
Synthesis and photophysical properties of phthalocyanines having calixpyrrole units
Zeynep Neslihan Erolab,
Pedro Atienzara,
Yasin Arslanoğluc,
Esin Hamuryudanb and
Hermenegildo García*a
aInstituto Universitario de Tecnología Química CSIC-UPV, Universidad Politécnica de Valencia, Av. de los Naranjos, s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es
bDepartment of Chemistry, Istanbul Technical University, Maslak, 34469 Istanbul, Turkey
cMaritime Faculty, Istanbul Technical University, 34940 Tuzla, Turkey
First published on 19th June 2015
Abstract
Zn2+ and Co2+ metallated phthalocyanines having four calixpyrrole units have been prepared from a calixpyrrole phthalonitrile using Zn2+ or Co2+ salts as templates of a phthalocyanine macrocycle. Host–guest complexes between the anions (Cl−, F− and H2PO4−) and the calixpyrrole units are formed as evidenced by NMR, the shift in the reduction potential of the calixpyrrole units by cyclic voltammetry and by observation of two straight lines in the plot of the conductivity versus the amount of halide. While halides do not form complexes with Zn2+ or Co2+ ions of the phthalocyanine core in the ground state as evidenced by the lack of variation in the position of the Q-band in optical spectroscopy, upon 355 nm excitation, the apical metal–halide complex should be formed as a transient as evidenced by the comparison of the transient absorption spectra in the presence and absence of halides.
Introduction
Metal phthalocyanines are one important type of pigment that finds many applications for the preparation of paints and in optoelectronic devices.1–3 The color of phthalocyanines is due to the presence of an intense absorption band in the visible region whose λmax depends on the medium, the state of aggregation and can be influenced by the interaction of the metal ion with different ligands.4 Metal phthalocyanines also exhibit interesting photophysical properties with intense emission and with the possibility to participate in photoinduced electron transfer processes with electron acceptor or donor quenchers.5 In fact, one possible use of metal phthalocyanines has been as light harvesters in dye synthesized solar cells (DSCCs) and in photoresponsive materials.6,7Due to the structural flexibility, there has been reported a number of dyads in which phthalocyanines have been covalently attached to fullerenes or other electron acceptor moieties or even triads in order to obtain photoresponsive materials.8,9 Continuing with this line of research, one of the possibilities to alter and gain control on the physical and photophysical properties of metal phthalocyanines would be a peripheral functionalization with units that could interact with some chemical species by forming complexes. Formation of these complexes could be reflected in the properties of the metal phthalocyanine core and in this way the response of the phthalocyanine could be modulated by addition of the chemical species capable to interact with the peripheral side groups. These novel metal phthalocyanines may have application for sensing or extraction of anions due to their functionalization at the peripheral positions of the phthalocyanine core. In the present work, we report the synthesis and characterization of two novel metal phthalocyanines (Zn and Co) that have covalently attached four calixpyrrole units. One of the merits of the present work is the design and successful synthesis of the dyad having a phthalocyanine core and the peripheral calixpyrrole. Calixpyrroles and other type of calixarenes are currently attracting considerable interest due to their ability to form complexes with anions or even to form an assembly with anion/cation pairs.10–12 We will show that while absorption spectroscopy does not allow assessing the interaction of the calixpyrrole armed phthalocyanine with halides, NMR, electrochemistry and photophysical measurements provide experimental support in favor of complexation of these calixpyrrole-substituted metal phthalocyanines with Cl−, H2PO4− and F− with the calixpyrrole units. In contrast to the behavior in the ground state, the metal-to-ligand charge transfer excited state of these phthalocyanines form an apical metal–halide complex in the excited state as evidenced by submillisecond transient absorption spectroscopy.
Results and discussion
Synthesis and characterization
The synthesis of metal phthalocyanines (M = Zn2+ 7, M = Co2+ 8) substituted with four calixpyrrole arms are indicated in Scheme 1. As it can be there seen, we have used a convergent synthetic route in which the phthalocyanine macroring is obtained in the last step by metal ion templated cyclotetramerization of substituted o-dicyanobenzene compound 6 (see Experimental section). The preparation of the starting material 6 was carried out by coupling under basic conditions of a phenol substituted calixpyrrole 4 with 4-nitrophthalonitrile 5.13 Calixpyrrole 4 having the hydroxyphenyl substituent was obtained by random cyclotetramerization of pyrrole (1), acetone (2) and 4-hydroxyacetophenone (3) in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 molar ratio in methanol under acid catalysis.14 Flash column chromatography with proper eluents was used in the purification of the compound 4 (see Experimental section) and column chromatography was used for purification of compounds 7 and 8. Synthesized compounds were characterized by their spectroscopic data that were in agreement with the proposed structure.
:3:1 molar ratio in methanol under acid catalysis.14 Flash column chromatography with proper eluents was used in the purification of the compound 4 (see Experimental section) and column chromatography was used for purification of compounds 7 and 8. Synthesized compounds were characterized by their spectroscopic data that were in agreement with the proposed structure.
 | ||
| Scheme 1 Synthetic route for the preparation of metal-phthalocyanines 7 and 8. | ||
The IR spectrum of compound 4 indicates the presence of the hydroxyl groups by the broad-OH stretching band at 3322 cm−1 and NH stretching peak at 3436 cm−1. In the 1H-NMR spectrum of compound 4, a sharp singlet at δ 1.49 ppm is assigned to the seven methyl groups, a doublet at δ 6.61 ppm to the eight pyrrole ring β-protons and a broad peak at δ 5.70 ppm is assigned to the four NH protons. Also, the H is bonded –OH group in the phenol is seen at δ 9.28 ppm.
Compound 6 exhibits in the IR spectrum the characteristic nitrile peak at 2231 cm−1 while the broad-OH stretching band present in compound 4 disappeared. In 1H-NMR spectroscopy the peak associated to the phenolic hydrogen in compound 4 disappeared and there is a triplet between δ 7.93 and δ 8.43 ppm due to the hydrogen in the benzene ring near the nitrile moiety.
In the IR spectra of both compound 7 and 8, the nitrile peak is absent. In the UV-vis spectra of metal phthalocyanines the characteristic Q-band for compound 7 appears at 683 nm and for compound 8 at 676 nm. It is not possible to record the NMR spectrum of compound 8, due to the paramagnetic properties of Co2+. In contrast, complex 7 has NMR spectra. In the 1H-NMR spectrum of compound 7 the chemical shift for hydrogens in the benzenes in the phthalocyanine rings was 7.93 and 8.08 ppm. Also the pyrrole groups exhibit a peak at 7 ppm and aliphatic hydrogens appeared at 1.21 ppm. Note that in the synthesis of 7 or 8 by cyclotetramerization of compound 6 by the templating effect of the metal ion up to 5 positional isomers can possibly form.
Anion bonding properties
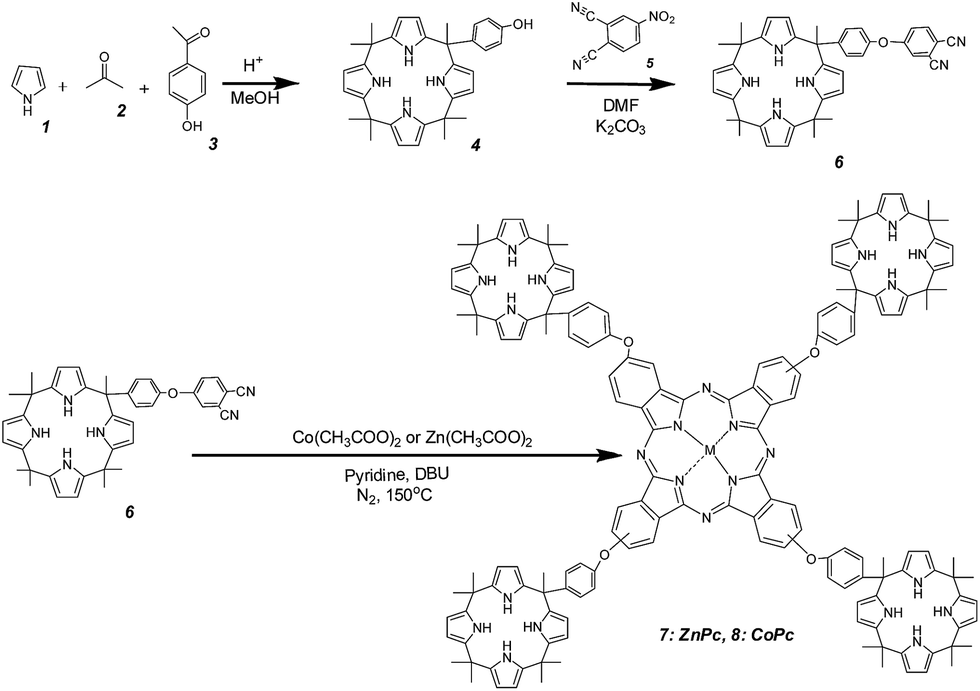
As commented in the introduction the main purpose of this work was to determine the possibility of complex formation between calixpyrrole units and halides (Cl− and F−) and H2PO4− and how the formation of these complexes at the periphery can influence some of the properties of the metal phthalocyanine core in the dyad. To address this issue, we performed spectroscopic measurements, as well as experiments of electrochemistry and conductivity. Addition of increasing amounts, in the range from 0 to 0.5 mM, of TBAX (TBA: tetrabutylammonium, X−: halide) did not lead to detectable changes in UV-vis spectrum of DMSO solutions of 7 and 8. In the literature, there are precedents showing that complexation of nucleophiles such as pyridine or aniline to the metal ion in metal phthalocyanines can lead to a small, but significant, variation of the Q-band.15 Changes about 5 nm have been reported for the complex of ZnPc and pyridine.15,16 Therefore, it should also be expected that in the present case, the interaction between the metal ions (Zn2+, Co2+) and X− should also have been detectable by some shift in the position of Q-band. In contrast, if the complexes are formed between X− anions and calixpyrrole units, then, it is reasonable to assume that the Q-band located on the phthalocyanine core could probably be insensitive to this type of complexes.Emission spectroscopy has also frequently been used as a tool to detect the formation of complexes in metal phthalocyanines. The metal phthalocyanines exhibit a relatively intense emission that corresponds to the decay of their electronic singlet excited state.17,18 The intensity of this emission has been reported to be quenched by the presence of nucleophiles and anions that form complexes with the central metal cation.19,20 Also in the present case, the emission of compounds 7 and 8 was not sensitive to the presence of X− or H2PO4− in the range of concentrations of a few mM or lower, but in excess with respect to the concentration of the metal phthalocyanines. In addition, the temporal profile of the signal also did not change in the absence or in the presence of X− or H2PO4−. This indicates not only that no apical complex is formed between the phthalocyanine metal ion and the anion in the ground state (static quenching resulting less intense emission), but also that these anions do not interact with the metal ion in the lifetime of phthalocyanine singlet excited state in the ns time scale. Fig. 1 shows selected UV-vis and emission spectra as well as temporal profiles of the photoluminescence in the absence and presence of anions to illustrate the previous points.
 | ||
| Fig. 1 (A) UV-vis spectra of compound 7 (3.9 × 10−5 M) in DMSO in the absence and presence of different anions (a: no anion, b: 50 μL TBACl (2 × 10−2 M), c: 50 μL NaH2PO4 (2 × 10−2 M), d: 50 μL NH4F (2 × 10−2 M)), (B) emission spectra of compound 7 (3.9 × 10−5 M) in the presence or absence of anions (a: NaH2PO4 (50 μL, 2 × 10−2 M), b: NH4F (50 μL, 2 × 10−2 M), c: TBACl (50 μL, 2 × 10−2 M), d: no anion), (C) temporal profile of the fluorescence of the compound 7 in the absence or presence of anions (a: lamp, b–f: with TBACl, NaH2PO4, NH4F, MeOH and only the compound, respectively at the concentrations indicated for (B)) in DMSO. | ||
In contrast to UV-vis and emission spectroscopy, evidence in support of the complexation of calixpyrrole units 4 with H2PO4− and F− in CDCl3 were obtained by 31P and 1H NMR spectroscopy. In 31P NMR spectroscopy there was a clear shift of the 31P peak from −19 to −70 ppm for H2PO4− in the absence and in the presence of stoichiometric amounts of calixpyrrole 4, respectively. Similarly in 1H NMR spectroscopy, the singlet signal corresponding to the eight pyrrole protons of 4 shifts from 5.674 to 5.439 ppm in the absence or presence of an excess of F−, respectively.
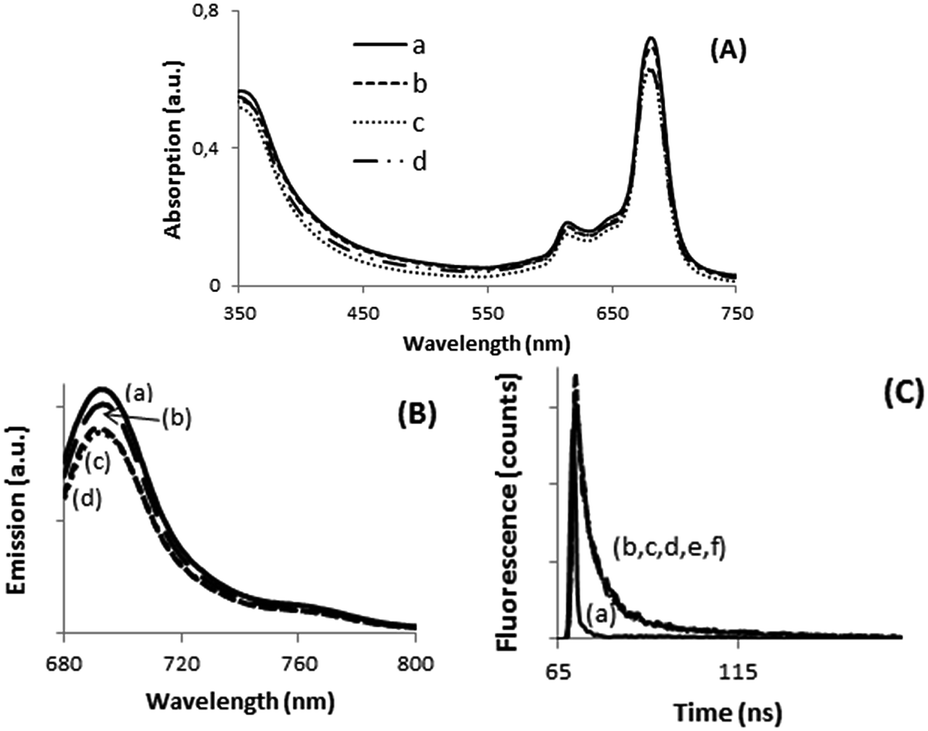
Electrochemical measurements by cyclic voltammetry are also in agreement with the formation of host–guest complexes with the calixpyrrole unit of dyads 7 and 8 and some anions. Initial electrochemical measurements were carried out in solution using DMSO as solvent, since compounds 7 and 8 are more soluble in DMSO than in DMF or other solvents. However, the electrochemical peaks could not be resolved, probably due to the insufficient concentration of phthalocyanines. For this reason, further electrochemical measurements were performed using films of phthalocyanines 7 and 8 supported on conductive FTO electrode. Preliminary controls show no electrochemical response for FTO in the potential window under study (see Fig. 2c, plot a). In contrast films of phthalocyanines showed higher current and were different for 7 and 8 (Fig. 2c, plots b and c) In the latter case, the appearance of a reduction peak due to CoII/CoI electrochemical pair was observed (see Fig. 2c, plot c). In contrast, dyads 7 and 8 did not exhibit peaks in the oxidation redox of the voltammogram.
 | ||
| Fig. 2 Cyclic voltammetry (CV) of 8 (A) and 7 (B), and influence of the presence of different salts (a: TBAH2PO4 (50 μL, 2 × 10−2 M), b: TBACl (50 μL, 2 × 10−2 M), c: NH4F (50 μL, 2 × 10−2 M), (C) CV of: (a) bare FTO substrate; (b) compound 7, and (c) compound 8 in TBAPF6 0.1 M. (D) Photograph of phthalocyanine film electrodes on FTO used for the electrochemical measurements. | ||
It was observed that the reduction part of the cyclic voltammetry of the dyads in the absence and in the presence of anions exhibits significant shifts. Table 1 summarizes the reduction potential measured as the semi sum of the forward and backward reduction peaks. In order to provide some information about the electrochemical processes taking place in the reduction and the influence by the presence of halides, we also submitted to electrochemical studies calixpyrrole 6 that corresponds closely to the moiety present in the dyads 7 and 8. It was observed that also calixpyrrole 6 exhibits a quasi-reversible reduction peak at similar potential values to those measured for dyads 7 and 8. Accordingly, we assume that the calixpyrrole unit should be the one responsible for the electrochemical response in compounds 7 and 8. Moreover, calixpyrrole 6 also behaves in the presence of X− and H2PO4− similarly to phthalocyanine dyads 7 and 8, suggesting again that the influence of the halide on the electrochemistry is due to the interaction with these units. Concerning the values of the shifts in the reduction peak potentials as a function of the nature of the halide anion, it follows the order Cl− < F−. This order suggests that the binding constants of calixpyrrole and X− should follow the same trend. In fact, in the case of F−, the reduction peak is moved outside the electrochemical window available in the measurements with DMSO. To illustrate these points, Fig. 2 shows selected voltammograms in the absence and in the presence of anions showing the influence of the presence of halides in the electrochemical response.
| Sample | Reduction potentiala (V) | Oxidation potentiala (V) | Homo energyb (eV) | Lumo energyb (eV) |
|---|---|---|---|---|
| a Reduction and oxidation peak potential versus Ag/AgCl in saturated KCl.b Energy levels referred to vacuum. The energy values were estimated using LUMO level = [Ered]onset + 4.4 and HOMO level = Eg − LUMO (Eg (optical band gap) of 7: 1.74 eV and 8: 1.68 eV). | ||||
| 7/TBAH2PO4 | −0.41 | — | 5.77 | −4.03 |
| 8/TBAH2PO4 | −0.42 | 1.16 | 5.71 | −4.03 |
| 7/TBACl | −0.6 | — | 5.72 | −3.98 |
| 8/TBACl | −0.61 | — | 5.64 | −3.96 |
| 7/NH4F | −0.47 | 1.12 | 5.65 | −3.91 |
| 8/NH4F | −0.6 | 1.23 | 5.48 | −3.8 |
| 7/NaH2PO4 | −0.31 | 0.81 | 5.8 | −4.06 |
| 8/NaH2PO4 | −0.31 | — | 5.23 | −3.55 |
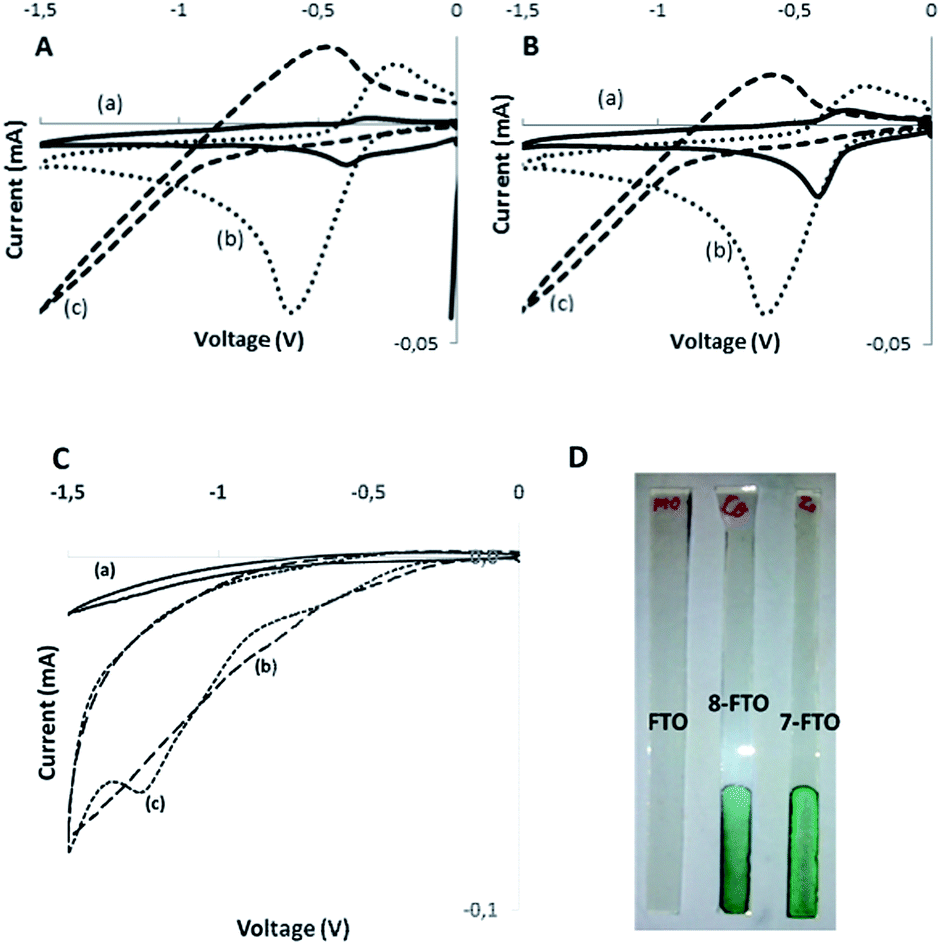
Conductivity measurements also evidence the formation of complexes between X− and the dyads 7 and 8. Thus, the conductivity of a solution of phthalocyanines in DMSO was measured upon addition of increasing amounts of TBACl in the range from 0 to 10−4 M. It was observed that the addition of TBACl increases initially the conductivity of the solution with a slope that was smaller than the slope of the straight line beyond the equivalence point for the formation of a 1:1 Pc:Cl− complex. Fig. 3 shows the concentration–conductivity plot for these measurements. The point in which two straight lines intercept should correspond to the titration point in which all the phthalocyanines form a 1:1 complex with Cl−. Further addition should result in the formation of a 1:2 complex (7-2Cl− or 8-2Cl−) and some free chloride in equilibrium. Up to 4 chlorides could bind with a phthalocyanine molecule, but complete shift of the equilibrium towards the formation of 1:4 complexes with phthalocyanine and chloride could require an excess of chloride that drives the conductivity far from the available measurement window. Thus, titration with 1:1 phthalocyanine complex was more appropriate from the experimental point of view.
 | ||
| Fig. 3 Conductivity of (a) compound 7 (2.89 × 10−5 M), (b) compound 8 (2.89 × 10−5 M) as a function of the concentration of Cl− ion in DMSO. | ||
All together, the characterization data presented above are consistent with X− and H2PO4− forming complexes with the calixpyrrole unit of the dyads 7 and 8, but not with the metal of the phthalocyanine. Furthermore, complexation of calixpyrrole at the periphery of compounds 7 and 8 does not seem to alter those properties of the molecule that are localized on the phthalocyanine core such as light absorption and emission.
Transient absorption spectroscopy is a powerful technique to detect species generated upon light absorption and to monitor their kinetics.21 Transient spectroscopy of phthalocyanines in particular can be sensitive to the presence of quenchers that can interact with the phthalocyanine chromophore. In the previous sections, we have commented that it has not been possible to detect any influence on phthalocyanine spectroscopic properties as consequence of the binding with anions. Therefore, we were interested in studying the transient absorption spectra of compounds 7 and 8 in the absence and in the presence of X− to detect possible transients that could provide evidence for these interactions occurring in the photogenerated transients in the submilliseconds time scale.
For the transient spectroscopy study, the third harmonic of a Nd-YAG operating at 355 nm was used. Upon laser excitation of DMSO solutions of 8, no transients were observed in the submillisecond time scale. This failure to detect any transient for 8 is in accordance with the fact that metal complexes having metal ions with partially occupied d orbitals such as Co2+ undergo ultra-fast relaxation that would not be detectable in our nanosecond laser system. In contrast, laser flash photolysis of 7 having a Zn+2 ion in d10 configuration exhibits upon laser excitation under Ar a transient signal. The transient signal monitored for 7 decays in hundreds of microseconds. The transient spectrum recorded at different delay times is shown in Fig. 4. As it can be seen there, these transient spectra are characterized by a broad absorption band from 400 to 600 nm accompanied by negative signals below 400 nm and between 600 and 700 nm. Considering the shape and the position of the negative signals, they have to be attributed to the bleaching of the phthalocyanine ground state. Analysis of the temporal profile at different wavelengths for the broad band from 400 to 600 nm indicates that these wavelengths should correspond to the same species based on the coincidence of the signal decay. In addition, as it can be seen in the inset of Fig. 4, the decay of the transient absorption monitored at 500 nm has the same profile as the bleaching recovery monitored at 680 nm. This coincidence in the decay and the recovery indicates also that the transient species relaxes to the ground state without the formation of any other intermediate.
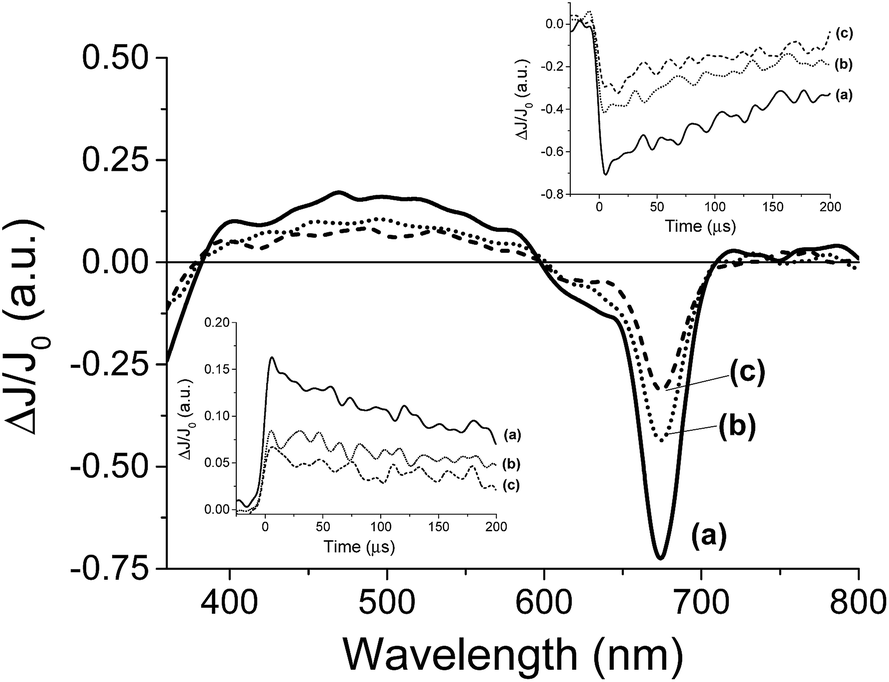 | ||
| Fig. 4 Transient absorption spectra recorded at 5 μs after 355 nm laser excitation of a N2-purged DMSO solution of 7 (3.9 × 10−5 M) (a), after purging with N2O (b) and after adding 100 μL of MeOH (c). The insets show the temporal profiles monitored at 520 nm (left) and 680 nm (right) for (a) N2 purged solution, (b) an N2O purged solution and (c) after adding 100 μL of MeOH. | ||
In order to assign the absorption band, we studied the behaviour of the transient signal in the presence of quenchers that can accept (N2O) or that can give electrons (MeOH). Selection of these quenchers is based on the general known photochemical behavior of metal phthalocyanines and other metal complexes that upon photochemical excitation generate excited electronic states with charge transfer character due to the migration of electrons from the ligand to the metal (LMCT state) or from the metal to the ligand (MLCT state).22 As it can be seen in Fig. 4 the transient signal decreases considerably in intensity either in the presence of N2O or upon addition of MeOH. These observations are in agreement with a charge transfer excited state upon excitation of 7. Thus, while emission spectroscopy follows the temporal profile of the singlet excited state of 7 that lives in the ns timescale; laser flash photolysis monitors the triplet excited states and allows us to follow the interaction of the excited state with anions in much longer time scale, reaching hundreds of microseconds.
Once preliminary transient measurements have established the generation of transient species that can be monitored in the time window available to our nanosecond laser system for 7, we were interested in determining the influence that the presence of Cl− and F− can exert on this transient. Two types of effects were observed, both on the transient spectra recorded at delay times longer than 5 μs and on the temporal profile of the signals. Fig. 5 presents the transient spectra monitored at 5 μs after the laser flash in the presence and absence of anion, showing a significant shift in the position of the Q-band in the presence of 0.95 mM concentration of Cl−. As it can be seen in Fig. 5, the presence of Cl− or F− alters the position of the negative band with respect to the Q band in the absence of these anions. This observation contrast with the fact that, as commented earlier, ground state UV-vis spectroscopy is not affected by the presence of these anions. However, upon laser excitation, the position of the Q-band bleaching clearly changes when Cl− or F− are present about 10 nm red-shifted with respect to that in the absence of Cl− or F−. In other words, while in the absence of Cl− or F− the position of the Q-band, appearing as a negative signal in the transition spectra, coincides with that observed in steady-state UV-vis spectroscopy, when Cl− or F− are present, a red-shift in the position of the Q-band is observed. As commented earlier when discussing the UV-vis spectra in the absence and in the presence of X− and H2PO4−, we were expecting a red-shift of the Q-band in the range of a few nm if complexation occurred at the central metal ion of the phthalocyanine core, but we were unable to observe this shift in the ground state. Now, in the transient spectra, we have monitored that this shift of the Q-band evolves upon generation of the excited state.
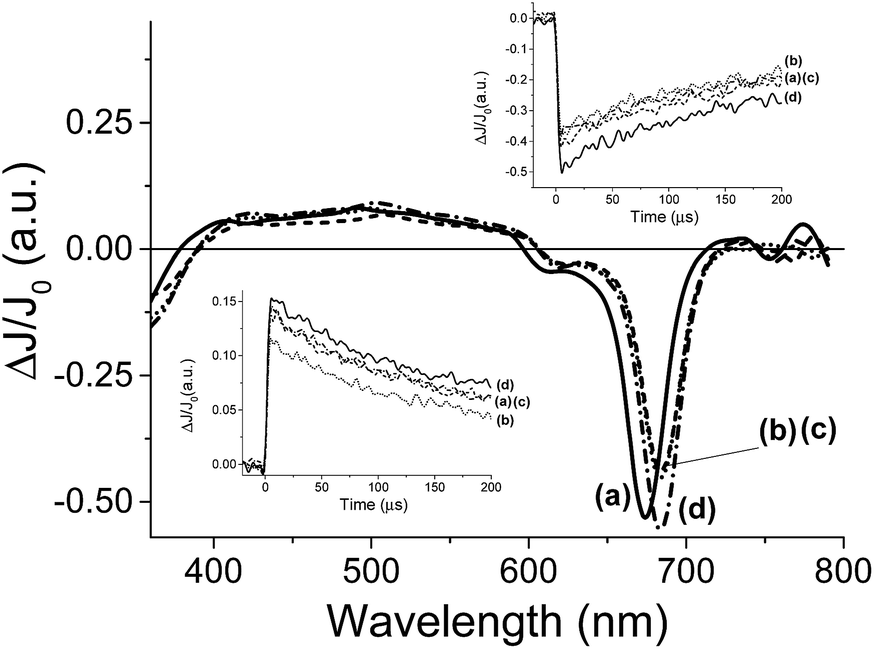 | ||
| Fig. 5 Transient absorption spectra recorded at 5 μs after 355 nm laser excitation for compound 7 (3.9 × 10−5 M) in DMSO in the absence and in the presence of different anions (a: no anion, b: Cl− (50 μL, 2 × 10−2 M), c: F− (50 μL, 2 × 10−2 M), d: H2PO4− (50 μL, 2 × 10−2 M)). Left inset: Transient signal recorded for compound 7 (3.9 × 10−5 M) in DMSO monitored at 490 nm in the absence and in the presence of different anions (a: no anion, b: Cl−, c: F− and d: H2PO4−, at the concentrations indicated above). Right inset: Temporal profile of the signal recorded for compound 7 (3.9 × 10−5 M) in DMSO monitored at 680 nm upon 355 nm excitation in the absence and in the presence of different anions (a: no anion, b: Cl−, c: F−, d: H2PO4−, at the concentrations indicated above). | ||
In agreement with the evolution of the Q-band in the transient spectra, the temporal profile of the transient signal monitored 680 nm shows that the maximum ΔJ/J0 value is reached not instantaneously after the laser pulse, but with a delay of a few μs, indicating that there is a process that is taking place in this timescale that finally is reflected in the shift in the position of the Q-band. In the literature there are precedents showing that complexation triggered by generation of excited states takes place in the μs time scale and this allows determining the rate constant of complex association.23 One of these examples was reported by Scaiano and co-workers who showed that while xanthone in the ground state forms an association complex with γ-cyclodextrin,24 upon photon absorption and generation of the corresponding xanthone triplet excited state, the much polar nature of this excited state determines the dissociation of the host–guest complex based on hydrophobic interactions and the migration of xanthone from the cavity of cyclodextrin to water.25 This process was accompanied, as in the case observed here, by a shift in the position of the λmax of the xanthone triplet that is sensitive to the polarity of the medium.26
Based on this precedent and also with the known shift in the Q-band when the metal ion of the phthalocyanine interacts with nucleophiles,27 we propose that the observation of the shift in the position of the Q-band can be interpreted in a similar way. Thus, initially Cl− or F− would not form a complex with Zn+2 ion, since otherwise a shift in the Q-band should have been observed by steady-state UV-vis spectroscopy. Complexation for the ground state occurs only on the calixpyrrole units. In contrast upon excitation of 7, and generation of the triplet excited state complexation between Zn+2 and Cl− and F− could occurs due to the different electronic density of this transient excited state.28 Accordingly the complex will be formed in a few μs after the laser pulse leading to this shift in the Q band. Scheme 2 summarises our proposal to rationalize the data obtained by transient absorption spectroscopy.
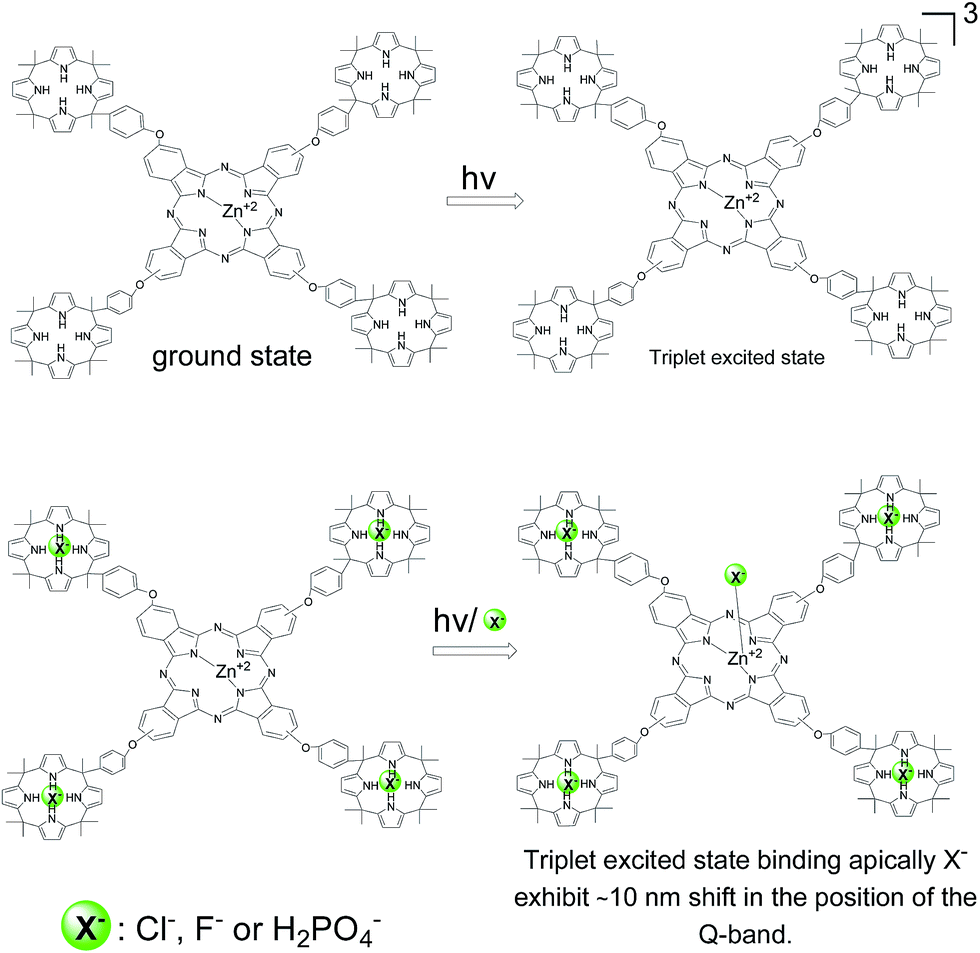 | ||
| Scheme 2 Proposal to rationalize the transient spectroscopy data explaining the red shift (+10 nm) in the position of the Q-band. The higher positive charge density on Zn+2 cation in the triplet excited state would favor apical complexation of X− to this position. | ||
Conclusions
In the present work we have synthesized tetracalixpyrrole-armed phthalocyanines that can bind X− and H2PO4− through the calixpyrrole units without influencing the spectroscopic properties intrinsic of the metal phthalocyanine core. Upon laser excitation, bleaching of the ground state and generation of triplet excited state takes place. The triplet excited state forms in tens of microseconds a new complex in which X− is bonded to the metal ion of the phthalocyanine that can be monitored by the 10 nm shifts of the Q-band.Experimental section
Materials
All reagents and solvents were purchased from commercial suppliers. The solvents were dried using standard techniques. 4-Nitrophthalonitrile was synthesized as reported in the literature.13 The progress of the reactions were monitored by TLC (SiO2) and column chromatography was performed on silica-gel 60 (0.04–0.063 mm). IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer (ATR sampling accessory), UV-vis spectra were recorded on Scinco Neosys-2000 double-beam ultraviolet visible (UV-vis) spectrophotometer using 1 cm path length cuvettes at room temperature. 1H NMR spectra were recorded on a Varian 500 MHz spectrometer using TMS as internal reference. Elemental analyses were performed by the Instrumental Analysis Laboratory of the TUBITAK Marmara Research Center. Mass spectra were performed on Bruker Micro TOF-ESI/MS and Bruker microflex LT MALDI-TOF MS spectrometer. Fluorescence excitation and emission spectra were recorded using Varian Cary Eclipse Fluorescence Spectrophotometer.:1, DCM/MeOH 9:1) gave the compound as white solid. Yield: 20%; FT-IR νmax/cm−1; 3322 (phenol), 3109–2959 (Ar–H), 2880–2871 (aliphatic-H), 1651(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1225(CN); 1H-NMR (DMSO-d6) δ/ppm; 9.28 ppm (s, 1H, Ar–OH), 6.61–6.62 (d, 4H, Ar–H), 5.70 (s, 4H, N–H), 5.66 (s, 8H, Ar–H), 2.49 (s, 3H, CH3),1.49 (s, 18H, CH3). Anal. calcd for C33H38N4O; C 78.23, H 7.56, N 11.06; found: C 78.14, H 7.49, N 11.02. MS: m/z: calcd: 506.30 amu; found: [M + H]+ 507.5 amu.
C), 1225(CN); 1H-NMR (DMSO-d6) δ/ppm; 9.28 ppm (s, 1H, Ar–OH), 6.61–6.62 (d, 4H, Ar–H), 5.70 (s, 4H, N–H), 5.66 (s, 8H, Ar–H), 2.49 (s, 3H, CH3),1.49 (s, 18H, CH3). Anal. calcd for C33H38N4O; C 78.23, H 7.56, N 11.06; found: C 78.14, H 7.49, N 11.02. MS: m/z: calcd: 506.30 amu; found: [M + H]+ 507.5 amu.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1596 (CC), 1251 (CN); 1H-NMR (DMSO-d6) δ/ppm; 7.93–8.43 (t, 3H, Ar–H), 6.91 (t, 4H, Ar–H), 5.56–5.74 (t, 8H, C–H), 1.70–1.75 (m, 21H, aliphatic-H). Anal. calcd for C41H40N6O; C 77.82, H 6.37, N 13.28; found: C 77.77, H 6.31, N 13.20. MS: m/z: calcd: 632.33 amu; found: [M + H]+ 633.4 amu.:2) and washed with MeOH, toluene, n-hexane, and diethyl ether several times until crude green product precipitated. The dark green product was purified by column chromatography over silica gel using ethyl acetate/chloroform (1:1) for compound 7 and toluene/methanol (1:3) for compound 8. For compound 7: yield: 17%; FT-IR νmax/cm−1; 2968 (C–H), 1596 (CC); 1H-NMR (DMSO-d6) δ/ppm; 7.93–8.08 (d, 12H, Ar–H), 7.01 (s, 16H, Ar–H), 4.63 (s, 32H, C–H), 1.21 (s, 84H, aliphatic-H); UV-vis λmax (nm) (logε) in DMSO; 350 and 683 nm. Anal. calcd for C164H160N24O4Zn; C 75.86, H 6.21, N 12.95; found: C 75.79, H 6.18, N 12.90. MS: m/z: calcd: 2593.23 amu; found: [M + H]+ 2594.5 amu.
N), 1596 (CC), 1251 (CN); 1H-NMR (DMSO-d6) δ/ppm; 7.93–8.43 (t, 3H, Ar–H), 6.91 (t, 4H, Ar–H), 5.56–5.74 (t, 8H, C–H), 1.70–1.75 (m, 21H, aliphatic-H). Anal. calcd for C41H40N6O; C 77.82, H 6.37, N 13.28; found: C 77.77, H 6.31, N 13.20. MS: m/z: calcd: 632.33 amu; found: [M + H]+ 633.4 amu.:2) and washed with MeOH, toluene, n-hexane, and diethyl ether several times until crude green product precipitated. The dark green product was purified by column chromatography over silica gel using ethyl acetate/chloroform (1:1) for compound 7 and toluene/methanol (1:3) for compound 8. For compound 7: yield: 17%; FT-IR νmax/cm−1; 2968 (C–H), 1596 (CC); 1H-NMR (DMSO-d6) δ/ppm; 7.93–8.08 (d, 12H, Ar–H), 7.01 (s, 16H, Ar–H), 4.63 (s, 32H, C–H), 1.21 (s, 84H, aliphatic-H); UV-vis λmax (nm) (logε) in DMSO; 350 and 683 nm. Anal. calcd for C164H160N24O4Zn; C 75.86, H 6.21, N 12.95; found: C 75.79, H 6.18, N 12.90. MS: m/z: calcd: 2593.23 amu; found: [M + H]+ 2594.5 amu.For compound 8: yield: 21%; FT-IR νmax/cm−1; 2963 (C–H), 1590 (CC); UV-vis λmax (nm) (logε) in DMSO; 330 and 676. Anal. calcd for C164H160N24O4Co; C 76.05, H 6.23, N 12.98; found: C 76.02, H 6.19, N 12.93. MS: m/z: calcd: 2588.25 amu; found: [M + H]+ 2589.5 amu.
Electrochemical measurements
Cyclic voltammetry measurements were performed using a three electrode standard configuration with a platinum wire as counter electrode and Ag/AgCl in saturated KCl as reference electrode in a 0.05 M TBABF4 (tetra-n-butylammonium tetrafluoroborate) acetonitrile solution as electrolyte. A FTO conducting glass substrate coated by a thin film of compound 7 or 8 in DMF (5 mg mL−1) deposited by drop casting was used as a working electrode (estimated film thickness 1 μm). The thickness of the films was measured by a MicroXAM-100 3D surface profilometer. The solution was purged with N2 for at least 30 min before measurements. Scan rate of 50 mV s−1. These electrodes were connected to an AMEL 7050 potentiostate that allows scanning the voltage while measuring the current.Conductivity measurements
Conductivity experiments were carried out using a conductimeter Crison GLP31.Photoluminescence measurements
Steady-state photoluminescence measurements were recorded in a Photon Technology International (PTI) 220B spectrofluorimeter having a Xe arc lamp light excitation and Czerny–Turner monochromator, coupled to a photomultiplier. Time-resolved measurements were performed using a PTI Easylife with 375 nm diode.Optical characterization
Steady-state absorption spectra in the visible and near-IR regions were measured on a Shimadzu UV 3600 spectrometer. Steady-state fluorescence spectra were measured on a Shimadzu RF-5300PC spectrofluorophotometer.Laser flash photolysis spectroscopy (LFP)
Laser flash photolysis experiments were carried out using the third (355 nm) harmonic of a Q-switched Nd:YAG laser (Spectron Laser Systems, UK; pulse width ca. 9 ns and 35 mJ per pulse). The signal from the monochromator/photomultiplier detection system was captured by a Tektronix TDS640A digitizer and transferred to a PC that controlled the experiment and provided suitable processing and data storage capabilities.Acknowledgements
Financial support from the Ministry of Science and Innovation of Spain, (CTQ 2012–2013) is gratefully acknowledged. P.A. also thanks the Spanish Ministry of Science and Innovation by a Ramon y Cajal research associate contract (RYC-2012-10702) and the UPV for the grant PAID-06-12.References
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- J. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS PubMed.
- G. de la Torre, P. Vaquez, F. Agullo-Lopez and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef CAS PubMed.
- P. A. Liddell, D. Kuciauskas, J. P. Sumida, B. Nash, D. Nguyen, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1997, 119, 1400–1405 CrossRef CAS.
- J. J. Cid, M. Garcia-Iglesias, J. H. Yum, A. Forneli, J. Albero, E. Martinez-Ferrero, P. Vazquez, M. Gratzel, M. K. Nazeeruddin, E. Palomares and T. Torres, Chem.–Eur. J., 2009, 15, 5130–5137 CrossRef CAS PubMed.
- T. Morimume, H. Kajii and Y. Ohmori, IEEE Photonics Technol. Lett., 2006, 18, 2662–2664 CrossRef.
- T. A. Moore, D. Gust, P. Mathis, J.-C. Mialocq, C. Chachaty, R. V. Bensasson, E. J. Land, D. Doizi, P. A. Liddell, W. R. Lehman, G. A. Nemeth and A. L. Moore, Nature, 1984, 307, 630–632 CrossRef CAS PubMed.
- S. Sürgün, Y. Arslanoğlu and E. Hamuryudan, Dyes Pigm., 2014, 100, 32–40 CrossRef PubMed.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS PubMed.
- J. L. Sessler, V. Král, T. V. Shishkanova and P. A. Gale, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4848–4853 CrossRef CAS PubMed.
- C.-H. Lee, H.-K. Na, D.-W. Yoon, D.-H. Won, W.-S. Cho, V. M. Lynch, S. V. Shevchuk and J. L. Sessler, J. Am. Chem. Soc., 2003, 125, 7301–7306 CrossRef CAS PubMed.
- J. G. Young and W. Onyebuagu, J. Org. Chem., 1990, 55, 2155–2159 CrossRef CAS.
- A. Aydogan and A. Akar, Chem.–Eur. J., 2012, 18, 1999–2005 CrossRef CAS PubMed.
- M. Alvaro, P. Atienzar, P. de la Cruz, J. L. Delgado, V. Troiani, H. Garcia, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6626–6635 CrossRef CAS PubMed.
- F. D'Souza, P. M. Smith, S. Gadde, A. L. McCarty, M. J. Kullman, M. E. Zandler, M. Itou, Y. Araki and O. Ito, J. Phys. Chem. B, 2004, 108, 11333–11343 CrossRef.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS PubMed.
- J. W. Owens, R. Smith, R. Robinson and M. Robins, Inorg. Chim. Acta, 1998, 279, 226–231 CrossRef CAS.
- M. Durmuş and T. Nyokong, Spectrochim. Acta, Part A, 2008, 69, 1170–1177 CrossRef PubMed.
- M. Idowu and T. Nyokong, J. Photochem. Photobiol., A, 2009, 204, 63–68 CrossRef CAS PubMed.
- J. C. Scaiano, CRC handbook of organic photochemistry, ed. J. C. Scaiano, CRC Press, Boca Raton, Fla, 1989 Search PubMed.
- J. Mack and M. J. Stillman, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Amsterdam, 2003, pp. 43–116, DOI:10.1016/b978-0-08-092390-1.50008-4.
- C. Bohne, Chem. Soc. Rev., 2014, 43, 4037–4050 RSC.
- M. Barra, C. Bohne and J. C. Scaiano, Photochem. Photobiol., 1991, 54, 1–5 CrossRef CAS PubMed.
- M. Barra, Supramol. Chem., 1997, 8, 263–266 CrossRef CAS PubMed.
- J. C. Scaiano, J. Am. Chem. Soc., 1980, 102, 7747–7753 CrossRef CAS.
- A. Ogunsipe and T. Nyokong, J. Mol. Struct., 2004, 689, 89–97 CrossRef CAS PubMed.
- F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410–1422 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |