
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombinatorial experimental and DFT theoretical investigation over the formation mechanism of a binuclear phthalocyanine dimer†
Chiming Wanga,
Xin Chena,
Qingyun Liu b,
Dongdong Qi*a,
Kang Wang*a and
Jianzhuang Jiang*a
b,
Dongdong Qi*a,
Kang Wang*a and
Jianzhuang Jiang*a
aBeijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry, University of Science and Technology Beijing, Beijing 100083, China. E-mail: jianzhuang@ustb.edu.cn
bCollege of Chemical and Environmental Engineering, Shandong University of Science and Technology, Qingdao 266510, Shandong, China
First published on 16th November 2017
Abstract
Herein, a density functional calculation method was used to explore the formation mechanism of a metal-free, homobinuclear phthalocyanine dimer. The result not only well rationalizes the Pc dimer formation starting from 1,2-diamino-9,10,16,17,23,24-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrin (1-OR) through an intricate pathway but, more importantly, also predicts the formation of ammonia during the reaction process; this results in the successful experimental detection of NH4+ in the acidic reaction system. Further experimental efforts have facilitated the isolation of one of the theoretically revealed reaction intermediates di-[2-amino-7,8,14,15,21,22-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrinato]-amine (9-OR); this indicates the significance of combining experimental with DFT calculation method towards clarifying the reaction mechanisms.
Introduction
In the past few decades, quantum chemistry, particularly in combination with density functional theory (DFT), has become a remarkable tool for chemists towards understanding the reaction mechanism or designing new type of reactions.1 First, quantum chemistry enables to elaborately study the modeling elementary reactions on the basis of reactivity and selectivity.2 In addition, overall reaction pathways can be elucidated by computational chemistry.3 Unambiguously, a combination of computational investigation and experimental studies can lead to a better understanding of mechanistic details (Schemes 1–3).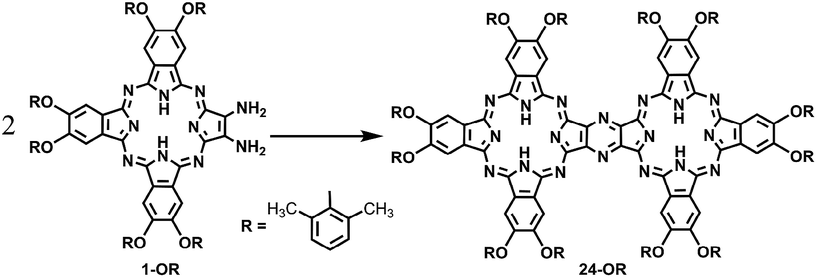 | ||
Scheme 1 Schematic showing the synthesis of the metal-free homobinuclear phthalocyanine 24-OR in CH2Cl2 at room temperature in the mixed solvent of CH2Cl2 and TFA (in the ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20), with 1-OR as the starting reagent. :20), with 1-OR as the starting reagent. | ||
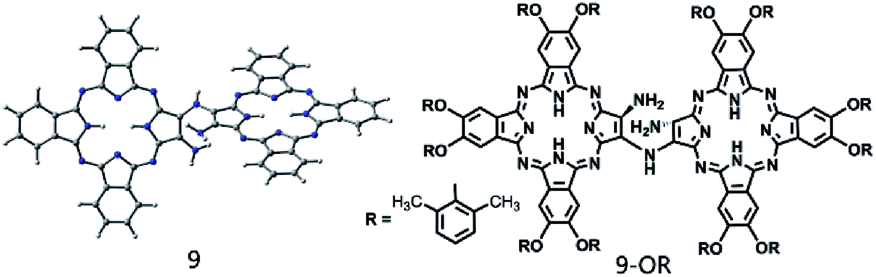 | ||
| Scheme 2 Schematic of the molecular structures of the relatively stable di-tribenzotetrazaporphyrinato-amine intermediates 9 and (9-OR). | ||
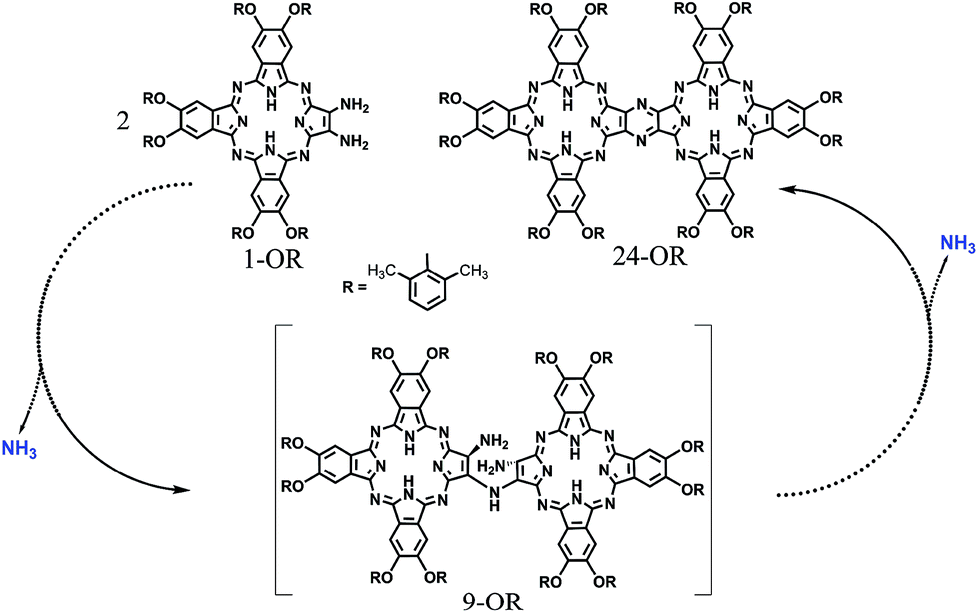 | ||
| Scheme 3 Schematic of the reaction pathway of the formation of 24-OR with 1-OR as the starting reagent though 9-OR together with the formation of ammonia. | ||
On the other hand, phthalocyanines (Pcs) have found a wide range of novel applications as advanced materials in addition to their traditional industrial applications in the past few decades.4 For the purpose of developing new Pc-based molecular materials, phthalocyanine oligomers, in particular, phthalocyanine dimers with conjugated electronic structures, have received extensive attention.5 Despite the great efforts being paid in this direction, the synthesis, especially the isolation of conjugated phthalocyanine dimers, still remains a significant challenge due to the statistical mixed product obtained from the random cyclotetramerization of 1,2,4,5-tetracyanobenzene with a second phthalonitrile, as shown in Scheme S1 (ESI†).6 However, recently, a novel post-cyclotetramerization strategy towards the efficient synthesis of binuclear Pcs in an impressively high yield under very mild reaction conditions has been developed by Jiang et al.7 For further extending the potential of this newly developed method to the synthesis of novel conjugated Pc oligomers, insight into the reaction mechanism of the new binuclear Pcs will surely be helpful.
In the present study, a DFT method was employed to investigate the mechanistic details of the Pc dimer formation via a post-cyclotetramerization reaction with 1,2-diamino-9,10,16,17,23,24-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrin (1-OR) as the starting reagent. A detailed reaction pathway and energy profile were obtained from the calculation results. In particular, the calculations predicted the formation of ammonia during the reaction process, which corresponded to the experimental detection of NH4+ in the acidic reaction system. Moreover, di-[2-amino-7,8,14,15,21,22-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrinato]-amine (9-OR), one of the reaction intermediates, as revealed through the computations, was successfully isolated and spectroscopically characterized. The present result clearly reveals the significance of integrating theory and experiment in understanding the reaction mechanisms.
Results and discussion
To gain insight into the reaction mechanism for the formation of the phthalocyanine dimer 24, DFT calculations were carried out at the level of M06-2X-D3/6-31+G(d,p)/6-311+G(2d,2p) in the SMD solvent mode8–11 using Gaussian 09 D.01 (ref. 12) for more computational details and the validity of M06-2X for organic mechanistic studies, please see the ESI.† All aryloxy substituents were replaced with hydrogen atoms to simplify the model and reduce the computational costs. As a result, the compounds in the energy profile were named based on their experimental counterparts, but without – OR. According to the calculations, in the absence of TFA, the self-condensation reaction is actually initiated by hydrogen transfer (H-transfer) between two starting reagent molecules (1) via the transition state TS2 with a moderate energy barrier of 20.48 kcal mol−1. The proton transfer nature of this elementary reaction is clarified by the NPA charge calculation, Fig. S1 (ESI†). After the formation of the charge-separated species, the unsaturated N atom in the 4-anion will nucleophilically attack the C-atom bonded – to the –NH3 substituent group in the 3-cation to form a σ(C–N) bond between two molecules with an energy barrier of 39.86 kcal mol−1. However, this energy barrier is too high to overcome under the reaction conditions. Interestingly, as shown by the black pathway in Fig. 1, an acid can enormously promote the reaction: a typical SN2 reaction can easily proceed to form the dimer intermediate 9 together with formation of ammonia, with an overall energy barrier of 26.77 kcal mol−1, which is in good agreement with the fact that the phthalocyanine dimer 24-OR can only be produced in the presence of TFA.7 Moreover, TFA can facilitate the reaction as a hydrogen-transfer (H-transfer) shuttle (vide infra). Thus, the key role of TFA catalyst in the formation of dimer 24-OR is unambiguously revealed.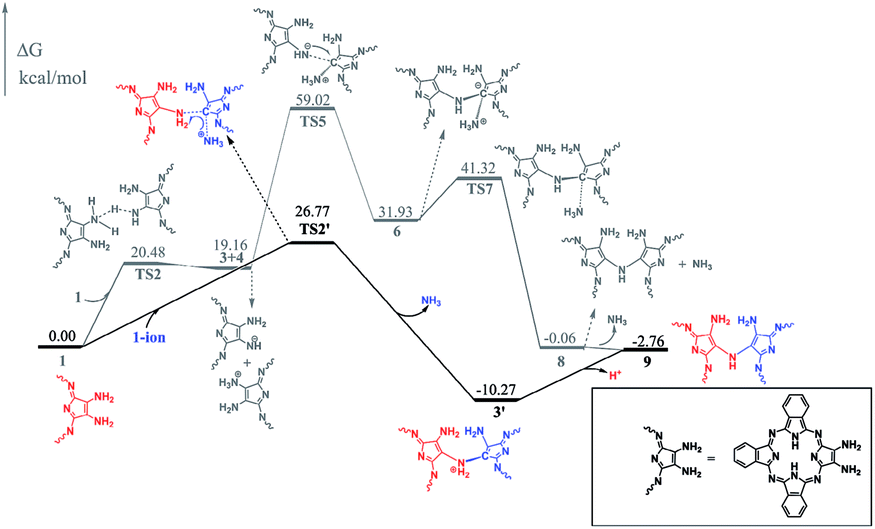 | ||
| Fig. 1 Energy profile for the dimerization of 1 promoted by TFA, along with the relative energies. | ||
Along with the detachment of the ammonia molecule from the dimer intermediate 9, dehydrogenation must proceed via an aerobic dehydrogenation reaction since no additional oxidizing agent is added to the reaction system. Fig. 2 illustrates the details for the reaction between 9 and O2, leading to the intermediate 17. Notably, some of the O2-involved triplet states of the intermediates are more stable due to the paramagnetic nature of oxygen. The triplet energies of all the intermediates formed between the two minimum energy crossing points were calculated, which are shown as the blue and black bars in Fig. 2. Additionally, two minimum energy crossing points (MECP) were also calculated using the program developed by the Harvey's group, as shown in Fig. 2 and S2 (ESI†).13,14 In the first elementary reaction in this process, the σ(C–O) bond forms via the transition state TS11t with an energy barrier of 24.21 kcal mol−1. After the oxygen molecule bonds with the intermediate 9, two dehydrogenation reactions proceed in turn. The intermediate 14 is slightly higher in energy than the transition state TS13s due to the thermodynamic correction factor. The second dehydrogenation reaction can only proceed by the assistance of TFA, which acts as a H-transfer shuttle and yields the intermediate 17 together with the dehydrogenation product, actually hydrogen peroxide. However, hydrogen peroxide can easily react with the intermediates due to its high reactivity and finally leads to the production of water molecules, as shown in Fig. S3 (ESI†).
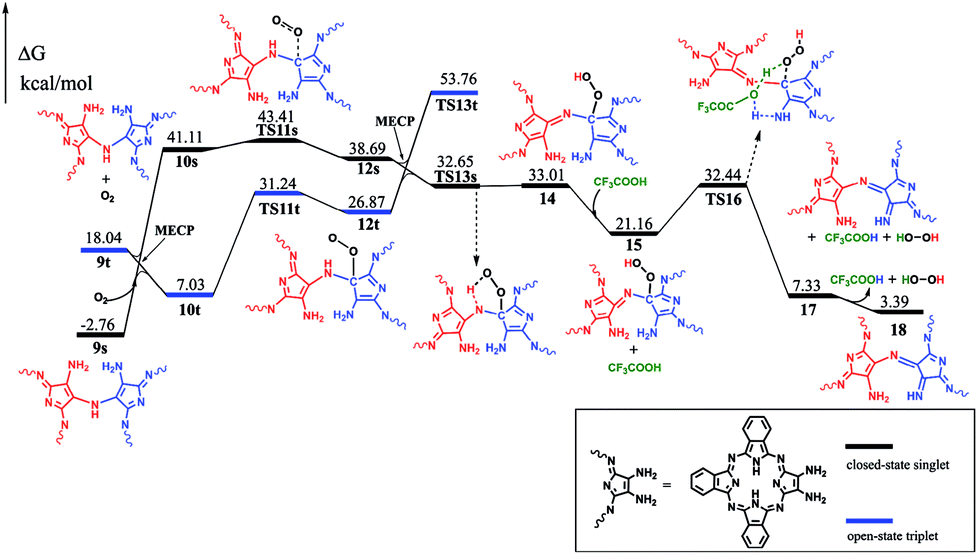 | ||
| Fig. 2 Energy profile for the dehydrogenation of intermediate 9, along with the relative energies. | ||
After dehydrogenation, the intermediate 17 can easily form an intramolecular σ(C–N) bond with an energy barrier of 21.28 kcal mol−1 and reaction exergonicity of 8.54 kcal mol−1. However, the further direct H-transfer reaction can only proceed with a high energy barrier of 38.16 kcal mol−1, which is surely thermodynamically unfavorable. On the other hand, the formation of the final product can be facilitated by TFA, which actually also acts as a H-transfer shuttle. As shown in Fig. 3, the concerted H-transfer reaction assisted by TFA, along with the formation of the target dimeric Pc 24 as well as ammonia, with a slight energy barrier of 11.00 kcal mol−1, is undoubtedly favorable under the experimental condition. Actually, the effect of the shuttle can be easily understood: TFA can transfer the two hydrogen atoms linearly; thus, the elementary reaction via the transition state TS22 is favorable in energy. In contrast, the tension of the four-membered ring in the pericyclic reaction in TS21′ results in a very large energy barrier.
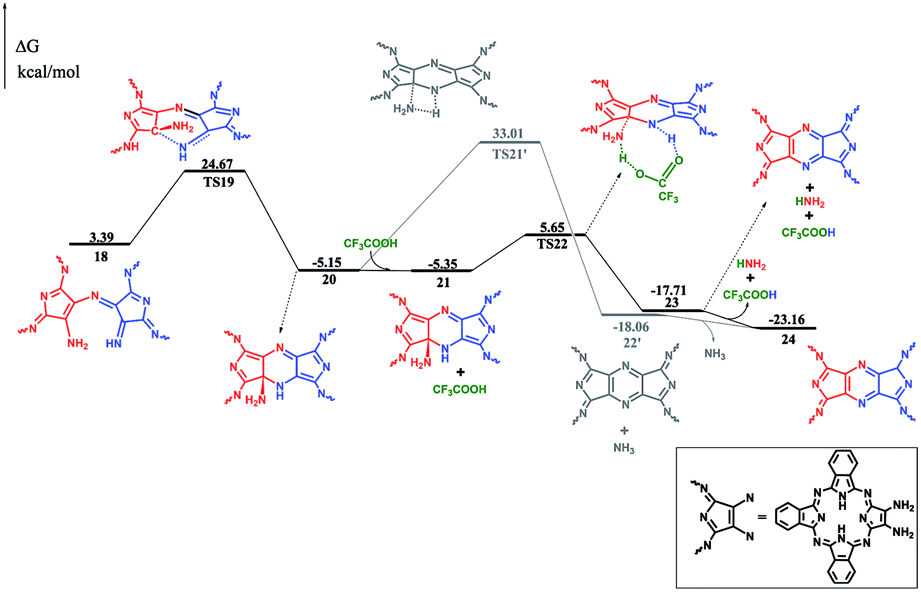 | ||
| Fig. 3 Energy profile for the formation of the final product 24, along with the relative energies. | ||
Reviewing the whole reaction pathway, the crucial catalytic role of TFA for the self-condensation reaction is unambiguously disclosed by the DFT calculation. At the early stage, it is necessary to activate the initial reagent by acid. However, it is worth noting that a very strong acid, such as sulfuric acid or hydrochloric acid, in particular, in excess amount would protonate all the initial reagents; this then prevents the reaction from further proceeding since two protonated reagents cannot react with each other due to electronic repulsion. On the other hand, a weak acid, such as acetic acid or propanoic acid (even in excess amount) is not able to protonate a sufficient amount of the initial reagents to initiate the reaction. Both results are fully in line with the experimental findings.7 As a result, only TFA with suitable acidic intensity and in an appropriate quantity could lead to excellent synthesis result for the binuclear Pcs from 1,2-diamino-9,10,16,17,23,24-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrin (1-OR). Moreover, the two elementary reactions are facilitated by TFA as an H-transfer shuttle. As can be seen in Fig. S4 (ESI†), the structure of TFA perfectly fits the hydrogen transfer orientation and thus can significantly decrease the energy barrier. Overall, TFA with appropriate acidity and geometric configuration and in a suitable quantity can act as a good catalyst for this reaction.
To validate the abovementioned reaction mechanism, effort is paid towards the detection of ammonia formed in the reaction system. The reaction mixture was extracted using water. When treated with the Nessler's reagent, the separated aqueous solution turns from light yellow to orange15 [see Fig. S5 (ESI†)]; this confirms the existence of NH4+ in this acidic reaction system and indicates the production of NH3 during the formation of 24-OR in the self-condensation of 1-OR, corresponding well with the calculation prediction.
However, since DFT calculations also predicted a series of reaction intermediates with relatively good stabilities (as shown by their relatively low Gibbs energies), trials have been carried out to isolate them during the self-condensation reaction of 1-OR in CH2Cl2 and TFA in the ratio of 80:20 by monitoring the progress using thin layer chromatography (TLC). TLC monitoring indicated that after the reaction progresses for only 0.5 h, the dimeric phthalocyanine compound 24-OR starts to appear. In addition, the monomeric phthalocyanine precursor 1-OR completely disappears after 3 h reaction. However, a new species with green color and larger polarity than the dimeric compound 24-OR starts to appear 1 h after the reaction starts, which completely disappears after the reaction proceeds for 4 h; this suggests the possible reaction intermediate nature of this newly observed species during the formation procedure of 24-OR. Fortunately, this compound is isolated from the reaction system after 1.5 h reaction by column chromatography in 13% yield in addition to the unreacted monomeric phthalocyanine precursor 1-OR and target phthalocyanine dimer 24-OR. MALDI-TOF in combination with the NMR spectroscopic result, as shown in Fig. S6 and S7 (ESI†), reveals the dimeric phthalocyanine nature of this compound, which is actually the monoamine-substituted binuclear phthalocyanine intermediate species 9-OR, the counterpart of the intermediate 9 shown in Fig. 2. Further support for the intermediate nature of this species originates from the transformation of 9-OR into the target homo-binuclear phthalocyanine dimer 24-OR in CH2Cl2 and TFA (80:20) at room temperature in a yield of 82% after 5 h reaction. This result provides solid experimental validation for the formation mechanism of Pc-dimer, revealed on the basis of DFT calculation.
At the end of this section, it is worth noting that the successful isolation of 9-OR can also be well rationalized on the basis of the DFT calculations. As shown in Fig. 2, the reaction between the intermediate 9 and the oxygen molecule is an endergonic process, indicating its thermodynamically unfavorable nature. In addition, the intermediate 9 is a thermodynamically stable species (with the reaction exergonicity of 0.87 kcal mol−1 relative to that of the reactant 1). Overall, the intermediate 9 would accumulate and therefore is able to be isolated during the reaction process.
Conclusions
In conclusion, the mechanistic details of the formation of a metal-free homo-binuclear phthalocynine dimer by the self-condensation of 1,2-diamino-9,10,16,17,23,24-hexa-(2,6-dimethylphenoxy)-tribenzotetrazaporphyrin have been clarified via a combination of experiments and density functional calculations. The appropriate acidity and geometric configuration of TFA play an important role in activating the precursor and acting as an H-transfer shuttle to promote the reaction. The production of ammonia was initially predicted by the theoretical study and then confirmed experimentally. Furthermore, one of the theoretically determined reaction intermediates was successfully isolated during the reaction process; this demonstrated the validity of the theoretical calculation. The present result clearly demonstrates the significance of combining theoretical calculations with experiments.Experimental detail
Synthesis and isolation of the intermediate 9-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :20
:20
Compound 1-OR (37.7 mg, 0.0310 mmol) was dissolved in 3.0 mL of the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20 and stirred at room temperature for 1.5 h. The reaction mixture was poured into water (10 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic solution was then washed with water (3 × 5 mL) and dried with Na2SO4/K2CO3. After evaporating the solvent, the residue was chromatographed on a silica gel column with CH2Cl2 as an eluent to obtain a green band containing the intermediate 9-OR, and then, another green band containing compound 24-OR was obtained. Repeated chromatography followed by recrystallization from CH2Cl2 and CH3OH provided the intermediate 9-OR as a green powder in a yield of 13% (along with 24-OR in a yield of 36%). 1H NMR (400 MHz, CDCl3) for 9-OR = 9.32 (s, 1H), 8.40 (s, 1H), 8.34 (m, 5H), 8.08 (br, 5H), 7.37 (br, 30H), 7.00–7.06 (m, 6H), 2.25–2.58 (m, 72H), −0.16 ppm (m, 4H); MS (MALDI-TOF): Calcd. For C152H129N19O12 [M]+ 2413.0; found m/z 2413.1.
Reaction of the intermediate 9-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20
By employing the abovementioned synthesis procedure of compound 24-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20 with 9-OR instead of compound 1-OR as the starting material with the reaction time of 5 h, the compound 24-OR was isolated in the yield of 82%.
Detection of NH4+ during the formation of 24-OR from the self-condensation reaction of 1-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20
Compound 1-OR (62.8 mg, 0.0516 mmol) was dissolved in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20 (5.0 mL) and stirred at room temperature for 5 h. Then, the reaction mixture was poured in water (10 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic solution was then washed with water (3 × 10 mL). The aqueous phases were combined, and the pH value of the resulting solution adjusted to 7.0 by adding 0.1 M KOH solution. Then, 0.1 mL of the aqueous solution was added to 2 mL Nessler's reagent, and the solution immediately changed from light yellow to orange.
Detection of NH4+ during the formation of 24-OR from the intermediate 9-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20
The abovementioned detection procedure of NH4+ during the formation of 24-OR from the self-condensation reaction of 1-OR in the mixed solvent of CH2Cl2 and TFA in the ratio of 80:20 with the intermediate 9-OR instead of compound 1-OR was employed, and the Nessler's reagent changed from light yellow to orange.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support received from the National Key Basic Research Program of China (Grant No. 2013CB933402), Natural Science Foundation of China (No. 21290174 and 21401009), Beijing Municipal Commission of Education, and University of Science and Technology Beijing is gratefully acknowledged.Notes and references
- (a) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532–9586 CrossRef CAS PubMed; (b) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed; (c) R. G. Parr, Annu. Rev. Phys. Chem., 1983, 34, 631–656 CrossRef CAS; (d) E. R. Davidson, Chem. Rev., 2000, 100, 351–352 CrossRef CAS PubMed; (e) T. B. Demissie, K. Ruud and J. H. Hansen, Organometallics, 2015, 34, 4218–4228 CrossRef CAS.
- (a) Y. Lam, M. Grayson, M. Holland, A. Simon and K. N. Houk, Acc. Chem. Res., 2016, 49, 750–762 CrossRef CAS PubMed; (b) M. Grayson, J. Org. Chem., 2017, 82, 4396–4401 CrossRef CAS PubMed; (c) Y. Wang, M. Tang, Y. Wang and D. Wei, J. Org. Chem., 2016, 81, 5370–5380 CrossRef CAS PubMed; (d) G. Jindal, H. K. Kisan and R. B. Sunoj, ACS Catal., 2015, 5, 480–503 CrossRef CAS.
- (a) Y. Dang, S. Qu, J. W. Nelson, H. Pham, Z. Wang and X. Wang, J. Am. Chem. Soc., 2015, 137, 2006–2014 CrossRef CAS PubMed; (b) M. Grayson, J. Org. Chem., 2017, 82, 4396–4401 CrossRef CAS PubMed; (c) D. A. Sharon, D. Mallick, B. Wang and S. Shaik, J. Am. Chem. Soc., 2016, 138, 9597–9610 CrossRef CAS PubMed; (d) S. Qu, Y. Dang, C. Song, M. Wen, K. Huang and Z. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed; (e) C. Wang, X. Chen, D. Qi, S. Bi and J. Jiang, Inorg. Chem. Commun., 2017, 85, 9–15 CrossRef CAS.
- (a) K. M. Kadish, K. M. Smith and R. Guilard, in Handbook of Porphyrin Science, World Scientific, Singapore, 2010, vol. 1–35 Search PubMed; (b) K. M. Kadish, K. M. Smith and R. Guilard, in The Porphyrin handbook, Academic Press, San Diego, CA, 2000 and 2003, vol. 1–20 Search PubMed; (c) D. M. P. Mingos and J. Jiang, Functional Phthalocyanine Molecular Materials, in Structure and Bonding, Springer-Verlag, Heidelberg, Germany, 2010. Vol. 135 Search PubMed; (d) N. B. McKeown, in Phthalocyanines Materials: Synthesis Structure and Function, Cambridge University Press, New York, 1998 Search PubMed; (e) A. B. P. Lever and C. C. Leznoff, in Phthalocyanine: Properties and Applications, VCH, Weinheim, 1989–1996, vol. 1–4 Search PubMed; (f) Y. Bian and J. Jiang, Recent Advances in Phthalocyanine-Based Functional Molecule Materials, in Structure and Bonding, Springer-Verlag, Heidelberg, Germany, 2016, vol. 172, pp. 159–199 Search PubMed; (g) J. Jiang, K. Kasuga and D. P. Arnold, in Supramolecular Photosensitive and Electroactive Materials, Academic Press, New York, 2001, pp. 113–210 Search PubMed; (h) H. Wang, B. Wang, Y. Bian, S. Gao and J. Jiang, Coord. Chem. Rev., 2016, 306, 195–216 CrossRef CAS; (i) J. Jiang, M. Bao, L. Rintoul and D. P. Arnold, Coord. Chem. Rev., 2006, 250, 424–448 CrossRef CAS; (j) J. Jiang and D. K. P. Ng, Acc. Chem. Res., 2009, 42, 79–88 CrossRef CAS PubMed.
- (a) N. Kobayashi, H. Lam, W. A. Nevin, P. Janda, C. C. Leznoff, T. Koyama, A. Monden and H. Shirai, J. Am. Chem. Soc., 1994, 116, 879–890 CrossRef CAS; (b) E. M. Maya, P. Vázquez and T. Torres, Chem. Commun., 1997, 1175 RSC; (c) S. G. Makarov, A. V. Piskunov, O. N. Suvorova, G. Schnurpfeil, G. A. Domrachev and D. Wöhrle, Chem.–Eur. J., 2007, 13, 3227 CrossRef CAS PubMed; (d) K. Wang, D. Qi, H. Wang, W. Cao, W. Li, T. Liu, C. Duan and J. Jiang, Chem.–Eur. J., 2013, 19, 11162–11166 CrossRef CAS PubMed; (e) C. Huang, K. Wang, J. Sun and J. Jiang, Dyes Pigm., 2014, 109, 163–168 CrossRef CAS; (f) Y. Shi and X. Li, Org. Electron., 2014, 15, 286–293 CrossRef CAS.
- (a) S. Makarov, C. Litwinski, E. A. Ermilov, O. Suvorova, B. Rçder and D. Wöhrle, Chem.–Eur. J., 2006, 12, 1468–1474 CrossRef CAS PubMed; (b) Y. Asano, J. Sato, T. Furuyama and N. Kobayashi, Chem. Commun., 2012, 4365–4367 RSC; (c) J. Yang and M. Mark, Tetrahedron Lett., 1993, 34, 5223–5226 CrossRef CAS; (d) M. Handa, N. Kataoka, Y. Ito, T. Tonomura, I. Hiromitsu, T. Sugimori, K. Sogabe and K. Kasuga, Bull. Chem. Soc. Jpn., 2004, 77, 1647–1648 CrossRef CAS.
- K. Wang, C. Huang, H. Pan, N. Kobayashi and J. Jiang, Inorg. Chem. Front., 2017, 4, 110–113 RSC.
- Y. Zhao and D. Truhlar, Acc. Chem. Res., 2008, 14, 157–167 CrossRef PubMed.
- (a) T. Clark, J. Chandrasekhar, G. Spitznagel and P. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS; (b) M. Frisch, J. Pople and J. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- (a) S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed; (b) S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (d) S. Grimme, S. Ehrlich and L. Georigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Frisch, et al., Gaussian, Inc., Wallingford CT, 2009.
- J. N. Harvey, M. Aschi, H. Schwarz and W. Koch, Theor. Chem. Acc., 1998, 99, 95 CrossRef CAS.
- Y. Dang, X. Deng, J. Guo, C. Song, W. Hu and Z. Wang, J. Am. Chem. Soc., 2016, 138, 2712–2723 CrossRef CAS PubMed.
- A. Vanselow, Ind. Eng. Chem., Anal. Ed., 1940, 12, 516–517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational detail, spectroscopic characterization, Cartesian coordinates for the optimized structures. See DOI: 10.1039/c7ra10678e |
| This journal is © The Royal Society of Chemistry 2017 |