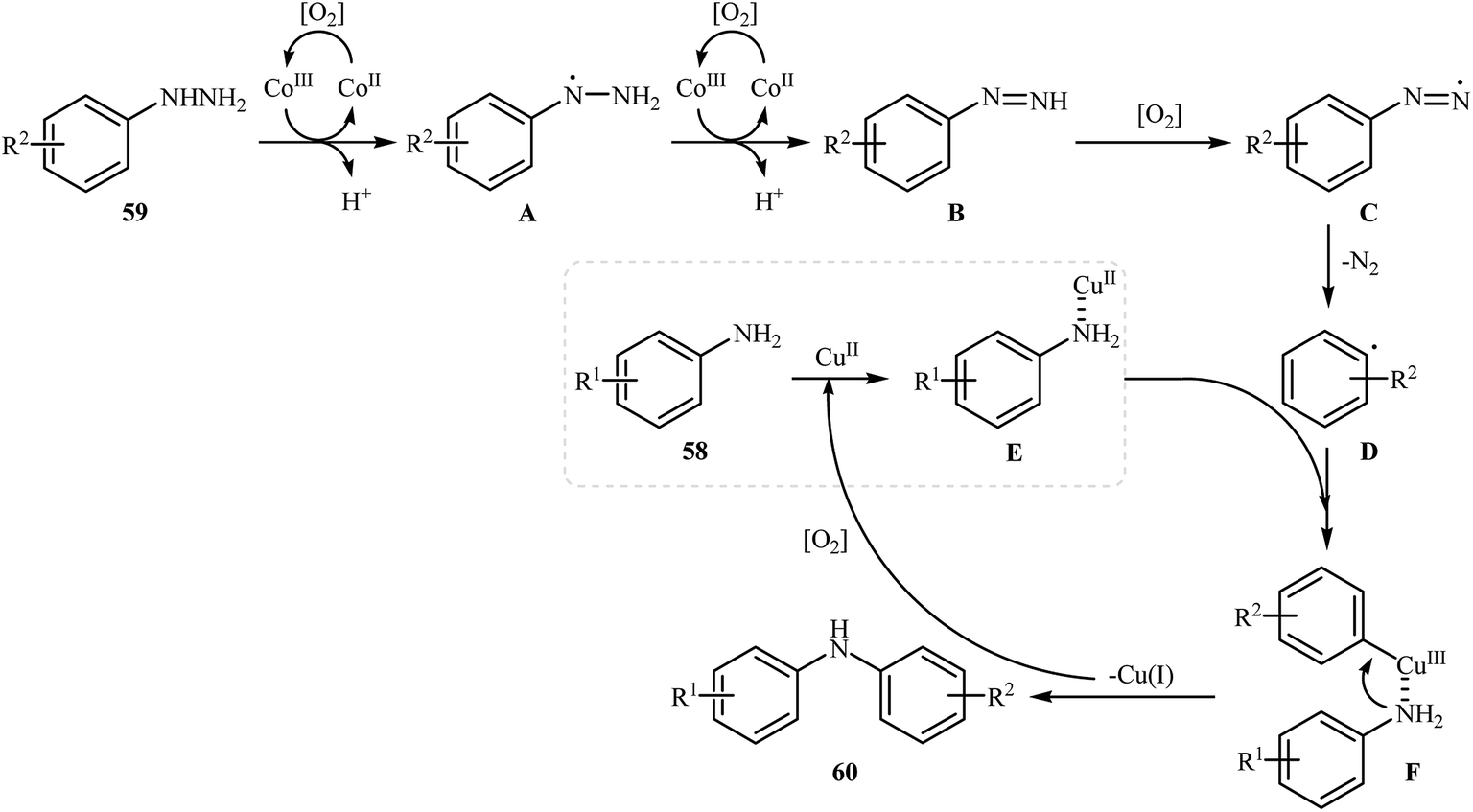
Three years later, in 2016, Zou and co-workers reported another interesting synthetic route for the denitrogenative cross-coupling of aromatic amines 58 with a diverse range of arylhydrazines 59 using CoPc and Cu(OAc)2 as catalysts in MeCN as solvent at 0 °C for 13 h, affording reasonable yields of the corresponding diarylamines 60 (Scheme 27).46 A variety of sensitive functional groups including nitro, cyano, amino, acetyl, fluoro, chloro, bromo, methoxy were well tolerated. Thus this procedure offers scope for further manipulation of products. Furthermore, CoPc/Cu(OAc)2 demonstrated very high catalytic activity toward the C–N cross-coupling of various primary heteroaromatic, bicyclic aromatic, benzylic and aliphatic amines with arylhydrazines. However, amides and secondary amines failed to participate in this reaction. The author proposed mechanism for this N-arylation, which is outlined in Scheme 28.
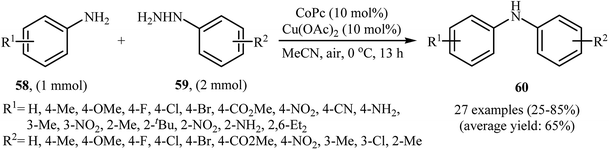 |
| | Scheme 27 Co/Cu-catalyzed C–N coupling of anilines 58 with arylhydrazines 59. | |
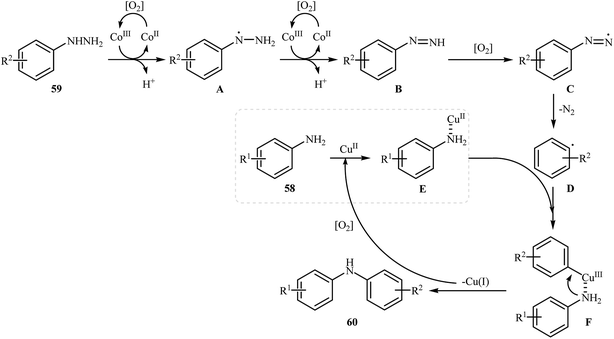 |
| | Scheme 28 Mechanistic proposal for the reactions in Scheme 27. | |
3.2 C–P bond formation
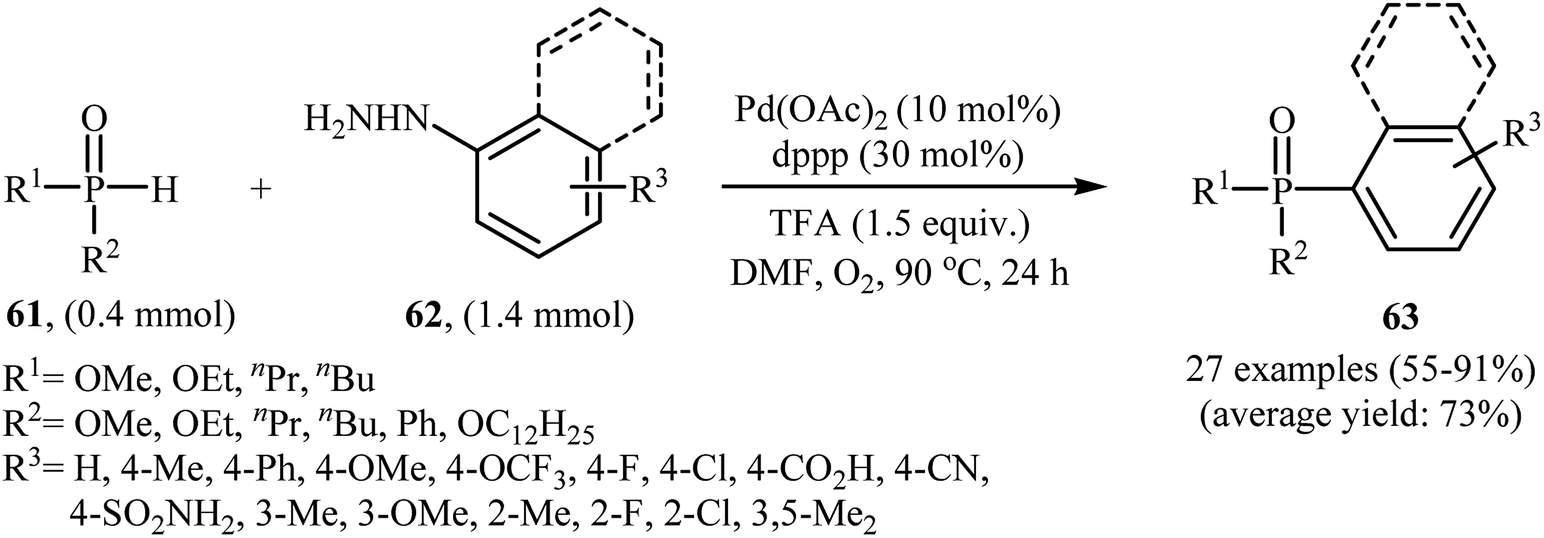
Gao and Yin along with their co-workers have recently reported the synthesis of arylphosphorus compounds 63, via the palladium-catalyzed oxygen-based oxidative cross-coupling of various P(O)H compounds 61 with arylhydrazines 62 at 90 °C within 24 h (Scheme 29).47 Of the palladium catalysts screened [e.g., Pd(OAc)2, Pd(PPh3)4, Pd2(dba)3, PdCl2], Pd(OAc)2 proved to be optimal. Furthermore, the reaction is sensitive to the ligand used, the best result was obtained when dppp ligand was used. Among the various solvents like PhCl, DMF, DMSO, NMP, 1,4-dioxane; DMF was the most efficient for the transformation. In their optimization study, the authors also found that the addition of 1.5 equiv. of trifluoroacetic acid (TFA) as an additive to the reaction mixture significantly improved the yield. Under optimized conditions, the reaction tolerated various electron-rich and electron-poor arylhydrazines and a range of H-phosphinates and H-phosphonates and gave the expected products in moderate to very good yields. However, under similar reaction conditions easily oxidizable secondary phosphine oxides R2P(O)H did not afford the desired coupling products since they were oxidized to R2P(O)OH by the present catalytic system. In addition, alkylhydrazines were not suitable for this reaction, and no coupling products were observed under similar conditions.
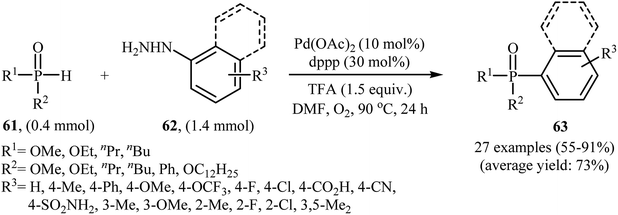 |
| | Scheme 29 Pd-catalyzed C–P cross-coupling of P(O)H compounds 61 with arylhydrazines 62 developed by Gao and Yin. | |
3.3 C–Se bond formation
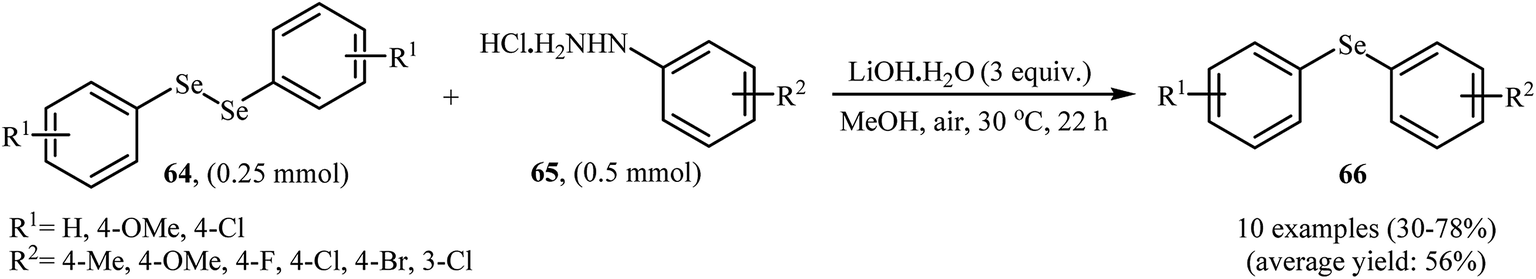
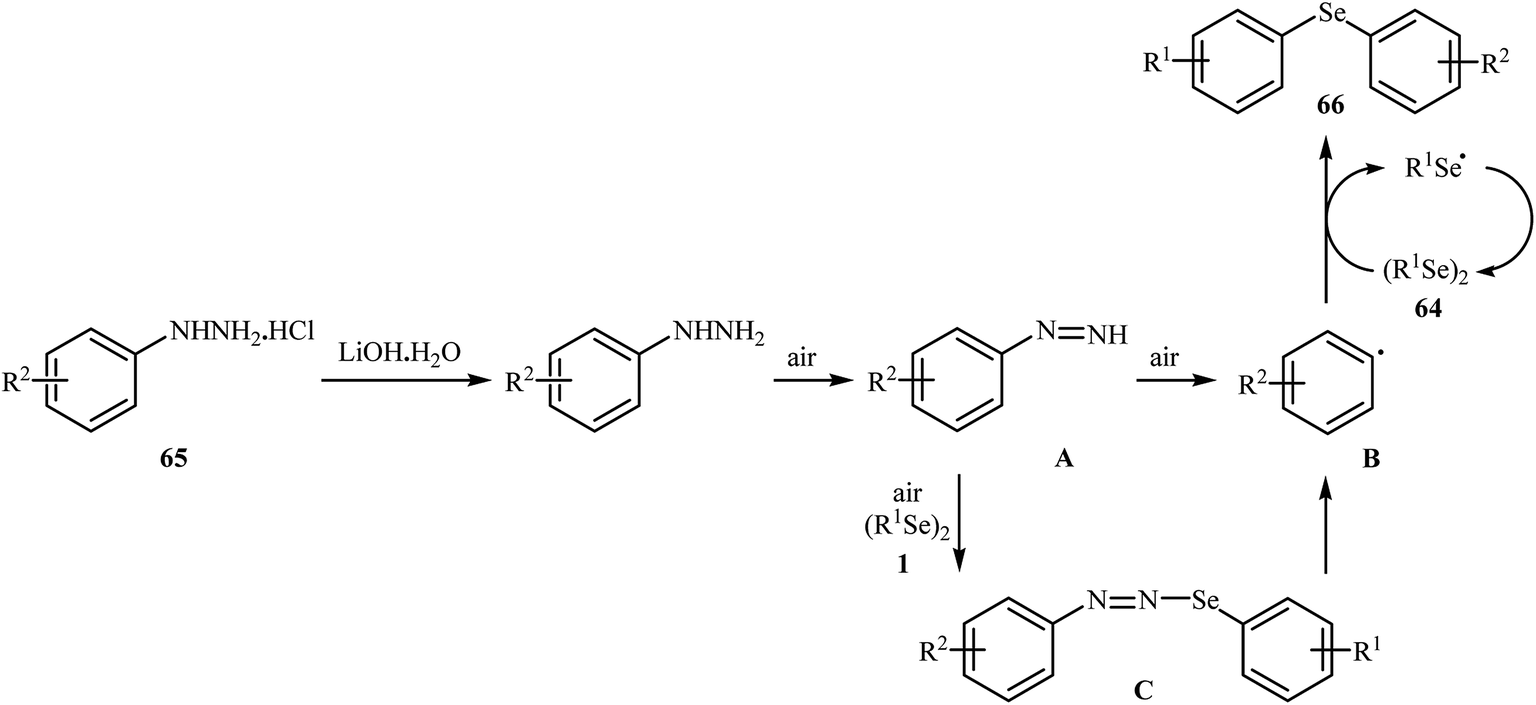
In 2017, Taniguchi and co-workers reported the first example of base-promoted direct oxidative cross-coupling of diaryl diselenides with arylhydrazines under transition-metal-free conditions, which allows for preparation of numerous unsymmetrical diaryl selenides.48 They showed that the coupling of symmetrical diaryl diselenides 64 with both electron-rich and electron-poor arylhydrazines 65 in the presence of 3 equiv. of commercially available LiOH·H2O in MeOH under air gave corresponding diaryl selenides 66 in modestly to good yields (Scheme 30). In addition, satisfactory yields were achieved in gram-scale reactions. However, dialkyl disulfides did not work well under this reaction conditions. In the optimization study, the authors found that other base also promoted the reaction (e.g., K2CO3, Li2CO3, Na2CO3, NaOH, Et3N); however, the use of LiOH·H2O gave the best results. In the absence of any base, only trace amounts of expected product was obtained. On the basis of experimental observations, the authors proposed a mechanism (Scheme 31) that involves the initial formation of diazene A via the oxidation of arylhydrazine 65 by air, which further oxidizes with air oxygen to form radical intermediate B. Another route to form intermediate B is through diazoselenide C. Subsequently, trapping of this radical B by diaryl disulfide 64 affords desired diaryl selenide 66 and arylseleno radical D. Finally, homocoupling of radical D regenerates starting diaryl disulfide 64.
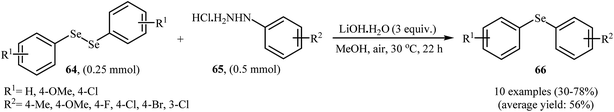 |
| | Scheme 30 Synthesis of unsymmetrical diaryl selenides 66 from diaryl diselenides 64 and arylhydrazines 65 reported by Taniguchi. | |
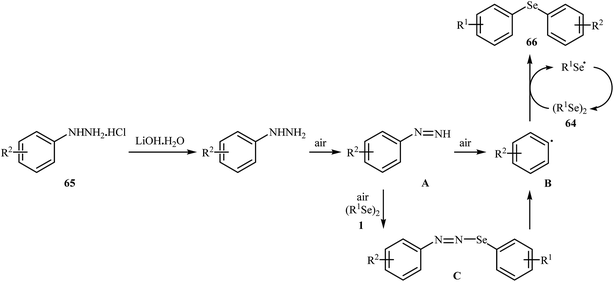 |
| | Scheme 31 Mechanism that accounts for the formation of diaryl selenides 66. | |
3.4 C–S bond formation
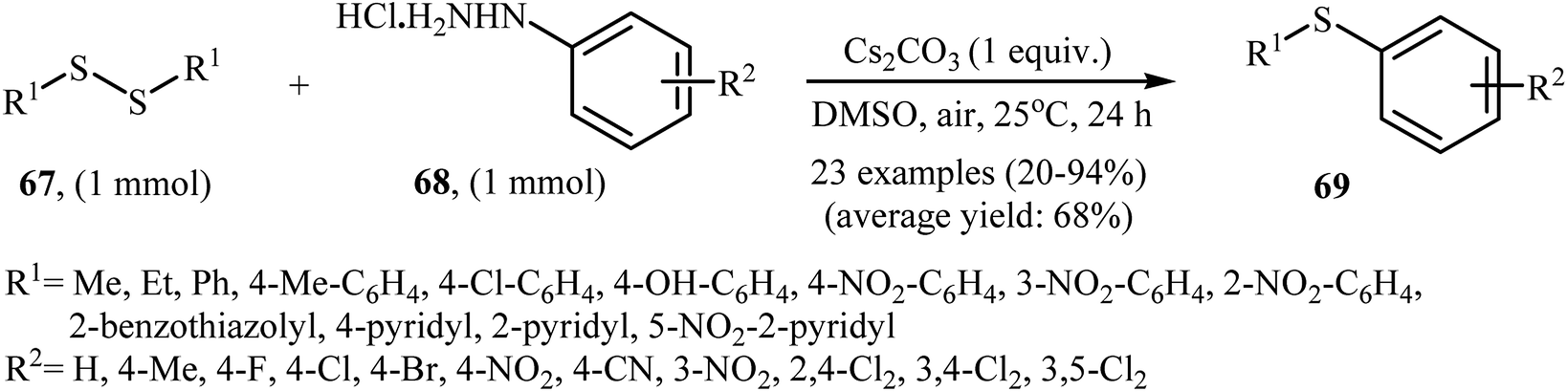
In 2017, Taniguchi and co-workers have developed an efficient and straightforward approach for the synthesis of unsymmetrical thioethers 69 via Cs2CO3-mediated aerobic oxidative cross-coupling of disulfides 67 and arylhydrazines 68 (Scheme 32).49 The reaction was performed in DMSO at room temperature, tolerated various functional groups (e.g., F, Cl, Br, NO2, CN, OH), and generally provided corresponding thioethers in moderate to high yields. Various aliphatic, aromatic, and heteroaromatic disulfides were used to establish the general applicability of the method. In addition, a gram-scale reaction was also successfully performed.
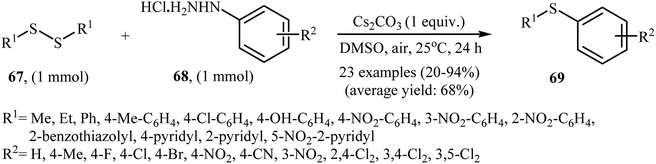 |
| | Scheme 32 Synthesis of unsymmetrical thioethers 69 via the Cs2CO3-mediated cross-coupling of disulfides 67 with arylhydrazines 68. | |
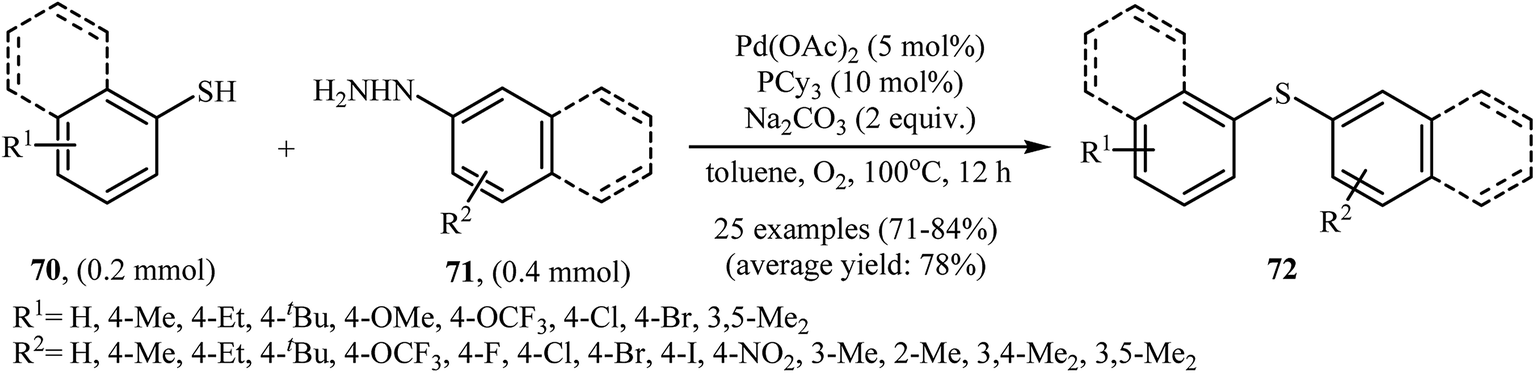
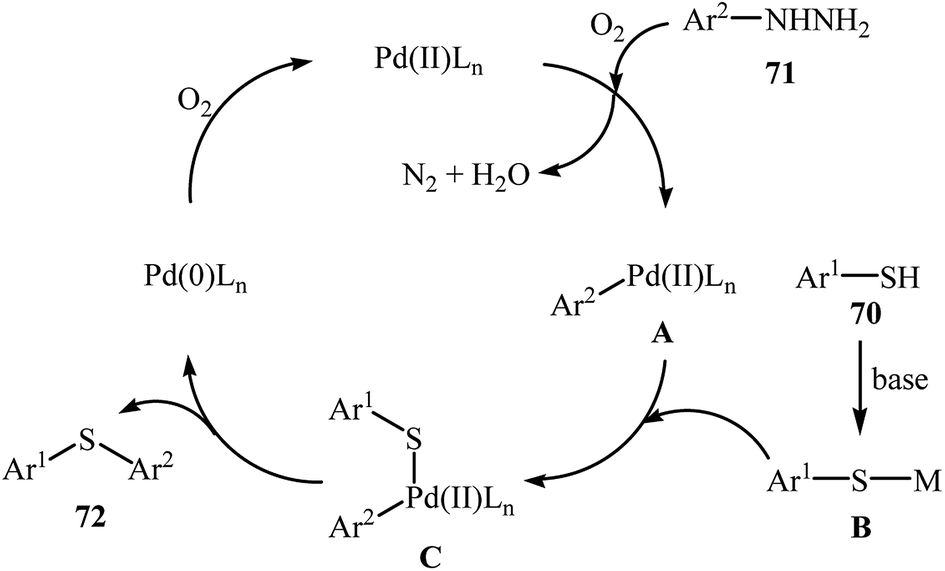
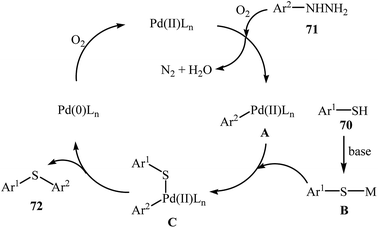
Very recently, the group of Zhao realized an unprecedented palladium-catalyzed oxidative cross-coupling of aromatic thiols 70 with arylhydrazines 71 under nitrogen atmosphere at 100 °C, to form the corresponding thioethers 72 (Scheme 33).50 This denitrogenative cross-coupling reaction afforded the optimum yield in the presence of Pd(OAc)2/PCy3/Na2CO3 combination as a catalytic system in toluene. In Scheme 34, a reasonable mechanistic pathway for this palladium-catalyzed oxidative cross-coupling reaction is suggested. It consists of the following key steps: (i) oxidative C–N bond cleavage of arylhydrazine 71 with Pd(OAc)2 under O2 forms the aryl palladium species A; (ii) reaction of thiol 70 with base generates thiolate intermediate B; (iii) transmetalation of species A with intermediate B gives the active palladium species C; and (iv) reductive elimination of palladium species C affords the expected coupling product 72 and regenerates the Pd catalyst.
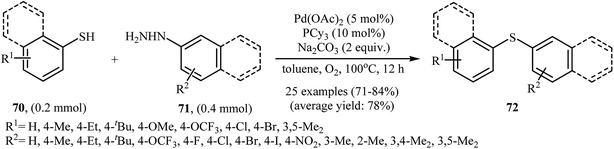 |
| | Scheme 33 Pd-catalyzed S-arylation of aromatic thiols 70 with various arylhydrazines 71. | |
 |
| | Scheme 34 Mechanistic proposal for the formation of thioethers 72. | |
4 Conclusion
Cross-coupling reactions comprise one of the most efficient and versatile methods for the construction of carbon–carbon and carbon–heteroatom bonds by combining two molecular fragments. Over the past decades, various carbon and heteroatom-centered nucleophilic partners (e.g., boronic acids, organosilanes, organotins, organozincs, alkynes, alkenes, aromatic compounds, alcohols, thiols, amines and amides) have been developed in this chemistry, however, electrophilic counterparts have been substantially limited to organic (pseudo) halides. Thus, the development of new electrophilic counterparts for these reactions is highly desirable. Arylhydrazines as novel, commercially available, non-toxic, environmentally friendly, and highly reactive arylating agents have drawn great attention in the past several years. As illustrated, these compounds have been successfully utilized as arylation agents in various C–C and C–X cross-coupling reactions such as Suzuki coupling, Sonogashira coupling, Hiyama coupling, Heck coupling, direct arylation, and Ullman type coupling reactions. Interesting is the fact that most of the reactions covered in this review have been carried out under transition-metal-free and very mild conditions. Furthermore, they could be scaled up to provide multigram quantities of coupling products. This results clearly show the potential application of these reactions in industry. However, the number of reported examples in some reactions (e.g., Sonogashira and Hiyama couplings) are narrow and there is still further need to study the scope and limitations of these reactions. In addition, some important coupling reactions which were found well-developed for (pseudo) halides are still silent for the aryl hydrazines such as Negishi, Stille, and Kumada couplings. We conclude this review by hoping that it will stimulate researchers to further exploration and research in this arena of cross-coupling reactions.
Arylhydrazines have evolved into environmentally friendly arylation agents for palladium-catalyzed oxidative cross-coupling reactions because the only byproducts are water and nitrogen.
Conflicts of interest
There are no conflicts of interest to declare.
References
-
(a) C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed;
(b) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC;
(c) B. V. Varun, J. Dhineshkumar, K. R. Bettadapur, Y. Siddaraju, K. Alagiri and K. R. Prabhu, Tetrahedron Lett., 2017, 58, 803–824 CrossRef CAS.
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed.
- C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963–4972 RSC.
- M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS.
- S. Kovács, Á. I. Csincsi, T. Z. Nagy, S. Boros, G. Timári and Z. Novák, Org. Lett., 2012, 14, 2022–2025 CrossRef PubMed.
- F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
- J. Hofmann and M. R. Heinrich, Tetrahedron Lett., 2016, 57, 4334–4340 CrossRef CAS.
- A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed.
- G. Coleman, Org. Synth. Coll., 1941, 1, 432–435 Search PubMed.
-
(a) D. G. Holland, G. J. Moore and C. Tamborski, J. Org. Chem., 1964, 29, 1562–1565 CrossRef CAS;
(b) M. Schlosser, T. Rausis and C. Bobbio, Org. Lett., 2005, 7, 127–129 CrossRef CAS PubMed;
(c) D.-D. Liang and M.-X. Wang, Org. Chem. Front., 2017, 4, 1425–1429 RSC.
-
(a) D. V. Kurandina, E. V. Eliseenkov, P. V. Ilyin and V. P. Boyarskiy, Tetrahedron, 2014, 70, 4043–4048 CrossRef CAS;
(b) S. V. Kumar and D. Ma, Chin. J. Chem., 2018 DOI:10.1002/cjoc.201800326.
-
(a) E. Vessally, K. Didehban, R. Mohammadi, A. Hosseinian and M. Babazadeh, J. Sulfur Chem., 2018, 39, 332–349 CrossRef CAS;
(b) E. Vessally, R. Mohammadi, A. Hosseinian, K. Didehban and L. Edjlali, J. Sulfur Chem., 2018, 39, 443–463 CrossRef CAS;
(c) A. Hosseinian, L. Zare Fekri, A. Monfared, E. Vessally and M. Nikpassand, J. Sulfur Chem., 2018 DOI:10.1080/17415993.2018.1471142;
(d) K. Didehban, E. Vessally, A. Hosseinian, L. Edjlali and E. S. Khosroshahi, RSC Adv., 2018, 8, 291–301 RSC;
(e) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(f) K. Nejati, S. Ahmadi, M. Nikpassand, P. D. K. Nezhad and E. Vessally, RSC Adv., 2018, 8, 19125–19143 RSC;
(g) A. Hosseinian, F. A. H. Nasab, S. Ahmadi, Z. Rahmani and E. Vessally, RSC Adv., 2018, 8, 26383–26398 RSC;
(h) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(i) E. Vessally, A. Hosseinian, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, J. Iran. Chem. Soc., 2017, 14, 2339–2353 CrossRef CAS;
(j) E. Vessally, M. Babazadeh, A. Hosseinian, S. Arshadi and L. Edjlali, J. CO2 Util., 2017, 21, 491–502 CrossRef CAS;
(k) E. Vessally, K. Didehban, M. Babazadeh, A. Hosseinian and L. Edjlali, J. CO2 Util., 2017, 21, 480–490 CrossRef CAS;
(l) M. Babazadeh, S. Soleimani-Amiri, E. Vessally, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 43716–43736 RSC;
(m) S. Farshbaf, L. Zare Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(n) E. Vessally, R. Mohammadi, A. Hosseinian, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 24, 361–368 CrossRef CAS;
(o) A. Hosseinian, S. Farshbaf, R. Mohammadi, A. Monfared and E. Vessally, RSC Adv., 2018, 8, 17976–17988 RSC.
-
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS;
(b) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC.
- Z. Peng, G. Hu, H. Qiao, P. Xu, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, 2733–2738 CrossRef CAS PubMed.
- J.-B. Liu, H.-P. Zhou and Y.-Y. Peng, Tetrahedron Lett., 2014, 55, 2872–2875 CrossRef CAS.
- G. Wang, M. Meng, L. Deng, K. Cheng and C. Qi, Appl. Organomet. Chem, 2018, 32, e4203 CrossRef.
- H. Zhang, C. Wang, Z. Li and Z. Wang, Tetrahedron Lett., 2015, 56, 5371–5376 CrossRef CAS.
- F. Christoffel and T. R. Ward, Catal. Lett., 2018, 148, 489–511 CrossRef CAS.
- M.-K. Zhu, J.-F. Zhao and T.-P. Loh, Org. Lett., 2011, 13, 6308–6311 CrossRef CAS PubMed.
- Y. Bai, L. M. H. Kim, H. Liao and X.-W. Liu, J. Org. Chem., 2013, 78, 8821–8825 CrossRef CAS PubMed.
- J.-B. Liu, F.-J. Chen, N. Liu and J. Hu, RSC Adv., 2015, 5, 45843–45846 RSC.
- P. Patil, A. Nimonkar and K. G. Akamanchi, J. Org. Chem., 2014, 79, 2331–2336 CrossRef CAS PubMed.
-
(a) H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed;
(b) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- Y. Zhao and Q. Song, Chem. Commun., 2015, 51, 13272–13274 RSC.
- A. S. Demir, Ö. Reis and E. Özgül-Karaaslan, J. Chem. Soc., Perkin Trans. 1, 2001, 3042–3045 RSC.
- A. S. Demir and H. Findik, Tetrahedron, 2008, 64, 6196–6201 CrossRef CAS.
- H. Jasch, J. Scheumann and M. R. Heinrich, J. Org. Chem., 2012, 77, 10699–10706 CrossRef CAS PubMed.
- J. Hofmann, H. Jasch and M. R. Heinrich, J. Org. Chem., 2014, 79, 2314–2320 CrossRef CAS PubMed.
- T. Jiang, S.-Y. Chen, H. Zhuang, R.-S. Zeng and J.-P. Zou, Tetrahedron Lett., 2014, 55, 4549–4552 CrossRef CAS.
- T. Jiang, S.-Y. Chen, G.-Y. Zhang, R.-S. Zeng and J.-P. Zou, Org. Biomol. Chem., 2014, 12, 6922–6926 RSC.
- M. Li and Y. Ye, ChemCatChem, 2015, 7, 4137–4142 CrossRef CAS.
- T. Taniguchi, M. Imoto, M. Takeda, T. Nakai, M. Mihara, T. Mizuno, A. Nomoto and A. Ogawa, Synthesis, 2017, 49, 1623–1631 CrossRef CAS.
- A. S. Demir, Ö. Reis and M. Emrullahoğlu, Tetrahedron, 2002, 58, 8055–8058 CrossRef CAS.
- P. Chauhan, M. Ravi, S. Singh, P. Prajapati and P. P. Yadav, RSC Adv., 2016, 6, 109–118 RSC.
- J.-W. Yuan, W.-J. Li, L.-R. Yang, P. Mao and Y.-M. Xiao, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 1115–1123 CAS.
- S. Paul and A. K. Bhattacharya, Org. Biomol. Chem., 2018, 16, 444–451 RSC.
- Y. Chen, S. Guo, K. Li, J. Qu, H. Yuan, Q. Hua and B. Chen, Adv. Synth. Catal., 2013, 355, 711–715 CrossRef CAS.
- Y. Li, W. Liu and C. Kuang, Chem. Commun., 2014, 50, 7124–7127 RSC.
- T. Taniguchi, M. Imoto, M. Takeda, F. Matsumoto, T. Nakai, M. Mihara, T. Mizuno, A. Nomoto and A. Ogawa, Tetrahedron, 2016, 72, 4132–4140 CrossRef CAS.
- M. Ravi, P. Chauhan, R. Kant, S. K. Shukla and P. P. Yadav, J. Org. Chem., 2015, 80, 5369–5376 CrossRef CAS PubMed.
- J.-W. Yuan, W.-J. Li and Y.-M. Xiao, Tetrahedron, 2017, 73, 179–186 CrossRef CAS.
- S. Paul, J. H. Ha, G. E. Park and Y. R. Lee, Adv. Synth. Catal., 2017, 359, 1515–1521 CrossRef CAS.
- E. Kocaoğlu, M. A. Karaman, H. Tokgöz and O. Talaz, ACS Omega, 2017, 2, 5000–5004 CrossRef.
- S. Jana, S. Samanta, A. K. Bagdi, V. Z. Shirinian and A. Hajra, RSC Adv., 2018, 8, 12360–12367 RSC.
- R. R. Jadhav, S. N. Huddar and K. G. Akamanchi, Eur. J. Org. Chem., 2013, 6779–6783 CrossRef CAS.
- W.-B. Sun, P.-Z. Zhang, T. Jiang, C.-K. Li, L.-T. An, A. Shoberu and J.-P. Zou, Tetrahedron, 2016, 72, 6477–6483 CrossRef CAS.
- W. Xu, G. Hu, P. Xu, Y. Gao, Y. Yin and Y. Zhao, Adv. Synth. Catal., 2014, 356, 2948–2954 CrossRef CAS.
- T. Taniguchi, A. Murata, M. Takeda, T. Mizuno, A. Nomoto and A. Ogawa, Eur. J. Org. Chem., 2017, 4928–4934 CrossRef CAS.
- T. Taniguchi, T. Naka, M. Imoto, M. Takeda, T. Nakai, M. Mihara, T. Mizuno, A. Nomoto and A. Ogawa, J. Org. Chem., 2017, 82, 6647–6655 CrossRef CAS PubMed.
- C. Wang, Z. Zhang, Y. Tu, Y. Li, J. Wu and J. Zhao, J. Org. Chem., 2018, 83, 2389–2394 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Robab Mohammadi
b,
Sheida Ahmadi
b,
Aazam Monfared
b and
Zahra Rahmani
c
a,
Robab Mohammadi
b,
Sheida Ahmadi
b,
Aazam Monfared
b and
Zahra Rahmani
c