
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVibrational properties of heme-nitrosoalkane complexes in comparison with those of their HNO analogs, and reactivity studies towards nitric oxide and Lewis acids†
Jill B.
Harland
a,
Ashley B.
LaLonde
 a,
Diamond J.
Thomas
a,
Daniel G.
Castella
a,
Jeff W.
Kampf
a,
Matthias
Zeller
b,
E. Ercan
Alp
c,
Michael Y.
Hu
c,
Jiyong
Zhao
c and
Nicolai
Lehnert
*a
a,
Diamond J.
Thomas
a,
Daniel G.
Castella
a,
Jeff W.
Kampf
a,
Matthias
Zeller
b,
E. Ercan
Alp
c,
Michael Y.
Hu
c,
Jiyong
Zhao
c and
Nicolai
Lehnert
*a
aDepartment of Chemistry and Department of Biophysics, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: lehnertn@umich.edu
bDepartment of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA
cAdvanced Photon Source, Argonne National Laboratory, Illinois 60439, USA
First published on 4th July 2024
Abstract
C-Nitroso compounds (RNO, R = alkyl and aryl) are byproducts of drug metabolism and bind to heme proteins, and their heme-RNO adducts are isoelectronic to ferrous nitroxyl (NO−/HNO) complexes. Importantly, heme-HNO compounds are key intermediates in the reduction of NO to N2O and nitrite to ammonium in the nitrogen cycle. Ferrous heme-RNO complexes act as stable analogs of these species, potentially allowing for the investigation of the vibrational and electronic properties of unstable heme-HNO intermediates. In this paper, a series of six-coordinate ferrous heme-RNO complexes (where R = iPr and Ph) were prepared using the TPP2− and 3,5-Me-BAFP2− co-ligands, and tetrahydrofuran, pyridine, and 1-methylimidazole as the axial ligands (bound trans to RNO). These complexes were characterized using different spectroscopic methods and X-ray crystallography. The complex [Fe(TPP)(THF)(iPrNO)] was further utilized for nuclear resonance vibrational spectroscopy (NRVS), allowing for the detailed assignment of the Fe–N(R)O vibrations of a heme-RNO complex for the first time. The vibrational properties of these species were then correlated with those of their HNO analogs, using DFT calculations. Our studies support previous findings that RNO ligands in ferrous heme complexes do not elicit a significant trans effect. In addition, the complexes are air-stable, and do not show any reactivity of their RNO ligands towards NO. So although ferrous heme-RNO complexes are suitable structural and electronic models for their HNO analogs, they are unsuitable to model the reactivity of heme-HNO complexes. We further investigated the reaction of our heme-RNO complexes with different Lewis acids. Here, [Fe(TPP)(THF)(iPrNO)] was found to be unreactive towards Lewis acids. In contrast, [Fe(3,5-Me-BAFP)(iPrNO)2] is reactive towards all of the Lewis acids investigated here, but in most cases the iron center is simply oxidized, resulting in the loss of the iPrNO ligand. In the case of the Lewis acid B2(pin)2, the reduced product [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)] was identified by X-ray crystallography.
Introduction
The oxidative metabolism of drugs (or xenobiotics) containing amine or hydroxylamine functional groups produces C-nitroso compounds (RNO, R = alkyl and aryl) as byproducts:| RNH2 → RNHOH → RNO | (1) |
These harmful species can also be formed through the reduction of nitroalkanes/arenes (RNO2).1–4C-Nitroso compounds bind to heme proteins, including myoglobin/hemoglobin (Mb/Hb), cytochrome (Cyt.) P450s, and soluble guanylate cyclase (sGC), by coordination to ferrous heme, thereby inhibiting their function. This inhibition contributes to the general toxicity of nitro-containing compounds to cells, since they can be reduced to C-nitroso derivatives.5 Interestingly, thiols like glutathione in red blood cells react with nitrosoarenes to form sulfonamides, before they are metabolized in the liver, which actually constitutes a defense mechanism against these toxic molecules.6 In addition, RNOs react with cysteine residues in proteins.7–11
Ferrous heme-RNO complexes are isoelectronic to ferrous nitroxyl (NO−/HNO) complexes, or ls-{FeN(H)O}8 species in the Enemark–Feltham notation (the superscript “8” represents the number of iron(d) plus NO(π*) electrons; ls = low-spin).12 In the denitrification and DNRA pathways in the nitrogen cycle, heme-nitroxyl compounds are proposed as important intermediates in the reduction of NO to N2O and nitrite to ammonium, respectively.1 For example, it has been proposed based on quantum-chemical calculations that in the catalytic cycle of Cyt. P450nor (fungal NO reductase), a ferrous heme-HNO intermediate is formed (termed “Intermediate I”) that subsequently reacts with NO directly by N–N bond formation, producing a ferric hyponitrite species in the central step of Cyt. P450nor catalysis (see Scheme 1).13–15 Recently, experimental data have been obtained that further support these claims.16–19
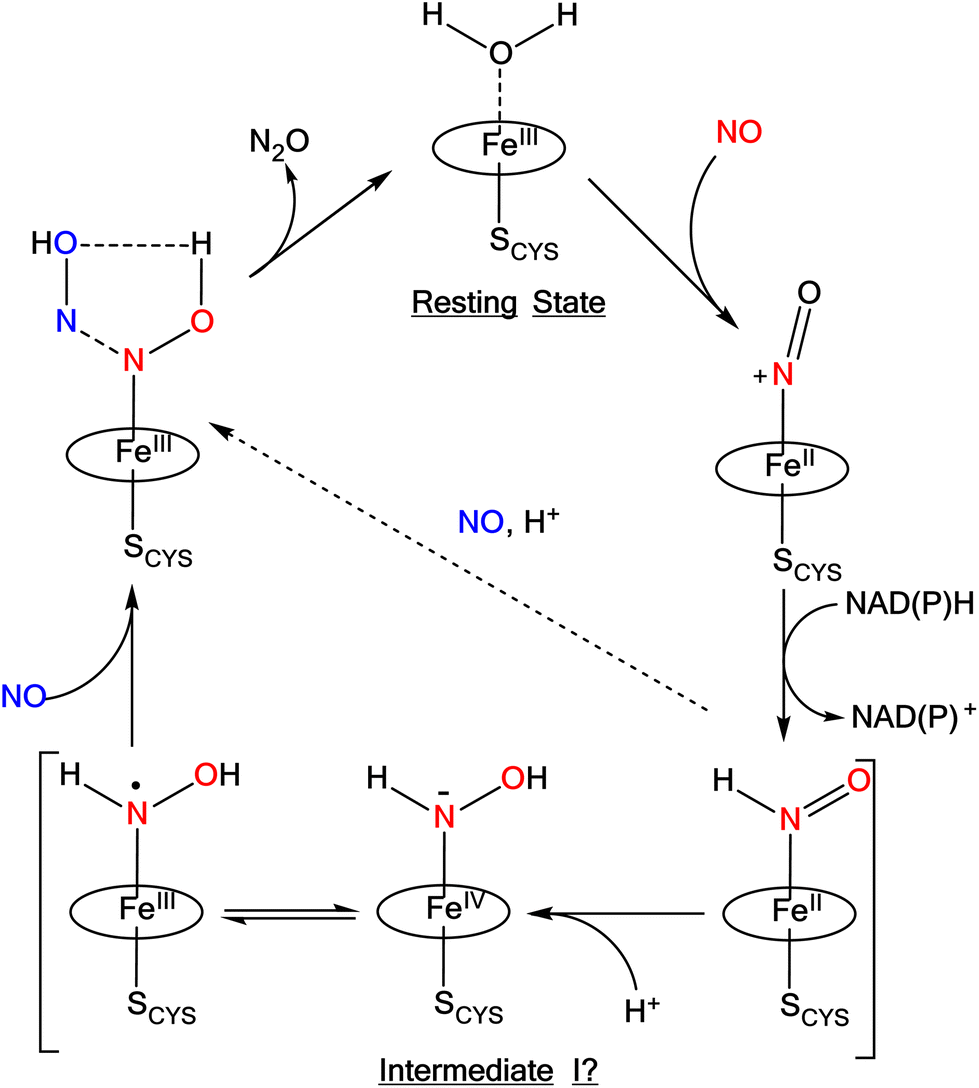 | ||
| Scheme 1 Proposed mechanism for N2O formation in Cyt. P450nor. The oval represents a generic porphyrin ligand. Reproduced with permission from ref. 1. Copyright 2021 American Chemical Society. | ||
Several heme-HNO adducts, in proteins and model complexes, have been reported in the literature. Farmer and coworkers prepared a heme-HNO complex in Mb, which was characterized in depth using different spectroscopic methods.20,21 Computational work on Mb(II)–NHO shows that hydrogen bonds formed in the distal pocket between the coordinated HNO, His64 and a H2O molecule likely aid in stabilizing the complex.22 Until recently, there have been no heme model complexes available that could generate a stable ls-{FeNHO}8 species, allowing for further reactivity studies.23 This is due to the disproportionation of the bound HNO ligand in simple [Fe(Porph)(NHO)] (Porph2− = generic porphyrin dianion) complexes, as first reported by Ryan and coworkers.24 This yields H2 and the corresponding ls-{FeNO}7 complex:
| 2 [Fe(Porph)(NHO)] → 2 [Fe(Porph)(NO)] + H2 | (2) |
Recently, Ryan's group was able to generate the complex [Fe(OEP)(NHO)] (OEP2− = octaethylporphyrin dianion) by reacting the corresponding nitroxyl complex [Fe(OEP)(NO)]− with phenols (PhOH) as weak acids. Here, the [Fe(OEP)(NHO)] complex seems to form an equilibrium with the hydrogen-bonded complex [Fe(OEP)(NO⋯HOPh)]−.25 Richter-Addo and coworkers were able to model the key hydride transfer step in the Cyt. P450nor mechanism.26 Direct hydride transfer from [NBu4](BH4) to the six-coordinate (6C) ls-{FeNO}6 model complex [Fe(OEP)(5-MI)(NO)]+ (5-MI = 5-methylimidazole) to form the corresponding 6C HNO complex, [Fe(OEP)(5-MI)(NHO)], was demonstrated.26 Formation of this species was monitored by solution IR spectroscopy at −20 °C. This assignment was further confirmed by 1H-NMR spectroscopy in conjunction with 15NO isotope labeling. Even at low temperature, this FeII–NHO complex disproportionates, forming the corresponding five-coordinate (5C) ls-{FeNO}7 complex [Fe(OEP)(NO)].26 The total yield of [Fe(OEP)(5-MI)(NHO)] was only 11%, which precluded further characterization of this species. In a follow-up study by the Richter-Addo group, it was shown that the addition of NO to [Fe(OEP)(5-MI)(NHO)] leads to the generation of N2O.19 In summary, the fast disproportionation of ferrous heme-HNO complexes in organic solvents has been a major obstacle for the preparation of these types of species.24,27,28 In contrast, the corresponding, deprotonated heme ls-{FeNO}8 complexes are stable.27,29–31
Recently, we prepared a long-lived 6C ls-{FeNHO}8 heme complex, [Fe(3,5-Me-BAFP)(MI)(NHO)], which is stabilized by steric bulk around the iron center, utilizing a bis-picket fence porphyrin co-ligand (3,5-Me-BAFP2−). In this case, the picket fence greatly slows down the disproportionation reaction of the bound HNO ligand. This complex has a half-life of 56 min at −30 °C, and was fully characterized by UV-Vis, 1H- and 15N-NMR, IR, and nuclear resonance vibrational spectroscopy (NRVS). Upon reaction with NO, this complex produces quantitative amounts of N2O.17 This result indicates that the initially formed HNO complex in the catalytic cycle of Cyt. P450nor (see Scheme 1) is catalytically competent and could react with the second equivalent of NO under N–N bond formation. Hence, intermediate I could actually correspond to the heme-HNO complex.1
We rationalized that we might obtain further insight into the vibrational and electronic properties of ferrous heme-HNO complexes and their reactivity by studying the much more stable ferrous heme-RNO analogs, as they have similar geometric and electronic structures.22 In this regard, note that a decent number of ferrous heme-RNO complexes with R = iPr, Ph, and Ph derivatives have been reported previously and characterized using different spectroscopic methods (IR, NMR, Raman, and UV-Vis) and X-ray crystallography, starting with pioneering studies by Mansuy and coworkers in the 1970s and 1980s.32,33 Reviews are further available that provide a summary of the previous studies.34–38
In this work, we have prepared a series of six-coordinate, ferrous heme-RNO complexes (where R = iPr, Ph) with the TPP2− and 3,5-Me-BAFP2− co-ligands, with the goal to (a) interrogate the vibrational properties of ferrous heme-RNO complexes, especially the low-energy Fe–N(R)O stretching and Fe–N–O bending modes, and (b) to study the reactivity of these complexes with NO (following the mechanism of Cyt. P450nor; see Scheme 2) and Lewis acids. The latter investigation was inspired by previous work by the Richter-Addo group. In their study, they found that when [Fe(OEP)(NO)] was reacted with several Lewis acids (BF3, B(C6F5)3 and K+), the Lewis acid coordinated to the nitrosyl O atom, activating the NO ligand toward N–N bond formation to generate N2O.39 The 6C ferrous heme-RNO complexes prepared here were fully characterized using UV-Vis, IR, and 1H-NMR spectroscopy and X-ray crystallography. Although crystal structures of ferrous heme-RNO complexes have been reported before (see Results and discussion), X-Ray crystallography was used here to affirm the exact identity of our complexes, for example with respect to axial ligation (ligands bound trans to the RNO group). Typical axial ligands used here are tetrahydrofuran, pyridine, and 1-methylimidazole. Important results include the full vibrational characterization of the complex [Fe(TPP)(THF)(iPrNO)] using IR and nuclear resonance vibrational spectroscopy (NRVS), and the correlation of these results with the vibrational properties of the HNO analog, using DFT calculations. Interestingly, 6C ferrous heme-RNO complexes react with NO by simple displacement of the RNO ligand, but without undergoing N–N bond formation, in contrast to the HNO analogs. On the other hand, reactions of our heme-RNO complexes with different boron-based Lewis acids led merely to redox reactions, but no stable adduct formation was observed.
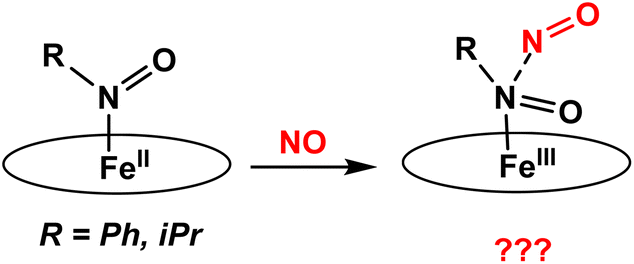 | ||
| Scheme 2 Proposed reactions of iron(II)-RNO complexes with NO to form a ferric hyponitrite-type species. The oval represents a generic porphyrin ligand. | ||
Experimental procedures
All reactions were performed under inert conditions using Schlenk techniques. Preparation and handling of air sensitive materials was carried out under a dinitrogen atmosphere in an MBraun glovebox equipped with a circulating purifier (O2, H2O < 0.1 ppm). All solvents (including deuterated solvents) and 1-methylimidazole (MI) were distilled from CaH2 under dinitrogen, then degassed via five freeze–pump–thaw cycles. The purified solvents were stored over appropriately sized activated molecular sieves in the glovebox until used. Nitric oxide (Cryogenic Gases Inc., 99.5%) was purified by passage through an ascarite II column (NaOH on silica), followed by a cold trap at −80 °C to remove higher order nitrogen oxide impurities. N-Isopropylhydroxylamine hydrochloride was purchased from TCI America and used without further purification. N-Phenylhydroxylamine, 15N-aniline, and 15N-sodium nitrite were purchased from Sigma Aldrich and used without any further purification. [Fe(TPP)(Cl)],40 [Fe(TPP)(X)] (where X = BF4−, PF6−, and SbF6−),41 [Fe(TPP)],27 [Fe(TPP)(PhNO)2],42 [Fe(3,5-Me-BAFP)(Cl)],27 [Fe(3,5-Me-BAFP)],27 and [Fe(3,5-Me-BAFP)(NO)]27 were synthesized as previously reported. Analogous 57Fe complexes were synthesized in a similar manner using 57FeCl2 dimethanol salt as the iron source.Physical measurements
Infrared spectra were obtained from KBr disks on Perkin-Elmer BX or GX spectrometers at room temperature. Solution IR spectra were recorded on a Bruker Alpha-E FTIR spectrometer. Solution samples were measured in a thin-layer solution cell equipped with CaF2 windows. Solid and gas-phase IR spectra were obtained on a Thermo-Nicolet IS-50 benchtop IR spectrometer. Gas samples were measured using a Pike HT gas cell (10 cm) with CaF2 windows. Electronic absorption spectra were measured using an Analytical Jena Specord S600 instrument at room temperature. In situ UV-Vis measurements were conducted with a Hellma quartz immersion probe with a 10 mm path length. 1H- and 13C-NMR spectra were recorded on a Varian MR 400 MHz instrument or a Varian NMRS 500 MHz spectrometer at room temperature. The resonance Raman (rRaman) experiments were performed using an INNOVA-301K* Krypton Ion gas laser system at an excitation wavelength of 407 nm with 20 mW power. Samples were transferred to a liquid nitrogen bath inside an EPR cold finger to cool the samples during the measurements. The scattered light from the samples was focused onto an Acton two-stage TriVista 555 monochromator and detected by a liquid N2-cooled Princeton Instruments Spec-10:400B/LN CCD camera. The accumulation and exposure times for the samples were 5 acquisitions for 60 seconds. The spectral resolution was 0.3 cm−1. Spectra were then plotted and processed using the OriginPro 9.0.0 (64-bit) software for baseline correction. Nuclear Resonance Vibrational Spectroscopy (NRVS) measurements were carried out as previously described43 at beamline 3-ID-XOR at the Advanced Photon Source (APS) at Argonne National Laboratory. This beamline provides about 2.5 × 109 photons per s in ∼1 meV bandwidth (8 cm−1) at 14.4125 keV in a 0.5 mm (vertical) × 0.5 mm (horizontal) spot. Samples were loaded into 4 × 7 × 1 mm copper cells. The final spectra represent averages of 4 scans. The Phoenix program was used to convert the NRVS raw data into the Vibrational Density of States (VDOS).44,45Synthesis of nitrosobenzene (PhNO)
Nitrosobenzene was synthesized using a modified literature procedure.46 A 4.6 mL portion of aniline (50.4 mmol) was stirred in a mixture of 15 mL methanol and 23 mL water in an ice bath. To this solution, 28 mL H2O2 (30% w/v) was slowly added and the reaction mixture was stirred for 10 minutes. Then, 0.76 g MoO3 (5.28 mmol) was added followed by 5 mL KOH (1 M), which caused the solution to turn orange-brown in color. The solution was allowed to stir for 6 hours in an ice bath. When the reaction was complete the solid was filtered off and washed with water (15 mL) and cold methanol (15 mL) to give a light yellow solid. The light yellow solid was dissolved in 50 mL absolute ethanol and allowed to precipitate in the −33 °C freezer overnight. The following day the solution was vacuum filtered through a frit to give a white solid. Yield: 4.23 g (82%). 1H-NMR (CD2Cl2, 400 MHz): δ = 7.621–7.906 (m, 5H). 13C-NMR: (CDCl3, 126 MHz) δ = 121.2 (s), 129.7 (s), 136.1 (s), 166.4 (s). IR ν(N–O) = 1589 cm−1. UV-Vis (CH2Cl2): 282, 304, 750 nm.The corresponding 15N-labeled nitrosobenzene compound was synthesized in the same way starting from 15N-labeled aniline.
Synthesis of N-isopropylhydroxylamine
1.0 g N-isopropylhydroxylamine hydrochloride (9 mmol) and 0.694 g potassium methoxide (9.9 mmol) were dispersed in 30 mL dichloromethane and allowed to stir for several hours. The next day the white precipitate (KCl) was filtered off and the filtrate was dried via rotary evaporation to give a white solid. Yield: 0.686 g (100%) 1H-NMR (CDCl3, 400 MHz): δ: 3.379–3.412 (sep, 1H); 1.276, 1.292 (d, 6H); 13C-NMR (CDCl3, 126 MHz): δ: 53.240 (s), 17.522 (s).Synthesis of 15N-isopropylhydroxylamine
The first step of this reaction was carried out according to a previous report.47 Reaction of 2-iodopropane and 15N-sodium nitrite delivers a crude product that contains 15N-2-nitropropane which, according to the literature, is purified via vacuum distillation to remove impurities. We used the crude product for the following reaction based on a modified patent procedure, as described next.48 ∼0.5 g of crude 15N-iPrNO2 (∼5.6 mmol), 96 μL acetic acid (16.8 mmol), and 0.550 g Zn powder (8.4 mmol) were added to 100 mL 95% ethanol and allowed to stir for 3 hours at 0 °C. Then, the solvent was evaporated under vacuum to give a grey powder. The grey solid was washed with dichloromethane (to dissolve the product) and the filtrate was dried by rotary evaporation. Crude Yield: ∼0.357 g (∼89%). The 1H-NMR spectrum showed that a large amount of acetic acid was present in the solid, so the product was further purified. For this purpose, ∼0.375 g of crude 15N-isopropylhydroxylamine (∼5 mmol) and 0.351 g KOMe (5 mmol) were stirred in 10 mL dichloromethane for 2 hours. Then, the reaction mixture was filtered through a cotton plug and the solvent was evaporated via rotary evaporation. The resulting oil was redissolved in 2 mL dichloromethane and layered with 48 mL hexanes and placed in the −33 °C freezer. The next day the solution was filtered through a frit to give a white needle-like microcrystalline solid. The product was dried under vacuum for a few hours before further use. 1H-NMR (CDCl3, 400 MHz): δ: 3.150–3.118 (sep, 1H); 1.044, 1.067 (dd, 6H).Synthesis of 2-nitrosopropane (iPrNO)
2-Nitrosopropane was synthesized according to a modified literature procedure.49 Under an inert atmosphere, 0.10 g (1.3 mmol) N-isopropylhydroxylamine was dissolved in 10 mL of dry methylene chloride, and 0.404 g Ag2CO3 (1.46 mmol) was added, leading to the formation of a black precipitate. Then, the reaction was brought outside the glovebox and stirred for one hour. The solution was filtered through a Celite pad in a frit and the resulting filtrate was dried to an oil via rotary evaporation. The crude material was chromatographed on silica gel with CH2Cl2 as the eluent (in a 9 inch Pasteur pipet). The solvent was removed via rotary evaporation and the product was further dried under vacuum to give a white solid (the solid is assumed to be in the dimer form based on previous literature reports).50,51 Yield: 32 mg (15%). 1H-NMR (CDCl3, 400 MHz): δ: 5.315–5.347 (sep, 1H); 1.35, 1.36 (d, 6H).Synthesis of [Fe(TPP)(THF)(PhNO)]
There are two literature methods32,52 reported to synthesize ferrous porphyrin RNO complexes: (1) reaction of a ferric porphyrin chloride complex with excess RNHOH, and (2) reaction of a ferrous porphyrin complex with excess RNO ligand. In this work, we synthesized our ferrous heme-RNO complexes using modified versions of these methods as described below to obtain the ferrous PhNO/Ph15NO complexes. For the complexes of natural abundance isotopes (n.a.i.), a 132 mg (0.175 mmol) portion of [Fe(TPP)(BF4)] (also works with the PF6− or SbF6− counterions) and 95 mg N-phenylhydroxylamine (0.880 mmol) were dissolved in 5 mL tetrahydrofuran. The solution was allowed to stir in the glovebox overnight and the reaction progress was monitored by UV-Vis spectroscopy. The following day, the solution was layered with 40 mL hexanes and placed in the −33 °C glovebox freezer. The next day, the reaction mixture was vacuum filtered through a frit and washed with hexanes to give a dark purple powder. Yield: 120 mg (0.208 mmol, 89%). UV-Vis (CH2Cl2): 420, 507 nm. UV-Vis (THF): 420, 535 nm. 1H-NMR (CD2Cl2, 400 MHz): δ = 8.58 (s); 8.014 (s); 7.72 (s); 6.339–6.509 (m); 6.014 (s). Anal. calcd for C50H33FeN5O: C, 76.50; H, 4.87; N, 8.26. Found: C, 75.57; H, 4.59; N, 9.42. IR (KBr): ν(NO) = 1350, 1368 cm−1.Single crystals suitable for X-ray diffraction were grown by vapor diffusion of hexanes into a concentrated THF solution of [Fe(TPP)(THF)(PhNO)] in the −33 °C glovebox freezer.
Synthesis of [Fe(TPP)(THF)(Ph15NO)]
A 14 mg (0.021 mmol) portion of [Fe(TPP)] and 9 mg 15N-nitrosobenzene (0.083 mmol) were dissolved in 1 mL tetrahydrofuran and the resulting solution was allowed to stir in the glovebox. The reaction progress was monitored by UV-Vis spectroscopy. After one hour, the solution was layered with 15 mL hexanes and left in the −33 °C freezer. After 2 days, the solution was filtered through a frit to give a dark purple powder. Yield: 11 mg (0.0142 mmol, 68%). The UV-Vis data are identical to those of the n.a.i complex in THF: 420, 535 nm. ν(15NO) = 1328, 1348 cm−1.Synthesis of [Fe(TPP)(Pyr)(PhNO)]
First [Fe(TPP)(Pyr)2] was synthesized by reacting [Fe(TPP)] (1 mmol) with excess pyridine (19 mmol) in methylene chloride. Under an inert atmosphere, 30 mg [Fe(TPP)(Pyr)2] (0.036 mmol) and 4 mg of PhNO (0.037 mmol) were dissolved in 2 mL dry methylene chloride and stirred. The reaction mixture was then layered with 48 mL heptanes and placed in the −33 °C glovebox freezer. The following day, the reaction mixture was filtered through a frit and the resulting solid was washed with cold heptanes to give a dark purple solid.Synthesis of [Fe(TPP)(MI)(PhNO)]
Under an inert atmosphere, 60 mg of [Fe(TPP)(MI)2] (1 mmol) and 10 mg PhNO (1.2 mmol) was stirred overnight. The reaction mixture was then layered with 48 mL heptanes and placed in the −33 °C glovebox freezer. The following day, the reaction mixture was filtered through a frit and the resulting solid was washed with cold heptanes to give a quantitative yield. UV-Vis (CH2Cl2): 426, 540 nm.Synthesis of [Fe(TPP)(THF)(iPrNO)]
Under an inert atmosphere, 100 mg [Fe(TPP)] (0.15 mmol) and 21 mg iPrNO (0.3 mmol) were dissolved in 2 mL dry tetrahydrofuran. The reaction was stirred for one hour, and then layered with 48 mL heptanes and placed in the −33 °C glovebox freezer. The following day, the reaction mixture was filtered through a frit and the resulting solid was washed with cold heptanes to give a dark purple solid. Yield: 81.7 mg (67%). UV-Vis (CH2Cl2): 415, 525 nm. UV-Vis (THF): 417, 535 nm. 1H-NMR (CDCl3, 400 MHz): δ = 8.731 (s); 8.108 (s); 7.704 (s); −0.61 (s) ; −2.065 (s). Anal. calcd for C51H43N5O2: C, 75.27; H, 5.33; N, 8.61. Found: C, 74.99; H, 5.11; N, 8.43.Single crystals were grown by dissolving ∼18 mg [Fe(TPP)(THF)(iPrNO)] in 0.2 mL THF and placing the solution in a 5 mm diameter glass tube. The solution was carefully layered with 2 mL hexanes, the tube was sealed with a septum, and left at room temperature. After 4 days crystals suitable for X-ray analysis were collected.
Synthesis of [Fe(TPP)(THF)(iPr15NO)]
70 mg [Fe(TPP)(SbF6)] (0.077 mmol) was dissolved in 5 mL THF in the glovebox. To this solution, 29 mg (0.39 mmol) 15N-isopropylhydroxylamine was added and the reaction mixture was allowed to stir overnight. The reaction progress was monitored by UV-Vis spectroscopy. When the reaction was complete, the solution was layered with heptanes and placed in the glovebox freezer at −33 °C. The following day, the solution was filtered through a frit and the resulting solid was washed with cold heptanes to give a dark purple solid. Yield: 62 mg (100%). The UV-Vis and 1H-NMR spectra are analogous to those of the n.a.i complex [Fe(TPP)(THF)(iPrNO)].The 57Fe-labeled complexes, [57Fe(TPP)(THF)(iPrNO)] and [57Fe(TPP)(THF)(iPr15NO)], were synthesized using the same methods, except that the precursor complex [57Fe(TPP)(Cl)] was used.
Synthesis of [Fe(3,5-Me-BAFP)(iPrNO)2]
Under an inert atmosphere, 30 mg [Fe(3,5-Me-BAFP)] (0.15 mmol) and 2.7 mg iPrNO (0.3 mmol) were dissolved in 2 mL dry tetrahydrofuran. The reaction mixture was stirred for one hour, and then layered with 20 mL hexanes and placed in the −33 °C glovebox freezer. The following day, the reaction mixture was filtered through a frit and the resulting solid was washed with cold hexanes to give a dark purple solid. Yield: 13.1 mg (41%). UV-Vis (THF): 425 nm. IR: N–O stretch at 1459 cm−1. EA was calculated based on the crystal structure with 20% THF and 80% iPrNO as the 6th ligand: [Fe(3,5-Me-BAFP)(iPrNO)2] Anal calcd for C114.2H106N5.8O10Fe·CH2Cl2·C6H14: C, 74.67; H, 6.12; N, 3.36. Found: C, 74.78; H, 6.32; N, 4.17.Single crystals of [Fe(3,5-Me-BAFP)(iPrNO)2] were grown by dissolving 2 mg of the complex in 0.2 mL of THF in a 5 mm glass tube layered with 0.8 mL of hexanes. The tube was sealed and left at room temperature in the glovebox for 5 days to grow crystals suitable for X-ray diffraction.
Reactions with NO gas
For the reactions of our RNO complexes with NO, we first equilibrated a solution of dichloromethane with NO gas. Then, we titrated a solution of cobalt tetraphenylporphyrin [Co(TPP)] in THF with the CH2Cl2/NO solution to form the corresponding complex [Co(TPP)(NO)], to determine the concentration of dissolved NO gas. From these data we approximated the NO concentration of the solution (typically ∼1 mM), and then titrated [Fe(TPP)(THF)(PhNO)], [Fe(TPP)(MI)(PhNO)] and [Fe(TPP)(THF)(iPrNO)] in THF with the calibrated CH2Cl2/NO solution.Lewis acid reactions
Solutions of [Fe(TPP)(THF)(iPrNO)] and [Fe(3,5-Me-BAFP)(iPrNO)2] were prepared by dissolving the compounds in minimum dry THF in the glovebox. The Lewis acids (2.5 equiv.) were added to the solutions either by micro-syringe (for liquids), or were weighed out and then added (for solids). The reactions were monitored by UV-Vis spectroscopy. The solutions were then layered with 4X hexanes and left in the −33 °C glovebox freezer to precipitate overnight. The resulting solids were filtered off and collected the next day. In the case of the solutions to which BF3·OEt2 was added, excess BF3·OEt2 was difficult to remove and could be seen coating the solids, even after several washes with hexanes.Single crystals of [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)] were grown by adding 2 mg [Fe(3,5-Me-BAFP)(iPrNO)2], dissolved in 0.2 mL THF, to a 5 mm glass tube. 2.5 equiv. of B2(pin)2 were added to the solution. The solution was carefully layered with 0.8 mL of hexanes and suitable crystals for X-ray crystallography grew in 5 days.
DFT calculations
The structures of the PhNO and iPrNO complexes and their HNO analogs were fully optimized for their singlet (S = 0) ground states, using the BP86 functional53,54 and the TZVP basis set.55,56 In these calculations, the simple porphine (P2−) ligand was used. Vibrational frequencies calculated for the structures show no imaginary frequencies. In all calculations, convergence was reached when the relative change in the density matrix between subsequent iterations was less than 1 × 10−8. All of these calculations were performed using the program package Gaussian 09.57Results and discussion
Preparation and spectroscopic characterization of [Fe(TPP)(L)(RNO)]
For the synthesis of ferrous porphyrin RNO complexes, two different methods have been reported in the literature.32,52 The first method is based on the reaction of the ferric complexes, [Fe(Porph)(Cl)] (where Porph2− = dianion of a generic porphyrin ligand), with excess N-(alkyl/aryl)hydroxylamines (RNHOH, R = alkyl, aryl), leading to a disproportionation reaction to give the ferrous RNO complexes, as shown in eqn (3). The second method of preparation is the direct reaction of the ferrous porphyrins, [Fe(Porph)], with nitrosoalkane ligands (RNO, R = alkyl, aryl) to give the corresponding ferrous RNO complexes. These reactions are usually carried out in the presence of excess neutral ligands (L), such as the N-donor ligands 1-methylimidazole (MI) or pyridine (Pyr), to form the corresponding six-coordinate (6C) complexes, [Fe(Porph)(L)(RNO)], as shown in eqn (4). In this work, we used modified versions of both literature methods to synthesize our ferrous porphyrin RNO model complexes (where R = iPr and Ph; see the Experimental section).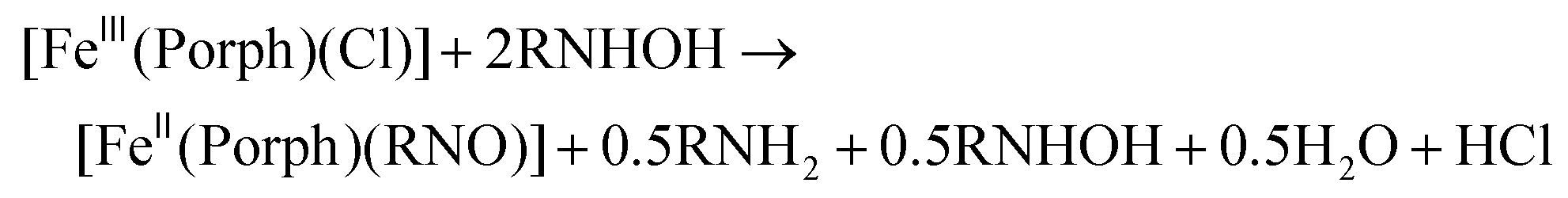 | (3) |
| [FeII(Porph)] + RNO + excess L → [FeII(Porph)(L)(RNO)] | (4) |
We modified the reaction shown in eqn (3) by starting from a ferric complex with a weakly coordinating anion, [Fe(TPP)(X)], where TPP2− = tetraphenylporphyrin dianion, and X = BF4−, PF6− or SbF6−. For example, we reacted [Fe(TPP)(BF4)] with excess N-phenylhydroxylamine (PhNHOH) in tetrahydrofuran (THF) to yield the corresponding ferrous PhNO complex in ∼90% yield. The UV-Vis spectrum of the reaction product (in CH2Cl2) exhibits a Soret band at 412 nm and the main Q band at 523 nm, indicative of a ferrous species as shown in Fig. S4.† Next, we further characterized the product by 1H-NMR spectroscopy. The iron-PhNO complex shows a multiplet of peaks from 6.04–6.51 ppm that originate from the bound PhNO ligand, and the porphyrin ligand peaks range from 7.72–8.58 ppm; see Fig. S4,† bottom. These chemical shifts indicate that the product is pure, and they are characteristic of a diamagnetic, low-spin (ls) ferrous porphyrin complex (the precursor is paramagnetic). It should be noted that these peaks were assigned based on the reported 1H-NMR spectrum of [Fe(TPP)(PhNO)2].42
Despite the available, thorough structural and spectroscopic characterization of ferrous heme-RNO complexes,34,35 the vibrational characterization of these complexes is incomplete. In particular, N–O stretching frequencies are often not confirmed by isotope labeling, and little is known about the Fe–N(R)O stretching and bending vibrations. In the case of PhNO complexes,32,42,52,58,59 N–O stretching frequencies have been reported in the 1340–1370 cm−1 range (see Table 1). To determine the N–O stretch of our mono-PhNO complex, we synthesized the corresponding Ph15NO complex using the reaction given in eqn (4). Surprisingly, the IR spectra in Fig. 1, (top panel) show the presence of two isotope sensitive features at 1350 and 1368 cm−1, where the latter band is significantly lower in intensity than the 1350 cm−1 feature. This observation prompted us to investigate whether the ferrous PhNO complex obtained in this way could be a mixture of different species. The reported N–O stretching frequency for [Fe(TPP)(PhNO)2] is 1353 cm−1,42 but unfortunately, the IR spectrum of this complex is not shown in the previous report, so it is not clear how intense this IR band is. The IR spectrum of [Fe(TPP)(PhNO)2] prepared by us, following the reported method of preparation,42 shows a very intense N–O stretching band at 1353 cm−1 that shifts to 1336 cm−1 with Ph15NO (Δ = 17 cm−1), as shown in Fig. 1, bottom panel. The 1353 cm−1 band (and its relative intensity) in [Fe(TPP)(PhNO)2] matches one of the bands in the IR spectrum of our “mono-PhNO” preparation (see Fig. S5†), which implies that our product contains a significant amount of the ferrous bis-PhNO complex. Additionally, the IR spectra in Fig. S6† (top panel) show that it makes little difference for the relative amounts of the iron-PhNO products formed whether one equivalent or excess PhNO is used in the reaction with [Fe(TPP)]. Richter-Addo and coworkers also reported the crystal structure of a mono-PhNO analog, [Fe(TPP)(MI)(o-tolNO)], which has an N–O stretching frequency of 1366 cm−1.59 This value is very close to the other N–O band at 1368 cm−1 in our PhNO complex. Based on this result, we conclude that the bis- and mono-PhNO complexes have in fact different N–O stretching frequencies, and that the “mono-PhNO” product that we obtained is in fact a mixture of [Fe(TPP)(THF)(PhNO)] and [Fe(TPP)(PhNO)2].
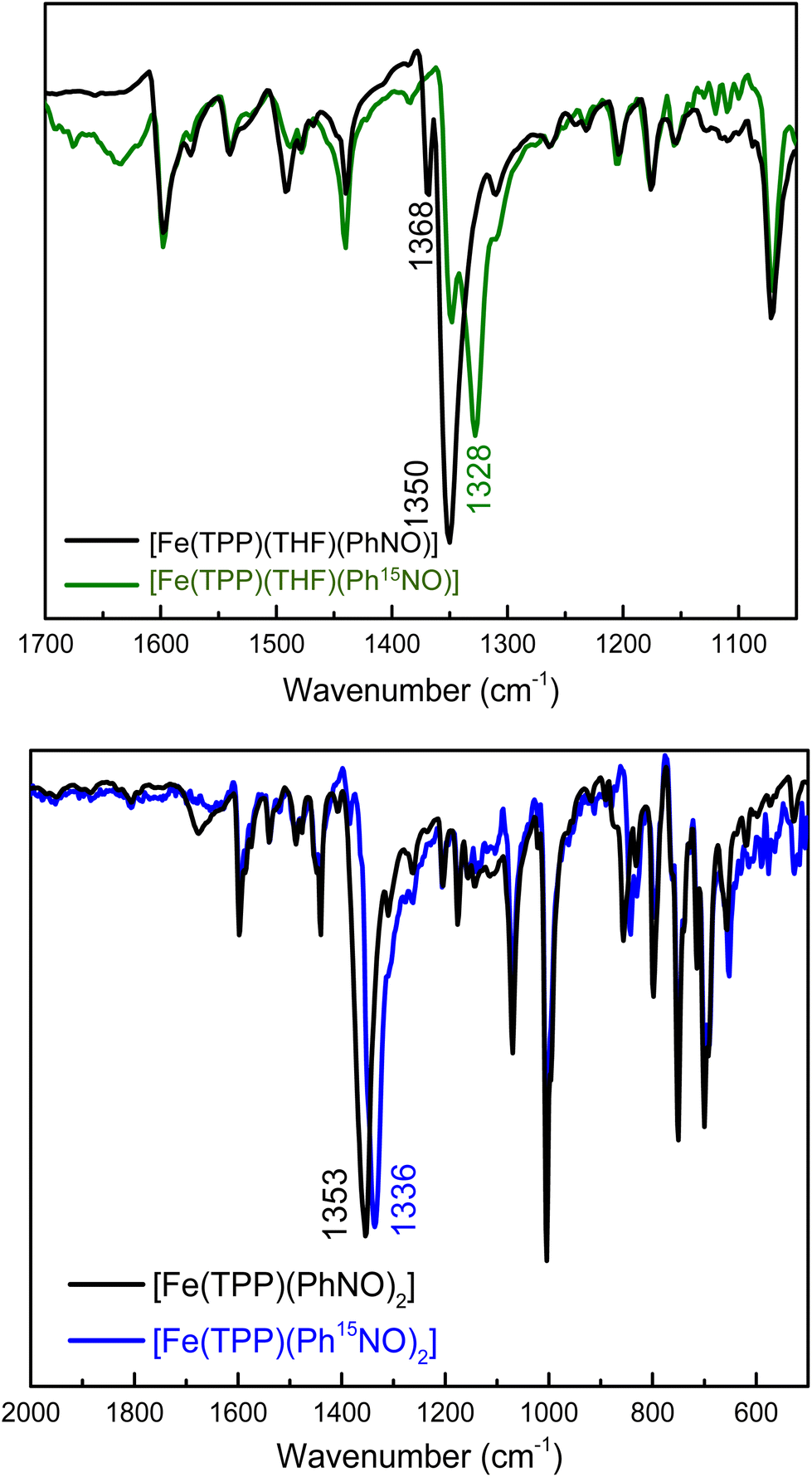 | ||
| Fig. 1 Top panel: Overlay of the IR spectra of [Fe(TPP)(THF)(PhNO)] (black), and of the 15N-labeled complex, [Fe(TPP)(THF)(Ph15NO)] (green), measured in KBr pellets (expanded view of the N–O stretching frequency region). Bottom panel: Overlay of the IR spectra of [Fe(TPP)(PhNO)2] (black) and the 15N-labeled complex, [Fe(TPP)(Ph15NO)2] (blue), measured in KBr pellets. See text for further discussion. | ||
| Complex | ΔFe–Np![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
ΔFe–N(NO) | ΔFe–X (X = O, N) | ΔN–O | <Fe–N–O | ν(N–O) | Ref. |
|---|---|---|---|---|---|---|---|
| a All values are given in Å and °. b Average value. c Data from the disordered structural components. d Two different molecules in the unit cell. e NODMA = 4-nitroso-N,N-dimethylaniline. f Likely from an impurity of the bis-PhNO complex. | |||||||
| Crystal structures | |||||||
| [Fe(TPP)(THF)(PhNO)]c,d | 1.998 | 1.813 | 2.141 | 1.253 | 122 | 1368 | t.w. |
| [Fe(3,5-Me-BAFP)(2-MeTHF)(PhNO)] | 2.002 | 1.906 | 2.088 | 1.259 | 125 | n.d | t.w. |
| [Fe(TPP)(PhNH2)(PhNO)]c,d | 2.002 | 1.994, 1.963 | 1.994, 1.963 | 1.172,1.194 | 119, 120 | 1350f | t.w. |
| [Fe(TPP)(PhNO)2]c | 1.998 | 1.874, 1.899 | — | 1.237, 1.227 | 124, 123 | 1353 | 42 |
| [Fe(TPP)(Pyr)(PhNO)] | 1.990 | 1.819 | 2.106 | 1.249 | 124 | 1342 | 58 |
| [Fe(TPP)(MI)(PhNO)]c,d | 1.986 | 1.8 | 2.029 | 1.254 | 125 | 1336 | 58 |
| 2.004 | 1.802, 1.807, 1.812 | 2.03 | 1.267, 1.263, 1.26 | 123, 124, 124 | n.d. | ||
| [Fe(TPP)(MI)(o-tolNO)] | 2.007 | 1.841 | 2.065 | 1.257 | 123 | 1366 | 59 |
| [Fe(TPP)(Pyr)(NODMA)]e | 1.981 | 1.859 | 2.095 | 1.252 | 120 | n.d. | 58 |
| [Fe(OEP)(MI)(PhNO)]c | 2.002 | 1.809, 1.802 | 2.092, 2.094 | 1.269, 1.258 | 123, 124 | n.d. | 58 |
| Calculated structures | |||||||
| [Fe(P)(PhNO)] | — | 1.779 | — | 1.245 | 123 | 1383 | t.w. |
| [Fe(P)(THF)(PhNO)] | — | 1.795 | 2.220 | 1.245 | 124 | 1386 | t.w. |
| [Fe(P)(MI)(PhNO)] | — | 1.830 | 2.084 | 1.246 | 124 | 1380 | t.w. |
| [Fe(P)(PhNO)2] | — | 1.936 | 1.936 | 1.239 | 121 | 1383 | t.w. |
| [Fe(P)(PhNHOH)(PhNO)] | — | 1.814 | 2.223 | 1.245, 1.456 | 110, 123 | 1386 | t.w. |
Finally, we synthesized the Pyr- and MI-bound complexes, [Fe(TPP)(Pyr)(PhNO)] and [Fe(TPP)(MI)(PhNO)], first reported by Oldfield and coworkers,58 which show the N–O stretch at 1342 cm−1 (1319 cm−1 with Ph15NO (Δ = 23 cm−1), see Fig. S7†) and 1336 cm−1 (1313 cm−1 with Ph15NO (Δ = 23 cm−1), see Fig. S8†), respectively. Interestingly, in the presence of the stronger ligands Pyr and MI (compared to THF), formation of the bis-PhNO complex is not observed.
To gain further insight into the vibrational properties of these PhNO complexes, we employed DFT calculations. We optimized a series of ferrous porphyrin-PhNO complexes as shown in Table 1 using BP86/TZVP. Comparison of the optimized structures to the available crystal structures shows good agreement, with deviations in bond lengths <0.06 Å. Unfortunately, the experimental difference in the N–O stretching frequencies (15–20 cm−1) between the mono- and bis-PhNO complexes is not reproduced by DFT calculations. The calculated N–O stretching frequencies of [Fe(P)(MI)(PhNO)], [Fe(P)(THF)(PhNO)] and [Fe(P)(PhNO)2] (P2− = porphine) are 1380, 1386 and 1383 cm−1, respectively (P2− = porphine dianion). It should be noted that the 1383 cm−1 band of the [Fe(P)(PhNO)2] complex is the antisymmetric combination of the N–O stretching coordinates, while the symmetric mode is not IR active (predicted at 1403 cm−1). Based on our combined experimental and theoretical results we conclude that the 1368 cm−1 band in the IR spectrum of our PhNO product belongs to the ferrous mono-PhNO complex (with a THF molecule bound to the iron center) and that the 1350 cm−1 band belongs to the ferrous bis-PhNO complex, [Fe(TPP)(PhNO)2].
We envisioned that changing the R group of the RNO ligand to an isopropyl group would result in a better model for a ferrous heme-HNO complex, due to the strong conjugation of the nitroso group with the aromatic phenyl ring in PhNO. Using the method shown in eqn (4) we reacted [Fe(TPP)] with a slight excess of iPrNO to form [Fe(TPP)(THF)(iPrNO)] in THF with a 67% yield. In the UV-Vis spectra shown in Fig. 2, the Soret band of the precursor, [Fe(TPP)], shifts from 426 to 417 nm upon coordination of iPrNO but remains very symmetric, which is a feature characteristic of ferrous porphyrins. At the same time, the main Q band of [Fe(TPP)] shifts from 550 to 535 nm. The resulting UV-Vis spectrum of [Fe(TPP)(THF)(iPrNO)] is similar to that of [Fe(TPP)(THF)(PhNO)]. The 1H-NMR spectrum of the isolated product is assigned based on a previous literature report.32 We observe the porphyrin resonances from 7.70 to 8.73 ppm, indicative of a diamagnetic ferrous heme complex (note that the ferrous precursor is paramagnetic), and two peaks shifted upfield to −0.61 and −2.07 ppm, which originate from the bound iPrNO ligand (due to the ring current of the porphyrin), as shown in Fig. S9.† X-ray crystallography (see below) further supports that [Fe(TPP)(THF)(iPrNO)] is obtained as a pure compound.
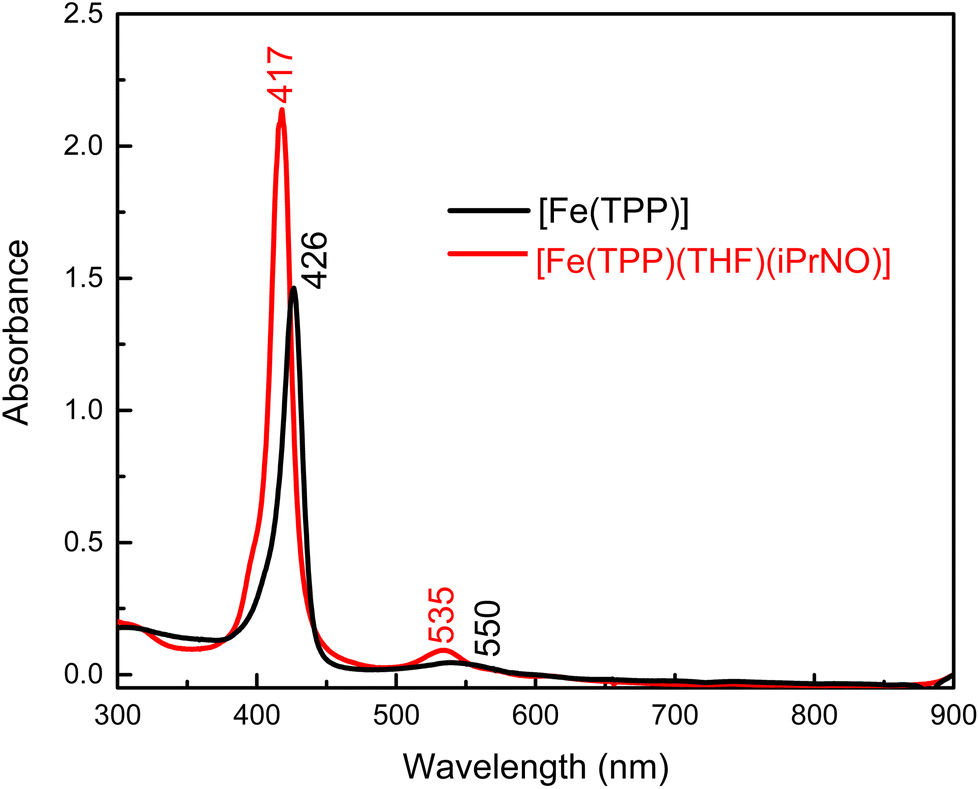 | ||
| Fig. 2 UV-Vis spectra of the precursor, [Fe(TPP)] (black), and of the isolated reaction product, [Fe(TPP)(THF)(iPrNO)] (red), in THF at room temperature. | ||
N–O stretching frequencies have previously been reported for 6C ferrous heme-iPrNO complexes with MI, Pyr, or methanol as axial ligands, which range from 1420 to 1440 cm−1 (see Table 2).32,52,59 We determined the N–O stretching frequency of [Fe(TPP)(THF)(iPrNO)] to be 1440 cm−1, which shifts to 1417 cm−1 with 15N-labeling (Δ = 23 cm−1), as shown in Fig. 3, consistent with these reports. Based on the closer similarity of the iPrNO ligand to HNO, and the fact that [Fe(TPP)(THF)(iPrNO)] can be obtained as a pure compound, we chose this complex for further detailed NRVS studies (see below).
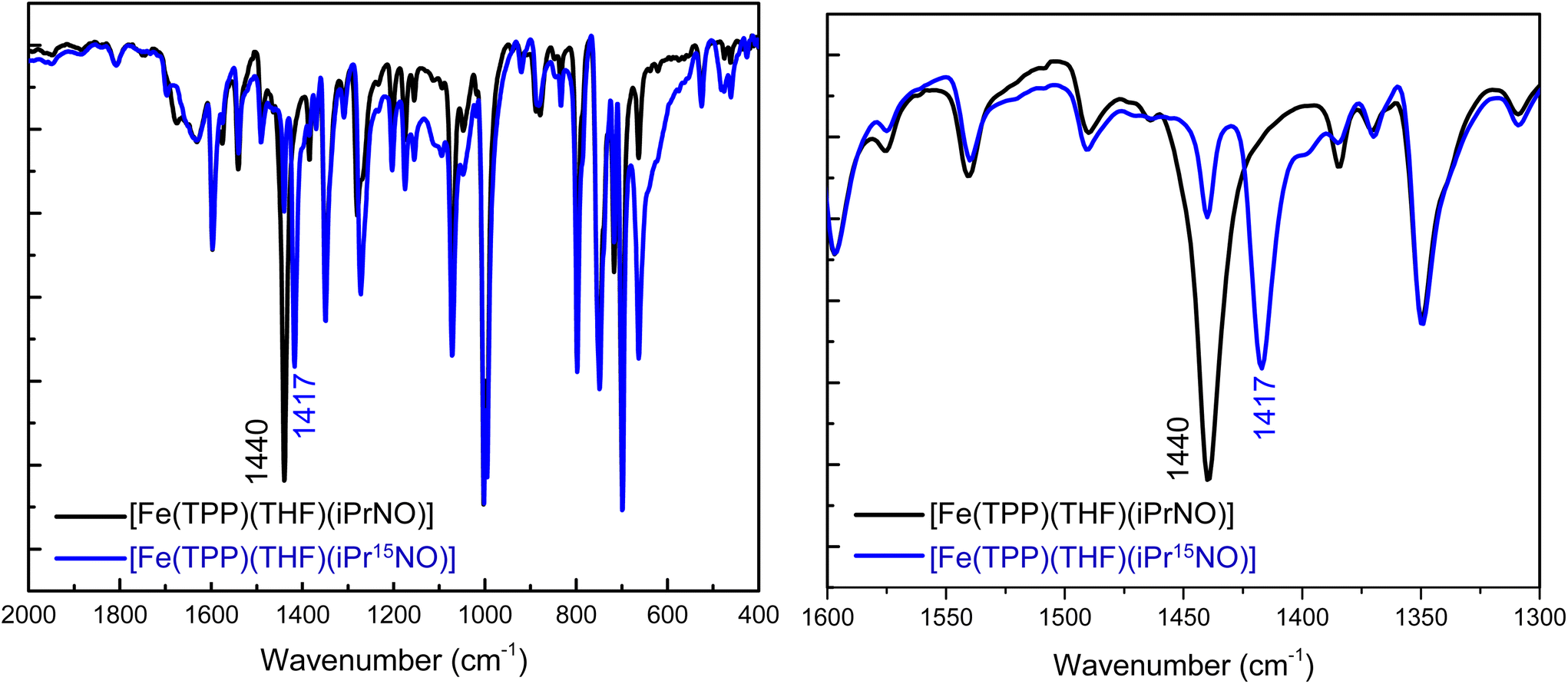 | ||
| Fig. 3 Left panel: Overlay of the IR spectra of [Fe(TPP)(THF)(iPrNO)] (black) and of the 15N-labeled complex, [Fe(TPP)(THF)(iPr15NO)] (blue). The data were obtained in KBr pellets. Right panel: Expanded region of the N–O stretching frequency. The natural abundance isotopes (n.a.i.) complex [Fe(TPP)(THF)(iPrNO)] has one isotope sensitive feature at 1440 cm−1 that shifts to 1417 cm−1 upon 15N-labeling. | ||
| Complex | ΔFe–Npb |
ΔFe–N(NO) | ΔFe–X (X = O, N) | ΔN–O | <Fe–N–O | ν(N–O) | Ref. |
|---|---|---|---|---|---|---|---|
| a All values are given in Å and °. b Average value. c Data from the disordered structural components. d Other vibrations that are coupled to the N–O stretching mode. | |||||||
| Crystal structures | |||||||
| [Fe(TPP)(THF)(iPrNO)]c | 1.998 | 1.853 | 2.090 | 1.212 | 121 | 1440 | t.w. |
| [Fe(3,5-Me-BAFP)(iPrNO)2]c | 2.000 | 1.920 | — | 1.239 | 122 | 1449 | t.w. |
| [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)]c | 2.000 | 1.920 | 1.959 | 1.239 | 122 | 1449 | t.w. |
| [Fe(TPP)(MI)(iPrNO)] | 1.996 | 1.81 | 2.050 | 1.24 | 122 | 1433 | 32 |
| [Fe(TPP)(iPrNH2)(iPrNO)]c | 1.993 | 1.86, 1.87 | 2.105, 2.094 | 1.26 | 124 | 1432 | 52 |
| [Fe(TTP)(MI)(iPrNO)]c | 1.997 | 1.81, 1.86 | 2.05, 2.03 | 1.25, 1.28 | 123, 121 | 1428 | 32 |
| [Fe(OEP)(MeOH)(iPrNO)] | 2.010 | 1.89 | 2.15 | 1.26 | 116 | 1433 | 32 |
| [Fe(OEP)(MI)(iPrNO)]c | 2.014 | 1.8 | 2.03, 2.11 | 1.26 | 123 | 1423 | 32 |
| [Fe(OEP)(Pyr)(iPrNO)]c | 2.009 | 1.84 | 2.12 | 1.23 | 124 | 1429 | 32 |
| Calculated structures | |||||||
| [Fe(P)(iPrNO)] | — | 1.770 | — | 1.235 | 124 | 1464/(1468, 1261)d | t.w. |
| [Fe(P)(THF)(iPrNO)] | — | 1.795 | 2.245 | 1.236 | 124 | 1461/(1466, 1265)d | t.w. |
| [Fe(P)(MI)(iPrNO)] | — | 1.831 | 2.089 | 1.237 | 124 | 1457/(1464, 1268)d | t.w. |
| [Fe(P)(NHO)] | — | 1.741 | — | 1.235 | 132 | 1466 | t.w. |
| [Fe(P)(THF)(NHO)] | — | 1.762 | 2.219 | 1.235 | 132 | 1466 | t.w. |
| [Fe(P)(MI)(NHO)] | — | 1.788 | 2.083 | 1.236 | 132 | 1459 | t.w. |
Crystallographic studies of [Fe(TPP)(L)(RNO)]
Experimental details about the structural studies are provided in the Experimental section. X-ray quality crystals of [Fe(TPP)(THF)(PhNO)] were obtained by vapor diffusion of hexanes into a concentrated THF solution of the complex at −33 °C in the glovebox freezer. This resulted in a crystal with two different PhNO complexes in the unit cell, as shown in Fig. 4 (top panel), but surprisingly, not the bis-PhNO complex, as anticipated from the IR results (see above). One of the species in the unit cell is the target complex [Fe(TPP)(THF)(PhNO)], which has a THF molecule bound to the iron center. The crystal structure exhibits an Fe–N(Ph)O bond length of 1.813 Å and an Fe–N–O angle of 122°. These geometric parameters are very similar to those of [Fe(TPP)(MI)(PhNO)] which has an Fe–N(Ph)O bond length of 1.80 Å and an Fe–N–O bond angle of 125° (see Table 1).58 The other heme complex in the unit cell is the phenylamine (PhNH2) bound complex, [Fe(TPP)(PhNH2)(PhNO)]. The phenylamine is formed during the disproportionation reaction of the ferric heme complex with excess PhNHOH, reflected in eqn (3). This was a surprise, as we did not expect PhNH2 to be bound in the presence of the coordinating solvent THF. This structure shows disorder of the bound PhNO and PhNH2 ligands above and below the heme plane, resulting in corresponding Fe–N bond lengths of 1.963 and 1.994 Å from the structural fit. However, these bond lengths are significantly longer than the Fe–N(Ph)O and Fe–O(THF) bond lengths of [Fe(TPP)(THF)(PhNO)]. They are also much longer than all of the other reported Fe–N(Ph)O distances in ferrous heme-PhNO complexes (1.80–1.90 Å), as shown in Table 1.32,42,52,58,59 On the other hand, the Fe–N(amine) bond length is shorter than that in bis(primary amine) ferrous porphyrins, where the Fe–NH2R bond lengths range from 2.028 to 2.045 Å.60 Based on these considerations, we feel that the Fe–N bond distances in [Fe(TPP)(PhNH2)(PhNO)] are less well defined due to the disorder, and should be treated with caution.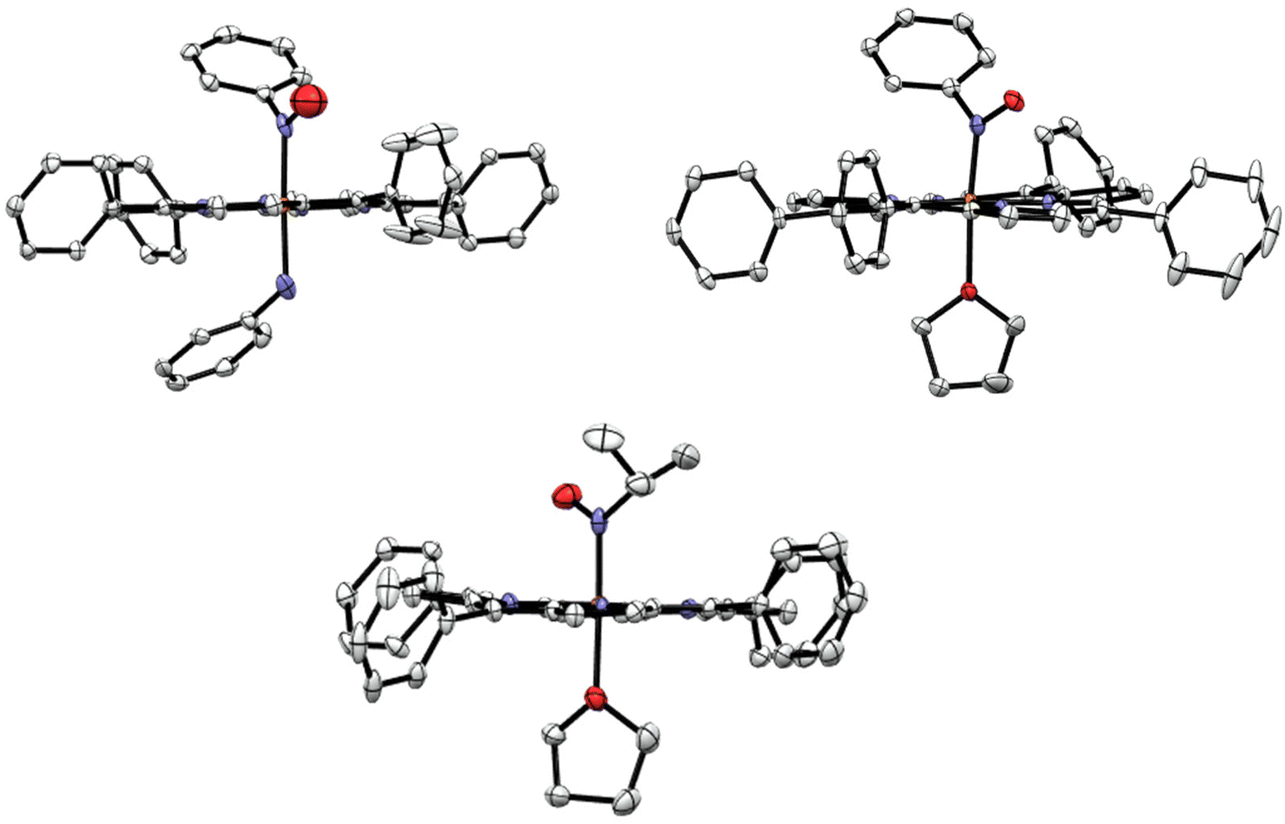 | ||
| Fig. 4 Top panel: Crystal structures of the two unique ferrous PhNO complexes in the unit cell of the co-crystallized compounds [Fe(TPP)(PhNH2)(PhNO)] (left panel) and [Fe(TPP)(THF)(PhNO)] (right panel). Bottom panel: Crystal structure of [Fe(TPP)(THF)(iPrNO)]. In the structures, the hydrogen atoms, disorder, and solvent molecules are omitted for clarity. Thermal ellipsoids are shown at 50% probability. | ||
X-ray quality crystals of [Fe(TPP)(THF)(iPrNO)] were obtained by layering a concentrated solution of the complex with hexanes at room temperature. The structure of [Fe(TPP)(THF)(iPrNO)] has a THF molecule bound to the iron center as shown in Fig. 4, as expected based on the synthesis conditions used. In the structure, the Fe–N(iPr)O bond length is 1.853 Å and the Fe–N–O bond angle of 121°. This is in good agreement with other 6C heme-iPrNO complexes that have Fe–N(iPr)O bond lengths between 1.80 and 1.89 Å and Fe–N–O bond angles ranging from 116 to 124°, as shown in Table 2.32,52 The Fe–N(iPr)O bond length of this complex is slightly longer than that of the MI adduct, [Fe(TPP)(MI)(iPrNO)] at 1.81 Å, but the Fe–N–O angle is quite similar (122°). These geometric parameters are also consistent with the crystal structure of the ls-{FeNO}8 complex [Fe(TFPPBr8)(NO)]− where TFPPBr8 = dianion of 2,3,7,8,12,13,17,18-octabromo-5,10,15,20-(tetra-(pentafluorophenyl)porphyrin), which exhibits an Fe–NO bond length of 1.814 Å and an Fe–N–O angle of 122°.61 Also, all of these values compare well with the Mb(II)–NHO adduct where EXAFS fitting determined the Fe–NHO bond length to be 1.82 Å.62 In contrast, ls-{FeNO}7 complexes, such as [Fe(TPP)(NO)] and [Fe(TPP)(MI)(NO)], exhibit much shorter Fe–NO bond lengths of 1.72 (ref. 63) and 1.750 Å,64 respectively.
The heme is completely planar in all of our RNO crystal structures, in agreement with previous reports,58 except in [Fe(TPP)(THF)(PhNO)] where porphyrin shows some ruffling distortion as illustrated in Fig. 5. This distortion is unusual for 6C ferrous porphyrins since the iron is located in the heme plane. A ruffling distortion is characterized by the rotation of trans pyrrole rings in the opposite direction around the Fe–Npyrrole bonds of the porphyrin co-ligand. This out-of-plane distortion is measured by the root mean square deviation (RMSD) of the porphyrin atoms from the heme plane. The 25-atom core displacement of [Fe(TPP)(THF)(PhNO)] is 0.15 Å, whereas planar hemes are defined by a 25-atom core displacement of less than 0.10 Å.65 For example, the completely planar complex [Fe(TMP)(MI)2](ClO4) (TMP = dianion of tetramesitylporphyrin) has a 25-atom core displacement of only 0.02 Å,66 which is 7.5 times smaller than that of [Fe(TPP)(THF)(PhNO)]. The RMSD for the 4-atom meso carbon displacement in [Fe(TPP)(THF)(PhNO)] is 0.15 Å.
 | ||
| Fig. 5 View looking down the meso carbon atoms of heme, taken from the crystal structure of [Fe(TPP)(THF)(PhNO)]. The graph clearly shows the out-of-plane distortion of the heme plane. All of the hydrogen atoms and phenyl groups of the porphyrin ligand are omitted for clarity. | ||
Lastly, 6C ferrous heme-RNO complexes typically exhibit average Fe–Npyrrole bond distances of ∼2.0 Å (see Tables 1 and 2). This bond length is indicative of low-spin ferrous hemes, which exhibit average Fe–Npyrrole bond distances between 1.981 and 2.008 Å.67
Characterization of [Fe(3,5-Me-BAFP)(iPrNO)2] and crystallographic studies
The iPrNO complex with our bulky bis-picket fence porphyrin, 3,5-Me-BAFP2−, was prepared under identical conditions to [Fe(TPP)(THF)(iPrNO)], where the reduced porphyrin was stirred with a slight excess of iPrNO in THF, and the product was precipitated with hexanes to yield a purple solid. The resulting complex was characterized by UV-Vis, IR, and rRaman spectroscopy, elemental analysis, and X-ray crystallography. It is interesting to note that unlike the TPP2− version, this reaction forms the bis-iPrNO complex almost exclusively. For example, UV-Vis titration of [Fe(3,5-Me-BAFP)] with iPrNO in THF shows that upon the addition of 1 equiv. of iPrNO, the bis-iPrNO complex forms along with the starting material, whereas the mono-iPrNO complex is not present in any significant amounts (Fig. S18†). The crystal structure of [Fe(3,5-Me-BAFP)(iPrNO)2] is shown in Fig. 6, where crystals were grown in a mixture of THF and hexanes. Two crystals were analyzed, but only one is discussed in the main text. It was found that in these crystals, 80–90% of the bis-iPrNO species is present, whereas 10–20% of the complex has THF as the 6th ligand. The crystal displays the iPrNO ligand disordered with a THF molecule. The occupancy ratio refined to 0.778(4) to 0.222(4). This indicates that Fe(3,5-Me-BAFP) has a stronger propensity towards the iPrNO ligand compared to Fe(TPP). We hypothesize that this could be due to the steric bulk in the bis-picket fence porphyrin, which increases the binding constant for the smaller iPrNO ligand compared to THF. Both porphyrins are completely planar. The crystal structures exhibit average Fe–Npyrrole bond distances of ∼2.0 Å, again indicative of low-spin ferrous hemes.51 On comparing the geometric properties of [Fe(TPP)(THF)(iPrNO)] and [Fe(3,5-Me-BAFP)(iPrNO)2] (Table 2), most of the distances and angles were found to be relatively similar except that the Fe–N(iPr)O distance in the 3,5-Me-BAFP system (1.920 Å) is longer than that in the TPP system (1.853 Å) and most of the porphyrin iPrNO complexes reported to date (see Table 2). [Fe(3,5-Me-BAFP)(iPrNO)2] shows the Soret band at 425 nm and has an N–O stretching frequency of 1449 cm−1, determined by IR spectroscopy (Fig. S18†).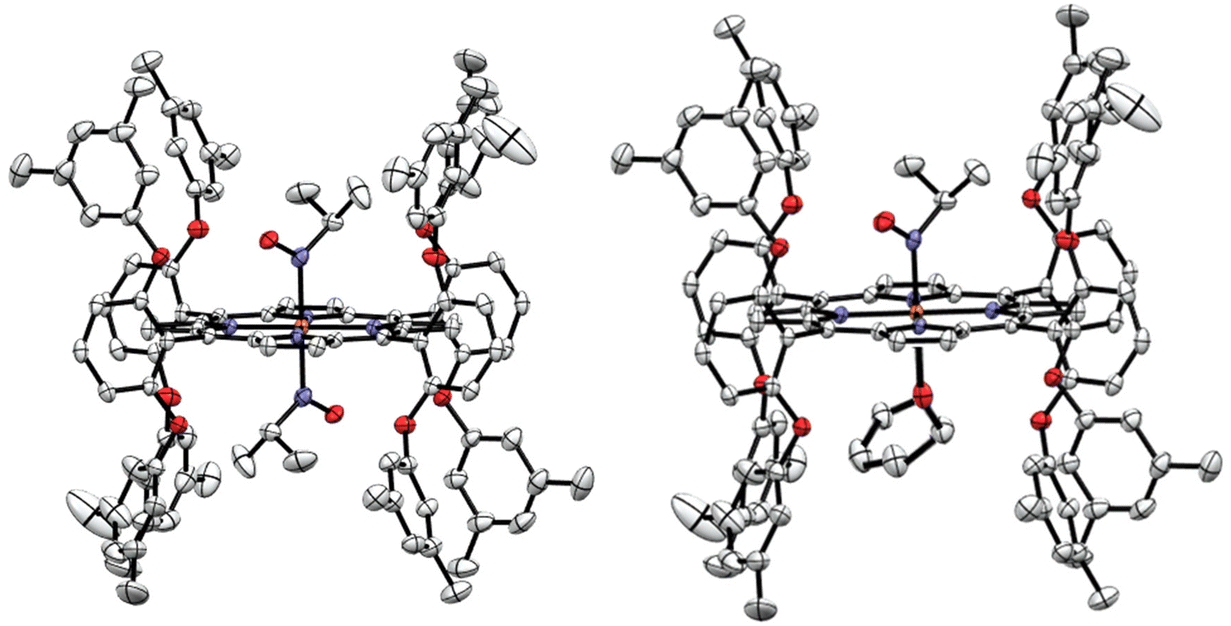 | ||
| Fig. 6 Crystal structures of [Fe(3,5-Me-BAFP)] reacted with iPrNO. Left panel: [Fe(3,5-Me-BAFP)(iPrNO)2]. Right panel: [Fe(3,5-Me-BAFP)(THF)(iPrNO)]. In the crystal, 10–20% of the 6th ligand is THF as shown on the right and 80–90% is the bis-iPrNO complex, shown on the left. | ||
Vibrational analysis of [Fe(TPP)(THF)(iPrNO)]
Detailed vibrational assignments of ferrous heme-nitrosoalkane complexes that include metal-N(R)O stretching and bending modes are lacking. On the other hand, for the ferrous heme-HNO adduct in myoglobin (Mb), Farmer and coworkers reported the Fe–NHO stretch at 651 cm−1, but the exact nature of this vibration was not further investigated (the assignment is simply based on the fact that this is the only low-energy mode observed by resonance Raman (rRaman) that is isotope sensitive).62 Recently, we showed that in the model complex [Fe(3,5-Me-BAFP)(MI)(NHO)], the Fe–NHO stretch and the Fe–N–O bend are located at 644 and 461 cm−1, respectively, using nuclear resonance vibrational spectroscopy (NRVS) coupled to 15N-isotope labeling.68 To investigate this further, we now measured the Fe–N(iPr)O stretching and bending frequencies of [Fe(TPP)(THF)(iPrNO)] using NRVS, and then analyzed these data using DFT calculations. The NRVS data of the 57Fe-labeled iPrNO/iPr15NO complexes are shown in Fig. 7, top panel. Two isotope sensitive bands are identified in the NRVS data. The first feature is observed at 426 cm−1, which shifts to 418 cm−1 with iPr15NO (Δ = 8 cm−1). The second band is significantly lower in intensity and is found at 633 cm−1. This feature shifts to 627 cm−1 with iPr15NO (Δ = 6 cm−1). To determine the nature of these FeNO-based vibrations we employed DFT computations. The BP86/TZVP calculated geometric parameters of the ferrous iPrNO complexes are listed in Table 2. Overall, the calculated structure of [Fe(P)(THF)(iPrNO)] is in good agreement with the crystal structure of [Fe(TPP)(THF)(iPrNO)] determined here. The N–O stretching frequency is also within ∼20 cm−1 of the experimental value for this complex. With this, we predicted the NRVS data of the [Fe(P)(iPrNO)] and [Fe(P)(THF)(iPrNO)] complexes, as shown in Fig. 7, bottom panel. Interestingly, the five-coordinate (5C) complex [Fe(P)(iPrNO)] is predicted to show an FeN(R)O-based feature at 608 cm−1, which increases in energy (646 cm−1) in the THF-bound complex. The calculated NRVS data of [Fe(P)(THF)(iPrNO)], shown in Fig. 7, bottom panel, exhibit isotope sensitive features at 646 and 447 cm−1, which nicely reproduce the intensity pattern of the 633 and 426 cm−1 bands observed in our experimental data (Fig. 7, top).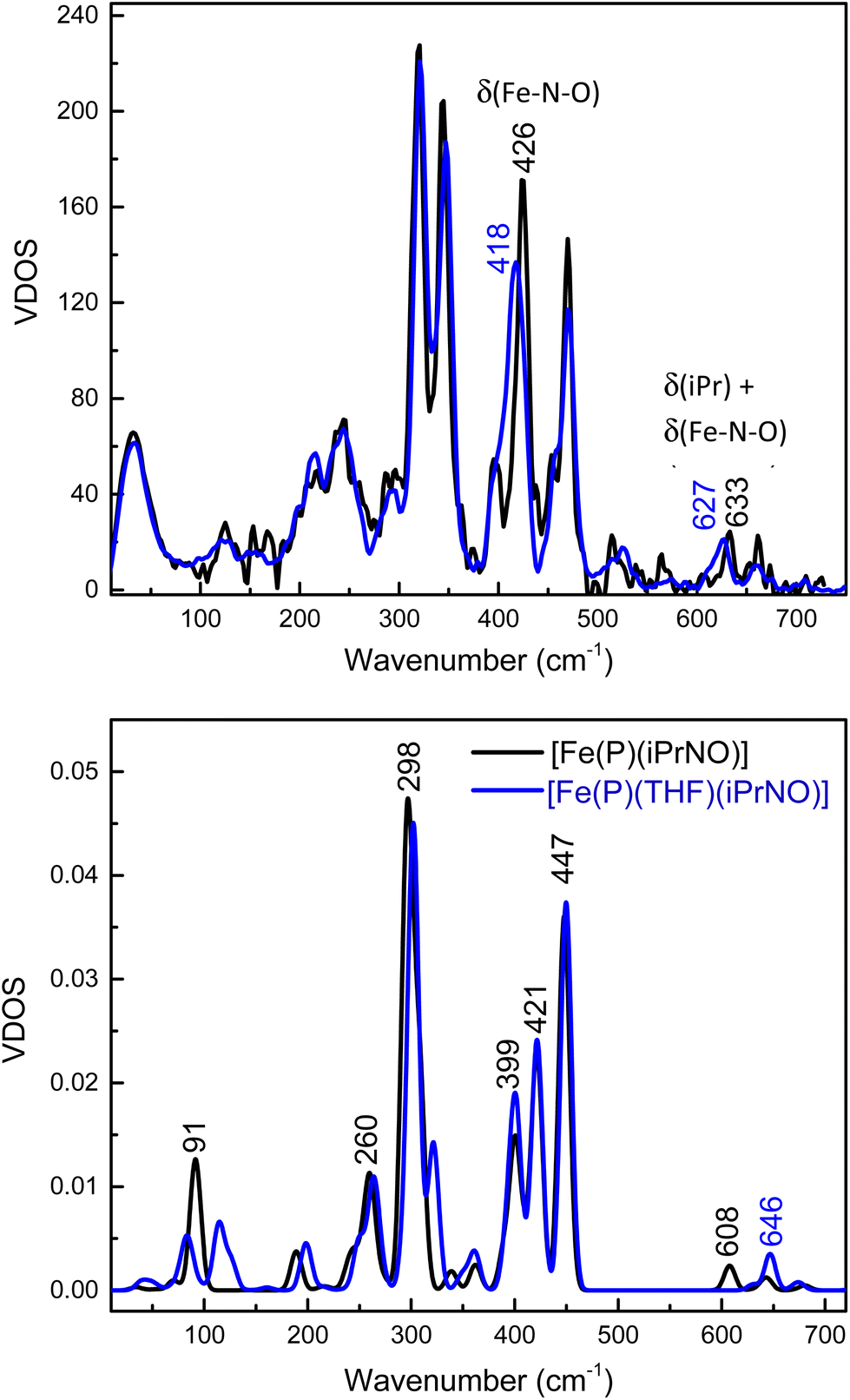 | ||
| Fig. 7 Top panel: NRVS-derived vibrational density of states (VDOS) for [57Fe(TPP)(THF)(iPrNO)] (black) and the 15N-labeled iPrNO complex (blue). Bottom panel: BP86/TZVP calculated NRVS VDOS for [Fe(P)(iPrNO)] (black; P2− = porphine dianion) and [Fe(P)(THF)(iPrNO)] (blue). | ||
In comparison, for RNO adducts of ferrous heme proteins (sGC, Hb, and Mb, where R = Me, pentyl, and octyl), Fe–N(R)O stretching modes have been assigned to rRaman features in the 540–560 cm−1 range in the literature.69–74 However, these assignments are merely based on the similarity of these vibrational energies to those of the supposed Fe–NO stretches of 6C ls-{FeNO}7 porphyrin complexes (545–565 cm−1), but no further analysis of the vibrational data was provided. Note that it was later determined, using NRVS, that the vibrational feature around 545–565 cm−1 in 6C ls-{FeNO}7 porphyrin complexes (for example, at 551 cm−1 in Mb(II)-NO and 563 cm−1 in [Fe(TPP)(MI)(NO)]) has a primary Fe–N–O bending character.43,75,76
To make further comparisons between the analogous heme-HNO and -RNO complexes, we also optimized the structures of [Fe(P)(L)(NHO)], where L = THF and MI, using MI is a model for histidine (see Table 2). The calculated structure of [Fe(P)(MI)(NHO)] agrees well with the EXAFS data for the Mb(II)-NHO species. However, the N–O stretching frequency for the HNO complex is overestimated by ∼75 cm−1. It is possible that the hydrogen bonding interactions between the HNO ligand and the distal histidine in the active site of Mb need to be included in order to accurately predict the N–O stretch. In this regard, Zhang and coworkers have shown that such hydrogen bonding interactions are important for reproducing experimental data of the Mb(II)–NHO complex with computational models.77 On the other hand, the experimental Fe–NHO stretching vibrations in Mb(II)–NHO (651 cm−1) and [Fe(3,5-Me-BAFP)(MI)(NHO)] (644 cm−1) are very close in energy to the DFT calculated feature at 653 cm−1. The calculated NRVS data of the HNO adducts are shown in Fig. S10.† The calculated vibrational frequencies of the iron(II)–NHO complex, with the Fe–NHO stretch and the Fe–N–O bend predicted at 653 cm−1 and 447 cm−1, respectively, are therefore in very good agreement with the experimental results for [Fe(3,5-Me-BAFP)(MI)(NHO)], where the Fe–NHO stretch was assigned to the mode at higher frequency (644 cm−1; also confirmed by Farmer's work on Mb(II)–NHO as mentioned above62), whereas the Fe–N–O bend was assigned to the peak at 461 cm−1.
Note that in the bent geometry of the FeNHO unit (with an Fe–N–O angle of about 130°), significant mixing between the Fe–NO stretching and Fe–N–O bending internal coordinates can be expected, as we have previously investigated in detail for ls-{FeNO}7 complexes that contain bent FeNO units.43,75,78 Vibrational analysis of our DFT results for [Fe(P)(NHO)]-type complexes shows indeed that the vibrational features at ∼650 and ∼460 cm−1 contain significant Fe–NO stretching, and Fe–N–O and Fe–N–H bending character, with the higher frequency mode best assigned to the Fe–NO stretch, whereas the Fe–N–O bend resembles the lower energy mode. On the other hand, based on our DFT model [Fe(P)(THF)(iPrNO)], we conclude that the low-intensity feature at 633 cm−1 in [Fe(TPP)(THF)(iPrNO)] is predominantly an isopropyl bending mode with some Fe–N–O bending character, whereas the intense feature at 426 cm−1 is the Fe–N–O bending mode, with some admixture of Fe–NO stretching character (calculated at ∼450 cm−1). The main component of the Fe–NO stretch is calculated as a weaker feature at 422 cm−1, but no corresponding peak could be unequivocally identified in the experimental NRVS data of [Fe(TPP)(THF)(iPrNO)]. The distinct differences in vibrational properties between the analogous Fe–N(H)O and Fe–N(iPr)O complexes are due to (a) a weaker Fe–N(R)O bond in the iPrNO complex (calculated Fe–N(R)O force constants are 3.1 mdyn Å−1 for the HNO compared to 2.7 mdyn Å−1 for the iPrNO complex), and (b) strong coupling between the low-energy modes of the FeNO unit and bending modes of the isopropyl substituent in the iPrNO complex. These two effects combined result in the observed differences between vibrational features in the iPrNO and HNO model complexes.
Reactivity with NO
To the best of our knowledge, the reactivity of ferrous heme-RNO complexes with NO gas has not been previously studied. However, as pointed out in the Introduction section, the reaction of the Fe(II)–NHO adduct in Cyt. P450nor with NO is one possible route for N–N bond formation and N2O generation in this enzyme.17 Hence, we decided to investigate whether our RNO complexes would react with NO, and what products would result from this reaction. The UV-Vis spectra in Fig. 8 show the titration of [Fe(TPP)(THF)(PhNO)] in THF with an NO-saturated CH2Cl2 solution (typically ∼1 mM) at room temperature. The reaction is complete with ∼1 equiv. of NO, and the UV-Vis data of the reaction product exhibit features at 412, 476, and 540 nm that are identical to those of the ls-{FeNO}7 complex, [Fe(TPP)(NO)]. We hypothesize that this ls-{FeNO}7 complex is generated by the formation of the intermediate, [Fe(TPP)(NO)(PhNO)], which, due to the strong thermodynamic σ-trans effect of NO (also referred to as a trans “interaction”, since it is a ground state effect), causes the PhNO ligand to dissociate, as shown in Scheme 3.78–80 It is important to point out that the proposed intermediate, [Fe(TPP)(PhNO)(NO)], would likely not be observed at room temperature, due to the large binding constant of NO to a ferrous heme (Keq = 1010–12 M−1).81–83 With this result, we hypothesized that binding of an N-donor ligand, MI, to the iron center, leading to the corresponding 6C complex, [Fe(TPP)(MI)(PhNO)], could force the NO to react with the PhNO ligand, rather than simply binding to the iron center in the trans position to PhNO, and then, due to the σ-trans effect of NO, causing the dissociation of the RNO ligand. The complex [Fe(TPP)(THF)(PhNO)] in dichloromethane binds ∼1 equivalent of MI to form the 6C complex, [Fe(TPP)(MI)(PhNO)], as shown in Fig. S11.† The UV-Vis spectra show that the Soret band shifts from 412 to 426 nm and the Q band at 523 shifts to 540 nm upon MI binding. Interestingly, this implies that RNO ligands in heme complexes have essentially no trans effect, in contrast to NO. This result compares well with a previous DFT study from our laboratory that predicts that heme-HNO complexes have a significantly weaker σ-trans effect compared to NO, more comparable to that of CO.84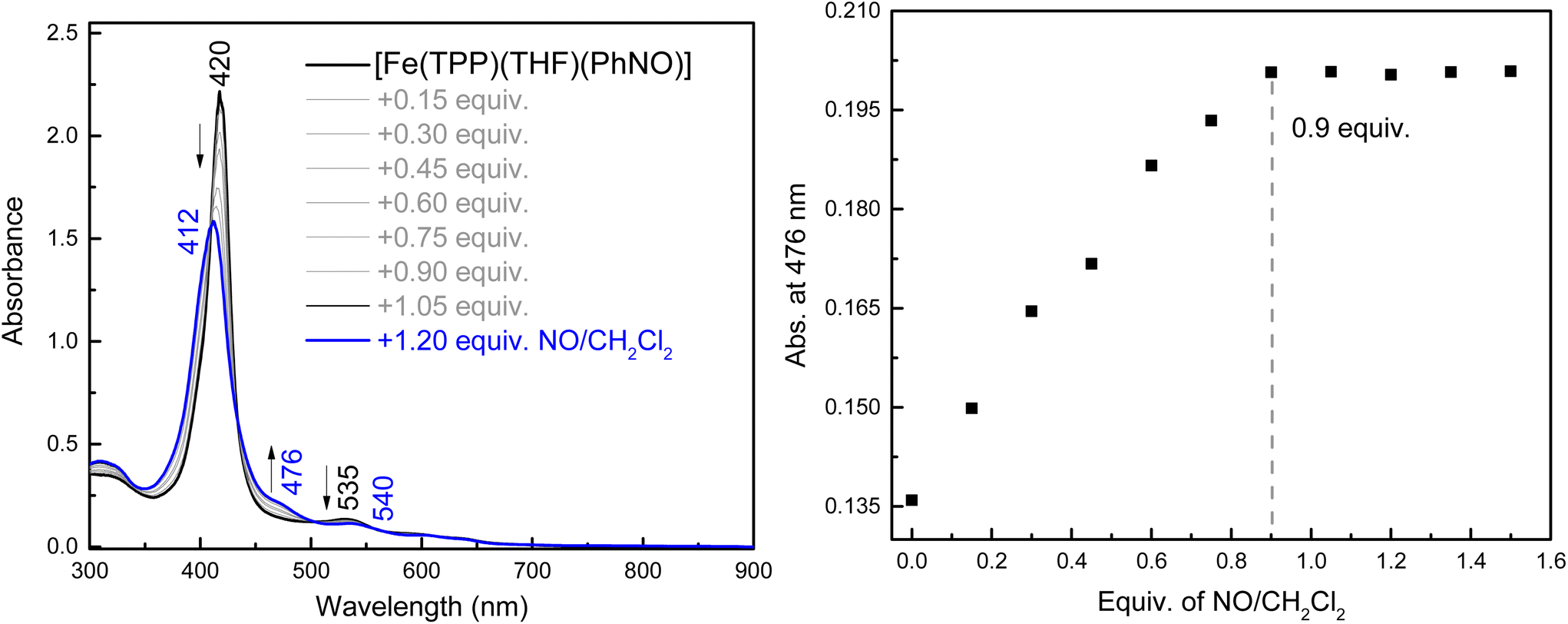 | ||
| Fig. 8 UV-Vis spectra of the titration of ∼14 μM [Fe(TPP)(THF)(PhNO)] (black) in THF with a NO-saturated solution of dichloromethane, forming the ferrous NO complex [Fe(TPP)(NO)] (blue). Right: Changes in absorbance at 476 nm upon addition of the NO solution, showing that the reaction is complete after the addition of ∼1 equivalent of the NO-saturated dichloromethane solution. | ||
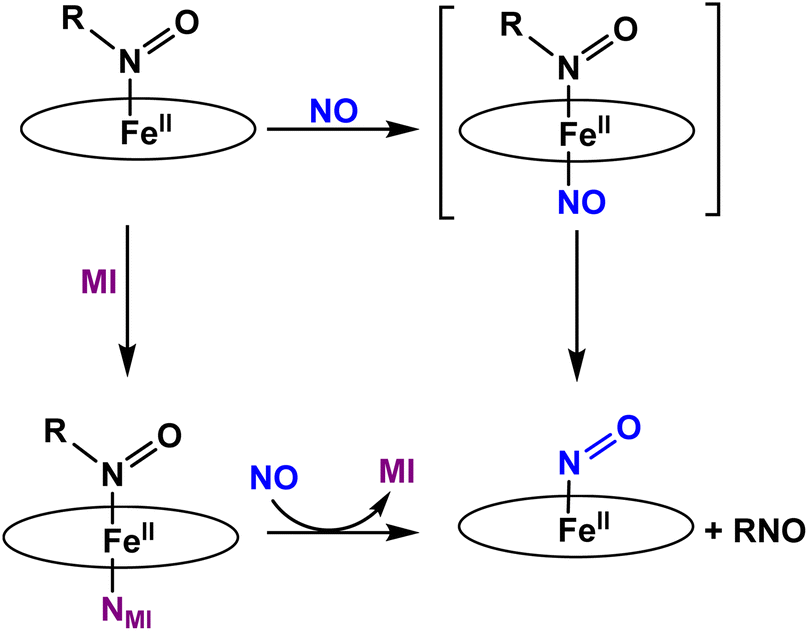 | ||
| Scheme 3 Summary of the reactivity observed for five- and six-coordinate ferrous heme-RNO (R = IPr and PhNO) complexes with the TPP2− coligand with NO. | ||
We then reacted [Fe(TPP)(MI)(PhNO)] with low equivalents of NO gas in dichloromethane at room temperature. The UV-Vis spectra of the products formed, shown in Fig. S12,† are again identical to that of [Fe(TPP)(NO)]. We envisioned that this could arise from (1) a weak Fe-RNO bond, or (2) the NO ligand having a higher binding constant than MI. Hypothesis (1) was tested by adding excess MI to a solution of [Fe(TPP)(THF)(PhNO)] in dichloromethane. In Fig. S13,† the UV-Vis spectra show that the addition of 1 equiv. or 50 equiv. of MI results in no change in the UV-Vis spectrum. We expected the formation of a ferrous bis-imidazole complex if the Fe–RNO bond is weak, but this was not the case (the ferrous bis-imidazole adduct has UV-Vis bands at 430 and 536 nm). In summary, ferrous RNO complexes have strong Fe–N(R)O bonds like ferrous heme-nitrosyls (ls-{FeNO}7) and ferrous heme-CO complexes. Based on these findings, we can rule out hypothesis (1) and we believe that the NO ligand first displaces the imidazole (MI) ligand, and then again forces out the RNO ligand via the σ-trans effect (see Scheme 3).
Finally, we investigated the reactivity of [Fe(TPP)(THF)(iPrNO)] with an NO-saturated CH2Cl2 solution at room temperature. Unfortunately, we observed the same general findings as with the ferrous PhNO complexes. In summary, even in the presence of MI we observe [Fe(TPP)(NO)] as the product of both reactions (see Fig. S14 and S15†). Overall, this shows that heme-RNO complexes with the TPP2− porphyrin are generally unreactive toward NO and do not lead to N–N bond formation, in contrast to heme-HNO complexes.17
Reactivity with Lewis acids
iPrNO is a better structural and isoelectronic mimic of HNO than PhNO, due to the strong conjugation of the nitroso group with the aromatic phenyl ring in PhNO. Thus, the following studies focused on iPrNO. The reactivity of [Fe(TPP)(THF)(iPrNO)] with Lewis acids was first explored. BF3·OEt2 (boron trifluoride dietherate) and bis(pinacolato)diboron (B2(pin)2) were chosen as the Lewis acids. BF3·OEt2 is a much stronger Lewis acid compared to B2(pin)2. BF3·OEt2 was also used successfully by the Richter-Addo group to obtain Lewis acid adducts of [Fe(OEP)(NO)].39 Another important factor in choosing Lewis acids is size. While Fe(TPP) is a completely open scaffold and can interact with Lewis acids of any size, the sterically bulky Fe(3,5-Me-BAFP) complex has a limited pocket size. Both of the Lewis acids, BF3·OEt2 and B2(pin)2, are small enough to fit into the pocket of 3,5-Me-BAFP2− (results described below). [Fe(TPP)(THF)(iPrNO)] was stirred in THF with both Lewis acids and the reaction was monitored by UV-Vis spectroscopy over the course of 2 h. No reaction was observed. No shift in the Soret band in UV-Vis spectroscopy was observed and there was no shift in the N–O stretching band by IR spectroscopy. Fe(3,5-Me-BAFP) was investigated next, to see if adding a non-polar, sterically protected pocket would help promote the binding of a Lewis acid to the corresponding ferrous iPrNO complex.[Fe(3,5-Me-BAFP)(iPrNO)2] was reacted with 2.5 equiv. of BF3·OEt2, B(OEt)3 (triethoxy borate), B(C6F5)(OH)2 (pentafluorophenylboronic acid), B(Mes)2Br (dimesitylboron bromide), and B2(pin)2 in THF and the reaction mixtures were monitored by UV-Vis spectroscopy. The reaction of [Fe(3,5-Me-BAFP)(iPrNO)2] with BF3·OEt2 caused a shift in the Soret band to 407 nm, whereas the reaction with B2(pin)2 shifted the Soret band to 416 nm (Fig. S19†). Both reactions were monitored over the course of 2 h by UV-Vis spectroscopy, but no further changes occurred after the initial 2 min. B(C6F5)(OH)2 and B(Mes)2Br reacted much slower, and over the course of 90 min, the Soret band shifted to 403 nm (Fig. S19†), which is similar to that observed for the product obtained with BF3·OEt2. B2(pin)2 therefore forms a unique product, compared to other Lewis acids, with the Soret band at 416 nm. B(OEt)3, which is a very weak Lewis acid, did not fully convert to a new product (Fig. S19†).
The products of these reactions were then further characterized. In the case of BF3·OEt2, the UV-Vis spectrum of the product shows the Soret and Q band at 403 and 527 nm, which matches with the absorption spectrum of [FeIII(3,5-Me-BAFP)(THF)2]. This indicates that the Lewis acids (BF3·OEt2, B(C6F5)(OH)2 and B(Mes)2Br) oxidize the iron center and this causes the iPrNO ligand to dissociate. This is in line with previous observations that oxidation of ferrous heme-RNO adducts leads to RNO release.32,72 In order to test this hypothesis, B(Mes)2Br was reacted with [FeII(3,5-Me-BAFP)] and the progress of the reaction was monitored by UV-Vis spectroscopy. Upon the addition of the Lewis acid, the Soret band of [FeII(3,5-Me-BAFP)] shifted to 403 nm, verifying the oxidizing ability of these Lewis acids.
In order to identify the product of the reaction of [Fe(3,5-Me-BAFP)(iPrNO)2] with B2(pin)2, we crystallized the resulting complex as shown in Fig. 9. The structure was solved by isomorphous replacement using the [Fe(3,5-Me-BAFP)(iPrNO)2] crystal structure. The disorder model was adjusted (two methylene chloride moieties, no THF molecule, and partial oxygen loss). The isopropyl nitrosyl ligand was found to be disordered with an isopropyl amine ligand, i.e. the oxygen atom at the nitrogen was partially replaced by two hydrogen atoms. No disorder was refined for the N atom or the isopropyl group. The occupancy ratio refined to 0.522(6) to 0.478(6) for iPrNO:iPrNH2. A solvate occupied area was refined as major hexanes and minor methylene chloride (two moieties). The crystal structure in Fig. 9 depicts the ∼50% disorder of iPrNO:iPrNH2 as the mixed complex, where iPrNO is coordinated on one side of the heme and iPrNH2 is bound in the trans position, on the other side of the heme. Just based on the crystallographic data, however, it also possible that the 50% disorder is represented as half bis-iPrNO and half bis-iPrNH2 complexes.
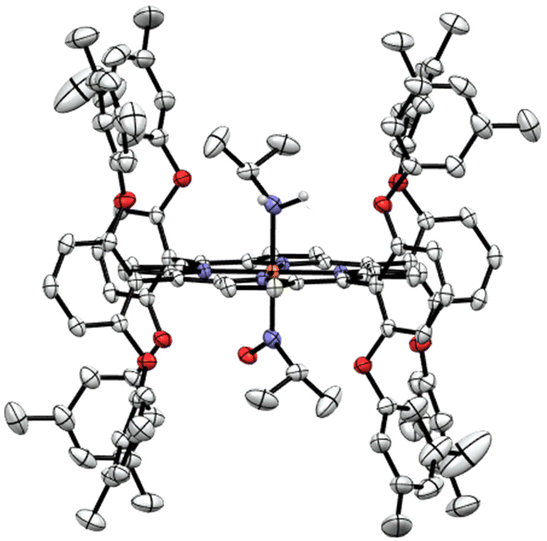 | ||
| Fig. 9 Crystal structure of [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)]. Hydrogen atoms, except for the amine group of iPrNH2, are omitted for clarity. Thermal ellipsoids are shown at 50% probability. | ||
The reaction product was also characterized by rRaman spectroscopy (Fig. S20†), which shows the oxidation state marker band at 1369 cm−1 and the spin state marker band at 1568 cm−1.85–87 There are some slight shifts compared to the rRaman spectrum of the starting complex [Fe(3,5-Me-BAFP)(iPrNO)2], indicating that the oxidation state and spin state remain the same upon the addition of the Lewis acid, and that the complex is in fact the mixed-ligand compound, [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)], as shown in Fig. 9. Further evidence for this conclusion comes from UV-Vis analysis, showing that upon the addition of the Lewis acid, the Soret band shifts into the region of the mono-iPrNO species. This indicates that the Lewis acid not only attacks the iPrNO group, but is also able to abstract an oxygen atom from the iPrNO ligand, followed by reduction of the remaining ligand fragment to the amine (iPrNH2). The required electrons likely originate from the Lewis acid B2(pin)2, which explains why this reaction is not observed with any of the other Lewis acids studied here.
Summary and conclusions
In this work, we prepared and fully characterized several ferrous heme-RNO complexes, where R is an isopropyl or phenyl group, using the TPP2− and 3,5-Me-BAFP2− coligands. Although a significant number of complexes of this type have been reported in the literature, as reviewed in ref. 34 and 35, their vibrational characterization is incomplete, and reactivity studies of these complexes with NO and Lewis acids have not been reported. These complexes are isoelectronic to ls-{FeNHO}8 complexes; however, they do not suffer from the same stability problems as these reactive species. In ferrous heme-RNO complexes, iron is in the low-spin (ls) state, resulting in complexes that are diamagnetic with a closed-shell ground state. Previously, Mansuy and coworkers noted that five-coordinate (5C) [Fe(Porph)(RNO)] complexes bind nitrogenous bases, L, with high affinity to form the corresponding six-coordinate (6C) complexes [Fe(Porph)(L)(RNO)].32 Our work shows that [Fe(TPP)(THF)(RNO)] complexes (R = iPr and Ph) generate the corresponding 6C complexes, [Fe(TPP)(MI)(RNO)], in the presence of ∼1 equiv. of 1-methylimidazole (MI). Therefore, RNO ligands in ferrous heme complexes do not elicit a significant thermodynamic σ-trans effect (interaction), unlike the NO ligand in ferrous heme-nitrosyl complexes.64,75,78,88,89 This is further supported by crystal structures of [Fe(TPP)(MI)(RNO)] (R = iPr, PhNO), [Fe(TPP)(MI)(NO)], and [Fe(TPP)(MI)(CO)], which exhibit Fe–NMI bond lengths of 2.05/2.03 Å, 2.173 Å, and 2.071 Å, respectively.58,59,64,90 By comparison, in [Fe(TPP)(MI)2], where no trans interaction exists, the Fe–NMI bond length is 1.997 Å.91 Hence, ferrous heme-RNO complexes are most similar to ferrous heme-CO complexes that bind to ferrous hemes predominately via strong π-backbonds, which do not result in a strong trans effect. This provides strong support for previous, computational work on ferrous heme-HNO complexes.22,84,92,93Using 15N isotope labeling, we further assigned the N–O stretching modes in our PhNO and iPrNO complexes. Our data show that the 6C [Fe(TPP)(L)(PhNO)] complexes with L = THF, pyridine (Pyr) and MI have N–O stretching frequencies of 1368, 1342 and 1336 cm−1, respectively. In the presence of a weak axial ligand like THF, the complexes further have a propensity to form the bis-PhNO analog, which shows the (antisymmetric) N–O stretch as a very intense feature at ∼1350 cm−1 in the IR spectrum.42 In the case of iPrNO, N–O stretches are located at 1440, 1429 and 1423 cm−1 for the 6C [Fe(TPP)(L)(iPrNO)] complexes with L = THF, Pyr and MI ligands, respectively. Interestingly, the N–O stretch of [Fe(TPP)(MI)(iPrNO)] at 1423 cm−1 is close to the N–O stretch of ferrous heme-HNO complexes, observed in the 1380–1400 cm−1 range.17,18,32,62
In order to further compare the vibrational properties of heme-iPrNO and -HNO complexes, we then obtained NRVS data for the [57Fe(TPP)(THF)(iPrNO)] complex. In this case, two 15N-isotope sensitive features are observed at 426 and 633 cm−1, which are assigned to the Fe–N–O bending mode and a primarily isopropyl bending mode with some Fe–N–O bending character, respectively. NRVS data on the heme-HNO model complex [Fe(3,5-Me-BAFP)(MI)(NHO)] show similar features, in this case at 461 and 644 cm−1, but interestingly, the vibrational assignments are quite different, with the Fe–N(H)O stretching mode at higher frequency (644 cm−1) and the Fe–N–O bending mode at lower frequency (461 cm−1).17 DFT calculations further confirm these assignments. These results indicate that the Fe–N(R)O bond is weaker than the Fe–NHO bond, causing the majority of the observed differences.
Another important difference between ferrous heme-HNO and -RNO complexes is their overall stability and reactivity. Whereas heme-RNO complexes are completely air stable,32 the HNO complexes are sensitive to O2. We therefore wondered how the reactivity of these complexes compares when it comes to NO. Previous studies have shown that heme-HNO complexes react with NO under N–N bond formation giving a quantitative N2O yield, and are therefore possible candidates for the key Intermediate I in Cyt. P450nor catalysis.17,26 In contrast, in this study we found that ferrous heme-RNO complexes are generally unreactive toward NO, resulting in the formation of ls-{FeNO}7 complexes rather than N–N bond formation. Based on these findings, we conclude that the presence of the R group greatly stabilizes the RNO ligand and corresponding ferrous heme complexes compared to HNO, and hence, although ferrous heme-RNO complexes are good structural and electronic models for their HNO analogs,22 they do not model the reactivity of the heme-HNO complexes at all. For future studies, a ferrous RNO complex with an axial anionic thiolate ligand could be synthesized, to examine this point further.
In addition, the Lewis acid reactivity of heme-RNO complexes was investigated to determine if these compounds would form Lewis acid adducts and could be activated in this way for reaction with NO. This work was in part inspired by a previous report in the literature, where the formation of Lewis acid adducts of ls-{FeNO}7 heme complexes was reported.39 We found that [Fe(TPP)(L)(RNO)] complexes are unreactive towards all Lewis acids tested here: BF3·OEt2, B(OEt)3, B(C6F5)(OH)2, B(Mes)2Br, and B2(pin)2 in THF. We then changed the porphyrin to a bulkier scaffold, the bis-picket fence porphyrin 3,5-Me-BAFP2−. Surprisingly, the complex [Fe(3,5-Me-BAFP)(iPrNO)2] is reactive towards Lewis acids, in contrast to the TPP2− complexes. With BF3·OEt2, B(OEt)3, B(C6F5)(OH)2, and B(Mes)2Br, simple oxidation of the iron center to the ferric oxidation state was observed, likely followed by loss of the iPrNO ligand. In contrast, the reaction of [Fe(3,5-Me-BAFP)(iPrNO)2] with B2(pin2) leads to O-atom abstraction from one of the iPrNO ligands, followed by reduction, to yield the corresponding amine, iPrNH2. From these reactions, the mixed ligand complex [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)] was isolated and characterized using different spectroscopic methods and X-ray crystallography. These results provide the foundation for further studies to be developed to ultimately capture a Lewis-acid appended heme-RNO species.
Data availability
The crystallographic data have been deposited in the Cambridge Crystallographic Data Centre, and the corresponding CCDC entry numbers are provided in the manuscript. UV-Vis, NMR and IR spectroscopic data are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Dr Benedict J. Elvers for his support with X-Ray crystallography. This work was supported by a grant from the National Science Foundation (CHE-1464696 to NL). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility at Argonne National Laboratory and is based on research supported by the U.S. DOE Office of Science-Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.References
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. Sosa Alfaro, The biologically relevant coordination chemistry of iron and nitric oxide: electronic structure and reactivity, Chem. Rev., 2021, 121(24), 14682–14905 CrossRef CAS PubMed
.
- D. Mansuy, E. Rouer, C. Bacot, P. Gans, J. C. Chottard and J. P. Leroux, Interaction of aliphatic N-hydroxylamines with microsomal cytochrome P450: Nature of the different derived complexes and inhibitory effects on monoxygenases activities, Biochem. Pharmacol., 1978, 27(8), 1229–1237, DOI:10.1016/0006-2952(78)90456-2
- C. Bensoussan, M. Delaforge and D. Mansuy, Particular ability of cytochromes P450 3A to form inhibitory P450-iron-metabolite complexes upon metabolic oxidation of aminodrugs, Biochem. Pharmacol., 1995, 49(5), 591–602, DOI:10.1016/0006-2952(94)00477-4
- M. Delaforge, M. Jaouen and D. Mansuy, Dual effects of macrolide antibiotics on rat liver cytochrome P-450: Induction and formation of metabolite-complexes: A structure-activity relationship, Biochem. Pharmacol., 1983, 32(15), 2309–2318, DOI:10.1016/0006-2952(83)90178-8
- P. J. O'Brien, W. C. Wong, J. Silva and S. Khan, Toxicity of nitrobenzene compounds towards isolated hepatocytes: dependence on reduction potential, Xenobiotica, 1990, 20(9), 945–955, DOI:10.3109/00498259009046910
- M. A. Belisario, R. Pecce, A. Garofalo, N. Sannolo and A. Malorni, Erythrocyte enzymes catalyze 1-nitropyrene and 3-nitrofluoranthene nitroreduction, Toxicology, 1996, 108(1), 101–108, DOI:10.1016/0300-483X(95)03293-O
- D. Ringe, R. J. Turesky, P. L. Skipper and S. R. Tannenbaum, Structure of the single stable hemoglobin adduct formed by 4-aminobiphenyl in vivo, Chem. Res. Toxicol., 1988, 1(1), 22–24, DOI:10.1021/tx00001a003
- P. Eyer, Reactions of nitrosobenzene with reduced glutathione, Chem.-Biol. Interact., 1979, 24(2), 227–239, DOI:10.1016/0009-2797(79)90011-5
- B. Dölle, W. Töpner and H. G. Neumann, Reaction of arylnitroso compounds with mercaptans, Xenobiotica, 1980, 10(7–8), 527–536, DOI:10.3109/00498258009033787
- H. Klehr, P. Eyer and W. Schäfer, On the mechanism of reactions of nitrosoarenes with thiols. Formation of a common intermediate “semimercaptal”, Biol. Chem. Hoppe-Seyler, 1985, 366(8), 755–760 CrossRef CAS PubMed
- P. Eyer and E. Lierheimer, Biotransformation of nitrosobenzene in the red cell and the role of glutathione, Xenobiotica, 1980, 10(7–8), 517–526, DOI:10.3109/00498258009033786
- J. H. Enemark and R. D. Feltham, Principles of structure, bonding, and reactivity for metal nitrosyl complexes, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS
- C. Riplinger and F. Neese, The reaction mechanism of cytochrome P450 NO reductase: A detailed quantum mechanics/molecular mechanics study, ChemPhysChem, 2011, 12, 3192–3203 CrossRef CAS PubMed
- A. B. McQuarters, N. E. Wirgau and N. Lehnert, Model complexes of key intermediates in fungal cytochrome P450 nitric oxide reductase (P450nor), Curr. Opin. Chem. Biol., 2014, 19, 82–89, DOI:10.1016/j.cbpa.2014.01.017
- N. Lehnert, V. K. K. Praneeth and F. Paulat, Electronic structure of iron(II)–porphyrin nitroxyl complexes: Molecular mechanism of fungal nitric oxide reductase (P450nor), J. Comput. Chem., 2006, 27(12), 1338–1351 CrossRef CAS PubMed
- T. Nomura, T. Kimura, Y. Kanematsu, D. Yamada, K. Yamashita, K. Hirata, G. Ueno, H. Murakami, T. Hisano and R. Yamagiwa, Short-lived intermediate in N2O generation by P450 NO reductase captured by time-resolved IR spectroscopy and XFEL crystallography, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(21), e2101481118 CrossRef CAS PubMed
- E. C. Manickas, A. B. LaLonde, M. Y. Hu, E. E. Alp and N. Lehnert, Stabilization of a Heme-HNO Model Complex Using a Bulky Bis-Picket Fence Porphyrin and Reactivity Studies with NO, J. Am. Chem. Soc., 2023, 145(42), 23014–23026 CrossRef CAS PubMed
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, Hydride Attack on a Coordinated Ferric Nitrosyl: Experimental and DFT Evidence for the Formation of a Heme Model–HNO Derivative, J. Am. Chem. Soc., 2016, 138(1), 104–107, DOI:10.1021/jacs.5b12008
- E. G. Abucayon, R. L. Khade, D. R. Powell, M. J. Shaw, Y. Zhang and G. B. Richter-Addo, Over or under: hydride attack at the metal versus the coordinated nitrosyl ligand in ferric nitrosyl porphyrins, Dalton Trans., 2016, 45(45), 18259–18266, 10.1039/C6DT03860C
- P. J. Farmer and F. Sulc, Coordination chemistry of the HNO ligand with hemes and synthetic coordination complexes, J. Inorg. Biochem., 2005, 99(1), 166–184 CrossRef CAS PubMed
- M. R. Kumar, J. M. Fukuto, K. M. Miranda and P. J. Farmer, Reactions of HNO with heme proteins: New routes to HNO− Heme complexes and insight into physiological effects, Inorg. Chem., 2010, 49(14), 6283–6292 CrossRef CAS PubMed
- Y. Ling, C. Mills, R. Weber, L. Yang and Y. Zhang, NMR, IR/Raman, and Structural Properties in HNO and RNO (R = Alkyl and Aryl) Metalloporphyrins with Implication for the HNO−Myoglobin Complex, J. Am. Chem. Soc., 2010, 132(5), 1583–1591, DOI:10.1021/ja907342s
- A. Mazzeo, J. Pellegrino and F. Doctorovich, Water-Soluble Nitroxyl Porphyrin Complexes FeIITPPSHNO and FeIITPPSNO– Obtained from Isolated FeIITPPSNO•, J. Am. Chem. Soc., 2019, 141, 18521–18530 CrossRef CAS PubMed
- I.-K. Choi, Y. Liu, D. Feng, K.-J. Paeng and M. D. Ryan, Electrochemical and spectroscopic studies of iron porphyrin nitrosyls and their reduction products, Inorg. Chem., 1991, 30, 1832–1839 CrossRef CAS
- M. H. Rahman and M. D. Ryan, Redox and spectroscopic properties of iron porphyrin nitroxyl in the presence of weak acids, Inorg. Chem., 2017, 56(6), 3302–3309 CrossRef CAS PubMed
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, Hydride Attack on a Coordinated Ferric Nitrosyl: Experimental and DFT Evidence for the Formation of a Heme Model–HNO Derivative, J. Am. Chem. Soc., 2016, 138(1), 104–107, DOI:10.1021/jacs.5b12008
- L. E. Goodrich, S. Roy, E. E. Alp, J. Zhao, M. Y. Hu and N. Lehnert, Electronic Structure and Biologically Relevant Reactivity of Low-Spin {FeNO}8 Porphyrin Model Complexes: New Insight from a Bis-Picket Fence Porphyrin, Inorg. Chem., 2013, 52(13), 7766–7780, DOI:10.1021/ic400977h
- M. H. Rahman, Y. Liu and M. D. Ryan, Proton transfer versus hydrogen bonding in a reduced iron porphyrin nitrosyl complex, Inorg. Chem., 2019, 58(20), 13788–13795 CrossRef CAS PubMed
- J. Pellegrino, S. E. Bari, D. E. Bikiel and F. Doctorovich, Successful Stabilization of the Elusive Species {FeNO}8 in a Heme Model, J. Am. Chem. Soc., 2010, 132(3), 989–995, DOI:10.1021/ja905062w
- N. Kundakarla, S. Lindeman, M. H. Rahman and M. D. Ryan, X-ray Structure and Properties of the Ferrous Octaethylporphyrin Nitroxyl Complex, Inorg. Chem., 2016, 55(5), 2070–2075, DOI:10.1021/acs.inorgchem.5b02384
- B. Hu and J. Li, One Electron Makes Differences: From Heme {FeNO}7 to {FeNO}8, Angew. Chem., Int. Ed., 2015, 54(36), 10579–10582, DOI:10.1002/anie.201505166
- D. Mansuy, P. Battioni, J. C. Chottard, C. Riche and A. Chiaroni, Nitrosoalkane complexes of iron-porphyrins: analogy between the bonding properties of nitrosoalkanes and dioxygen, J. Am. Chem. Soc., 1983, 105(3), 455–463, DOI:10.1021/ja00341a027
- D. Mansuy, P. Beaune, J. C. Chottard, J. F. Bartoli and P. Gans, The nature of the “455 nm absorbing complex” formed during the cytochrome P450 dependent oxidative metabolism of amphetamine, Biochem. Pharmacol., 1976, 25(5), 609–612, DOI:10.1016/0006-2952(76)90398-1
- J. Lee, L. Chen, A. H. West and G. B. Richter-Addo, Interactions of Organic Nitroso Compounds with Metals, Chem. Rev., 2002, 102(4), 1019–1066, DOI:10.1021/cr0000731
-
N. Xu and G. B. Richter-Addo, Interactions of Nitrosoalkanes/arenes, Nitrosamines, Nitrosothiols, and Alkyl Nitrites with Metals, in Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2014, vol. 59, pp. 381–446 Search PubMed
- E. G. Abucayon, D. R. Powell and G. B. Richter-Addo, Carbon–Nitrogen and Nitrogen–Nitrogen Bond Formation from Nucleophilic Attack at Coordinated Nitrosyls in Fe and Ru Heme Models, J. Am. Chem. Soc., 2017, 139(28), 9495–9498, DOI:10.1021/jacs.7b05209
- K. A. Emhoff, L. Balaraman, A. M. H. Salem, K. I. Mudarmah and W. C. Boyd, Coordination chemistry of organic nitric oxide derivatives, Coord. Chem. Rev., 2019, 396, 124–140, DOI:10.1016/j.ccr.2019.06.011
- V. E. Herrera, T. P. Charles, T. G. Scott, K. Y. Prather, N. T. Nguyen, C. D. Sohl, L. M. Thomas and G. B. Richter-Addo, Insights into Nitrosoalkane Binding to Myoglobin Provided by Crystallography of Wild-Type and Distal Pocket Mutant Derivatives, Biochemistry, 2023, 62(8), 1406–1419, DOI:10.1021/acs.biochem.2c00725
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, Lewis acid activation of the ferrous heme–NO fragment toward the N–N coupling reaction with NO to generate N2O, J. Am. Chem. Soc., 2018, 140(12), 4204–4207 CrossRef CAS PubMed
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, On the preparation of metalloporphyrins, J. Inorg. Nucl. Chem., 1970, 32(7), 2443–2445, DOI:10.1016/0022-1902(70)80535-8
- A. B. McQuarters, L. E. Goodrich, C. M. Goodrich and N. Lehnert, Disproportionation of O-Benzylhydroxylamine Catalyzed by a Ferric Bis-Picket Fence Porphyrin Complex, Z. Anorg. Allg. Chem., 2013, 639(8–9), 1520–1526, DOI:10.1002/zaac.201300125
- L.-S. Wang, L. Chen, M. A. Khan and G. B. Richter-Addo, The first structural studies of nitrosoarene binding to iron-(II) and -(III) porphyrins, Chem. Commun., 1996,(3), 323–324, 10.1039/CC9960000323
- F. Paulat, T. C. Berto, S. DeBeer George, L. Goodrich, V. K. K. Praneeth, C. D. Sulok and N. Lehnert, Vibrational Assignments of Six-Coordinate Ferrous Heme Nitrosyls: New Insight from Nuclear Resonance Vibrational Spectroscopy, Inorg. Chem., 2008, 47(24), 11449–11451, DOI:10.1021/ic801626w
- J. T. Sage, C. Paxson, G. R. A. Wyllie, W. Sturhahn, S. M. Durbin, P. M. Champion, E. E. Alp and W. R. Scheidt, Nuclear resonance vibrational spectroscopy of a protein active-site mimic, J. Condens. Matter Phys., 2001, 13(34), 7707 CrossRef CAS
- W. Sturhahn, CONUSS and PHOENIX: Evaluation of nuclear resonant scattering data, Hyperfine Interact., 2000, 125(1), 149–172, DOI:10.1023/A:1012681503686
- A. Defoin, Simple Preparation of Nitroso Benzenes and Nitro Benzenes by Oxidation of Anilines with H2O2 Catalysed with Molybdenum Salts, Synthesis, 2004,(05), 706–710, DOI:10.1055/s-2004-815964
- F. Leinisch, J. Jiang, L. J. Deterding and R. P. Mason, Simplified Synthesis of Isotopically Labeled 5,5-Dimethyl-pyrroline N-Oxide, Molecules, 2011, 16(10), 8428–8436 CrossRef CAS PubMed
-
M. B. Sherwin and P. Pichaichanarong, Hydrogenation of nitroalkanes to hydroxylamines, Google Pat., EP0321219A1, 1994 Search PubMed
- J. A. Maassen and T. J. de Boer, Silver carbonate, a convenient reagent for preparing C-nitroso compounds from hydroxylamines, Recl. Trav. Chim. Pays-Bas, 1971, 90(4), 373–376, DOI:10.1002/recl.19710900405
- B. G. Gowenlock and W. Luttke, Structure and properties of C-nitroso-compounds, Q. Rev., Chem. Soc., 1958, 12(4), 321–340, 10.1039/QR9581200321
- W. D. Emmons, The Synthesis of Nitrosoalkane Dimers, J. Am. Chem. Soc., 1957, 79(24), 6522–6524, DOI:10.1021/ja01581a043
- C. D. Sohl, J. Lee, S. S. Alguindigue, M. A. Khan and G. B. Richter-Addo, Synthesis and solid-state molecular structures of nitrosoalkane complexes of iron porphyrins containing methanol, pyridine, and 1-methylimidazole ligands, J. Inorg. Biochem., 2004, 98(7), 1238–1246, DOI:10.1016/j.jinorgbio.2004.04.008
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. A, 1986, 33(12), 8822–8824 Search PubMed
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS PubMed
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97(4), 2571–2577, DOI:10.1063/1.463096
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100(8), 5829–5835, DOI:10.1063/1.467146
-
Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009, (accessed) Search PubMed
- N. Godbout, L. K. Sanders, R. Salzmann, R. H. Havlin, M. Wojdelski and E. Oldfield, Solid-State NMR, Mössbauer, Crystallographic, and Density Functional Theory Investigation of Fe–O2 and Fe–O2 Analogue Metalloporphyrins and Metalloproteins, J. Am. Chem. Soc., 1999, 121(16), 3829–3844, DOI:10.1021/ja9832820
- E. G. Abucayon, D. Awasabisah, D. R. Powell and G. B. Richter-Addo, (1-Methyl-1H-imidazole-κN3)(1-methyl-2-nitrosobenzene-[kappa]N)(5,10,15,20-tetraphenylporphyrinato-κ4N)iron(II) dichloromethane monosolvate, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70(2), m51–m52, DOI:10.1107/S160053681400083X
- O. Q. Munro, P. S. Madlala, R. A. F. Warby, T. B. Seda and G. Hearne, Structural, Conformational, and Spectroscopic Studies of Primary Amine Complexes of Iron(II) Porphyrins, Inorg. Chem., 1999, 38(21), 4724–4736, DOI:10.1021/ic990178q
- B. Hu and J. Li, One Electron Makes Differences: From Heme {FeNO}7 to {FeNO}8, Angew. Chem., Int. Ed., 2015, 54(36), 10579–10582, DOI:10.1002/anie.201505166
- C. E. Immoos, F. Sulc, P. J. Farmer, K. Czarnecki, D. F. Bocian, A. Levina, J. B. Aitken, R. S. Armstrong and P. A. Lay, Bonding in HNO-Myoglobin as Characterized by X-ray Absorption and Resonance Raman Spectroscopies, J. Am. Chem. Soc., 2005, 127(3), 814–815, DOI:10.1021/ja0433727
- W. R. Scheidt and M. E. Frisse, Nitrosylmetalloporphyrins. II. Synthesis and molecular stereochemistry of nitrosyl-.alpha.,.beta.,.gamma.,.delta.-tetraphenylporphinatoiron(II), J. Am. Chem. Soc., 1975, 97(1), 17–21, DOI:10.1021/ja00834a005
- G. R. A. Wyllie, C. E. Schulz and W. R. Scheidt, Five- to Six-Coordination in (Nitrosyl)iron(II) Porphyrinates: Effects of Binding the Sixth Ligand, Inorg. Chem., 2003, 42(18), 5722–5734, DOI:10.1021/ic034473t
- F. A. Walker, Nitric oxide interaction with insect nitrophorins and thoughts on the electron configuration of the {FeNO}6 complex, J. Inorg. Biochem., 2005, 99(1), 216–236, DOI:10.1016/j.jinorgbio.2004.10.009
- M. K. Safo, F. A. Walker, A. M. Raitsimring, W. P. Walters, D. P. Dolata, P. G. Debrunner and W. R. Scheidt, Axial Ligand Orientation in Iron(III) Porphyrinates: Effect of Axial π-Acceptors. Characterization of the Low-Spin Complex [Fe(TPP)(4-CNPy)2]ClO4, J. Am. Chem. Soc., 1994, 116(17), 7760–7770, DOI:10.1021/ja00096a037
- W. R. Scheidt and C. A. Reed, Spin-state/stereochemical relationships in iron porphyrins: implications for the hemoproteins, Chem. Rev., 1981, 81(6), 543–555, DOI:10.1021/cr00046a002
- A. B. McQuarters, E. J. Blaesi, J. W. Kampf, E. E. Alp, J. Zhao, M. Hu, C. Krebs and N. Lehnert, Synthetic model complex of the key intermediate in cytochrome P450 nitric oxide reductase, Inorg. Chem., 2019, 58(2), 1398–1413 CrossRef CAS PubMed
- E. R. Derbyshire, R. Tran, R. A. Mathies and M. A. Marletta, Characterization of Nitrosoalkane Binding and Activation of Soluble Guanylate Cyclase, Biochemistry, 2005, 44(49), 16257–16265, DOI:10.1021/bi0515671
- J. W. Denninger and M. A. Marletta, Guanylate cyclase and the ˙NO/cGMP signaling pathway, Biochim. Biophys. Acta, Bioenerg., 1999, 1411(2–3), 334–350, DOI:10.1016/S0005-2728(99)00024-9
- D. S. Karow, D. Pan, J. H. Davis, S. Behrends, R. A. Mathies and M. A. Marletta, Characterization of Functional Heme Domains from Soluble Guanylate Cyclase, Biochemistry, 2005, 44(49), 16266–16274, DOI:10.1021/bi051601b
- D. Mansuy, J. C. Chottard and G. Chottard, Nitrosoalkanes as Fe(II) Ligands in the Hemoglobin and Myoglobin Complexes Formed from Nitroalkanes in Reducing Conditions, Eur. J. Biochem., 1977, 76(2), 617–623, DOI:10.1111/j.1432-1033.1977.tb11632.x
- J. R. Stone and M. A. Marletta, The Ferrous Heme of Soluble Guanylate Cyclase: Formation of Hexacoordinate Complexes with Carbon Monoxide and Nitrosomethane, Biochemistry, 1995, 34(50), 16397–16403, DOI:10.1021/bi00050a021
- R. W. Romberg and R. J. Kassner, Nitric oxide and carbon monoxide equilibriums of horse myoglobin and (N-methylimidazole)protoheme. Evidence for steric interaction with the distal residues, Biochemistry, 1979, 18(24), 5387–5392, DOI:10.1021/bi00591a020
- N. Lehnert, J. T. Sage, N. Silvernail, W. R. Scheidt, E. E. Alp, W. Sturhahn and J. Zhao, Oriented Single-Crystal Nuclear Resonance Vibrational Spectroscopy of [Fe(TPP)(MI)(NO)]: Quantitative Assessment of the trans Effect of NO, Inorg. Chem., 2010, 49(15), 7197–7215, DOI:10.1021/ic1010677
- W. Zeng, N. J. Silvernail, D. C. Wharton, G. Y. Georgiev, B. M. Leu, W. R. Scheidt, J. Zhao, W. Sturhahn, E. E. Alp and J. T. Sage, Direct probe of iron vibrations elucidates NO activation of heme proteins, J. Am. Chem. Soc., 2005, 127(32), 11200–11201 CrossRef CAS PubMed
- L. Yang, Y. Ling and Y. Zhang, HNO Binding in a Heme Protein: Structures, Spectroscopic Properties, and Stabilities, J. Am. Chem. Soc., 2011, 133(35), 13814–13817, DOI:10.1021/ja204072j
- V. K. K. Praneeth, C. Näther, G. Peters and N. Lehnert, Spectroscopic Properties and Electronic Structure of Five- and Six-Coordinate Iron(II) Porphyrin NO Complexes: Effect of the Axial N-Donor Ligand, Inorg. Chem., 2006, 45(7), 2795–2811, DOI:10.1021/ic050865j
- T. C. Berto, V. K. K. Praneeth, L. E. Goodrich and N. Lehnert, Iron-porphyrin NO complexes with covalently attached N-donor ligands: Formation of a stable six-coordinate species in solution, J. Am. Chem. Soc., 2009, 131(47), 17116–17126 CrossRef CAS PubMed
-
N. Lehnert, W. R. Scheidt and M. W. Wolf, Structure and Bonding in Heme–Nitrosyl Complexes and Implications for Biology, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine II, ed. D. M. P. Mingos, Springer, Berlin, Heidelberg, 2014, pp. 155–223 Search PubMed
-
N. Lehnert, T. C. Berto, M. G. I. Galinato and L. E. Goodrich, The Role of Heme-Nitrosyls in the Biosynthesis, Transport, Sensing, and Detoxification of Nitric Oxide (NO) in Biological Systems: Enzymes and Model Complexes, in The Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, New Jersey, 2011, vol. 14, pp. 1–247 Search PubMed
- C. E. Cooper, Nitric oxide and iron proteins, Biochim. Biophys. Acta, 1999, 1411, 290–309 CrossRef CAS PubMed
- M. Hoshino, K. Ozawa, H. Seki and P. C. Ford, Photochemistry of nitric oxide adducts of water-soluble iron(III) porphyrin and ferrihemoproteins studied by nanosecond laser photolysis, J. Am. Chem. Soc., 1993, 115, 9568–9575 CrossRef CAS
- L. E. Goodrich and N. Lehnert, The trans, effect of nitroxyl (HNO) in ferrous heme systems: Implications for soluble guanylate cyclase activation by HNO, J. Inorg. Biochem., 2013, 118, 179–186 CrossRef CAS PubMed
- T. G. Spiro and T. C. Strekas, Resonance Raman spectra of heme proteins. Effects of oxidation and spin state, J. Am. Chem. Soc., 1974, 96(2), 338–345, DOI:10.1021/ja00809a004
-
T. G. Spiro, The Resonance Raman Spectroscopy of Metalloporphyrins and Heme Proteins, in Iron Porphyrins (Part Two), ed. A. B. P. Lever and H. B. Gray, Addison-Wesley Publishing Company, 1983, pp. 89–159 Search PubMed
- F. Paulat, V. K. K. Praneeth, C. Näther and N. Lehnert, Quantum Chemistry-Based Analysis of the Vibrational Spectra of Five-Coordinate Metalloporphyrins [M(TPP)Cl], Inorg. Chem., 2006, 45, 2835–2856 CrossRef CAS PubMed
- T. G. Traylor and V. S. Sharma, Why NO?, Biochemistry, 1992, 31, 2847–2849 CrossRef CAS PubMed
- L. E. Goodrich, F. Paulat, V. K. K. Praneeth and N. Lehnert, Electronic Structure of Heme-Nitrosyls and Its Significance for Nitric Oxide Reactivity, Sensing, Transport, and Toxicity in Biological Systems, Inorg. Chem., 2010, 49(14), 6293–6316, DOI:10.1021/ic902304a
- N. J. Silvernail, A. Roth, C. E. Schulz, B. C. Noll and W. R. Scheidt, Heme Carbonyls: Environmental Effects on νC-O and Fe−C/C−O Bond Length Correlations, J. Am. Chem. Soc., 2005, 127(41), 14422–14433, DOI:10.1021/ja053148x
- J. Li, S. M. Nair, B. C. Noll, C. E. Schulz and W. R. Scheidt, Relative Axial Ligand Orientation in Bis(imidazole)iron(II) Porphyrinates: Are “Picket Fence” Derivatives Different?, Inorg. Chem., 2008, 47(9), 3841–3850, DOI:10.1021/ic702498c
- D. P. Linder and K. R. Rodgers, Structural, Electronic, and Vibrational Characterization of Fe−HNO Porphyrinates by Density Functional Theory, Inorg. Chem., 2005, 44(23), 8259–8264, DOI:10.1021/ic0504745
- L. Yang, W. Fang and Y. Zhang, Metal centre effects on HNO binding in porphyrins and the electronic origin: metal's electronic configuration, position in the periodic table, and oxidation state, Chem. Commun., 2012, 48(32), 3842–3844, 10.1039/C2CC31016C
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis, NMR, and FT-IR spectra for [Fe(TPP)(L)(PhNO)] where L = THF, MI, and pyridine, and [Fe(TPP)(THF)(iPrNO)]; UV-Vis, IR, and rRaman spectra for [Fe(3,5-Me-BAFP)(iPrNO)2] and for products from the reaction with Lewis acids; crystallographic information on [Fe(TPP)(THF)(PhNO)]/[Fe(TPP)(PhNH2)(PhNO)], [Fe(TPP)(THF)(iPrNO)], [Fe(3,5-Me-BAFP)(iPrNO)2], [Fe(3,5-Me-BAFP)(iPrNH2)(iPrNO)], and [Fe(3,5-Me-BAFP)(2-MeTHF)(PhNO)]. CCDC 2340714, 2341278, 2339812, 2339811, and 2357458. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01632g |
| This journal is © The Royal Society of Chemistry 2024 |