
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
E. coli-based semi-artificial photosynthesis: biocompatibility of redox mediators and electron donors in [FeFe] hydrogenase driven hydrogen evolution†
Mira T.
Gamache‡
 a,
Larissa
Kurth‡
a,
Dawit T.
Filmon
b,
Nicolas
Plumeré
b and
Gustav
Berggren
*a
a,
Larissa
Kurth‡
a,
Dawit T.
Filmon
b,
Nicolas
Plumeré
b and
Gustav
Berggren
*a
aDepartment of Chemistry – Ångström, Molecular Biomimetics, Uppsala University, Box 523, 75120 Uppsala, Sweden. E-mail: Gustav.berggren@kemi.uu.se
bTUM Campus Straubing for Biotechnology and Sustainability – Technical University of Munich, Uferstrasse 53, 94315 Straubing, Germany
First published on 6th November 2023
Abstract
Semi-artificial photosynthesis aims to harness the power of biocatalysis while breaking away from the limitations of Nature's photosynthetic machinery, by merging artificial light harvesters with enzyme catalysts. However, the artificial photocatalytic components are generally toxic towards the biological components. In this study, we investigate a system wherein Escherichia coli cells, heterologously expressing an [FeFe] hydrogenase, act as hydrogen evolution catalyst in combination with an artificial photosensitizer, sacrificial electron donor, and redox mediator. Previously, the use of artificial components or their reaction products was found to be toxic to E. coli cells. To overcome this challenge, we examined alternative electron donors and redox mediators, achieving turnover numbers (TON, 39.6 μmol H2 per 1 mL sample with OD600 = 5) and turnover frequencies (TOF, 812 nmol H2 h−1 per 1 mL sample with OD600 = 5) on par with previously reported high performing E. coli-based systems while greatly reducing cytotoxic effects. Transient absorption spectroscopy revealed how the choice of photosensitizer, electron donor, and redox mediator affects the observed photocatalytic TOFs. Following optimization of the redox mediator and electron donor the biocatalyst demonstrated remarkable stability throughout the experiments. We identified the availability of electrons from the electron donor as the primary limiting factor, with approximately 85% of electrons being effectively utilized for hydrogen production. Furthermore, the observed activity with different [FeFe] hydrogenases verified the broad applicability of the identified photocatalytic components to promote light-driven catalysis in bio-hybrid systems.
Introduction
Semi-artificial photosynthesis strives to merge the strengths of artificial light harvesters with the power of biocatalysts. In particular whole-cell-based biohybrid systems are highly promising in a solar-fuel context. Indeed, such assemblies can harness the cellular component's capacity for self-replication and efficient catalysis, while circumventing the limitations of Nature's photosynthetic apparatus. Still, compatibility between artificial and biological components remains a key challenge. The hydrogenase enzymes serve as a striking example of the efficiency of enzyme catalysis in a solar-fuel context.1 These metalloenzymes are Nature's premier catalyst for hydrogen production, capable of achieving catalytic rates of 105 s−1, with minimal energy losses.2 Previous research has shown that it is possible to employ hydrogenases for both in vitro3–9 and in vivo10–12 semi-artificial photosynthesis systems. In an in vivo context, a wide range of combinations of hydrogenase-containing microorganisms and photosensitizers (PSs) have been reported for light-driven hydrogen gas production.13–20Considering molecular photosensitizers, we and others have shown that Eosin Y (EY) can enter Escherichia coli and serve as an intracellular PS to drive heterologously expressed [FeFe] hydrogenase in the presence of triethanolamine (TEOA) as a sacrificial electron donor (SED).19–22 The addition of a redox mediator (RM) such as methyl viologen (MV) improves the overall efficiency of the photochemistry, and also facilitates transmembrane electron transfer and thus enables the use of extracellular PSs such as ruthenium tris-bipyridine ([Ru(bpy)3]2+).19 However, the active photosystem, or components thereof, exhibits cytotoxicity, hence decreasing E. coli's viability or ability to reproduce. Our previous results suggested that both MV and by-products of the hydrogen evolution could lead to this observed cytotoxicity.19 As a case in point, oxidation of the common SED TEOA can lead to the formation of aldehydes such as glycoaldehyde.23–26 Due to the high reactivity of such aldehydes, TEOA is potentially toxic for E. coli. Indeed, our previous studies showed that cell growth was entirely inhibited already within hours for samples that were photocatalytically producing hydrogen with TEOA as the SED.19 The issue of toxicity is not restricted to molecular PSs or specific SEDs, and cell-toxicity has been observed also with e.g. CdS nanoparticles which are commonly employed in semi-artificial photosynthesis assemblies.27,28 Identifying and tuning biocompatible photocatalytic components is critical to fully realize the advantages of these potentially self-healing systems.
In this study, we focus on optimizing the artificial photocatalytic components of E. coli-based semi-artificial systems, examining both productivity and cytotoxicity. Different SEDs and RMs are explored for whole-cell-based photocatalytic hydrogen evolution in combination with intra- and extracellular PSs. Distinct differences in catalytic rates and cell viability are observed between the various systems. Variations in hydrogen production activities observed with different combinations of PSs, SEDs, and RMs are rationalized through spectroscopic investigations. Critically, the study shows that high productivity does not have to be associated with high toxicity. Following optimization of the photocatalytic components, a detailed study on the limiting factors showcases the robustness and longevity of the [FeFe] hydrogenase-driven hydrogen production in E. coli.
Results and discussion
Photocatalytic assays
The semi-artificial system was assembled as described previously.19 A detailed description is outlined in the ESI,† but in short photocatalytic samples contained E. coli bacteria which heterologously expressed the [FeFe] hydrogenase enzyme HydA1 from Chlamydomonas reinhardtii (CrHydA1), unless otherwise stated. The active form of the enzyme was prepared through artificial maturation to yield a high intracellular concentration at a defined time-point.29,30 The cell density was subsequently set to give an optical density at 600 nm (OD600) of 5. EY or [Ru(bpy)3]2+ were used as PSs, and an RM and an SED were added. The samples (1 mL sample volume in 8 mL vials) were prepared under inert-gas atmosphere and exposed to continuous white light irradiation (4000 lx, main emission 400–650 nm, Fig. S.I. 5, ESI†) at 30 °C, and hydrogen evolution was quantified by gas chromatography measurements of the headspace at specific timepoints. We have previously shown that using EY as PS and TEOA as SED under such conditions yield photocatalytic systems with apparent quantum yields of ≈1.5%, and a TOF of 138 ± 19 nmol H2 mL−1 h−1 OD600−1 when employing MV as a redox mediator.19Here, TEOA and other potential SEDs, which have been previously utilized in the context of artificial photosynthesis,26 were tested for their ability to drive hydrogen production in the whole-cell system. All SEDs were tested with both EY and [Ru(bpy)3]2+ as PSs, with and without MV as RM, and at both pH 6.5 and pH 7.5. The different components tested herein are depicted in Scheme 1 and the results are summarized in Table 1.
 | ||
| Scheme 1 Overview of the PSs, RMs and SEDs tested in this study. | ||
| PS | SED | RM | t /h | TOFmaxc/nmol h−1 OD600−1 | TONmaxc/μmol OD600−1 |
|---|---|---|---|---|---|
| a Samples contain 100 μM PS, 100 mM SED, 1 mM RM, and cells (OD600 = 5) in 100 mM PBS (pH 6.5 for L-cysteine samples, pH 7.5 for TEOA samples). Experiments are conducted under an inert gas atmosphere. b Time-point at which TONmax is determined. c Calculated for a 1 mL suspension of cells at an OD600 = 5. d Higher SED concentration (200 mM). | |||||
| EY | TEOA | MV | 168 | 88.6 ± 7.4 | 8.62 ± 0.04 |
| EY | L-Cysteine | MV | 168 | 75.8 ± 5.6 | 4.42 ± 0.26 |
| EY | TEOA | DQ-OH | 120 | 319.6 ± 32.4 | 8.74 ± 0.64 |
| [Ru(bpy)3]2+ | TEOA | MV | 168 | 30.6 ± 2.4 | 3.42 ± 0.08 |
| [Ru(bpy)3]2+ | L-Cysteine | MV | 72 | 162.4 ± 4.8 | 7.92 ± 0.2 |
| [Ru(bpy)3]2+ | L-Cysteine | DQ-OH | 120 | 60 ± 3.6 | 6.5 ± 0.18 |
| EY | TEOAd | MV | 96 | 182.4 ± 26.2 | 7.7 ± 0.66 |
| [Ru(bpy)3]2+ | L-Cysteined | MV | 192 | 125.8 ± 2 | 14.9 ± 1.46 |
L-Ascorbic acid (E0 = 0.71 V vs. SHE31,32) and lactic acid (E0 = −0.19 V vs. SHE33) have both been previously used as SEDs in in vitro systems and are expected to be non-toxic towards E. coli.34,35 However, as compared to samples employing TEOA, the two acids appeared non-functional as SEDs in whole-cell photocatalytic assays and yielded only trace amounts of H2 (see Fig. S.I. 7 and S.I. 8, ESI†). This is likely attributable to the fact that both L-ascorbic and lactic acid can be fermentatively metabolized in E. coli under the applied anaerobic conditions.36–40
Conversely, substantial amounts of hydrogen were produced with both EY and [Ru(bpy)3]2+ using L-cysteine (E0 = 0.92 V vs. SHE41,42) as SED in the presence of MV (see Fig. 1a). Furthermore, we found that hydrogen production in the presence of L-cysteine is more efficient at the lower pH of 6.5 (see Fig. S.I. 10, ESI†). Cysteine has been reported to be a faster quencher of excited ruthenium complexes at higher pH, but the low cage escape yield of the deprotonated species could potentially explain the better performance at a lower pH.43,44 In contrast, systems employing TEOA as SED worked better at more alkaline pH conditions (i.e. pH = 7.5, see Fig. S.I. 9, ESI†), in agreement with earlier reports.26,45 When [Ru(bpy)3]2+ is used as PS with MV as RM and L-cysteine as the SED at pH = 6.5, the maximum TOF and TON are comparable to that of the highest performing TEOA-based systems, i.e. EY with an RM and TEOA at pH = 7.5. L-Cysteine can hence act as an effective SED under the photocatalytic conditions.
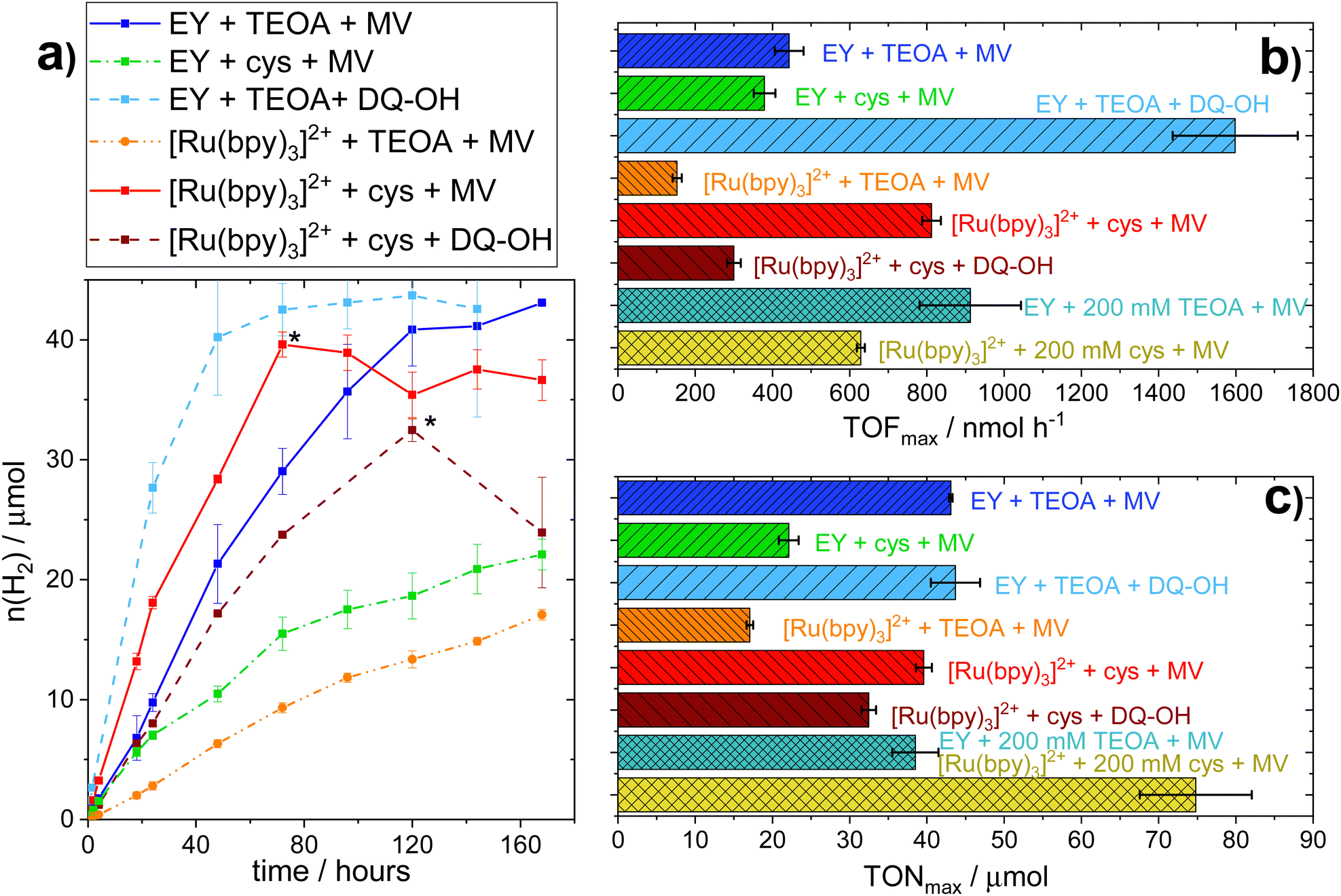 | ||
| Fig. 1 Photocatalytic assays of H2-production upon variation of photocatalytic components. H2-Production of functional photocatalytic whole cell-based biohybrid systems under continuous illumination. Samples contain 100 μM PS (EY or [Ru(bpy)3]2+), 100 mM SED (TEOA at pH 7.5 or L-cysteine at pH 6.5), and 1 mM RM (MV or DQ-OH) in 100 mM PBS. (a) Cumulative hydrogen production * apparent decreases in hydrogen production are due to experimental artefacts, e.g. leakage of the sample vials (b) maximum TOF, including samples with higher SED concentration; (c) maximum TON reached, including samples with higher SED concentration. | ||
To take advantage of the potential self-healing properties of the whole-cell system, it is crucial that the system does not exhibit high toxicity towards cell growth. Indeed, the herein used system is not capable of generating fully functional hydrogenase by itself, as emphasis was placed on generating well-defined systems for comparative studies. Yet, future applications using E. coli cells expressing the entire hydrogenase maturation machinery, should be able to regenerate decomposed catalyst for long-term hydrogen production. To verify the viability of the cells, i.e. their ability to replicate, photocatalytic samples were plated on agar plates after different durations of hydrogen production. The results are summarized in Table 2, while a detailed explanation of the experimental conditions and the scale can be found in the ESI† (Section III).
| Cells+ | 3 h | 5 h | 20 h | 24 h | 72 h | 120 h |
|---|---|---|---|---|---|---|
| a E. coli cell suspensions (OD600 = 5), with holo-[FeFe] hydrogenase, in 100 mM PBS buffer with different other components added. Samples were plated after being kept under constant light irradiation at 30 °C for different periods as indicated. Rating: − no cell growth; + some well isolated cell colonies are observed; ++ usually well isolated cell colonies covering most of the plate; +++ most of the plate is covered in cell colonies; ++++ the plate is completely overgrown with no distinguishable cell colonies. For an illustration of the rating see ESI (Fig. S.I. 13). b pH 7.5. c pH 6.5. | ||||||
| EY + TEOA + MVb | ++ | + | − | |||
| EY + TEOA + DQ-OHb | +++ | ++ | − | |||
| EY + L-cysteine + MVc | + | + | − | |||
| EY + L-cysteine + DQ-OHc | ++++ | ++++ | ||||
| [Ru(bpy)3]2+ + TEOA + MVb | ++++ | + | + | − | ||
| [Ru(bpy)3]2+ + TEOA + DQ-OHb | ++++ | +++ | ++ | − | ||
| [Ru(bpy)3]2++ L-cysteine + MVc | ++ | + | + | |||
| [Ru(bpy)3]2++ L-cysteine + DQ-OHc | +++ | +++ | +++ | |||
As observed before,19 samples using TEOA as the SED show a strong decrease in cell viability within the first 24 hours. Notably, the less active [Ru(bpy)3]2+ samples show a slower decrease of cell viability relative to systems using EY as PS. This is in line with the lower hydrogen evolution rate and subsequently lower TEOA oxidation rate, leading to less toxic by-products. On the other hand, samples using L-cysteine as SED exhibit cell viability, albeit limited, even after being exposed to light for up to 120 hours. The observation that L-cysteine increases both H2 production and viability when [Ru(bpy)3]2+ is used as PS, shows that there is no strict correlation between productivity and cell viability.
In parallel to the SED, MV in isolation exhibits toxicity to the E. coli cells. We hence tested different diquat derivatives, shown in Scheme 1 (see ESI,† Sections I.3, IV and V for a full characterization) for their suitability as RM and identified DQ-OH as the most active of the diquats when L-cysteine is used (see Fig. 11 and 12, ESI†). E. coli cells under these photocatalytic conditions are able to replicate even after six days of constant light irradiation, during which time substantial amounts of hydrogen are produced. As can be seen in Fig. 1, changing the RM from MV to DQ-OH does not equally affect systems with EY+TEOA or [Ru(bpy)3]++L-cysteine. These differences can be rationalized from different electron transfer mechanisms depending on the PS.
Spectroscopic investigations
To understand the differences in observed hydrogen evolution rates under photocatalytic conditions, we studied the best-performing MV systems, i.e. EY and [Ru(bpy)3]2+ in combination with TEOA (at pH = 7.5) and L-cysteine (at pH = 6.5), using transient absorption spectroscopy. Kinetic traces as well as a summary of the results can be found in the ESI,† Section VI. As expected when considering the redox properties of EY and the SEDs, EY can be reductively quenched by both TEOA and L-cysteine.19,46 With TEOA, efficient electron transfer can be observed, forming long-lived one-electron reduced EY (EYred) with a second-order rate constant of (1.3 ± 0.2) × 107 M−1 s−1 (see Fig. S.I. 18, ESI†). When EY and L-cysteine are used, we also observe reductive quenching with a second-order rate constant of (5.7 ± 0.5) × 106 M−1 s−1 (see Fig. S.I. 20, ESI†). However, the electron transfer is reversible and the cage escape yield of the reductively quenched EYred species is only 5%. Consequently, under continuous illumination in the presence of MV (20 μM), systems using EY and TEOA reach a steady-state concentration of one-electron reduced MV (MVred) of 17.9 μM, corresponding to the reduction of 90% of the total MV amount, while in an analogous system with EY and L-cysteine, only 63% of MV (12.7 μM) is reduced.The slightly more negative excited-state reduction potential (1.01 V vs. SHE),47 as well as the shorter excited-state lifetime (570 ns) of [Ru(bpy)3]2+ compared to EY (1.07 V vs. SHE48 and 80 μs), make it harder to be reductively quenched. Indeed, no definite indication of reductive quenching was observed neither with TEOA (1.06 V vs. SHE) nor L-cysteine (0.92 V vs. SHE)45,49 (see Fig. S.I. 22 and 23, ESI†). Conversely, in the presence of MV (30 mM), oxidative quenching of the excited ruthenium complex could be observed with a second-order rate constant of (1.5 ± 0.3) × 109 M−1 s−1 (see Fig. S.I. 24, ESI†). However, the cage escape yield was only 13%, in line with previous reports on the low cage escape efficiency in this system.50,51 This leads to a relatively low steady-state concentration of MVred under continuous illumination, i.e. 6.8 μM MVred (34% of total MV) in the presence of TEOA, and 11.7 μM (59% of total MV) in the presence of L-cysteine. The spectroscopic data shows that under the used conditions, oxidative quenching is clearly the dominant process. Hence, the first electron transfer step involves only [Ru(bpy)3]2+ and MV, regardless of the SED used.
This is in agreement with literature reports showing that more electron withdrawing ligands such as bipyrimidine or bipyrazine are needed to allow for reductive quenching of the excited ruthenium complex.43,44,52,53 The differences in observed MVred accumulation under continuous irradiation for TEOA and L-cysteine presumably stem from the different driving forces for the subsequent electron transfer step from the SED to the oxidized PS. Furthermore, it has been reported that MV forms charge-transfer (CT) complexes with different SEDs, which was found to alter the cage escape yield when excited [Ru(bpy)3]2+ is quenched.54 Differences between TEOA and L-cysteine regarding CT complex formation could thus also influence the observed MV reduction efficiency.
The preference for oxidative quenching with [Ru(bpy)3]2+ as PS can also rationalize the strong difference in RMred accumulation observed when either MV or DQ-OH are used (Table 3). The lower yield of DQ-OH correlates with its more negative redox potential (−0.594 V vs. SHE). Comparing this to the excited-state oxidation potential of the ruthenium complex (−0.57 V vs. SHE55), it becomes apparent that oxidative quenching is not thermodynamically favourable. It can therefore be assumed that in this system, the electron transfer follows a reductive quenching mechanism, which is not efficient (Fig. S.I. 23, ESI†).
| PS | SED | RM | Cells | c(RMred)/μM | Ratio RMred/RM | E/mV vs. SHE | TOFmax/nmol h−1 OD600−1 |
|---|---|---|---|---|---|---|---|
| EY | TEOA | MV | − | 17.9 ± 0.9 | 0.90 | −0.504 ± 0.014 | |
| EY | TEOA | MV | + | 16.7 ± 0.6 | 0.84 | −0.488 ± 0.006 | 88.6 ± 7.4 |
| EY | TEOA | DQ-OH | − | 22.7 ± 0.2 | 0.91 | −0.653 ± 0.002 | |
| EY | TEOA | DQ-OH | + | 15.5 ± 2.2 | 0.62 | −0.607 ± 0.010 | 319.6 ± 32.4 |
| EY | Cys | MV | − | 12.7 ± 1.7 | 0.63 | −0.460 ± 0.009 | |
| EY | Cys | MV | + | 9.8 ± 2.5 | 0.49 | −0.445 ± 0.013 | 75.8 ± 5.6 |
| Ru | TEOA | MV | − | 6.8 ± 0.7 | 0.34 | −0.429 ± 0.004 | |
| Ru | TEOA | MV | + | 5.3 ± 0.3 | 0.27 | −0.420 ± 0.002 | 30.6 ± 2.4 |
| Ru | Cys | MV | − | 11.7 ± 0.5 | 0.59 | −0.456 ± 0.002 | |
| Ru | Cys | MV | + | 8.7 ± 0.5 | 0.43 | −0.439 ± 0.003 | 162.4 ± 4.8 |
| Ru | Cys | DQ-OH | − | 2.4 ± 0.1 | 0.10 | −0.537 ± 0.001 | |
| Ru | Cys | DQ-OH | + | 1.9 ± 0.1 | 0.07 | −0.529 ± 0.002 | 60 ± 3.6 |
In the presence of fresh cells, a decrease in steady-state RMred concentration under illumination is observed in all cases (see Table 3), in line with consumption of the RMred species by the hydrogenase or other competing cellular electron acceptors. The relative decrease, as compared to RMred concentrations in the absence of cells, is ≈20–30% for all samples except when EY, TEOA, and MV are used. In the latter case, the steady-state MVred concentration in the presence of cells is only 7% lower as compared to the analogous sample without cells. We assume that this is due to very efficient overall electron transfer from TEOA to EY and then to MV, as compared to the electron transfer step MVred to the cellular electron acceptor. Indeed, earlier studies have reported this latter step to be rather slow.19,56,57
In addition to the time-resolved spectroscopy, we have previously introduced the solution potential as a measure to rationalize variations in hydrogen evolution activity between different combinations of photocatalytic components. The solution potential, calculated through the Nernst equation, includes both the impact of RMred concentration and the driving force in the form of the redox potential of the RM (for details see Section VI.2, ESI†).19 We note that the comparison between solution potential and hydrogen evolution should only be considered qualitative due to the different experimental conditions which had to be employed for the RMred accumulation studies as compared to the hydrogen evolution assays. Still, we expect general trends to hold true across both conditions. Moreover, a direct comparison between systems employing [Ru(bpy)3]2+ and EY as respective PS is complicated by differences in irradiation conditions employed for the two PSs in the RMred accumulation experiments. Thus, the datasets for the two separate PSs should be considered independently.
For systems employing EY as PS, hydrogen evolution clearly correlates with the solution potential as previously noted,19i.e. systems with a more negative solution potential show higher TOFs (Table 3). However, when considering the [Ru(bpy)3]2+ system, it becomes evident that this correlation only holds true when comparing systems employing the same RM. Similarly, it is clear that neither the concentration of RMred nor the redox potential of the RM alone can account for the differences in hydrogen evolution rates observed during photocatalysis. We speculate that this difference in correlation between solution potential and hydrogen production between the PSs is due to their different localizations. The extracellular nature of [Ru(bpy)3]2+, as compared to the intracellular localization of EY, introduces additional kinetic barriers in the electron transfer process in the former case. It follows that the different RMs could display distinctly different efficiencies in their transmembrane transport, which in turn impacts overall system performance.
Limiting factors
All different combinations of photocatalytic components resulted in distinct TOFs, but this trend did not hold true when considering the TONs. Indeed, those photocatalytic samples that reach a plateau of hydrogen evolution do so at similar TONs (≈40 μmol), regardless of PS or SED used. In all systems studied here, the addition of PS, SED or whole-cell catalyst after the plateau is reached does not result in reactivation of the H2 production (see Fig. S.I. 27, ESI†).However, collecting the cells through centrifugation after the plateau is reached and resuspending them in fresh buffer containing all required photocatalytic supplements (100 mM SED, 100 μM PS, 1 mM MV), results in reactivation as evident in Fig. 2a. This effect was observed for both EY+TEOA+MV and [Ru(bpy)3]2++L-cysteine+MV systems. Reactivation could be performed twice, yielding approximately another equivalent of ≈40 μmol H2 before the activity plateaus again. From this, we can infer that the biocatalyst is still active for a period of more than 10 days, and that the initial inactivation of the systems is reversible by a complete buffer and head-space atmosphere exchange. A shift in pH and product (H2) inhibition could be ruled out. The pH remained very stable over the course of the photocatalysis (±0.1 pH units comparing t = 0 h and t = 160 h); and an exchange of 30% of the headspace atmosphere for neat argon once the H2 production had plateaued did not yield any apparent reactivation of the system (Fig. S.I. 27c, ESI†).
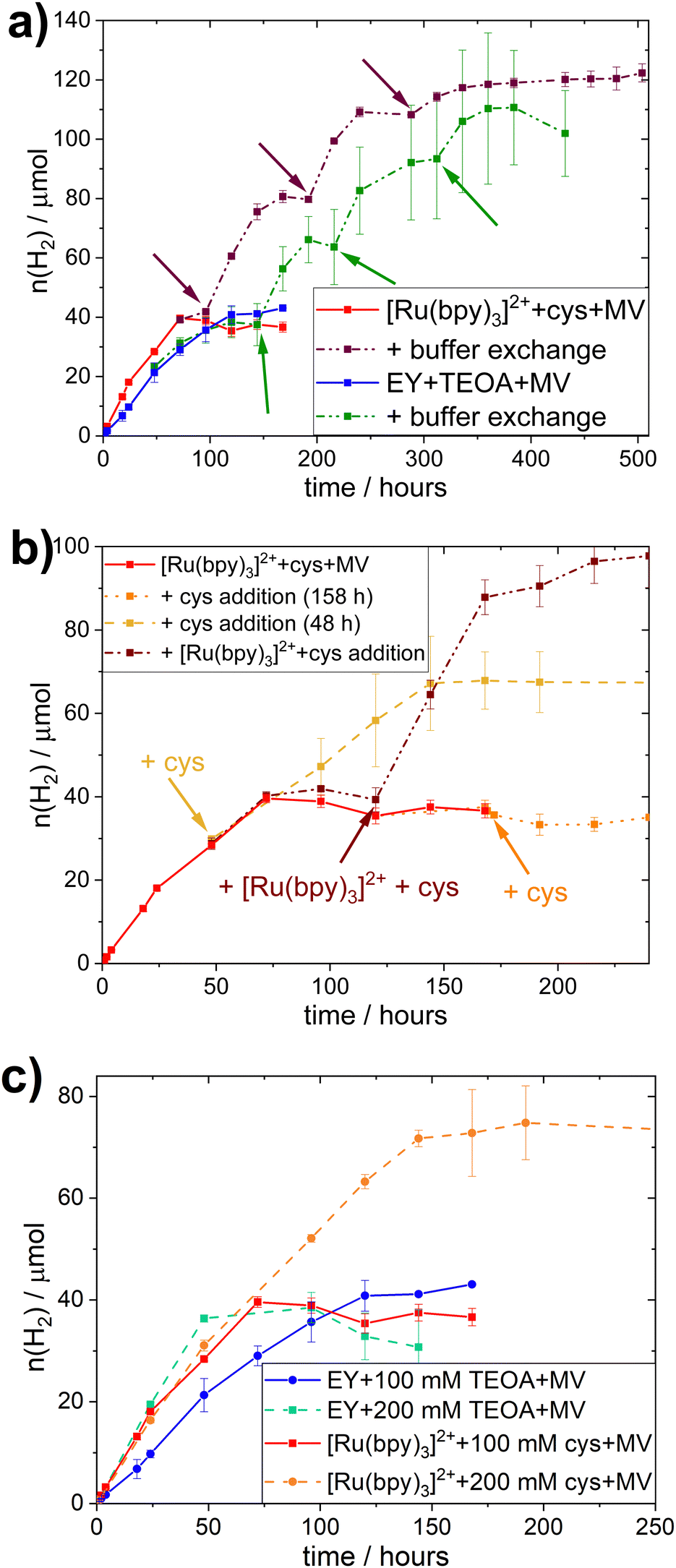 | ||
| Fig. 2 Limiting factor study during photocatalytic H2-production: (a) buffer exchange assays upon centrifugation and resuspending of cell pellets in fresh buffer (100 μM PS, 1 mM MV, 100 mM SED) at designated time points indicated with arrows; (b) addition assays for L-cysteine + [Ru(bpy)3]2+ + MV systems; addition timepoints indicated with arrows; (c) photocatalytic H2-production assay with different SED starting concentrations (100 or 200 mM SED) with 100 μM PS, and 1 mM MV. All experiments are carried out under inert gas atmosphere at 30 °C under continuous illumination. | ||
The stability of the whole-cell catalyst implies that the long-term hydrogen evolution is limited by the photocatalytic system(s). To identify the limiting factor, we added single photocatalytic components at earlier timepoints, i.e. before the plateau was reached; or combinations of the different components before and after the plateau was reached (Fig. 2b and Fig. S.I. 28, ESI†). In the case of the EY+TEOA+MV system, the addition experiments did not give a clear indication on the exact mechanism limiting the system (Fig. S.I. 28, ESI†) and starting with higher TEOA concentrations (Fig. 2c) did not yield significantly higher TONs. A more detailed discussion of possible degradation of the photoactive system can be found in the ESI† (Section VI.3).
In the [Ru(bpy)3]2++-L-cysteine+MV system however, while no reactivation by late L-cysteine addition was possible, the system could be kept active by adding more L-cysteine before the plateau was reached (Fig. 2b). When both L-cysteine and [Ru(bpy)3]2+ were added after the plateau was reached, a similar reactivation of the system was observed. We hence conclude that there is nothing in the system that inhibits hydrogen evolution. Instead, supply of electrons from L-cysteine is the primary limiting factor in this system and, once this electron supply runs out, the photo-oxidized [Ru(bpy)3]3+ decomposes, e.g. via hydrolysis of the bipyridine ligands.58,59 Thus, degradation of the Ru-based PS can occur in the whole-cell assembly, it is not the primary limiting factor. This notion is further supported by the comparison of photocatalytic experiments with different starting concentrations of L-cysteine (200 vs. 100 mM), where the TON clearly correlated with SED concentration in the [Ru(bpy)3]2++L-cysteine+MV system (Fig. 2c).
Under typical conditions (i.e. 100 mM SED, 100 μM PS, and 1 mM MV) this system reaches a TON of 39.6 μmol of H2 at OD600 = 5, and the amount of L-cysteine in the system is 93 μmol, meaning that 85% of available electrons from the SED are used for hydrogen evolution.
In comparison, samples with 200 mM L-cysteine as SED yield a maximum of 74.8 μmol of H2, which would be equivalent to 79% of the available electrons being used for the reduction of H+ to produce H2 assuming that two electrons are needed for the production of one equivalent of H2. This is similar to the value obtained for 100 mM L-cysteine samples and implies an equal efficiency in using the SED regardless its concentration. It further gives evidence that the SED is indeed the limiting factor in this biohybrid system. Nevertheless, the slightly lower electron to H2 efficiency presumably stems from the limited solubility of L-cysteine. At 200 mM, some of the L-cysteine precipitated, yielding a lower actual concentration of L-cysteine than the one used for this calculation and consequently, the percentage of electrons used for H2-production appears to be lower.
The stability and long-time performance of the E. coli-based biocatalyst with [Ru(bpy)3]2+-photosensitizers is assumed to extend to other stable PSs with similar quenching mechanisms. In contrast, organic fluorescent dyes, such as EY, are prone to photobleaching, emphasizing one of their major shortcomings in photocatalytic applications.
The benefit of applying the assembly with [Ru(bpy)3]2+ is underscored by the buffer exchange experiments. Here we also note that the active [FeFe] hydrogenase is generated through artificial maturation in cells lacking intrinsic capacity for enzyme re-activation. Thus, a stability of 10 days is achieved even before allowing self-regeneration of the catalyst.
Lastly, to verify that the applicability of these whole-cell photocatalytic systems is not limited to one specific enzyme, we assayed analogous E. coli systems heterologously expressing alternative [FeFe] hydrogenases (Fig. S.I. 29 and 30, ESI†). CrHydA1 was replaced with two [FeFe] hydrogenases incorporating more complex compositions of metal cofactors. More specifically, hydrogenase 1 from Clostridium pasteurianum (CpI) and the putative sensory [FeFe] hydrogenase from Thermoanaerobacter mathranii (TamHydS), both of which feature multiple [4Fe–4S] clusters in addition to the catalytic H-cluster. Similar to CrHydA1, the CpI enzyme is a so-called Group A (or “prototypical”) [FeFe] hydrogenase capable of high catalytic rates at negligible overpotential.60–62 Conversely, TamHydS is a relatively sluggish proton reduction catalyst, requiring a significant overpotential.63,64 Albeit differences both with regards to TOFs and TONs were observed, hydrogen gas production was evident from E. coli cells expressing all three different hydrogenases in combination with the herein established photosystems (compare Fig. 1 and Fig. S.I. 29 and 30, ESI†). To some extent the variations in H2 production are likely influenced by intrinsic differences in enzyme properties, but they are arguably also reflecting variations in intracellular enzyme concentrations.
Conclusions
Herein we explored E. coli-based systems for semi-artificial photosynthesis and addressed the issue of compatibility between the artificial photocatalytic components and the biocatalyst. Critically, the work shows that the photocatalytic system can be tuned to allow for high TOF and TON without exposing significant toxicity to the E. coli host. Indeed, the optimized biohybrid system becomes limited by electron supply from the SED, while the [FeFe] hydrogenase containing E. coli cells allow for long-lived hydrogen evolution. The fact that the biocatalyst component remains active for several days shows that the system should be highly suitable for artificial enzymes that lack re-activation capacity. Moreover, it suggests that further optimization with regards to system longevity should focus on the photocatalytic components. One of the remaining key challenges is evidently the recycling of the thiol-based SED.65 The observed activity with different [FeFe] hydrogenases also highlights the broad applicability of this kind of system. The observation that the TamHydS/E. coli system is generating hydrogen reveals that the photocatalytic system is capable of driving processes at more negative potentials than the H+/H2 redox couple, underscoring its potential for producing a broader range of solar fuels. One can also readily envision introducing other enzymatic pathways and employ the produced H2 as an intermediate in the biosynthesis of more complex products.Author contributions
G. B. and M. G. conceived the project. M. G. and L. K. carried out photocatalytic assays as well as studies on cell viability and integrity assays, and handled data analysis under the guidance of G. B. D. F. carried out the synthesis and analysis of the diquat derivative under the supervision of N. P. M. G. prepared the first manuscript draft with support from L. K. and all authors were involved in analysis and revision of the manuscript. All authors have given approval to the final version of the manuscript.Abbreviations
| CT | Charge transfer |
| E. coli | Escherichia coli |
| Cp | Clostridium pasteurianum |
| Tam | Thermoanaerobacter mathranii |
| Cr | Chlamydomonas reinhardtii |
| PS | Photosensitizer |
| TEOA | Triethanolamine |
| SED | Sacrificial electron donor |
| TOF | Turn-over frequency |
| TON | Turn-over number |
| EY | Eosin Y |
| EYred | One-electron reduced form of Eosin Y |
| MV | Methyl viologen (1,1′-dimethyl-4,4′-bipyridinium) |
| MVred | One electron reduced form of methyl viologen |
| RM | Redox mediator |
| DQ-OH | Diquat derivative 7-hydroxy-3,11-dimethyl-7,8-dihydro-6H-dipyrido[1,2-a:2′,1′-c][1,4]diazepine-5,9-diium |
| [Ru(bpy)3]2+ (Ru, in Table 3) | Ruthenium tris-2-2′-bipyridine |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swedish Research Council (VR, grant no. 2021-04471 to G. B.), the Carl Trygger Foundation (grant no. 20:39 to G. B. and M. G.), and the ANR-DFG “solar driven chemistry” (grant no. PL746/5-1 to N. P.) are gratefully acknowledged for funding. Dr. Sagar Ganguli at Uppsala University is gratefully acknowledged for collecting and analyzing the cyclic voltammetry data.References
- R. M. Evans, B. Siritanaratkul, C. F. Megarity, K. Pandey, T. F. Esterle, S. Badiani and F. A. Armstrong, Chem. Soc. Rev., 2019, 48, 2039–2052 RSC.
- (a) M. Lorenzi and G. Berggren, Compr. Coord. Chem. II, 2021, 731–756 Search PubMed; (b) W. Lubitz, H. Ogata and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- K. Holá, M. V. Pavliuk, B. Németh, P. Huang, L. Zdražil, H. Land, G. Berggren and H. Tian, ACS Catal., 2020, 10, 9943–9952 CrossRef.
- B. L. Greene, ACS Catal., 2021, 11(23), 14635–14650 CrossRef CAS.
- E. Reisner, Eur. J. Inorg. Chem., 2011, 1005–1016 CrossRef CAS.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- Y. Zhao, N. C. Anderson, M. W. Ratzloff, D. W. Mulder, K. Zhu, J. A. Turner, N. R. Neale, P. W. King and H. M. Branz, ACS Appl. Mater. Interfaces, 2016, 8, 14481–14487 CrossRef CAS PubMed.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, J. Am. Chem. Soc., 2012, 134, 5627–5636 CrossRef CAS PubMed.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 3, 944–951 CrossRef CAS.
- N. Kosem, M. Watanabe, J. T. Song, A. Takagaki and T. Ishihara, Appl. Catal., A, 2023, 651, 119019 CrossRef CAS.
- S. Shi, C. Zeng, T. Si, B. Wang and P. K. Wong, ACS ES&T Eng., 2022, 2(6), 989–1000 Search PubMed.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed.
- Z. Yi, S. Tian, W. Geng, T. Zhang, W. Zhang, Y. Huang, H.-N. Barad, G. Tian and X.-Y. Yang, Chem. – Eur. J., 2023, 29, e202203662 CrossRef CAS PubMed.
- M. Martins, C. Toste and I. A. C. Pereira, Angew. Chem., Int. Ed., 2021, 60, 9055–9062 CrossRef CAS PubMed.
- N. Kosem, Y. Honda, M. Watanabe, A. Takagaki, Z. P. Tehrani, F. Haydous, T. Lippert and T. Ishihara, Catal. Sci. Technol., 2020, 10, 4042–4052 RSC.
- Y. Honda, Y. Shinohara and H. Fujii, Catal. Sci. Technol., 2020, 10, 6006–6012 RSC.
- Y. Honda, H. Hagiwara, S. Ida and T. Ishihara, Angew. Chem., Int. Ed., 2016, 55, 8045–8048 CrossRef CAS PubMed.
- W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu and J. Zhao, Sci. Adv., 2018, 4, eaap9253 CrossRef PubMed.
- M. T. Gamache, R. Charaf, L. Kurth, D. T. Filmon, M. Senger, N. Plumeré, L. Hammarström and G. Berggren, ACS Catal., 2023, 9476–9486 CrossRef CAS.
- M. Lorenzi, M. T. Gamache, H. J. Redman, H. Land, M. Senger and G. Berggren, ACS Sustainable Chem. Eng., 2022, 10, 10760–10767 CrossRef CAS PubMed.
- Y. Honda, Y. Shinohara, M. Watanabe, T. Ishihara and H. Fujii, ChemBioChem, 2020, 21, 3389–3397 CrossRef CAS PubMed.
- Z. Wang, D. Gao, H. Geng and C. Xing, J. Mater. Chem. A, 2021, 9, 19788–19795 RSC.
- P. J. DeLaive, T. K. Foreman, C. Giannotti and D. G. Whitten, J. Am. Chem. Soc., 1980, 102, 5627–5631 CrossRef CAS.
- D. G. Whitten, Acc. Chem. Res., 1980, 13, 83–90 CrossRef CAS.
- P. J. Smith and C. K. Mann, J. Org. Chem., 1969, 34, 1821–1826 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim, 2017, 20, 283–295 CrossRef CAS.
- S. T. Hossain and S. K. Mukherjee, J. Hazard. Mater., 2013, 260, 1073–1082 CrossRef CAS PubMed.
- D. Cui, J. Wang, H. Wang, Y. Yang and M. Zhao, J. Hazard. Mater., 2021, 409, 124485 CrossRef CAS PubMed.
- L. S. Mészáros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 4608–4617 RSC.
- N. Khanna, C. Esmieu, L. S. Meszaros, P. Lindblad and G. Berggren, Energy Environ. Sci., 2017, 10, 1563–1567 RSC.
- D. H. Macartney and N. Sutin, Inorg. Chim. Acta, 1983, 74, 221–228 CrossRef CAS.
- C. Creutz, Inorg. Chem., 1981, 20, 4449–4452 CrossRef CAS.
- R. H. Garrett, C. M. Grisham and M. Sabat, Biochemistry, Saunders College Pub. Fort Worth, 1999 Search PubMed.
- S. Pandit, V. Ravikumar, A. M. Abdel-Haleem, A. Derouiche, V. R. S. S. Mokkapati, C. Sihlbom, K. Mineta, T. Gojobori, X. Gao, F. Westerlund and I. Mijakovic, Front. Microbiol., 2017, 8, 2599–2610 CrossRef PubMed.
- T. Warnecke and R. T. Gill, Microb. Cell Fact., 2005, 4(25), 1475–1483 Search PubMed.
- Z. Zhang, M. Aboulwafa, M. H. Smith and M. H. Saier, Jr., J. Bacteriol., 2003, 185, 2243–2250 CrossRef CAS PubMed.
- W. B. Esselen and J. E. Fuller, J. Bacteriol., 1939, 37, 501–521 CrossRef CAS PubMed.
- H. E. Richter, J. Switala and P. C. Loewen, Can. J. Microbiol., 1988, 34, 822–824 CrossRef CAS PubMed.
- D. P. Clark, FEMS Microbiol. Lett., 1989, 63, 223–234 CrossRef CAS.
- K. A. Presser, D. A. Ratkowsky and T. Ross, Appl. Environ. Microbiol., 1997, 63, 2355–2360 CrossRef CAS PubMed.
- P. Surdhar and D. A. Armstrong, J. Phys. Chem., 1986, 90, 5915–5917 CrossRef CAS.
- G. R. Buettner, Arch. Biochem. Biophys., 1993, 300, 535–543 CrossRef CAS PubMed.
- G. Neshvad and M. Z. Hoffman, J. Phys. Chem., 1989, 93, 2445–2452 CrossRef CAS.
- G. Neshvad, M. Z. Hoffman, Q. G. Mulazzani, M. Venturi, M. Ciano and M. D'Angelantonio, J. Phys. Chem., 1989, 93, 6080–6088 CrossRef CAS.
- K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2720–2730 CrossRef CAS.
- S. F. Rowe, G. Le Gall, E. V. Ainsworth, J. A. Davies, C. W. J. Lockwood, L. Shi, A. Elliston, I. N. Roberts, K. W. Waldron, D. J. Richardson, T. A. Clarke, L. J. C. Jeuken, E. Reisner and J. N. Butt, ACS Catal., 2017, 7, 7558–7566 CrossRef CAS.
- C. Creutz and N. Sutin, Inorg. Chem., 1976, 15, 496–499 CrossRef CAS.
- T. Lazarides, T. McCormick, P. Du, G. Luo, B. Lindley and R. Eisenberg, J. Am. Chem. Soc., 2009, 131, 9192–9194 CrossRef CAS PubMed.
- J. R. Milligan, J. A. Aguilera, A. Ly, N. Q. Tran, O. Hoang and J. F. Ward, Nucleic Acids Res., 2003, 31, 6258–6263 CrossRef CAS PubMed.
- M. Georgopoulos and M. Z. Hoffman, J. Phys. Chem., 1991, 95, 7717–7721 CrossRef CAS.
- J. R. Darwent and K. Kalyanasundaram, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 373–382 RSC.
- S. Tazuke, N. Kitamura and Y. Kawanishi, J. Photochem., 1985, 29, 123–138 CrossRef CAS.
- H. Sun and M. Z. Hoffman, J. Phys. Chem., 1994, 98, 11719–11726 CrossRef CAS.
- D. R. Prasad and M. Z. Hoffman, J. Phys. Chem., 1984, 88, 5660–5665 CrossRef CAS.
- C. R. Bock, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 2909–2911 CrossRef CAS.
- T. Hiraishi, T. Kamachi and I. Okura, J. Photochem. Photobiol., A, 1996, 101, 45–47 CrossRef CAS.
- M. V. Pavliuk, M. Lorenzi, D. R. Morado, L. Gedda, S. Wrede, S. H. Mejias, A. Liu, M. Senger, S. Glover, K. Edwards, G. Berggren and H. Tian, J. Am. Chem. Soc., 2022, 144, 13600–13611 CrossRef CAS PubMed.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772–4783 CrossRef CAS.
- B. Limburg, E. Bouwman and S. Bonnet, ACS Catal., 2016, 6, 5273–5284 CrossRef CAS.
- M. W. Adams and L. E. Mortenson, J. Biol. Chem., 1984, 259, 7045–7055 CrossRef CAS PubMed.
- J.-S. Chen and L. E. Mortenson, Biochim. Biophys. Acta, Protein Struct., 1974, 371, 283–298 CrossRef CAS PubMed.
- J. H. Artz, O. A. Zadvornyy, D. W. Mulder, S. M. Keable, A. E. Cohen, M. W. Ratzloff, S. G. Williams, B. Ginovska, N. Kumar, J. Song, S. E. McPhillips, C. M. Davidson, A. Y. Lyubimov, N. Pence, G. J. Schut, A. K. Jones, S. M. Soltis, M. W. W. Adams, S. Raugei, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2020, 142, 1227–1235 CrossRef CAS PubMed.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, J. Am. Chem. Soc., 2021, 143, 20320–20325 CrossRef CAS PubMed.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Németh, N. Polidori, L. S. Mészáros, M. Senger, S. T. Stripp and G. Berggren, Chem. Sci., 2020, 11, 12789–12801 RSC.
- K. K. Sakimoto, S. J. Zhang and P. Yang, Nano Lett., 2016, 16, 5883–5887 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00462g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |