
Transient control of lytic activity via a non-equilibrium chemical reaction system†
Kohei
Sato‡§
 *a,
Yume
Nakagawa‡
a,
Miki
Mori
a,
Masahiro
Takinoue
abc and
Kazushi
Kinbara
*ac
*a,
Yume
Nakagawa‡
a,
Miki
Mori
a,
Masahiro
Takinoue
abc and
Kazushi
Kinbara
*ac
aSchool of Life Science and Technology, International Research Frontiers Initiative, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan. E-mail: ksato@kwansei.ac.jp; kkinbara@bio.titech.ac.jp
bDepartment of Computer Science, International Research Frontiers Initiative, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
cLiving Systems Materialogy Research Group, International Research Frontiers Initiative, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
First published on 2nd March 2024
Abstract
The development of artificial non-equilibrium chemical reaction systems has recently attracted considerable attention as a new type of biomimetic. However, due to the lack of bioorthogonality, such reaction systems could not be linked to the regulation of any biological phenomena. Here, we have newly designed a non-equilibrium reaction system based on olefin metathesis to produce the Triton X-mimetic non-ionic amphiphile as a kinetic product. Using phospholipid vesicles encapsulating fluorescent dyes and red blood cells as cell models, we demonstrate that the developed chemical reaction system is applicable for transient control of the resulting lytic activity.
 Kohei Sato | Kohei Sato received his PhD from the University of Tokyo in 2014 under the supervision of Professor Takuzo Aida. He then conducted postdoctoral research in the group of Professor Samuel I. Stupp at Northwestern University. In 2018, he joined the group of Professor Kazushi Kinbara as an assistant professor. In 2023, he started his independent career as an associate professor in the Department of Chemistry at Kwansei Gakuin University. His current research focuses on the development of bioactive supramolecular materials. |
 Yume Nakagawa | Yume Nakagawa was born in Mie, Japan in 1999. She completed her undergraduate course from the Tokyo Institute of Technology and is currently continuing her research as a postgraduate student in the Kinbara group. Her research interests are non-equilibrium chemical reactions, particularly in Systems Chemistry. |
 Masahiro Takinoue | Masahiro Takinoue is a biophysicist studying artificial cell engineering and molecular computing based on DNA nanotechnology. He received a B.Sc. in Physics in 2002, a M.Sc. in Physics in 2004, and a Ph.D. in Physics in 2007 from The University of Tokyo (UT), Japan. After serving as a postdoctoral fellow at Kyoto University and a project assistant professor at UT, he served as an associate professor at Tokyo Institute of Technology (Tokyo Tech), Japan (2011-2022). He has been a full professor at the Department of Computer Science and Living Systems Materialogy (LiSM) Research Group at Tokyo Tech (2022-). |
 Kazushi Kinbara | Dr. Kazushi Kinbara received a Ph.D. degree in organic chemistry from the University of Tokyo in 1996 under the direction of Professor Kazuhiko Saigo. From 2001, he joined Professor Takuzo Aida’s group at School of Engineering, the University of Tokyo as a lecturer and associate professor. In 2008, he was promoted to a professor of the Institute of Multidisciplinary Research for Advanced Materials, Tohoku University. In 2015, he moved to Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology. His research interests include (1) bioinpired chemistry and (2) supramolecular chemistry. |
Introduction
The development of life-like architectures has long been the ultimate goal in organic chemistry.1–3 To date, remarkable advances in synthetic and supramolecular chemistry have enabled the construction of complex molecular assemblies reminiscent of natural biomacromolecules such as proteins4–15 and nucleic acids.16–22 More recently, the development of artificial systems that mimic the non-equilibrium chemical reactions in living organisms, known as systems chemistry, has attracted considerable attention as a new type of biomimetic.23–32 Furthermore, such reactions have been associated with functional outcomes, for example, drug release,33,34 modulation of chemical reactions,35,36 and control of the microscopic movement of colloids.37 However, to the best of our knowledge, such reaction systems have never been involved in interacting directly with biomolecules to regulate any biological phenomena. Undoubtedly, non-equilibrium chemical reaction systems are highly advantageous for their promising potential to precisely and temporally control biological activities. However, conventional systems chemistry was mostly based on the reactions of polar functional groups,38 including carboxylic acids for carbodiimide-fueled reactions,39,40 amines for imine formation,41,42 and thiols for thiol–ester and thiol–disulfide exchange reactions.43–45 Because such polar functional groups are abundant in biomolecules, these reactions would most likely show interference under physiological conditions.Recently, Fletcher and coworkers have developed non-equilibrium chemical reaction systems based on the ruthenium-catalyzed cross-metathesis of hydrophilic and hydrophobic alkenes.46 Here, we took advantage of the bioorthogonality of the metathesis reaction47–50 and sought to introduce a new molecular design strategy into Fletcher's system to promote direct interaction between the resulting product and biomolecules. The chemical structure of a well-known commercial synthetic surfactant, Triton X (Fig. 1), provided us with a clue. Triton X is a non-ionic amphiphile consisting of a hydrophobic aliphatic tail, an aromatic ring, and a hydrophilic poly(ethylene glycol) (PEG) chain, known for its high propensity to interact with lipid molecules to disrupt biomembranes while preventing protein denaturation.51,52 Therefore, we designed a non-equilibrium chemical reaction system based on olefin metathesis to produce a Triton-mimetic amphiphile as a kinetic product. Through this study, we expected to establish a proof-of-concept system to realize the temporal control of biological activity synchronous to an artificial non-equilibrium chemical reaction. In particular, we focused on controlling the interaction with cell membranes (Fig. 1), the vital components of life.3,53,54
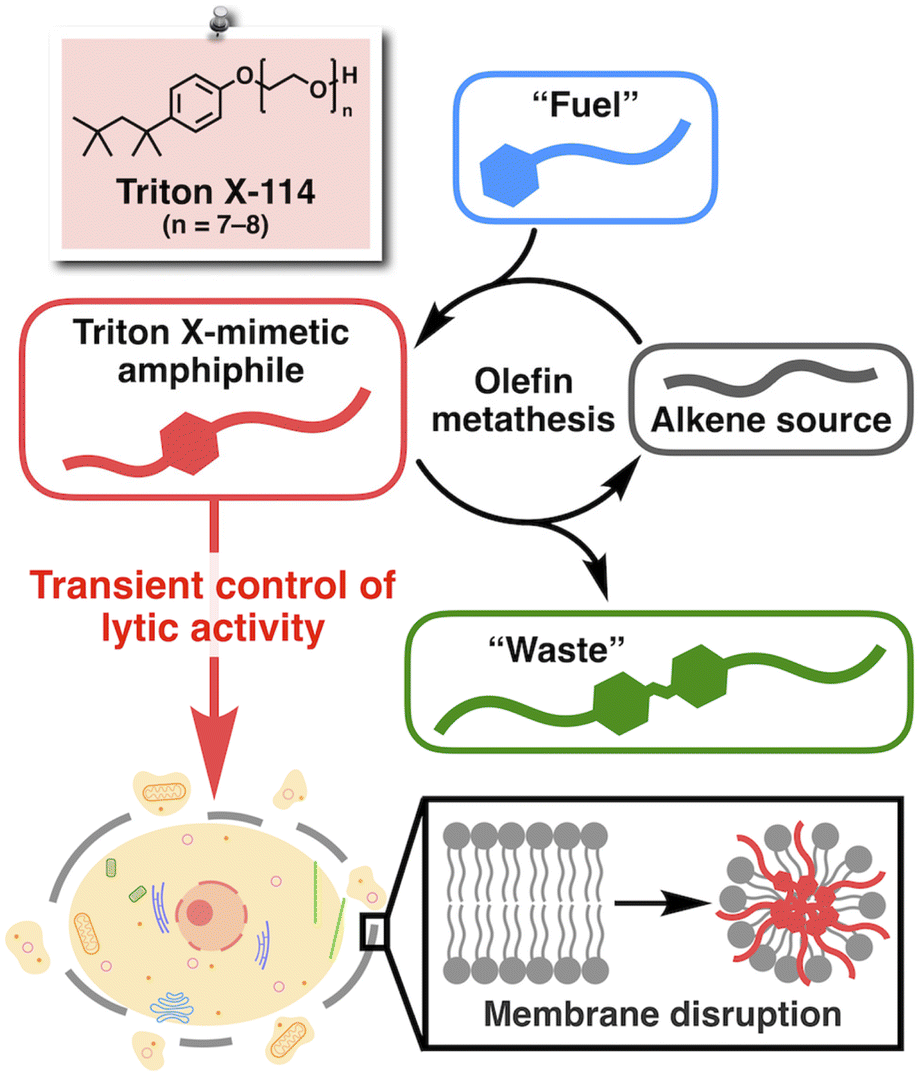 | ||
| Fig. 1 Schematic illustrations of the chemical structure of Triton X-114 and a non-equilibrium chemical reaction system that produces the Triton-mimetic amphiphile as a kinetic product through olefin metathesis. The amphiphile was designed to interact with cell membranes to induce their disruption in a transient manner. | ||
Results and discussion
The chemical structures of the reactants and products, and an overview of the chemical reaction system are shown in Fig. 2a. We selected 7-tetradecene as the alkene source of the hydrophobic moiety and designed a fuel alkene FuelPEG so that the resulting amphiphilic metathesis product AmpPEG would possess the same hydrophobic–hydrophilic balance as Triton X-114 (Fig. 1) in terms of its constituent atoms. The synthesized 7-tetradecene, FuelPEG, AmpPEG, and HydPEG (vide infra) were unambiguously characterized by NMR and high-resolution mass spectrometry (Fig. S1–S13, see the ESI†).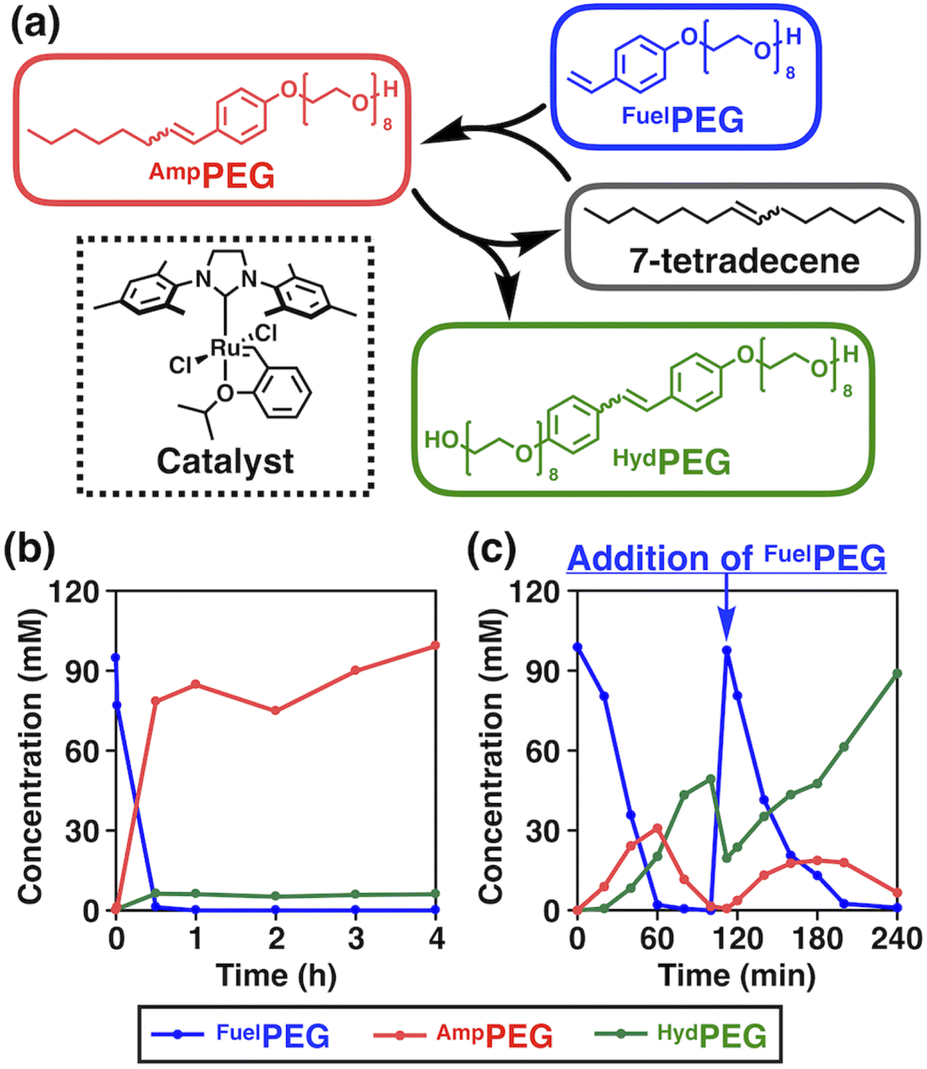 | ||
| Fig. 2 (a) Schematic illustration of the designed non-equilibrium chemical reaction system based on olefin metathesis. (b) Concentration vs. time plot of the homogeneous reaction mixture using CH2Cl2 as a solvent at 25 °C (480 rpm). (c) Concentration vs. time plot of the biphasic reaction mixture using H2O as a solvent at 50 °C (480 rpm). At 100 min, FuelPEG was added as an additional “fuel”. | ||
First, we mixed 7-tetradecene, FuelPEG, and Hoveyda–Grubbs catalyst 2nd generation (HG-II)46,55,56 in CH2Cl2 and monitored the reaction progress using ultra-performance liquid chromatography (UPLC) (Fig. S14a, see the ESI†). Note that all reagents were uniformly dissolved so that the reaction was performed under homogeneous conditions. As shown in Fig. 2b, the concentration of AmpPEG (Fig. 2b, red) increased monotonically in response to the consumption of FuelPEG (Fig. 2b, blue), representing the conventional reaction kinetics. Next, we used water instead of CH2Cl2 as a solvent to perform the same reaction under biphasic conditions (Fig. S14b, see the ESI†). Intriguingly, the reaction showed non-equilibrium kinetics where the concentration of AmpPEG (Fig. 2c, red) reached its maximum at 60 min and then gradually decreased. In addition, the hydrophilic metathesis product HydPEG (Fig. 2c, green) started to appear after 20 min, following the generation of AmpPEG. At 100 min, we observed the complete consumption of AmpPEG and the exclusive formation of HydPEG. These results demonstrate that AmpPEG was formed as a transient product, while HydPEG was formed as an accumulating “waste” product. We also added FuelPEG to the reaction mixture as an additional “fuel” after the complete consumption of AmpPEG at 100 min and again observed the transient formation of AmpPEG (Fig. 2c, red), clearly demonstrating the repeatability of the designed reaction system. Note that the apparent decrease in HydPEG at 100 min (Fig. 2c, green) is due to the dilution of the reaction mixture upon addition of the aqueous solution of FuelPEG (see the ESI†).
In the previous study by Fletcher and coworkers, they reported that the ruthenium catalyst was compartmentalized within the droplet of hydrophobic alkene, and cross-metathesis reactions of hydrophobic and hydrophilic alkene occurred at the interface with water.36,46 We hypothesized that a similar reaction mechanism also took place in our system and performed qualitative kinetic simulations. In fact, the simulation for the biphasic conditions showed that the concentration of AmpPEG increased and then decreased while that for the homogeneous conditions did not (Fig. S15, see the ESI†).
Regarding the reaction mechanism, we also tested the emulsifying properties of FuelPEG and HydPEG by mixing their aqueous solution with 7-tetradecene. As shown in Fig. 3a, FuelPEG formed turbid dispersions, while HydPEG resulted in the much-less turbid phase-separated mixture (Fig. 3b). The difference in turbidity was also quantified by measuring the optical density (O.D.) at 600 nm (Fig. S17†). These results demonstrated that FuelPEG could stabilize the emulsions due to its amphiphilicity, while HydPEG could not, likely because HydPEG is too hydrophilic. In fact, using dynamic light scattering (DLS, Fig. 3c), transmission electron microscopy (Fig. 3d), and surface tension measurements (Fig. 3e), we confirmed that FuelPEG can self-assemble into spherical aggregates in water while HydPEG cannot (Fig. S19b†). In addition, we performed an optical microscopic observation of the emulsions formed by 7-tetradecene and FuelPEG in the presence of HG-II and found that the interior of the emulsions were colored green (Fig. S20†), which is characteristic of the ruthenium catalyst.57 This result indicates that HG-II was compartmentalized within the droplets of the emulsions as proposed by Fletcher and coworkers in their earlier study.46 Considering all these results, it seems that the cross-metathesis reaction between 7-tetradecene and FuelPEG occurred predominantly to generate AmpPEG in the early stage (Fig. 3f-i). Later, HydPEG was generated but then diffused out of the interface due to its hydrophilicity, and as a result, HydPEG was excluded from the reaction system, and the reaction equilibrium was further shifted to lead to the complete consumption of AmpPEG (Fig. 3f-ii).
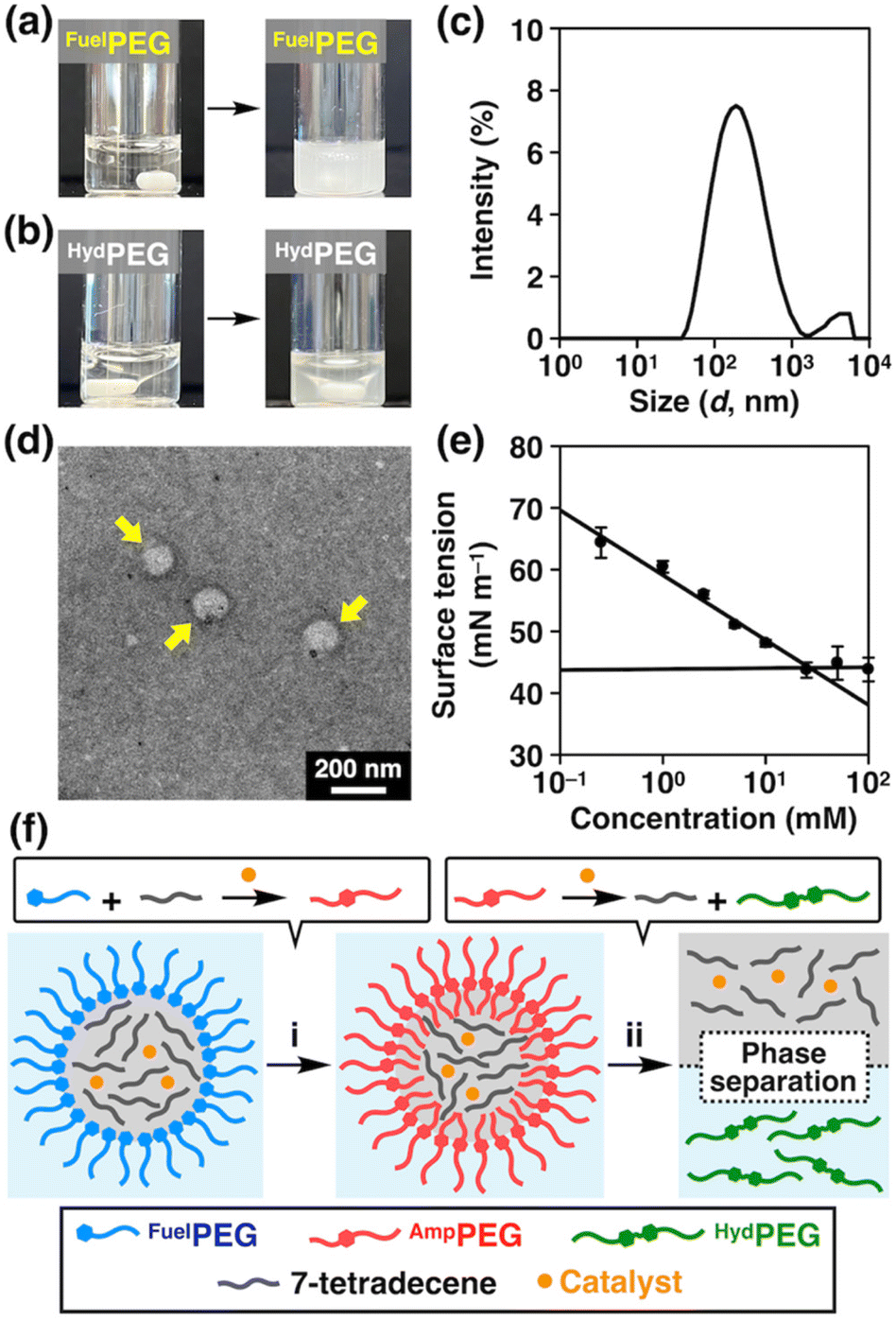 | ||
| Fig. 3 (a and b) Photographs of the mixtures of 7-tetradecene and (a) aqueous FuelPEG (50 mM) and (b) aqueous HydPEG (50 mM) before (left) and after (right) stirring for 4 h at 25 °C. (c) An intensity-based particle size distribution profile of aqueous FuelPEG (30 mM) at 20 °C, analyzed by dynamic light scattering. The mean hydrodynamic diameter of the particle: 222.2 nm. (d) Transmission electron micrograph of aqueous FuelPEG stained with gadolinium acetate. (e) Surface tension vs. concentration plot of aqueous FuelPEG. The critical aggregation concentration (CAC) was determined from the intersection of the fitted lines ([FuelPEG]CAC = 26.7 mM). (f) Schematic illustration of the mechanism of the out-of-equilibrium chemical reaction. First, hydrophobic 7-tetradecene and the catalyst are surrounded by FuelPEG in water. Then, the cross-metathesis reaction between 7-tetradecene and FuelPEG occurs at the interface to form AmpPEG. Subsequently, hydrophilic HydPEG is formed and diffuses out of the interface. As a result, the reaction equilibrium is shifted, leading to the complete consumption of AmpPEG. Note that 7-tetradecene acts as both a reactant and an organic phase. | ||
With the newly developed non-equilibrium chemical reaction system in hand, we then investigated its potential to exhibit the biologically relevant properties arising from the structural similarity of AmpPEG to Triton X-114. As model cell membranes, we prepared giant unilamellar vesicles (GUVs) formed by 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) by the electroformation method (see the ESI†) and added an aliquot of the reaction mixture at different time points. We used DilC18(3) to label the GUVs to observe their morphologies by fluorescence microscopy. Strikingly, we observed a distinct time-dependent effect; we clearly observed GUVs when we added the reaction mixture with reaction times of 1 min (Fig. 4a, right) and 120 min (Fig. 4a, left), but we did not observe any GUVs with a reaction time of 60 min (Fig. 4a, middle). To quantitatively analyze this phenomenon, we prepared DOPC large unilamellar vesicles (LUVs) encapsulating 5-carboxyfluorescein (5-CF) by the extrusion method and examined the release of 5-CF (see the ESI†). Note that fluorescence quenching of 5-CF occurs when its concentration is high, so an increase in fluorescence intensity will be observed when it is released from inside the LUVs.15,58–60 Using authentic samples of FuelPEG, AmpPEG, and HydPEG, we confirmed that AmpPEG effectively ruptured the LUVs, while FuelPEG and HydPEG did not (Fig. S24b†). We then measured the fluorescence spectra of 5-CF at different reaction time points (Fig. 4b) and plotted their intensity. As shown in Fig. 4c, the fluorescence intensity change showed a good correlation with the concentration of AmpPEG generated during the reaction. We also performed DLS measurements of the mixtures and observed the transient size change from LUVs to smaller aggregates with a diameter of 9.0 nm at a reaction time of 60 min (Fig. S25†), similar to the size of spherical micelles formed by AmpPEG alone (Fig. S18, S19a and S26†). These results clearly demonstrated the time-dependent disruption of LUVs caused by the transient generation of AmpPEG, which co-assembled with DOPC phospholipids to form micelles (Fig. 4a middle, inset).61,62
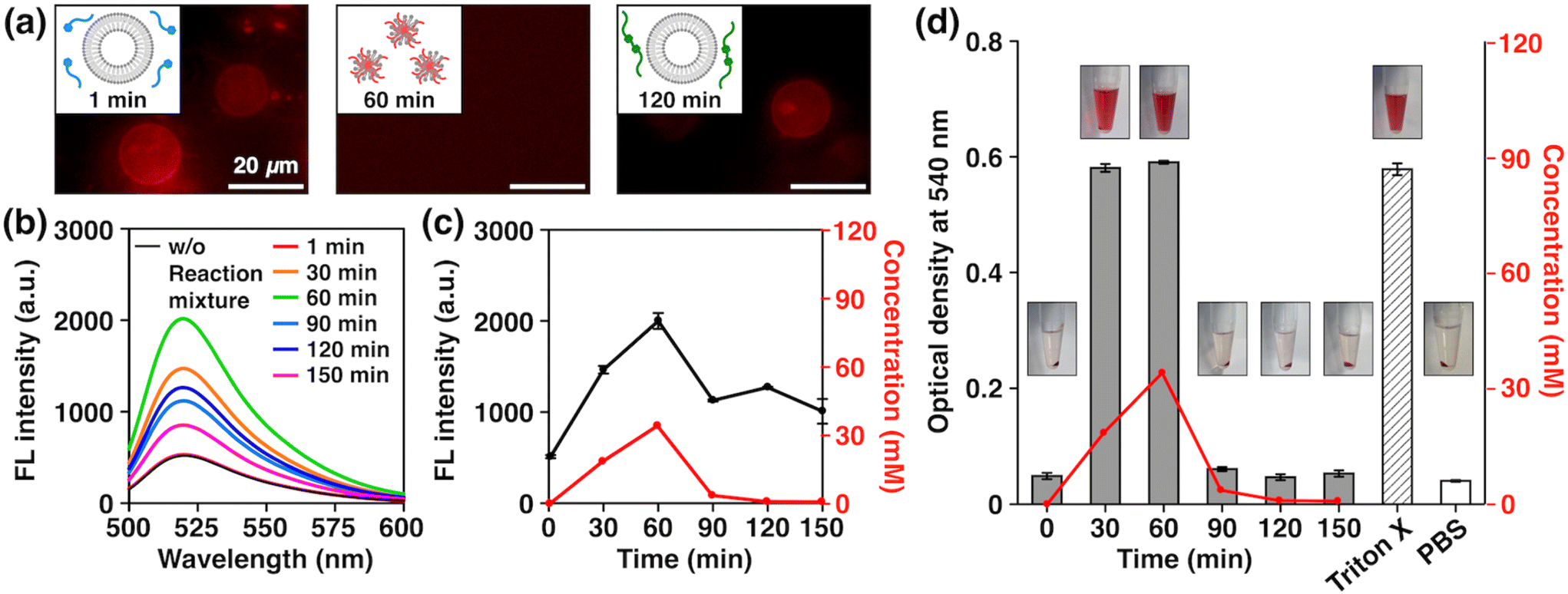 | ||
| Fig. 4 (a) Fluorescence micrographs of DiIC18(3)-labeled GUVs ([DOPC] = 600 μM, [DiIC18(3)] = 600 nM) in aqueous glucose (2.0 mM) after the addition of the reaction mixtures at reaction times of 1 (left), 60 (middle), and 120 min (right) at 25 °C (λex = 520–550 nm, λobsd > 580 nm). (b) Representative fluorescence intensity changes of 5-CF encapsulated in DOPC LUVs ([DOPC] = 100 μM, [5-CF] = 50 mM) in HEPES buffer ([HEPES] = 20 mM, [NaCl] = 50 mM, pH 7.5) at 20 °C after the addition of the reaction mixtures at different reaction times (λex = 490 nm, λem = 520 nm). (c) Overlay of fluorescence intensity changes of 5-CF at 520 nm (left axis, black) and concentration changes of AmpPEG (right axis, red) as a function of time. Error bars represent standard deviations (n = 3). (d) Overlay of optical density changes of the red blood cell (RBC) suspensions at 540 nm (left axis, black) and concentration changes of AmpPEG in the reaction mixture (right axis, red) as a function of time. Error bars represent standard deviations (n = 5). The insets show the photographs of the mixtures of RBC suspensions and reaction mixtures at different reaction times. | ||
To further extend the properties of our non-equilibrium chemical reaction system, we investigated the possibility of transient hemolytic activity, the disruption of red blood cells (RBCs), which is highly associated with the maintenance of homeostasis in the body.63 We added an aliquot of the reaction mixture at different time points to the suspensions of RBCs, incubated the mixture for 30 min, centrifuged, and measured the O.D. of the supernatant at 540 nm to quantify the amount of hemoglobin released by hemolysis (see the ESI†).64 As shown in Fig. 4d, the O.D. values showed a clear correlation with the concentration of AmpPEG generated during the reaction, where high hemolytic activity was observed at the reaction time points of 30 and 60 min. Using authentic samples of FuelPEG, AmpPEG, and HydPEG, we confirmed that AmpPEG is the only hemolytically active compound besides FuelPEG and HydPEG (Fig. S27†). These results clearly demonstrated the first example of transient control of lytic activity by a non-equilibrium reaction system. We also analyzed the absorption spectra of the mixture and found that the denaturation of hemoglobin did not occur (Fig. S28†), which nicely corresponds to the membrane-specific activity of Triton X as mentioned earlier.65
Conclusions
We have newly developed a non-equilibrium chemical reaction system that generates a Triton X-mimetic amphiphile AmpPEG as a kinetic product. Through a quantitative assay using dye-embedded LUVs, we confirmed that the generation of AmpPEG enabled time-dependent membrane disruption through interaction with phospholipids. Furthermore, we demonstrated that the chemical reaction system is applicable to transient control of the resulting lytic activity. In fact, we successfully achieved time-controlled hemolysis of RBCs without denaturing hemoglobin. Although further optimization is needed to realize in situ control of biological activity, we believe this work has opened the door to the development of next-generation functional supramolecular materials that can precisely and temporally modulate biological events.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Engine” (18H05418, 18H05419, and 23H05418 to K.K.), a Grant-in-Aid for Challenging Research (Pioneering) (23K17363 to K.K.), a Grant-in-Aid for Scientific Research (B) (23H02080 to K.S.), a Grant-in-Aid for Challenging Research (Exploratory) (23K17973 to K.S.), and a Grant-in-Aid for Transformative Research Areas “Molecular Cybernetics” (23H04408 to K. S.). K. S. also thanks The Salt Science Research Foundation, Iketani Science and Technology Foundation, The Ube Foundation, and The Mitsubishi Foundation for their financial support. The authors thank Suzukakedai Materials Analysis Division, Open Facility Center, Tokyo Institute of Technology, for NMR spectroscopy, EI mass spectrometry, ESI-TOF mass spectrometry, TEM observations, and surface tension measurements. In addition, we acknowledge Yuka Akimoto for her assistance with TEM. We thank the Open Research Facilities for Life Science and Technology, Tokyo Institute of Technology, for the use of a plate reader and an autoclave machine. We also thank Prof. Atsushi Maruyama for the use of an ultracentrifuge.References
- L. Milanesi and S. Tomas, Annu. Rep. Prog. Chem., Sect. B, 2010, 106, 477–469 RSC.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 144, 285–366 CrossRef PubMed.
- B. C. Buddingh’ and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef PubMed.
- P. L. Anelli, N. Spencer and J. F. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Magonov and S. A. Vinogradov, Nature, 2004, 430, 764–768 CrossRef CAS PubMed.
- M. S. Masar, N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149–10158 CrossRef CAS PubMed.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J.-C. Chang, S.-H. Tseng, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Nat. Chem., 2016, 9, 128–134 CrossRef.
- G. De Bo, M. A. Y. Gall, S. Kuschel, J. De Winter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381–385 CrossRef CAS PubMed.
- S.-P. Zheng, L.-B. Huang, Z. Sun and M. Barboiu, Angew. Chem., Int. Ed., 2021, 60, 566–597 CrossRef CAS PubMed.
- K. Sato, R. Sasaki, R. Matsuda, M. Nakagawa, T. Ekimoto, T. Yamane, M. Ikeguchi, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2022, 144, 11802–11809 CrossRef CAS PubMed.
- U. Koert, M. M. Harding and J.-M. Lehn, Nature, 1990, 346, 339–342 CrossRef CAS PubMed.
- B. Hasenknopf and J.-M. Lehn, Helv. Chim. Acta, 1996, 79, 1643–1650 CrossRef CAS.
- J. H. K. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed.
- H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176–7178 CrossRef CAS PubMed.
- Q. Gan, C. Bao, B. Kauffmann, A. Grélard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715–1718 CrossRef CAS PubMed.
- W. Makiguchi, S. Kobayashi, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 5275–5279 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- A. K. Dambenieks, P. H. Q. Vu and T. M. Fyles, Chem. Sci., 2014, 5, 3396–3403 RSC.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- S. Maiti, I. Fortunati, C. Ferrante, P. Scrimin and L. J. Prins, Nat. Chem., 2016, 8, 725–731 CrossRef CAS PubMed.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef PubMed.
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC.
- E. A. J. Post and S. P. Fletcher, Chem. Sci., 2020, 11, 9434–9442 RSC.
- J. Deng and A. Walther, Adv. Mater., 2020, 32, 2002629 CrossRef CAS PubMed.
- K. Das, L. Gabrielli and L. J. Prins, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed.
- J. Flores, R. J. Brea, A. Lamas, A. Fracassi, M. Salvador-Castell, C. Xu, C. R. Baiz, S. K. Sinha and N. K. Devaraj, Angew. Chem., Int. Ed., 2022, 61, e202200549 CrossRef CAS PubMed.
- A. Sharko, D. Livitz, S. De Piccoli, K. J. M. Bishop and T. M. Hermans, Chem. Rev., 2022, 122, 11759–11777 CrossRef CAS PubMed.
- S. Bai, X. Niu, H. Wang, L. Wei, L. Liu, X. Liu, R. Eelkema, X. Guo, J. H. van Esch and Y. Wang, Chem. Eng. J., 2021, 414, 128877 CrossRef CAS.
- P. S. Schwarz, L. Tebcharani, J. E. Heger, P. Müller-Buschbaum and J. Boekhoven, Chem. Sci., 2021, 12, 9969–9976 RSC.
- M. A. Cardona and L. J. Prins, Chem. Sci., 2020, 11, 1518–1522 RSC.
- I. Colomer, A. Borissov and S. P. Fletcher, Nat. Commun., 2020, 11, 176 CrossRef CAS PubMed.
- D. Babu, R. J. H. Scanes, R. Plamont, A. Ryabchun, F. Lancia, T. Kudernac, S. P. Fletcher and N. Katsonis, Nat. Commun., 2021, 12, 2959 CrossRef CAS PubMed.
- N. Singh, G. J. M. Formon, S. De Piccoli and T. M. Hermans, Adv. Mater., 2020, 32, 1906834 CrossRef CAS PubMed.
- M. Tena-Solsona, B. Rieß, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS PubMed.
- P. S. Schwarz, M. Tena-Solsona, K. Dai and J. Boekhoven, Chem. Commun., 2022, 58, 1284–1297 RSC.
- R. Nguyen, L. Allouche, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2009, 48, 1093–1096 CrossRef CAS PubMed.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- M. Colomb-Delsuc, E. Mattia, J. W. Sadownik and S. Otto, Nat. Commun., 2015, 6, 7427 CrossRef CAS PubMed.
- S. M. Morrow, I. Colomer and S. P. Fletcher, Nat. Commun., 2019, 10, 1011 CrossRef PubMed.
- I. Colomer, S. M. Morrow and S. P. Fletcher, Nat. Commun., 2018, 9, 2239 CrossRef PubMed.
- J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS PubMed.
- B. Bhushan, Y. A. Lin, M. Bak, A. Phanumartwiwath, N. Yang, M. K. Bilyard, T. Tanaka, K. L. Hudson, L. Lercher, M. Stegmann, S. Mohammed and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 14599–14603 CrossRef CAS PubMed.
- V. Sabatino, J. G. Rebelein and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 17048–17052 CrossRef CAS PubMed.
- I. Nasibullin, I. Smirnov, P. Ahmadi, K. Vong, A. Kurbangalieva and K. Tanaka, Nat. Commun., 2022, 13, 39 CrossRef CAS PubMed.
- A. Helenius and K. Simons, Biochim. Biophys. Acta, 1975, 415, 29–79 CrossRef CAS PubMed.
- S. Kalipatnapu and A. Chattopadhyay, IUBMB Life, 2005, 57, 505–512 CrossRef CAS PubMed.
- D. L. Abel, Life, 2012, 2, 106–134 CrossRef PubMed.
- D. E. Koshland Jr., Science, 2002, 295, 2215–2216 CrossRef PubMed.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- T. Matsuo, Catalysts, 2021, 11, 359 CrossRef CAS.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- R. Sasaki, K. Sato, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2021, 143, 1348–1355 CrossRef CAS PubMed.
- M. Mori, K. Sato, T. Ekimoto, S. Okumura, M. Ikeguchi, K. V. Tabata, H. Noji and K. Kinbara, Chem. – Asian J., 2021, 16, 147–157 CrossRef CAS PubMed.
- O. López, A. de la Maza, L. Coderch, C. López-Iglesias, E. Wehrli and J. L. Parra, FEBS Lett., 1998, 426, 314–318 CrossRef PubMed.
- A. Pizzirusso, A. De Nicola, G. J. A. Sevink, A. Correa, M. Cascella, T. Kawakatsu, M. Rocco, Y. Zhao, M. Celino and G. Milano, Phys. Chem. Chem. Phys., 2017, 19, 29780–29794 RSC.
- R. E. Mebius and G. Kraal, Nat. Rev. Immunol., 2005, 5, 606–616 CrossRef CAS PubMed.
- J.-B. Wang, Y.-M. Wang and C.-M. Zeng, Biochem. Biophys. Res. Commun., 2011, 415, 675–679 CrossRef CAS PubMed.
- W. Li, L. Lin and G. Li, Anal. Methods, 2014, 6, 1082–1089 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06626f |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Chemistry, School of Science, Kwansei Gakuin University, 1 Gakuen Uegahara, Sanda-shi, Hyogo 669–1330, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |