
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater splitting with polyoxometalate-treated photoanodes: enhancing performance through sensitizer design†
John
Fielden‡
*ab,
Jordan M.
Sumliner
a,
Nannan
Han
a,
Yurii V.
Geletii
a,
Xu
Xiang
ac,
Djamaladdin G.
Musaev
*a,
Tianquan
Lian
*a and
Craig L.
Hill
*a
aDepartment of Chemistry, Cherry L. Emerson Center for Scientific Computation, Emory University, Atlanta, GA 30322, USA. E-mail: chill@emory.edu
bWestCHEM, School of Chemistry, University of Glasgow, G12 8QQ, UK
cState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 1000029, P. R. China
First published on 11th June 2015
Abstract
Visible light driven water oxidation has been demonstrated at near-neutral pH using photoanodes based on nanoporous films of TiO2, polyoxometalate (POM) water oxidation catalyst [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− (1), and both known photosensitizer [Ru(bpy)2(H4dpbpy)]2+ (P2) and the novel crown ether functionalized dye [Ru(5-crownphen)2(H2dpbpy)](H22). Both triads, containing catalyst 1, and catalyst-free dyads, produce O2 with high faradaic efficiencies (80 to 94%), but presence of catalyst enhances quantum yield by up to 190% (maximum 0.39%). New sensitizer H22 absorbs light more strongly than P2, and increases O2 quantum yields by up to 270%. TiO2-2 based photoelectrodes are also more stable to desorption of active species than TiO2–P2: losses of catalyst 1 are halved when pH > TiO2 point-of-zero charge (pzc), and losses of sensitizer reduced below the pzc (no catalyst is lost when pH < pzc). For the triads, quantum yields of O2 are higher at pH 5.8 than at pH 7.2, opposing the trend observed for 1 under homogeneous conditions. This is ascribed to lower stability of the dye oxidized states at higher pH, and less efficient electron transfer to TiO2, and is also consistent with the 4th1-to-dye electron transfer limiting performance rather than catalyst TOFmax. Transient absorption reveals that TiO2–2–1 has similar 1st electron transfer dynamics to TiO2–P2–1, with rapid (ps timescale) formation of long-lived TiO2(e−)–2–1(h+) charge separated states, and demonstrates that metallation of the crown ether groups (Na+/Mg2+) has little or no effect on electron transfer from 1 to 2. The most widely relevant findings of this study are therefore: (i) increased dye extinction coefficients and binding stability significantly improve performance in dye-sensitized water splitting systems; (ii) binding of POMs to electrode surfaces can be stabilized through use of recognition groups; (iii) the optimal homogeneous and TiO2-bound operating pHs of a catalyst may not be the same; and (iv) dye-sensitized TiO2 can oxidize water without a catalyst.
Introduction
Efficient water oxidation remains a key challenge in the development of systems for artificial photosynthesis. The last five years have witnessed dramatic progress in water oxidation catalyst (WOC) speed and stability,1,2 with leading catalysts now showing turnover frequencies (TOFs) comparable to that of the biological OEC.1r,2j Even so, efforts to develop complete devices for efficient solar fuel production are still in their infancy.3 Any such system is likely to be centred on a water-oxidizing photoelectrode that serves as a man-made analogue of nature's light-driven water splitting agent, photosystem II.Prior to development of the dye-sensitized solar cell (DSSC), dye sensitized TiO2 (ds-TiO2) was pioneered as a material for light-driven water oxidation.4 Many ruthenium-based dyes have, in their Ru3+ oxidation state, sufficient potential to oxidize water and thanks to two decades of research into DSSCs, efficient, mesoporous TiO2 photoelectrodes are easily accessible.5,6 Consequently, incorporation of WOCs into ds-TiO2 has become an important approach to water-oxidizing photoanodes.7 Although many of these devices are hampered by inefficient electron transfer (ET),7a,d or insufficient catalyst speed,7i single-wavelength quantum efficiencies as high as 14% were recently reported with a fast ruthenium polypyridyl-based catalyst.7j Successful engineering of practical devices will require not only fast stable WOCs, efficient stable sensitizers, and rapid, directional ET from the WOC to the oxidized sensitizer, but also sensitizer and catalyst binding that resists aqueous buffers and electrolytes. Even so, there has been little work addressing the influence of sensitizers on performance: Meyer et al. have developed strategies to mitigate dye desorption,7k,8 and covalent linkages have been engineered between Ru tris-bipyridyls and water oxidation catalysts,7a,d,g,h,l but to our knowledge there are no systematic studies into the effect of light absorption and catalyst binding properties.
We, and other groups, are investigating the incorporation of polyoxometalate (POM) based WOCs into dye-sensitized TiO2,7i,9 and their deposition onto other electrode surfaces.10 These oxidatively stable, molecular all-inorganic WOCs can achieve efficient, fast homogeneous light driven water oxidation using [Ru(bpy)3]2+ as photosensitizer, but turnover numbers (TONs) are limited by degradation of dye.2,11 As oxidative degradation of Ru-polypyridyls occurs primarily through attack of hydroxide and hydroxyl radicals on the oxidized, Ru3+ state,12 placing the dye and catalyst on a surface (e.g. TiO2) can be expected to mitigate degradation. Close association on the surface should speed ET from catalyst-to-dye, reducing time spent by the dye in the vulnerable Ru3+ state, and also reduce exposure of the dye to nucleophilic species. Recently, we found that vs. solution, ET from the catalyst [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− (1) to the oxidized state of the [Ru(bpy)2(H4dpbpy)]2+ (P2) dye is dramatically accelerated on TiO2 and SnO2, and that long-lived charge separated states are generated.7i,9b Furthermore, enhancement of visible-light photocurrents by 1 suggested water oxidation. However, this was not confirmed, and rapid losses of active species occurred under turnover conditions – an increasingly recognized problem in ds-TiO2 based water splitting.7k,8 POM-based catalysts present a particular challenge, as catalytically useful, transition metal substituted POMs such as 1 cannot be derivatized through the strategies used for other POM anions,13 and their terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are both poor hydrogen bond acceptors and very weak ligands. Consequently, binding predominantly depends on size and surface charge.
O groups are both poor hydrogen bond acceptors and very weak ligands. Consequently, binding predominantly depends on size and surface charge.
In this study, our focus moves from ET dynamics, to quantifying photoelectrochemical water oxidation by 1 at ds-TiO2 photoanodes based both on P2, and new, crown ether decorated dye [Ru(5-crownphen)2(H2dpbpy)](H22, Fig. 1) under visible light. While both systems show high (80–90%) faradaic efficiency, H22 increases maximum quantum yields almost 4×, while also dramatically reducing the loss of 1 when pH > TiO2 pzc. This provides the first clear demonstration that major performance gains can be achieved through sensitizer design, that non-covalent recognition groups can stabilize binding of POMs at surfaces, and also facilitates an in-depth understanding that may guide further studies of POM-based photoelectrodes. In addition, we quantify water oxidation by catalyst-free ds-TiO2 for the first time, finding a high faradaic efficiency, and low but appreciable quantum yield for this under-recognized phenomenon.
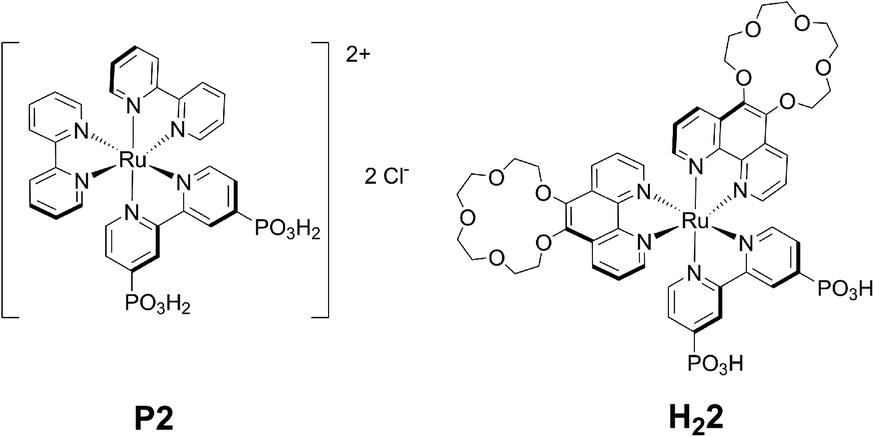 | ||
| Fig. 1 Molecular structures of P2, isolated as its chloride salt, and 2, obtained in its doubly deprotonated, neutral form H22. | ||
Results and discussion
Sensitizer design, synthesis and structure
The design of H22 was inspired by previous reports of 5-crown phenanthroline derivatized Ru-polypyridyl sensitizers and rhenium carbonyls.14,15 The Ru-based systems14 bind alkali and alkaline earth metals, but retain very similar photophysical and redox properties to underivatized [Ru(bpy)3]2+, [Ru(bpy)2(phen)]2+ and [Ru(bpy)(phen)2]2+, while [(5-crown-phen)Re(CO)3Cl] has shown an ability to associate with [PW12O40]3− in solid state and solution.15 Therefore, 5-crown-phen appeared an obvious way to increase the strength of sensitizer–catalyst interactions, while retaining similar photophysical and redox properties to P2. In addition, phenanthroline ligands increase extinction coefficients compared to bipyridines.165-crown-phen is easily accessed from phenanthroline, using established methods.14b Subsequently, H22 was synthesized in moderate yields starting from this ligand and Ru(DMSO)4Cl2, using methods described in the ESI (Scheme S1†). H22's molecular structure and purity has been confirmed by 1H-NMR, ESI+-MS, elemental analysis and X-ray crystallography. A thermal ellipsoid plot of its crystal structure (space group Pnna) is displayed in Fig. 2 (excluding crystallographically located water and acetone); selected bond lengths and angles can be found in Table S1 (ESI†). The coordination geometry of the pseudo-octahedral ruthenium atom is very similar to that observed in [Ru(phen)2(bpy)]2+,16 with distortion resulting from the small bite angles (ca. 80°) of the ligands. Notably, the presence of –PO3H groups, crown ethers and water molecules leads to a 3D hydrogen bonded network based on chains of Et22 molecules linked via –PO3H (Fig. S1, ESI†).
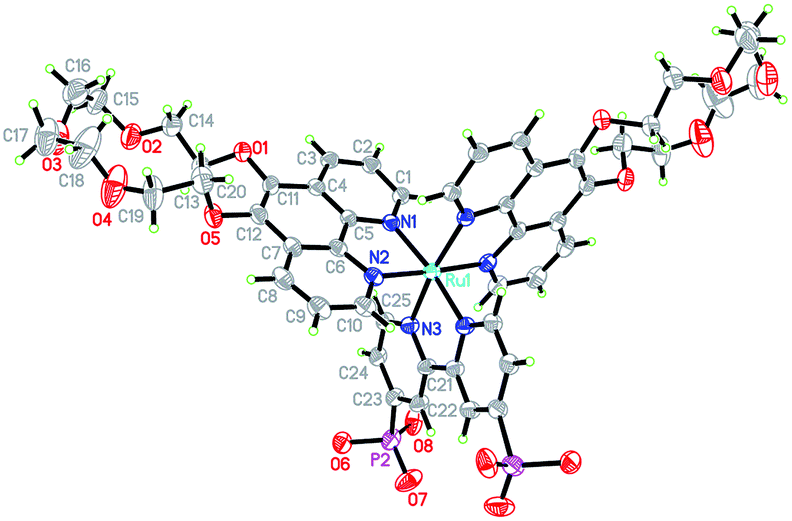 | ||
| Fig. 2 Representation of the molecular structure of H22 (30% probability ellipsoids). Symmetry equivalent atoms are unlabelled. C atoms are grey; N, blue; O, red; P, magenta; Ru, light blue; H, green circles of arbitrary radii. | ||
Electronic spectroscopy, cyclic voltammetry and DFT calculations: the effect of cations on 2
The new sensitizer H22 has a similar UV-vis spectrum to P2 in water (Fig. 3), but with higher extinction coefficients for both ligand centred and MLCT transitions. This is consistent with typical observations for phenanthroline,16vs. bipyridine complexes and results from the larger π-conjugated system. The aqueous electrochemistry of the two sensitizers is also very similar, with H22 showing a pseudo-reversible Ru2+/Ru3+ couple at +1.35 V vs. Ag/AgCl. Ligand reductions, however are obscured by the onset of proton reduction in aqueous media.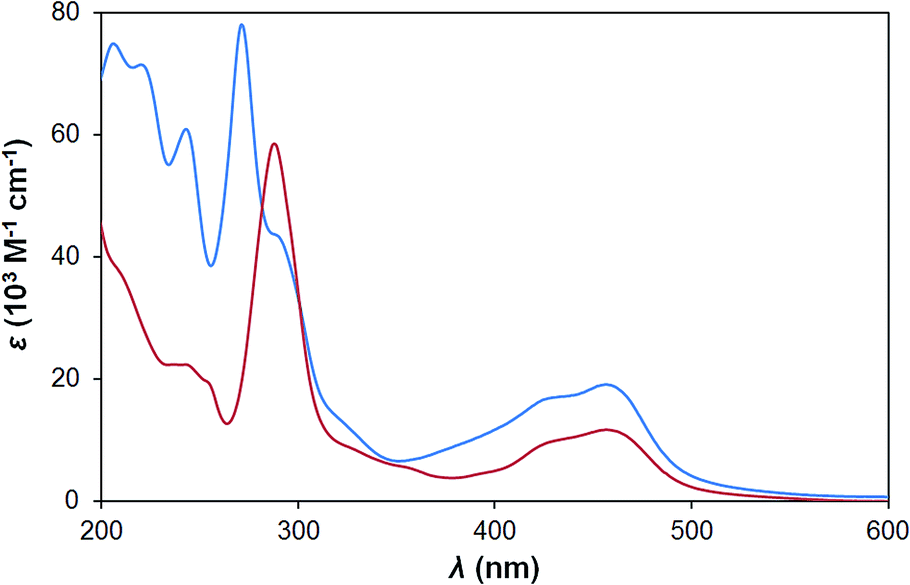 | ||
| Fig. 3 UV-vis spectra of H22 (blue) and P2 (red) in H2O at 298 K. | ||
Alkali and alkaline earth metal cations bind strongly to mono-5-crown-phen and 5-crown-dppz ruthenium complexes in MeCN, but have only small effects on the spectral and electrochemical properties.14 While H22 is insoluble in organic media, study of the esterified precursor [Et42][PF6]2 has produced results (Table 1 and Fig. S2†) consistent with this literature. Cation binding has only a minimal influence on the photophysical properties of the bis-5-crown-phen system [Et42]2+, with almost no change seen in the MLCT absorption, and a 1 nm red shift in 3MLCT emission, upon addition of 500 equivalents of Na+. A more significant change (ca. 5% fall) is observed in the intensity of the 272 nm ligand centred absorption band. Addition of the same concentration of non-binding NBu4PF6 had no effect on 3MLCT emission and led to a 23% increase in the intensity of the 272 nm band, indicating that Na+ binding and not a just simple increase in ionic strength is responsible for the change. Greater changes are seen when Mg2+ is added: a slight broadening and flattening of the MLCT absorption, a 7 nm red shift in 3MLCT emission, and a 14% increase in the intensity of the 272 nm band. Even so, for both cations, these results imply that the energy of the MLCT excited state barely changes upon metallation.
| Compound/solvent | Salt added | λ abs/nm (ε/10−4 M−1 cm−1) | λ em (nm) |
|---|---|---|---|
| H22/H2O | No salt | 206 (74.9), 220 (71.4), 243 (60.9), 271 (78.0), 457 (19.1) | 594 |
| [Et42][PF6]2/MeCN | No salt | 207 (75.5), 245 (71.2), 272 (79.9), 380 (13.7), 449 (16.8) | 635 |
| 0.01 M NaClO4 | 207 (75.5), 245 (70.2), 272 (76.3), 381 (13.7), 449 (16.9) | 636 | |
| 0.01 M Mg(ClO4)2 | 206 (84.0), 246 (75.5), 272 (91.7), 379 (14.5), 449 (16.4) | 642 | |
| 0.01 M NBu4PF6 | 207 (77.3), 246 (78.2), 272 (98.5), 380 (13.8), 449 (16.9) | 635 |
Cyclic voltammetry (Table 2 and Fig. S3†) echoes the electronic spectra, by showing no change in the Ru2+/3+ electrode potential upon addition of 75 equivalents of Na+ or Mg2+, but some effects on the ligand-based reductions. In the presence of Na+, there are positive shifts of 20, 70 and 70 mV in E1/2 for the 1st, 2nd and 3rd reductions. Addition of Mg2+ produces a 70 mV positive shift in the 1st reduction, while the 2nd and 3rd waves merge into an irreversible process with an EPC midway between the 2nd and 3rd reduction of cation free Et42.
| Compound/electrolyte | Salt added | L/L−˙, E1/2/EPC, (ΔEp, mV) | Ru2+/3+, E1/2, V (ΔEp, mV) |
|---|---|---|---|
| Et42/0.1 M NBu4PF6 in MeCN | No salt | −0.96 (105), −1.39 (81), −1.58 (95) | 1.46 (83) |
| 0.05 M NaClO4 | −0.94 (113), −1.32 (93), −1.51 (105) | 1.46 (95) | |
| 0.05 M Mg(ClO4)2 | −0.891 (130), −1.501 | 1.46 (116) | |
| H22/0.1 M HClO4 in MeCN/H2O | No salt | Obscured by proton reduction | 1.35 (82) |
| P2/0.1 M HClO4 in MeCN/H2O | No salt | Obscured by proton reduction | 1.34 (80) |
Electronic structure calculations (DFT, B3LYP/{Lanl2dz(Ru) + 6-31G(d) (P, N, C, O, H)}) conducted on H422+ also indicate that changes upon addition of Na+ and Mg2+ are small (Table 3). In both cases, the cation lowers the energy of the 5-crown-phen based LUMO+1 level (by ca. 0.14 eV), while raising the energy of the dpbpy based LUMO (ca. 0.06 eV for Na+, 0.115 eV for Mg2+). However, the lowest energy LUMO orbitals remain on the dpbpy ligand, favouring electron injection to TiO2. In summary, electronic spectroscopy, cyclic voltammetry and DFT calculations show that Et42/H42 and their Na+/Mg2+ complexes have similar electronic structures to P2 in solution. This indicates a high degree of electronic isolation between the Ru-centre and metalla-crown, so that ground and excited state potentials of the 2 manifold relevant to electron injection into TiO2, and oxidation of catalyst 1, change only minimally upon metallation. The new sensitizer can therefore be expected to show similar behaviour to P2 on the TiO2 surface.
| Molecule | ΔES–Ta | ΔE[HOMO−Ru − LUMO+x]b | |
|---|---|---|---|
| LUMO (dpbpy) | LUMO (crown/bpy) | ||
| a Separation between ground state singlet and triplet excited state. b Separation between ruthenium based HOMO and LUMO of relevant ligand. | |||
| [Ru(bpy)2(dpbpy)]2+ (P2) | 2.000 | 3.324 | 3.712 |
| [Ru(5-crown-phen)2(dpbpy)]2+ (H42) | 2.051 | 3.367 | 3.731 |
| [Ru(Na-5-crown-phen)2(dpbpy)]4+ (H42–Na2) | 2.111 | 3.503 | 3.663 |
| [Ru(Mg-5-crown-phen)2(dpbpy)]4+ (H42–Mg2) | 2.190 | 3.510 | 3.616 |
Film assembly and characterization
TiO2 films were sensitized with P2 and 2 (H22 is assumed to deprotonate on the TiO2 surface) in acidic conditions,7 and where required TiO2–2 was metallated by soaking in solutions of NaClO4 or MgClO4. For photoelectrochemical (PEC) measurements, the TiO2-dye films were treated with toluene solutions of hydrophobic THpA8.5H1.5[1] salt, as deposition of THpA+ (tetraheptylammonium) at the electrode surface helps stabilize binding of 1 in aqueous buffers. For ultrafast spectroscopic studies, 1 was deposited from its tetrabutylammonium, TBA7H3[1], as this gave better transparency without introducing undesired metal cations.In our previous work,7i loadings of 1 were seen to depend upon the point-of-zero charge (pzc), and hence the surface positive charge, of the metal oxides TiO2, SnO2 and ZrO2. Charge introduced the dye appeared to have less influence. This is confirmed in our current study. Loadings of catalyst used for PEC experiments, estimated by UV-vis (ESI, Fig. S4/Table S2†), are typically in the range of 10 to 14 nmol cm−2 with no clear dependence on the loading or metallation (charge) of dye (which varies between TiO2 batches). Ratios of dye-to-catalyst are lower for 2 simply because loadings of 2 are generally lower, due to its greater size. Although assembly conditions (e.g. soaking time) were not strictly controlled, the results indicate that the charge of the dye is not a key factor in determining the loading of 1. Films assembled under controlled conditions for desorption experiments confirm that the dye has at most a marginal influence (vide infra). For ultrafast photophysical measurements, we found that much higher catalyst loadings could be achieved with extended soaking times and higher concentration soaking solutions (Table S3†).
Transient absorption spectroscopy (TAS)
Electron injection from 2 into TiO2, with and without 1, has been studied by time-resolved mid-IR transient absorption (pump 515 nm, probe 5000 nm). Measurements on TiO2–2, by comparison with a TiO2–N3 reference, indicate that near 100% of photons absorbed by 2 result in electron injection to the metal oxide (Fig. 4a).17h However, like other phosphonate binding dyes, 2's injection kinetics are substantially slower than those of carboxylate based N3 – injection from excited 2 occurs over ca. 100 ps, instead of completing within 2 ps as for N3. This is considered a consequence of weak electronic communication across the tetrahedral phosphorus center,17i giving similar biphasic injection kinetics to P2,7i with an ultrafast component due to injection from the 1MLCT excited state of the dye and a slower component arising from the 3MLCT. In the presence of 1, electron injection to TiO2 still occurs in high yield from the 1MLCT state of 2 (Fig. 4b), but the slower 3MLCT component is significantly suppressed. This indicates that ET from excited 2 to TiO2 is still the main quenching pathway when 1 is present, but as with P2, presence of the POM appears to introduce a competing deactivation mechanism for the 3MLCT.
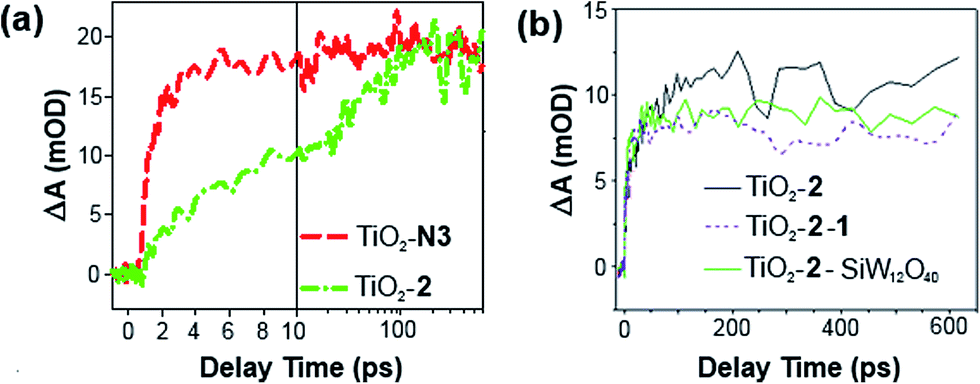 | ||
| Fig. 4 Mid-IR comparison of the kinetics of electron injection in (a) TiO2–2 and TiO2–N3, and; (b) TiO2–2–1 and TiO2–2–SiW12O40. Data have been scaled to the absorbance of the supporting dyad film. | ||
Treatment of a TiO2–2 photoelectrode with the colourless, less charged POM [SiW12O40]4− (SiW12) confirms that the competing quenching pathway is electron transfer to 1, rather than energy transfer or an effect of its high (10−) negative charge on the TiO2 conduction band edge potential. This is because SiW12 produces a similar decrease in injection to TiO2 from the 3MLCT excited state (Fig. 4b), but SiW12 has no absorption bands overlapping with emission from 2, precluding energy transfer and suggesting there must be electron transfer to this species. Interestingly, this occurs even though the W-based LUMO of SiW12 is significantly higher in energy than the first accessible Ru-based reduction of 1. Similar quenching of the 3MLCT excited state of Ru(bpy)32+ by electron transfer to 1 has been observed in solution.19
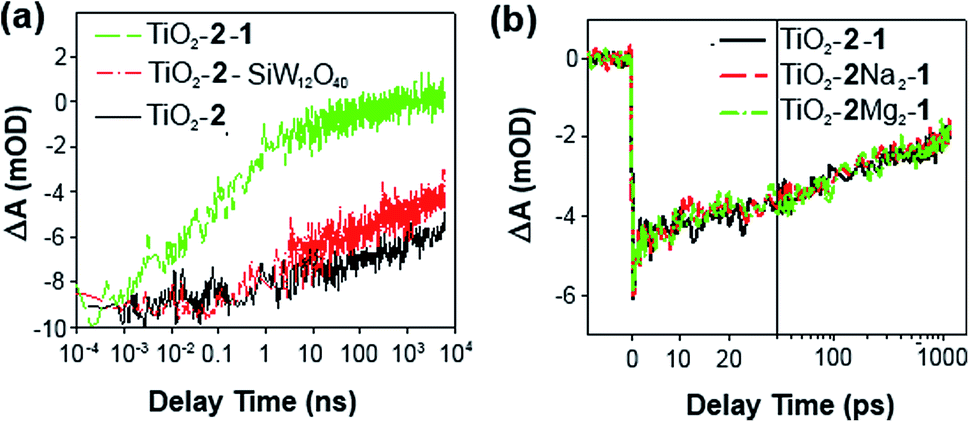 | ||
| Fig. 5 (a) Ground state bleach recovery kinetics of TiO2–2 (black), TiO2–2–SiW12O40 (red) and TiO2–2–1. (b) Ground state bleach recovery kinetics of TiO2–2–1 (black), TiO2–2–Na2/1 (red) and TiO2–2–Mg2–1 (green). | ||
The positive charge on 2 can be increased by metallation, by 2+ by adding Na+, and by 4+ with Mg2+. Strong electronic interactions are observed between ion paired Ru(bpy)32+ and POMs in solution,20 so we speculated that increased charge in 2–Na2 and 2–Mg2 might strengthen such interactions and enhance ET from 1 to oxidized 2. However, measurements on TiO2–2–1, TiO2–2–Na2–1 and TiO2–2–Mg2–1 at a controlled dye-to-catalyst ratio show that this is not the case (Fig. 5b), as the kinetics of bleach recovery for the three systems are superimposable. This implies that once catalyst and dye are closely associated on the film, increasing charge on the dye does not significantly affect donor–acceptor coupling between the two species. It is also consistent with the observation that the crown and Ru-centre are electronically isolated from one another (vide supra), so that strengthened interaction between 5-crown-phen and 1 does not feed through to the Ru-centre. However, metallation, combined with increased soaking times and catalyst concentration has helped access triads with very high loadings of 1 (Table S3 and Fig. S5a, ESI†), confirming that bleach recovery is faster when more electron donor 1 is present.9a This effect saturates once there are around 1.5 catalyst molecules (1) per sensitizer (2).
Lastly, we studied the effect of pH. Compound 1 has the widest pH range of any known POM WOC (active even at pH 1),2b but is typically faster at pH >6.2c Rinsing the films in different pH solutions before measurement (Fig. S5b†), shows that at pH 1.1 bleach recovery is significantly slower than at pH 2.3 or 3.1. This suggests that increased protonation of 1 disfavours ET. However, the minimal change between pH 3.1 and 6.5 implies that either the TiO2 film buffers the system, or factors other than the first ET are responsible for the increased speed of 1 at higher pH. This is consistent with our established mechanism for water oxidation by 1.2c,d
In summary, TAS shows that the electron transfer dynamics of TiO2–2–1 are similar to TiO2–P2–1, and that metallation of 2 does not affect 1-to-2 ET, but pH does. It also confirms that high loadings of 1 speed the recovery of the oxidized sensitizer, and that the main inefficiency is initial ET to 1 instead of TiO2, which reduces the injection yield from the dye 3MLCT state.
Photoelectrochemistry and oxygen evolution
Photocurrent transients for TiO2–P2 and TiO2–2 films are shown in Fig. 6. Upon illumination, both films (at pH 5.8 and 7.2) show an initial spike in generator current, resulting from electron injection to TiO2. An instantaneous response occurs at the collector, before generator and collector currents decline towards a steady state. Photocurrent density and ηF are higher at pH 7.2 (Table 4), where water oxidation is more thermodynamically favourable, and TiO2–2 films show far greater photocurrents than TiO2–P2 (by 290% at pH 5.8, 190% at pH 7.2). Detector currents increase almost in line with photocurrents, so ηF remains high for 2, and similar increases are seen in water oxidation internal quantum efficiency (IQE).
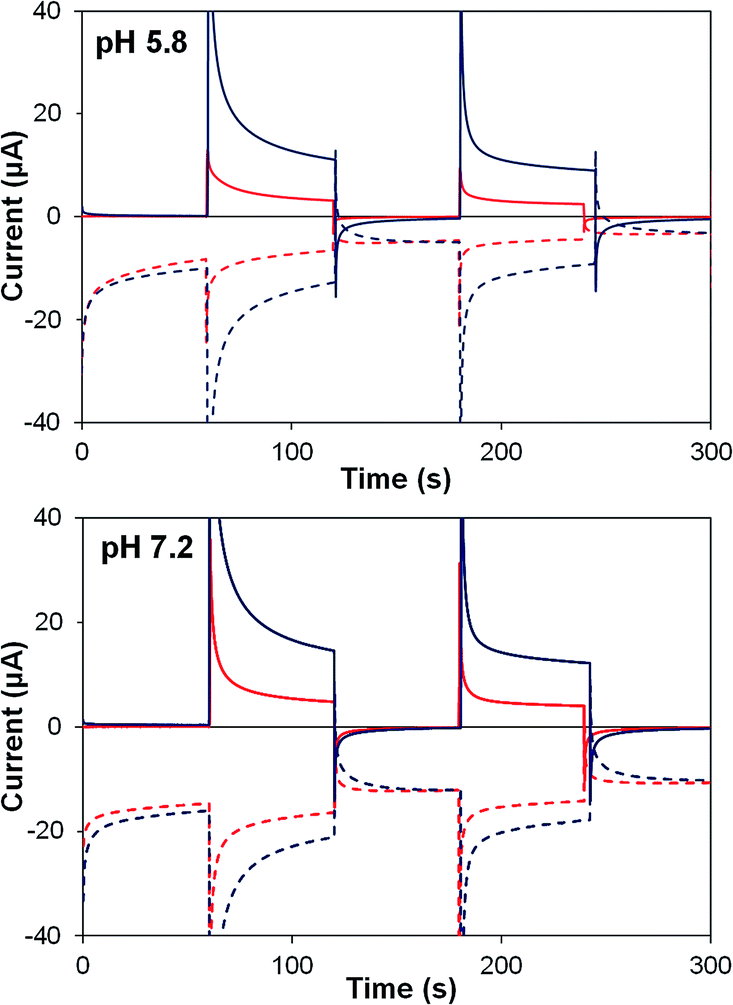 | ||
| Fig. 6 Photoelectrochemical generator–collector measurements (chronoamperometry) of TiO2–P2 (red) and TiO2–2 (dark blue) at pH 5.8 and pH 7.2. Solid lines are the photoanode current; dashed lines, collector current; collector efficiency ca. 60%. Applied bias 0 mV vs. Ag/AgCl, illuminated with a 455 nm LED (33 mW cm−2). Photocurrent densities are reported in Table 4. | ||
| Photoanode | pH 5.8 Na2SiF6/NaHCO3a | pH 7.2 lutidine/HClO4a | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Photo-currentb, μA | Photocurrent density, μA cm−2 | η F , % | IQEd, % | IQE gain with 1e | IQE gain for 2f | Photo-currentb, μA | Photocurrent density, μA cm−2 | ηFc % | IQEd, % | IQE gain with 1e | IQE gain for 2f | |
| a NaClO4 added so [Na+] = 200 mM. b At end of first 60 s transient, average of at least two films. c Faradaic efficiency (ηF), average of 5 to 8 transients (≥2 films). d Internal quantum efficiency (IQE) for water oxidation, taking into account ηF. e IQE gain on addition of 1. f IQE gain on replacing P2 with 2. | ||||||||||||
| TiO2–P2 | 3 | 5 | 88 | 0.037 | 4.3 | 7.2 | 94 | 0.058 | ||||
| TiO2–P2–1 | 8.5 | 14.2 | 90 | 0.106 | 186% | 8.3 | 13.8 | 81 | 0.094 | 62% | ||
| TiO2–2 | 11.8 | 19.7 | 80 | 0.134 | 262% | 12.6 | 21 | 88 | 0.157 | 170% | ||
| TiO2–2Na2–1 | 32.9 | 54.8 | 86 | 0.392 | 193% | 270% | 20.5 | 34.2 | 80 | 0.228 | 45% | 143% |
Despite the earlier literature, we were surprised that catalyst-free ds-TiO2 oxidizes water. The low rate of O2 production has made GC detection impractical, but it is difficult to envisage a source of the detector current other than reduction of O2, based upon the following lines of evidence:
(1) There is no steady-state detector response if aqueous buffers are replaced by an acetonitrile electrolyte (Fig. S6†).
(2) Steady state detector responses are tiny (<0.1 μA) if Pt@FTO is replaced with plain FTO glass (Fig. S7†).
(3) Plain FTO glass responds strongly to injection of H2O2, but very weakly to air (Fig. S8†).
(4) Pt@FTO responds strongly to air (Fig. S9†).
(5) Photocurrents and faradaic efficiencies for the inorganic, and lutidine-based buffers are broadly similar.
(6) Irradiating solution-based Ru-polypyridyl dyes (Fig. S10a†) produces no detector response.
Thus, appreciable detector currents require presence of water, and do not result from Ru-dyes or organic buffer. Furthermore, the lack of response from plain FTO indicates that the detector currents do not result from H2O2 or other easily reduced species. Therefore, the photoelectrodes must be producing O2, by a mechanism beyond the scope of this study – the weak (0.3 μA) response from bare TiO2 photoanodes (Fig. S10b†) confirms that direct bandgap excitation cannot be primarily responsible. Catalysis by small Ru impurities cannot be excluded, however low levels of O2 production are consistently observed from catalyst-free [Ru(bpy)3]2+ in homogeneous work,11 and previous failure to detect O2 from TiO2–P2 may be due to use of Nafion coatings.3c Nafion (used to segregate O2 and H2) has low permeability to O2, and O2 adheres strongly to TiO2 surfaces.22
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ru2+ ratio (increased probability of excitation for each dye molecule) must increase the potential felt by the catalyst.
:Ru2+ ratio (increased probability of excitation for each dye molecule) must increase the potential felt by the catalyst.
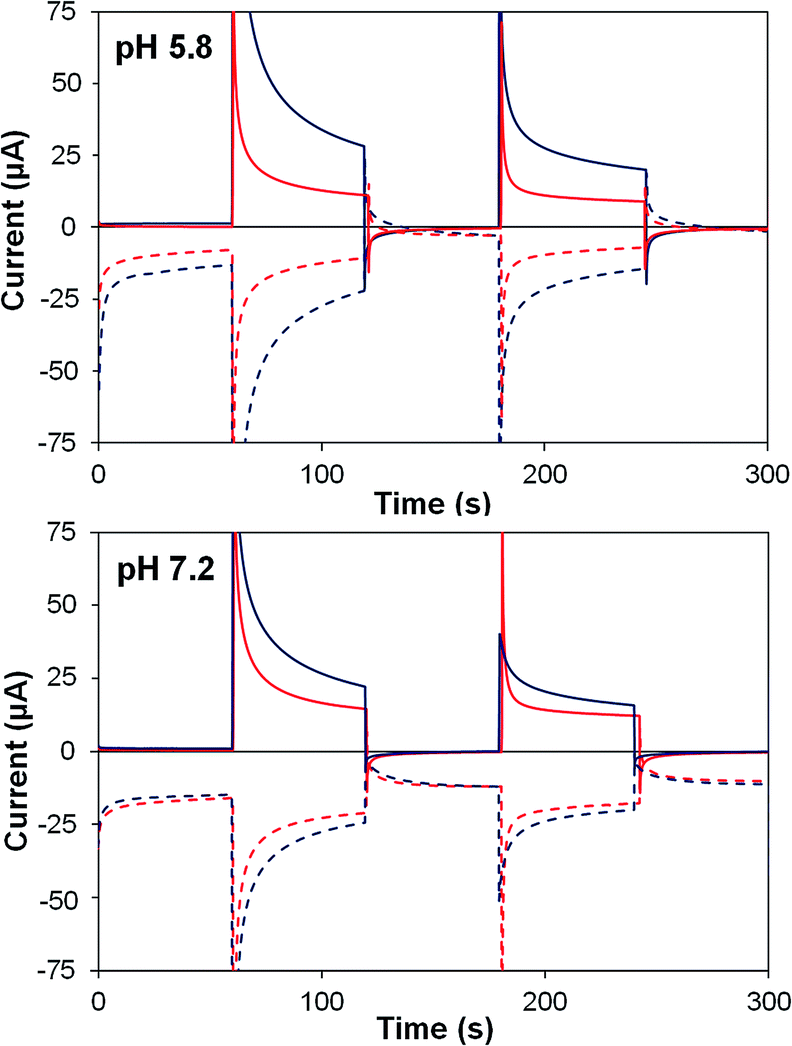 | ||
| Fig. 7 Photoelectrochemical generator–collector measurements (chronoamperometry) of TiO2–2 (red) and TiO2–2Na2–1 (dark blue) at pH 5.8 and pH 7.2. Solid lines are the photoanode current; dashed lines, collector current; collector efficiency ca. 60%. Applied bias 0 mV vs. Ag/AgCl, illuminated with a 455 nm LED (33 mW cm−2). Photocurrent densities are reported in Table 4. | ||
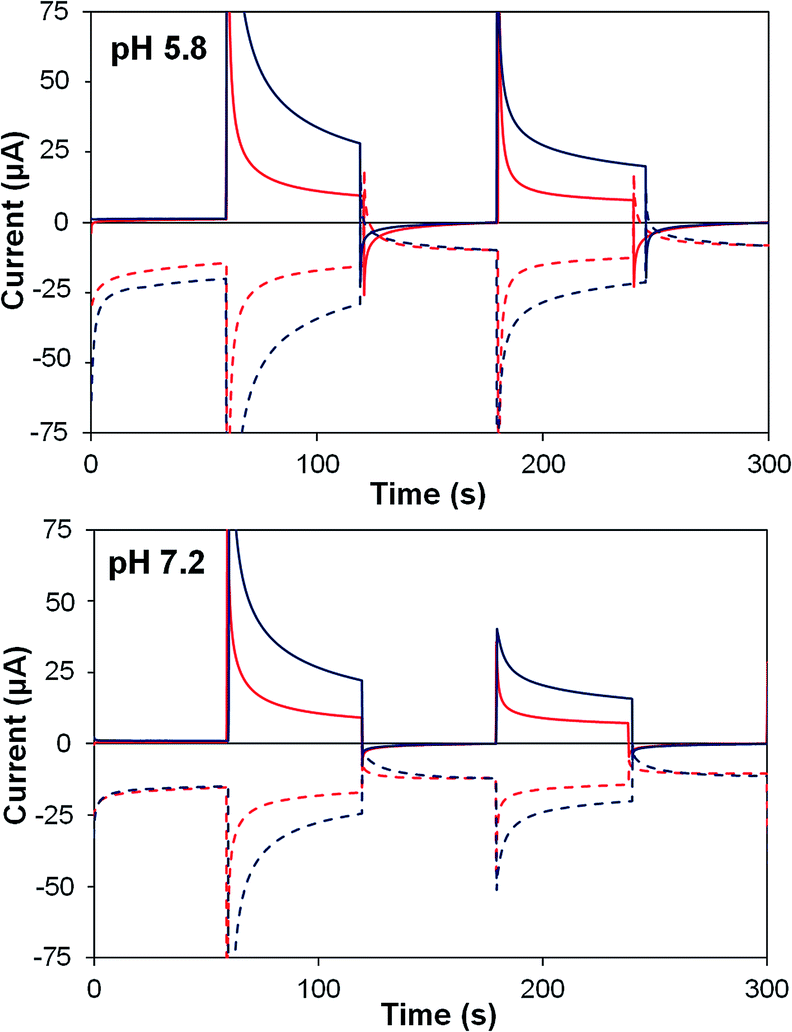 | ||
| Fig. 8 Photoelectrochemical generator–collector measurements (chronoamperometry) of TiO2–P2–1 (red) and TiO2–2Na2–1 (dark blue) at pH 5.8 and pH 7.2. Solid lines, photoanode current; dashed lines, collector current; collector efficiency ca. 60%. Applied bias 0 mV vs. Ag/AgCl, illuminated with a 455 nm LED (33 mW cm−2). Photocurrent densities are reported in Table 5. | ||
In extended experiments (Fig. S11–S13†) photocurrents fall below 1 μA (1.7 μA cm−2) within ca. 3 h for TiO2–P2–1 at pH 5.8, 4 h for TiO2–2Na2–1 at pH 5.8, and 5 h for TiO2–P2–1 at pH 7.2. For TiO2–2Na2–1 at pH 7.2, the photocurrent is still above 1.5 μA (2.5 μA cm−2) after 5 h. Stronger long term performance in the pH 7.2 buffer may be due to reduced dye desorption, however it is difficult to comment on the competing effects of stronger catalyst binding by 2, and its reduced oxidative stability vs.P2 (vide infra). TiO2–2Na2–1 at pH 5.8 passed most charge, due to its high performance early in the experiment, with 0.056 C equating to 124 nmol of O2 assuming initial ηF is maintained. This corresponds to TONs of 3 for dye 2, and 14 for catalyst 1 after 4 hours (based on a 0.6 cm2 active area). At pH 7.2, a similar photoelectrode passed 0.053 C after 5 h (110 nmol O2), but due to lower loadings, TONDye = 3 and TONCat = 22. With P2 based photoelectrodes, less than half this total charge was passed, at similar loadings of dye and catalyst. As Pt@FTO detectors degrade noticeably with use, needing cleaning after each short run,21 we cannot confirm the quantity of O2 produced. However, using a detector does show qualitatively that O2 is still being produced after 5 h (Fig. S13†).
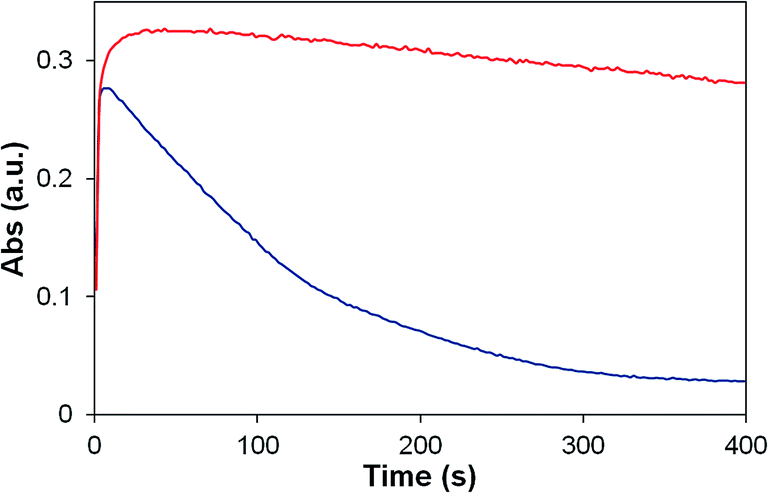 | ||
| Fig. 9 UV-vis stopped flow experiment showing the self-decomposition of oxidized P2 (red) and 2 (blue) at pH 1. Monitored at 670 nm (P2), 674 nm (2). | ||
Photocurrents and IQEs found here are lower than for some comparable systems with other catalysts.7a,d,i Brief analysis suggests the bottleneck is likely the 4th electron transfer from catalyst to dye: TAS measurements indicate a fast 1st ET, but give no information on later, less thermodynamically favourable ET events. Using 0.25 s−1 as a conservative estimate of 1's TOF (measured at ∼0.1 to 0.82 s−1),2c,14b and loadings of ∼10 nmol cm−2, steady state photocurrents of ∼5 × 10−4 A might be expected – over ten times higher than those observed. In other words, 1 is operating well below its maximum TOF. The Ru2+/3+ potentials of P2 and 2 are very close to those of [Ru(bpy)3]2+,23 and the driving force for the fourth ET from 1 to [Ru(bpy)3]2+ is small.2c,n Therefore, homogeneous systems need large [Ru(bpy)3]2+:1 ratios (200:1) and generation of high Ru3+:Ru2+ ratios to work efficiently.11a In our photoelectrodes, smaller excesses of sensitizer and a slow 4th ET prevent 1 from attaining its maximum TOF, leading to photo-bleaching and reduced light absorption – the situation is improved by 2, which due to higher ε attains higher Ru3+:Ru2+ ratios (vide supra), favouring the 4th ET. Further improvement may therefore come through (i) catalysts with lower overpotential or (ii) sensitizers with higher oxidation potentials24 and extinction coefficients.
Stability of the sensitizers and photoelectrodes
Two processes – desorption of active species and oxidative damage to the sensitizers – cause the loss of activity seen in TiO2-dye-1 photoanodes. On short timescales, desorption seems to be more important: in our previous work7i similar losses of absorption (at 455 nm) occurred over 20 minutes whether films were subjected to photoelectrochemical conditions, or left in ambient light (pH 5.8 Na2SiF6/NaHCO3). Other groups have found that illumination under unbuffered aqueous conditions promotes desorption of dye but does not cause noticeable photochemical degradation.8a Even so, a vital consideration for any sensitizer in light driven water oxidation is the stability of its oxidized (Ru3+) state towards self-decomposition under turnover conditions. Oxidized Ru-polypyridyls are vulnerable, as nucleophilic attack of species such as H2O, [OH]− and [OOH]− on the electron deficient pyridyl ring produces O-substituted species with oxidation potentials too low to continue driving water oxidation.12 Therefore, dye stability limits TONs for POM WOCs in homogeneous light-driven water oxidation.11 Below, we describe the loss of 1, P2 and 2 from the photoelectrodes at pH 5.8 and 7.2, and compare the oxidative stability of the two dyes, giving insight into the photoelectrochemical performance of the two photoanode systems.000 at pH >5, but 52000 at pH 1)2c allow their contributions to be distinguished by acidification once desorbed into the buffer (ESI, Fig. S15–S18†). In all experiments, the desorbed species found in the soaking buffer solutions, plus the pH driven change in ε for 1 remaining on the TiO2 surface, account for 70 to 95% of the observed absorbance loss of the films. The results, summarized in Table 5, demonstrate that, depending on pH, 2 mitigates loss of catalyst or dye. The evolution of absorbance loss over time (ESI, Fig, S19†) shows that in all cases losses are rapid initially, but are slowing by the end of the one hour experiment.
| Film | pH 5.8 Na2SiF6/NaHCO3/NaClO4 | pH 7.2 lutidine/HClO4/NaClO4 | ||||
|---|---|---|---|---|---|---|
| TiO2–P2–1a | TiO2–2Na2–1a | TiO2–P2–1 | TiO2–2–1 | TiO2–2Na2–1 | TiO2–2Mg2–1 | |
| a Loadings are based on flat surface areas, ignoring TiO2 porosity. b Calculated from UV-vis absorbances of triad, dyad and parent TiO2 film. c Calculated from absorbances of soaking solutions at 455 nm, at buffer pH and pH 1, and absorbances of the species on the TiO2 film. d Predicted loss of film absorbance at 455 nm, after subtracting TiO2 background, based on solution measurements. e Differences between calculated and actual film absorbance losses may be accounted for by unknowns such as the precise ε of the dyes on the TiO2 surface, other pH driven changes in film absorbance, and inhomogeneities in the films. | ||||||
| Loading of dye (nmol cm−2)a | 37 | 29 | 93 | 65 | 65 | 62 |
| Loading of 1 (nmol cm−2)a | 10 | 8 | 10 | 8 | 10 | 11 |
| Dye:1 ratiob |
37:10 |
38:10 |
92:10 |
84:10 |
63:10 |
59:10 |
| Dye lost to bufferc | 69% | 53% | 22% | 20% | 22% | 31% |
| 1 lost to bufferc | 0% | 0% | 30% | 12% | 15% | 17% |
| Calcd. Loss film Abs455d | ||||||
| From: dye loss | 39% | 41% | 17% | 17% | 17% | 24% |
| Loss of 1 | 0% | 0% | 7% | 2% | 3% | 4% |
| pH effect on ε of 1 | 29% | 21% | 11% | 10% | 12% | 12% |
| Total calcd. loss film Abs455 | 68% | 62% | 35% | 29% | 32% | 40% |
| Actual loss film Abs455e | 76% | 77% | 48% | 32% | 40% | 42% |
The films in Table 5 were assembled using constant soaking times and concentrations of 1, so it is clear that the surface properties of TiO2 are more important for catalyst loading than the charge of the dye. Metallating 2 only weakly influences the amount of catalyst bound during film assembly. This fits with previous findings that metal oxide pzc (TiO2, SnO2 or ZrO2) critically influences 1 loading in P2-based triads.7i Upon soaking at pH 5.8, neither TiO2–P2–1 nor TiO2–2–1 leaches catalyst, but significant loss occurs at pH 7.2, which is reasonable given the pzc of TiO2 (ca. 6) and the hydrophobicity of the THpA{1} salt. Importantly, these catalyst losses are cut dramatically (by 50%) for TiO2–2Na2–1vs. TiO2–P2–1, showing that the crown derivative stabilizes binding of 1 to TiO2 when pH > pzc. Interestingly, however, metallating with Mg2+ instead of Na+ destabilizes the photoelectrodes – most likely by increasing the aqueous solubility of the dye – while the result for un-metallated 2 is similar (within likely experimental errors) to that of 2Na2. This suggests that the strengthened catalyst binding of 2 is as much a result of hydrophobic surface interactions that exclude water and buffer, as electrostatics.
Surprisingly, losses of the dyes do not follow the expected pH dependence and are much more severe at pH 5.8. We surmise that carbonate from the buffer competes with phosphonate for the TiO2 surface, whilst lutidine does not. However, less 2 is lost than P2 (53% vs. 69%), presumably because it associates more strongly with the catalyst that remains on the surface. Total % losses of absorbance from the 2 and P2 photoanodes are similar though, which is explained by a lower dye contribution to the TiO2–P2–1 absorbance due to P2's lower ε, and a higher loading of 1. Thus, it appears that introducing a simple recognition group can help significantly in stabilizing the binding of polyoxometalates to ds-TiO2.
Conclusions
We have confirmed that ds-TiO2 treated with POM WOCs is able to oxidize water, at neutral pH and below. This is the first time that visible light-driven water oxidation has been demonstrated with a surface-immobilized POM species. Performance is higher at pH 5.8 than at pH 7.2, opposing the homogeneous trend: this is ascribed to dye stability, catalyst binding and electron transfer efficiency. It is also consistent with photoanode performance being limited by the 4th electron transfer rather than catalyst TOFmax, and indicates that optimum conditions for immobilized WOCs may vary from homogeneous use. We have also found that ds-TiO2 itself oxidizes water, with high faradaic efficiency but substantially lower quantum efficiency than the catalysed systems. This suggests that care should be taken to run catalyst-free controls in related work, particularly if photocurrents are low (<10 μA).This study also shows that increasing the extinction coefficient of the dye, across the range of 400 to 550 nm, is a very effective way to increase water oxidation photocurrents and quantum yields, and incorporating a simple supramolecular recognition motif (crown ether) stabilizes binding of POM WOCs at pH >TiO2 pzc. However, it also indicates that oxidative stability of the dye under water oxidation conditions is a key consideration, and modifications which can reduce the stability of the dye to nucleophilic attack are best avoided. Adequate loadings and stable binding of the POM WOC can be achieved with unmodified dyes provided the operating pH remains below the pzc of the metal oxide substrate. It therefore appears that there are three profitable future directions for this work: (i) development of fast POM WOCs that operate efficiently below pH 6, with lower overpotential; (ii) producing strongly absorbing dyes, with high binding and oxidative stability and more positive redox potentials; and (iii) testing metal oxides with high points of zero charge, such as ZnO.
Lastly, we have shown that lutidine is a good, oxidation resistant organic buffer for near-neutral conditions and better tolerated by ds-TiO2 than potentially surface-binding inorganic anions such as phosphate, borate and carbonate.
Experimental
General
Full details of materials, synthetic procedures, and instruments and methods used for primary structural characterization are available in the ESI.† UV-vis spectra were obtained using Agilent 8453 and Cary 60 spectrophotometers. Fluorescence spectra were acquired on a Perkin Elmer LS 55 fluorimeter. Electrochemical data (cyclic voltammetry) were obtained using BASi CV-50W and Autolab PGSTAT302N potentiostats in a three electrode configuration with Ag/AgCl reference, glassy carbon working and platinum counter electrodes. The electrolyte was 0.1 M HClO4 in MeCN/H2O (for H22), or 0.1 M NBu4PF6 (for [Et42][PF6]2). UV-visible stopped flow measurements were performed at 25 °C using a Hi-Tech KinetAsyst Stopped Flow SF-61SX2 equipped with a tungsten lamp and diode array detector (400–700 nm). The oxidant, (NH4)4Ce(SO4)4·2H2O was dissolved in 0.1 M H2SO4 (0.4 M in LiClO4) and the sensitizer was dissolved in 0.1 M H2SO4 alone.Film preparation and characterization
Transparent TiO2 films were prepared from colloidal suspensions by the doctor-blade technique, using Scotch tape to control area and thickness. The films were calcined at 400 °C for 90 minutes, soaked in acidic solutions of P2 or H22 (0.2 mM in 0.1 M HClO4(aq)) for 24 hours, and soaked 24 hours in 0.1 M HClO4(aq) to remove free or weakly-adsorbed dye molecules, before rinsing with water and drying in air. Metallation of TiO2–2 was achieved by soaking for 30 minutes in 0.1 M solutions of NaClO4 or Mg(ClO4)2 in acetonitrile. TiO2–2–1 and TiO2–2M2–1 triads for ultrafast spectroscopy were prepared by soaking the dye-sensitized films in acetone solutions of TBA7H3[1] (0.15 to 1 mM) for 10 to 30 minutes, rinsing with acetone and air drying. For photoelectrochemistry, triads were instead prepared by soaking TiO2–P2 and TiO2–2Na2 films in a 0.28 mM toluene solution of hydrophobic THpA8.5H1.5[1] for 5 minutes, rinsing with toluene and air drying. Dye-to-1 ratios were estimated from UV-vis spectra (Fig. S4†) by using the extinction coefficients measured for H22 (19100 M−1 cm−1 at 455 nm) and P2 (11700 M−1 cm−1) to quantify the dye, and the estimated extinction coefficient of 32000 M−1 cm−1 at 455 nm for 1 on the TiO2 surface.7i
Computational details
Geometries and energetics of P2, H42, H42–Na2 and H42–Mg2 were calculated at their lower-lying electronic states, in aqueous solution with no geometry constraints. Vibrational analyses were performed to ensure that all converged structures are true minima. In these calculations we used the spin-unrestricted DFT method (the hybrid B3LYP functional)26 in conjunction with the lanl2dz basis set with the associated Hay-Wadt ECPs27 for Ru atoms, and the 6-31G(d) basis sets for all other atoms. Solvent effects were incorporated at the polarizable continuum model (PCM).28 All calculations were carried out with the Gaussian 09 software package.29Laser photophysical measurements
For ultrafast measurements, the femtosecond transient absorption spectrometer is based on a Ti:sapphire laser (coherent Legend, 800 nm, 150 fs, 3 mJ per pulse, 1 kHz repetition rate) and Helios spectrometer (Ultrafast Systems LLC). To probe the IR, a Clark IR optical parametric amplifier was used, generating two tunable near-IR pulses in the 1.1 to 2.5 μm range (signal and idler, respectively). Nanosecond to microsecond measurements were carried out in an EOS spectrometer (Ultrafast Systems LLC), with pump pulses generated from the above laser. Full details are in the ESI.†Dye and catalyst desorption experiments
TiO2–P2–1, TiO2–2–1 and TiO2–2M2–1 films (ca. 1.4 cm2) were immersed in 3 mL of buffer solution. The films were removed after 2 seconds, and then 1, 5, 15, 30 and 60 minutes, and UV-vis spectra measured to assess the extent of desorption. UV-vis spectra of the soaking solutions were measured at the same intervals. At 60 minutes, the soaking solutions were acidified to ca. pH 1 by addition of a drop of concentrated H2SO4. As the 455 nm extinction coefficients of both dyes fall at low pH, while that of 1 dramatically increases, the change in absorbance can be used to assess relative contribution of the two species. See the ESI for further details (Fig. S15–S18†).Photoelectrochemical measurements and oxygen detection
Photoelectrochemical measurements (chronoamperometry) were obtained in a two compartment electrochemical cell, with a flat fronted working compartment for the photoanode, under a constant stream of Ar gas. The potentiostat was an Autolab PGSTAT302N with bipotentiostat module, in four electrode configuration (WE1, WE2, reference and counter), and the light source a 455 nm LED (Osram) at 20 mW power output focused onto ca. 0.6 cm2 with a fused silica lens. We used the generator/collector methods of Mallouk et al.,21 with a platinized FTO detector film held ca. 1 mm from the working surface of the back-side illuminated photoanode, the sides of the assembly were sealed with wax and the bottom left open for ingress of buffer. The photoanode (generator) was set to an applied bias of 0 V vs. Ag/AgCl, and the detector to −0.5 V (pH 5.8) or −0.584 V (pH 7.2), and the photoanode exposed to the light for several 60 s transients. At pH 5.8 we used 38 mM Na2SiF6, adjusted to pH 5.8 using NaHCO3 (final concentration ca. 80 mM), with NaClO4 added for a total Na+ concentration of 200 mM. At pH 7.2, we used 50 mM lutidine, adjusted to pH 7.2 with HClO4, and 200 mM NaClO4 supporting electrolyte. The collector efficiency was determined to be ca. 60% in both buffers, by calibrating with a platinised FTO generator film set to +1.1 or +1.2 V (depending on pH) vs. Ag/AgCl. For further details, including photographs and calibration methods, see the ESI (Fig. S20 and S21†).Acknowledgements
We thank Dr Jie Song for help with synthesis of P2, Dr Kenneth Hardcastle for assisting with X-ray diffraction, and Dr Gregory G. Wildgoose for access to a bipotentiostat. Mass spectra for [Et42](PF6)2 and [(Et4dpbpy)2Ru(5-crown-phen)](PF6)2 were obtained by the UK EPSRC National Mass Spectrometry facility in Swansea. Our work was funded by the U.S. Department of Energy, Office of Basic Energy Sciences, Solar Photochemistry Program (DE-FG02-07ER-15906) to C.L.H., T.L. and D.G.M.; the EU through a Marie Curie IOF to J.F. (POMHYDCAT, contract no. 254339), and by the University of East Anglia. X. X. thanks the China Scholarship Council and NSFC (#21376020) for financial support.Notes and references
- (a) R. A. Binstead, C. W. Chronister, J. Ni, C. M. Hartshorn and T. J. Meyer, J. Am. Chem. Soc., 2000, 122, 8464 CrossRef CAS; (b) M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21 CrossRef CAS PubMed; (c) J. K. Hurst, Coord. Chem. Rev., 2005, 249, 313 CrossRef CAS; (d) R. Zong and R. Thummel, J. Am. Chem. Soc., 2005, 127, 12802 CrossRef CAS PubMed; (e) R. Eisenberg and H. B. Gray, Inorg. Chem., 2008, 47, 1697 CrossRef CAS PubMed; (f) N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed; (g) R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Angew. Chem., Int. Ed., 2008, 47, 7335 CrossRef CAS PubMed; (h) J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954 CrossRef CAS PubMed; (i) J. K. Hurst, J. L. Cape, A. E. Clark, S. Das and C. Qin, Inorg. Chem., 2008, 47, 1753 CrossRef CAS PubMed; (j) T. A. Betley, Y. Surendranath, M. V. Childress, G. E. Alliger, R. Fu, C. C. Cummins and D. G. Nocera, Philos. Trans. R. Soc., B, 2008, 363, 1293 CrossRef CAS PubMed; (k) J. T. Muckerman, D. E. Polyansky, T. Wada, K. Tanaka and E. Fujita, Inorg. Chem., 2008, 47, 1787 CrossRef CAS PubMed; (l) X. Sala, I. Romero, M. Rodríguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842 CrossRef CAS PubMed; (m) R. Nakamura and H. Frei, J. Am. Chem. Soc., 2006, 128, 10668 CrossRef CAS PubMed; (n) F. Jiao and H. Frei, Angew. Chem., Int. Ed., 2009, 48, 1841 CrossRef CAS PubMed; (o) J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed; (p) L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson and L. Sun, Chem.–Eur. J., 2010, 16, 4659 CrossRef CAS PubMed; (q) S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182 CrossRef CAS; (r) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed; (s) Z. Chen and T. J. Meyer, Angew. Chem., Int. Ed., 2013, 52, 700 CrossRef CAS PubMed.
- (a) Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896 CrossRef CAS PubMed; (b) A. Sartorel, M. Carraro, G. Scorrano, R. D. Zorzi, S. Geremia, N. D. McDaniel, S. Bernhard and M. Bonchio, J. Am. Chem. Soc., 2008, 130, 5006 CrossRef CAS PubMed; (c) Y. V. Geletii, C. Besson, Y. Hou, Q. Yin, D. G. Musaev, D. Quinonero, R. Cao, K. I. Hardcastle, A. Proust, P. Kögerler and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 17360 CrossRef CAS PubMed; (d) A. E. Kuznetsov, Y. V. Geletii, C. L. Hill, K. Morokuma and D. G. Musaev, J. Am. Chem. Soc., 2009, 131, 6844 CrossRef CAS PubMed; (e) A. Sartorel, P. Miro, E. Salvadori, S. Romain, M. Carraro, G. Scorrano, M. D. Valentin, A. Llobet, C. Bo and M. Bonchio, J. Am. Chem. Soc., 2009, 131, 16051 CrossRef CAS PubMed; (f) C. Besson, Z. Huang, Y. V. Geletii, S. Lense, K. I. Hardcastle, D. G. Musaev, T. Lian, A. Proust and C. L. Hill, Chem. Commun., 2010, 46, 2784 RSC; (g) Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342 CrossRef CAS PubMed; (h) R. Cao, H. Ma, Y. V. Geletii, K. I. Hardcastle and C. L. Hill, Inorg. Chem., 2009, 48, 5596 CrossRef CAS PubMed; (i) F. Song, Y. Ding, B. Ma, C. Wang, Q. Wang, X. Du, S. Fu and J. Song, Energy Environ. Sci., 2013, 6, 1170 RSC; (j) H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Basca, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed; (k) S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707 CrossRef PubMed; (l) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC; (m) R. Al-Oweini, A. Sartorel, B. S. Bassil, M. Natali, S. Berardi, F. Scandola, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2014, 53, 11182 CrossRef CAS PubMed; (n) Y. Liu, S.-X. Guo, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Inorg. Chem., 2013, 52, 11986 CrossRef CAS PubMed.
- (a) W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966 CrossRef CAS PubMed; (b) L. Duan, L. Tong, Y. Xu and L. Sun, Energy Environ. Sci., 2011, 4, 3296 RSC; (c) W. Song, Z. Chen, C. R. K. Glasson, K. Hanson, H. Luo, M. R. Norris, D. L. Ashford, J. J. Concepcion, M. K. Brennaman and T. J. Meyer, ChemPhysChem, 2012, 13, 2882 CrossRef CAS PubMed; (d) D. G. Nocera, Acc. Chem. Res., 2012, 45, 767 CrossRef CAS PubMed.
- (a) S. Anderson, E. C. Constable, M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett, K. R. Seddon and R. D. Wright, Nature, 1979, 280, 571 CrossRef CAS; (b) M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett, K. R. Seddon and R. D. Wright, Faraday Discuss. Chem. Soc., 1980, 70, 285 RSC; (c) R. Memming and F. Schröppel, Chem. Phys. Lett., 1979, 62, 207 CrossRef CAS; (d) R. Memming, F. Schröppel and U. Bringmann, J. Electroanal. Chem., 1979, 100, 307 CrossRef CAS.
- A. Juris and V. Balzani, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- (a) W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926 CrossRef CAS PubMed; (b) R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892 CrossRef CAS PubMed; (c) L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307 RSC; (d) Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612 CrossRef CAS PubMed; (e) G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389 RSC; (f) W. Song, C. R. K. Glasson, H. Luo, K. Hanson, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Phys. Chem. C, 2011, 115, 7081 CrossRef CAS; (g) W. Song, C. R. K. Glasson, H. Luo, K. Hanson, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Phys. Chem. Lett., 2011, 2, 1808 CrossRef CAS; (h) G. Li, E. M. Sproviero, W. R. McNamara, R. C. Snoeberger, R. H. Crabtree, G. W. Brudvig and V. S. Batista, J. Phys. Chem. B, 2010, 114, 14214 CrossRef CAS PubMed; (i) X. Xiang, J. Fielden, W. Rodríguez-Córdoba, Z. Huang, N. Zhang, Z. Luo, D. G. Musaev, T. Lian and C. L. Hill, J. Phys. Chem. C, 2013, 117, 918 CrossRef CAS; (j) Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219 CrossRef CAS PubMed; (k) D. L. Ashford, A. M. Lapides, A. K. Vannucci, K. Hanson, D. A. Torelli, D. P. Harrison, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6578 CrossRef CAS PubMed; (l) L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008 CrossRef CAS PubMed.
- (a) K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462 CrossRef CAS PubMed; (b) A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918 CrossRef CAS PubMed; (c) K.-R. Wee, M. K. Brennaman, L. Alibabaei, B. H. Farnum, B. Sherman, A. M. Lapides and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 13514 CrossRef CAS PubMed; (d) K. Hanson, M. D. Losego, B. Kalanyan, D. L. Ashford, G. N. Parsons and T. J. Meyer, Chem. Mater., 2013, 25, 3 CrossRef CAS; (e) A. M. Lapides, D. L. Ashford, K. Hanson, D. A. Torelli, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 15450 CrossRef CAS PubMed.
- (a) M. Orlandi, R. A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and F. Scandola, Chem. Commun., 2010, 46, 3152 RSC; (b) Z. Huang, Y. V. Geletii, D. G. Musaev, C. L. Hill and T. Lian, Ind. Eng. Chem. Res., 2012, 51, 11850 CrossRef CAS.
- (a) S.-X. Guo, Y. Liu, C.-Y. Lee, A. M. Bond, J. Zhang, Y. V. Geletii and C. L. Hill, Energy Environ. Sci., 2013, 6, 2654 RSC; (b) S.-X. Guo, C.-Y. Lee, J. Zhang, A. M. Bond, Y. V. Geletii and C. L. Hill, Inorg. Chem., 2014, 53, 7561 CrossRef CAS PubMed; (c) N. Anwar, A. Sartorel, M. Yaqub, K. Wearen, F. Laffir, G. Armstrong, C. Dickinson, M. Bonchio and T. McCormac, ACS Appl. Mater. Interfaces, 2012, 6, 8022 CrossRef PubMed; (d) J. Soriano-López, S. Goberna-Ferrón, L. Vigara, J. J. Carbó, J. M. Poblet and J. R. Galán-Mascarós, Inorg. Chem., 2013, 52, 4753 CrossRef PubMed; (e) J. Wu, L. Liao, W. Yan, Y. Xue, Y. Sun, X. Yan, Y. Chen and Y. Xi, ChemSusChem, 2012, 5, 1207 CrossRef CAS PubMed; (f) F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. R. Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826 CrossRef CAS PubMed.
- (a) Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522 CrossRef CAS PubMed; (b) F. Puntoriero, G. L. Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725 RSC; (c) Z. Huang, Z. Luo, Y. V. Geletii, J. W. Vickers, Q. Yin, D. Wu, Y. Hou, Y. Ding, J. Song, D. G. Musaev, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2011, 133, 2068 CAS; (d) J. W. Vickers, H. Lv, J. M. Sumliner, G. Zhu, Z. Luo, D. G. Musaev, Y. V. Geletii and C. L. Hill, J. Am. Chem. Soc., 2013, 135, 14110 CrossRef CAS PubMed; (e) H. Lv, J. A. Rudd, P. F. Zhuk, J. Y. Lee, E. C. Constable, C. E. Housecroft, C. L. Hill, D. G. Musaev and Y. V. Geletii, RSC Adv., 2013, 3, 20647 RSC; (f) J. M. Sumliner, H. Lv, J. Fielden, Y. V. Geletii and C. L. Hill, Eur. J. Inorg. Chem., 2014, 635 CrossRef CAS; (g) G. Zhu, E. Glass, C. Zhao, H. Lv, J. W. Vickers, Y. V. Geletii, D. G. Musaev, J. Song and C. L. Hill, Dalton Trans., 2012, 13043 RSC.
- (a) C. Creutz and N. Sutin, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2858 CrossRef CAS PubMed; (b) P. K. Ghosh, B. S. Brunschwig, M. C. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772 CrossRef CAS.
- (a) A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605 RSC; (b) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, V. W.-M. Lee, F. Ke and K.-W. M. Siu, Inorg. Chem., 1997, 36, 2124 CrossRef CAS PubMed; (b) C. R. Rice, A. Guerrero, Z. R. Bell, R. L. Paul, G. R. Motson, J. C. Jeffery and M. D. Ward, New J. Chem., 2001, 25, 185 RSC.
- J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2011, 133, 188 CrossRef CAS PubMed.
- P. U. Maheswari, V. Rajendran, M. Palaniandavar, R. Thomas and G. U. Kulkarni, Inorg. Chim. Acta, 2006, 359, 4601 CrossRef CAS.
- Selected examples: (a) J. B. Asbury, R. J. Ellingson, H. N. Ghosh, S. Ferrere, A. J. Nozik and T. Lian, J. Phys. Chem. B, 1999, 103, 3110 CrossRef CAS; (b) J. B. Asbury, Y. Q. Wang and T. Lian, J. Phys. Chem. B, 1999, 103, 6643 CrossRef CAS; (c) T. A. Heimer and E. J. Heilweil, J. Phys. Chem. B, 1997, 101, 10990 CrossRef CAS; (d) Y. Trachibana, S. A. Haque, I. P. Mercer, J. R. Durrant and D. R. Klug, J. Phys. Chem. B, 2000, 104, 1198 CrossRef; (e) R. C. Snoeberger, K. J. Young, J. Tang, L. J. Allen, R. H. Crabtree, G. W. Brudvig, P. Coppens, V. S. Batista and J. B. Benedict, J. Am. Chem. Soc., 2012, 134, 8911 CrossRef CAS PubMed; (f) J. Huang, D. Stockwell, A. Boulesbaa, J. Guo and T. Lian, J. Phys. Chem. C, 2008, 112, 5203 CrossRef CAS; (g) D. Stockwell, Y. Yang, J. Huang, C. Anfuso, Z. Huang and T. Lian, J. Phys. Chem. C, 2010, 114, 6560 CrossRef CAS; (h) K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837 CrossRef CAS; (i) R. Ernstorfer, L. Gundlach, S. Felber, W. Storck, R. Eichberger and F. Willig, J. Phys. Chem. B, 2006, 110, 25383 CrossRef CAS PubMed.
- J. M. Gardner, M. Beyler, M. Karnahl, S. Tschierlei, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 19322 CrossRef CAS PubMed.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324 CrossRef CAS PubMed.
- (a) T. E. Keyes, E. Gicquel, L. Guerin, V. M. Hultgren, A. M. Bond and A. G. Wedd, Inorg. Chem., 2003, 42, 7897 CrossRef CAS PubMed; (b) M. K. Seery, L. Guerin, R. J. Forster, E. Gicquel, V. M. Hultgren, A. M. Bond, A. G. Wedd and T. E. Keyes, J. Phys. Chem. A, 2004, 108, 7399 CrossRef CAS; (c) N. Fay, V. M. Hultgren, A. G. Wedd, T. E. Keyes, R. J. Forster, D. Leane and A. M. Bond, Dalton Trans., 2006, 4218 RSC; (d) M. K. Seery, N. Fay, T. McCormac, E. Dempsey, R. J. Forster and T. E. Keyes, Phys. Chem. Chem. Phys., 2005, 7, 3426 RSC.
- S.-H. A. Lee, Y. Zhao, E. A. Hernandez-Pagan, L. Blasdel, W. J. Youngblood and T. E. Mallouk, Faraday Discuss., 2012, 155, 165 RSC.
- D. Duonghong, E. Borgarello and M. Grätzel, J. Am. Chem. Soc., 1981, 103, 4685 CrossRef CAS.
- A. Juris and V. Balzani, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- D. L. Ashford, M. K. Brennaman, R. J. Brown, S. Keinan, J. J. Concepcion, J. M. Papanikolas, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2015, 54, 460 CrossRef CAS PubMed.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- For ECP see: (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef; (c) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1997, 107, 3032 CrossRef.
- For PCM method see: (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS, and (b) R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of materials, synthetic procedures, crystallographic data; details of ultrafast and nanosecond transient absorption spectrometers and photoelectrochemistry set-up; supporting UV-vis, cyclic voltammetry and transient absorption measurements. CCDC 1059343. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01439e |
| ‡ Current address: School of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: John.Fielden@uea.ac.uk |
| This journal is © The Royal Society of Chemistry 2015 |