
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn overview of the recent advances and future prospects of three-dimensional particle electrode systems for treating wastewater
Mingyue Piao
ab,
Hongxue Du
 *a and
Honghui Teng
*a
*a and
Honghui Teng
*a
aKey Laboratory of Environmental Materials and Pollution Control, Education Department of Jilin Province, Jilin Normal University, 1301 Haifeng Road, Siping 136000, China. E-mail: duhx_jlsd@163.com; tenghonghui@163.com
bCollege of Engineering, Jilin Normal University, Siping, China
First published on 2nd September 2024
Abstract
Three-dimensional (3D) electrochemical technology is considered a very effective industrial wastewater treatment method for its high treatment efficiency, high current efficiency, low energy consumption, and, especially, ability to completely mineralize nonbiodegradable organic contaminants. Particle electrodes, which are the fundamental components of 3D electrochemical technology, have multiple functions in the electrochemical reaction process. Various types of particle electrodes have been created and applied for wastewater treatment. Herein, we present a thorough analysis of the research and development of particle electrodes used for electrocatalyzing pollutants. Initially, reactor designs, factors affecting the removal efficiency of pollutants and degradation mechanisms are introduced. In particular, a detailed investigation is conducted into the selection of particle electrode materials and the roles they play in the 3D electrochemical treatment of wastewater. Subsequently, the degradation efficiency and energy consumption associated with 3D electrochemical technology for different pollutants are investigated. Finally, the directions and outlook for further studies on particle electrodes are discussed. We believe that this review will offer a useful perspective on the development and application of particle electrodes for wastewater purification.
 Mingyue Piao | Mingyue Piao received her PhD in Environmental Engineering from Jilin University in 2019. Her current research focuses on the design and application of electrocatalysts for environmental pollution remediation and hydrogen production. |
 Hongxue Du | Hongxue Du received his ME degree from Jilin Normal University in 2020. His current research focuses on the design, fabrication, and application of advanced electrocatalysts for wastewater treatment. |
 Honghui Teng | Honghui Teng, supervisor of doctoral students, received his PhD in Analytical Chemistry from Northeastern University in 2015. His research interests are mainly focused on environmental monitoring, environmental pollution control and resource utilization. |
1. Introduction
The discharge of various contaminants into water bodies has led to serious issues concerning both the ecological environment and human health, and thus, millions of people worldwide lack access to safe drinking water.1,2 Especially, persistent organic pollutants, including pesticides, antibiotics, pharmaceuticals, and personal care products, as well as endocrine disrupting chemicals, pose a significant challenge as they are not effectively broken down or eliminated by traditional wastewater treatment systems.3 Therefore, there is a pressing need for adaptable water platforms and technologies that can cater to refractory wastewater treatment. Electrocatalytic processes, which combine redox reactions and adsorption, are particularly promising owing to their high efficiency in generating multiple oxidants in situ, minimal space requirements, and ease of operation and management. Thus, through the proper design of reactors and optimization of electrodes, electrocatalytic processes can effectively degrade refractory pollutants.4Electrodes used in electrocatalytic oxidation can be classified as 2D or 3D based on their structural makeup. Conventional planar 2D electrodes have a smaller effective electrode area and a smaller reactor with a lower unit processing capacity. However, the low current efficiency of this type of electrode prevents it from being widely used in the industry.5 In early 1969, Backhurst et al. introduced the concept of a 3D electrode, incorporating a conductive filler into a 2D-ER to create a third electrode between the cathode and anode.6 The presence of particle electrodes can address the limitation of a confined reaction zone by polarizing particle electrodes into charged entities, enabling the reaction to occur across the entire reactor and expanding the reaction zone. Additionally, the rough surface, porous structure, and large specific surface area of particle electrodes can enhance the unit space-time yield and current efficiency, thereby improving electrochemical oxidation efficiency. As a result, there has been a growing interest in studies focusing on the treatment of different types of wastewaters using 3D electrodes in recent years.7–11
Several studies have shown that utilizing a 3D-ER with particle electrodes can significantly enhance pollutant removal efficiency and reduce EEC compared to a traditional 2D-ER. Shen et al. found that the expanded reaction area and improved mass transfer in particle electrodes led to a higher removal efficiency and reduced EEC.12 Alighardashi et al. demonstrated that a 3D-ER with particle electrodes operated as micro-electrolysis cells, resulting in higher efficiency compared to 2D-ER.13 Wei et al. showed that combining GAC and PCP as particle electrodes improved the COD removal efficiency and reduced the EEC.14 Can et al. reported that the removal efficiency of COD in the 3D-ER was about 50%, while it was 20% in the 2D-ER. Also, the EEC was about 180 kW h kg−1 COD, which was much lower than that in the 2D-ER (300 kW h kg−1 COD).5 Cho et al. developed a continuous-flow 3D-ER with GAC electrodes for the removal of IBP. The IBP removal efficiency was 98% in 4 h, which was 2.5-times higher than that in the 2D-ER.11 Shi et al. demonstrated that GAC in the 3D system reduced the mass transfer resistance, with a total organic carbon (TOC) removal efficiency and reaction rate constant of 47.6% and 0.22 h−1, which are much higher than that in the 2D system (30.7% and 0.12 h−1), respectively.15 Using PAC as particle electrodes, the TOC removal efficiency in the 3D-ER was 94.1% after 180 min electrolysis, which was 10–19% higher than that in the 2D-ER.16 Overall, using particle electrodes in 3D electrode systems can enhance the pollutant removal efficiency and reduce the EEC.
Particle electrodes are regarded as critical in the performance of electrocatalytic oxidation in 3D electrochemical systems. Metals, metal oxides, AC, and other carbon-based materials are the most commonly used particle electrodes in current studies. Precious metal-coated electrodes such as ruthenium (Ru), palladium (Pd), and platinum (Pt) demonstrate excellent electrocatalytic activity and stability. However, their high cost, scarcity, and limited durability hinder their practical application.17,18 In contrast, transition metals such as iron (Fe), cobalt (Co), nickel (Ni), copper (Cu), and manganese (Mn) are relatively inexpensive and more abundant compared to precious metals.19,20 Nevertheless, their industrial application is still limited due to their short lifespan, narrow pH range, and low cost-effectiveness. Additionally, metal or metal oxide electrodes may release metal ions into solution under an electric field, leading to secondary pollution. In this case, carbonaceous materials offer advantages such as resistance to acids and alkalis, absorbability, catalytic activity, and environmental compatibility, making them promising green materials in electrocatalytic systems. Various carbon materials, including graphite,21 AC,22 and BC,23 have been employed as particle electrodes in electrochemical reactors. However, the electrochemical efficiency of these carbon materials remains unsatisfactory due to their poor electron transfer capability and electrical reactivity.
This study highlights the advantages of particle electrodes in terms of pollutant removal efficiency and EEC. It provides a detailed review of particle electrodes used in 3D-ER, analyzes their roles in the electrochemical treatment of pollutants, and investigates the materials for the fabrication of particle electrodes, factors influencing the electrochemical degradation process, and degradation mechanisms of pollutants. Future prospects for the development of particle electrodes are proposed to enhance their applicability in practical wastewater treatment.
2. Brief introduction to 3D electrochemical technology
2.1 Reactor structure of 3D-ER
Based on the polarity of reactors, they can be categorized into two types, monopolar and bipolar.24 In a monopolar reactor, a diaphragm separates the main electrode, creating a cathode zone and an anode zone. When the anode area is filled with particles (as shown in Fig. 1a), these particles acquire the same charge distribution as the main electrode, essentially extending the main electrode and increasing the reaction area. Simultaneously, electrochemical reactions occur on both the main electrode and the particle electrodes, with the anode zone oxidizing organic pollutants and the cathode zone reducing metal ions. In a bipolar reactor (Fig. 1b), the particle electrodes are charged through electrostatic induction induced by an electric field. The charged particles on the surface become independent 3D electrodes, leading to electrochemical oxidation and reduction reactions at both ends of the particles. Each charged particle effectively forms a ‘micro electrolysis cell’, significantly enhancing the area for the electrochemical reaction.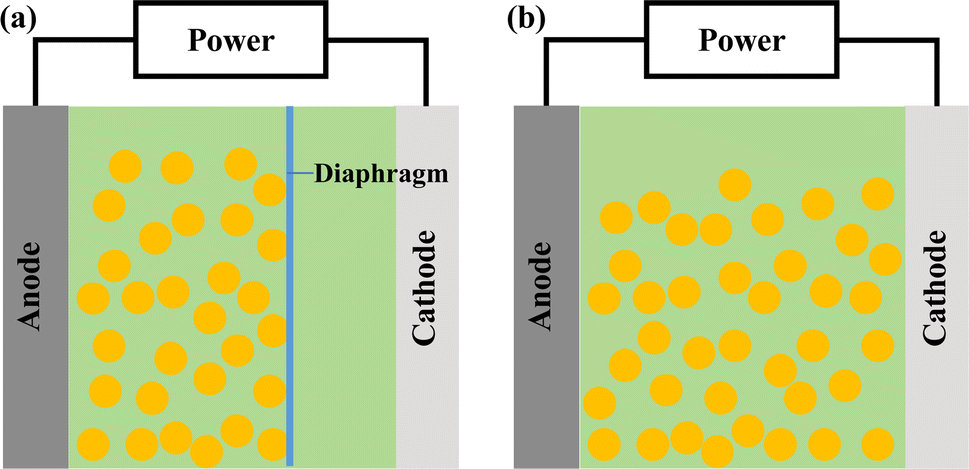 | ||
| Fig. 1 Working structure of a 3D-ER: (a) monopolar; (b) bipolar. | ||
Current research primarily focuses on bipolar reactors. As is illustrated in Fig. 2, the bipolar reactor operates through three types of current, as follows: (1) bypass current, which is generated by charge movement in the electrolyte without passing through the particle electrodes; (2) reaction current, which is created by electric charge entering the particle electrode from the electrolyte, and then returning to the electrolyte; and (3) short-circuit current, where electric charge enters the particle electrodes directly from the main electrode without going through the electrolyte, moving along the adjacent particle electrodes. The degradation of compounds primarily depends on the reaction current, and therefore reducing the short-circuit current and bypass current can enhance the reaction current, leading to an improved electrochemical oxidation efficiency.25
 | ||
| Fig. 2 Current model in a 3D-ER. | ||
The reactor can be classified into two types based on the states of the particle electrodes, i.e., fixed bed and fluidized bed. In a fixed bed reactor, the particle electrodes remain in a stationary state, known as a 3D fixed bed reactor. Conversely, in a fluidized bed reactor, the particle electrodes are in a flowing state, including a moving bed, eddy current bed, and jet bed configuration.
 | ||
| Fig. 3 3D-ER with particle electrodes of different densities. | ||
2.2 Influencing factors
In 3D electrode systems, the degradation of pollutants is influenced by various factors, including the properties of the pollutants, external environmental factors, and the characteristics of the particle electrodes.31 The external environmental factors include pH, applied potential, particle electrode dosage, current density, and treatment time. For instance, Piao et al. designed a 3D-ER with stainless steel as the cathode and anode and modified BC as a particle electrode and found that under neutral condition, an increase in applied potential can accelerate the chemical reaction; however, an excessively high value can hinder the process.32,33 Increasing the current density within an appropriate range can enhance the pollutant removal efficiency because a higher current density boosts the repolarization degree of particle electrodes, thereby increasing the effective electrode area for electrooxidation reactions.34 Time is a crucial factor, given that an excessively long duration incurs additional costs, while very short duration results in a poor degradation efficiency.35,36 The impact of pH varies for different substances, with some being significantly affected, while others are not sensitive. The research by Song et al. revealed that the removal efficiency of NOR using sulfur–zinc-modified kaolin–steel slag particle electrodes was 100% under acidic condition, above 90% under neutral condition, and more than 80% under alkaline condition.37 In the case of certain pollutants such as drugs and herbicides, a wide pH range (pH 3–10) can yield optimal degradation outcomes.21,38,39The properties of electrode materials affect the degradation efficiency of contaminants, including the loaded active material, surface area, porosity, particle size, and even shape. For example, Gedam and Neti treated recalcitrant chemical industry wastewater using a GAC particle electrode. According to the results, the apparent Faradaic efficiency and specific electrical EEC were estimated to be 3.42% and 6.59 kW h kg−1 COD for the GAC with a higher surface area and 0.78% and 28.65 kW h kg−1 COD for the GAC with a lower surface area, respectively.40 The PAP degradation results showed that the activity of the particle electrodes (Ti–Cu–Ni–Zn–Sb–Mn@AC) supported by block honeycomb AC was much better than that of the particle electrodes supported by granular AC.41 Pd supported multi-walled carbon nanotubes (Pd@MWCNTs) were synthesized for the removal of 4-CP, and the removal efficiency was significantly affected by the size of the loaded Pd nanoparticles, following the order of Pd-6.4 nm (100%) > Pd-9.5 nm (60%) > Pd-13.1 nm (29%).42 Xie et al. designed BC-loaded particle electrodes, and the effects of different loaded catalyst on the removal of 4-CP were studied. The removal efficiencies were found to follow the order of Mn@BC (99.93%) > Sn@BC (99.0%) > Sb@BC (91.5%).43
To investigate the impact of particle position on the electrocatalytic reaction rate, a study was conducted using spherical graphite particles as particle electrodes and MB as the model pollutant. The results obtained through the COMSOL Multiphysics software revealed that the degradation efficiency of MB by the particle electrodes within the electrocatalytic reactor was not uniform. The closer the particle electrode was to the anode, the stronger the pollutant degradation ability. When the particle electrode was positioned near the anode, the potential difference in the particle electrode increased, thereby facilitating the electrocatalytic reaction. Additionally, the proximity of the particles to the anode reduced the mass transfer distance for electron convection and pollutant diffusion, which enhanced the rate of the electrocatalytic reaction. Moreover, the central position of the particle electrodes can enhance the electrocatalytic efficiency, primarily due to the lower central potential of the spherical particle electrodes. This characteristic increased the potential difference by interacting with the potential of the external electrolyte, thereby improving the electrocatalytic efficiency at the central position of the particle electrodes. A study also noted that an increase in electric field intensity and particle size led to a reduction in the disparity in the reaction rates.44
2.3 Electrocatalysis mechanism
The mechanism for the 3D electrolytic treatment of pollutants is intricate and multifaceted, involving factors such as wastewater composition, electrode materials, and particle electrodes. Fig. 4 illustrates the key principles of this mechanism, with most pollutants being degraded through oxidation processes. The electrochemical oxidation of pollutants can be categorized into direct oxidation, where pollutants are degraded through electrochemical reactions on the anode, and indirect oxidation, where intermediate substances with strong oxidizing properties are produced through electrode reactions. The most common intermediate is ·OH, a potent oxidant with an oxidation potential of 2.8 V, which can effectively degrade components.19,45–47 Another method involves the indirect oxidation of pollutants using the anions present in the water to generate strong oxidizing substances such as peroxydisulfate (S2O82−) and active chlorine.48,49 Additionally, the reversible cycle of high- and low-valence metal ions, such as Fe3+/Fe2+,50 Cu2+/Cu+,48 Ni3+/Ni2+,47 Ce4+/Ce3+,51 and Co3+/Co2+,52,53 is utilized to continuously oxidize and remove pollutants. This cyclic process involves the oxidation of low-valence cations or metal oxides to high-valence oxides, and then reduced back to the low-valence state after pollutant degradation, ensuring effective pollutant removal. | ||
| Fig. 4 General electrocatalysis mechanism in a 3D-ER. | ||
In addition to active substances, non-active substances can also degrade contaminants. For example, Xu et al. found that the indirect electrochemical degradation of TC was dominated by the non-radical pathway, where singlet oxygen (1O2), rather than free radicals (·OH and superoxide radical (·O2−)), played a key function.54 Based on trapping experiments and electron spin resonance (ESR) analyses, Wang et al. determined that ·SO4−, ·OH, and 1O2 were all involved in the 3D-PMS process, while 1O2 was the dominant ROS for NOR degradation.55 Quenching experiments, probe experiments, and electron paramagnetic resonance (EPR) analyses suggested that ·OH and 1O2 were the main ROS during the removal of CBZ by N–Mn@BC as particle electrodes, and the non-radical oxidation played a key role.20 The degradation mechanism of CBZ is shown in Fig. 5. Zhao et al. found that the surface-bound ·O2− radicals and 1O2 generated by N–Fe@BC played a crucial role in the decomplexation and removal of Ni–EDTA, and the contribution of different free radicals to its degradation was different.23 Ni–Fe@GAC was used as a particle electrode for the degradation of SMA in a 3D-ER with sulfite hetero activator. Quenching experiments and EPR demonstrated that the degradation of SMA was the result of both free and non-free radicals, with ·OH, ·SO4− and 1O2 contributing 51%, 12% and 37% to SMA removal, respectively.56
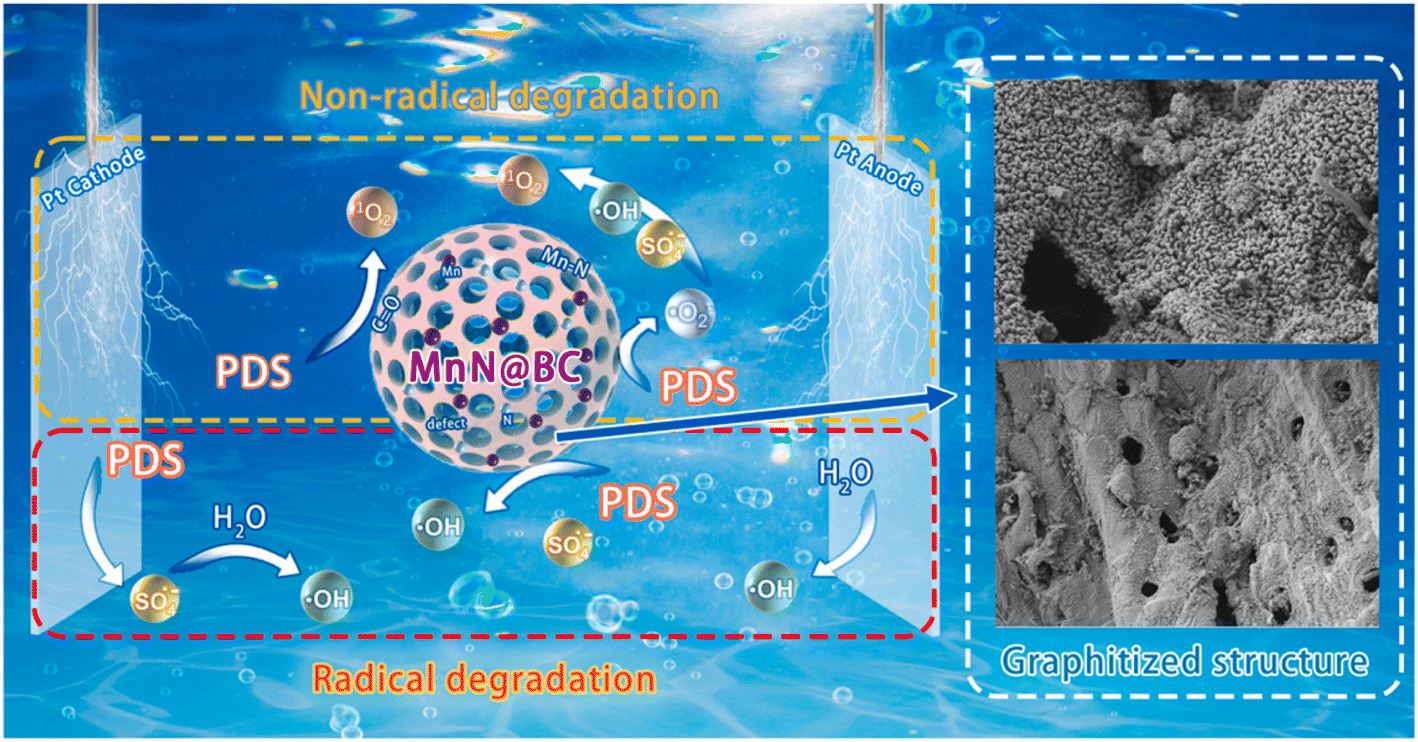 | ||
| Fig. 5 Degradation mechanism of CBZ by non-radical active substances and radical active substances. Reproduced from ref. 20 with permission from [Elsevier], copyright [2023]. | ||
The removal mechanism of some pollutants not only involves oxidation but also reduction. For example, Co@AC was used in a continuous electrochemical reactor to degrade HA. The ESR signals indicated that both H· and ·OH were catalytically generated by Co@AC. The degradation of HA was achieved by both electro-oxidation and electro-reduction.57 During the degradation of 4-CP, reduction by atomic H· was responsible for the dechlorination of 4-CP.42
3. Current research direction of 3D electrochemical technology
3.1 Materials of particle electrodes
Electrode materials are crucial components in 3D-ER. The primary factors to consider when selecting electrode materials include cost, durability, electrolysis efficiency, and the physical and chemical properties of the wastewater being treated. Many studies have utilized GAC as a particle electrode due to its excellent chemical stability and biocompatibility. For example, Zhang et al. incorporated GAC as the third electrode in a 3D-ER setup to degrade SMX and TC, achieving impressive removal efficiencies of 88.9–93.5% and 89.3–95.6% for SMX and TC, respectively.58 Cho et al. developed a continuous 3D-ER with GAC as the third electrode for the removal of IBP.11 Furthermore, researchers have modified GAC-based 3D-ER to enhance their performance. For instance, Bu et al. enhanced the current efficiency of a 3D-ER for the degradation of SA by utilizing Co–Mn@GAC.59 Li et al. treated SMA wastewater in a 3D-ER using Ni–Fe-loaded GAC as a particle electrode, which improved the oxygen evolution potential (OEP) and suppressed side reactions, facilitating the electrochemical oxidation of SMA.56 Although some studies have shown the use of alternative materials such as pyrolusite,60 mud,61 carbon nanotubes,50 clay,17,62–64 graphene,65 aerogels,66–68 and steel slag69 as particle electrodes for 3D-ER, GAC remains a popular choice due to its efficiency and cost-effectiveness.56,70–73In recent decades, there has been increasing focus on synthesizing component particle electrodes using various materials to enhance their properties.43 The commonly used loading elements include precious metals, rare metals, and transition metals. Precious metals and rare metals are known for their high activity, wide applicability, and long lifespan, but their high cost limits their widespread industrial use.74,75 Transition metals also exhibit strong catalytic activity, but they are prone to loss as the active components and have a relatively short service life. Previous studies have shown that precious metals (such as Ru, iridium, Pt, and lead) and rare metals (such as tungsten and tantalum) are less commonly used compared to transition metals.76 Transition metal oxides, including Co,77 Cu,61 Fe,78 Mn,79 Ni,80 titanium (Ti),81 and tin (Sn),82 are frequently utilized. Among them, Fe-containing materials are often chosen due to their dual role as a carrier for active materials and as active materials, releasing Fe2+ to generate highly oxidized ·OH for electrocatalytic reactions.83–86 Additionally, Fe and aluminum (Al) have electrocoagulation effects, which are crucial for pollutant removal.87,88 SnO2 is a semiconductor composite metal oxide with high OEP and good conductivity.64 When SnO2 is doped with metal-based cations, it forms a semiconductor solid solution with crystal defects, creating a surface with numerous holes and active sites, which produce crystal oxygen with stronger oxidation potential than undoped SnO2.22,89,90
In addition to metal elements, graphene can also be used to improve the catalytic properties of the base material. BC was decorated by graphene to synthesize a carbon composite (graphene@BC) as a particle electrode. The graphene@BC electrode had a higher OEP (1.76 V) and lower Tafel slope (0.013 V dec−1) than that of the bare BC. Coating graphene improved the electrical conductivity and electron aggregation on surface of BC, resulting in the greater generation of ROS.54 In addition, some scholars used organic catalysts for electrocatalytic oxidation. The conductive polyurethane–polypyrrole@graphene was prepared via an in situ oxidative polymerization method and used it as a particle electrode to degrade LEV in a 3D-ER. The results showed that more than 90% of LEV was degraded and the EEC was 20.12 kW h g−1 LEV under the condition of pH of 7, voltage of 6 V, aeration volume of 2.0 L min−1, initial LEV concentration of 20 mg L−1, and Na2SO4 of 7 mM. The toxicity evaluation using luminescent bacteria showed that the toxicities of some intermediates were higher than the parent compounds, but the toxicity of the degradation process for LEV was decreased effectively.91 An Ni3(HITP)2@graphene-based composite aerogel particle electrode was developed for the oxidation removal of phenol by 3D electrode technology. The graphene layer converted H2O2 into ·OH using the 1e− excitation of Ni3(HITP)2 and the microelectrode action of the particle electrodes. Furthermore, the simultaneous generation of ·OH and ·O2− in the system greatly reduced the dependence on an acidic environment and broadened the scope of utilization.92
3.2 The roles of particle electrodes
To accurately understand the mechanism of the 3D electrochemical process and develop particle electrodes with excellent performances, it is essential to thoroughly investigate the roles played by these electrodes in the 3D-ER. A schematic diagram of the roles played by particle electrodes in the 3D-ER is shown in Fig. 6. | ||
| Fig. 6 Roles played by particle electrodes in a 3D-ER. | ||
Electrosorption is a phenomenon that occurs on the surface of particle electrodes during electrolysis. It involves the adsorption of dissolved chemical species on an electrode through surface binding induced by an electric field. Electrosorption occurs when the electrode is polarized by an applied voltage.96 In a 3D-ER system, positive and negative charges accumulate on both sides of the particle electrodes due to the external voltage. This leads to the charged ions in the electrolyte solution moving towards the oppositely charged side of the particle electrodes under the influence of Coulomb force, resulting in electrosorption.96,97 The combination of electrosorption and electrooxidation effectively enhances the degradation of pollutants. When the voltage applied to the reactor is low, the removal efficiency of pollutants is primarily influenced by the adsorption and electrosorption.98 Andrés García et al. constructed a 3D-ER for treating greywater, and demonstrated that the synergistic effect of electrosorption and electrooxidation using GAC particle electrodes significantly improved the removal efficiency of COD and TOC in the wastewater.99
Particle electrodes exhibit the capability to remove contaminants such as heavy metals and nitrate nitrogen through reductive processes. The research conducted by Zhang et al. demonstrated a significant decrease in wastewater toxicity when Cr(VI) was reduced to Cr(III) using the high electrocatalytic reduction ability of P@rGO particle electrodes.68 Ye et al. investigated the electro-reduction of nitrate utilizing particle electrodes with Co as a catalyst. Their findings indicated that nitrate in the wastewater was either directly reduced by Co at the cathode side of the electrode, or indirectly through reaction with the H· radical catalytically produced by Co. Furthermore, the utilization of particle electrodes extended the oxidation–reduction reactions of contaminants, leading to a reduction in the mass transfer distance between the reactants and electrodes, thereby significantly increasing the effective surface area for the reactions. Consequently, this enhancement resulted in an improved electrolysis efficiency and current efficiency and reduced the EEC in the reaction system.100
3.3 Coupling of 3D-ER with other methods
In combination with other methods, such as biodegradation, photocatalysis, ultrasonic, and ozone, the degradation efficiency can be further improved and the EEC be reduced.101,102 One common method, 3D-BER, involves combining biological processes with electricity.103–105 The synergy between electricity and microbes in 3D-BER is primarily due to the fact that appropriate electrical stimulation can enhance microbial degradation because electrochemical processes can help to transform refractory organic pollutants into intermediate products that can be utilized by microorganisms.106,107 Additionally, more CO2 is generated in 3D-BER, providing an inorganic carbon source for autotrophic microorganisms and serving as a pH buffer for the reaction process. Li et al. designed a 3D-BER with GAC as a particle electrode, which promoted biofilm formation due to its high specific surface area and good conductivity. They achieved over 95% removal of TBBPA in 120 min at 5 V. The synergy of electricity and the biofilm in TBBPA degradation was significant, given that electrical stimulation enhanced the microbial activity, accelerating debromination and the decomposition of TBBPA.108 Guo et al. designed polyaniline-loaded AC as a 3D particle electrode in a 3D-BER for removing DEX from micro-polluted oligotrophic groundwater. This system achieved a DEX removal efficiency of 95.7%, which was 14.1% higher than that in the 2D bio-electrochemical system. This improvement was attributed to the abundance of functional microbes with electron transfer ability and reductive dehalogenating genera in the 3D system, leading to a higher copy number of functional genes.109 Sun and Zhu demonstrated that the MNZ removal kinetics in a 3D-BER system followed the pseudo-first-order model, with a maximum rate constant of 0.853 h−1. This value was significantly higher compared to that of pure microorganisms and pure electrochemical reactors by 4.1 and 2.8 times, respectively.110 They further developed a 3D biofilm electrode magnetism reactor (3D-BEMR) for the removal of NPX. The results showed that the average removal efficiency of NPX in 3D-BEMR was 88.36%, marking a substantial improvement over the 3D-biofilm reactor, 3D biofilm magnetism reactor, and 3D-BER by 75.24%, 65.03%, and 12.36%, respectively. This enhancement was attributed to the effects of electro-magnetic adsorption, electro-oxidation catalysis, and electro-magnetic biodegradation, as well as the presence of high-abundance genera in 3D-BEMR.111 The possible degradation mechanism of coking water by GAC in the 3D-BER is shown in Fig. 7. | ||
| Fig. 7 Possible degradation mechanism of coking water by GAC in the 3D-BER. Reproduced from ref. 104 with permission from [Elsevier], copyright [2023]. | ||
The study conducted by An et al. demonstrated that a photoelectrochemical process utilizing MnO2@GAC as particle electrodes and TiO2 as the photocatalyst outperformed electrochemical oxidation alone in terms of MB degradation. Specifically, the photoelectrochemical process achieved a decolorization efficiency of 95% and COD reduction of 87%, whereas the single electrochemical process only achieved a decolorization efficiency of 78% and COD reduction of 68%.112 This team also studied the photoelectrocatalytic degradation of oxalic acid. Their findings indicated that oxalic acid could be degraded more effectively through the photoelectrocatalytic process compared to degradation through photocatalytic or electrochemical oxidation individually.113 BiFe@Bent particle electrodes were utilized in a 3D-EF system to degrade EX under visible light. This study revealed that EX was degraded into CO2, H2O, and SO42− through photoelectrocatalytic degradation. The exceptional oxidative capacity of BiFe@Bent was attributed to its efficient generation of H2O2, multiple sources of active species, and involvement of all active species (·OH, h+ and ·O2−). The possible mechanism is shown in Fig. 8.114 CuFe2O4@RM was synthesized and utilized for the photoelectrochemical activation of persulfate in the degradation of CIP. The highest removal efficiency of CIP (71.39%) was observed after 60 min under the optimized conditions of CuFe2O4@RM dosage of 1.0 g L−1, persulfate concentration of 9 mM, pH of 7, and applied current of 640 mA. Additionally, the maximum removal efficiencies of COD and TOC in the CIP solution were approximately 49.21% and 35.7% after 60 min, respectively. This study revealed the synergistic effects of visible light, electric field, and persulfate in the photoelectrochemical-persulfate system.115,116
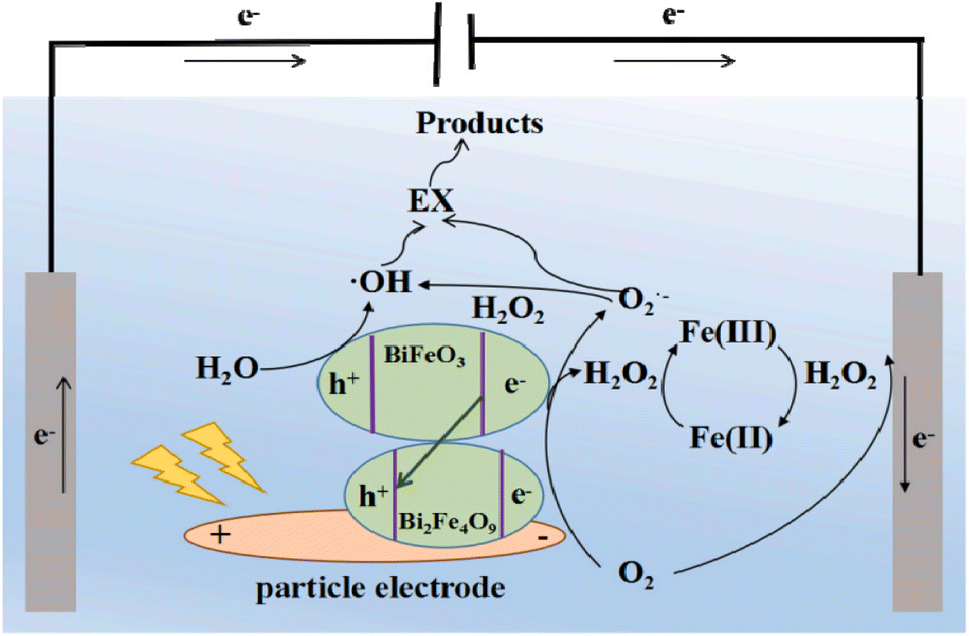 | ||
| Fig. 8 Possible mechanism for the photoelectrocatalytic degradation of EX by BiFe/Bent particle electrodes. Reproduced from ref. 114 with permission from [Elsevier], copyright [2021]. | ||
A significant synergistic effect was also observed when electrolysis, ozone, and GAC were combined in the treatment of NB, resulting in the remarkable removal of TOC (95.58%) within 120 min due to the abundant production of ·OH radicals. This combined process also demonstrated a higher energy efficiency ratio for ·OH production compared to individual electrochemical processes. The results indicated that ·OH electrochemical oxidation, peroxone reaction, GAC-catalyzed ozone reaction, and electro-reduction of ozone reactions contributed to 12.50%, 37.50%, 8.75%, and 31.25% of ·OH generation, respectively. The possible degradation mechanism of NB by GAC in the 3D-ER/O3 is shown in Fig. 9.45 The treatment of pharmaceutical production wastewater using electrolysis with 3D electrodes, ozonation, and their combined process (3D-ER/O3) was investigated by Zhan et al.116 The wastewater had a high organic concentration (TOC of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 475 mg L−1) and high acute toxicity (100% inhibition of luminescent bacterial Vibrio fischeri). After 6 h of treatment, individual ozonation and 3D-ER with GAC as particle electrodes removed approximately 23% and 43% of TOC, respectively. However, the acute toxicity of the wastewater was minimally reduced by these individual processes. In contrast, the 3D-ER/O3 process increased the TOC abatement to around 71% and reduced the luminescent bacterial inhibition to less than 70%. These improvements were primarily due to the enhanced ·OH production in the 3D-ER/O3 process through various reaction mechanisms, such as the reaction of sparged O3 with in situ-generated H2O2, electrochemical reduction of O3, and GAC-catalyzed O3 decomposition. Consequently, this enhanced the oxidation of organics in the bulk and that adsorbed on the GAC particle electrodes.
475 mg L−1) and high acute toxicity (100% inhibition of luminescent bacterial Vibrio fischeri). After 6 h of treatment, individual ozonation and 3D-ER with GAC as particle electrodes removed approximately 23% and 43% of TOC, respectively. However, the acute toxicity of the wastewater was minimally reduced by these individual processes. In contrast, the 3D-ER/O3 process increased the TOC abatement to around 71% and reduced the luminescent bacterial inhibition to less than 70%. These improvements were primarily due to the enhanced ·OH production in the 3D-ER/O3 process through various reaction mechanisms, such as the reaction of sparged O3 with in situ-generated H2O2, electrochemical reduction of O3, and GAC-catalyzed O3 decomposition. Consequently, this enhanced the oxidation of organics in the bulk and that adsorbed on the GAC particle electrodes.
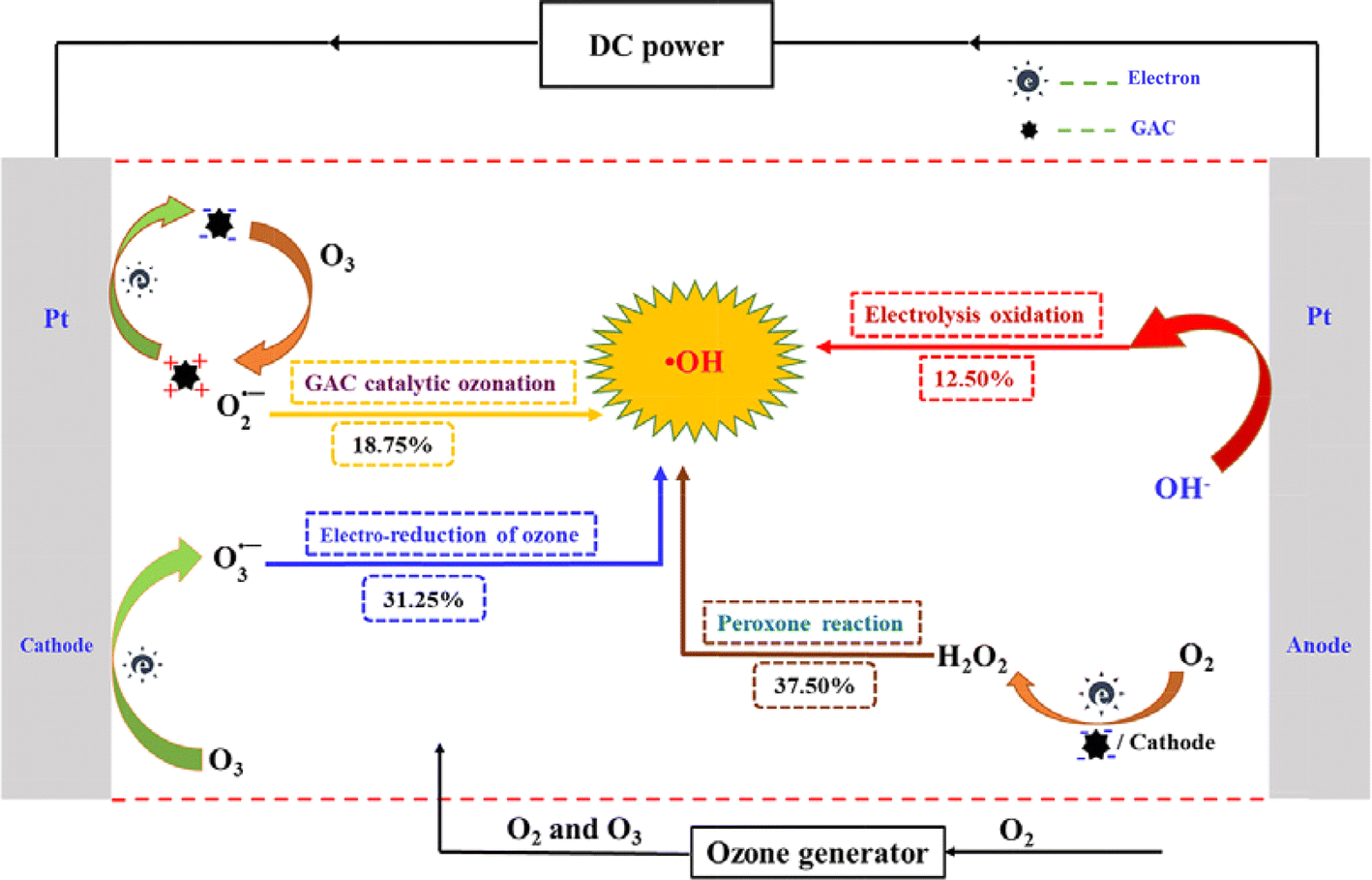 | ||
| Fig. 9 Possible degradation mechanism of NB by GAC in 3D-ER/O3. Reproduced from ref. 45 with permission from [Elsevier], copyright [2020]. | ||
4. Applications for typical pollutants
4.1 3D-ER used for medicine treatment
Antibiotics are one of the most commonly used and successful drugs in human and animal medicine. Nevertheless, the extensive use of antibiotics has led to the spread of antibiotic-resistant bacteria, presenting a great threat to the human body and environment. As can be seen in Table 1, many medicines can be treated by 3D electrochemical systems. Ti–Sn–Sb@γ-Al2O3 particle electrodes were prepared and utilized for the degradation of OTC. The removal efficiencies of OTC and TOC achieved were approximately 92.0% and 41.0%, respectively, under the optimal operating conditions. Furthermore, it was observed that the main role played by the Ti–Sn–Sb@γ-Al2O3 particle electrodes in the 3D electrode electrolysis process was the potent oxidizing capability of ·OH.90 CuFe2O4 magnetic nanoparticles were utilized in a 3D electrochemical process for the degradation of ATZ. The highest efficiency in ATZ degradation (>99%) and removal of TOC (22.1%) were observed after 35 min with a CuFe2O4 dosage of 3.0 g L−1, Na2S2O8 concentration of 4.0 mM, current density of 4 mA cm−2, and initial pH of 6.3. The CuFe2O4 electrodes demonstrated good stability over five runs under the optimal conditions. The primary reactive radicals involved in the ATZ degradation process were sulfate radicals (·SO42−).49 Song et al. discovered that volcanic rocks could efficiently degrade NOR at a low voltage (4 V) within 40 min across a wide pH range (3–11). In acidic environments, the removal of NOR exceeded 85%. After six cycles, the weight loss rate of the volcanic rocks was measured to be 3.87%. The volcanic rock 3D system generated ·OH and O2−.123 The surface of the volcanic rock was identified as the primary site for NOR degradation.123 Rahmani et al. investigated the electrocatalytic performance of porous carbon felt/PbO2 and planar Ti/PbO2 anodes in a 3D-ER system with GAC particles for the synergistic degradation of DIU herbicide. Under the optimal conditions (pH = 4.7, current density = 13.5 mA cm−2, Na2SO4 concentration = 0.04 M, and DIU concentration = 40 mg L−1), the 3D-ER system with carbon felt/PbO2 and Ti/PbO2 anodes achieved TOC mineralization efficiencies of 100% and 63.8% after 50 min, respectively. In the absence of GAC particles, the electrocatalytic efficiencies of the carbon felt/PbO2 and Ti/PbO2 anodes for TOC removal were 85.5% and 42.7%, respectively. Furthermore, the COD removal efficiencies after 300 min in the 3D-ER system with carbon felt/PbO2 and Ti/PbO2 electrodes were 95% and 62.5%, respectively.7 Zhou et al. investigated the degradation of BH using Fe3O4–SnO2@GO particle electrodes. The Fe3O4–SnO2@GO system exhibited superior electrocatalytic activity and impressive stability compared to 2D systems, achieving a BH removal efficiency of 94.8% within 90 min. Notably, even after five cycles, the ternary composite retained its strong pollutant oxidation capability. Quenching experiments indicated that ·OH served as the primary active species in the 3D-EF system, leading to the oxidation of BH into CO2, H2O, or other byproducts.125 Chen et al. investigated the electrochemical degradation of DOX in combination with PMS activation using VER powder as particle electrodes in a 3D-ER. The results demonstrated that the introduction of VER particles enhanced the electrochemical conversion efficiency of the 3D-ER by boosting the OEP and preventing oxygen evolution side reactions. Quenching experiments revealed that ·SO42− was the primary reactive oxygen species in the system.126| Main electrodes | Particle electrodes | Pollutants | Process parameters | Results | Ref. |
|---|---|---|---|---|---|
| A: Zr/Ti4O7, C: Ti | CuFe2O4 | Atenolol = 40 mg L−1, florfenicol = 40 mg L−1, diclofenac sodium = 40 mg L−1, COD = 432 mg L−1 | CPE = 0.1 g L−1, pH = 6.3, t = 120 min, j = 25 mA cm−2, Na2SO4 = 100 mM | DEAtenolol = 98.8%, DEFlorfenicol = 93.4%, DEDiclofenac sodium = 85.5%, EEC = 25.67 kW h m−3 COD | 117 |
| A: Ti/IrO2/RuO2, C: Ti | C-PTFE and Fe–Co@C | CBZ = 70 mg L−1 | CPE = 5 g L−1 of C-PTFE and 4 g L−1 of Fe–Co@C, pH = 3, t = 60 min, j = 10 mA cm−2, Na2SO4 = 50 mM | DECBZ = 100%, DECOD = 80%, EEC = 0.0077 kW h g−1 COD | 118 |
| A: Pt/Ti, C: Pt–Ti | N–Mn@BC | CBZ = 0.042 mM | CPE = 0.1 g L−1, pH = 7.9, t = 60 min, j = 20 mA cm−2, Na2SO4 = 50 mM | DE = 96.84%, EEC = 0.99 kW h g−1 CBZ | 20 |
| A: graphite, C: carbon | Fe@BC | SDS = 20 mg L−1 | CPE = 1.0 g L−1, pH = 3, t = 120 min, j = 4.66 mA cm−2 | DE = 92.7 ± 3.1%, EEC = 0.31 US per m3 | 119 |
| A: Ti/RuO2/IRO2/Ta2O5, C: SS | Co–Mn@GAC | SA = 50 mg L−1 | CPE = 194.4 g L−1, pH = 6.2, t = 120 min, U = 15 V, Na2SO4 = 1.67 g L−1 | DESA = 99.86%, DETOC = 89.68% | 59 |
| A: Ti/RuO2/IRO2/Ta2O5, C: SS | Cu–Mn@Halloysite | SA = 50 mg L−1 | CPE = 194.4 g L−1, pH = 6.0, t = 90 min, U = 12 V, Na2SO4 = 1.67 g L−1 | DESA = 99.84%, DETOC = 88.95% | 120 |
| A: Ti/IrO2/RuO2, C: graphite | Ni–Fe@GAC | SMA = 1 mg L−1 | CPE = 80 g L−1, pH = 7, t = 30 min, U = 5 V, Na2SO4 = 1 mM | DE = 96.91% | 56 |
| A: Ti/IrO2/RuO2, C: graphite | Ni–Fe@GAC | SMA = 1 mg L−1 | CPE = 3 g L−1, t = 30 min, U = 5 V, Na2SO4 = 10 mM | DE = 90.89% | 80 |
| A: graphite, C: graphite | MMT–Fe3O4@GH | ACV = 20 mg L−1, ARB = 20 mg L−1 | CPE = 5 g L−1, pH = 3.0–11.0, t = 15 min, U = 10 V, j = 1.39 mA cm−2, Na2SO4 = 20 mM | DEACV = 91–96%; DEARB = 96–98% | 121 |
| A: graphite, C: graphite | MMT–Fe3O4@rGO | ACV = 20 mg L−1 | CPE = 40 g L−1, pH = 3.0–11.0, t = 120 min, U = 20 V, Na2SO4 = 20 mM | DE = 100% | 38 |
| A: Ti/RuO2/IrO2, C: graphite | Steel slag@kaolin | NOR = 20 mg L−1 | CPE = 150 g L−1, pH = 3.0, t = 30 min, U = 4 V, Na2SO4 = 100 mM | DENOR = 96.02%, DECOD = 93.45%, EEC = 0.99 kW h m−3 | 122 |
| A: Pt/Ti, C: Pt/Ti | BC | NOR = 10 mg L−1 | CPE = 5 g L−1, pH = 7.2, t = 120 min, j = 18 mA cm−2, Na2SO4 = 50 mM, PMS = 10 mM | DE = 87%, EEC = 29.7 kW h g−1 TOC | 55 |
| A: Ti/RuO2/IrO2, C: graphite | Sulfur–zinc modified steel slag@kaolin | NOR = 20 mg L−1 | pH = 3–10, t = 90 min, U = 4 V, Na2SO4 = 100 mM | DE = 100% in acidic environment, DE > 90% in neutral environment, and DE > 80% in alkaline environment | 37 |
| A: Ti/RuO2/IrO2, C: graphite rod | Volcanic rocks | NOR = 20 mg L−1 | CPE = 150 g L−1, pH = 3–11, t = 40 min, U = 4 V, Na2SO4 = 100 mM | DE > 85% in acidic environment | 123 |
| A: Ti/RuO2/IrO2, C: Ti | N–Fe@carbon | m-cresol, MB, and CAP (concentration not mentioned) | CPE = 10 g L−1, pH = 3–10, t = 35 min, U = 5 V, j = 0.08 A cm−2 | DEm-cresol = 100%, DEMB = 100%, DECAP = 100% | 124 |
| A: Ti/RuO2/IrO2, C: SS | Graphene-BC | TC = 5 mg L−1 | CPE = 2 g L−1, pH = 3–11, t = 120 min, U = 10 V, Na2SO4 = 100 mM | DETC = 99.48%, DECOD = 66.1%, EEC = 7.56 kW h mg−1 TC | 54 |
| Not mentioned | Bi–Sn–Sb@γ-Al2O3 | TC = 100 mg L−1 | CPE = 15 g in continuous-flow reactor, pH = 5.9, HRT = 180 min, I = 0.1 A, σ = 4000 μs cm−1 | DE = 78.9% | 89 |
| A: graphite, C: graphite | Fe-coal fly ash@AC | Ofloxacin = 100 mg L−1 | CPE = 100 g L−1, pH = 2–9, t = 90 min, j = 12 mA cm−2, Na2SO4 = 100 mM | DECOD = 63.6%, DETOC = 51.4%, cost = $1.71 per ton | 39 |
| A: graphite, C: graphite | CuFe2O4@red mud | CIP = 100 mg L−1 | CPE = 1.0 g L−1, pH = 7, t = 60 min, I = 640 mA, Na2SO4 = 3 g L−1, KPS = 9 mM | DECIP = 71.39%, DECOD = 49.21%, DETOC = 35.7% | 115 |
| A: graphite, C: graphite | CuO@red mud | CIP = 100 mg L−1 | CPE = 4 g/3 L, pH = 7, t = 80 min, U = 10 V, Na2SO4 = 3 g L−1 | DE = 80.66% | 61 |
| A: RuO2/IrO2/SnO2/Ti, C: gas diffusion electrode | Fe3O4–SnO2@GO | BH = 40 M | CPE = 0.2 g L−1, pH = 3, t = 90 min, j = 15 mA cm−2, Na2SO4 = 50 mM | DE = 94.8% | 125 |
| A: Ti/RuO2, C: Ti/RuO2 | Mn–Co@GAC | AMX = 150 mg L−1 | CPE = 31.14 g, t = 120 min, j = 5.68 mA cm−2, NaCl = 127 mM | DETOC = 85.24%, DEAMX = 100%, EEC = 0.073 kW h g−1 TOC | 79 |
| A: Ti/RuO2, C: Ti/RuO2 | AC | AMX = 200 mg L−1 | pH = 5.56, t = 120 min, j = 5 mA cm−2, NaCl = 17 mM | DE = 98.98% | 15 |
| A: Ti/IrO2/Ta2O5, C: Ti | VER | DOX = 50 mg L−1 | CPE = 2.5 g L−1, pH = 7.2, t = 80 min, j = 44.8 mA cm−2, PMS = 50 mg L−1, Na2SO4 = 35 mM | DE = 79.8%, EEC = 71.55 kW h m−3 | 126 |
| A: SS, C: SS | CPU-PPy-Gr | LEV = 20 mg L−1 | pH = 7, t = 180 min, U = 6 V, Na2SO4 = 7 mM | DE = 90%, EEC = 20.12 kW h g−1 LEV | 91 |
| A: Ti/IrO2, C: SS | GAC | IBP = 73 μM | CPE = 20.65 g in continuous-flow reactor, HRT = 240 min, j = 20 mA cm−2, Na2SO4 = 17 mM | DE = 98%, EEC = 16.1 kW h mg−1 IBP | 11 |
| A: Al, C: Al | AC | CBZ = 5 mg L−1 | CPE = 0.5 g L−1, t = 10 min, j = 9 mA cm−2, NaCl = 500 mg L−1 | DE = 89.8%, EEC = 2.52 kW h m−3, CE = 15.65% | 13 |
| Not mentioned | Ti–Sn@γ-Al2O3 | CAP = 100 mg L−1 | pH = 6, t = 180 min, I = 0.1 A, σ = 6000 μS cm−1 | DE = 70%, EEC = 57.1 kW h kg−1 CAP | 82 |
| A: graphite, C: Ti | Ti–Sb@AP | CAP = 100 mg L−1 | CPE = 50% of filling degree in fixed bed reactor, pH = 1, t = 270 min, j = 20 mA cm−2, σ = 5000 μS cm−1 | DE = 73.7%, EEC = 191.3 kW h kg−1 CAP | 34 |
| Not mentioned | Ti–Sn–Sb@γ-Al2O3 | OTC = 100 mg L−1 | CPE = 40 g L−1, pH = 6.0, t = 120 min, j = 0.5 mA cm−2, σ = 6.0 mS cm−1 | DEOTC = 92.0%, DETOC = 41.0%, EEC = 66 kW h kg−1 OTC | 90 |
4.2 3D-ER used for dye treatment
Dyes, particularly azo dyes with chromophores, are significant components in wastewater. Ren et al. utilized the combination of a 3D-ER system and γ-Fe2O3-CNT particle electrodes for the treatment of RhB dye wastewater. The results demonstrated an impressive COD removal efficiency of over 99.61% within just 7 min. The enhanced catalytic activity of the system was primarily attributed to the charge transfer pathway within the γ-Fe2O3-CNT structure, which facilitated electron mass transfer. The degradation of RhB was observed to progress through five stages including deethylation, chromophore cracking, debenzylation and deamination, ring opening, and mineralization, with ·OH playing a crucial role in the process. Following the reaction, the majority of RhB was converted into CO2 and H2O. Furthermore, this study highlighted that the Fe3+/Fe2+ circulating system established between the anode and cathode significantly contributed to the high degradation efficiency of RhB. Remarkably, even after five cycles of experiments, a degradation efficiency of 92.55% was maintained.50 Zhou et al. developed a CuFeO@kaolin particle electrode for the efficient removal of Orange G, achieving a high electrochemical degradation efficiency of 99.48% with an EEC of 0.88 kW h m−3 and CE of 11.91% after 60 min under neutral condition. The interaction between CuFeO and kaolin resulted in the formation of a stable Fe–O–Al bond. Radical scavenging experiments confirmed the generation of both ·OH and ·O2−, with ·OH being the predominant species responsible for the efficient degradation of Orange G through various mechanisms such as desulfonation, decolorization, naphthalene opening, and benzene opening. The CuFeO@kaolin catalysts exhibited high stability, maintaining the Orange G removal efficiency of 92.18% after 8 cycles with minimal ion leaching.19 The use of Fe3O4@carbon black for MO removal resulted in the removal efficiency of 93.75% of MO and 75.02% of COD. Even after 5 cycles, the MO degradation efficiency remained at 83.83%, demonstrating the good reusability of Fe3O4@carbon black.83 Table 2 provides a comprehensive list of the dyes treated in a 3D-ER by different particle electrodes.| Main electrodes | Particle electrodes | Pollutants | Process parameters | Results | Ref. |
|---|---|---|---|---|---|
| A: SS, C: Ti | Steel converter slag and zeolite slag | RhB = 10 mg L−1 | pH = 5.86, t = 16 d, I = 1 A, Na2SO4 = 100 mM | DERhB = 91.68%, DECOD = 87.63%, DENH4+–N = 90.54% | 127 |
| Not mentioned | γ-Fe2O3@CNT | RhB = 100 mg L−1 | CPE = 1 g L−1, pH = 7.0, t = 7 min, j = 20 mA cm−2 | DE = 99.16% | 50 |
| A: Ni, C: graphite | Fe–Cu@antimony tailing | RhB = 100 mg L−1 | CPE = 1.5 g L−1, pH = 5.0, t = 15 min, j = 20 mA cm−2, Na2SO4 = 20 g L−1 | DERhB = 99.40%, ETOC = 98.81%, EEC = 53.72 kW h m−3 | 84 |
| A: SS, C: SS | Mn@steel slag | RhB = 20 mg L−1 | CPE = 1.0 g L−1, pH = 3, t = 15 min, j = 57.14 mA cm−2, H2O2 = 5 mM | CE = 9.36%, DE = 99.52%, EEC = 10.41 W h L−1 | 69 |
| A: SS, C: Ti | Sn–Mn@steel slag zeolite | RhB = 10 mg L−1 | pH = 5.86, t = 180 min, I = 0.5 A, Na2SO4 = 125 mM | DE = 95% | 128 |
| A: graphite, C: graphite | Fe@C | Reactive red = 1.1 g L−1 | CPE = 10 g L−1, pH = 6.0, t = 180 min, j = 20 mA cm−2, Na2SO4 = 50 mM | DEChroma = 99.63% ± 1%, DECOD = 79.86 ± 2%, EEC = 0.31 kW h g−1 COD | 129 |
| A: Ti/RuO2/IrO2/SnO2, C: AC fiber | CuFeO@kaolin | Orange G = 50 mg L−1 | CPE = 2 g L−1, pH = 6.71, t = 60 min, j = 10 mA cm−2, Na2SO4 = 50 mM | DEOG = 99.48%, DETOC = 58.13%, EEC = 0.88 kW h g−1 TOC, CE = 11.91% | 19 |
| A: graphite, C: graphite | Fe3O4@carbon black | MO = 400 mg L−1 | CPE = 2.0 g L−1, pH = 5, t = 90 min, U = 5 V, j = 15 mA cm−2, Na2SO4 = 50 mM | DECOD = 75.02%, DEMO = 93.75% | 83 |
| A: graphite, C: graphite | Fe2(MoO4)3@kaolin | MO = 100 mg L−1 | CPE = 6.6 g L−1, pH = 4.34, t = 60 min, I = 2.1 A, Na2SO4 = 50 mM | DE = 92.48% | 63 |
| A: Ti/RuO2/IrO2, C: graphite | Steel slag@kaolin | MB = 20 mg L−1 | CPE = 150 g L−1, pH = 3.0, t = 90 min, U = 11 V, Na2SO4 = 100 mM | DE = 87% | 9 |
| A: SS, C: graphite | Slag | MB = 20 mg L−1 | CPE = 100 g L−1, pH = 6, t = 80 min, U = 10 V, Na2SO4 = 50 mM | DE = 88.51%, EEC = 31.21 kW h m−3 | 130 |
| A: Ti/RuO2, C: SS | Lithium-modified rectorite | MB = 400 mg L−1 | CPE = 5 g L−1, t = 180 min, U = 9 V, Na2SO4 = 60 mM | DE = 32.49% | 17 |
| A: Ti/RuO2/IrO2, C: Ti | Zn–Fe@GSC | MB = 100 mg L−1 | pH = 7, HRT = 6 min, U = 8 V, Na2SO4 = 100 mM | DE = 80% | 131 |
| A: Pt, C: SS | Steel slag | MB = 0.01 mM | t = 80 min, U = 3 V, Na2SO4 = 100 mM | DE = 93.22% | 132 |
| A: SS, C: SS | Eucalyptus globulus seeds | Malachite green = 500 mg L−1 | CPE = 30 g L−1, pH = 10, t = 15 min, U = 12 V | DE = 99.8% | 95 |
4.3 3D-ER used for nitrogen treatment
3D electrodes have been demonstrated for the effective treatment of NH4+–N and TN pollution. Li et al. conducted a study utilizing a 3D electrode for NH4+–N removal in water, achieving a removal efficiency of over 99%. Furthermore, it was observed that increasing the salt content in the reaction system could enhance the overall reaction efficiency.133 A composite particle electrode was developed utilizing Co as the catalyst, AC as the carrier, and acetylene black as the conductor. The electrode contained three valence states of Co including Co0, Co2+, and Co3+. The experimental results indicated that 95% of TN removal was achieved at a current of 0.4 A, pH of 7, HRT of 60 min, and initial TN concentration of 20 mg L−1. The presence of Cl− was found to enhance the removal of TN, while HCO3−, PO43−, CO32−, and dissolved organic matter were identified as inhibitors. Cyclic voltammetry and ESR analysis revealed that nitrate was directly reduced by Co0 and indirectly reduced by H·.1004.4 3D-ER used for EDC treatment
3D electrolysis has been effectively utilized in the treatment of various wastewater. However, most studies have primarily focused on treating highly concentrated organic wastewater, with only a limited number of investigations exploring the removal of trace-level pollutants. Shen et al. discovered that when treating estriol with granular graphite as particle electrodes, the reaction rate per unit area in batch mode was 3.23–5.75 times higher than that in the conventional 2D-ER, while the EEC was only 1/7–1/5 of the latter. Progesterone and 3à-hydroxy-5á-androstane-17-one were identified as degradation intermediates of estriol. The mechanism analysis revealed that indirect oxidation was the primary contributor to estriol degradation, with the repolarization of particle electrodes playing a role in the degradation process.12 Chen et al. investigated the use of an N-GH aerogel as particle electrodes in a 3D-ER for treating BPA. Under the optimal condition, the degradation efficiency of 15 mg L−1 BPA solution surpassed 90% after only 30 min of treatment. Additionally, the COD removal efficiency was measured to be 85%. Furthermore, after over 50 cycles of reuse, the degradation efficiency of BPA remained above 85%, while the COD removal efficiency stabilized at approximately 73%. The mechanisms involved the continuous attack of ·OH generated by water electrolysis on the aromatic ring, leading to the formation of various intermediates such as hydroxylated-BPA, isopropylphenol, hydroquinone, phenol, butantetraol, maleic acid, and oxalic acid. Ultimately, these compounds were mineralized into CO2 and H2O.67 Zn–Fe-rich GSC was proven to be a typical macroporous material with a large specific surface area and superior electrochemical properties. It showed good electrocatalytic activity in degrading BPA, with 89.56% of BPA being degraded in a continuous-flow 3D-ER. This degradation was attributed to the collaboration between the iron oxide and zinc oxide components.1314.5 3D-ER used for the treatment of other pollutants
In addition to the above-mentioned four common pollutants, 3D-ER has also been successfully used to treat phenol, pesticides, and other organic pollutants. Table 3 demonstrates the effectiveness of treating various organic pollutants. The CB-PTFE material showed enhanced and longer-lasting activity. Treatment of a 100 mg L−1 phenol solution resulted in the removal of 100% of phenol and 80% of COD after 120 min, with a low EEC of 0.08 kW h g−1 COD. In the CB-PTFE-based 3D system, a current density of 10 mA cm−2 was sufficient for all the CB-PTFE particles to reach a cathodic potential of −0.67 V/SCE, promoting the production of H2O2 and ·OH in the presence of Fe2+. The degradation of phenol was mainly attributed to the ·OH-mediated indirect oxidation process.134 Column granular electrodes were fabricated using Co-loaded PAC, and subsequently utilized in a continuous 3D-ER for the removal of HA. This process resulted in a significant removal efficiency of 95.3% for COD under the specific operating conditions of current of 0.3 A, HRT of 8 min, pH of 7.0, electrolyte concentration of 100 mM Na2SO4, and initial HA concentration of 200 mg L−1. The presence of a substantial amount of H˙ in the electrochemical reactions was confirmed through ESR spectroscopy. Moreover, the 3D electrochemical system effectively decomposed HA into smaller molecular fragments.57 Chen et al. focused on the degradation of salicylic acid using Mn-loaded Cu–Fe particle electrodes. The efficiency of salicylic acid degradation reached 76.9% under the optimal conditions. Salicylic acid underwent decarboxylation and substitution reactions before being mineralized through a ring-opening reaction facilitated by the presence of ·OH.138 A bimetallic aerogel-like carbon material composed of Cu, Fe, and sodium alginate was synthesized and utilized as catalytic particle electrodes for the removal of FA. The removal efficiency for FA ranged from 74.4% to 82.9% across a wide pH range of 3 to 7 under an applied voltage of 2.5 V. The comparative experimental data revealed that the 3D-EF system exhibited higher efficiency in both FA degradation and mineralization compared to the 2D-EF system. Trapping experiments indicated that the primary contributor to FA degradation and mineralization was the ·OH radical generated through a heterogeneous Fenton-like reaction. The proposed mechanism likely involved processes such as adsorption, anode oxidation, and heterogeneous/homogeneous Fenton-like reactions.139 BFD particle electrodes were specifically designed as particle electrodes for the treatment of saline PVA wastewater. The degradation efficiency of PVA reached 89.33% within 120 min under the specific conditions of a current density of 30 mA cm−2, electrode distance of 30 mm, pH of 7.0, and particle electrode filling degree of 50%. The degradation of PVA was achieved through direct catalytic oxidation. The scavenger experiments indicated that free radicals, specifically ·OH and HClO, played significant roles in the degradation of PVA wastewater.140 In a continuous-flow 3D-EF system packed with Fe–Cu@carbon black particles, an average p-NP removal efficiency of 88% and an average COD removal efficiency of over 76% were achieved after 6 h. This system exhibited minimal sensitivity to solution pH in the range of 4.0 to 10.0, with a specific EEC of approximately 0.089 kW h g−1 COD.136| Main electrodes | Particle electrodes | Pollutants | Process parameters | Results | Ref. |
|---|---|---|---|---|---|
| A: Ti/RuO2/IrO2, C: Ti | Ti4O7 | Phenol = 100 mg L−1 | CPE = 250 g L−1, pH = 4.0, t = 120 min, j = 30 mA cm−2 | DEPhenol = 88%, DECOD = 51%, EEC = 0.668 kW h g−1 COD | 134 |
| A: Ti/RuO2/IrO2, C: Ti | C-PTFE | Phenol = 100 mg L−1 | CPE = 250 g L−1, pH = 4.0, t = 120 min, j = 30 mA cm−2 | DEPhenol = 100%, DECOD = 80%, EEC = 0.08 kW h g−1 COD | 134 |
| A: Ti/RuO2/IrO2, C: SS | Sn–Mn–Ce@GAC | Phenol = 500 mg L−1 | CPE = 2.0 g L−1, pH = 6.6, t = 90 min, U = 12 V, Na2SO4 = 2.0 g L−1 | DECOD = 75.6%, EEC = 7.6 kW h kg−1 COD, CE = 72.6% | 135 |
| A: Ru/Ir/Ti/Sn/Ti, C: Ti | Sn–Sb@GAC | Phenol = 250 mg L−1 | t = 120 min, I = 0.6 A, Na2SO4 = 50 mM | DEPhenol = 91.9%, DECOD = 74.5%, EEC = 28.7 kW h kg−1 COD, CE = 60% | 22 |
| A: β-PbO2/Ti, C: Ti | SnO2–Sb@AP | Phenol = 0.22 g L−1 | pH = 4.95, t = 120 min, U = 6.1 V, Na2SO4 = 43 g L−1 | DECOD = 100%, EEC = 98 kW h kg−1 COD | 64 |
| A: Ti/RuO2/IrO2, C: graphite | AC | Phenol = 100 mg L−1 | pH = 7, t = 180 min, I = 0.6 A | DE = 90% | 71 |
| A: Ti/RuO2/IrO2, C: Ti | Fe–Cu@carbon black | p-NP = 200 mg L−1 | pH = 3.0, t = 360 min, j = 20 mA cm−2, Na2SO4 = 50 mM | DEp-NP = 88%, DECOD = 76%, EEC = 0.089 kW h g−1 COD | 136 |
| A: PbO2/graphite, C: SS | Fe@SBA-15 | 2,4-DNP = 50 mg L−1 | CPE = 5 g L−1, pH = 5, t = 60 min, j = 5.0 mA cm−2, Na2SO4 = 0.3 g/250 mL | DE2,4-DNP = 96.3%, DECOD = 88.28%, DETOC = 83.82% | 101 |
| A: graphite, C: graphite | Pd–CuFe2O4@BC | PCP = 50 mg L−1 | CPE = 0.25 g L−1, t = 240 min, U = 0.8 V, Na2SO4 = 100 mM | DE = 99.0%, CE = 28.1% | 76 |
| A: Ti/RuO2/IrO2/Ta2O5, C: SS | Ti–Cu–Ni–Zn–Sb–Mn@AC | PAP = 50 mg L−1 | pH = 7, t = 120 min, Na2SO4 = 2 g L−1 | DE = 99.87% | 41 |
| A: Pb, C: RuO2/IrO2/Ti | Pd–Co@Fe2O4 | 2,4-DCP = 200 mg L−1 | CPE = 0.08 g L−1, pH = 11.2, t = 30 min, j = 2.5 mA cm−2, Na2SO4 = 50 mM | DE = 100%, CE = 64.4% | 18 |
| A: DSA, C: Ti | Mn@AC | 4-CP = 500 mg L−1 | t = 60 min, I = 1 A, Na2SO4 = 2 g L−1 | DE = 99.93%, EEC = 0.2 kW h g−1 | 43 |
| A: Ti, C: Ti | Pd@MWCN | 4-CP = 0.2 mM | CPE = 0.1 g L−1, pH = 2.6–8.6, t = 30 min, j = 4.0 mA cm−2, Na2SO4 = 5 mM | DE = 100%, CE = 68% | 42 |
| A: PbO2/SS, C: SS | Fe3O4@AC | 2,4-D = 50 mg L−1 | CPE = 5 g L−1, pH = 3, t = 60 min, j = 5 mA cm−2, H2O2 = 0.2 mL L−1, Na2SO4 = 0.3 g/250 mL | DE2,4-D = 96.2%, DECOD = 92.31%, DETOC = 86.5% | 137 |
| A: Ti/RuO2/IrO2, C: graphite | Al-sludge-graphite powder | Diuron = 40 mg L−1 | CPE = 125 g L−1, pH = 3–11, t = 40 min, U = 5 V, Na2SO4 = 100 mM | DE = 81.2–93.6% | 21 |
| A: PbO2, C: carbon | GAC | Diuron = 40 mg L−1 | CPE = 40 g L−1, pH = 4.7, t = 50 min, j = 13.5 mA cm−2, Na2SO4 = 40 mM | DE = 100%, EEC = 0.7 kW h g−1 TOC | 7 |
| A: Ti/RuO2/IrO2, C: SS | CuFe2O4 | ATZ = 46 μmol L−1 | CPE = 3.0 g L−1, pH = 6.3, t = 35 min, j = 4 mA cm−2, Na2S2O8 = 4.0 mM | DE = 99%, EEC = 0.21 kW h g−1 ATZ | 49 |
| A: RuO2/Ti, C: Ti | N–C@AC | Pyridazine = 100 mg L−1, pyrimidine = 100 mg L−1, pyrazine = 100 mg L−1, pyridine = 100 mg L−1 | pH = 7.0, t = 40, I = 0.3 A, Na2SO4 = 50 mM | DE = 90.2–93.7%, EEC = 9.1 kW h m−3 | 31 |
| A: Ti, C: carbon | Manganese slag (CuO and Fe2O3) | Salicylic acid = 100 mM | pH = 3.0, HRT = 300 min, U = 10 V, Na2SO4 = 50 mM | DE = 76.9% | 138 |
| A: Ti/RuO2, C: Ti | Co@AC | HA (COD = 200 mg L−1) | CPE = 250 g L−1, pH = 7.0, t = 20 min, I = 0.1 A, Na2SO4 = 10 mM | DECOD = 88–94%, DETOC = 87.3–93.1%, EEC = 0.46 kW h g−1 COD | 77 |
| A: Ti/RuO2, C: Ti | Co@AC | HA (COD = 200 mg L−1) | pH = 7.0, HRT = 8 min, I = 0.3 A, Na2SO4 = 10 mM | DECOD = 95.3%, EEC = 34.2 kW h kg−1 COD | 57 |
| A: RuO2/IrO2/Ti, C: AC | Cu–Fe@sodium alginate carbon | FA (COD = 300 mg L−1) | CPE = 4 g L−1, pH = 5.4, t = 150 min, U = 2.5 V, Na2SO4 = 50 mM | DE = 82.9%, CE = 30.54% | 139 |
| A: graphitic, C: graphitic | Iron foam | FA (COD = 10000 mg L−1) |
CPE = 4 g/150 mL, pH = 3, t = 360 min, I = 0.3 A | DECOD = 43.5%, DETN = 70.4% | 87 |
| A: Ti/RuO2/IrO2, C: graphite | Blast furnace dust | PVA (COD = 665.2 mg L−1) | CPE = 50%, pH = 7.0, t = 120 min, j = 30 mA cm−2, Cl− = 6274.6 ± 300.0 mg L−1 | DEPAV = 89.33%, DETOC = 58.17%, EEC = 8.96 kW h kg−1 TOC, CE = 31.6% | 140 |
| A: graphite, C: graphite | BC | Cu(II) = 50 mg L−1 | CPE = 20 g L−1, pH = 7, t = 180 min, I = 40 mA, Na2SO4 = 50 mM | DE = 90.7% | 141 |
| A: BDD, C: Ti | Fe–N@BC | Ni–EDTA = 50 mg L−1 | CPE = 2 g L−1, pH = 3.3, t = 120 min, j = 20 mA cm−2, Na2SO4 = 50 mM, H2O2 = 10 mM | DENi(II) = 94.1%, DETOC = 76.2%, cost = 15.26 RMB per m3 | 23 |
| A: DSA, C: SS | Polypyrrole@rGO | Cr(VI) = 80 mg L−1, BPA = 20 mg L−1 | pH = 3.0, t = 30 min, U = 25 V | DECr(VI) = 98.52% ± 1.48%, DEBPA = 98.00% ± 1.17% | 68 |
4.6 3D-ER used for real wastewater treatment
Some studies have investigated the practical industrial applications of particle electrodes in various wastewater, such as cyanide wastewater, coal chemical water, coking wastewater, and especially for landfill leachate, as shown in Table 4. Yu and Pei examined the efficiency of a 3D electrode dynamic reactor with iron carbon granules in removing pollutants from landfill leachate. Their study found that increasing the current density or decreasing the flow rate improved the pollutant removal efficiency. The optimal process parameters were determined to be a current density of 16 mA cm−2 and flow rate of 0.75 L h−1, resulting in the removal of 60.02% of TOC, 96.50% of chroma, 64.98% of COD, and 99.46% of NH4+–N. Additionally, the characteristic peaks of refractory organic pollutants were reduced by 97.95%, and the BOD/COD increased from 0.24 to 0.32 after the reaction. Notably, 85.90% of IBU, an emerging trace organic in landfill leachate, was successfully removed.144 Song et al. demonstrated that a 3D-ER exhibited a higher efficiency in removing NH4+–N compared to COD in landfill leachate. Specifically, the 3D electrode composed of Fe@BC achieved a removal efficiency of 98.6% for NH4+–N and 95.5% for COD.104 Li et al. utilized Al particles as granular electrodes within a 3D electrode system, resulting in a significant mineralization efficiency for landfill leachate. At a current density of 5.5 A m−2, the EEC was measured to be 42.46 kW h kg−1 COD, indicating that the 3D electrode required a lower energy input, while effectively treating landfill leachate.88 Hu et al. investigated the efficiency of a 3D electrode system utilizing Fe–Cu@needle coke particle electrodes for the removal of COD, NH4+–N, TN, and chromaticity in landfill leachate. The results showed that 94.2% of COD, 93.8% of NH4+–N, 90.3% of TN, and 99% of chromaticity were successfully eliminated.86 In a separate study on recalcitrant chemical industry wastewater treatment using GAC as particle electrodes, 49% of COD was removed at an EEC of 6.59 kW h kg−1. However, it was observed that GAC experienced slow attrition during electrolysis, indicating a potential challenge for scaling up operations. This issue can lead to a gradual decrease in the liquid holding capacity of the carbon bed due to stratification and the accumulation of carbon fine dust.40| Main electrodes | Particle electrodes | Pollutants | Process parameters | Results | Ref. |
|---|---|---|---|---|---|
| A: Ti/RuO2/IrO2, C: SS | Fe2O3@PAC | Domestic wastewater (NH4Cl = 64.5 mg L−1) | CPE = 0.3 g/85 mL, t = 20 min, U = 20 V | DE = 95.3% | 93 |
| A: coal-based electrode, C: coal-based electrode | AC | Cyanide wastewater (CNT = 1872.60 mg L−1, CN− = 383.40 mg L−1, Cu2+ = 593.20 mg L−1, Zn2+ = 468.80 mg L−1, SCN− = 262.15 mg L−1) | CPE = 40 g L−1, t = 150 min, U = 4 V | DECNT = 97.03%, DECu2+ = 95.79%, DEZn2+ = 99.82%, DECN− = 94.42%, DESCN− = 94.19% | 9 |
| Not mentioned | W–Ag–Ti@γ-Al2O3 | Coal chemical water (total phenol = 1036–1140 mg L−1, COD = 3500–3800 mg L−1, NH4+–N = 720–750 mg L−1) | CPE = 50% of filling degree in fixed bed reactor, pH = 3, t = 180 min, I = 0.1 A | DECOD = 67.6%, DENH4+–N = 42.8%, DETotal phenol = 51.7% | 74 |
| A: Ti/RuO2/IrO2, C: Ti | Coal gasification fine slag | m-cresol = 200 mg L−1 | CPE = 8 g L−1, t = 60 min, j = 60 mA cm−2 | DE = 100% | 142 |
| A: Ti/IrO2/RuO2, C: Ti | VTM | Coking wastewater (COD = 180–225 mg L−1) | CPE = 33.3 g L−1, pH = 8.6–9.3, t = 30 min, j = 15 mA cm−2, KPS = 4 mM | DECOD = 94.15%, DEDOC = 57.67%, DEUV254 = 86.10% | 143 |
| A: DSA, C: Ni | Fe@needle coke | Coking wastewater (COD = 1730 mg L−1, NH4+–N = 57.34 mg L−1, NO3−–N = 50.35 mg L−1, TN = 146.46 mg L−1, and chroma = 400 folds) | CPE = 12.23 g L−1, pH = 2.62, t = 360 min, U = 11.15 V | DECOD = 87.5%, DENH4+–N = 100%, DENO3−–N = 72.2%, DETN = 84.8%, DEChroma = 95%, DEUV254 = 72.4%, EEC = 0.069 W h mg−1 COD | 85 |
| A: DSA, C: Ni activated carbon | Fe–Cu@needle coke | Landfill leachate (COD = 1370 mg L−1, NH4+–N = 1226.4 mg L−1, TN = 1373.77 mg L−1, chroma = 200 folds) | CPE = 25 g L−1, pH = 2, t = 360 min, U = 9 V | DECOD = 94.2%, DENH4+–N = 93.8%, DETN = 90.3%, DEChroma = 99% | 86 |
| A: graphite, C: SS | Fe@BC | Landfill leachate (COD = 700–900 mg L−1, NH4+–N = 130–150 mg L−1) | pH = 7.0–7.2, t = 45 days, I = 1 V | DECOD = 95.5%, DENH4+–N = 98.6% | 104 |
| A: GAC, C: GAC | Al particles | Landfill leachate (concentration not mentioned) | t = 120 min, j = 5.5 A m−2 | DEChroma = 98.94%, DEUV254 = 84.72%, DETOC = 67.46%, DECOD = 51.93%, cost = 1.307 USD per m3, EEC = 42.46 kW h kg−1 COD | 88 |
| A: Ti/RuO2/IrO2, C: Ti/RuO2/IrO2 | Fe@GAC | Landfill leachate (COD = 1041.38 mg L−1, TOC = 632.65 mg L−1) | t = 360 min, j = 16 mA cm−2, Na2S2O8 = 28 mM | DETOC = 60.02%, DEChroma = 96.5%, DECOD = 64.98%, DENH4+–N = 99.46%, CE = 35.42% | 144 |
| A: Ti/RuO2/IrO2, C: Ti/RuO2/IrO2 | Fe@C | Landfill leachate (COD = 1041.38 mg L−1, NH4+–N = 444.39 mg L−1) | CPE = 1 g/60 mL, t = 120 min, U = 5 V, Na2S2O8 = 28 mM | DECOD = 72.9%, DENH4+–N = 99.9% | 78 |
| Not mentioned | MnO2–TiO2@GAC | Landfill leachate (concentration not mentioned) | CPE = 200 g L−1, pH = 3, j = 15 mA cm−2, Fe2+ = 1 mM | DECOD = 84.68%, DENH4+–N = 81.3% | 81 |
| A: graphite, C: graphite | Pyrolusite | Sulphonated phenolic resin (COD = 500 mg L−1) | CPE = 50 g L−1, pH = 7.0, t = 40 min, I = 0.9 A, σ = 3600 μS cm−1 | DECOD = 49.56%, EEC = 78% | 60 |
| A: Ti/PbO2, C: SS | GAC and quartz sand | Secondary effluent of an industrial wastewater treatment plant (COD = 92 mg L−1) | pH = 8.5, t = 60 min, j = 5 mA cm−2, σ = 7.5 mS cm−1 | DECOD = 50%, EEC = 180 kW h kg−1 COD | 5 |
| A: Ti/SnO2, C: active carbon | Fe@AC | Biologically pretreated coal gasification wastewater (COD = 173.3 ± 14.1 mg L−1) | CPE = 5.0 g L−1, pH = 6.8, t = 120 min, j = 15 mA cm−2 | DECOD = 65.5% | 145 |
| A: carbon, C: SS | GAC | Recalcitrant chemical industry wastewater (COD = 10358 mg L−1) |
pH = 8.1, HRT = 2 h, I = 3 A, NaCl = 2 g L−1 | DECOD = 49%, EEC = 6.59 kW h kg−1 COD, CE = 3.42% | 40 |
| A: graphite, C: graphite | AC | Pharmaceutical wastewater (TOC = 890.5 mg L−1) and steel wastewater (TOC < 2 mg L−1) with the ratio of 3:1 |
CPE = 62 g L−1, pH = 3.0, t = 180 min, j = 17 mA cm−2, Cl− = 20 mM | DETOC = 94.1%, EEC = 14.47 kW h m−3, CE = 2.5% | 16 |
5. Future scope
5.1 Appropriate selection of particle electrodes
Electrode materials are crucial for 3D-ER, particularly particle electrodes, which play significant roles in pollutant degradation. Ideally, particle electrode materials should exhibit good conductivity, high corrosion resistance, non-toxicity, and good biocompatibility. Currently, GAC is a widely used material due to its favorable electrochemical performance, large specific surface area, and biocompatibility. However, GAC has some drawbacks. For example, during the electrolysis process, GAC may be crushed, resulting in the presence of ash particles in the treated wastewater, necessitating further separation. Moreover, when selecting electrode materials, the principle of ‘waste utilization’ should be followed to prevent secondary pollution and reduce costs.145 Meng et al. developed GSC particle electrodes converted from waste sludge, which not only aid in degradation but also enable sludge recycling.94 Additionally, certain metal slag can serve as particle electrodes, helping to avoid the wastage of metal resources.128,130,132,146 Particle materials should be conducive to adsorption, electron transfer, and ROS formation, thereby enhancing the electrocatalytic performance in pollutant decomposition.20,21 Hence, it is crucial to consider factors such as increasing the active sites of particle electrodes, improving the OEP, suppressing side reactions such as oxygen evolution, and enhancing the production of active substances such as ·OH and active chlorine.45,60,1265.2 In-depth investigation of mechanisms
Past research has predominantly focused on the factors influencing the degradation process, with limited exploration of the underlying mechanisms. Particle electrodes typically possess adsorption capacity, raising questions on how the adsorption process impacts electro-degradation. When 3D-ER is combined with other methods such as microbial degradation, photocatalytic degradation, or other forms of oxidation, it is essential to clarify the respective contributions of each method to the degradation efficiency. For instance, in a 3D-BER system, the large pore diameter and specific surface area of the particle electrodes can promote microbial growth. However, the potential competitive relationship between microbial growth and electrochemical degradation remains unclear. Thus, investigating how current and microorganisms collaborate can be a crucial area for future research.5.3 Consideration of economic feasibility and toxicity of the end products
In addition to the removal efficiency of pollutants, the cost and recyclability of particle electrodes should also be considered when evaluating the economic feasibility of 3D-ER. Factors such as reactor design, electrode selection, parameter optimization, and EEC should be considered. It is also important to assess whether the degradation products become less toxic. Some studies have shown a significant reduction in toxicity compared to the original pollutants, but the toxicity results are often not mentioned.91,118,119 Therefore, the key challenge in the 3D electrocatalytic oxidation process is the development of electrode materials that are cost-effective, lower the EEC, and exhibit significant degradation effects and decreased toxicity.6. Conclusion
3D-ER, as a type of advanced oxidation technology, offers the advantage of not requiring an additional oxidiser. It has shown promising progress in wastewater treatment and holds a positive application outlook. Herein, we presented a thorough investigation into the reactor structure, particle electrodes, key process parameters, and EEC. In comparison to the traditional 2D-ER, 3D electrode technology is cost-effective and allows easy expansion of the reaction area throughout the reactor. However, challenges such as efficiency loss due to mass transfer limitations and side reactions, the formation of toxic by-products, and electrode durability and stability need to be addressed for its successful large-scale implementation. Future research should focus on selecting appropriate particle electrodes, optimizing process parameters for electrocatalysis, and resolving issues related to particle electrode replacement to optimize the use of 3D electrode systems in wastewater treatment.Abbreviations
Particle electrode
| AC | Activated carbon |
| AP | Attapulgite |
| BC | Biochar |
| BiF@Bent | Bentonite-based bismuth ferrites |
| BDD | Boron-doped diamond |
| BFD | Blast furnace dust |
| CB-PTFE | Carbon black-polytetrafluoroethylene |
| CPU-PPy-GH | Graphene–polyurethane–graphene |
| CuFe2O4@RM | Red mud-based CuFe2O4 |
| DSA | Dimension-stable anode |
| GAC | Granular activated carbon |
| GH | Graphene |
| GSC | Granular sludge carbon |
| MMT | Montmorillonite |
| MWCN | Multi-walled carbon nanotubes |
| N-GH | N-doped graphene |
| PAC | Pallet activated carbon |
| PCP | Porous ceramsite particle |
| RM | Red mud |
| SS | Stainless steel |
| VER | Vermiculite |
| VTM | Vanadium–titanium magnetite |
Experimental parameters
| A | Anode |
| C | Cathode |
| CE | Current efficiency |
| DE | Degradation efficiency |
| EEC | Electrode energy consumption |
| HRT | Hydraulic retention time |
| j | Current density |
| KPS | Potassium persulfate |
| PE | Particle electrode |
| PMS | Peroxymonosulfate |
| ROS | Reactive oxygen species |
| σ | Electric conductivity |
| 2D-ER | Two-dimensional electrode reactor |
| 3D-ER | Three-dimensional electrode reactor |
| 3D-BER | Three-dimensional biofilm electrode reactor |
Pollutants
| ACV | Acyclovir |
| AMX | Amoxicillin |
| ATZ | Atrazine |
| ARB | Arbidol |
| BH | Berberine hydrochloride |
| CAP | Chloramphenicol |
| CBZ | Carbamazepine |
| CIP | Ciprofloxacin |
| COD | Chemical oxygen demand |
| DEX | Dexamethasone |
| DIU | Diuron herbicide |
| DOX | Doxycycline |
| EX | Ethyl xanthate |
| FA | Fulvic acid |
| HA | Humic acid |
| IBP | Ibuprofen |
| LEV | Levofloxacin |
| MO | Methyl orange |
| MNZ | Metronidazole |
| NB | Nitrobenzene |
| NOR | Norfloxacin |
| NPX | Naproxen |
| OTC | Oxytetracycline |
| p-NP | p-nitrophenol |
| PAP | p-aminophenol |
| PCP | Pentachlorophenol |
| PVA | Polyvinyl alcohol |
| RhB | Rhodamine B |
| SA | Sulfanilamide |
| SDS | Sodium dodecyl sulphate |
| SMA | Sulfamethazine |
| SMX | Sulfamethazole |
| TBBPA | Tetrabromobisphenol |
| TC | Tetracycline |
| 2,4-D | 2,4-Dichlorophenoxyacetic acid |
| 2,4-DCP | 2,4-Dichlorophenol |
| 2,4-DNP | 2,4-Dinitrophenol |
| 4-CP | 4-Chlorophenol |
Data availability
No new data were created or analyzed in this study.Author contributions
Mingyue Piao (writing – original draft, and funding acquisition), Honghui Teng (investigation), Hongxue Du (writing – review & editing, and funding acquisition).Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by Education Department of Jilin Province of China (JJKH20240575KJ), Department of Science and Technology of Jilin Province of China (20240601063RC), and the Bureau of Science and Technology of Siping (2023079).References
- Y. Liu, P. Wang, B. Gojenko, J. J. Yu, L. Z. Wei, D. G. Luo and T. F. Xiao, Environ. Pollut., 2021, 291, 118209 CrossRef CAS PubMed.
- J. O. Ighalo, A. G. Adeniyi, J. A. Adeniran and S. Ogunniyi, J. Cleaner Prod., 2021, 283, 124566 CrossRef CAS.
- Y. G. Yu, Z. Wang, B. Yao and Y. Y. Zhou, Sci. Total Environ., 2024, 923, 171388 CrossRef CAS PubMed.
- X. C. Li, S. Lu and G. Zhang, J. Hazard. Mater., 2023, 445, 130524 CrossRef CAS PubMed.
- W. Can, H. Yao-Kun, Z. Qing and J. Min, Chem. Eng. J., 2014, 243, 1–6 CrossRef.
- J. R. Backhurst, J. M. Coulson, F. Goodridg, R. E. Plimley and M. Fleischm, J. Electrochem. Soc., 1969, 116, 1600–1607 CrossRef.
- A. Rahmani, M. Leili, A. Seid-mohammadi, A. Shabanloo, A. Ansari, D. Nematollahi and S. Alizadeh, J. Cleaner Prod., 2021, 322, 129094 CrossRef CAS.
- Y. J. Niu, L. Q. Chen, S. X. Guo, J. Xu, H. C. Li, F. H. Guo, Y. X. Zhang and J. J. Wu, Fuel, 2024, 358, 130139 CrossRef CAS.
- B. Song, Z. Y. Wang, J. F. Li and Y. N. Ma, Catal. Commun., 2021, 148, 106177 CrossRef CAS.
- X. L. Teng, Z. Y. Wang, X. R. Wu, Y. Jiang, J. F. Li, C. Zhao and X. L. He, Sci. Adv. Mater., 2019, 11, 88–92 CrossRef CAS.
- S. Cho, C. Kim and I. Hwang, Chemosphere, 2020, 259, 127382 CrossRef CAS PubMed.
- B. Shen, X. H. Wen and X. Huang, Chem. Eng. J., 2017, 327, 597–607 CrossRef CAS.
- A. Alighardashi, R. S. Aghta and H. Ebrahimzadeh, Int. J. Environ. Res., 2018, 12, 451–458 CrossRef CAS.
- L. Y. Wei, S. H. Guo, G. X. Yan, C. M. Chen and X. Y. Jiang, Electrochim. Acta, 2010, 55, 8615–8620 CrossRef CAS.
- H. M. Shi, Q. H. Wang, J. Ni, Y. S. Xu, N. Song and M. Gao, J. Water Process Eng., 2020, 38, 101656 CrossRef.
- H. H. Phan Quang, T. P. Nguyen, D. D. Duc Nguyen, L. T. Ngoc Bao, D. C. Nguyen and V.-H. Nguyen, Chemosphere, 2022, 297, 134074 CrossRef CAS PubMed.
- J. Huang, Y. A. Ming, Y. Du, Y. R. Wang and C. Wang, J. Anal. Methods Chem., 2016, 2016 Search PubMed.
- Y. Z. Xue and C. H. Feng, Appl. Surf. Sci., 2021, 545, 149007 CrossRef CAS.
- C. Y. Zhou, X. E. Peng, X. L. Li, K. Qi and L. L. Gao, J. Environ. Chem. Eng., 2023, 11, 109562 CrossRef CAS.
- Z. Liu, R. Y. Chen, M. Y. Li, S. S. Yang, J. Zhang, S. C. Yuan, Y. Z. Hou, C. Li and Y. Chen, J. Hazard. Mater., 2023, 459, 132089 CrossRef CAS PubMed.
- Y. L. Yang, J. F. Li, W. Y. Qu, C. X. Ma, X. T. Feng, Y. Guo, J. J. He and X. L. He, Sep. Purif. Technol., 2024, 330, 125326 CrossRef CAS.
- R. Y. Zhao, H. J. Zhao, J. Li, Y. M. Wang, Q. Zhang, F. Liu, C. S. Liu and X. F. Lan, Chem. Eng. J. Adv., 2023, 16, 100549 CrossRef CAS.
- Z. L. Zhao, Y. T. Ren, S. Y. Qi, Z. G. Ning, X. Wang, W. Y. Dong and H. J. Wang, Chem. Eng. J., 2023, 471, 144717 CrossRef CAS.
- F. C. Ban in Study on the Treatment of Phenol Wastewater by Three-Dimensional Electrode-Fenton Reagent Method, China, 2010 Search PubMed.
- M. Li, Y. F. Guo and H. C. Pang, Coal Chem. Ind., 2004, 6, 63–66 Search PubMed.
- J. F. Li, X. L. Teng, Z. Y. Wang, Y. Xu, J. Yang, Y. L. Yang, J. H. Tao, C. X. Ma, D. B. Song, K. J. Li, J. P. Zhang, P. Chen and B. Song, China Patent No. CN201910359182.4, 2019.
- Y. H. Xu, H. Zhao, L. Chen and X. Y. Yang. China Patent No. CN1986434A, 2007.
- J. F. Zhang, J. Xia, Y. Ma, Y. Huang and J. Cao, China Patent No. CN107162119A, 2017.
- J. P. Hu, S. Gao, X. K. Zhou, L. Ma, X. P. Chu, H. J. Hou, B. C. Liu, J. K. Yang, Z. F. Pan and L. Y. Wu, China Patent No. CN207861964U, 2018.
- J. X. Cheng, H. T. Yang, C. L. Fan, R. X. Li, X. H. Yu and H. T. Li, J. Solid State Electrochem., 2020, 24, 2199–2217 CrossRef CAS.
- Y. Liu, L. L. Qin, Y. M. Qin, T. Yang, H. R. Lu, Y. L. Liu, Q. Q. Zhang and W. Y. Liang, J. Hazard. Mater., 2024, 463, 132899 CrossRef CAS PubMed.
- M. Y. Piao, J. Zhang, H. S. Du, H. X. Du and H. H. Teng, Desalin. Water Treat., 2023, 316, 317–326 CrossRef CAS.
- M. Y. Piao, J. Zhang, H. S. Du, H. X. Du, Y. W. Sun and H. H. Teng, Environ. Technol., 2023, 1–9 DOI:10.1080/09593330.2023.2260121.
- Y. J. Sun, A. W. Chen, S. C. Zhu, W. Q. Sun, K. J. Shah and H. L. Zheng, Water Environ. Res., 2019, 91, 756–769 CrossRef CAS PubMed.
- R. X. Hao, S. M. Li, J. B. Li and C. C. Meng, Bioresour. Technol., 2013, 143, 178–186 CrossRef CAS PubMed.
- Q. Tang, Y. Q. Sheng, C. Y. Li, W. J. Wang and X. Z. Liu, Bioresour. Technol., 2020, 318, 124096 CrossRef CAS PubMed.
- B. Song, Z. Y. Wang, J. F. Li, M. Q. Luo, P. W. Cao and C. Zhang, Chemosphere, 2021, 284, 131328 CrossRef CAS PubMed.
- N. Cai, G. Bai, T. Zhang, Y. Q. Lei, P. R. Guo, Z. L. Chen and J. W. Xu, Chin. Chem. Lett., 2024, 35, 108514 CrossRef CAS PubMed.
- N. N. Wang, L. W. Li, W. H. Zou and P. Wang, J. Environ. Chem. Eng., 2023, 11, 109561 CrossRef CAS.
- N. Gedam and N. R. Neti, J. Environ. Chem. Eng., 2014, 2, 1527–1532 CrossRef CAS.
- J. Wan, F. P. Zhao, Y. Meng, M. Y. Guo, C. J. Tang, Y. Shi, Y. Ke and R. Hu, Sep. Purif. Technol., 2021, 267, 118662 CrossRef CAS.
- X. Y. Shu, Q. Yang, F. B. Yao, Y. Zhong, W. C. Ren, F. Chen, J. Sun, Y. H. Ma, Z. Y. Fu, D. B. Wang and X. M. Li, Chem. Eng. J., 2019, 358, 903–911 CrossRef CAS.
- S. M. Xie, M. Li, Y. X. Liao, Q. Qin, S. X. Sun and Y. H. Tan, Chemosphere, 2021, 273, 128506 CrossRef CAS PubMed.
- K. T. Lu and Y. G. Zhang, China Pet. Process. Petrochem. Technol., 2021, 23, 30–39 CAS.
- T. Wang, Y. Q. Song, H. J. Ding, Z. Liu, A. Baldwin, I. Wong, H. Li and C. Zhao, Chem. Eng. J., 2020, 394, 124852 CrossRef CAS.
- Z. W. Jiang, Y. C. Wang, H. Yu, N. Yao, J. H. Shen, Y. Li, H. Y. Zhang and X. Bai, Chemosphere, 2023, 313, 137446 CrossRef CAS PubMed.
- W. Liu, Z. H. Ai and L. Z. Zhang, J. Hazard. Mater., 2012, 243, 257–264 CrossRef CAS PubMed.
- M. M. Ta, T. Wang, J. Guo, Y. Wang, J. Zhang, C. L. Zhao, S. S. Liu, G. H. Liu and H. X. Yang, Sep. Purif. Technol., 2023, 310, 123067 CrossRef CAS.
- J. Li, J. F. Yan, G. Yao, Y. H. Zhang, X. Li and B. Lai, Chem. Eng. J., 2019, 361, 1317–1332 CrossRef CAS.
- Y. C. Ren, J. Wang, G. F. Qu, N. Q. Ren, P. Lu, X. P. Chen, Z. L. Wang, Y. Y. Yang and Y. Hu, Sep. Purif. Technol., 2023, 314, 123616 CrossRef CAS.
- M. Zhang, L. Zhang, H. Wang and Z. Y. Bian, Sci. Total Environ., 2020, 724, 138170 CrossRef CAS PubMed.
- K. Zhao and Y. G. Zhang, Sep. Purif. Technol., 2023, 308, 122962 CrossRef CAS.
- Z. J. Zhang and Y. G. Zhang, Mol. Catal., 2024, 553, 113778 CrossRef CAS.
- X. T. Xu, X. L. Zeng, C. K. Zhang, R. L. Huang and W. C. Ding, J. Cleaner Prod., 2023, 419, 138257 CrossRef CAS.
- T. Wang, M. M. Ta, J. Guo, L. E. Liang, C. Bai, J. Zhang and H. J. Ding, Sep. Purif. Technol., 2023, 304, 122354 CrossRef CAS.
- S. W. Li, H. B. Yu, Y. Z. Lin, S. Y. Zhu, G. Liu and C. Y. Shi, Process Saf. Environ. Prot., 2023, 171, 176–187 CrossRef CAS.
- W. W. Zhang, D. Xie, X. Li, W. J. Ye, X. X. Jiang, Y. Y. Wang and W. Y. Liang, Appl. Catal., A, 2018, 559, 75–84 CrossRef CAS.
- S. Zhang, H.-L. Song, X. L. Yang, K. Y. Yang and X. Y. Wang, Chemosphere, 2016, 164, 113–119 CrossRef CAS PubMed.
- J. Q. Bu, Z. W. Deng, H. Liu, T. H. Li, Y. J. Yang and S. A. Zhong, J. Cleaner Prod., 2021, 324, 129256 CrossRef CAS.
- H. Liang, Y. Qiu, L. Y. Zhao, Y. Y. Chen and B. Wang, Water Sci. Technol., 2018, 78, 1427–1437 CrossRef CAS PubMed.
- C. P. Lu, G. T. Wei, J. S. Ba, Q. Y. Li, L. Y. Zhang, Z. M. Li, L. F. Cen, J. H. Ma and Y. Song, J. Environ. Chem. Eng., 2022, 10, 107151 CrossRef CAS.
- W. P. Kong, B. Wang, H. Z. Ma and L. Gu, J. Hazard. Mater., 2006, 137, 1532–1537 CrossRef CAS PubMed.
- W. Y. He, Q. L. Ma, J. Wang, J. Yu, W. R. Bao, H. Z. Ma and A. Amrane, Appl. Clay Sci., 2014, 99, 178–186 CrossRef CAS.
- J. Hou, S. Y. Shen and L. Z. Wang, Environ. Sci. Pollut. Res., 2023, 30, 102363–102373 CrossRef CAS PubMed.
- Y. M. Zhang, Z. Chen, P. P. Wu, Y. X. Duan, L. C. Zhou, Y. X. Lai, F. Wang and S. Li, J. Hazard. Mater., 2020, 393, 120448 CrossRef CAS PubMed.
- G. F. Lv, D. C. Wu and R. W. Fu, J. Hazard. Mater., 2009, 165, 961–966 CrossRef CAS PubMed.
- Z. Chen, Y. M. Zhang, L. C. Zhou, H. Zhu, F. Wan, Y. Wang and D. D. Zhang, J. Hazard. Mater., 2017, 332, 70–78 CrossRef CAS PubMed.
- Y. M. Zhang, D. D. Zhang, L. C. Zhou, Y. L. Zhao, J. Chen, Z. Chen and F. Wang, Chem. Eng. J., 2018, 336, 690–700 CrossRef CAS.
- X. Ren, P. X. Tang, B. Hou, Z. Y. Yu, J. Huang, Q. Y. Wang and K. Song, J. Environ. Chem. Eng., 2023, 11, 109475 CrossRef CAS.
- L. N. Xu, H. Z. Zhao, S. Y. Shi, G. Z. Zhang and J. R. Ni, Dyes Pigm., 2008, 77, 158–164 CrossRef CAS.
- T. T. Pang, Y. Wang, H. Yang, T. L. Wang and W. F. Cai, Chemosphere, 2018, 206, 107–114 CrossRef CAS PubMed.
- L. Li, X. Y. Fang, D. F. Zhang and Y. X. Huang, Int. J. Electrochem. Sci., 2015, 10, 4083–4089 CrossRef CAS.
- F. Qi, Z. Q. Zeng, Q. Wen and Z. G. Huang, J. Hazard. Mater., 2021, 416, 125810 CrossRef CAS PubMed.
- W. Q. Sun, S. B. Zhou, Y. J. Sun, Y. H. Xu and H. L. Zheng, J. Environ. Chem. Eng., 2021, 9, 104681 CrossRef CAS.
- J. Yu, J. Zou, P. Xu and Q. L. He, J. Cleaner Prod., 2020, 251, 119744 CrossRef CAS.
- Y. Wang, C. Y. Cui, G. D. Zhang, Y. J. Xin and S. X. Wang, Sep. Purif. Technol., 2021, 258, 118017 CrossRef CAS.
- W. W. Zhang, W. J. Ye, X. X. Hu and W. Y. Liang, Chemosphere, 2021, 263, 128200 CrossRef CAS PubMed.
- D. Y. Yu, J. Cui, X. Q. Li, H. Zhang and Y. S. Pei, Chemosphere, 2020, 243, 125438 CrossRef CAS PubMed.
- J. S. Ma, M. Gao, Q. Liu and Q. H. Wang, Environ. Res., 2022, 209, 112728 CrossRef CAS PubMed.
- S. W. Li, Y. Z. Lin, S. Y. Zhu and G. Liu, Environ. Sci. Pollut. Res., 2022, 29, 57112–57126 CrossRef CAS PubMed.
- J. X. Fu, X. He, J. H. Zhu, W. Y. Gu and X. X. Li, Int. J. Electrochem. Sci., 2018, 13, 5872–5887 CrossRef CAS.
- Y. J. Sun, P. Li, H. L. Zheng, C. Zhao, X. F. Xiao, Y. H. Xu, W. Q. Sun, H. F. Wu and M. J. Ren, Chem. Eng. J., 2017, 308, 1233–1242 CrossRef CAS.
- M. M. Sun, Y. Z. Zhao and H. H. Ge, Int. J. Electrochem. Sci., 2022, 17, 220822 CrossRef CAS.
- Y. C. Ren, P. Lu, G. F. Qu, P. Ning, N. Q. Ren, J. Wang, F. H. Wu, X. P. Chen, Z. L. Wang, T. Zhang, M. H. Cheng and X. M. Chu, Water Res., 2023, 244, 120487 CrossRef CAS PubMed.
- Y. Hu, F. Z. Yu, Z. T. Bai, Y. Q. Wang, H. Zhang, X. Y. Gao, Y. X. Wang and X. Li, Chemosphere, 2022, 308, 136544 CrossRef CAS PubMed.
- Y. Hu, Y. Q. Wang, L. H. Kou, X. Y. Gao, Y. X. Wang, Y. T. Guo and X. Li, Chem. Eng. J., 2024, 481, 148427 CrossRef CAS.
- Y. S. Zheng, S. Qiu, F. X. Deng, Y. S. Zhu, G. J. Li and F. Ma, Sep. Purif. Technol., 2019, 224, 463–474 CrossRef CAS.
- J. Li, L. Wang, J. B. Lu, W. L. Peng, J. Chen, G. M. Jiang and D. F. Liu, Waste Manage., 2024, 173, 118–130 CrossRef CAS PubMed.
- W. Q. Sun, Y. J. Sun, K. J. Shah, P.-C. Chiang and H. L. Zheng, J. Hazard. Mater., 2019, 370, 24–32 CrossRef CAS PubMed.
- W. Q. Sun, Y. J. Sun, K. J. Shah, H. L. Zheng and B. Ma, J. Environ. Manage., 2019, 241, 22–31 CrossRef CAS PubMed.
- C. C. Guo, H. Y. Liu, C. Z. Wang, J. C. Zhao, W. J. Zhao, N. Lu, J. Qu, X. Yuan and Y. N. Zhang, Environ. Pollut., 2020, 260, 114101 CrossRef CAS PubMed.
- S. J. Gao, W. F. Liu, M. L. Wang, Z. B. Zhao and X. G. Liu, Appl. Catal., B, 2024, 340, 123276 CrossRef CAS.
- M. Yuan, F. R. Yan, Y. G. Chen, J. J. Luo and Z. Y. Li, RSC Adv., 2020, 10, 8773–8779 RSC.
- H. S. Meng, C. Chen, Z. R. Yan, X. Y. Li, J. Xu and G. P. Sheng, Water Res., 2019, 165, 115016 CrossRef CAS PubMed.
- A. A. Renita, K. H. Vardhan, P. S. Kumar, P. T. Ngueagni, A. Abilarasu, S. Nath, P. Kumari and R. Saravanan, Chemosphere, 2021, 273, 129634 CrossRef CAS PubMed.
- R. Chen, T. Sheehan, J. L. Ng, M. Brucks and X. Su, Environ. Sci. Water Resour., 2020, 6, 258–282 RSC.
- K. Y. Foo and B. H. Hameed, J. Hazard. Mater., 2009, 170, 552–559 CrossRef CAS PubMed.
- Y. H. Song, S. M. Lei, N. Yin and W. J. He, Environ. Technol., 2021, 42, 1693–1702 CrossRef CAS PubMed.
- E. Andrés García, M. Agulló-Barceló, P. Bond, J. Keller, W. Gernjak and J. Radjenovic, Chem. Eng. J., 2018, 352, 405–411 CrossRef.
- W. J. Ye, W. W. Zhang, X. X. Hu, S. Yang and W. Y. Liang, Sci. Total Environ., 2020, 732, 139245 CrossRef CAS PubMed.
- A. Dargahi, H. Rahimzadeh Barzoki, M. Vosoughi and S. Ahmad Mokhtari, Arabian J. Chem., 2022, 15, 103801 CrossRef CAS.
- M. Zhang, H. Wang, J. G. Wu and H. Wang, Int. J. Electrochem. Sci., 2020, 15, 5698–5711 CrossRef CAS.
- Z. Y. Wu, Y. Liu, S. Y. Wang, P. Peng, X. Y. Li, J. Xu and W. H. Li, Bioresour. Technol., 2019, 284, 222–230 CrossRef CAS PubMed.
- Z. Song, R. F. Liao, X. L. Su, X. Zhang, Z. L. Zhao and F. Y. Sun, Sci. Total Environ., 2023, 903, 166980 CrossRef CAS PubMed.
- Q. Y. Wu, Y. Chen, Y. He, Q. M. Cheng, Q. Wu, Z. Liu, Y. Q. Li, Z. M. Yang, Y. Q. Tan and Y. Yuan, J. Environ. Manage., 2023, 348, 119346 CrossRef CAS PubMed.
- X. X. Li, X. Li, Y. Feng, X. W. Wang, N. Suo, S. M. Yang, Y. Y. Long and S. B. Zhang, Chemosphere, 2021, 282, 131020 CrossRef CAS PubMed.
- L. Feng, X. Y. Li, L. H. Gan and J. Xu, Electrochim. Acta, 2018, 290, 165–175 CrossRef CAS.
- X. Y. Li, P. Peng, W. K. Wang, S. Y. Wang, L. Feng, Y. C. Zhang and J. Xu, Environ. Res., 2021, 197, 111089 CrossRef CAS PubMed.
- Y. T. Guo, E. R. Rene, B. Y. Han and W. F. Ma, J. Hazard. Mater., 2021, 416, 126197 CrossRef CAS PubMed.
- Q. Sun and G. C. Zhu, Environ. Res., 2022, 210, 112856 CrossRef CAS PubMed.
- Z. C. Li, Y. Feng, L. Chang, Y. Y. Long, N. Suo, Z. W. Wang and Y. Z. Yu, Bioresour. Technol., 2022, 346, 126653 CrossRef CAS PubMed.
- T. C. An, X. H. Zhu and Y. Xiong, Chemosphere, 2002, 46, 897–903 CrossRef CAS PubMed.
- T. C. An, G. Y. Li, X. H. Zhu, J. M. Fu, G. Y. Sheng and Z. Kun, Appl. Catal., A, 2005, 279, 247–256 CrossRef CAS.
- Y. J. Yang, L. Y. Zhang, Y. L. Zhu, G. T. Wei, Z. M. Li and R. L. Mo, J. Environ. Chem. Eng., 2021, 9, 105559 CrossRef CAS.
- J. C. Gu, G. T. Wei, Y. L. Zhu, C. P. Lu, L. Y. Zhang, Z. J. Huang, Q. F. Su and S. J. Pan, J. Environ. Chem. Eng., 2023, 11, 109137 CrossRef CAS.
- J. H. Zhan, Z. X. Li, G. Yu, X. J. Pan, J. L. Wang, W. Zhu, X. Han and Y. J. Wang, Sep. Purif. Technol., 2019, 208, 12–18 CrossRef CAS.
- A. Wang, X. Liu, Y. Wen, Y. Qiu, S. Lv, M. Xu, C. Meng, K. Wang, F. Lin, S. Xie and Q. Zhuo, Water Res., 2023, 245, 120596 CrossRef CAS PubMed.
- Z. K. Zhao, Y. J. Hao, J. L. Wu, Z. Y. Feng, F. Feng, Y. F. Li, Q. P. Yang and B. Jiang, Sep. Purif. Technol., 2023, 317, 123960 CrossRef CAS.
- A. Ahmad, M. Priyadarshini, S. Yadav, M. M. Ghangrekar and R. Y. Surampalli, Environ. Res., 2024, 245, 117998 CrossRef CAS PubMed.
- J. Q. Bu, Z. W. Deng, H. Liu, T. H. Li, Y. J. Yang and S. Zhong, J. Environ. Manage., 2022, 312, 114975 CrossRef CAS PubMed.
- T. Zhang, G. Bai, N. Cai, Y. Q. Lei, P. R. Guo and J. W. Xu, Appl. Clay Sci., 2023, 243, 107056 CrossRef CAS.
- Z. Y. Wang, B. Song, J. F. Li and X. L. Teng, Chemosphere, 2021, 270, 128652 CrossRef CAS PubMed.
- B. Song, Z. Y. Wang, J. F. Li, M. Q. Luo, P. W. Cao and C. Zhang, Chem. Eng. J., 2021, 426, 131940 CrossRef CAS.
- Y. J. Niu, J. Xu, L. Q. Chen, S. X. Guo, F. H. Guo, Y. X. Zhang and J. J. Wu, Fuel, 2024, 355, 129449 CrossRef CAS.
- T. H. Zhou, X. X. Huang, T. J. Zhai, K. Ma, H. W. Zhang and G. Z. Zhang, Chemosphere, 2022, 287, 132397 CrossRef CAS PubMed.
- Z. H. Chen, L. B. Yang, X. W. Luo, D. L. Xie, C. Zhao, R. L. Qiu and Z. J. Huang, Sep. Purif. Technol., 2022, 296, 121402 CrossRef CAS.
- Y. Feng, Z. J. Zhang, Y. H. Zhao, L. Song, X. W. Wang, S. M. Yang, Y. Y. Long, C. H. Zhao and L. P. Qiu, Bioresour. Technol., 2019, 283, 1–9 CrossRef CAS PubMed.
- Z. J. Zhang, Y. Feng, N. Liu, Y. H. Zhao, X. W. Wang, S. M. Yang, Y. Y. Long and L. P. Qiu, J. Water Process Eng., 2020, 37, 101417 CrossRef.
- Y. Yao, Q. Y. Chen, Z. L. Huang and J. Zhou, Environ. Technol. Innovation, 2021, 21, 101321 CrossRef CAS.
- S. Liu, Z. Y. Wang, J. F. Li, C. Zhao, X. L. He and G. Yang, Chemosphere, 2018, 213, 377–383 CrossRef CAS PubMed.
- J. Ji, X. Y. Li, J. Xu, X. Y. Yang, H. S. Meng and Z. R. Yan, Electrochim. Acta, 2018, 284, 587–596 CrossRef CAS.
- Z. Y. Wang, J. Y. Qi, Y. Feng, K. Li and X. Li, J. Ind. Eng. Chem., 2014, 20, 3672–3677 CrossRef CAS.
- L. Li, X. Fang, X. Jin and Y. Mu, J. Water Resour. Water Eng., 2015, 26, 40–50 CrossRef.
- H. J. Xiao, Y. J. Hao, J. L. Wu, X. Z. Meng, F. Feng, F. Q. Xu, S. Y. Luo and B. Jiang, Chemosphere, 2023, 325, 138423 CrossRef CAS PubMed.
- Z. Z. Jia, X. Zhao, C. Y. Yu, Q. Wan and Y. F. Liu, Environ. Technol. Innovation, 2021, 23, 101554 CrossRef CAS.
- X. Z. Meng, K. Li, Z. K. Zhao, Y. F. Li, Q. P. Yang and B. Jiang, Sep. Purif. Technol., 2022, 298, 121672 CrossRef CAS.
- A. Dargahi, K. Hasani, S. A. Mokhtari, M. Vosoughi, M. Moradi and Y. Vaziri, J. Environ. Chem. Eng., 2021, 9, 105889 CrossRef CAS.
- H. Chen, Y. Feng, N. Suo, Y. Y. Long, X. Li, Y. L. Shi and Y. Z. Yu, Chemosphere, 2019, 216, 281–288 CrossRef CAS PubMed.
- M. R. Li, X. Qin, J. X. Cui, R. Guo, C. R. Guo, Z. Y. Wang and T. H. Li, J. Environ. Chem. Eng., 2021, 9, 105573 CrossRef CAS.
- Y. Wang, X. X. Qi, Y. T. Qin, C. Y. An, J. H. Guo and J. Wang, Environ. Pollut., 2023, 337, 122574 CrossRef CAS PubMed.
- X. Wang, Z. L. Zhao, H. J. Wang, F. Wang and W. Y. Dong, J. Environ. Sci., 2023, 124, 630–643 CrossRef CAS PubMed.
- Y. J. Niu, J. Xu, Z. K. Miao, F. H. Guo, Y. X. Zhang and J. J. Wu, Chemosphere, 2022, 303, 135159 CrossRef CAS PubMed.
- W. D. Wang, L. L. Wang, K. Wang, T. T. Zhang, Y. B. Sun and W. P. Li, J. Cleaner Prod., 2021, 322, 128683 CrossRef CAS.
- D. Y. Yu and Y. S. Pei, J. Environ. Manage., 2022, 321, 115890 CrossRef CAS PubMed.
- B. L. Hou, H. J. Han, S. Y. Jia, H. F. Zhuang, P. Xu and K. Li, J. Taiwan Inst. Chem. Eng., 2016, 60, 352–360 CrossRef CAS.
- Y. Feng, M. Y. Guo, X. Q. Jia, N. Liu, X. X. Li, X. Li, L. Song, X. W. Wang, L. P. Qiu and Y. Z. Yu, Bioresour. Technol., 2020, 309, 123346 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |